
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 22 May 2019
Sec. Experimental Pharmacology and Drug Discovery
Volume 10 - 2019 | https://doi.org/10.3389/fphar.2019.00531
This article is part of the Research Topic Targeting Monocytes/Macrophages to Treat Atherosclerotic Inflammation View all 10 articles
 Emiel P. C. van der Vorst1,2,3,4*
Emiel P. C. van der Vorst1,2,3,4* Linsey J. F. Peters1
Linsey J. F. Peters1 Madeleine Müller1
Madeleine Müller1 Selin Gencer1
Selin Gencer1 Yi Yan1
Yi Yan1 Christian Weber1,4,5
Christian Weber1,4,5 Yvonne Döring1,4*
Yvonne Döring1,4*Atherosclerosis, the underlying cause of the majority of cardiovascular diseases (CVDs), is a lipid-driven, inflammatory disease of the large arteries. Gold standard therapy with statins and the more recently developed proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors have improved health conditions among CVD patients by lowering low density lipoprotein (LDL) cholesterol. Nevertheless, a substantial part of these patients is still suffering and it seems that ‘just’ lipid lowering is insufficient. The results of the Canakinumab Anti-inflammatory Thrombosis Outcome Study (CANTOS) have now proven that inflammation is a key driver of atherosclerosis and that targeting inflammation improves CVD outcomes. Therefore, the identification of novel drug targets and development of novel therapeutics that block atherosclerosis-specific inflammatory pathways have to be promoted. The inflammatory processes in atherosclerosis are facilitated by a network of immune cells and their subsequent responses. Cell networking is orchestrated by various (inflammatory) mediators which interact, bind and induce signaling. Over the last years, G-protein coupled receptors (GPCRs) emerged as important players in recognizing these mediators, because of their diverse functions in steady state but also and specifically during chronic inflammatory processes – such as atherosclerosis. In this review, we will therefore highlight a selection of these receptors or receptor sub-families mainly expressed on myeloid cells and their role in atherosclerosis. More specifically, we will focus on chemokine receptors, both classical and atypical, formyl-peptide receptors, the chemerin receptor 23 and the calcium-sensing receptor. When information is available, we will also describe the consequences of their targeting which may hold promising options for future treatment of CVD.
Cardiovascular diseases (CVDs), with myocardial infarction (MI) and stroke as most common clinical manifestation, remain the leading cause of death worldwide (Hansson, 2005), underpinning the importance of further research into and development of novel therapeutic approaches. Atherosclerosis, a lipid-driven chronic inflammatory disease, has been recognized as the main underlying cause of CVD (Ross, 1999; Hansson, 2005; Braunersreuther et al., 2007a). Endothelial damage by hemodynamic shear stress is a main initiator of atherosclerosis formation, resulting in increased endothelial permeability and hence increased susceptibility for lipid infiltration (Hansson et al., 2015). This damage enables the infiltration of various lipids, like low-density lipoprotein (LDL), into the intima where it is subsequently modified into oxidized LDL (oxLDL) (Ross, 1999; Braunersreuther et al., 2007a). Together with the endothelial damage, accumulation of these modified lipids triggers an inflammatory response, resulting in progressive inflammatory cell infiltration into the sub-endothelial layer (Ross, 1999; Braunersreuther et al., 2007a). During this mobilization stage (Figure 1), predominately monocytes will bind to adhesion molecules on the activated endothelium and subsequently infiltrate into the vessel wall by transmigration (Schumski et al., 2018). Infiltrated monocytes subsequently differentiate into macrophages which phagocyte cell debris and oxLDL, resulting in the formation of foam cells (Hansson, 2005; Charo and Ransohoff, 2006). As foam cells also trigger inflammation by releasing cytokines and chemokines, a vicious circle is created resulting in the continued recruitment and mobilization of leukocytes to the vascular wall. This results in the formation of so called fatty streak lesions which will continue to develop and grow over time. During this progression stage, activated lesional macrophages also secrete matrix metalloproteinases that can digest extracellular matrix components, leading to plaque destabilization. In the end, macrophages become apoptotic due to the continued lipid accumulation and contribute to the formation of necrotic cores (Moore and Tabas, 2011). Besides monocytes and macrophages, also neutrophils have been described to play an important role in the development of atherosclerotic lesions. It has been shown that neutrophils can influence almost every step of this pathology, including endothelial dysfunction, monocyte recruitment, foam cell formation and plaque destabilization (Döring et al., 2015). Eventually, plaque growth or the rupture of lesions resulting in atherothrombosis can cause the artery to occlude. This occlusion will cause ischemia in downstream tissues, resulting in cardiovascular events like stroke or MI (Hansson, 2005).
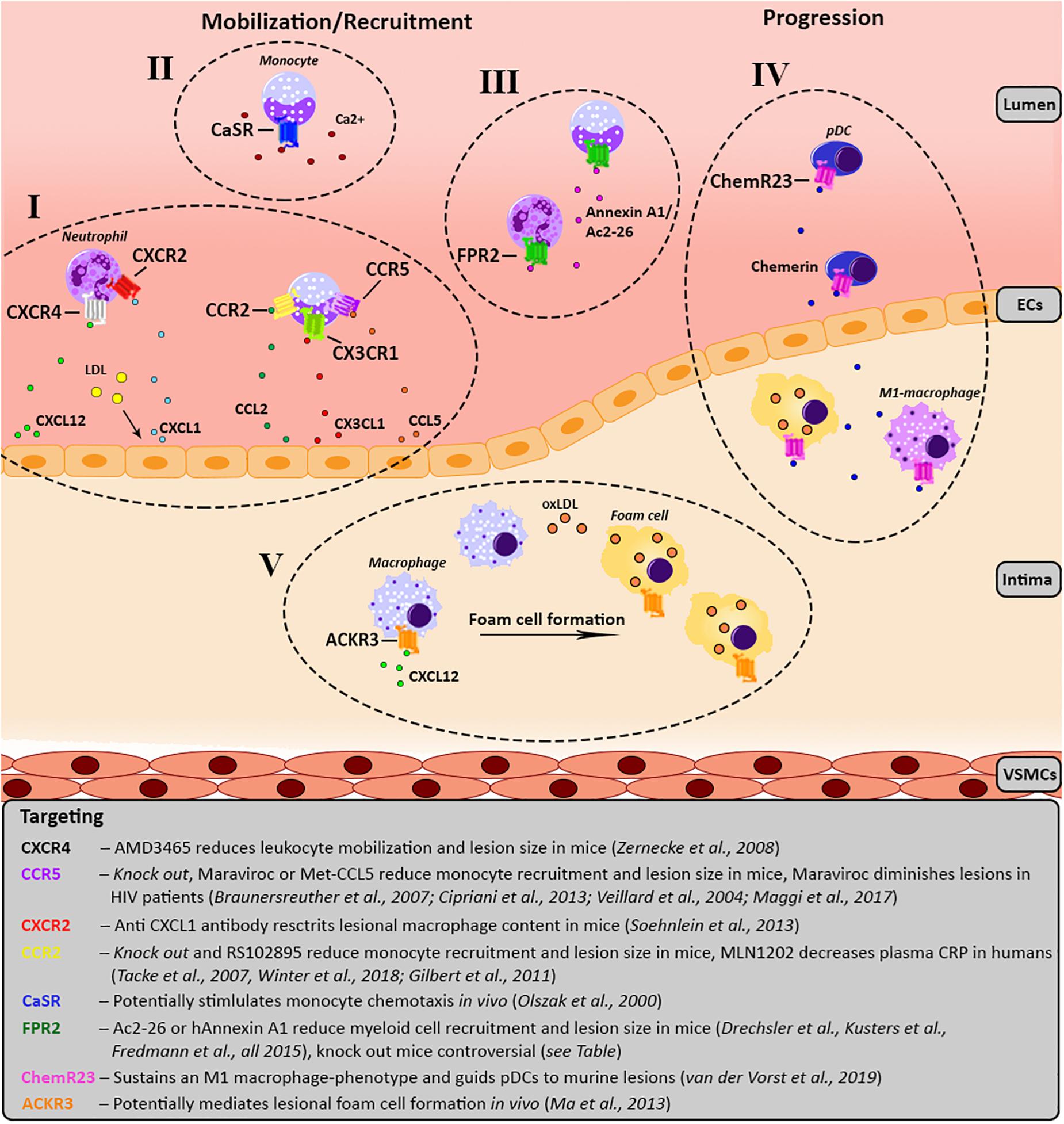
Figure 1. Schematic overview of the involvement of the various GPCRs in atherosclerosis development. For each receptor the key processes, as well as agonists/antagonists are summarized and depicted over three main phases of atherosclerosis development; mobilization, leukocyte recruitment and plaque progression. Receptors of the same GPCR-subfamily are clustered together and categorized from I till V. (I) Chemokine receptor CXCR4 causes migration of leukocytes toward its ligand CXCL12. Additionally, upon LDL stimulation CXCL1 is released by endothelial cells causing myeloid cells, which carry CXCR2 on their surface to migrate toward the endothelium. CCL2 and CX3CL1 mediate the recruitment of monocytes expressing CCR2 and CX3CR1, respectively. In line with this, monocytes expressing CCR5 are recruited to the lesion by CCL5. (II) Monocytes show a CaSR-dependent increase of chemotaxis toward CCL2 upon stimulation with calcium. (III) FPR2 is mostly expressed on myeloid cells and has several contradictory effects, please see Table 1. FPR2-agonists like Ac2-26, an Annexin A1 peptide, and Annexin A1 reduce monocyte/neutrophil recruitment. (IV) ChemR23 maintains a M1 macrophage phenotype and stimulates pDC migration and infiltration into atherosclerotic plaques. (V) ACKR3 expression is upregulated in lesional macrophages which engulf modified lipids resulting in foam cell formation. ACKR3, atypical chemokine receptor 3; CAD, coronary artery disease; CaSR, calcium-sensing receptor; CCR, C-C chemokine receptor; CCL, C-C chemokine ligand; ChemR23, chemerin receptor 23; CRP, C-reactive protein; CXCR, C-X-C chemokine receptor; CX3CR1, CX3C chemokine receptor 1; CXCL, C-X-C chemokine ligand; CX3CL1, CX3C chemokine ligand 1; FPR2, formyl-peptide receptor 2; HIV, human immunodeficiency virus; LDL, low-density lipoprotein; oxLDL, oxidized LDL; pDC, plasmacytoid dendritic cell.
Cardiovascular disease-therapy is mostly focussing on mitigation of hyperlipidemia (statins) and management of thrombotic factors (aspirin) to prevent further progression of the disease. Statins are inhibitors of the HMG-CoA reductase, thereby reducing the production of cholesterol and the current golden CVD-therapy (Okopien et al., 2016). A recent meta-analysis of several statin clinical trials indeed confirmed that statin use clearly reduces plasma LDL levels (up to 55–60%) and thereby also resulted in significant reductions in cardiovascular risk (Boekholdt et al., 2014). However, as with a lot of therapies there are also off-target side effects due to the use of statins. It has been shown that statin treatment results in a striking 9% increased risk for the development of diabetes (Preiss et al., 2011). This has led to a debate about the use of statins and especially fuelled the development of adequate alternatives. One of these intriguing new players in the field of hyperlipidemia therapy is monoclonal antibodies against proprotein convertase subtilisin/kexin type 9 (PCSK9). In a physiological condition, PCSK9 interacts with the LDL receptor in the liver to stimulate its degradation and additionally prevention its recycling to the cell membrane (Cohen et al., 2005). Inhibiting PCSK9 thus results in an increased surface expression of LDL receptors that are capable of binding and internalizing LDL particles, thereby reducing the plasma LDL levels. The great potential is demonstrated by the fact that PCSK9 inhibition can cause a 60% reduction of LDL, even on top of the LDL lowering due to statin use, without any indications of serious side effects (Robinson et al., 2015; Stone and Lloyd-Jones, 2015; Zhang et al., 2015). The only major drawback of these monoclonal antibodies is the fact that the production is still very costly and therefore wide-scale usage is not yet feasible.
Besides above described therapies focussing on lipid modulation, immunomodulation has emerged during the last decades as a promising therapeutic option. Accumulating evidence especially supports the beneficial role of interleukin-1β (IL-1β), tumor necrosis factor (TNF) and IL-6 inhibition (Ridker and Luscher, 2014). All of these cytokines are part of a common pathway. IL-1β is initially produced as an inactive precursor and therefore requires proteolytic cleavage which is mediated by the nucleotide-binding leucine-rich repeat-containing pyrin receptor 3 (NLRP3) inflammasome (Strowig et al., 2012). Inhibition of IL-1β using the monoclonal antibody canakinumab results in the significant reduction of plasma IL-6 and high-sensitivity C-reactive protein (hsCRP) levels, without lowering LDL cholesterol (Ridker et al., 2012). The effect of IL-1β targeting on cardiovascular risk has recently been evaluated in the Canakinumab Anti-inflammatory Thrombosis Outcome Study (CANTOS) trial. This randomized, double-blind, placebo-controlled trial involving stable patients with previous MI showed that canakinumab was effective in reducing plasma hsCRP levels and preventing adverse cardiac events (Ridker et al., 2017). Although this study shows great promise of immunomodulatory therapies, the use of canakinumab was associated with an increased risk of fatal infection or sepsis, despite the exclusion of patients with chronic or recurrent infection. Therefore, more elaborate studies are needed to elucidate the mechanism behind these adverse side effects in order to develop a more specific targeting approach.
Although several novel therapies have been explored over the last years, atherosclerosis still cannot be fully reversed by medical treatment, warranting the necessity of innovative therapeutic approaches. Recently, G-protein coupled receptors (GPCRs) have emerged as promising pharmacological targets because of their diverse functions. This is also highlighted by the fact that several recent reviews discussed the targeting of GPCRs in atherosclerosis in a rather general setting (Desimine et al., 2018; Pirault and Back, 2018; Tang et al., 2018; Gencer et al., 2019; Noels et al., 2019). Therefore, in this review we will restrict ourselves to the discussion of the role of GPCRs on myeloid cells in atherosclerosis and CVD and their potential targeting (please also refer to Figure 1).
G-protein coupled receptors, also known as seven-transmembrane domain receptors or heptahelical receptors constitute, with at least 800 members, the largest family of cell surface receptors (Gloriam et al., 2007; Trzaskowski et al., 2012). These various names can be explained by the fact that these receptors all pass the cell membrane seven times and couple to G proteins to activate internal signal transduction upon activation. GPCRs can bind a wide variety of endogenous ligands, including neuropeptides, amino acids, ions, hormones, chemokines, lipid-derived mediators and ions (Marinissen and Gutkind, 2001; Lagerstrom and Schioth, 2008; Hazell et al., 2012). However, the exact ligands of several receptors remain to be identified, making them orphan GPCRs (Chung et al., 2008).
Classically, the GPCR family was divided into three main classes (A, B, and C) with no detectable sequence homology between these classes (Bjarnadottir et al., 2006). Over the years several subgroups emerged to create a more detailed classification, for example class A which accounts for almost 85% of all GPCRs has been further subdivided into 19 subgroups (A1–A19) (Joost and Methner, 2002). Additionally, the main classification also increased in diversity, creating six main classes based on sequence homology and functional similarity (A-F system) (Foord et al., 2005). Also alternative classification systems have been created, for example the GRAFS system that subdivides the receptors based on phylogenetic analysis into five groups called Glutamate, Rhodopsin, Adhesion, Frizzled/Taste2, and Secretin (Fredriksson et al., 2003).
G-protein coupled receptors have distinct binding sites, whereas the main binding site is called the orthosteric binding site, several distinct sites are also susceptible to ligand-binding and are called allosteric binding sites. Depending on the binding site that is used, the ligands can be given the corresponding term orthosteric or allosteric ligand. Binding of allosteric ligands to the receptor will induce a conformational change that influences the affinity or binding potential of orthosteric ligands in a positive (positive allosteric modulator) or a negative (negative allosteric modulator) manner. Before going into detail by discussing the specific GPCRs or sub-families and their respective ligands, it is important for the comprehension of the reader to have a proper definition of several pharmacological concepts (IUPHAR/BPS; Wacker et al., 2017).
An agonist is a ligand or drug that binds to a receptor and alters the receptor state resulting in a biological response. Conventional agonists increase the receptor activity either to the maximum extent (full agonist) or to less than 100% of the maximal response (partial agonist). In contrast, an inverse agonist reduces the receptor activity. For this, the receptor must elicit intrinsic or basal activity already in the absence of the ligand, as the inverse agonist can only then decrease the activity below this basal level. Antagonists do not produce a biological response upon binding to a receptor, but reduces the action of another drug, generally an agonist. Also here, there are two different subtypes being competitive or non-competitive antagonists. Competitive antagonists bind to the same site as the agonist (usually the orthosteric site) on the receptor without causing activation, but thereby blocking the binding of the agonist. This kind of antagonism is reversible by increasing the concentration of agonist to outcompete the antagonist. However, non-competitive antagonists do not compete directly with the binding of the agonist as they bind to an allosteric site on the receptor, resulting in an irreversible effect.
In the inactive state, GPCRs are bound to a guanosine diphosphate (GDP) associated heterotrimeric G protein complex (Gαβγ) (Rosenbaum et al., 2009). Upon activation, the GPCR will undergo a conformational change which induces cytoplasmic signal transduction by influencing the Gα subunit via protein domain dynamics (Hilger et al., 2018). The activated Gα subunit subsequently exchanges guanosine triphosphate (GTP) in place of GDP, triggering the dissociation of the Gα subunit from the Gβγ dimeric subunit and from the receptor. These two dissociated subunits can then interact with other intracellular effector proteins to further activate various signaling cascades (Digby et al., 2006). Gα subunits especially target effectors like adenylyl cyclases, cGMP phosphodiesterase, phospholipase C (PLC), and RhoGEFs (Kristiansen, 2004; Milligan and Kostenis, 2006), while Gβγ recruit kinases to the membrane and regulate potassium channels, voltage-dependent Ca2+ channels, adenylyl cyclases, PLC, phosphoinositide 3 kinase and mitogen-activated protein kinases (Smrcka, 2008; Khan et al., 2013). As Gα has intrinsic GTPase activity, the cellular response is terminated once this subunit hydrolyses GTP again to GDP resulting in the reassociation with Gβγ. The induced signaling and thus functional consequences of GPCR activation are highly variable and largely depend on the nature and binding efficacy of the ligand (Maudsley et al., 2005; Woehler and Ponimaskin, 2009; Kenakin and Miller, 2010; Zheng et al., 2010; Ambrosio et al., 2011). Currently, there are at least 20 different Gα subunits identified, which based on structural and functional similarities can be divided into four families, i.e., Gi, Gs, Gq, and G12/13 (Simon et al., 1991). Members of the Gi family (e.g., Gαi, Gαt, Gαz) mediate primarily the inhibition of adenylyl cyclase or the activation of phosphodiesterase 6, while members of the Gs family (e.g., Gαs, Gαalf) facilitate the activation of adenylyl cyclase. Furthermore, the Gq family members (e.g., Gαq, Gα11, Gα14) are known to activate the kinase PLC, while the G12/13 family members (Gα12 and Gα13) activate the Rho family of GTPases. Additionally, also G protein-independent interactions have been demonstrated for GPCRs, mainly with β-arrestins (Beaulieu et al., 2005; Lefkowitz and Shenoy, 2005), resulting in the internalization of the receptor into endosomes followed by degradation or recycling of the receptor (Daaka, 2012). Arrestin coupling can also induce activation of downstream effector proteins like mitogen-activated protein kinases or SRC kinases. Interestingly, some GPCRs are even able to activate both G protein-dependent as well as G protein-independent signaling (Feng et al., 2005).
The classical view is that the binding of an agonist to a particular GPCR elicits its effects through a single mechanism of activation, suggesting a single activated confirmational state of the receptor (Stephenson, 1956; Black and Leff, 1983). Recently, by measuring broader networks of signals stimulated by agonists, it has become clear that agonists do not only show quantitative differences (e.g., partial or full agonist, fitting in the classical view) but also functional selectivity (e.g., one ligand selectively stimulates one signal whereas another ligand selectively stimulates a second signal via the same receptor) which is not fitting with this classical view (DeWire and Violin, 2011). This gave rise to the concept of ligand bias or also termed biased agonism (Michel and Charlton, 2018), which especially during the past decade received more appreciation and support. The concept of functional selectivity and ligand bias has been comprehensively reviewed elsewhere (Kenakin and Miller, 2010), even in the context of cardiovascular pharmacology (DeWire and Violin, 2011).
Due to the large variety in functional effects mediated by GPCRs, they have been implicated in a multitude of processes that play a crucial role in atherosclerosis development. GPCRs are the most “druggable” receptor class, as a striking 30–35% of all medicines currently on the market target one of these receptors (Hauser et al., 2017; Sriram and Insel, 2018). This is mainly caused by the fact that most GPCRs have small ligands and thus the corresponding binding pockets in these receptors are also small and therefore relatively easy to target. However, especially within the chemokine receptor family difficulties arise when a single receptor can bind multiple ligands. A small molecule blocking the binding site of one of these ligands does not necessary also blocks the binding of all others (Wells et al., 2006). Besides the use of small molecules also different targeting approaches are being used or at least evaluated, like the modification of ligands or antibodies against specific receptors. As Hauser et al. (2017) recently published a very elegant review of trends in GPCR drug discovery further elucidating the various GPCR drugs and agents that are in clinical trials, we will keep the discussion of this rather limited.
In this review, we will highlight a selection of GPCRs or receptor sub-families mainly expressed on myeloid cells and clearly linked to atherosclerosis. The chemokine receptors, both classical and atypical, formyl-peptide receptors (FPRs), chemerin receptor 23 and the calcium-sensing receptor (CaSR) will be described in detail as they have been shown to play an important role in chronic inflammation and atherosclerosis (Figure 1). When information is available, we will also describe the consequences of their (therapeutic) targeting in CVD.
Chemokines (small chemotactic cytokines) and their receptors are multifunctional operators of the immune system facilitating many vital steps of an immune response, such as leukocyte activation, migration, differentiation, phagocytosis and adhesion in addition to their homeostatic roles, such as leukocyte homing (Johnston and Butcher, 2002; Kim, 2004). Chemokines are classified according to their conserved cysteine residues and bind to two types of seven transmembrane receptors: conventional (GPCRs) and atypical chemokine receptors (ACKRs). The main difference between the two types of receptors is the structural inability of ACKRs to couple and thus signal through G proteins (Griffith et al., 2014). With approximately 50 different ligands and 20 receptors, the chemokine/chemokine-receptor family comprises a very complex and also highly dynamic system. Based on the crucial role of this system in various processes that are important in atherosclerosis development and CVD, targeting specific chemokine–chemokine receptors dyads are promising approaches for CVD-treatment (Weber and Noels, 2011).
As described before, the accumulation of oxLDL in the vessel is one of the initiating steps of atherogenesis. The oxidation of LDL generates lysophosphatidylcholine, which is the main substrate for the enzyme autotaxin. This enzyme subsequently transforms lysophosphatidylcholine into lysophosphatidic acid (LPA). This LDL-derived LPA will induce the release of CXCL1 from endothelial cells (Zhou et al., 2011). Subsequently, CXCL1 interacts with CXCR2 on neutrophils and classical monocytes, thereby stimulating their mobilization into the blood stream and migration to sites of inflammation. In line with this, systemic absence of CXCL1 or hematopoietic CXCR2-deficiency has been shown to be protective against atherosclerosis in mice by reducing the intra-plaque macrophage accumulation (Soehnlein et al., 2013).
Another chemokine-axis that has been shown to play an important role during these initial phases of lesion development is the CCL2/CCR2-axis, especially by mediating the mobilization of classical, inflammatory monocytes. Accordingly, CCR2-deficient mice show reduced atherosclerotic lesion formation due to an attenuation of monocytosis (Swirski et al., 2007; Tacke et al., 2007; Weber and Noels, 2011). Recently, it has been shown that there is a striking circadian control of endothelial and myeloid cell activities. This circadian control is part of the daily rhythms, which are controlled by key proteins like CLOCK and BMAL1 (Zhang et al., 2014). A recent study by Winter et al. (2018) could show that such rhythmic control is also present in chronic inflammatory processes of large vessels, thereby mediating rhythmic myeloid cell recruitment. The recruitment of neutrophils and monocytes to atherosclerotic lesions oscillates with a peak during the transition from the activity to the resting phase (Winter et al., 2018). They could show that this oscillating recruitment is regulated by the rhythmic release of myeloid cell-derived CCL2, as blockage of this signaling abolished the oscillatory leukocyte adhesion. Interestingly, the adhesion of myeloid cells to the microvasculature is different than the previously discussed macrovascular effects as here the adhesion peak was reached during the early activity phase. This opens up novel opportunities of well-timed pharmacological targeting of CCL2 in order to modulate the effects on atherosclerosis formation, without disturbing the microvascular cell recruitment (Winter et al., 2018).
Interestingly, deletion of both CCL2 and CX3CR1, or CCR2 and CX3CL1 even further decreased atherosclerosis development compared with single deficiencies in the proteins, which could be attributed to a strongly attenuated monocytosis and hence reduced plaque macrophage accumulation (Combadiere et al., 2008; Saederup et al., 2008). Pharmacological targeting could further confirm these results, as administration of a non-agonistic CCL2-competing mutant (PA508) with increased proteoglycan affinity, or siRNA-mediated silencing of CCR2 in mouse models of MI resulted in reduced recruitment of classical monocytes to the infarcted areas (Liehn et al., 2010; Majmudar et al., 2013). Targeting the CCL2–CCR2 axis has already been evaluated in a phase 2 human clinical trial, where blockage of CCR2 with MLN1202, a specific humanized monoclonal antibody that inhibits CCL2 binding, resulted in reduced plasma CRP levels in patients at risk for CVD (Gilbert et al., 2011). Thereby, targeting of this chemokine-axis remains a promising approach for future CVD therapies.
Besides recruitment, chemokines and their receptors also play an important role in leukocyte arrest on the endothelium by integrin activation. For example, activated platelets release CCL5 which is subsequently immobilized on the surface of inflamed endothelium, triggering leukocyte arrest (von Hundelshausen et al., 2001). This CCL5-mediated myeloid cell recruitment has been shown to be dependent on sialylation of the receptors CCR1 or CCR5, as deficiency of sialyltransferase St3Gal-IV in mice resulted in decreased monocyte and neutrophil recruitment and reduced atherosclerotic lesion size in a CCL5-related manner (Döring et al., 2014). The potential of targeting CCL5 receptors as therapeutic approach was further validated by studies where CCR5 deficiency (Braunersreuther et al., 2007b), inhibition of CCR5 with maraviroc (Cipriani et al., 2013) or general blockage of CCL5 receptors using Met-CCL5 (Veillard et al., 2004) all showed clearly reduced atherosclerotic lesion size and lesional macrophage content in mice. As maraviroc is an FDA-approved HIV-entry inhibitor, it is already used in the clinic, where it could be observed that treatment of HIV-patients with maraviroc seemed to lower atherosclerotic lesion growth (Maggi et al., 2017). Furthermore, it is interesting to note that there is a correlation between plasma CCL5 levels and the progression of atherosclerosis in patients after acute coronary syndrome (Blanchet et al., 2014). All by all, the CCL5–CCR5 chemokine axis seems a promising therapeutic target, especially as an inhibitor is already in clinical use and proven reduce the atherosclerotic risk.
With respect to the development of pharmacological targeting of chemokine receptors, it is intriguing to note that chemokines can also form higher-order complexes with themselves (homomers) or with other proteins (heteromers). For example, CCL5 can form a heteromeric complex with CXCL4 and thereby augmenting the CCL5-stimulated arterial monocyte adhesion (von Hundelshausen et al., 2005). This also has clear implications for atherosclerosis development as selective disruption of the CCL5-CXCL4 heteromer by the cyclic peptide MKEY results in reduced plaque formation in mice (Koenen et al., 2009). Administration of MKEY did not interfere with systemic immune responses, like T cell proliferation of clearance of viral infections, clearly highlighting the potential and specificity of this peptide. Additionally, treatment with MKEY has been shown to preserve heart function and decrease the infarct size in a model of myocardial ischemia/reperfusion injury. Moreover, MKEY treatment resulted in a reduced inflammatory reaction in response to injury, demonstrated by the attenuation of monocyte and neutrophil recruitment. Interestingly, there was also a significant reduction of citrullinated histone 3 in the infarcted tissue, showing that MKEY can also prevent NETosis (Vajen et al., 2018). Another example of a heteromer that stimulates leukocyte adhesion is the complex between neutrophil-borne human neutrophil peptide 1 (HNP1) and platelet-derived CCL5 (Alard et al., 2015). Disruption of this complex with the specific peptide SKY resulted in decreased recruitment of classical monocytes in a murine MI model (Alard et al., 2015). The continued elucidation of the precise physiological and especially pathological functions of various chemokine–chemokine interactions (von Hundelshausen et al., 2017) will further identify novel and interesting targets with clinical potential.
Another important chemokine axis in cell homeostasis, mobilization and immunity is the CXCL12–CXCR4 axis (van der Vorst et al., 2015). For example, systemic treatment of atherosclerosis prone mice with the biglycan CXCR4 antagonist AMD3465 resulted in increased atherosclerosis lesion size compared to untreated controls due to increased neutrophil mobilization (Zernecke et al., 2008). Using cell-specific genetic ablation of CXCR4, endothelial CXCR4 has been shown to promote re-endothelialization after vascular injury and prevent neointimal hyperplasia (Noels et al., 2014) and to limit atherosclerosis development by maintaining the endothelial integrity (Döring et al., 2017). This endothelial barrier integrity was mainly promoted by the signaling of CXCL12–CXCR4 to Akt/WNT/β-catenin resulting in enhanced VE-cadherin expression thereby stabilizing the cellular junctions. Additionally, CXCR4 was shown to be crucial in the maintenance of a normal contractile SMC phenotype. In sharp contrast to the clearly atheroprotective role of vascular CXCR4, its ligand CXCL12 seems to be atheroprogressive as endothelial derived CXCL12 promotes lesion development (Döring et al., 2019). Since the current studies only focused on the role of vascular CXCL12 and CXCR4, it remains to be identified whether and which hematopoietic cells play an important role in the modulation of inflammation by CXCL12–CXCR4. In humans, it has already been shown that both CXCL12 and CXCR4 are associated with CVD. For example, regression analysis demonstrated that the C-allele at rs2322864 in the CXCR4 locus is associated with an increased risk for coronary heart disease (Döring et al., 2017). Additionally, expression of both CXCR4 and CXCL12 was increased in human carotid atherosclerotic lesions compared to healthy vessels (Merckelbach et al., 2018). Genome-wide association studies further confirmed the importance of CXCL12 by showing that a single nucleotide polymorphism at 10q11 near the CXCL12 locus is independently associated with the risk for coronary artery disease (CAD) (Mehta et al., 2011; Döring et al., 2019). Furthermore, the causal role of CXCL12 as mediator of CAD has been confirmed in the ORIGIN and CARDIoGRAM populations by a mendelian randomization study (Sjaarda et al., 2018). All by all, these data clearly support an important role for the CXCL12–CXCR4 chemokine axis in atherosclerosis development and CVD occurrence.
Classical chemokine receptors and their corresponding ligands play a key role in the immune system and have been shown to be drivers and regulators of CVD (please refer to Table 1 for a summary of important studies and their key findings and to Table 2 for an overview of ligand types involved). Interference with this system seems like a very promising therapeutic approach, although this should be carefully designed and has to be context-specific to avoid unwanted, but almost unavoidable, side-effects.
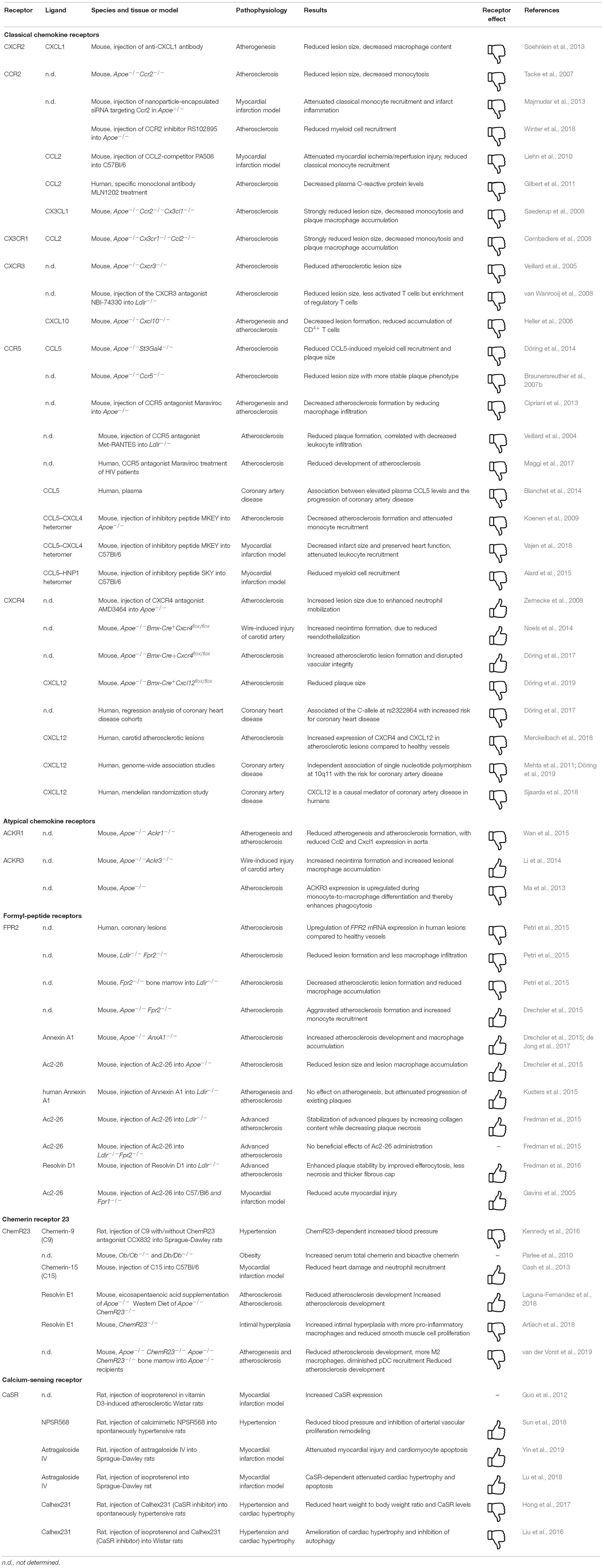
Table 1. Consequences of GPCRs targeting in cardiovascular disease in vivo.
As mentioned before, ACKRs are unable to signal through G proteins but are known to recruit β-arrestin upon ligand binding and are thereby key directors of chemokine driven immune responses as they regulate the bioavailability, internalization, localization as well as the gradient establishment of chemokines (Patel et al., 2009; Ulvmar et al., 2011; Graham et al., 2012; Cancellieri et al., 2013; Bonecchi and Graham, 2016). Moreover, ACKRs can modify the signaling activity of other chemokine receptors via heterodimer formation, thus may also ultimately influence G-protein signaling pathways (Decaillot et al., 2011). Due to their broad-spectrum immunological functions, ACKRs are promising therapeutic targets for the treatment of inflammatory diseases, such as atherosclerosis (Gencer et al., 2019). So far, four types of ACKRs are well recognized: ACKR1 (DARC), ACKR2 (D6), ACKR3 (CXCR7 or RDC-1) and ACKR4 (CCRL1), whereas new members are subject to further investigation: ACKR5 (CCRL2) and ACKR6 (PITPNM3) (Ulvmar et al., 2011). Three members of this family, ACKR1, ACKR2, and ACKR3, are critical for inflammatory responses and will therefore be discussed in greater detail, whereas ACKR4 seems to be primarily involved in homeostatic processes.
ACKR1 is expressed on erythrocytes as well as venular endothelial cells and binds plentiful inflammatory chemokines. It is well known that the absence of ACKR1 on erythrocytes causes a Duffy-negative phenotype in African people (Howes et al., 2011; Novitzky-Basso and Rot, 2012; Horuk, 2015). A study by Duchene et al. (2017) showed that Duffy negative individuals exhibited an altered neutrophil phenotype by CCR2, CD16, and CD45 overexpression in comparison to Duffy positive individuals, indicating an amplified defense mode of neutrophils as a result of the lack of ACKR1 on erythrocytes. Considering that ACKR1 binds a wide range of inflammatory chemokines in addition to the characteristic scavenging activity of ACKRs, it is concluded that erythrocyte-specific ACKR1 is a decoy receptor regulating the levels of circulating inflammatory chemokines, such as CCL2 and CXCL8 (Jenkins et al., 2017). Endothelial ACKR1, on the other hand, mediates the internalization of extracellular chemokines and allows their presentation on the cell surface (Novitzky-Basso and Rot, 2012). This process enhances leukocyte recruitment and supports leukocyte-endothelium adhesion, augmenting inflammation. Due to its contrasting roles in different cell types, it is difficult to gauge the impact of systemic ACKR1 deficiency in the context of atherosclerosis. One possibility is that it may lead to a rise in circulating inflammatory myeloid cells, such as monocytes, through an increase in circulating inflammatory chemokines, which would be considered a pro-atherosclerotic event. On the other hand, it may result in a reduction of myeloid cell adhesion to the endothelium, which may in turn decrease lesional macrophage accumulation and thereby limit the development of lesions. Wan et al. (2015) reported an atheroprotective role of ACKR1 deficiency in an apolipoprotein E deficient (ApoE−/−) mouse model. This was shown to be a result of decreased lesion sizes observed with a decreased inflammatory phenotype in circulating monocytes and macrophages in addition to decreased T-cells in the aortic vessel wall (Wan et al., 2015). This finding highlights a detrimental role of ACKR1 in atherosclerosis. Another study investigating ACKR1 in the context of inflammation through a bone fracture model in mice reported a significant reduction in macrophage numbers around the fractures in ACKR1 deficient mice (Rundle et al., 2013). This outcome was observed with a concomitant decrease in inflammatory markers, such as IL-1β, IL-6 as well as monocyte chemotactic protein-1, confirming a detrimental role for ACKR1 in macrophage recruitment and inflammation. Taken these findings into account, the inhibition of this receptor might be a therapeutic approach in atherosclerosis treatment.
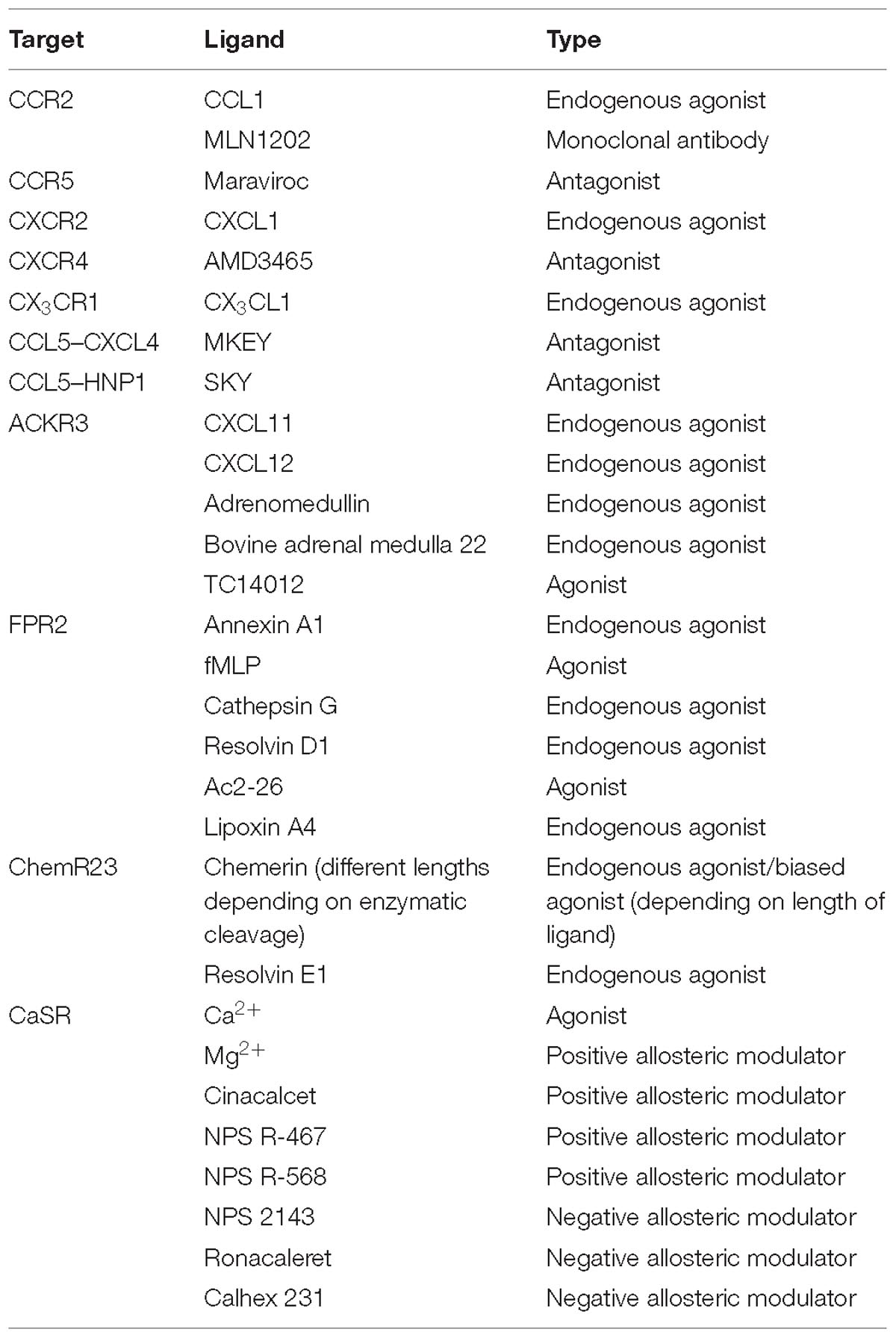
Table 2. Types of GPCR-ligands discussed in the review.
Similar to ACKR1, ACKR2 also binds numerous inflammatory chemokines. It is expressed on lymphatic endothelial cells, innate-like B cells and some macrophage subsets (Bonecchi and Graham, 2016). Growing evidence discloses an anti-inflammatory profile for ACKR2 with a central role in the resolution of inflammation (Bonavita et al., 2016; Bideak et al., 2018; Massara et al., 2018). ACKR2 is defined as a scavenger receptor for inflammatory chemokines, because ACKR2 deficient mice reproducibly showed increased levels of inflammatory chemokines, like CCL2 (Jamieson et al., 2005; Martinez de la Torre et al., 2005; Whitehead et al., 2007; Collins et al., 2010; Vetrano et al., 2010). The anti-inflammatory properties of ACKR2 are not only limited to its scavenging activities; this receptor is also involved in the regulation of monocyte and macrophage dependent immune responses. For example, ACKR2 deficiency in a murine zymosan A-initiated peritonitis mouse model was shown to promote macrophage efferocytosis, suggesting an important potential function of ACKR2 in atherosclerotic plaques with regards to the efficiency of foam cell efferocytosis (Pashover-Schallinger et al., 2012). Additionally, a Mycobacterium tuberculosis disease model lead to rapid death in ACKR2 deficient mice with concomitant increased infiltration of mononuclear cells, e.g., macrophages, into inflamed tissues as well as lymph nodes (Di Liberto et al., 2008). Macrophage infiltration and accumulation in atherosclerotic lesions leads to the progression and eventually growth of plaques. It is therefore of great interest to inhibit these key processes in order to treat atherosclerosis. Considering its roles in macrophage efferocytosis and immune cell infiltration, ACKR2 may be a novel therapeutic target in the research of atherosclerosis treatment. A study conducted by Savino et al. (2012) reported a CCR2-dependent, selective increase in Ly6Chigh monocyte numbers in circulation as well as secondary lymphoid organs of mice lacking ACKR2 in the non-hematopoietic fragment. This outcome was observed with a delayed graft versus host disease development due to the immunosuppressive activity of the Ly6Chigh monocytes pointing toward a contrasting role of the receptor in the context of adaptive immune responses. Nevertheless, in the context of atherosclerosis, a rise in inflammatory monocytes in circulation may lead to increased monocyte infiltration and intra-plaque macrophage accumulation, thus result in more advanced lesions. Hence, ACKR2 is a significant immunomodulatory candidate and its roles shall be scrutinized in a cell type and disease model specific manner.
ACKR3 is expressed in endothelial cells, marginal B cells, neurons as well as mesenchymal and some hematopoietic cells (Massara et al., 2016). It binds two well-known chemokine ligands, CXCL11 and CXCL12, in addition to adrenomedullin and bovine adrenal medulla 22 (BAM22) (Wang et al., 2018). ACKR3 can signal through β-arrestin and activate extracellular signal-regulated kinase (ERK) as well as phosphoinositide 3-kinase (PI3K)-Akt signaling pathways (Ma et al., 2013). Moreover, ACKR3 can control CXCL12 signaling by either regulating its concentrations or heterodimerization with its alternative receptor CXCR4 (Levoye et al., 2009). Although ACKR3 is crucial in vascular and cardiac development, a number of studies demonstrated its detrimental effects in the context of inflammation.
Research suggests that inflammation caused an increased expression of ACKR3 on immune cells, especially myeloid cells. Infiltrating monocytes in a mouse peritonitis model as well as lesional macrophages in aortic atheroma of mice showed increased ACKR3 expression, pointing toward an inflammatory role of ACKR3 (Ma et al., 2013; Chatterjee et al., 2015). Ma et al. (2013) showed that ACKR3 expression was detected in the macrophage positive area defined by F4/80 positivity within atherosclerotic lesions, whereas this was not observed in the vessel wall of healthy aortas. Moreover, this study showed that whilst undifferentiated THP-1 cells expressed CXCR4 but not ACKR3 mRNA, phorbol 12-myristate 13-acetate (PMA) treatment in THP-1 cells (promoting macrophage differentiation) induced the expression of ACKR3 mRNA whilst downregulating CXCR4 mRNA. Further functional analysis of macrophages with regards to ACKR3 activity was assessed by ACKR3 agonists, such as CXCL12 and TC14012. Treatment of macrophages with these agonists showed increased uptake of FITC-labeled E. coli, demonstrating a significant increase in cellular phagocytosis. This effect was abolished by siRNA silencing of ACKR3, confirming that the observed phagocytosis was a result of ACKR3 activity. These findings were endorsed by increased uptake of acetylated LDL by the macrophages stimulated with the same ACKR3 agonists. Another study by Chatterjee et al. (2015) showed that monocytes in the peritoneal fluid of mice with peritonitis showed enhanced CXCR4, ACKR3, and CXCL12 expression, also suggesting that this axis plays an important role in monocyte function during inflammation. Furthermore, ACKR3 was shown to promote monocyte survival and adhesion onto a CXCL12 rich platelet surface as well as the phagocytic activity and foam cell formation of macrophages (Chatterjee et al., 2015). In line with these results, ACKR3 is suggested to support monocyte to macrophage differentiation through CXCL12 activity (Sanchez-Martin et al., 2011). This was supported by the significant reduction of CD136 expression of human monocytes upon both CXCR4 and ACKR3 antagonist treatment. In the same study, monocyte differentiation into CD136+ macrophages was shown to be inhibited by means of CXCL12 neutralization as well as CXCR4 and ACKR3 blocking. Moreover, exogenous CXCL12 dependent M-CSF production by the monocytes was partially inhibited by CXCR4 and ACKR3 antagonists, further confirming CXCL12, CXCR4, and ACKR3 dependent regulation of monocyte to macrophage differentiation. Altogether, these findings suggest that ACKR3 promotes atherosclerosis by supporting monocyte and macrophage driven inflammatory processes. Therefore, its inhibition might be a valuable therapeutic target in order to interfere with key events driving atherosclerosis.
Without a doubt, ACKRs play crucial roles in the regulation of immune responses and therefore offer significant therapeutic targets in order to control the inflammatory processes. Nevertheless, their wide array of functions establishes a great complexity, making it very difficult to determine individual targets. Thus, it is of great importance to scrutinize and understand the biology of ACKRs CVD (please refer to Table 1 for a summary of important studies and their key findings and to Table 2 for an overview of ligand types involved).
Formyl-peptide receptors (FPRs) belong to the group of pattern recognition receptors (PRRs) and comprise a family of chemoattractant GPCRs involved in host defense against bacterial infections and clearance of cell debris. FPRs are well conserved among mammals (Ye et al., 2009) and are mainly present on myeloid cells such as neutrophils (except FPR3) and monocytes (He and Ye, 2017). In addition to myeloid cells, astrocytes, microglia, hepatocytes, and immature dendritic cells express FPR1, whereas FPR2 is also expressed on epithelial cells, hepatocytes, microvascular endothelial cells, and smooth muscle cells (He and Ye, 2017). FPRs were originally discovered as receptors that bind highly conserved N-formyl methionine-containing protein and peptide sequences of bacterial and mitochondrial origin (Forsman et al., 2015). For example, one of the most potent agonists for FPR1 is the Escherichia coli-derived peptide N-formyl methionyl-leucyl-phenylalanine (fMLF) (Ye et al., 2009), while the most prominent bacterial FPR2 ligands are the staphylococcal-derived phenol-soluble modulins (PSMs) (Kretschmer et al., 2015). However, it has become evident that FPR1 and FPR2 recognize a variety of structurally diverse ligands including many host-derived endogenous agonists (see also Table 2) such as Annexin A1, Resolvin D1, Cathepsin G, and the cathelicidin LL37 (mouse: Cramp) all of which have been associated with inflammation and/or resolution in mice and man (He and Ye, 2017; Filep et al., 2018). Hence, FPRs may exert ambivalent effects during leukocyte recruitment and in (chronic) inflammatory conditions such as atherosclerosis.
Studies on human atherosclerotic plaque specimens supported the notion of the involvement of FPRs in lesion development by pointing at defective resolution within these lesions (Fredman et al., 2016). Additionally, FPR2 mRNA expression was upregulated in human samples from coronary lesions in comparison to healthy vessels (Petri et al., 2015). Similarly, mice deficient for the low-density lipoprotein receptor (LDLR) and FPR2 exhibited decreased atherosclerosis development and less monocyte infiltration and foam cell formation compared with control animals. Analogous results were obtained in Ldlr−/− mice transplanted with FPR2-deficient bone marrow, here dampened activation of lesional macrophages was also attributed to the lack of FPR2 (Petri et al., 2015). These findings support in vitro work from Lee et al. (2013, 2014) showing that oxLDL and serum amyloid-2 mediate foam cell formation via FPR2. Hence, one could argue that agonists, which mediate lesional macrophage activation via FPR2 disturb resolution. However, FPR2 expression on vascular smooth muscle cells (VSMCs) seems to stabilize atherosclerotic lesions suggesting a diverse role of FPR2 on hematopoietic versus vascular cell types. Still, specific agonists or antagonists, which mediate one or the other response, were not investigated in this study (Petri et al., 2015). In contrast, Apoe−/− mice which also lacked FPR2 or Annexin A1 showed enhanced atherosclerotic lesion development, increased myeloid cell recruitment and adhesion to the inflamed vessel wall. One explanation focusses on the observation that Annexin A1/FPR2 interaction seems to tightly control and inhibit integrin activation (Drechsler et al., 2015; de Jong et al., 2017). In line, treatment of Apoe−/− or Ldlr−/− mice with Annexin A1 or the Annexin A1 fragment Ac2-26 reduced atherogenesis by decreasing necrosis, mediating efferocytosis and supporting fibrous cap stability (Drechsler et al., 2015; Kusters et al., 2015). Equivalent results were obtained in Ldlr−/− mice with advanced atherosclerosis, which were treated with the agonist Ac2-26 packed into nanoparticles that targeted type IV collagen to ensure deposition in atherosclerotic lesions. Plaques of animals treated with Ac2-26 nanoparticles displayed reduced macrophage numbers, smaller necrotic core sizes, and higher amounts of anti-inflammatory interleukin 10 compared to control animals. On the contrary, when treating Ldlr−/− Fpr2−/− mice with Ac2-26, the protective effects were abolished suggesting an important role of FPR2 on myeloid cells in mediating arterial (lesional) resolution through interaction with Annexin A1 (Fredman et al., 2015).
Specialized pro-resolving lipid mediators (SPMs) including the resolvins are derived from the ω-3 PUFAs eicosapentaenoic acid (EPA) or docosahexaenoic acid (DHA). They have important roles in the resolution of inflammation, either via their own GPCRs or by modulating GPCRs for ω-6 PUFA. For example, resolvin E1 (RvE1) enhances the phagocytosis of apoptotic neutrophils via ChemR23 (please also see section “ChemR23 and Resolvin E1 – Mechanisms of Resolution”) and inhibits the infiltration of neutrophils by antagonizing LTB4 or leukotriene B4 receptor 1 (BLT1). Resolvin D1 (RvD1) instead has been shown to bind to two GPCRs, namely, the orphan receptor, GPR32, and the lipoxin receptor, FPR2/ALX through which it mediates its pro-resolving effects (Jannaway et al., 2018). In line with this, if the endogenous agonists RvD1 was administered to Ldlr−/− mice during the transition phase of atherosclerotic lesions from early into advanced plaques. Fredman et al. (2016) could show that RvD1 enhanced lesional efferocytosis, and decreased plaque necrosis compared with vehicle controls. Similarly, repetitive administration of endogenous agonists Resolvin D2 to Apoe−/− mice prevented atheroprogression, though most likely mediated via the G-protein coupled receptor 18 (Viola et al., 2016). These findings illustrate the therapeutic potential of pro-resolving FPR agonists to restore defective resolution, which is most likely mediated through myeloid cells in atherosclerotic lesions.
Consistently, a protective role of Annexin A1 and its mimetic peptides could also be demonstrated in experimental models of ischemia-reperfusion injury, e.g., in a mouse model of MI (Gavins et al., 2005). Moreover, Ferraro et al. (2019) for example examined to what extent endogenous control of inflammation resolution and its therapeutic stimulation enables improved cardiac function in the absence and presence of Annexin A1. They showed that myeloid cells infiltrating at early stages post MI deliver Annexin A1 hereby terminating inflammation and promoting healing through macrophages with an angiogenic phenotype with release of VEGF-A. They could further reveal similar protective functions of Annexin A1 in a model of MI in pigs, hence demonstrating that Annexin A1 facilitated cardiac angiogenesis and myocardial repair (Ferraro et al., 2019).
Other FPR agonists such as Cathepsin G (Ortega-Gomez et al., 2016) and LL37/Cramp (Döring et al., 2012; Wantha et al., 2013) clearly mediate pro-atherogenic effects by enhancing monocyte adhesion and recruitment, though one cannot exclude that these functions may partly be mediated by other receptors. As FPRs recognize both pro-inflammatory and pro-resolving signals, the question remains how one receptor can mediate opposing responses. In this context, Cooray et al. (2013) suggested that anti-inflammatory, but not pro-inflammatory signals activate homodimerization of FPR2, which, in turn trigger the release of anti-inflammatory mediators such as interleukin 10. Heterodimers instead can transduce, e.g., pro-apoptotic signals, explaining why the same receptor system may integrate diverse signals (Cooray et al., 2013). Another plausible option is the concept of biased agonism (please also see section “GPCR Signaling”) (Michel and Charlton, 2018) describing that agonists/antagonists might activate specific receptor domains, thereby promoting downstream responses, which at least in part do not overlap. As an example, the small lipid lipoxin A4 has been shown to activate FPR2 by interacting with its extracellular loop III (Chiang et al., 2000), while, e.g., serum amyloid A responses were reliant on extracellular loops I and II (Bena et al., 2012). Hence, all of the latter should be considered in the context of designing potential new therapeutics triggering resolution via FPRs.
Formyl-peptide receptors have evolved to be a class of receptors that recognize a broad range of structurally distinct ligands and are expressed by a variety of cell types. Many studies have also shown that FPR function is not restricted to host defense against microbes, but also impacts on chronic inflammatory disease such as atherosclerosis and autoimmune diseases or even cancer. Most interestingly, FPR2 does not only mediate pro-inflammatory but also resolution processes and return to homeostasis. While these findings greatly expanded the scope of the pharmacology and biology of FPRs, a better understanding of how FPRs recognize and respond to distinct ligands is needed to explore their further potential as therapeutic targets (please refer to Table 1 for a summary of important studies and their key findings and to Table 2 for an overview of ligand types involved).
The chemerin receptor 23 (ChemR23; chemokine-like receptor 1, CMKLR1) is a class A (rhodopsin-like) GPCR expressed on the surface of immune cells subtypes such as dendritic cells (Vermi et al., 2005), monocytes and macrophages (Herova et al., 2015). It is therefore expressed in spleen and lymph nodes, but also in the skin, adipose tissue (Goralski et al., 2007; Goralski and Sinal, 2009) and lung (Wittamer et al., 2003; Roh et al., 2007). Functionally, ChemR23 – mostly through its bona fide ligand chemerin- mediates immune cell activation and chemoattraction (Carlino et al., 2012; Rourke et al., 2013). The gene encoding for ChemR23 is called CMKLR1 (non-human = cmklr1), first cloned in 1996 by Gantz et al. (1996) and, under the name of ChemR23, in 1998 by Samson et al. (1998). Another gene encoding G-protein coupled receptor 1 (GPR1) was proved to share a common ancestor with CMKLR1 (Vassilatis et al., 2003) with a sequence identity of 37% (Kennedy and Davenport, 2018). Therefore, it is designated as chemerin receptor 2. The corresponding human sequence for ChemR23 and GPR1 share 80% sequence identity with its corresponding murine genes (Kennedy and Davenport, 2018). ChemR23 has two known ligands in mouse and human, namely chemerin and Resolvin E1 (RvE1). Based on its similarities with GMKLR1, Barnea et al. (2008) were the first to identify chemerin as a ligand for GPR1. GPR1 can act to modify glucose homeostasis during obesity, in line with known functions of chemerin (Rourke et al., 2015). However, as yet it is largely unknown what G protein pathway it activates, only few studies showed chemerin modestly induced calcium release (Barnea et al., 2008) or RhoA signaling (Rourke et al., 2015), more investigation are warranted to unveil the downstream pathways. In this review, we mainly discuss the role of ChemR23 in CVD.
The adipokine chemerin is encoded by the RARRES2 (non-human = rarres2) gene (Nagpal et al., 1997; Busmann et al., 2004). After being translated into the circulating pro-chemerin, the protein undergoes extensive enzymatic processing. It has been shown that the serine proteases, cathepsin G and elastase, are the main enzymes responsible for the conversion of pro-chemerin into its active form (Wittamer et al., 2005; Ortega-Gomez et al., 2016). The resulting protein variants differ in length and functional properties (Meder et al., 2003; Wittamer et al., 2003). Depending on the chemerin variant binding to ChemR23, the receptor couples to a different subtype of Gαi or isoform of Gαo (Wittamer et al., 2004). Regarding downstream-signaling via ChemR23, it could be shown that the variants C9 (or chemerin-9) and 13 were more potent in inhibiting G protein-dependent cAMP, but less potent in inducing β-arrestin compared with human chemerin 21-157. In summary injection of C9 into rats increased blood pressure via ChemR23 but not via GPR1 mediated signaling and could be inhibited by applying the ChemR23 specific antagonist CCX832 (Kennedy et al., 2016). This lead to the conclusion that shorter C-terminal fragments of chemerin seem to impose a strong bias toward activating G protein coupled signaling. Therefore, signaling via ChemR23 cannot be pinpointed to induce neither purely pro-inflammatory nor anti-inflammatory effects.
Studies in animal models of CAD for example showed that expression of both ChemR23 and chemerin were induced in mice which were fed a high fat diet (Roh et al., 2007). Human studies also show elevated plasma concentrations of chemerin and their association with an increased risk of hypertension (Kennedy et al., 2016; Watts et al., 2018), a higher body mass index and blood pressure in patients with type 2 diabetes mellitus compared to healthy controls (Yang et al., 2010). Increased expression levels of ChemR23 were also described in perivascular fat tissue and correlated with increased blood pressure (Neves et al., 2015). In line, augmented expression levels of both ChemR23 and chemerin have been shown in atherosclerotic plaques of human patients and in mouse models of vascular inflammation and a positive correlation between chemerin expression in perivascular adipose tissue and atheroprogression has already been demonstrated (Kostopoulos et al., 2014). Another hypothesis includes an influence of adipokine expression in the heart and vasculature and subsequent plaque progression (Spiroglou et al., 2010). Specifically, it was already found that chemerin expression in human epicardial adipose tissue was positively correlated with the severity of coronary atherosclerosis (Gao et al., 2011). In addition, plasma chemerin levels are associated with markers of inflammation and are significantly higher in CAD patients, which do not receive low dose aspirin treatment. The latter does also reduce pro-inflammatory cytokine secretion by macrophages, which may lead to reduced chemerin secretion by adipocytes and may be a reason for the lower chemerin levels in the circulation of CAD patients on low dose aspirin (Herova et al., 2014).
In contrast, Cash et al. (2013) reveal a protective role of certain chemerin variants in a model of acute MI by preventing excessive neutrophil infiltration. Chemerin-15 induced signaling via ChemR23 was also described to increase efferocytosis in macrophages in vitro and in an in vivo model of peritoneal inflammation (Cash et al., 2010) and was shown to reduce acute intravascular inflammatory events in murine cutaneous wounds (Cash et al., 2014). In a recently published dietary intervention study, anti-atherosclerotic effects of ChemR23 were outlined using a ChemR23 knock-out mouse model on an Apoe−/− background (Laguna-Fernandez et al., 2018). A deficiency in ChemR23 (Apoe−/− ChemR23−/−) seemed to accelerate atherogenic signaling in macrophages, induced cholesterol uptake and phagocytosis and lead to an increased lesion size and reduced plaque stability, hence claiming that a functional receptor mediates atheroprotective signaling (Laguna-Fernandez et al., 2018). Contradictory, in a very recent publication from our group we saw that hematopoietic ChemR23-deficiency increases the proportion of alternatively activated M2 macrophages in atherosclerotic lesions and attenuates pDC homing to lymphatic organs and recruitment to atherosclerotic lesions, which synergistically restricts atherosclerotic plaque formation and progression (van der Vorst et al., 2019). Nevertheless, ChemR23−/− VSMCs exhibited a significantly lower proliferation rate compared with VSMCs derived from ChemR23+/+ mice while ChemR23-deficient peritoneal macrophages from had significantly higher mRNA levels of pro-inflammatory cytokines compared with ChemR23+/+ macrophages. Finally, conditioned media (CM) transferred from ChemR23−/− macrophages to VSMCs significantly increased VSMC proliferation compared to treatment with CM from ChemR23+/+ macrophages at least in vitro. These results assert dual signaling effects to ChemR23 depending on cell type (VSMCs versus macrophages) expressing the receptor (Artiach et al., 2018), pointing at a diverse role of the receptor on hematopoietic versus vascular cells in atherosclerotic lesion development. An alternative hypothesis suggests that anti-inflammatory effects of chemerin in atherosclerosis are exerted via reduced adhesion to the affected vascular endothelium. One study could show a downregulation of vascular cell adhesion molecule – 1 following chemerin treatment in human umbilical vein endothelial cells and rat aorta and consequently, a reduced monocyte adhesion to the arterial wall (Yamawaki et al., 2012). In conclusion, while chemerin/ChemR23 seems to exert more pro-inflammatory effects on hematopoietic cells, its presence on vascular cells seems to point at an anti-inflammatory role of this ligand receptor pair. However, the relative abundance of pro- versus anti-inflammatory ligands which may also compete with each other and their highly tissue specific expression patterns are expected to further shape these diverse cellular responses in different stages of (chronic) inflammation such as atherosclerosis.
RvE1, a metabolite of EPA (a type of polyunsaturated fatty acids), plays an important role in the return to tissue homeostasis (Schwab et al., 2007; Gao et al., 2013; Hasturk et al., 2015) and is suggested to exhibit anti-inflammatory and pro-resolving effects via ChemR23 or leukotriene B4 receptor 1 (BLT1) (Arita et al., 2005b; Arita et al., 2007) (please also see section “Pro-resolving Lipid Mediators”). RvE1-dependent blockage of VSMC migration, a critical process in the progression of atherosclerosis, and switching into a protective anti-atherosclerotic phenotypic in VSMCs, confer an anti-inflammatory role of vascular ChemR23 signaling (Ho et al., 2010). Moreover, RvE1 rescues impaired neutrophil phagocytosis, oxidized LDL uptake and phagocytosis of macrophages, promotes phagocytosis-induced neutrophil apoptosis (El Kebir et al., 2012; Herrera et al., 2015; Artiach et al., 2018), and also attenuates APC functions targeting dendritic cell migration and reducing IL-12 production via ChemR23 (Arita et al., 2005a). Furthermore, RvE1 can restore inflammation induced mitochondrial dysfunction and reduce polymorphonuclear leukocyte infiltration in BLT1 dependent manner (Arita et al., 2007; Mayer et al., 2019), along with ChemR23-mediated counter regulatory actions to mediate the resolution of inflammation. It is also evident that RvE1 suppresses inflammatory cytokine release, facilitating the healing process, and inhibits macrophage migration by activating ChemR23 in a ligation model of acute MI (Liu et al., 2018). Moreover, supplementation of Apoe−/− mice with polyunsaturated fats as potentially beneficial intervention was supposed to enhanced interaction of RvE1 with ChemR23 in atherosclerosis prone mice and reduced their lesion size (Laguna-Fernandez et al., 2018). However, one study proposed that RvE1 and chemerin compete for the same recognition site on ChemR23, and RvE1 binding is blocked at the presence of the chemerin peptide (Arita et al., 2005a). Whether above summarized results hold true when RvE1 is not actively administered or otherwise supplemented, most likely exceeding competition for the recognition site on ChemR23 by chemerin, has yet to be fully elucidated.
Taken together, all these findings open a potential new avenue for the modulation of the magnitude of the local inflammatory responses also in chronic inflammatory disease such as atherosclerosis by fine tuning receptor specific responses in a cell and tissue specific manner.
The chemerin/ChemR23 axis is a complex network involved in the regulation of immune responses contributing to both the onset and the termination of inflammation. However, several studies show that the various chemerin isoforms may exert different actions downstream of ChemR23. Since chemerin has multiple and different actions, the possibility to selectively modulate its activity can become an attractive target for drug development. Thus, the use of substances boosting its anti-inflammatory properties could be a promising target in exploiting new strategies to treat atherosclerosis (please refer to Table 1 for a summary of important studies and their key findings and to Table 2 for an overview of ligand types involved).
The CaSR belongs to the metabotropic glutamate receptor GPCR subfamily and is most abundantly expressed in the parathyroid gland and kidney (Brown et al., 1993; Aida et al., 1995; Riccardi et al., 1995; Bockaert and Pin, 1999). Here it senses changes in extracellular Ca2+ concentrations and couples this to intracellular signaling pathways that modify parathyroid hormone (PTH) secretion and renal calcium reabsorption in order to maintain the Ca2+ homeostasis (Diez-Fraile et al., 2013).
Although Ca2+ is the main agonist for this receptor, CaSR responds to several other cations (e.g., Mg2+, Gd2+, Sr2+, La2+, and Ba2+) and a variety of other ligands (McLarnon and Riccardi, 2002). Interestingly, Mg2+ has the potency to augment CaSR signaling responses in the presence of Ca2+ and thus is a positive allosteric modulator, meaning that serum Mg2+ will affect Ca2+-CaSR signaling in clinical conditions (Ruat et al., 1996). Moreover, Mg2+ has been shown to also stimulate CaSR mRNA expression and protein levels whilst CaSR activation decreases Mg2+ levels (Ikari et al., 2001), indicating a negative feedback loop between Mg2+ and CaSR. Other, non-cation agonists of the receptor include polyamides, such as spermine and spermidine, and various amino acids (Quinn et al., 1997; Conigrave et al., 2000).
Upon ligand binding, CaSR activates several intracellular signal transduction pathways, mainly through Gαi, Gαq, and Gα12/13 G-protein subtypes. CaSR influences several effectors, such as PLC, adenylate cyclase (AC), cytosolic phospholipase A2 (cPLA2), phosphatidylinositol 4-kinase (PI4K), phospholipase D (PLD), and ERK (Kifor et al., 1997; Chang et al., 1998; Arthur et al., 2000; Huang et al., 2002; Huang et al., 2004).
Abnormal CaSR activity or expression contributes to the development of CVDs. Following the discovery that CaSR is expressed in the heart tissue of rats, Guo et al. (2012) investigated the relationship between CaSR and MI in atherosclerosis by inducing MI in atherosclerotic rats and non-atherosclerotic controls. Here the authors showed that CaSR expression was significantly increased in the atherosclerotic MI group compared to the MI controls, suggesting that CaSR plays an important role in MI caused by atherosclerosis (Guo et al., 2012). Besides its suspected role in atherosclerosis and MI, CaSR is also important in numerous other inflammatory diseases. For example, a population-wide study and Felderbauer et al. (2006) linked several CaSR polymorphisms and mutations to chronic pancreatitis (CP) and idiopathic CP (Muddana et al., 2008). Furthermore, a study focusing on asthma showed that asthmatic patients and allergen-sensitized mice have higher expression levels of CaSR (Yarova et al., 2015), linking abnormal CaSR expression to yet another inflammatory disease. Moreover, Cheng et al. (2014) reported a diminished intestinal barrier function and a more inflammatory immune response in intestinal epithelial-specific CaSR knockout mice, all of which increased their susceptibility to chemically induced colitis. Together, these studies clearly show a very broad involvement of CaSR in inflammatory processes.
CaSR is expressed on various inflammatory cells, such as monocytes and macrophages. Expression of CaSR on monocytes has been implicated in chemotaxis, a key process in inflammatory diseases. For example, one study indicated an interrelationship between CCR2 and CaSR and also showed that Ca2+ stimulates the chemotaxis of monocytes to CCL2 in a CaSR-dependent manner (Olszak et al., 2000). Paccou et al. (2013) followed up on this study by investigating CaSR expression on monocytes in response to several stimuli. Here, total CaSR expression increased in monocytes upon calcitriol, the biologically active form of vitamin D, stimulation whilst TNF decreased total CaSR expression in a dose-dependent manner (Paccou et al., 2013). Further connecting this receptor to inflammatory diseases, several studies report the activation of the NLRP3 inflammasome by CaSR in monocytes, where the review by Tang et al. (2018) provides an overview of all GPCRs involved in NLRP3 inflammasome activation and inhibition. NLRP3 inflammasome activation consequently leads to caspase-1 activation, which in turn cleaves pro IL-1β and pro IL-18 into their active forms mediating a pro-inflammatory response (Groslambert and Py, 2018). A study by Rossol et al. (2012) showed that monocytes sense changes in extracellular Ca2+ concentrations via CaSR signaling, which subsequently leads to the activation of the NLRP3 inflammasome (Rossol et al., 2012). However, another study showed that NLRP3 is also activated by decreased cAMP concentrations, which is in striking contrast to the study by Rossol et al. (2012) where no significant influence of cAMP levels on inflammasome activation could be detected (Lee et al., 2012). Also in a human setting, CaSR could already be linked to atherosclerosis development as Malecki et al. (2013) showed a 1.5-fold increased CaSR expression on peripheral blood monocytes of patients with peripheral artery disease (PAD). Overall, CaSR expression on monocytes seems to enhance pro-inflammatory responses via stimulation of chemotaxis and inflammasome activation.
Focusing more on macrophage-specific CaSR, stimulation of CaSR promotes the release of pro-inflammatory mediators, such as IL-1β and TNF-α, by monocyte derived macrophages (Xi et al., 2010). In line with this, Canton et al. (2016) reports that extracellular Ca2+ is sensed by CaSR which subsequently signals through PLC and PI3K to induce constitutive micropinocytosis. Interestingly, two studies also linked macrophage-specific CaSR to the activation of the NLRP3 inflammasome. One study investigated the causal role of CaSR in the activation of the NLRP3 inflammasome via proteolytic pathways. It showed that the receptor activates the NLRP3 inflammasome and proteolytic maturation of IL-1β in differentiated macrophages via a chaperone-assisted degradative pathway (Gutierrez-Lopez et al., 2018). Another study by Lee et al. (2012) focused on the role of extracellular cations in inflammasome activation. It showed that CaSR activates the NLRP3 inflammasome in bone marrow-derived macrophages via increased intracellular Ca2+ and decreased cAMP (Lee et al., 2012). Together, CaSR expressed on macrophages also promotes pro-inflammatory responses via increased release of cytokines and inflammasome activation.
As CaSR is implicated in various diseases, it has become an interesting pharmacological target to investigate. The compounds which target the receptor can be divided into two categories: the calcimimetics and calcilytics. Calcimimetic compounds are positive allosteric modulators and include Cinacalcet, NPS R-467 and NPS R-568 (Nemeth et al., 1998, 2004). On the other hand, calcilytics such as NPS 2143, Ronacaleret and Calhex 231 are negative allosteric modulators (Nemeth et al., 2001; Petrel et al., 2003; Balan et al., 2009). Cinacalcet was the first allosteric GPCR modulating compound to be approved for the market (Brauner-Osborne et al., 2007), showing promising results in several case studies. One study reports durable and robust effects of Cinacalcet therapy in patients with neonatal severe hyperparathyroidism (NSHPT). Cinacalcet was offered as an experimental alternative drug in a case of NSHPT, where Cinacalcet was successful in rapidly normalizing the patient’s serum calcium levels, thereby improving muscle tone and the overall clinical condition (Gannon et al., 2014). Another study reports an acute increase of urinary calcium excretion in renal transplant recipients with secondary hyperparathyroidism after treatment with Cinacalcet, without showing adverse effects on glomerular filtration rate or renal graft calcium deposits (Courbebaisse et al., 2012). Calcilytics are mostly researched as a potential treatment of osteoporosis, but with limited success. Although clinically safe, no calcilytic to this day has been approved to be used to treat osteoporosis in humans (Kiefer et al., 2011). However, research has suggested the use of calcilytics in other diseases than osteoporosis, as for example Yarova et al. (2015) showed that calcilytics abrogate airway hyperresponsiveness and inflammation in allergic asthma.
Overall, CaSR plays an important role in various chronic inflammatory diseases, which is underlined by the many pro-inflammatory mechanisms induced by CaSR signaling in monocytes and macrophages. Calcimimetics and calcilytics show great therapeutic potential in other disease types, suggesting a potential of these drugs in the treatment of chronic inflammatory diseases as well. Important to keep in mind is that certain mutations and polymorphisms of CaSR affect the binding affinity of its allosteric modulators (Leach et al., 2013), making various patients less sensitive to pharmacological intervention. The development of compounds which can overcome these obstacles should be a focus point in future pharmacological research (please refer to Table 1 for a summary of important studies and their key findings and to Table 2 for an overview of ligand types involved).
Research in recent decades has improved our understanding of the complex mechanisms of inflammatory processes within atherosclerosis. In addition, the CANTOS trial has clearly shown that the reduction of inflammatory processes has a positive effect on the outcome of CVD. The latter is particularly true for patients with pre-existing inflammatory conditions. In the context of these inflammatory processes, the interaction and activation of immune cells plays an important role. GPCRs are a group of receptors that play a central role in controlling these immune responses, but they are ubiquitously expressed and convey both pro- and anti-inflammatory signals. In addition, many of these receptors detect different ligands, which in turn deliver diverse immune responses depending on context (acute versus chronic), tissue and cell type involved. For example, the expression of CXCR4 seems atheroprotective by retention of neutrophils in the bone marrow and by maintaining arterial endothelial integrity while simultaneously endothelial-derived CXCL12 (bona fide ligand of CXCR4) appears pro-atherogenic. Data on FPR2 are similarly contradictory; here in vivo studies in mice show both more and less plaque progression in case of deletion of FPR2. In the context of an Annexin A1 supplementation, however, protective effects of the interaction of Annexin A1 and FPR2 on myeloid cells have been described. These examples, in turn, underline the significance of a specific ligand and cell type as part of a particular immune response. It is therefore crucial, in addition to the further characterization of receptor–ligand interactions and their consequences within chronic inflammation, to not draw generalized conclusions, but focus on individual conditions. With regard to therapeutic intervention aimed at GPCR-mediated immune responses, it is therefore also crucial to improve cell-specific drug delivery approaches and to identify other potentially impacting factors such as variation of genetic or epigenetic factors, which may influence therapeutic outcomes. Eventually, improvement of CVD therapy with respect to effective but safe therapeutics does clearly point in the direction of a cell-specific treatment tailored to the individual patient. Further elucidation and understanding of the concept of biased ligands, resulting in different signaling and thus effects of the binding of distinct ligands to the same receptor, could further improve the development of tailored treatment.
EvdV, LP, MM, SG, YY, and YD drafted the manuscript and made critical revisions. CW made critical revisions.
This research was supported by the DFG (SFB1123 TP A1) to YD and CW, by the European Research Council (AdG°692511) to CW, and by the Alexander von Humboldt Foundation, a grant from the Interdisciplinary Center for Clinical Research within the Faculty of Medicine at the RWTH Aachen University, the DZHK (German Centre for Cardiovascular Research), and the BMBF (German Ministry of Education and Research) to EvdV.
The reviewer RK declared a shared affiliation, with no collaboration, with the authors EvdV, CW to the handling Editor at the time of review.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer SL declared a past co-authorship with several of the authors to the handling Editor.
Aida, K., Koishi, S., Tawata, M., and Onaya, T. (1995). Molecular cloning of a putative Ca(2+)-sensing receptor cDNA from human kidney. Biochem. Biophys. Res. Commun. 214, 524–529. doi: 10.1006/bbrc.1995.2318
Alard, J. E., Ortega-Gomez, A., Wichapong, K., Bongiovanni, D., Horckmans, M., Megens, R. T., et al. (2015). Recruitment of classical monocytes can be inhibited by disturbing heteromers of neutrophil HNP1 and platelet CCL5. Sci. Transl. Med. 7:317ra196. doi: 10.1126/scitranslmed.aad5330
Ambrosio, M., Zurn, A., and Lohse, M. J. (2011). Sensing G protein-coupled receptor activation. Neuropharmacology 60, 45–51. doi: 10.1016/j.neuropharm.2010.08.006
Arita, M., Bianchini, F., Aliberti, J., Sher, A., Chiang, N., Hong, S., et al. (2005a). Stereochemical assignment, antiinflammatory properties, and receptor for the omega-3 lipid mediator resolvin E1. J. Exp. Med. 201, 713–722. doi: 10.1084/jem.20042031
Arita, M., Yoshida, M., Hong, S., Tjonahen, E., Glickman, J. N., Petasis, N. A., et al. (2005b). Resolvin E1, an endogenous lipid mediator derived from omega-3 eicosapentaenoic acid, protects against 2,4,6-trinitrobenzene sulfonic acid-induced colitis. Proc. Natl. Acad. Sci. U.S.A. 102, 7671–7676. doi: 10.1073/pnas.0409271102
Arita, M., Ohira, T., Sun, Y. P., Elangovan, S., Chiang, N., and Serhan, C. N. (2007). Resolvin E1 selectively interacts with leukotriene B4 receptor BLT1 and ChemR23 to regulate inflammation. J. Immunol. 178, 3912–3917. doi: 10.4049/jimmunol.178.6.3912
Arthur, J. M., Lawrence, M. S., Payne, C. R., Rane, M. J., and Mcleish, K. R. (2000). The calcium-sensing receptor stimulates JNK in MDCK cells. Biochem. Biophys. Res. Commun. 275, 538–541. doi: 10.1006/bbrc.2000.3226
Artiach, G., Carracedo, M., Claria, J., Laguna-Fernandez, A., and Back, M. (2018). Opposing effects on vascular smooth muscle cell proliferation and macrophage-induced inflammation reveal a protective role for the proresolving lipid mediator receptor ChemR23 in intimal hyperplasia. Front. Pharmacol. 9:1327. doi: 10.3389/fphar.2018.01327
Balan, G., Bauman, J., Bhattacharya, S., Castrodad, M., Healy, D. R., Herr, M., et al. (2009). The discovery of novel calcium sensing receptor negative allosteric modulators. Bioorg. Med. Chem. Lett. 19, 3328–3332.
Barnea, G., Strapps, W., Herrada, G., Berman, Y., Ong, J., Kloss, B., et al. (2008). The genetic design of signaling cascades to record receptor activation. Proc. Natl. Acad. Sci. U.S.A. 105, 64–69. doi: 10.1073/pnas.0710487105
Beaulieu, J. M., Sotnikova, T. D., Marion, S., Lefkowitz, R. J., Gainetdinov, R. R., and Caron, M. G. (2005). An Akt/beta-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell 122, 261–273. doi: 10.1016/j.cell.2005.05.012
Bena, S., Brancaleone, V., Wang, J. M., Perretti, M., and Flower, R. J. (2012). Annexin A1 interaction with the FPR2/ALX receptor: identification of distinct domains and downstream associated signaling. J. Biol. Chem. 287, 24690–24697. doi: 10.1074/jbc.m112.377101
Bideak, A., Blaut, A., Hoppe, J. M., Muller, M. B., Federico, G., Eltrich, N., et al. (2018). The atypical chemokine receptor 2 limits renal inflammation and fibrosis in murine progressive immune complex glomerulonephritis. Kidney Int. 93, 826–841. doi: 10.1016/j.kint.2017.11.013
Bjarnadottir, T. K., Gloriam, D. E., Hellstrand, S. H., Kristiansson, H., Fredriksson, R., and Schioth, H. B. (2006). Comprehensive repertoire and phylogenetic analysis of the G protein-coupled receptors in human and mouse. Genomics 88, 263–273. doi: 10.1016/j.ygeno.2006.04.001
Black, J. W., and Leff, P. (1983). Operational models of pharmacological agonism. Proc. R. Soc. Lond. B Biol. Sci. 220, 141–162. doi: 10.1098/rspb.1983.0093
Blanchet, X., Cesarek, K., Brandt, J., Herwald, H., Teupser, D., Kuchenhoff, H., et al. (2014). Inflammatory role and prognostic value of platelet chemokines in acute coronary syndrome. Thromb. Haemost. 112, 1277–1287. doi: 10.1160/th14-02-0139
Bockaert, J., and Pin, J. P. (1999). Molecular tinkering of G protein-coupled receptors: an evolutionary success. EMBO J. 18, 1723–1729. doi: 10.1093/emboj/18.7.1723
Boekholdt, S. M., Hovingh, G. K., Mora, S., Arsenault, B. J., Amarenco, P., Pedersen, T. R., et al. (2014). Very low levels of atherogenic lipoproteins and the risk for cardiovascular events: a meta-analysis of statin trials. J. Am. Coll. Cardiol. 64, 485–494.
Bonavita, O., Mollica Poeta, V., Setten, E., Massara, M., and Bonecchi, R. (2016). ACKR2: an atypical chemokine receptor regulating lymphatic biology. Front. Immunol. 7:691. doi: 10.3389/fimmu.2016.00691
Bonecchi, R., and Graham, G. J. (2016). Atypical chemokine receptors and their roles in the resolution of the inflammatory response. Front. Immunol. 7:224. doi: 10.3389/fimmu.2016.00224
Brauner-Osborne, H., Wellendorph, P., and Jensen, A. A. (2007). Structure, pharmacology and therapeutic prospects of family C G-protein coupled receptors. Curr. Drug Targets 8, 169–184. doi: 10.2174/138945007779315614
Braunersreuther, V., Mach, F., and Steffens, S. (2007a). The specific role of chemokines in atherosclerosis. Thromb. Haemost. 97, 714–721. doi: 10.1160/th07-01-0036
Braunersreuther, V., Zernecke, A., Arnaud, C., Liehn, E. A., Steffens, S., Shagdarsuren, E., et al. (2007b). Ccr5 but not Ccr1 deficiency reduces development of diet-induced atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 27, 373–379. doi: 10.1161/01.atv.0000253886.44609.ae
Brown, E. M., Gamba, G., Riccardi, D., Lombardi, M., Butters, R., Kifor, O., et al. (1993). Cloning and characterization of an extracellular Ca(2+)-sensing receptor from bovine parathyroid. Nature 366, 575–580. doi: 10.1038/366575a0
Busmann, A., Walden, M., Wendland, M., Kutzleb, C., Forssmann, W. G., and John, H. (2004). A three-step purification strategy for isolation of hamster TIG2 from CHO cells: characterization of two processed endogenous forms. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 811, 217–223. doi: 10.1016/s1570-0232(04)00738-x
Cancellieri, C., Vacchini, A., Locati, M., Bonecchi, R., and Borroni, E. M. (2013). Atypical chemokine receptors: from silence to sound. Biochem. Soc. Trans. 41, 231–236. doi: 10.1042/bst20120246
Canton, J., Schlam, D., Breuer, C., Gutschow, M., Glogauer, M., and Grinstein, S. (2016). Calcium-sensing receptors signal constitutive macropinocytosis and facilitate the uptake of NOD2 ligands in macrophages. Nat. Commun. 7:11284.
Carlino, C., Trotta, E., Stabile, H., Morrone, S., Bulla, R., Soriani, A., et al. (2012). Chemerin regulates NK cell accumulation and endothelial cell morphogenesis in the decidua during early pregnancy. J. Clin. Endocrinol. Metab. 97, 3603–3612. doi: 10.1210/jc.2012-1102
Cash, J. L., Bass, M. D., Campbell, J., Barnes, M., Kubes, P., and Martin, P. (2014). Resolution mediator chemerin15 reprograms the wound microenvironment to promote repair and reduce scarring. Curr. Biol. 24:1435. doi: 10.1016/j.cub.2014.06.010
Cash, J. L., Bena, S., Headland, S. E., Mcarthur, S., Brancaleone, V., and Perretti, M. (2013). Chemerin15 inhibits neutrophil-mediated vascular inflammation and myocardial ischemia-reperfusion injury through ChemR23. EMBO Rep. 14, 999–1007. doi: 10.1038/embor.2013.138
Cash, J. L., Christian, A. R., and Greaves, D. R. (2010). Chemerin peptides promote phagocytosis in a ChemR23- and Syk-dependent manner. J. Immunol. 184, 5315–5324. doi: 10.4049/jimmunol.0903378
Chang, W., Pratt, S., Chen, T. H., Nemeth, E., Huang, Z., and Shoback, D. (1998). Coupling of calcium receptors to inositol phosphate and cyclic AMP generation in mammalian cells and Xenopus laevis oocytes and immunodetection of receptor protein by region-specific antipeptide antisera. J. Bone Miner. Res. 13, 570–580. doi: 10.1359/jbmr.1998.13.4.570
Charo, I. F., and Ransohoff, R. M. (2006). The many roles of chemokines and chemokine receptors in inflammation. N. Engl. J. Med. 354, 610–621. doi: 10.1056/nejmra052723
Chatterjee, M., Von Ungern-Sternberg, S. N., Seizer, P., Schlegel, F., Buttcher, M., Sindhu, N. A., et al. (2015). Platelet-derived CXCL12 regulates monocyte function, survival, differentiation into macrophages and foam cells through differential involvement of CXCR4-CXCR7. Cell Death Dis. 6:e1989. doi: 10.1038/cddis.2015.233
Cheng, S. X., Lightfoot, Y. L., Yang, T., Zadeh, M., Tang, L., Sahay, B., et al. (2014). Epithelial CaSR deficiency alters intestinal integrity and promotes proinflammatory immune responses. FEBS Lett. 588, 4158–4166. doi: 10.1016/j.febslet.2014.05.007
Chiang, N., Fierro, I. M., Gronert, K., and Serhan, C. N. (2000). Activation of lipoxin A(4) receptors by aspirin-triggered lipoxins and select peptides evokes ligand-specific responses in inflammation. J. Exp. Med. 191, 1197–1208. doi: 10.1084/jem.191.7.1197
Chung, S., Funakoshi, T., and Civelli, O. (2008). Orphan GPCR research. Br. J. Pharmacol. 153(Suppl. 1), S339–S346.
Cipriani, S., Francisci, D., Mencarelli, A., Renga, B., Schiaroli, E., D’amore, C., et al. (2013). Efficacy of the CCR5 antagonist maraviroc in reducing early, ritonavir-induced atherogenesis and advanced plaque progression in mice. Circulation 127, 2114–2124. doi: 10.1161/circulationaha.113.001278
Cohen, J., Pertsemlidis, A., Kotowski, I. K., Graham, R., Garcia, C. K., and Hobbs, H. H. (2005). Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat. Genet. 37, 161–165. doi: 10.1038/ng1509
Collins, C. B., Mcnamee, E. N., Wermers, J. D., Lebsack, M. D., and Rivera-Nieves, J. (2010). Chemokine decoy receptor D6 in inflammatory bowel disease (IBD) and IBD-associated colon cancer. Gut 59, 151–152. doi: 10.1136/gut.2009.192708
Combadiere, C., Potteaux, S., Rodero, M., Simon, T., Pezard, A., Esposito, B., et al. (2008). Combined inhibition of CCL2, CX3CR1, and CCR5 abrogates Ly6C(hi) and Ly6C(lo) monocytosis and almost abolishes atherosclerosis in hypercholesterolemic mice. Circulation 117, 1649–1657. doi: 10.1161/circulationaha.107.745091
Conigrave, A. D., Quinn, S. J., and Brown, E. M. (2000). L-amino acid sensing by the extracellular Ca2+-sensing receptor. Proc. Natl. Acad. Sci. U.S.A. 97, 4814–4819. doi: 10.1073/pnas.97.9.4814
Cooray, S. N., Gobbetti, T., Montero-Melendez, T., Mcarthur, S., Thompson, D., Clark, A. J., et al. (2013). Ligand-specific conformational change of the G-protein-coupled receptor ALX/FPR2 determines proresolving functional responses. Proc. Natl. Acad. Sci. U.S.A. 110, 18232–18237. doi: 10.1073/pnas.1308253110
Courbebaisse, M., Diet, C., Timsit, M. O., Mamzer, M. F., Thervet, E., Noel, L. H., et al. (2012). Effects of cinacalcet in renal transplant patients with hyperparathyroidism. Am. J. Nephrol. 35, 341–348.
Daaka, Y. (2012). S-nitrosylation-regulated GPCR signaling. Biochim. Biophys. Acta 1820, 743–751. doi: 10.1016/j.bbagen.2011.03.007
de Jong, R. J., Paulin, N., Lemnitzer, P., Viola, J. R., Winter, C., Ferraro, B., et al. (2017). Protective aptitude of annexin A1 in arterial neointima formation in atherosclerosis-prone mice-brief report. Arterioscler. Thromb. Vasc. Biol. 37, 312–315. doi: 10.1161/atvbaha.116.308744
Decaillot, F. M., Kazmi, M. A., Lin, Y., Ray-Saha, S., Sakmar, T. P., and Sachdev, P. (2011). CXCR7/CXCR4 heterodimer constitutively recruits beta-arrestin to enhance cell migration. J. Biol. Chem. 286, 32188–32197. doi: 10.1074/jbc.m111.277038
Desimine, V. L., Mccrink, K. A., Parker, B. M., Wertz, S. L., Maning, J., and Lymperopoulos, A. (2018). Biased agonism/antagonism of cardiovascular GPCRs for heart failure therapy. Int. Rev. Cell Mol. Biol. 339, 41–61. doi: 10.1016/bs.ircmb.2018.02.007
DeWire, S. M., and Violin, J. D. (2011). Biased ligands for better cardiovascular drugs: dissecting G-protein-coupled receptor pharmacology. Circ. Res. 109, 205–216. doi: 10.1161/circresaha.110.231308
Di Liberto, D., Locati, M., Caccamo, N., Vecchi, A., Meraviglia, S., Salerno, A., et al. (2008). Role of the chemokine decoy receptor D6 in balancing inflammation, immune activation, and antimicrobial resistance in Mycobacterium tuberculosis infection. J. Exp. Med. 205, 2075–2084. doi: 10.1084/jem.20070608
Diez-Fraile, A., Lammens, T., Benoit, Y., and D’herde, K. G. (2013). The calcium-sensing receptor as a regulator of cellular fate in normal and pathological conditions. Curr. Mol. Med. 13, 282–295. doi: 10.2174/1566524011313020005
Digby, G. J., Lober, R. M., Sethi, P. R., and Lambert, N. A. (2006). Some G protein heterotrimers physically dissociate in living cells. Proc. Natl. Acad. Sci. U.S.A. 103, 17789–17794. doi: 10.1073/pnas.0607116103
Döring, Y., Drechsler, M., Soehnlein, O., and Weber, C. (2015). Neutrophils in atherosclerosis: from mice to man. Arterioscler. Thromb. Vasc. Biol. 35, 288–295. doi: 10.1161/atvbaha.114.303564
Döring, Y., Drechsler, M., Wantha, S., Kemmerich, K., Lievens, D., Vijayan, S., et al. (2012). Lack of neutrophil-derived CRAMP reduces atherosclerosis in mice. Circ. Res. 110, 1052–1056. doi: 10.1161/circresaha.112.265868
Döring, Y., Noels, H., Mandl, M., Kramp, B., Neideck, C., Lievens, D., et al. (2014). Deficiency of the sialyltransferase St3Gal4 reduces Ccl5-mediated myeloid cell recruitment and arrest: short communication. Circ. Res. 114, 976–981. doi: 10.1161/circresaha.114.302426
Döring, Y., Noels, H., Van Der Vorst, E. P. C., Neideck, C., Egea, V., Drechsler, M., et al. (2017). Vascular CXCR4 limits atherosclerosis by maintaining arterial integrity: evidence from mouse and human studies. Circulation 136, 388–403. doi: 10.1161/circulationaha.117.027646
Döring, Y., Van Der Vorst, E. P. C., Duchene, J., Jansen, Y., Gencer, S., Bidzhekov, K., et al. (2019). CXCL12 derived from endothelial cells promotes atherosclerosis to drive coronary artery disease. Circulation 139, 1338–1340. doi: 10.1161/circulationaha.118.037953
Drechsler, M., De Jong, R., Rossaint, J., Viola, J. R., Leoni, G., Wang, J. M., et al. (2015). Annexin A1 counteracts chemokine-induced arterial myeloid cell recruitment. Circ. Res. 116, 827–835. doi: 10.1161/circresaha.116.305825
Duchene, J., Novitzky-Basso, I., Thiriot, A., Casanova-Acebes, M., Bianchini, M., Etheridge, S. L., et al. (2017). Atypical chemokine receptor 1 on nucleated erythroid cells regulates hematopoiesis. Nat. Immunol. 18, 753–761. doi: 10.1038/ni.3763
El Kebir, D., Gjorstrup, P., and Filep, J. G. (2012). Resolvin E1 promotes phagocytosis-induced neutrophil apoptosis and accelerates resolution of pulmonary inflammation. Proc. Natl. Acad. Sci. U.S.A. 109, 14983–14988. doi: 10.1073/pnas.1206641109
Felderbauer, P., Klein, W., Bulut, K., Ansorge, N., Dekomien, G., Werner, I., et al. (2006). Mutations in the calcium-sensing receptor: a new genetic risk factor for chronic pancreatitis? Scand. J. Gastroenterol. 41, 343–348. doi: 10.1080/00365520510024214
Feng, Y. H., Ding, Y., Ren, S., Zhou, L., Xu, C., and Karnik, S. S. (2005). Unconventional homologous internalization of the angiotensin II type-1 receptor induced by G-protein-independent signals. Hypertension 46, 419–425. doi: 10.1161/01.hyp.0000172621.68061.22
Ferraro, B., Leoni, G., Hinkel, R., Ormanns, S., Paulin, N., Ortega-Gomez, A., et al. (2019). Annexin a1 induces a pro-angiogenic macrophage phenotype to promote myocardial repair. J. Am. Coll. Cardiol. doi: 10.1016/j.jacc.2019.03.503
Filep, J. G., Sekheri, M., and El Kebir, D. (2018). Targeting formyl peptide receptors to facilitate the resolution of inflammation. Eur. J. Pharmacol. 833, 339–348. doi: 10.1016/j.ejphar.2018.06.025
Foord, S. M., Bonner, T. I., Neubig, R. R., Rosser, E. M., Pin, J. P., Davenport, A. P., et al. (2005). International union of pharmacology. XLVI. G protein-coupled receptor list. Pharmacol. Rev. 57, 279–288. doi: 10.1124/pr.57.2.5
Forsman, H., Winther, M., Gabl, M., Skovbakke, S. L., Boulay, F., Rabiet, M. J., et al. (2015). Structural changes of the ligand and of the receptor alters the receptor preference for neutrophil activating peptides starting with a formylmethionyl group. Biochim. Biophys. Acta 1853, 192–200. doi: 10.1016/j.bbamcr.2014.10.021
Fredman, G., Hellmann, J., Proto, J. D., Kuriakose, G., Colas, R. A., Dorweiler, B., et al. (2016). An imbalance between specialized pro-resolving lipid mediators and pro-inflammatory leukotrienes promotes instability of atherosclerotic plaques. Nat. Commun. 7:12859.
Fredman, G., Kamaly, N., Spolitu, S., Milton, J., Ghorpade, D., Chiasson, R., et al. (2015). Targeted nanoparticles containing the proresolving peptide Ac2-26 protect against advanced atherosclerosis in hypercholesterolemic mice. Sci. Transl. Med. 7:275ra20. doi: 10.1126/scitranslmed.aaa1065
Fredriksson, R., Lagerstrom, M. C., Lundin, L. G., and Schioth, H. B. (2003). The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol. 63, 1256–1272. doi: 10.1124/mol.63.6.1256
Gannon, A. W., Monk, H. M., and Levine, M. A. (2014). Cinacalcet monotherapy in neonatal severe hyperparathyroidism: a case study and review. J. Clin. Endocrinol. Metab. 99, 7–11. doi: 10.1210/jc.2013-2834
Gantz, I., Konda, Y., Yang, Y. K., Miller, D. E., Dierick, H. A., and Yamada, T. (1996). Molecular cloning of a novel receptor (CMKLR1) with homology to the chemotactic factor receptors. Cytogenet. Cell Genet. 74, 286–290. doi: 10.1159/000134436
Gao, L., Faibish, D., Fredman, G., Herrera, B. S., Chiang, N., Serhan, C. N., et al. (2013). Resolvin E1 and chemokine-like receptor 1 mediate bone preservation. J. Immunol. 190, 689–694. doi: 10.4049/jimmunol.1103688
Gao, X., Mi, S., Zhang, F., Gong, F., Lai, Y., Gao, F., et al. (2011). Association of chemerin mRNA expression in human epicardial adipose tissue with coronary atherosclerosis. Cardiovasc. Diabetol. 10:87. doi: 10.1186/1475-2840-10-87
Gavins, F. N., Kamal, A. M., D’amico, M., Oliani, S. M., and Perretti, M. (2005). Formyl-peptide receptor is not involved in the protection afforded by annexin 1 in murine acute myocardial infarct. FASEB J. 19, 100–102. doi: 10.1096/fj.04-2178fje
Gencer, S., van der Vorst, E. P. C., Aslani, M., Weber, C., Döring, Y., and Duchene, J. (2019). Atypical chemokine receptors in cardiovascular disease. Thromb. Haemost. 119, 534–541. doi: 10.1055/s-0038-1676988
Gilbert, J., Lekstrom-Himes, J., Donaldson, D., Lee, Y., Hu, M., Xu, J., et al. (2011). Effect of CC chemokine receptor 2 CCR2 blockade on serum C-reactive protein in individuals at atherosclerotic risk and with a single nucleotide polymorphism of the monocyte chemoattractant protein-1 promoter region. Am. J. Cardiol. 107, 906–911. doi: 10.1016/j.amjcard.2010.11.005
Gloriam, D. E., Fredriksson, R., and Schioth, H. B. (2007). The G protein-coupled receptor subset of the rat genome. BMC Genomics 8:338. doi: 10.1186/1471-2164-8-338
Goralski, K. B., Mccarthy, T. C., Hanniman, E. A., Zabel, B. A., Butcher, E. C., Parlee, S. D., et al. (2007). Chemerin, a novel adipokine that regulates adipogenesis and adipocyte metabolism. J. Biol. Chem. 282, 28175–28188. doi: 10.1074/jbc.m700793200
Goralski, K. B., and Sinal, C. J. (2009). Elucidation of chemerin and chemokine-like receptor-1 function in adipocytes by adenoviral-mediated shRNA knockdown of gene expression. Methods Enzymol. 460, 289–312. doi: 10.1016/s0076-6879(09)05214-8
Graham, G. J., Locati, M., Mantovani, A., Rot, A., and Thelen, M. (2012). The biochemistry and biology of the atypical chemokine receptors. Immunol. Lett. 145, 30–38. doi: 10.1016/j.imlet.2012.04.004
Griffith, J. W., Sokol, C. L., and Luster, A. D. (2014). Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu. Rev. Immunol. 32, 659–702. doi: 10.1146/annurev-immunol-032713-120145
Groslambert, M., and Py, B. F. (2018). Spotlight on the NLRP3 inflammasome pathway. J. Inflamm. Res. 11, 359–374. doi: 10.2147/jir.s141220
Guo, J., Li, H. Z., Wang, L. C., Zhang, W. H., Li, G. W., Xing, W. J., et al. (2012). Increased expression of calcium-sensing receptors in atherosclerosis confers hypersensitivity to acute myocardial infarction in rats. Mol. Cell. Biochem. 366, 345–354. doi: 10.1007/s11010-012-1312-0
Gutierrez-Lopez, T. Y., Orduna-Castillo, L. B., Hernandez-Vasquez, M. N., Vazquez-Prado, J., and Reyes-Cruz, G. (2018). Calcium sensing receptor activates the NLRP3 inflammasome via a chaperone-assisted degradative pathway involving Hsp70 and LC3-II. Biochem. Biophys. Res. Commun. 505, 1121–1127. doi: 10.1016/j.bbrc.2018.10.028
Hansson, G. K. (2005). Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 352, 1685–1695. doi: 10.1056/nejmra043430
Hansson, G. K., Libby, P., and Tabas, I. (2015). Inflammation and plaque vulnerability. J. Intern. Med. 278, 483–493. doi: 10.1111/joim.12406
Hasturk, H., Abdallah, R., Kantarci, A., Nguyen, D., Giordano, N., Hamilton, J., et al. (2015). Resolvin E1 (RvE1) attenuates atherosclerotic plaque formation in diet and inflammation-induced atherogenesis. Arterioscler. Thromb. Vasc. Biol. 35, 1123–1133. doi: 10.1161/atvbaha.115.305324
Hauser, A. S., Attwood, M. M., Rask-Andersen, M., Schioth, H. B., and Gloriam, D. E. (2017). Trends in GPCR drug discovery: new agents, targets and indications. Nat. Rev. Drug Discov. 16, 829–842. doi: 10.1038/nrd.2017.178
Hazell, G. G., Hindmarch, C. C., Pope, G. R., Roper, J. A., Lightman, S. L., Murphy, D., et al. (2012). G protein-coupled receptors in the hypothalamic paraventricular and supraoptic nuclei–serpentine gateways to neuroendocrine homeostasis. Front. Neuroendocrinol. 33, 45–66. doi: 10.1016/j.yfrne.2011.07.002
He, H. Q., and Ye, R. D. (2017). The formyl peptide receptors: diversity of ligands and mechanism for recognition. Molecules 22:E455.
Heller, E. A., Liu, E., Tager, A. M., Yuan, Q., Lin, A. Y., Ahluwalia, N., et al. (2006). Chemokine CXCL10 promotes atherogenesis by modulating the local balance of effector and regulatory T cells. Circulation 113, 2301–2312. doi: 10.1161/CIRCULATIONAHA.105.605121
Herova, M., Schmid, M., Gemperle, C., and Hersberger, M. (2015). ChemR23, the receptor for chemerin and resolvin E1, is expressed and functional on M1 but not on M2 macrophages. J. Immunol. 194, 2330–2337. doi: 10.4049/jimmunol.1402166
Herova, M., Schmid, M., Gemperle, C., Loretz, C., and Hersberger, M. (2014). Low dose aspirin is associated with plasma chemerin levels and may reduce adipose tissue inflammation. Atherosclerosis 235, 256–262. doi: 10.1016/j.atherosclerosis.2014.05.912
Herrera, B. S., Hasturk, H., Kantarci, A., Freire, M. O., Nguyen, O., Kansal, S., et al. (2015). Impact of resolvin E1 on murine neutrophil phagocytosis in type 2 diabetes. Infect. Immun. 83, 792–801. doi: 10.1128/iai.02444-14
Hilger, D., Masureel, M., and Kobilka, B. K. (2018). Structure and dynamics of GPCR signaling complexes. Nat. Struct. Mol. Biol. 25, 4–12. doi: 10.1038/s41594-017-0011-7
Ho, K. J., Spite, M., Owens, C. D., Lancero, H., Kroemer, A. H., Pande, R., et al. (2010). Aspirin-triggered lipoxin and resolvin E1 modulate vascular smooth muscle phenotype and correlate with peripheral atherosclerosis. Am. J. Pathol. 177, 2116–2123. doi: 10.2353/ajpath.2010.091082
Hong, S., Zhang, X., Zhang, X., Liu, W., Fu, Y., Liu, Y., et al. (2017). Role of the calcium sensing receptor in cardiomyocyte apoptosis via mitochondrial dynamics in compensatory hypertrophied myocardium of spontaneously hypertensive rat. Biochem. Biophys. Res. Commun. 487, 728–733. doi: 10.1016/j.bbrc.2017.04.126
Horuk, R. (2015). The Duffy antigen receptor for chemokines DARC/ACKR1. Front. Immunol. 6:279. doi: 10.3389/fimmu.2015.00279
Howes, R. E., Patil, A. P., Piel, F. B., Nyangiri, O. A., Kabaria, C. W., Gething, P. W., et al. (2011). The global distribution of the Duffy blood group. Nat. Commun. 2:266.
Huang, C., Handlogten, M. E., and Miller, R. T. (2002). Parallel activation of phosphatidylinositol 4-kinase and phospholipase C by the extracellular calcium-sensing receptor. J. Biol. Chem. 277, 20293–20300. doi: 10.1074/jbc.m200831200
Huang, C., Hujer, K. M., Wu, Z., and Miller, R. T. (2004). The Ca2+-sensing receptor couples to Galpha12/13 to activate phospholipase D in Madin-Darby canine kidney cells. Am. J. Physiol. Cell Physiol. 286, C22–C30.
Ikari, A., Nakajima, K., Kawano, K., and Suketa, Y. (2001). Polyvalent cation-sensing mechanism increased Na(+)-independent Mg(2+) transport in renal epithelial cells. Biochem. Biophys. Res. Commun. 287, 671–674. doi: 10.1006/bbrc.2001.5644
Jamieson, T., Cook, D. N., Nibbs, R. J., Rot, A., Nixon, C., Mclean, P., et al. (2005). The chemokine receptor D6 limits the inflammatory response in vivo. Nat. Immunol. 6, 403–411. doi: 10.1038/ni1182
Jannaway, M., Torrens, C., Warner, J. A., and Sampson, A. P. (2018). Resolvin E1, resolvin D1 and resolvin D2 inhibit constriction of rat thoracic aorta and human pulmonary artery induced by the thromboxane mimetic U46619. Br. J. Pharmacol. 175, 1100–1108. doi: 10.1111/bph.14151
Jenkins, B. D., Hire, R., Howerth, E., Monteil, M., Martini, R., and Davis, M. B. (2017). “Atypical chemokine receptor 1 (ACKR1/DARC) expressing tumors are associated with distinct recruitment of immune cells and increased pro-inflammatory chemokines [abstract],” in Proceedings of the American Association for Cancer Research Annual Meeting 2017, Washington, DC.
Johnston, B., and Butcher, E. C. (2002). Chemokines in rapid leukocyte adhesion triggering and migration. Semin. Immunol. 14, 83–92. doi: 10.1006/smim.2001.0345
Joost, P., and Methner, A. (2002). Phylogenetic analysis of 277 human G-protein-coupled receptors as a tool for the prediction of orphan receptor ligands. Genome Biol. 3:RESEARCH0063.
Kenakin, T., and Miller, L. J. (2010). Seven transmembrane receptors as shapeshifting proteins: the impact of allosteric modulation and functional selectivity on new drug discovery. Pharmacol. Rev. 62, 265–304. doi: 10.1124/pr.108.000992
Kennedy, A. J., and Davenport, A. P. (2018). International union of basic and clinical pharmacology CIII: chemerin receptors CMKLR1 (Chemerin1) and GPR1 (Chemerin2) nomenclature, pharmacology, and function. Pharmacol. Rev. 70, 174–196. doi: 10.1124/pr.116.013177
Kennedy, A. J., Yang, P., Read, C., Kuc, R. E., Yang, L., Taylor, E. J., et al. (2016). Chemerin elicits potent constrictor actions via chemokine-like receptor 1 (CMKLR1), not G-protein-coupled receptor 1 (GPR1), in human and rat vasculature. J. Am. Heart Assoc. 5:e004421.
Khan, S. M., Sleno, R., Gora, S., Zylbergold, P., Laverdure, J. P., Labbe, J. C., et al. (2013). The expanding roles of Gbetagamma subunits in G protein-coupled receptor signaling and drug action. Pharmacol. Rev. 65, 545–577. doi: 10.1124/pr.111.005603
Kiefer, L., Leiris, S., and Dodd, R. H. (2011). Novel calcium sensing receptor ligands: a patent survey. Expert Opin. Ther. Pat. 21, 681–698. doi: 10.1517/13543776.2011.568479
Kifor, O., Diaz, R., Butters, R., and Brown, E. M. (1997). The Ca2+-sensing receptor (CaR) activates phospholipases C, A2, and D in bovine parathyroid and CaR-transfected, human embryonic kidney (HEK293) cells. J. Bone Miner. Res. 12, 715–725. doi: 10.1359/jbmr.1997.12.5.715
Kim, C. H. (2004). Chemokine-chemokine receptor network in immune cell trafficking. Curr. Drug Targets Immune Endocr. Metabol. Disord. 4, 343–361. doi: 10.2174/1568008043339712
Koenen, R. R., Von Hundelshausen, P., Nesmelova, I. V., Zernecke, A., Liehn, E. A., Sarabi, A., et al. (2009). Disrupting functional interactions between platelet chemokines inhibits atherosclerosis in hyperlipidemic mice. Nat. Med. 15, 97–103. doi: 10.1038/nm.1898
Kostopoulos, C. G., Spiroglou, S. G., Varakis, J. N., Apostolakis, E., and Papadaki, H. H. (2014). Chemerin and CMKLR1 expression in human arteries and periadventitial fat: a possible role for local chemerin in atherosclerosis? BMC Cardiovasc. Disord. 14:56. doi: 10.1186/1471-2261-14-56
Kretschmer, D., Rautenberg, M., Linke, D., and Peschel, A. (2015). Peptide length and folding state govern the capacity of staphylococcal beta-type phenol-soluble modulins to activate human formyl-peptide receptors 1 or 2. J. Leukoc. Biol. 97, 689–697. doi: 10.1189/jlb.2a0514-275r
Kristiansen, K. (2004). Molecular mechanisms of ligand binding, signaling, and regulation within the superfamily of G-protein-coupled receptors: molecular modeling and mutagenesis approaches to receptor structure and function. Pharmacol. Ther. 103, 21–80. doi: 10.1016/j.pharmthera.2004.05.002
Kusters, D. H., Chatrou, M. L., Willems, B. A., De Saint-Hubert, M., Bauwens, M., Van Der Vorst, E., et al. (2015). Pharmacological treatment with annexin A1 reduces atherosclerotic plaque burden in LDLR-/- mice on western type diet. PLoS One 10:e0130484. doi: 10.1371/journal.pone.0130484
Lagerstrom, M. C., and Schioth, H. B. (2008). Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat. Rev. Drug Discov. 7, 339–357. doi: 10.1038/nrd2518
Laguna-Fernandez, A., Checa, A., Carracedo, M., Artiach, G., Petri, M. H., Baumgartner, R., et al. (2018). ERV1/ChemR23 signaling protects against atherosclerosis by modifying oxidized low-density lipoprotein uptake and phagocytosis in macrophages. Circulation 138, 1693–1705. doi: 10.1161/circulationaha.117.032801
Leach, K., Wen, A., Cook, A. E., Sexton, P. M., Conigrave, A. D., and Christopoulos, A. (2013). Impact of clinically relevant mutations on the pharmacoregulation and signaling bias of the calcium-sensing receptor by positive and negative allosteric modulators. Endocrinology 154, 1105–1116. doi: 10.1210/en.2012-1887
Lee, G. S., Subramanian, N., Kim, A. I., Aksentijevich, I., Goldbach-Mansky, R., Sacks, D. B., et al. (2012). The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature 492, 123–127. doi: 10.1038/nature11588
Lee, H. Y., Kim, S. D., Baek, S. H., Choi, J. H., and Bae, Y. S. (2013). Role of formyl peptide receptor 2 on the serum amyloid A-induced macrophage foam cell formation. Biochem. Biophys. Res. Commun. 433, 255–259. doi: 10.1016/j.bbrc.2013.03.002
Lee, H. Y., Oh, E., Kim, S. D., Seo, J. K., and Bae, Y. S. (2014). Oxidized low-density lipoprotein-induced foam cell formation is mediated by formyl peptide receptor 2. Biochem. Biophys. Res. Commun. 443, 1003–1007. doi: 10.1016/j.bbrc.2013.12.082
Lefkowitz, R. J., and Shenoy, S. K. (2005). Transduction of receptor signals by beta-arrestins. Science 308, 512–517. doi: 10.1126/science.1109237
Levoye, A., Balabanian, K., Baleux, F., Bachelerie, F., and Lagane, B. (2009). CXCR7 heterodimerizes with CXCR4 and regulates CXCL12-mediated G protein signaling. Blood 113, 6085–6093. doi: 10.1182/blood-2008-12-196618
Li, X., Zhu, M., Penfold, M. E., Koenen, R. R., Thiemann, A., Heyll, K., et al. (2014). Activation of CXCR7 limits atherosclerosis and improves hyperlipidemia by increasing cholesterol uptake in adipose tissue. Circulation 129, 1244–1253. doi: 10.1161/CIRCULATIONAHA.113.006840
Liehn, E. A., Piccinini, A. M., Koenen, R. R., Soehnlein, O., Adage, T., Fatu, R., et al. (2010). A new monocyte chemotactic protein-1/chemokine CC motif ligand-2 competitor limiting neointima formation and myocardial ischemia/reperfusion injury in mice. J. Am. Coll. Cardiol. 56, 1847–1857. doi: 10.1016/j.jacc.2010.04.066
Liu, G., Liu, Q., Shen, Y., Kong, D., Gong, Y., Tao, B., et al. (2018). Early treatment with Resolvin E1 facilitates myocardial recovery from ischaemia in mice. Br. J. Pharmacol. 175, 1205–1216. doi: 10.1111/bph.14041
Liu, L., Wang, C., Lin, Y., Xi, Y., Li, H., Shi, S., et al. (2016). Suppression of calcium-sensing receptor ameliorates cardiac hypertrophy through inhibition of autophagy. Mol. Med. Rep. 14, 111–120. doi: 10.3892/mmr.2016.5279
Lu, M., Leng, B., He, X., Zhang, Z., Wang, H., and Tang, F. (2018). Calcium sensing receptor-related pathway contributes to cardiac injury and the mechanism of astragaloside IV on cardioprotection. Front. Pharmacol. 9:1163. doi: 10.3389/fphar.2018.01163
Ma, W., Liu, Y., Ellison, N., and Shen, J. (2013). Induction of C-X-C chemokine receptor type 7 (CXCR7) switches stromal cell-derived factor-1 (SDF-1) signaling and phagocytic activity in macrophages linked to atherosclerosis. J. Biol. Chem. 288, 15481–15494. doi: 10.1074/jbc.m112.445510
Maggi, P., Bruno, G., Perilli, F., Saracino, A., Volpe, A., Santoro, C., et al. (2017). Effects of therapy with maraviroc on the carotid intima media thickness in HIV-1/HCV Co-infected patients. In Vivo 31, 125–131.
Majmudar, M. D., Keliher, E. J., Heidt, T., Leuschner, F., Truelove, J., Sena, B. F., et al. (2013). Monocyte-directed RNAi targeting CCR2 improves infarct healing in atherosclerosis-prone mice. Circulation 127, 2038–2046. doi: 10.1161/circulationaha.112.000116
Malecki, R., Fiodorenko-Dumas, Z., Jakobsche-Policht, U., Malodobra, M., and Adamiec, R. (2013). Altered monocyte calcium-sensing receptor expression in patients with type 2 diabetes mellitus and atherosclerosis. J. Physiol. Pharmacol. 64, 521–527.
Marinissen, M. J., and Gutkind, J. S. (2001). G-protein-coupled receptors and signaling networks: emerging paradigms. Trends Pharmacol. Sci. 22, 368–376. doi: 10.1016/s0165-6147(00)01678-3
Martinez de la Torre, Y., Locati, M., Buracchi, C., Dupor, J., Cook, D. N., Bonecchi, R., et al. (2005). Increased inflammation in mice deficient for the chemokine decoy receptor D6. Eur. J. Immunol. 35, 1342–1346. doi: 10.1002/eji.200526114
Massara, M., Bonavita, O., Mantovani, A., Locati, M., and Bonecchi, R. (2016). Atypical chemokine receptors in cancer: friends or foes? J. Leukoc. Biol. 99, 927–933. doi: 10.1189/jlb.3mr0915-431rr
Massara, M., Bonavita, O., Savino, B., Caronni, N., Mollica Poeta, V., Sironi, M., et al. (2018). ACKR2 in hematopoietic precursors as a checkpoint of neutrophil release and anti-metastatic activity. Nat. Commun. 9:676.
Maudsley, S., Martin, B., and Luttrell, L. M. (2005). The origins of diversity and specificity in g protein-coupled receptor signaling. J. Pharmacol. Exp. Ther. 314, 485–494. doi: 10.1124/jpet.105.083121
Mayer, K., Sommer, N., Hache, K., Hecker, A., Reiche, S., Schneck, E., et al. (2019). Resolvin E1 improves mitochondrial function in human alveolar epithelial cells during severe inflammation. Lipids 54, 53–65. doi: 10.1002/lipd.12119
McLarnon, S. J., and Riccardi, D. (2002). Physiological and pharmacological agonists of the extracellular Ca2+-sensing receptor. Eur. J. Pharmacol. 447, 271–278. doi: 10.1016/s0014-2999(02)01849-6
Meder, W., Wendland, M., Busmann, A., Kutzleb, C., Spodsberg, N., John, H., et al. (2003). Characterization of human circulating TIG2 as a ligand for the orphan receptor ChemR23. FEBS Lett. 555, 495–499. doi: 10.1016/s0014-5793(03)01312-7
Mehta, N. N., Li, M., William, D., Khera, A. V., Derohannessian, S., Qu, L., et al. (2011). The novel atherosclerosis locus at 10q11 regulates plasma CXCL12 levels. Eur. Heart J. 32, 963–971. doi: 10.1093/eurheartj/ehr091
Merckelbach, S., Van Der Vorst, E. P. C., Kallmayer, M., Rischpler, C., Burgkart, R., Döring, Y., et al. (2018). Expression and cellular localization of CXCR4 and CXCL12 in human carotid atherosclerotic plaques. Thromb. Haemost. 118, 195–206. doi: 10.1160/th17-04-0271
Michel, M. C., and Charlton, S. J. (2018). Biased agonism in drug discovery-is it too soon to choose a path? Mol. Pharmacol. 93, 259–265. doi: 10.1124/mol.117.110890
Milligan, G., and Kostenis, E. (2006). Heterotrimeric G-proteins: a short history. Br. J. Pharmacol. 147(Suppl. 1), S46–S55.
Moore, K. J., and Tabas, I. (2011). Macrophages in the pathogenesis of atherosclerosis. Cell 145, 341–355. doi: 10.1016/j.cell.2011.04.005
Muddana, V., Lamb, J., Greer, J. B., Elinoff, B., Hawes, R. H., Cotton, P. B., et al. (2008). Association between calcium sensing receptor gene polymorphisms and chronic pancreatitis in a US population: role of serine protease inhibitor Kazal 1type and alcohol. World J. Gastroenterol. 14, 4486–4491.
Nagpal, S., Patel, S., Jacobe, H., Disepio, D., Ghosn, C., Malhotra, M., et al. (1997). Tazarotene-induced gene 2 (TIG2), a novel retinoid-responsive gene in skin. J. Invest. Dermatol. 109, 91–95. doi: 10.1111/1523-1747.ep12276660
Nemeth, E. F., Delmar, E. G., Heaton, W. L., Miller, M. A., Lambert, L. D., Conklin, R. L., et al. (2001). Calcilytic compounds: potent and selective Ca2+ receptor antagonists that stimulate secretion of parathyroid hormone. J. Pharmacol. Exp. Ther. 299, 323–331.
Nemeth, E. F., Heaton, W. H., Miller, M., Fox, J., Balandrin, M. F., Van Wagenen, B. C., et al. (2004). Pharmacodynamics of the type II calcimimetic compound cinacalcet HCl. J. Pharmacol. Exp. Ther. 308, 627–635. doi: 10.1124/jpet.103.057273
Nemeth, E. F., Steffey, M. E., Hammerland, L. G., Hung, B. C., Van Wagenen, B. C., Delmar, E. G., et al. (1998). Calcimimetics with potent and selective activity on the parathyroid calcium receptor. Proc. Natl. Acad. Sci. U.S.A. 95, 4040–4045. doi: 10.1073/pnas.95.7.4040
Neves, K. B., Nguyen Dinh, Cat, A., Lopes, R. A., Rios, F. J., Anagnostopoulou, A., et al. (2015). Chemerin regulates crosstalk between adipocytes and vascular cells through Nox. Hypertension 66, 657–666. doi: 10.1161/hypertensionaha.115.05616
Noels, H., Weber, C., and Koenen, R. R. (2019). Chemokines as therapeutic targets in cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 39, 583–592. doi: 10.1161/atvbaha.118.312037
Noels, H., Zhou, B., Tilstam, P. V., Theelen, W., Li, X., Pawig, L., et al. (2014). Deficiency of endothelial CXCR4 reduces reendothelialization and enhances neointimal hyperplasia after vascular injury in atherosclerosis-prone mice. Arterioscler. Thromb. Vasc. Biol. 34, 1209–1220. doi: 10.1161/atvbaha.113.302878
Novitzky-Basso, I., and Rot, A. (2012). Duffy antigen receptor for chemokines and its involvement in patterning and control of inflammatory chemokines. Front. Immunol. 3:266. doi: 10.3389/fimmu.2012.00266
Okopien, B., Buldak, L., and Boldys, A. (2016). Current and future trends in the lipid lowering therapy. Pharmacol. Rep. 68, 737–747. doi: 10.1016/j.pharep.2016.03.016
Olszak, I. T., Poznansky, M. C., Evans, R. H., Olson, D., Kos, C., Pollak, M. R., et al. (2000). Extracellular calcium elicits a chemokinetic response from monocytes in vitro and in vivo. J. Clin. Invest. 105, 1299–1305. doi: 10.1172/jci9799
Ortega-Gomez, A., Salvermoser, M., Rossaint, J., Pick, R., Brauner, J., Lemnitzer, P., et al. (2016). Cathepsin G controls arterial but not venular myeloid cell recruitment. Circulation 134, 1176–1188. doi: 10.1161/circulationaha.116.024790
Paccou, J., Boudot, C., Mary, A., Kamel, S., Drueke, T. B., Fardellone, P., et al. (2013). Determination and modulation of total and surface calcium-sensing receptor expression in monocytes in vivo and in vitro. PLoS One 8:e74800. doi: 10.1371/journal.pone.0074800
Parlee, S. D., Ernst, M. C., Muruganandan, S., Sinal, C. J., and Goralski, K. B. (2010). Serum chemerin levels vary with time of day and are modified by obesity and tumor necrosis factor-{alpha}. Endocrinology 151, 2590–2602. doi: 10.1210/en.2009-0794
Pashover-Schallinger, E., Aswad, M., Schif-Zuck, S., Shapiro, H., Singer, P., and Ariel, A. (2012). The atypical chemokine receptor D6 controls macrophage efferocytosis and cytokine secretion during the resolution of inflammation. FASEB J. 26, 3891–3900. doi: 10.1096/fj.11-194894
Patel, M., Mcinnes, I. B., and Graham, G. (2009). Atypical chemokine receptors in inflammatory disease. Curr. Mol. Med. 9, 86–93. doi: 10.2174/156652409787314480
Petrel, C., Kessler, A., Maslah, F., Dauban, P., Dodd, R. H., Rognan, D., et al. (2003). Modeling and mutagenesis of the binding site of Calhex 231, a novel negative allosteric modulator of the extracellular Ca(2+)-sensing receptor. J. Biol. Chem. 278, 49487–49494. doi: 10.1074/jbc.m308010200
Petri, M. H., Laguna-Fernandez, A., Gonzalez-Diez, M., Paulsson-Berne, G., Hansson, G. K., and Back, M. (2015). The role of the FPR2/ALX receptor in atherosclerosis development and plaque stability. Cardiovasc. Res. 105, 65–74. doi: 10.1093/cvr/cvu224
Pirault, J., and Back, M. (2018). Lipoxin and resolvin receptors transducing the resolution of inflammation in cardiovascular disease. Front. Pharmacol. 9:1273. doi: 10.3389/fphar.2018.01273
Preiss, D., Seshasai, S. R., Welsh, P., Murphy, S. A., Ho, J. E., Waters, D. D., et al. (2011). Risk of incident diabetes with intensive-dose compared with moderate-dose statin therapy: a meta-analysis. JAMA 305, 2556–2564.
Quinn, S. J., Ye, C. P., Diaz, R., Kifor, O., Bai, M., Vassilev, P., et al. (1997). The Ca2+-sensing receptor: a target for polyamines. Am. J. Physiol. 273, C1315–C1323.
Riccardi, D., Park, J., Lee, W. S., Gamba, G., Brown, E. M., and Hebert, S. C. (1995). Cloning and functional expression of a rat kidney extracellular calcium/polyvalent cation-sensing receptor. Proc. Natl. Acad. Sci. U.S.A. 92, 131–135. doi: 10.1073/pnas.92.1.131
Ridker, P. M., Everett, B. M., Thuren, T., Macfadyen, J. G., Chang, W. H., Ballantyne, C., et al. (2017). Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 377, 1119–1131.
Ridker, P. M., Howard, C. P., Walter, V., Everett, B., Libby, P., Hensen, J., et al. (2012). Effects of interleukin-1beta inhibition with canakinumab on hemoglobin A1c, lipids, C-reactive protein, interleukin-6, and fibrinogen: a phase IIb randomized, placebo-controlled trial. Circulation 126, 2739–2748. doi: 10.1161/circulationaha.112.122556
Ridker, P. M., and Luscher, T. F. (2014). Anti-inflammatory therapies for cardiovascular disease. Eur. Heart J. 35, 1782–1791. doi: 10.1093/eurheartj/ehu203
Robinson, J. G., Farnier, M., Krempf, M., Bergeron, J., Luc, G., Averna, M., et al. (2015). Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N. Engl. J. Med. 372, 1489–1499. doi: 10.1056/nejmoa1501031
Roh, S. G., Song, S. H., Choi, K. C., Katoh, K., Wittamer, V., Parmentier, M., et al. (2007). Chemerin–a new adipokine that modulates adipogenesis via its own receptor. Biochem. Biophys. Res. Commun. 362, 1013–1018. doi: 10.1016/j.bbrc.2007.08.104
Rosenbaum, D. M., Rasmussen, S. G., and Kobilka, B. K. (2009). The structure and function of G-protein-coupled receptors. Nature 459, 356–363.
Rossol, M., Pierer, M., Raulien, N., Quandt, D., Meusch, U., Rothe, K., et al. (2012). Extracellular Ca2+ is a danger signal activating the NLRP3 inflammasome through G protein-coupled calcium sensing receptors. Nat. Commun. 3:1329.
Rourke, J. L., Dranse, H. J., and Sinal, C. J. (2013). Towards an integrative approach to understanding the role of chemerin in human health and disease. Obes. Rev. 14, 245–262. doi: 10.1111/obr.12009
Rourke, J. L., Dranse, H. J., and Sinal, C. J. (2015). CMKLR1 and GPR1 mediate chemerin signaling through the RhoA/ROCK pathway. Mol. Cell. Endocrinol. 417, 36–51. doi: 10.1016/j.mce.2015.09.002
Ruat, M., Snowman, A. M., Hester, L. D., and Snyder, S. H. (1996). Cloned and expressed rat Ca2+-sensing receptor. J. Biol. Chem. 271, 5972–5975. doi: 10.1074/jbc.271.11.5972
Rundle, C. H., Mohan, S., and Edderkaoui, B. (2013). Duffy antigen receptor for chemokines regulates post-fracture inflammation. PLoS One 8:e77362. doi: 10.1371/journal.pone.0077362
Saederup, N., Chan, L., Lira, S. A., and Charo, I. F. (2008). Fractalkine deficiency markedly reduces macrophage accumulation and atherosclerotic lesion formation in CCR2-/- mice: evidence for independent chemokine functions in atherogenesis. Circulation 117, 1642–1648. doi: 10.1161/circulationaha.107.743872
Samson, M., Edinger, A. L., Stordeur, P., Rucker, J., Verhasselt, V., Sharron, M., et al. (1998). ChemR23, a putative chemoattractant receptor, is expressed in monocyte-derived dendritic cells and macrophages and is a coreceptor for SIV and some primary HIV-1 strains. Eur. J. Immunol. 28, 1689–1700. doi: 10.1002/(sici)1521-4141(199805)28:05<1689::aid-immu1689>3.0.co;2-i
Sanchez-Martin, L., Estecha, A., Samaniego, R., Sanchez-Ramon, S., Vega, M. A., and Sanchez-Mateos, P. (2011). The chemokine CXCL12 regulates monocyte-macrophage differentiation and RUNX3 expression. Blood 117, 88–97. doi: 10.1182/blood-2009-12-258186
Savino, B., Castor, M. G., Caronni, N., Sarukhan, A., Anselmo, A., Buracchi, C., et al. (2012). Control of murine Ly6C(high) monocyte traffic and immunosuppressive activities by atypical chemokine receptor D6. Blood 119, 5250–5260. doi: 10.1182/blood-2011-10-388082
Schumski, A., Winter, C., Döring, Y., and Soehnlein, O. (2018). The ins and outs of myeloid cells in atherosclerosis. J. Innate Immun. 10, 479–486. doi: 10.1159/000488091
Schwab, J. M., Chiang, N., Arita, M., and Serhan, C. N. (2007). Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature 447, 869–874. doi: 10.1038/nature05877
Simon, M. I., Strathmann, M. P., and Gautam, N. (1991). Diversity of G proteins in signal transduction. Science 252, 802–808. doi: 10.1126/science.1902986
Sjaarda, J., Gerstein, H., Chong, M., Yusuf, S., Meyre, D., Anand, S. S., et al. (2018). Blood CSF1 and CXCL12 as causal mediators of coronary artery disease. J. Am. Coll. Cardiol. 72, 300–310. doi: 10.1016/j.jacc.2018.04.067
Smrcka, A. V. (2008). G protein betagamma subunits: central mediators of G protein-coupled receptor signaling. Cell. Mol. Life Sci. 65, 2191–2214. doi: 10.1007/s00018-008-8006-5
Soehnlein, O., Drechsler, M., Döring, Y., Lievens, D., Hartwig, H., Kemmerich, K., et al. (2013). Distinct functions of chemokine receptor axes in the atherogenic mobilization and recruitment of classical monocytes. EMBO Mol. Med. 5, 471–481. doi: 10.1002/emmm.201201717
Spiroglou, S. G., Kostopoulos, C. G., Varakis, J. N., and Papadaki, H. H. (2010). Adipokines in periaortic and epicardial adipose tissue: differential expression and relation to atherosclerosis. J. Atheroscler. Thromb. 17, 115–130. doi: 10.5551/jat.1735
Sriram, K., and Insel, P. A. (2018). G protein-coupled receptors as targets for approved drugs: how many targets and how many drugs? Mol. Pharmacol. 93, 251–258. doi: 10.1124/mol.117.111062
Stephenson, R. P. (1956). A modification of receptor theory. Br. J. Pharmacol. Chemother. 11, 379–393. doi: 10.1111/j.1476-5381.1956.tb00006.x
Stone, N. J., and Lloyd-Jones, D. M. (2015). Lowering LDL cholesterol is good, but how and in whom? N. Engl. J. Med. 372, 1564–1565. doi: 10.1056/nejme1502192
Strowig, T., Henao-Mejia, J., Elinav, E., and Flavell, R. (2012). Inflammasomes in health and disease. Nature 481, 278–286. doi: 10.1038/nature10759
Sun, R., Zhang, W., Zhong, H., Wang, L., Tang, N., Liu, Y., et al. (2018). Calcimimetic R568 reduced the blood pressure and improved aortic remodeling in spontaneously hypertensive rats by inhibiting local renin-angiotensin system activity. Exp. Ther. Med. 16, 4089–4099. doi: 10.3892/etm.2018.6734
Swirski, F. K., Libby, P., Aikawa, E., Alcaide, P., Luscinskas, F. W., Weissleder, R., et al. (2007). Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J. Clin. Invest. 117, 195–205. doi: 10.1172/jci29950
Tacke, F., Alvarez, D., Kaplan, T. J., Jakubzick, C., Spanbroek, R., Llodra, J., et al. (2007). Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J. Clin. Invest. 117, 185–194. doi: 10.1172/jci28549
Tang, T., Gong, T., Jiang, W., and Zhou, R. (2018). GPCRs in NLRP3 inflammasome activation, regulation, and therapeutics. Trends Pharmacol. Sci. 39, 798–811. doi: 10.1016/j.tips.2018.07.002
Trzaskowski, B., Latek, D., Yuan, S., Ghoshdastider, U., Debinski, A., and Filipek, S. (2012). Action of molecular switches in GPCRs–theoretical and experimental studies. Curr. Med. Chem. 19, 1090–1109. doi: 10.2174/092986712799320556
Ulvmar, M. H., Hub, E., and Rot, A. (2011). Atypical chemokine receptors. Exp. Cell Res. 317, 556–568. doi: 10.1016/j.yexcr.2011.01.012
Vajen, T., Koenen, R. R., Werner, I., Staudt, M., Projahn, D., Curaj, A., et al. (2018). Blocking CCL5-CXCL4 heteromerization preserves heart function after myocardial infarction by attenuating leukocyte recruitment and NETosis. Sci. Rep. 8:10647.
van der Vorst, E. P., Döring, Y., and Weber, C. (2015). Chemokines and their receptors in Atherosclerosis. J. Mol. Med. 93, 963–971. doi: 10.1007/s00109-015-1317-8
van der Vorst, E. P. C., Mandl, M., Müller, M., Neideck, C., Jansen, Y., Hristov, M., et al. (2019). Hematopoietic ChemR23 (Chemerin Receptor 23) fuels atherosclerosis by sustaining an M1 macrophage-phenotype and guidance of plasmacytoid dendritic cells to murine lesions-brief report. Arterioscler. Thromb. Vasc. Biol. 39, 685–693. doi: 10.1161/ATVBAHA.119.312386
van Wanrooij, E. J., de Jager, S. C., van Es, T., de Vos, P., Birch, H. L., Owen, D. A., et al. (2008). CXCR3 antagonist NBI-74330 attenuates atherosclerotic plaque formation in LDL receptor-deficient mice. Arterioscler. Thromb. Vasc. Biol. 28, 251–257. doi: 10.1161/ATVBAHA.107.147827
Vassilatis, D. K., Hohmann, J. G., Zeng, H., Li, F., Ranchalis, J. E., Mortrud, M. T., et al. (2003). The G protein-coupled receptor repertoires of human and mouse. Proc. Natl. Acad. Sci. U.S.A. 100, 4903–4908.
Veillard, N. R., Kwak, B., Pelli, G., Mulhaupt, F., James, R. W., Proudfoot, A. E., et al. (2004). Antagonism of RANTES receptors reduces atherosclerotic plaque formation in mice. Circ. Res. 94, 253–261. doi: 10.1161/01.res.0000109793.17591.4e
Veillard, N. R., Steffens, S., Pelli, G., Lu, B., Kwak, B. R., Gerard, C., et al. (2005). Differential influence of chemokine receptors CCR2 and CXCR3 in development of atherosclerosis in vivo. Circulation 112, 870–878. doi: 10.1161/CIRCULATIONAHA.104.520718
Vermi, W., Riboldi, E., Wittamer, V., Gentili, F., Luini, W., Marrelli, S., et al. (2005). Role of ChemR23 in directing the migration of myeloid and plasmacytoid dendritic cells to lymphoid organs and inflamed skin. J. Exp. Med. 201, 509–515. doi: 10.1084/jem.20041310
Vetrano, S., Borroni, E. M., Sarukhan, A., Savino, B., Bonecchi, R., Correale, C., et al. (2010). The lymphatic system controls intestinal inflammation and inflammation-associated Colon Cancer through the chemokine decoy receptor D6. Gut 59, 197–206. doi: 10.1136/gut.2009.183772
Viola, J. R., Lemnitzer, P., Jansen, Y., Csaba, G., Winter, C., Neideck, C., et al. (2016). Resolving lipid mediators maresin 1 and resolvin D2 prevent atheroprogression in mice. Circ. Res. 119, 1030–1038. doi: 10.1161/circresaha.116.309492
von Hundelshausen, P., Agten, S. M., Eckardt, V., Blanchet, X., Schmitt, M. M., Ippel, H., et al. (2017). Chemokine interactome mapping enables tailored intervention in acute and chronic inflammation. Sci. Transl. Med. 9:eaah6650. doi: 10.1126/scitranslmed.aah6650
von Hundelshausen, P., Koenen, R. R., Sack, M., Mause, S. F., Adriaens, W., Proudfoot, A. E., et al. (2005). Heterophilic interactions of platelet factor 4 and RANTES promote monocyte arrest on endothelium. Blood 105, 924–930. doi: 10.1182/blood-2004-06-2475
von Hundelshausen, P., Weber, K. S., Huo, Y., Proudfoot, A. E., Nelson, P. J., Ley, K., et al. (2001). RANTES deposition by platelets triggers monocyte arrest on inflamed and atherosclerotic endothelium. Circulation 103, 1772–1777. doi: 10.1161/01.cir.103.13.1772
Wacker, D., Stevens, R. C., and Roth, B. L. (2017). How ligands illuminate GPCR molecular pharmacology. Cell 170, 414–427. doi: 10.1016/j.cell.2017.07.009
Wan, W., Liu, Q., Lionakis, M. S., Marino, A. P., Anderson, S. A., Swamydas, M., et al. (2015). Atypical chemokine receptor 1 deficiency reduces atherogenesis in ApoE-knockout mice. Cardiovasc. Res. 106, 478–487. doi: 10.1093/cvr/cvv124
Wang, C., Chen, W., and Shen, J. (2018). CXCR7 targeting and its major disease relevance. Front. Pharmacol. 9:641. doi: 10.3389/fphar.2018.00641
Wantha, S., Alard, J. E., Megens, R. T., Van Der Does, A. M., Doring, Y., Drechsler, M., et al. (2013). Neutrophil-derived cathelicidin promotes adhesion of classical monocytes. Circ. Res. 112, 792–801. doi: 10.1161/circresaha.112.300666
Watts, S. W., Darios, E. S., Mullick, A. E., Garver, H., Saunders, T. L., Hughes, E. D., et al. (2018). The chemerin knockout rat reveals chemerin dependence in female, but not male, experimental hypertension. FASEB J. doi: 10.1096/fj.201800479 [Epub ahead of print].
Weber, C., and Noels, H. (2011). Atherosclerosis: current pathogenesis and therapeutic options. Nat. Med. 17, 1410–1422. doi: 10.1038/nm.2538
Wells, T. N., Power, C. A., Shaw, J. P., and Proudfoot, A. E. (2006). Chemokine blockers–therapeutics in the making? Trends Pharmacol. Sci. 27, 41–47. doi: 10.1016/j.tips.2005.11.001
Whitehead, G. S., Wang, T., Degraff, L. M., Card, J. W., Lira, S. A., Graham, G. J., et al. (2007). The chemokine receptor D6 has opposing effects on allergic inflammation and airway reactivity. Am. J. Respir. Crit. Care Med. 175, 243–249. doi: 10.1164/rccm.200606-839oc
Winter, C., Silvestre-Roig, C., Ortega-Gomez, A., Lemnitzer, P., Poelman, H., Schumski, A., et al. (2018). Chrono-pharmacological targeting of the CCL2-CCR2 axis ameliorates atherosclerosis. Cell Metab. 28, 175–182.e5. doi: 10.1016/j.cmet.2018.05.002
Wittamer, V., Bondue, B., Guillabert, A., Vassart, G., Parmentier, M., and Communi, D. (2005). Neutrophil-mediated maturation of chemerin: a link between innate and adaptive immunity. J. Immunol. 175, 487–493. doi: 10.4049/jimmunol.175.1.487
Wittamer, V., Franssen, J. D., Vulcano, M., Mirjolet, J. F., Le Poul, E., Migeotte, I., et al. (2003). Specific recruitment of antigen-presenting cells by chemerin, a novel processed ligand from human inflammatory fluids. J. Exp. Med. 198, 977–985. doi: 10.1084/jem.20030382
Wittamer, V., Gregoire, F., Robberecht, P., Vassart, G., Communi, D., and Parmentier, M. (2004). The C-terminal nonapeptide of mature chemerin activates the chemerin receptor with low nanomolar potency. J. Biol. Chem. 279, 9956–9962. doi: 10.1074/jbc.m313016200
Woehler, A., and Ponimaskin, E. G. (2009). G protein–mediated signaling: same receptor, multiple effectors. Curr. Mol. Pharmacol. 2, 237–248. doi: 10.2174/1874467210902030237
Xi, Y. H., Li, H. Z., Zhang, W. H., Wang, L. N., Zhang, L., Lin, Y., et al. (2010). The functional expression of calcium-sensing receptor in the differentiated THP-1 cells. Mol. Cell. Biochem. 342, 233–240. doi: 10.1007/s11010-010-0489-3
Yamawaki, H., Kameshima, S., Usui, T., Okada, M., and Hara, Y. (2012). A novel adipocytokine, chemerin exerts anti-inflammatory roles in human vascular endothelial cells. Biochem. Biophys. Res. Commun. 423, 152–157. doi: 10.1016/j.bbrc.2012.05.103
Yang, M., Yang, G., Dong, J., Liu, Y., Zong, H., Liu, H., et al. (2010). Elevated plasma levels of chemerin in newly diagnosed type 2 diabetes mellitus with hypertension. J. Investig. Med. 58, 883–886. doi: 10.2310/jim.0b013e3181ec5db2
Yarova, P. L., Stewart, A. L., Sathish, V., Britt, R. D. Jr., Thompson, M. A., et al. (2015). Calcium-sensing receptor antagonists abrogate airway hyperresponsiveness and inflammation in allergic asthma. Sci. Transl. Med. 7:284ra60.
Ye, R. D., Boulay, F., Wang, J. M., Dahlgren, C., Gerard, C., Parmentier, M., et al. (2009). International union of basic and clinical pharmacology. LXXIII. Nomenclature for the formyl peptide receptor (FPR) family. Pharmacol. Rev. 61, 119–161. doi: 10.1124/pr.109.001578
Yin, B., Hou, X.-W., and Lu, M.-L. (2019). Astragaloside IV attenuates myocardial ischemia/reperfusion injury in rats via inhibition of calcium-sensing receptor-mediated apoptotic signaling pathways. Acta. Pharmacol. Sin. 40, 599–607. doi: 10.1038/s41401-018-0082-y
Zernecke, A., Bot, I., Djalali-Talab, Y., Shagdarsuren, E., Bidzhekov, K., Meiler, S., et al. (2008). Protective role of CXC receptor 4/CXC ligand 12 unveils the importance of neutrophils in atherosclerosis. Circ. Res. 102, 209–217. doi: 10.1161/circresaha.107.160697
Zhang, R., Lahens, N. F., Ballance, H. I., Hughes, M. E., and Hogenesch, J. B. (2014). A circadian gene expression atlas in mammals: implications for biology and medicine. Proc. Natl. Acad. Sci. U.S.A. 111, 16219–16224. doi: 10.1073/pnas.1408886111
Zhang, X. L., Zhu, Q. Q., Zhu, L., Chen, J. Z., Chen, Q. H., Li, G. N., et al. (2015). Safety and efficacy of anti-PCSK9 antibodies: a meta-analysis of 25 randomized, controlled trials. BMC Med. 13:123. doi: 10.1186/s12916-015-0358-8
Zheng, H., Loh, H. H., and Law, P. Y. (2010). Agonist-selective signaling of G protein-coupled receptor: mechanisms and implications. IUBMB Life 62, 112–119.
Keywords: G-protein coupled receptors, myeloid cells, cardiovascular disease, atherosclerosis, therapy
Citation: van der Vorst EPC, Peters LJF, Müller M, Gencer S, Yan Y, Weber C and Döring Y (2019) G-Protein Coupled Receptor Targeting on Myeloid Cells in Atherosclerosis. Front. Pharmacol. 10:531. doi: 10.3389/fphar.2019.00531
Received: 31 January 2019; Accepted: 29 April 2019;
Published: 22 May 2019.
Edited by:
Caroline Gaucher, University of Lorraine, FranceReviewed by:
Rory R. Koenen, Maastricht University, NetherlandsCopyright © 2019 van der Vorst, Peters, Müller, Gencer, Yan, Weber and Döring. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Emiel P. C. van der Vorst, RW1pZWwuVmFuX2Rlcl9Wb3JzdEBtZWQudW5pLW11ZW5jaGVuLmRl Yvonne Döring, eWRvZXJpbmdAbWVkLmxtdS5kZQ==; eXZvbm5lLmRvZXJpbmdAbWVkLnVuaS1tdWVuY2hlbi5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.