 Yi Wang1
Yi Wang1 Xiuli Wang1
Xiuli Wang1 Selena Chen2
Selena Chen2 Xiaoyu Tian1
Xiaoyu Tian1 Lulu Zhang1
Lulu Zhang1 Yaqian Huang1
Yaqian Huang1 Chaoshu Tang3,4
Chaoshu Tang3,4 Junbao Du1
Junbao Du1 Hongfang Jin1*
Hongfang Jin1*- 1Department of Pediatrics, Peking University First Hospital, Beijing, China
- 2Division of Biological Sciences, University of California, San Diego, La Jolla, CA, United States
- 3Department of Physiology and Pathophysiology, Peking University Health Science Center, Beijing, China
- 4Key Laboratory of Molecular Cardiology, Ministry of Education, Beijing, China
Sulfur dioxide (SO2) is a colorless and irritating gas. Recent studies indicate that SO2 acts as the gas signal molecule and inhibits vascular smooth muscle cell (VSMC) proliferation. Cell proliferation depends on intracellular pH (pHi). Transmembrane cystein mutation of Na+- independent Cl-/HCO3- exchanger (anion exchanger, AE) affects pHi. However, whether SO2 inhibits VSMC proliferation by reducing pHi is still unknown. Here, we investigated whether SO2 reduced pHi to inhibit the proliferation of VSMCs and explore its molecular mechanisms. Within a range of 50–200 μM, SO2 was found to lower the pHi in VSMCs. Concurrently, NH4Cl pre-perfusion showed that SO2 significantly activated AE, whereas the AE inhibitor 4,4′-diisothiocyanatostilbene- 2,20-disulfonic acid (DIDS) significantly attenuated the effect of SO2 on pHi in VSMCs. While 200 μM SO2 sulphenylated AE2, while dithiothreitol (DTT) blocked the sulphenylation of AE2 and subsequent AE activation by SO2, thereby restoring the pHi in VSMCs. Furthermore, DIDS pretreatment eliminated SO2-induced inhibition of PDGF-BB-stimulated VSMC proliferation. We report for the first time that SO2 inhibits VSMC proliferation in part by direct activation of the AE via posttranslational sulphenylation and induction of intracellular acidification.
Introduction
Aberrant proliferation of vascular smooth muscle cells (VSMCs) contributes to the pathological change of vascular diseases such as hypertension, diabetic angiopathy and atherosclerosis (Owens et al., 2004; Chistiakov et al., 2015; Bennett et al., 2016). Previous studies have shown that the intracellular pH (pHi) is an important factor involved in the regulation of cell proliferation. Additionally, cell proliferation of multiple species is dependent on the pHi. Mitogen stimulation promotes cell cycle progression and ultimately proliferation. DNA, RNA and protein synthesis all require an alkaline pHi (Schreiber, 2005; Flinck et al., 2018).
To maintain pHi homeostasis, cells utilize ionophores on the membrane to regulate the pHi within a narrow physiological range (Cardone et al., 2005; Boron et al., 2009; Casey et al., 2010). Those ionophores include channels, pumps, exchangers and cotransporters, all of which synergistically regulate the influx and outflux of H+/HCO3- ions (Concepcion et al., 2013; Chen et al., 2018). Among them, the Na+-independent and electroneutral Cl-/HCO3- exchanger (anion exchanger, AE) is encoded by the SLC4 gene family, including SLC4A1/AE1, SLC4A2/AE2, and SLC4A3/AE3, which help regulate the pHi, cell volume and membrane potential of various cell types (Alper, 2009; Liu et al., 2015). Genome-wide association analysis showed that AE2 exon deletion resulted in a loss of function of AE2 in osteoclast and cell alkalization, resulting in bone resorption lacunae disorder, the genetic cause of Angoras cattle and mouse osteopetrosis (Meyers et al., 2010; Coury et al., 2013). Transforming growth factor beta 1 promotes fibroblast cell membrane AE2 expression and HCO3- excretion, which can neutralize tumor microenvironment H+ ions to inhibit tumor cell invasion (Hulikova et al., 2016). Concepcion et al. (2014) found that compared with wild-type mice, the pHi of CD8+ T cells derived from AE2 knockout mice is significantly increased, and CD8+ T cell proliferation and activation levels are obviously enhanced after CD3 stimulation.
Reimold et al. (2013) constructed a mouse AE2 model devoid of transmembrane domain cysteine (Cys) residues to investigate structure-function relationships for AE2. They found that extracellular pH was alkaline-shifted by a minimum of 0.6-0.7 pH units, and the anion exchange rate was significantly decreased in the absence of transmembrane domain Cys residues (Reimold et al., 2013). Sulfur dioxide (SO2) was found to oxidize the -SH of Cys to -SOH, which changed the protein conformation and affected protein activity levels (Svoboda et al., 2012). We confirmed that SO2 suppressed the inflammatory response by sulphenylating NF-κB p65 at Cys38 in oleic acid-induced acute lung injury (Chen et al., 2017).
Recent studies have shown that aspartate aminotransferase can be catalyzed and produces SO2 in the metabolic pathway of sulfur-containing amino acids in mammalian organisms. Furthermore, SO2, which is considered the fourth gas signal molecule followed by NO, CO and H2S, plays an important role in the regulation of cardiovascular physiology and pathophysiology, such as vasodilation, inhibition of vascular calcification and inflammation, anti-oxidation and protection of the myocardium (Du et al., 2008; Sun et al., 2010; Zhang et al., 2011; Huang et al., 2016; Li et al., 2016; Yu et al., 2018).
Therefore, we hypothesized that SO2 might activate the AE in VSMCs, thereby lowering the pHi and further inhibiting VSMC proliferation. It has been reported that AE expression varies significantly among different tissues, and only SLC4A2/AE2 mRNA expression is detected in the VSMC cell line A7r5 cells (Brosius et al., 1997). Therefore, in this study, we aimed to determine whether SO2 impacts the pHi and further inhibits the proliferation of VSMCs, as well as explore underlying molecular mechanisms.
Materials and Methods
Chemicals and Drugs
Sodium sulfite and sodium bisulfite (Na2SO3/NaHSO3), 4,4′- diisothiocyanatostilbene -2,20-disulfonic acid (DIDS) and H89 were purchased from Sigma-Aldrich (St. Louis, MO, United States). Nigericin (N1495) and Fluo 4-AM (F14217) were purchased from Invitrogen (Eugene, OR, United States), and BCECF/AM was purchased from Thermo Scientific (Waltham, MA, United States). Dithiothreitol (DTT) was purchased from Roche Diagnostics GmbH (Mannheim, Germany). Bay K8644 was purchased from selleck (Houston, TX, United States). SO2 donor was freshly prepared using Na2SO3/NaHSO3 dissolved in deionized water in 3:1 mole ratio. DIDS, H89, BCECF/AM and Fluo 4-AM were dissolved in DMSO. Nigericin was dissolved in ethanol.
Cells and Cell Culture
A7r5 VSMCs were purchased from Kunming Cell Bank of Chinese Academy of Sciences (Kunming, China). Cell culture refers to previous literature (Liu et al., 2014). The cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum and 1% antibiotics (penicillin and streptomycin), and cells were placed in an incubator containing 5% CO2 at 37°C. To detect the pHi, cells were seeded in confocal dishes and experiments were performed once the cell density reached 90%.
Measurement of pHi in VSMCs
Measurement of pHi was performed according to the literature (Lee et al., 2007; Galifianakis et al., 2018). Fluorescent indicator BCECF/AM was used to monitor pHi changes. Cells were washed twice with Krebs’ bicarbonate buffer, followed by the incubation with Krebs’ buffer containing 0.5 μM BCECF/AM for 30 min at room temperature. Loaded cells were washed twice with fresh Krebs’ buffer to remove unbound dye and left at room temperature for another 30 min to allow the dye to be fully de-esterified in the cells. The pHi was monitored using a confocal scanning laser microscope (Leica TCS SP8 MP FLIM, Mannheim, Baden-Württember, Germany). The fluorescence intensity was measured at an excitation wavelength of 405 and 496 nm, and an emission wavelength of 535 nm was recorded. The fluorescence intensity ratio (F496/F405) was used to evaluate the pHi. pHi image analysis was performed using LAF-AS software (Leica). The Krebs’ buffer was prepared as follows (mM): 118 NaCl, 5.4 KCl, 1.3 CaCl2, 1.2 MgSO4, 1.2 KH2PO4, 25 NaHCO3 and 11.7 glucose, and the pH was adjusted to 7.4 with NaOH. During the measurement of pHi, drugs were added directly to the buffer and the fluorescence intensity was recorded. A high KCl solution (130 mM KCl, 10 mM Hepes buffer, pH 6.3–9.1) was prepared, and H+ was equilibrated with 20 μM cation ionophore nigericin to prepare a fluorescence intensity-pHi standard curve (Supplementary Figure S1). The different concentrations of SO2 in the study of pHi were grouped into control, SO2 (50 μM), SO2 (100 μM), and SO2 (200 μM). The change of pHi was calculated as the difference in pHi between the starting time point of SO2 treatment and 9 min after SO2 treatment.
Determination of AE Activity in VSMCs
AE activity was measured according to the method described in the previous literature (Simchowitz and roos, 1985; Xu and Spitzer, 1994; Lee et al., 2007). In brief, the activity of AE in A7r5 cells was evaluated by detecting the recovery rate of cells from an intracellular alkalinization. Cells were loaded with BCECF/AM as previously described. The Hepes buffer solution was prepared as follows (mM): 150 NaCl, 5.4 KCl, 2 CaCl2, 1 MgCl2, 5 Hepes and 10 glucose, adjusted to pH 7.4 with NaOH. At the first min, 20 mM NH4Cl was quickly added to the buffer. Once the cells were exposed to NH4Cl, NH3 rapidly diffused into the cells and combined with intracellular H+ to form NH4+, leading to rapid intracellular alkalinization. The pHi gradually decreases from the alkaline peak as HCO3- ions efflux during AE stimulation. The rate at which the pHi returns within the first minute (ΔpH/min) represents AE activity. For the SO2 and the AE activation experiment, the groups were divided as follows: control, SO2 (100 μM) and SO2 (200 μM), and SO2 donor administrated 10 min before pHi measurement. To verify that SO2 stimulated AE via sulphenylation of AE2, we divided the groups as follows: control, SO2, SO2 + DTT or PDGF-BB, PDGF-BB + SO2 and PDGF-BB + SO2 + DTT. One hour before the pHi measurement, cells were treated with 50 ng/ml PDGF-BB, and 50 min before pHi measurement, cells were pretreated with 200 μM SO2 and / or 0.4 mM DTT.
Western Blotting
Vascular smooth muscle cells were seeded in six-well plates and upon a cell density of 60–70%, were synchronized with DMEM containing 0.5% FBS for 24 h.
To confirm that AE was involved in the process of SO2 inhibition of PDGF-BB-induced VSMC proliferation, cell were either (1) untreated; (2) treated with 50 ng/ml PDGF-BBC for 24 h; (3) pretreated with 200 μM SO2 donor for 30 min, then 50 ng/ml PDGF-BB for 24 h; (4) treated with 200 μM SO2 donor for 30 min; (5) treated with 50 ng/ml PDGF-BB and 30 μM DIDS for 24 h; and (6) pretreated with 200 μM SO2 donor for 30 min, then 50 ng/ml PDGF-BB and 30 μM DIDS for 24 h.
To measure the phosphorylation level of PKA by SO2, cells were either (1) untreated; (2) treated with 20 μM H89 for 30 min; (3) pretreated with 20 μM H89 for 30 min, and 200 μM SO2 donor for 10 min; or (4) treated with 200 μM SO2 donor for 10 min.
All cells were collected and lysed in lysis buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1 mM EDTA, 1% NP40, 0.25% sodium deoxycholate, 1 mM PMSF, protease and phosphatase inhibitors) for 20 min at 4°C. They were then centrifuged at 12000 rpm for 10 min at 4°C, and 2x denatured protein loading buffer was added to the supernatant. The mixture was boiled at 100°C for 10 min and cooled at room temperature. Equal amounts of protein (30–60 μg) were run on an 8–10% SDS-PAGE gel. After protein separation, they were transferred to nitrocellulose membranes. The primary antibody dilutions were: 1:1000 for both PKA and p-PKA, and 1:500 for Ki67.
Measurement of VSMC Proliferation With Cell Counting Kit-8 (CCK8)
Cell proliferation was measured according to the reference (Liu et al., 2014). By using a CCK-8 kit, A7r5 cells were first seeded in 96-well plates at 2 × 103 cells / well, and divided into seven groups: (1) blank control (cell-free medium); (2) control (cell-containing medium); (3) treated with 50 ng/ml PDGF-BBC for 24 h; (4) pretreated with 200 μM SO2 donor for 30 min, then 50 ng/ml PDGF-BB for 24 h; (5) treated with 200 μM SO2 donor for 30 min; (6) treated with 50 ng/ml PDGF-BB and 30 μM DIDS for 24 h; and (7) pretreated with 200 μM SO2 donor for 30 min, then 50 ng/ml PDGF-BB and 30 μM DIDS for 24 h. Ten μl of CCK8 reagent was added into a 100 μl- medium, and incubated at 37°C for 4 h. The absorbance was measured at 450 nm using a microplate reader. The mean absorbance value of the blank control group was subtracted from each group as the corrected absorbance of each group.
Measurement of AE2 Sulphenylation in VSMCs
Sulphenic acid modification of AE2 was measured as described previously (Chen et al., 2017). The cells were divided into three groups: untreated, treated with 200 μM SO2 donor for 10 min, or simultaneously with SO2 donor and 0.4 mM DTT for 10 min. Cells were pretreated with 50 ng/ml PDGF-BB for 1 h to induce cell proliferation. DAz-2 is a specific sulphenic acid probe used to label proteins modified by sulphenic acid. The cells were lysed with non-denaturing lysis buffer containing 5 mM DAz-2 and centrifuged at 16000 g for 4 min at 4°C. The supernatant was divided into two parts. 10 μl was used as total protein, which was added to denatured protein loading buffer and boiled for 10 min at 100°C. The remaining supernatant was incubated at 37°C for 2 h with gentle shaking to extract the sulphenic acid modified protein. The mixture was then incubated with 250 μM p-biotin for 2 h at 37°C with gentle shaking to label the protein with biotin. Biotinylated proteins were precipitated with UltraLinkTM Immobilized NeutrAvidinTM (Thermo Fisher Scientific, Waltham, MA, United States) and incubated for 4 h on a shaker at 4°C. The beads were washed three times with PBS, and non-denatured protein loading buffer was added and boiled at 100°C for 10 min. Total protein and sulphenic acid modified protein were subjected to WB analysis. The primary antibody for AE2 was diluted 1:500. The secondary antibody was diluted 1:2000.
Imaging the Intracellular Calcium in VSMCs
The intracellular calcium in VSMCs was imaged with fluorescent calcium probe, Fluo 4-AM. A7r5 cells were divided into four groups: PDGF-BB, PDGF-BB + SO2, PDGF-BB + Bay K8644, and PDGF-BB + Bay K8644 + SO2. Cells were incubated with 50 ng/ml PDGF-BB for 1 h, 1 μM Bay K8644, an L-type calcium channel agonist, for 30 min, and 200 μM SO2 for 10 min. After the treatment, the cells were washed with Krebs’ buffer for twice and incubated with 5 μM Fluo 4-AM in the dark for 30 min at 37°C. The unincorporated dye was removed by washing the cells twice. Loaded cells were maintained at room temperature for another 30 min to allow Fluo 4-AM to de-esterify. Fluorescence image was obtained using a laser scanning confocal microscope (Olympus), at appropriate wavelength settings (excitation at 488 nm and emission at 520 nm).
Statistical Analysis
Data were processed using SPSS 17.0 software (SPSS Inc, Chicago, IL, United States). All data were expressed as mean ± standard error. To examine the effect of SO2 on AE, a t-test was performed to compare the difference between control and DIDS. To examine the effect of different concentrations of SO2 on AE, PKA phosphorylation and A7r5 cell proliferation, comparisons of three or more groups were analyzed by ANOVA, and the Bonferroni test or the Dunnett T3 test were used to compare the difference between two groups. p < 0.05 was considered statistically significant.
Results
SO2 Reduced pHi in VSMCs
By real-time monitoring of the pHi with the fluorescent probe, we found that the SO2 donor at the concentrations of 50, 100, and 200 μM decreased the pHi in VSMCs by 0.120 ± 0.012, 0.134 ± 0.011, and 0.200 ± 0.020, respectively (all p < 0.01, Figures 1A,B).
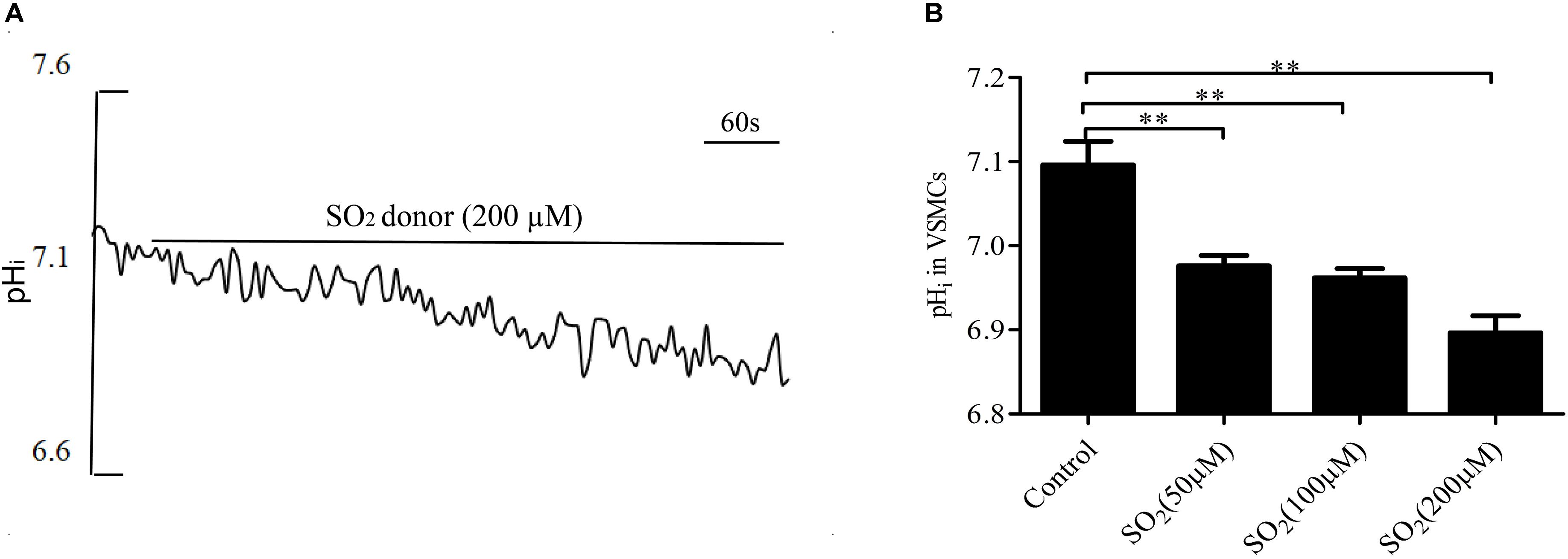
Figure 1. SO2 decreased the pHi in VSMCs. (A) Representative tracing showing 200 μM SO2 donor. (B) Effect of SO2 donor on pHi (n = 16–33). ∗∗p < 0.01 compared with pHi before SO2 donor application.
SO2 Activated AE to Reduce pHi in VSMCs
Anion exchanger is the main acid loader of VSMCs, which pumps out one HCO3- in exchange for one Cl- into the cells, maintaining intracellular Cl- concentration and lowering the pHi. To further validate the effect of SO2 on the AE, we tested the activity of the AE in VSMCs by using the widely accepted NH4Cl perfusion method. As shown in Figures 2A,B, the SO2 donor at varying concentrations of 100 and 200 μM activated the AE in VSMCs (p < 0.05 and p < 0.01, respectively).
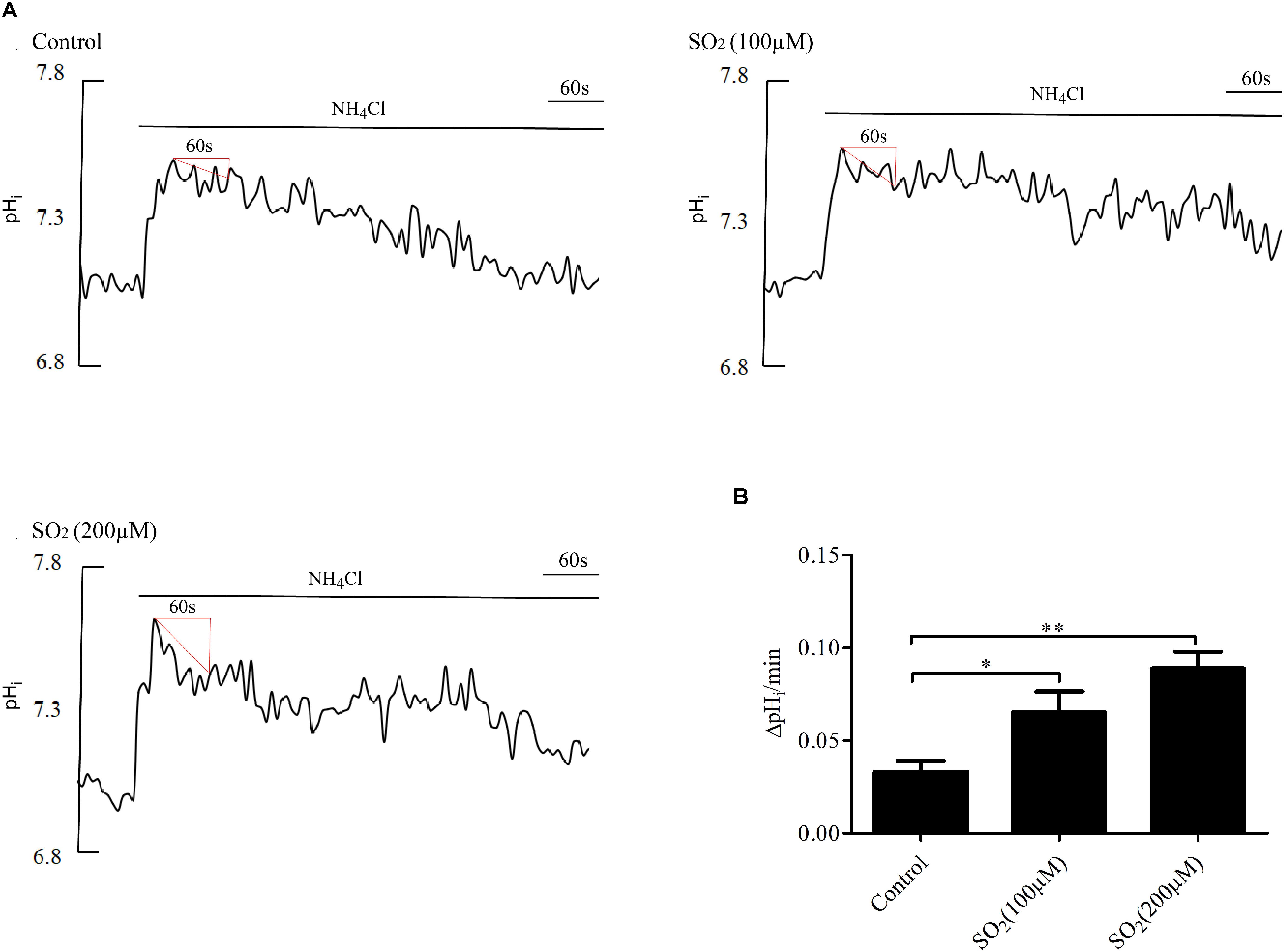
Figure 2. Effect of SO2 on AE activity. (A) Representative tracings of the rapid alkalinization of 20 mM NH4Cl and recovery from higher pHi at different concentrations. SO2 was given 10 min before measurement. (B) Results were shown in mean ± SEM (n = 25–39). ∗p < 0.05, ∗∗p < 0.01 compared with control.
Pretreatment with 30 μM DIDS, a Cl-/HCO3- exchanger inhibitor, for 20 min significantly attenuated the pHi reduction caused by SO2 (p < 0.01, Figures 3A,B), suggesting that SO2 donor reduced pHi by activating the AE in VSMCs.
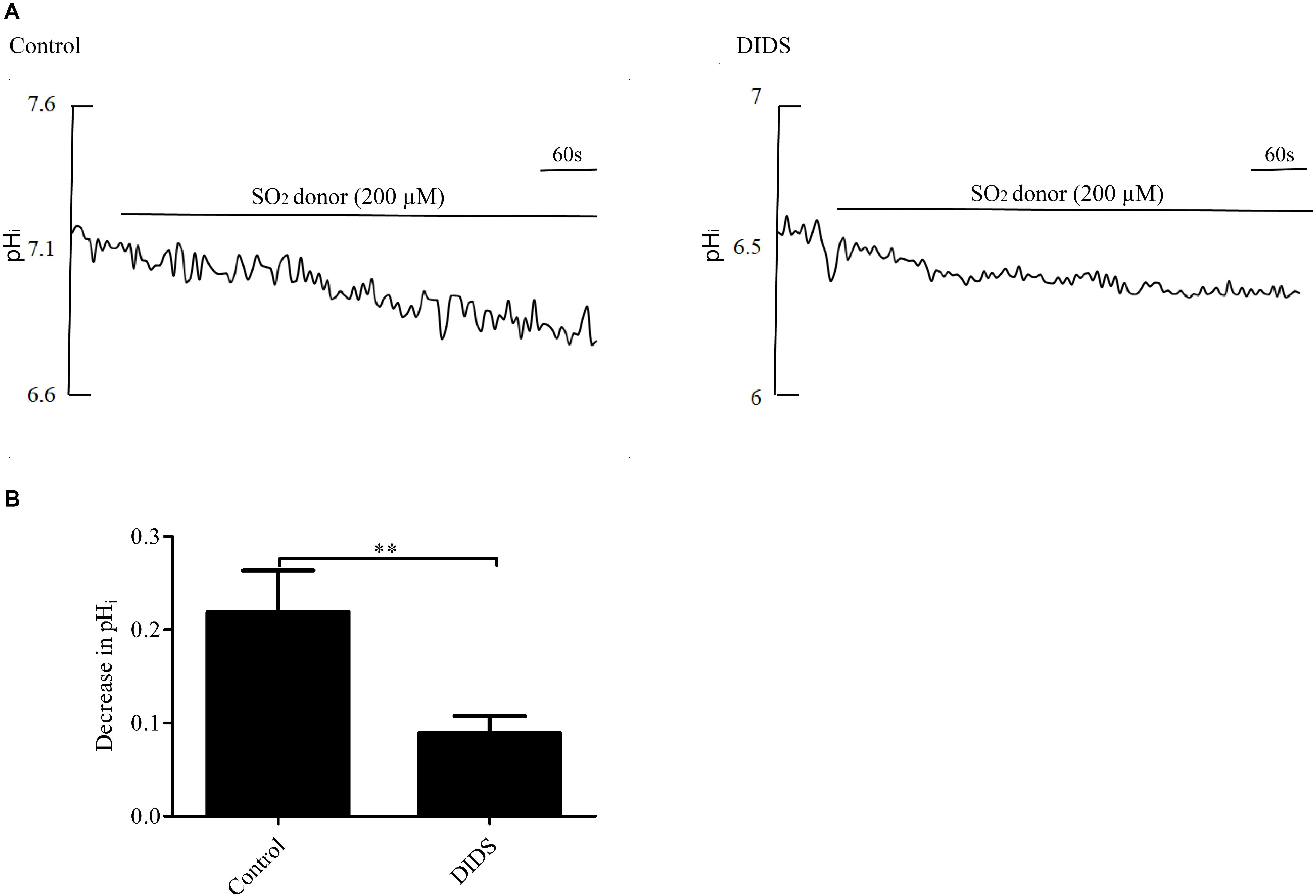
Figure 3. Effect of SO2 on pHi in the presence or absence of DIDS in VSMCs. (A) Representative tracings of 200 μM SO2 donor alone (n = 19) and in the presence of 30 μM DIDS (n = 19). (B) Results were shown in mean ± SEM. ∗∗p < 0.01 compared with control.
SO2 Activated AE by Sulphenylating AE2 in VSMCs
To investigate the mechanism by which SO2 activates AE activity in VSMCs, we tested whether SO2 oxidizes Cys of AE2 by AE2 sulphenylation. As shown in Figures 4A,B, SO2 promoted AE2 sulphenylation and AE activation (p < 0.05, p < 0.01, respectively), and reduced pHi (p < 0.05). Addition of 0.4 mM DTT, a thiol reductant, reversed SO2-induced AE2 sulphenylation and activation of the AE (both p < 0.05), restoring SO2-induced pHi reduction in VSMCs as well (p < 0.01, Figure 4C). This suggests that the thiol group is a likely target of SO2 for activation of the AE in VSMCs.
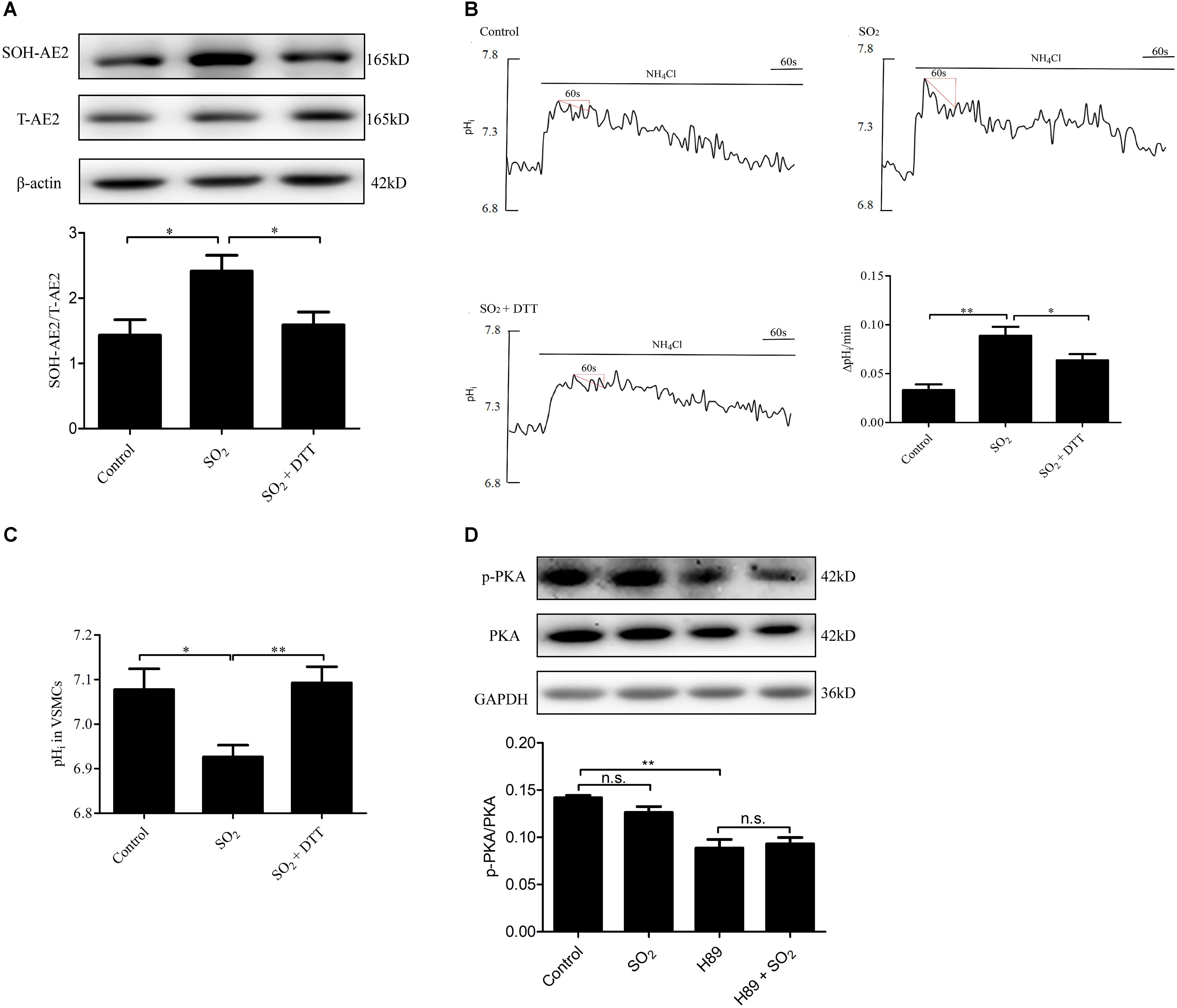
Figure 4. Mechanisms by which SO2 donor activated AE. (A) Sulphenylation of AE2 by 200 μM SO2, and 0.4 mM DTT reversed the process. SO2 and DTT were given 10 min before collecting cells (n = 6). (B) Representative tracings of AE activity in the absence or presence of 200 μM SO2 donor and 0.4 mM DTT. Results were shown as mean ± SEM (n = 25–39). (C) The change of pHi in VSMCs in each group. (D) Effect of SO2 donor on PKA phosphorylation. Cells were pretreated with H89 30 min, followed by SO2 donor in 10 min (n = 6). ∗p < 0.05, ∗∗p < 0.01, n.s.p > 0.05.
We also aimed to understand if SO2 affects the pHi by activation of PKA and further activation of the exchanger in VSMCs. As shown in Figure 4D, SO2 (200 μM) did not stimulate PKA within 10 min in VSMCs (p > 0.05). The results suggested that the PKA pathway did not mediate pHi reduction by SO2 in an acute phase.
SO2 Inhibited VSMC Proliferation Depending on AE Activation
To further elucidate if SO2 inhibit VSMC proliferation depending on AE activation, we constructed a PDGF-BB-induced VSMC proliferation cell model. In VSMCs pretreated with PDGF-BB and treated with SO2, levels of AE2 sulphenylation and AE activity were significantly increased (p < 0.05, p < 0.01, respectively, Figures 5A,B) and the pHi was significantly reduced (p < 0.05, Figure 5C). VSMC proliferation evaluated by Ki67 protein expression and CCK8 activity was significantly inhibited by SO2 (p < 0.05, p < 0.01, respectively, Figures 5D,E), as compared with those in VSMCs pretreated with PDGF-BB only. However, SO2 failed to inhibit PDGF-BB-induced VSMCs proliferation once AE was inhibited by DIDS (both p > 0.05, Figures 5D,E).
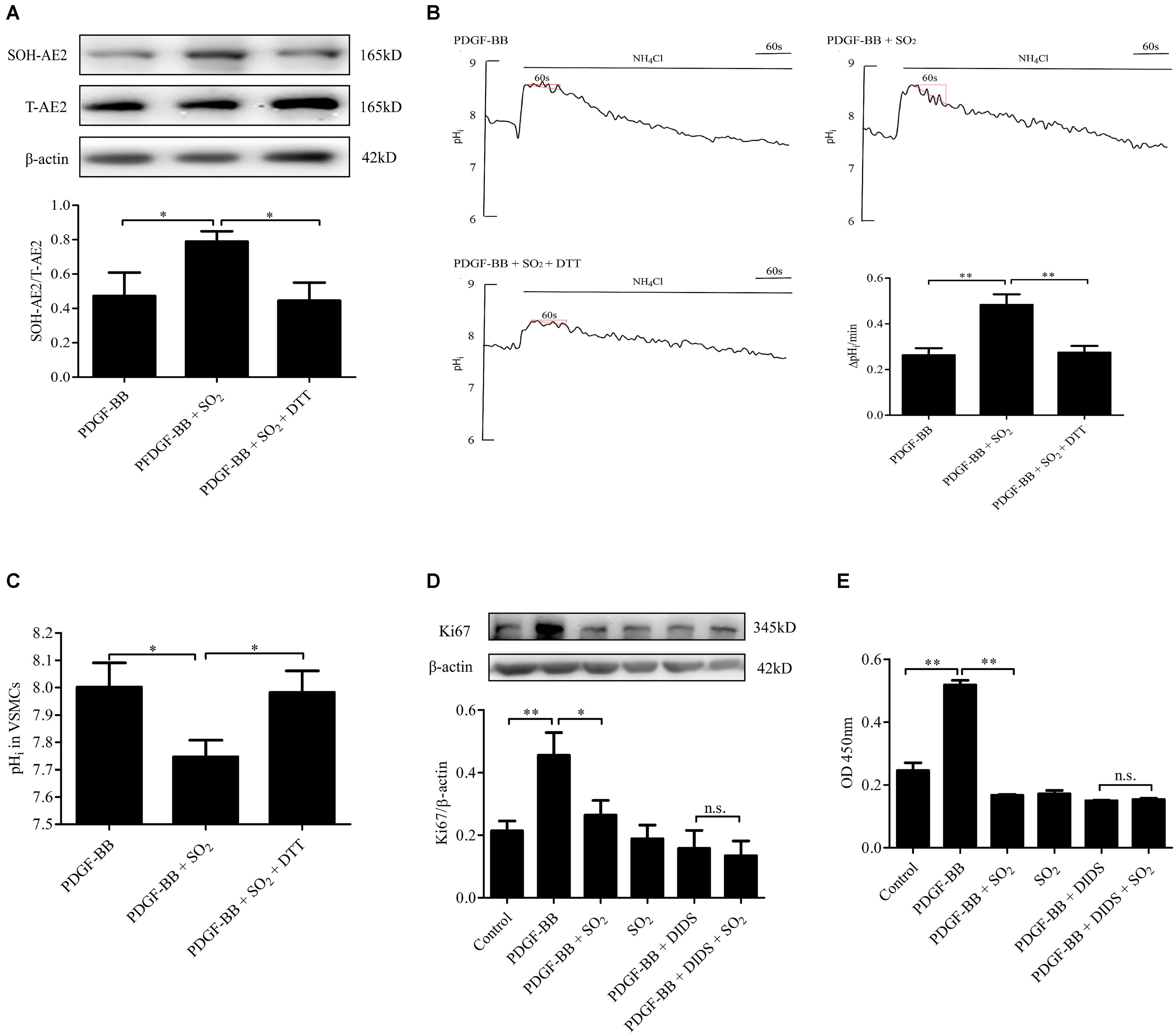
Figure 5. Activation of AE was responsible for SO2 to inhibit VSMC proliferation. (A) Sulphenylation of AE2 by SO2 in the presence of PDGF-BB. Cells were pretreated with 50 ng/ml PDGF-BB 1 h, followed by 200 μM SO2 and 0.4 mM DTT 10 min (n = 6). (B) Representative tracings of AE activity in the absence or presence of 200 μM SO2 donor and 0.4 mM DTT when cells were pretreated with 50 ng/ml PDGF-BB 1 h (n = 31–39). (C) pHi change in each group. (D,E) WB and CCK8 were used to measure the proliferation of VSMCs. 50 ng/ml PDGF-BB, 200 μM SO2 and 30 μM DIDS were administered for 24 h (n = 6). ∗p < 0.05, ∗∗p < 0.01, n.s.p > 0.05.
SO2 Reduced the Intracellular Calcium by Inhibiting L-Type Calcium Channel in VSMCs
To investigate the effects of SO2 on intracellular calcium in VSMCs, we used Fluo 4-AM to image the intracellular calcium in VSMCs. The results showed that SO2 reduced PDGF-stimulated intracellular calcium in VSMCs. While, Bay K8644, an L-type calcium channel agonist, blocked SO2-reduced intracellular calcium content in PDGF-stimulated VSMCs (Figure 6). The results indicated that SO2 might decrease the calcium level by inhibiting L-type calcium channel in VSMCs.
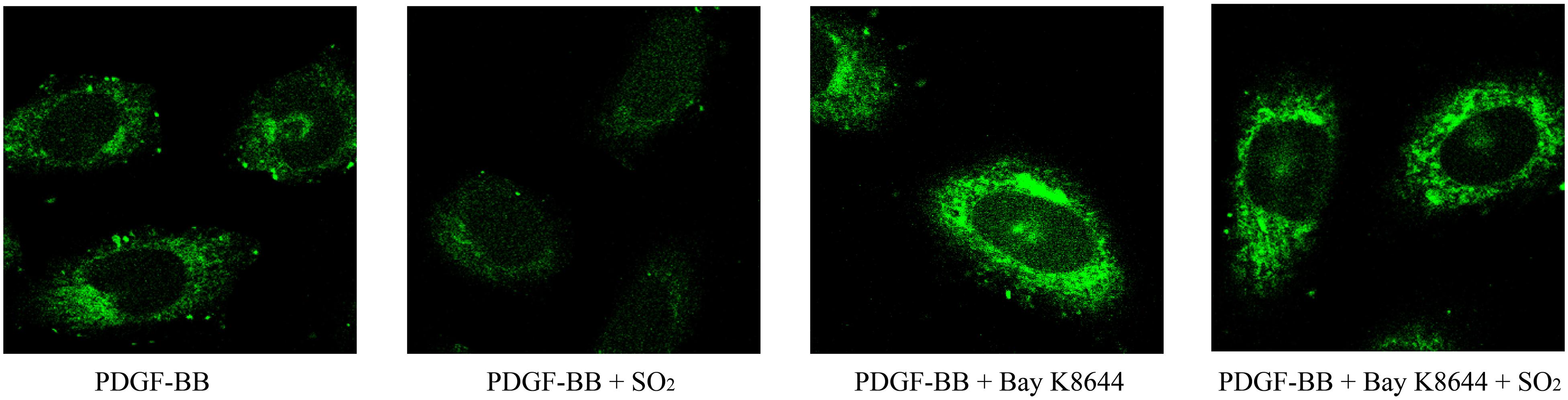
Figure 6. SO2 decreased the intracellular calcium level by inhibiting L-type calcium channel in VSMCs. Cells were incubated with 50 ng/ml PDGF-BB for 1 h, 1 μM Bay K8644 for 30 min and 200 μM SO2 for 10 min. Fluorescence intensity represents calcium levels.
Discussion
As well known, pHi is precisely controlled and needs to be maintained within physiological range. The imbalance of pHi is an important pathological basis for the abnormal cell metabolism and life activities. Therefore, pHi is a vital target for clinical treatment of diseases. In the present study, we firstly reported that SO2 donor decreased the pHi of VSMCs by enhancing AE2 sulphenylation to activate the AE, which might partially mediate the inhibitory effect of SO2 on VSMC proliferation.
At first, by monitoring pHi in real time, we found that the treatment of 50 ∼ 200 μM SO2 donor for 9 min decreased pHi in VSMCs by 1.7–2.8%, respectively. The mechanisms by which SO2 reduced pHi in VSMCs, however, have yet been unclear. AE is the main acid loader in VSMCs and plays an important role in the regulation of pHi (Vigne et al., 1988). Previous studies reported that pHi was increased significantly when NH4Cl loaded cells, and consequently AE was activated to extrude HCO3-, which eventually restored the pHi to normal levels (Xu and Spitzer, 1994; Lee et al., 2007). Therefore, we directly detected the activity of AE using NH4Cl stimulation as previously reported (Simchowitz and roos, 1985). The data showed that SO2 donor markedly steepened the recovery slope of pHi trace following the pHi peak due to NH4+-induced alkalinization, suggesting that SO2 activated AE in VSMCs as we expected. Subsequently, the fact that an inhibitor of Cl-/HCO3- exchanger, DIDS blunted the SO2-induced decrease in pHi in VSMCs supported the speculation that the activation of AE was involved in the effect of SO2 on the pHi in VSMCs.
However, up to now, the mechanisms by which SO2 activated the AE in VSMCs remain unclear. Cysteine thiol group (-SH) can be oxidized to sulphenic acids (-SOH) and the process is termed as protein sulphenylation, which is an important mechanism for regulating protein function. Hourihan et al. (2016) found that sulphenylation of inositol-requiring enzyme 1 could inhibit endoplasmic reticulum stress and activate antioxidant response. The sulphenylation of NF-κB p65 by SO2 resulted in an inactivation of NF-κB pathway (Chen et al., 2017). While Cys residues mutation within transmembrane domain of AE2 could affect the activity of AE2 (Reimold et al., 2013). Interestingly, our data showed that SO2 sulphenylated AE2 in VSMCs in association with the enhancement of the activity of AE. While, a thiol reductant DTT blocked the effect of SO2 on sulphenylation and the activity of AE2. Moreover, PKA pathway was also reported to participate the activation of AE (Puceat, 1999) and SO2 treatment for 30 min activated cAMP/PKA pathway (Liu et al., 2014). But, in the present study, SO2 incubation for 9 min could not enhance the phosphorylation of PKA, which excluded the possibility that SO2 indirectly promoted the activation of AE via PKA pathway. Those above results suggested that SO2 might directly activate the AE by inducing the sulphenylation of AE2 at the posttranslational level.
To investigate the significance of SO2-induced reduction of pHi in VSMCs in its inhibitory effect on VSMC proliferation, DIDS was used to block the SO2-induced reduction of pHi in VSMCs, and then Ki67 expression and CCK8 activity were analyzed as the markers of VSMC proliferation. We found that SO2 inhibited PDGF-BB-induced VSMC proliferation, while DIDS abolished the inhibitory effect of SO2 on PDGF-BB-induced VSMC proliferation, which suggested that SO2 inhibited VSMC proliferation at least partly through decreasing pHi in VSMCs.
However, we also found an interesting detached phenomenon that SO2 donor alone did not inhibit the proliferation of VSMC but inhibited PDGF-BB-induced VSMC proliferation. In fact, under physiological condition, VSMC-derived endogenous SO2 was sufficient enough to inhibit cell proliferation. Therefore, the supplement of additional exogenous SO2 donor to the VSMC on the basis of sufficient endogenous SO2 level would not further exert the anti-proliferative effect (Liu et al., 2014). However, under certain pathophysiological conditions, when VSMCs were insulted by the exogenous injury stimuli, endogenous SO2 production was decreased and the anti-proliferative effect was weakened, resulting to the excessive cell proliferation. In such a case, the supplement of SO2 donor, on the basis of the deficient endogenous SO2 level, would exert a markedly anti-proliferative effect on the proliferating VSMC (Sun et al., 2010; Liu et al., 2014; Wu et al., 2016; Yu et al., 2016). In brief, this discrepancy effect of SO2 donor on the VSMC proliferation on the different conditions might provide a novel idea for the treatment of vascular remodeling in vascular-injury diseases.
In addition to pHi, Ca2+ mobilization is another important stimulus for cell migration and proliferation (Yamamura, 2014; Luo et al., 2018). Endogenous SO2 and its derivates could inhibit L-type calcium channel, which might help explain the mechanism of vasorelaxant function (Du et al., 2008). Therefore, we observed the effect of SO2 on the L-type calcium channel. The data showed that 200 μM SO2 could inhibit the PDGF-BB-stimulated increase in the cytosolic Ca2+ concentration in VSMCs. However, pretreatment of Bay K8644, a specific activator of L-type calcium channel, could block the inhibitory effect of SO2 on the cytosolic Ca2+ concentration, suggesting that L-type calcium channel inhibition occurs under experimental conditions in the presence of SO2 donor.
However, our study still has some limitation. For example, we studied the effect of SO2 on pHi only in cultured VSMCs, but whether the phenomenon occurs in the complex vascular wall is still unknown. As we all know, vascular wall is composed of complex multicellular tissue. It needs to respond to various stimuli such as mechanical stress and neurological and humoral factors in a coordinate manner. The intercellular communication among the constituent cells of vessel wall plays an important role in the regulation of vascular structure and activity and is indispensable for the synchronous response by the wall of vessels. The gap junction was found to exert the abovementioned function of intercellular communication (Haefliger et al., 2004; Sorensen and Holstein-Rathlou, 2012; Yang et al., 2017). The gap junction is composed of a kind of transmembrane proteins termed connexin and form a direct conduit for the exchanges of intercellular signals such as ions and bioactive metabolites, which allows vessel cells to sense the functional and metabolic state of neighbor cells and rapidly modulate the activity by themselves, and therefore synchronously respond to the stimuli (Haefliger et al., 2004; Sorensen and Holstein-Rathlou, 2012; Yang et al., 2017). In addition to the gap junction, gasotransmitters also participate the intercellular communication. Since 1980s, endothelial nitric oxide as the first gasotransmitter was found to regulate the VSMC relaxation via a paracrine pathway, which is partly due to its unique properties including small molecular weight, rapid transmembrane diffusion and extensive action (Wang, 2014; Huang et al., 2016; Kimura, 2016; Nagpure and Bian, 2016). In the previous study, pulmonary artery smooth muscle cell-derived SO2 was found to inhibit the collagen accumulation in the pulmonary artery fibroblasts (Yu et al., 2016). Therefore, we speculated that SO2 might act as an intercellular signal molecule to transduce the massagers among the constitute cells in the vessels, which participated in the synchronicity of vascular function. However, more experiments are needed to extend the effect of SO2 on single channel activity to complex vascular wall.
Conclusion
We discovered the effect of SO2 on pHi of VSMCs and clarified the mechanism by which SO2 decreased pHi of VSMCs. Most importantly, we demonstrated that SO2-induced decrease in pHi of VSMCs might participate the inhibitory effect of SO2 donor on the VSMC proliferation stimulated by mitogen such as PDGF-BB. We expect that those interesting results maybe provide a new idea for the potential clinical prevention and treatment of vascular remodeling in vascular injury diseases such as hypertension.
Data Availability
All datasets generated for this study are included in the manuscript and/or the Supplementary Files.
Author Contributions
YW, JD, and HJ designed the study. YW performed the experiments, analyzed the data, and wrote the manuscript. HJ, JD, and SC revised the manuscript. CT and YH provided useful suggestion and comments to the design of the research. XW, XT, and LZ were involved in the experiments. All the authors read the manuscript and approved the final version.
Funding
This study was supported by National Natural Science Foundation of China (Nos. 31130030 and 81622004).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2019.00313/full#supplementary-material
FIGURE S1 | The fluorescence intensity-pHi standard curve obtained with nigericin.
References
Alper, S. L. (2009). Molecular physiology and genetics of Na+-independent SLC4 anion exchangers. J. Exp. Biol. 212, 1672–1683. doi: 10.1242/jeb.029454
Bennett, M. R., Sinha, S., and Owens, G. K. (2016). Vascular smooth muscle cells in atherosclerosis. Circ. Res. 118, 692–702. doi: 10.1161/CIRCRESAHA.115.306361
Boron, W. F., Chen, L., and Parker, M. D. (2009). Modular structure of sodium-coupled bicarbonate transporters. J. Exp. Biol. 212, 1697–1706. doi: 10.1242/jeb.028563
Brosius, F. C. III, Pisoni, R. L., Cao, X., Deshmukh, G., Yannoukakos, D., Stauart-tilley, A. K., et al. (1997). AE anion exchanger mRNA and protein expression in vascular smooth muscle cells, aorta, and renal microvessels. Am. J. Physiol. 273, F1039–F1047. doi: 10.1152/ajprenal.1997.273.6.F1039
Cardone, R. A., Casavola, V., and Reshkin, S. J. (2005). The role of disturbed pH dynamics and the Na+/H+ exchanger in metastasis. Nat. Rev. Cancer. 5, 786–795. doi: 10.1038/nrc1713
Casey, J. R., Grinstein, S., and Orlowski, J. (2010). Sensors and regulators of intracellular pH. Nat. Rev. Mol. Cell. Biol. 11, 50–61. doi: 10.1038/nrm2820
Chen, G. S., Lee, S. P., Huang, S. F., Chao, S. C., Chang, C. Y., Wu, G. J., et al. (2018). Functional and molecular characterization of transmembrane intracellular pH regulators in human dental pulp stem cells. Arch. Oral. Biol. 90, 19–26. doi: 10.1016/j.archoralbio.2018.02.018
Chen, S., Huang, Y., Liu, Z., Yu, W., Zhang, H., Li, K., et al. (2017). Sulphur dioxide suppresses inflammatory response by sulphenylating NF-κB p65 at Cys38 in a rat model of acute lung injury. Clin. Sci. 131, 2655–2670. doi: 10.1042/CS20170274
Chistiakov, D. A., Orekhov, A. N., and Bobryshev, Y. V. (2015). Vascular smooth muscle cell in atherosclerosis. Acta Physiol. 214, 33–50. doi: 10.1111/apha.12466
Concepcion, A. R., Lopez, M., Ardura-Fabregat, A., and Medina, J. F. (2013). Role of AE2 for pHi regulation in biliary epithelial cells. Front. Physiol. 4:413. doi: 10.3389/fphys.2013.00413
Concepcion, A. R., Salas, J. T., Sarvide, S., Saez, E., Ferrer, A., Lopez, M., et al. (2014). Anion exchanger 2 is critical for CD8+ T cells to maintain pHi homeostasis and modulate immune responses. Eur. J. Immunol. 44, 1341–1351. doi: 10.1002/eji.201344218
Coury, F., Zenger, S., Stewart, A. K., Stephens, S., Neff, L., Tsang, K., et al. (2013). SLC4A2-mediated Cl-/HCO3- exchange activity is essential for calpain-dependent regulation of the actin cytoskeleton in osteoclasts. Proc. Natl. Acad. Sci. U.S.A. 110, 2163–2168. doi: 10.1073/pnas.1206392110
Du, S. X., Jin, H. F., Bu, D. F., Zhao, X., Geng, B., Tang, C. S., et al. (2008). Endogenously generated sulfur dioxide and its vasorelaxant effect in rats. Acta Pharmacol. Sin. 29, 923–930. doi: 10.1111/j.1745-7254.2008.00845.x
Flinck, M., Kramer, S. H., and Pedersen, S. F. (2018). Roles of pH in control of cell proliferation. Acta Physiol. 223:e13068. doi: 10.1111/apha.13068
Galifianakis, N. V., Placantonakis, D. G., and Chesler, M. (2018). Intracellular pH measurements in glioblastoma cells using the pH-sensitive dye BCECF. Methods Mol. Biol. 1741, 103–109. doi: 10.1007/978-1-4939-7659-1_8
Haefliger, J. A., Nicod, P., and Meda, P. (2004). Contribution of connexins to the function of the vascular wall. Cardiovasc. Res. 62, 345–356. doi: 10.1016/j.cardiores.2003.11.015
Hourihan, J. M., Moronetti Mazzeo, L. E., Fernández-Cárdenas, L. P., and Blackwell, T. K. (2016). Cysteine sulfenylation directs IRE-1 to activate the SKN-1/Nrf2 antioxidant response. Mol. Cell. 63, 553–566. doi: 10.1016/j.molcel.2016.07.019
Huang, Y., Tang, C., Du, J., and Jin, H. (2016). Endogenous sulfur dioxide: a new member of gasotransmitter family in the cardiovascular system. Oxid. Med. Cell. Longev. 2016:8961951. doi: 10.1155/2016/8961951
Hulikova, A., Black, N., Hsia, L. T., Wilding, J., Bodmer, W. F., and Swietach, P. (2016). Stromal uptake and transmission of acid is a pathway for venting cancer cell-generated acid. Proc. Natl. Acad. Sci. U.S.A. 113, 5344–5353. doi: 10.1073/pnas.1610954113
Kimura, H. (2016). Hydrogen polysulfide (H2Sn) signaling along with hydrogen sulfide (H2S) and nitric oxide (NO). J. Neural Transm. 123, 1235–1245.
Lee, S. W., Cheng, Y., Moore, P. K., and Bian, J. S. (2007). Hydrogen sulphide regulates intracellular pH in vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 358, 1142–1147. doi: 10.1016/j.bbrc.2007.05.063
Li, Z., Huang, Y., Du, J., Liu, A. D., Tang, C., Qi, Y., et al. (2016). Endogenous sulfur dioxide inhibits vascular calcification in association with the TGF-beta/smad signaling pathway. Int. J. Mol. Sci. 17:266. doi: 10.3390/ijms17030266
Liu, D., Huang, Y., Bu, D., Liu, A. D., Holmberg, L., Jia, Y., et al. (2014). Sulfur dioxide inhibits vascular smooth muscle cell proliferation via suppressing the Erk/MAP kinase pathway mediated by cAMP/PKA signaling. Cell. Death Dis. 5:e1251. doi: 10.1038/cddis.2014.229
Liu, Y., Yang, J., and Chen, L. M. (2015). Structure and function of slc4 family transporters. Front. Physiol. 6:355. doi: 10.3389/fphys.2015.00355
Luo, L. M., Hong, X. Y., Diao, B., Chen, S. Y., and Hei, M. Y. (2018). Sulfur dioxide attenuates hypoxia-induced pulmonary arteriolar remodeling via Dkk1/Wnt signaling pathway. Biomed. Pharmacother. 106, 692–698. doi: 10.1016/j.biopha.2018.07.017
Meyers, S. N., McDaneld, T. G., Swist, S. L., Marron, B. M., Steffen, D. J., O’Toole, D., et al. (2010). A deletion mutation in bovine SLC4A2 is associated with osteopetrosis in red angus cattle. BMC. Genomics 11:337. doi: 10.1186/1471-2164-11-337
Nagpure, B. V., and Bian, J. S. (2016). Interaction of hydrogen sulfide with nitric oxide in the cardiovascular system. Oxid. Med. Cell. Longev. 2016:6904327. doi: 10.1155/2016/6904327
Owens, G. K., Kumar, M. S., and Wamhoff, B. R. (2004). Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 84, 767–801. doi: 10.1152/physrev.00041.2003
Puceat, M. (1999). pHi regulatory ion transporters: an update on structure, regulation and cell function. Cell. Mol. Life Sci. 55, 1216–1229. doi: 10.1007/s000180050368
Reimold, F. R., Stewart, A. K., Stolpe, K., Heneghan, J. F., Shmukler, B. E., and Alper, S. L. (2013). Substitution of transmembrane domain cys residues alters pHo-sensitive anion transport by AE2/SLC4A2 anion exchanger. Pflugers Arch. 465, 839–851. doi: 10.1007/s00424-012-1196-6
Schreiber, R. (2005). Ca2+ signaling, intracellular pH and cell volume in cell proliferation. J. Membr. Biol. 205, 129–137. doi: 10.1007/s00232-005-0778-z
Simchowitz, L., and roos, A. (1985). Regulation of intracellular pH in human neutrophils. J. Gen. Physiol. 85, 443–470. doi: 10.1085/jgp.85.3.443
Sorensen, C. M., and Holstein-Rathlou, N. H. (2012). Cell-cell communication in the kidney microcirculation. Microcirculation 19, 451–460. doi: 10.1111/j.1549-8719.2011.00149.x
Sun, Y., Tian, Y., Prabha, M., Liu, D., Chen, S., Zhang, R., et al. (2010). Effects of sulfur dioxide on hypoxic pulmonary vascular structural remodeling. Lab. Invest. 90, 68–82. doi: 10.1038/labinvest.2009.102
Svoboda, L. K., Reddie, K. G., Zhang, L., Vesely, E. D., Williams, E. S., Schumacher, S. M., et al. (2012). Redox-sensitive sulfenic acid modification regulates surface expression of the cardiovascular voltage-gated potassium channel Kv 1.5. Circ. Res. 111, 842–853. doi: 10.1161/CIRCRESAHA.111.263525
Vigne, P., Breittmayer, J. P., Frelin, C., and Lazdunski, M. (1988). Dual control of the intracellular pH in aortic smooth muscle cells by a cAMP-sensitive HCO3-/Cl- antiporter and a protein kinase C-sensitive Na+/H+ antiporter. J. Biol. Chem. 263, 18023–18029.
Wang, R. (2014). Gasotransmitters: growing pains and joys. Trends Biochem. Sci. 39, 227–232. doi: 10.1016/j.tibs.2014.03.003
Wu, H. J., Huang, Y. Q., Chen, Q. H., Tian, X. Y., Liu, J., Tang, C. S., et al. (2016). Sulfur dioxide inhibits extracellular signal-regulated kinase signaling to attenuate vascular smooth muscle cell proliferation in angiotensin II- induced hypertensive mice. Chin. Med. J. 129, 2226–2232. doi: 10.4103/0366-6999.189927
Xu, P., and Spitzer, K. W. (1994). Na-independent Cl-HCO3-exchange mediates recovery of pHi from alkalosis in guinea pig ventricular myocytes. Am. J. Physiol. 267, H85–H91. doi: 10.1152/ajpheart.1994.267.1.H85
Yamamura, A. (2014). Pathological function of Ca2+-sensing receptor in pulmonary arterial hypertension. J. Smooth Muscle Res. 50, 8–17. doi: 10.1540/jsmr.50.8
Yang, G. M., Peng, X. Y., Wu, Y., Li, T., and Liu, L. M. (2017). Involvement of connexin 43 phosphorylation and gap junctional communication between smooth muscle cells in vasopressin-induced ROCK-dependent vasoconstriction after hemorrhagic shock. Am. J. Physiol. Cell Physiol. 313, C362–C370. doi: 10.1152/ajpcell.00258.2016
Yu, W., Jin, H. F., Tang, C. S., Du, J. B., and Zhang, Z. R. (2018). Sulfur-containing gaseous signal molecules, ion channels and cardiovascular diseases. Br. J. Pharmacol. 175, 1114–1125. doi: 10.1111/bph.13829
Yu, W., Liu, D., Liang, C., Ochs, T., Chen, S., Du, S., et al. (2016). Sulfur dioxide protects against collagen accumulation in pulmonary artery in association with downregulation of the transforming growth factor β1/smad pathway in pulmonary hypertensive rats. J. Am. Heart Assoc. 5:e003910. doi: 10.1161/JAHA.116.003910
Keywords: sulfur dioxide, vascular smooth muscle cell, intracellular pH, Cl-/HCO3- exchanger, AE2 sulphenylation
Citation: Wang Y, Wang X, Chen S, Tian X, Zhang L, Huang Y, Tang C, Du J and Jin H (2019) Sulfur Dioxide Activates Cl-/HCO3- Exchanger via Sulphenylating AE2 to Reduce Intracellular pH in Vascular Smooth Muscle Cells. Front. Pharmacol. 10:313. doi: 10.3389/fphar.2019.00313
Received: 23 November 2018; Accepted: 14 March 2019;
Published: 27 March 2019.
Edited by:
Francesco Rossi, Università degli Studi della Campania Luigi Vanvitelli, ItalyReviewed by:
Antonella Liantonio, University of Bari Aldo Moro, ItalyJavier Angulo, Hospital Universitario Ramón y Cajal, Spain
Copyright © 2019 Wang, Wang, Chen, Tian, Zhang, Huang, Tang, Du and Jin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongfang Jin, amluaG9uZ2Zhbmc1MUAxMjYuY29t