
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol. , 22 March 2019
Sec. Translational Pharmacology
Volume 10 - 2019 | https://doi.org/10.3389/fphar.2019.00276
This article is part of the Research Topic Metabolomics, Pharmacometabonomics and Pharmacology View all 8 articles
 Kaili Fu1
Kaili Fu1 Conghui Wang2
Conghui Wang2 Yue Gao1Shicheng Fan1Huizhen Zhang1
Yue Gao1Shicheng Fan1Huizhen Zhang1 Jiahong Sun1Yiming Jiang1Conghui Liu1Lihuan Guan1Junling Liu2*Min Huang1
Jiahong Sun1Yiming Jiang1Conghui Liu1Lihuan Guan1Junling Liu2*Min Huang1 Huichang Bi1*
Huichang Bi1*Vascular protein sorting-associated protein 33B (VPS33B) plays important roles in hepatic polarity, which directly maintains the functional structure of the liver. It has reported that VPS33B has close association with arthrogryposis, renal dysfunction and cholestasis (ARC) syndrome. Unfortunately, no further studies were conducted to reveal the role of Vps33b in the homeostasis of bile acids. In the current study, hepatic Vps33b-depleted male mice were used to investigate the metabolomics and lipidomics profiles of hepatic Vps33b deficiency based on ultrahigh-performance liquid chromatography coupled with an electrospray ionization high-resolution mass spectrometry (UHPLC-ESI-HRMS) system. Hepatic Vps33b-depleted male mice displayed cholestasis and slight liver damage with increased serum levels of ALT, AST, ALP and T-Bili compared to wild-type mice. Targeted metabolomics analysis of bile acids revealed that increased taurine-conjugated bile acids accumulated in the serum of hepatic Vps33b-depleted mice, while unconjugated bile acids were prone to decrease, accompanied by the regulation of bile acid homeostasis-related genes. In addition, lipid profiles were significantly altered with the lack of Vps33b in the liver. A variety of lipids, such as triglycerides and sphingomyelins, were significantly decreased in the liver and increased in the serum of hepatic Vps33b-depleted mice compared to those in wild-type mice. Our study demonstrated that Vps33b influences the progress of liver metabolism both in bile acid circulation and lipid metabolism, which is involved in the progression of liver cholestasis in mice.
Arthrogryposis, renal dysfunction and cholestasis (ARC) syndrome (OMIM 208085), an autosomal recessive multiorgan disorder that was originally described in 1973 by Lutz-Richner typically presents with neurogenic arthrogryposis, multiplex congenita, renal tubular dysfunction and neonatal cholestasis with bile duct hypoplasia. This disorder mostly affects the offspring of consanguineous unions, where patients usually die within a few months of birth (Horslen et al., 1994). Until now, approximately 75% of ARC patients were diagnosed with a VPS33B (Vacuolar protein sorting-associated protein 33B) mutation (Cullinane et al., 2009), while others were characterized with a mutation in VIPAR, a VPS33B interacting protein, or, Vps16B (Ackermann et al., 2014; Rogerson and Gissen, 2018).
VPS33B, a 617 AA protein of the Sec-1/Muc18 (SM) family (Huizing et al., 2001), is involved in the vesicular intracellular trafficking process and protein interactions (Chen C.H. et al., 2017). SM proteins together with SNAREs are generally required for membrane fusion through regulating the formation of quaternary SNARE complexes, including GTPases and other tethering proteins containing long coiled domains. (Sudhof and Rothman, 2009; Lobingier and Merz, 2012). The general role of these proteins in membrane trafficking demonstrated the vital role of Vps33b in intracellular trafficking functions.
The function of VPS33B and its association with ARC disorder has been reported according to the many clinical features of this disease. For example, VPS33B is one of the first proteins found to be essential for a-granule biogenesis according to ARC syndrome. The absence of Vps33b causes a lack of a-granules, soluble cargo and p-selectin, a granule-specific membrane protein, in platelets leading to a bleeding diathesis, secondary to platelet dysfunction in ARC syndrome (Hayes et al., 2004; Dai et al., 2016). Second, Vps33b was reported to take participate in the maturation of phagosomes and endosomes following microbial antigen ingestion (Akbar et al., 2016). The lack of Vps33b in Drosophila contributes to accelerated inflammatory responses and microbial stimulation of pattern-recognition receptors such as toll-like receptors. Furthermore, severe liver histological change is one of the most important features of ARC syndrome, including cholestasis, bile duct hypoplasia and lipofuscin granule deposition (Eastham et al., 2001). Currently, although VPS33B function has been partly studied, there is no curative therapy for ARC syndrome, which is involved in multiple clinical features.
Clinical therapy for ARC simply relieves patient discomfort, such as ursodeoxycholate therapy, a promising drug for cholestasis that has been used to decrease pruritus mainly caused by the increased level of serum bilirubin. In that case, it suggests that bile acid metabolism disturbances might have a role in the etiopathogenesis of cholestasis in these ARC cases (Abdullah et al., 2000). Unfortunately, no further studies were conducted to reveal the role of Vps33b in the homeostasis of bile acids. Otherwise, it is reported that liver transplantation can significantly improve ARC patients’ clinical symptoms and prolong patient growth status (Dehghani et al., 2013). Therefore, dysregulated liver function is a severe sign among ARC patients who bear the VPS33B mutation. Thus, it is clear that VPS33B may play an important role in the liver, and mechanistic studies of VPS33B might be helpful to develop an ARC therapy or address other liver diseases.
Recently, the rise of omics studies, especially metabolomics and lipidomics, have become the preferred method to provide new insights into the etiology of liver diseases, new treatment modalities and a new understanding that may reveal potential therapeutic targets. For example, a metabolomics strategy has been used to map the bile acid profile in vivo and reveal how bile acid homeostasis is disrupted during the disease progression (Chen P. et al., 2017).
In the current study, hepatic Vps33b-depleted male mice were used to investigate the metabolomics and lipidomics profiles of hepatic Vps33b deficiency in order to map the bile acids deposition and lipid profile of Vps33b hepatic knockout mice. Furthermore, the role of Vps33b in bile acids homeostasis and lipid metabolism and the involved mechanisms were further studied to find out possible therapeutics targets of diagnostic markers.
Serum biochemistry measurement kits (ALT, AST, ALP, and TBILI) were purchased from Shanghai Kehua Bio-Engineering Co., Ltd. (China). Serum TBA measurement kits were purchased from Nanjing Jiancheng Bioengineering Institute (China). All other solvents and regents were of analytical or HPLC grade when appropriate.
Vps33b hepatic knockout mice (Vps33bflox/flox, alb-cre) and wild-type mice (Vps33bflox/flox) with a C57BL/6 genetic background were obtained from Junling Liu’s laboratory (Shanghai Jiao Tong University School of Medicine, China). Mice were housed in stainless exhaust-ventilated closed-system cages in a specific-pathogen-free environment. Mice were maintained under a standard 12 h light/12 h dark cycle with water and a normal diet provided ad libitum. Animal experiments were performed in accordance with the guidelines of the Institutional Animal Care and Use Committee at Sun Yat-sen University (Guangzhou, China).
The Vps33b floxed allele was generated as previous reported (Wang et al., 2018). Mice were bred by crossing Vps33bflox/flox, alb-cre and Vps33bflox/flox mice. A 1–2 mm portion of mouse tail was cut for genotyping at the age of 3 weeks. A one-step mouse genotyping kit was purchased from Vazyme Biotech Co., Ltd (China). The supernatant of tail lysis buffer was added into the PCR system with 2x Taq Plus Master Mix (Dye Plus), primers and DEPC water. The PCR procedure were performed by agarose gel electrophoresis. Vps33bflox/flox, alb-cre mice showed two bands at 300 and 606 bp, while Vps33bflox/flox mice only had only a 606 bp band. Primer sequence of genotyping are listed as follows: Cre-reverse primer 5′-ATTTGCCTGCATTACCGGTCG-3′ and Cre-forward primer 5′-CAGCATTGCTGTCACTTGGTC-3′; Vps33b-floxed allele gene primer A1 5′-CTGACTAGGGGAGGAGTAGAAGGT-3′, A2 5′-GTATCACTGAGTCACACACATCCA-3′ and A3 5′-ATAGAGACGTTAGCAATTCGATCC-3′.
Vps33bflox/flox, alb-cre mice were sacrificed at 3–4 months of age, and age-matched Vps33bflox/flox mice were used as wild-type controls. All procedures were undertaken with the approval of the Institutional Animal Care and Use Committee at Sun Yat-sen University (Guangzhou, China). Serum was obtained after centrifugation of the blood at 3000 rpm for 10 min at RT. Bile was transferred into a 1.5 mL Eppendorf tube and weighed. Livers, intestines and feces were collected for further study respectively.
Serum activities of ALT, AST, ALP, TBILI, and TBA were measured by commercially available kits on an automatic biochemical analyzer.
Samples preparation for metabolomic analysis were performed according to our previously reported methods with some slight modifications (Chen P. et al., 2017; Zeng et al., 2017; Zhang et al., 2017b). Ten microliters serum and five microliters bile were mixed with 67% aqueous acetonitrile in distilled water to remove the protein, followed by centrifugation at 18000 g for 20 min at 4°C to obtain the supernatant. Liver and intestinal tissues were homogenized with 50% aqueous acetonitrile in distilled water and centrifuged at 18000 g for 20 min at 4°C to precipitate the protein. Twenty microliters of feces-PBS homogenate (20 μg per 400 μL) were vortexed with 67% ACN, followed by centrifugation to obtain the supernatant.
The obtained supernatant was transferred to an UPLC vial. Five microliter aliquots of metabolic samples were injected into the UPLC-ESI-Q Exactive interfered system (Dionex Corporation, Sunnyvale, CA, United States, Thermo Fisher Scientific, Waltham, MA, United States). ACQUITY UPLC BEH C18 column 1.7 μm (2.1∗50 mm, Waters Corporation, Milford, MA, United States) was used to performed chromatography separation. Colum temperature was 60°C. The mobile phase consists of solvent A (0.1% formic acid in water) and solvent B (0.1% formic acid in acetonitrile) with the flow rate of 0.5 mL/min. The gradient program was as follows: 0 min 5% B, 0.5 min 5% B, 1 min 35% B, 6 min 60% B, 8 min 95% B, 9 min 95% B, 10 min 5% B, 10.5 min 5% B.
Electrospray negative ionization mode was used for analysis. The spray voltage was set to 3.5 kV. Capillary and auxiliary gas heater temperatures were set at 325 and 350°C respectively. Nitrogen was used as both the sheath gas at a flow rate of 60 arbitrary units and the auxiliary gas at a flow rate of 20 arbitrary units. Target SIM method which included the accurate Q/Z of bile acids was used to get higher sensitivity. The mass spectral data were aligned using SIEVE 2.2 (Thermo Fisher Scientific, Waltham, MA, United States). Then the extracted component was prepared for further analysis.
Multivariate data analysis was performed using SIMCA 13.0 software (Umetrics, Kinnelon, NJ, United States). Principal components analysis (PCA) and supervised orthogonal partial least squares discriminate analysis (OPLS-DA) models were used to analyze the data of tissues samples, such as the serum, liver, bile, intestine, and feces. Further identification of bile acids was conducted by comparing the retention time and fragmentation patterns with authentic standards according to our previously reported method (Chen P. et al., 2017).
Methyl tert butyl ether (MTBE) method (Matyash et al., 2008) was used to extract lipids from the serum and liver tissues. Twenty microliters serum was vortexed by adding prechill methanol, MTBE, and ultrapure water successively for 30 s respectively. Then the mixture was centrifuged at 3000 × rpm for 10 min. The supernatant was obtained for vacuum drying. Twenty micrograms liver tissues were homogenized with PBS and lipids were extracted as we mentioned before (Li et al., 2018). Before injection, samples were re-suspended in 200 μL mixture of methanol/isopropanol (1:1, v/v) and centrifuged at 18,000 × g for 5 min at 4°C.
Chromatography separation was performed using an Ascentis Express C18 2.7 μm column (100 mm × 2.1 mm, Sigma-Aldrich, St. Louis, MO, United States) on a Thermo Scientific Dionex Ultimate 3000 UPLC-ESI-Q Exactive system. The chromatograpy conditions, including the consistent of mobile phase and linear gradient, were similar with our previous report (Li et al., 2018). Mass spectrometry was performed with electrospray positive (ESI+) and negative (ESI-) ionization modes. The main parameters for MS/MS included the following parameters: AGC target 1e5, maximum IT 65 ms, isolation window 1.2 m/z, normalized collision energy 25, 35 eV in positive mode, 20, 30, and 40 eV in negative mode, apex trigger 5–10 s, and dynamic exclusion 10.0 s. Ionization conditions were operated at a spray voltage of 3.5 kV and a capillary temperature of 300°C.
Lipidomic data processing was performed according to our previous report (Zhang et al., 2017a) using Lipid Search software (Thermo Scientific, San Jose, CA, United States).
RNA isolation and qRT-PCR analysis of hepatic gene mRNA expression level was performed as described previously (Chen P. et al., 2017). The primer sequences were obtained from Primer Bank and synthesized by Thermo Fisher Scientific. Primer sequences were listed in Supplementary Table 1.
Liver total protein extraction and western blot analysis were performed as described previously. Bolts were incubated with primary antibody against BSEP (F-6) (Santa Cruz Biotechnology, Santa Cruz, CA, United States), E-CADHERIN1 (Gentex Corporation, Zeeland) and GAPDH (Cell Signaling Technologies, Danvers, MA, United States).
Liver specimens were fixed in 10% formalin solution and processed routinely for paraffin embedding. Sections (4 μm thick) were deparaffinized and then stained with hematoxylin and eosin solutions (H&E) and examined under a light microscope (Nikon 80i, Japan). Immunohistological staining was performed with a primary antibody against CLAUDIN-1 (Abcam, San Francisco, CA, United States).
Each group was consisted of six animals. All values are expressed as the means ± SD. Statistical analysis was performed by unpaired Students’ t-test or Mann–Whitney U-test with Prism 6 (GraphPad Software Inc., San Diego, CA, United States) or SPSS statistical software. A p-value of less than 0.05 was considered statistically significant.
Compared to the age-matched male wild-type mice, hepatic Vps33b-depleted mice fed a controlled diet were identified for Vps33b depletion efficiency in the liver. Vps33b mRNA and protein levels were significantly decreased in Vps33bflox/flox, alb-cre mice compared with those in the Vps33bflox/flox mice (Supplementary Figures 1A–C).
Furthermore, no significant differences in body weight and liver/body weight ratio were observed between Vps33bflox/flox and Vps33bflox/flox, alb-cre mice (Supplementary Figures 2A,B). Although no significant differences in bile volume were observed between Vps33bflox/flox and Vps33bflox/flox, alb-cre mice, Vps33bflox/flox, alb-cre mice displayed a distinctly smaller gall bladder. In addition, serum biochemistry analysis showed that ALP and serum total bilirubin levels in Vps33bflox/flox, alb-cre mice were higher than those in Vps33bflox/flox mice (Figures 1E,F). Liver damage indexes, such as serum ALT and AST levels, were also elevated in Vps33bflox/flox, alb-cre mice compared with those in Vps33bflox/flox mice (Figure 1). In addition, histological analysis showed hepatocyte degeneration, necrosis and ductal proliferation around the portal area in Vps33bflox/flox, alb-cre mice (Figure 2). These results indicated that hepatic Vps33b knockout mice displayed cholestasis and slight liver injury compared to Vps33bflox/flox mice.
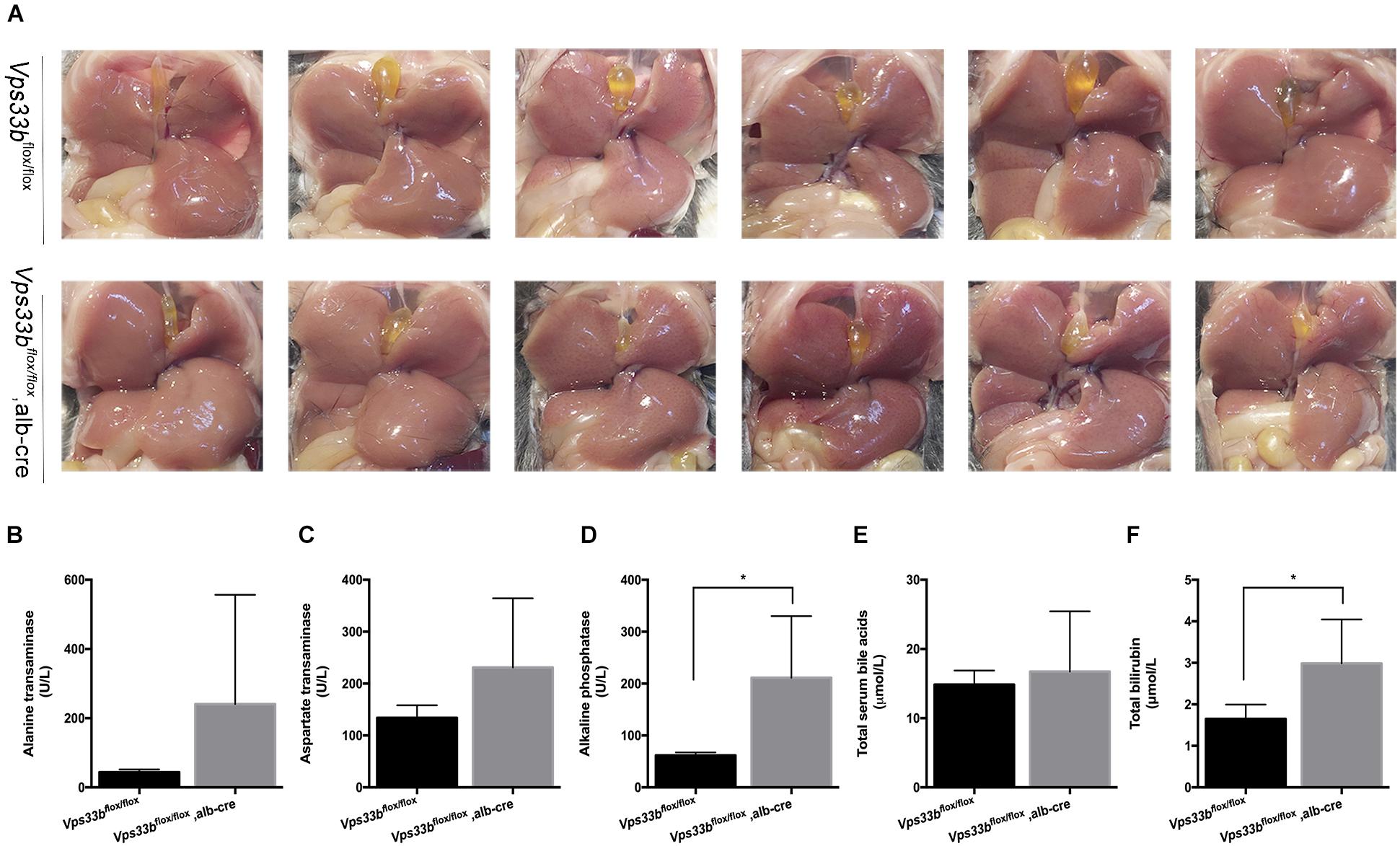
Figure 1. Vps33bflox/flox, alb-cre mice had distinctly smaller gallbladders (A) and higher serum biochemistry levels (B–F) compared to those of Vps33bflox/flox mice. (A) In situ view of gallbladders from the two groups; (B) ALT, (C) AST, (D) ALP, (E) total bile acids, and (F) total bilirubin levels in serum of adult male mice (3–4 months of age) with the indicated genotypes. Values are expressed as the mean ± standard deviation (n = 5 per group). ∗p < 0.05 vs. Vps33bflox/flox mice.
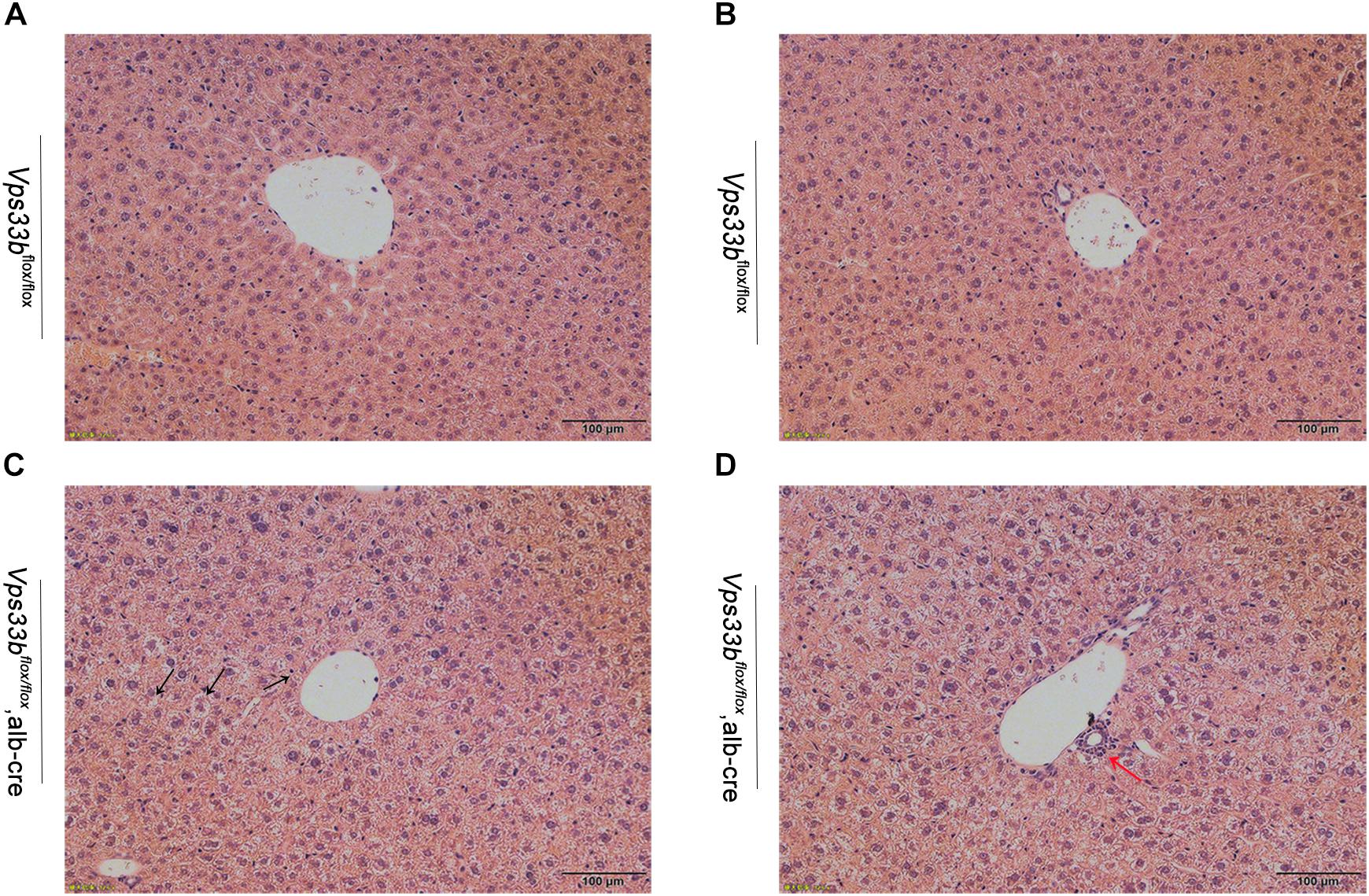
Figure 2. Representative H&E-stained livers from Vps33bflox/flox (A,B) and Vps33bflox/flox, alb-cre mice (C,D). Necrosis of hepatocytes (black arrow) and ductal proliferation (red arrow) were observed.
Metabolomics analysis was performed to examine dynamic changes of endogenous metabolites in the serum, liver, bile, intestine and feces. Unsupervised PCA was used to analyze the data sets from Vps33bflox/flox mice and Vps33bflox/flox, alb-cre mice. Each point represented a mouse in their group. Significant separation of serum, liver and bile samples was observed in the PCA model, as shown by the distribution of Vps33bflox/flox and Vps33bflox/flox alb-cre mice in different quadrants, indicating a significant difference in endogenous metabolomes between the two groups of mice (Figures 3A–C). However, both the intestine and feces maintained a cross-distribution of metabolites in the PCA score plot (Figures 3D,E). These results were in accordance with the trend observed in the serum biochemistry. We further performed targeted metabolomics analysis of bile acids to gain a full understanding of how hepatic Vps33b depletion influences bile acid homeostasis. The extracted ions from mass spectrum of mice samples were confirmed by comparing with authentic bile acids standard according to retention time and MS/MS (Supplementary Figure 3). Bile acids were classified as conjugated and unconjugated types, and their relative amount in the tissues of mice was calculated (Figures 3F–O).
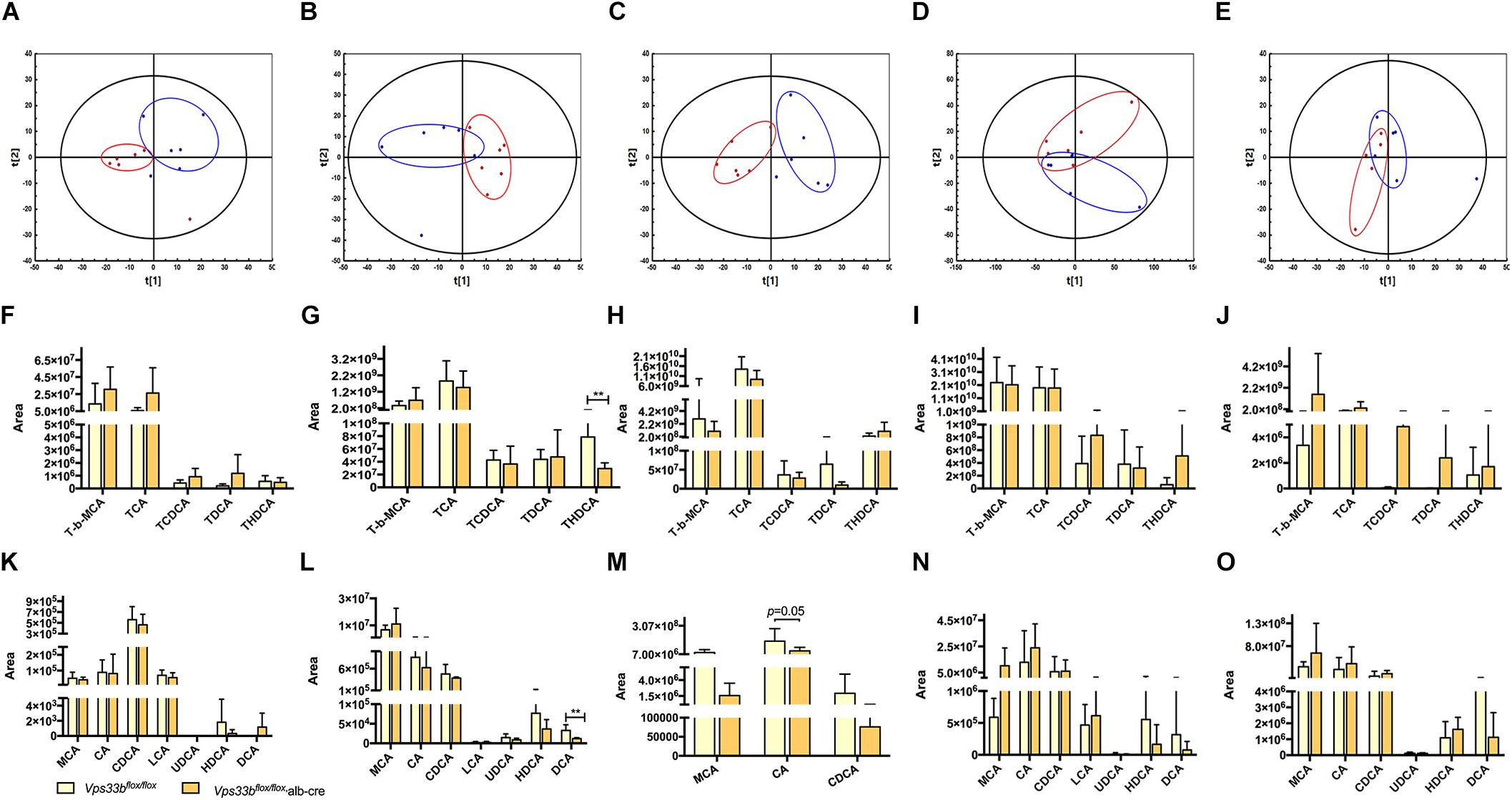
Figure 3. Principal component analysis (PCA) score plot (A–E) of Vps33bflox/flox mice (Blue) and Vps33bflox/flox, alb-cre mice (Red). ESI-negative mode; Serum (A); Liver (B), Bile (C); Intestine (D) and Feces (E). Bile acid components in serum (F,K), liver (G,L), bile (H,M), intestine (I,N) and feces (J,O) of mice. Conjugated bile acids (F–J); Unconjugated bile acids (K–O).
TCA and T-beta MCA consisted of a majority of conjugated bile acids in mouse tissue samples. Both were elevated in serum and reduced in bile by hepatic depletion of Vps33b. T-beta MCA was increased in the livers of Vps33bflox/flox, alb-cre mice, while TCA was prone to reduction compared with those in the Vps33bflox/flox mice. These major conjugated bile acid levels in the intestine were not changed by Vps33b deficiency in the liver. Overall, combined with other conjugated bile acids, such as TCDCA, TDCA and THDCA, conjugated bile acids were mostly increased in the serum of Vps33bflox/flox, alb-cre mice, while decreased in the liver and bile compared with those in the Vps33bflox/flox mice. However, unconjugated bile acids were generally reduced in the serum, liver and bile of Vps33bflox/flox, alb-cre mice. Furthermore, hepatic Vps33b depletion seemed to have no effect on bile acid distribution between the intestine and feces (Supplementary Figure 4). In summary, the bile acid distribution pattern was clearly changed in hepatic Vps33b-depleted mice compared with that in the Vps33bflox/flox mice, demonstrating that Vps33b plays an important role in sustaining bile acid homeostasis.
According to the disrupted bile acid metabolomics pattern in Vps33bflox/flox, alb-cre mice, we detected several bile acid homeostasis-related gene expression levels in the liver (Figure 4). Cyp7a1 converts cholesterol to 7a-hydroxycheolesterol, which is the rate-limiting enzyme of bile acid formation in the liver. The mRNA expression level of Cyp7a1 was downregulated in Vps33bflox/flox, alb-cre mice compared to that in Vps33bflox/flox mice. However, Cyp450s enzymes, such as Cyp2b10 and Cyp3a11, which function in the formation of hydrophilic bile acids, were significantly upregulated in the Vps33bflox/flox, alb-cre mice.
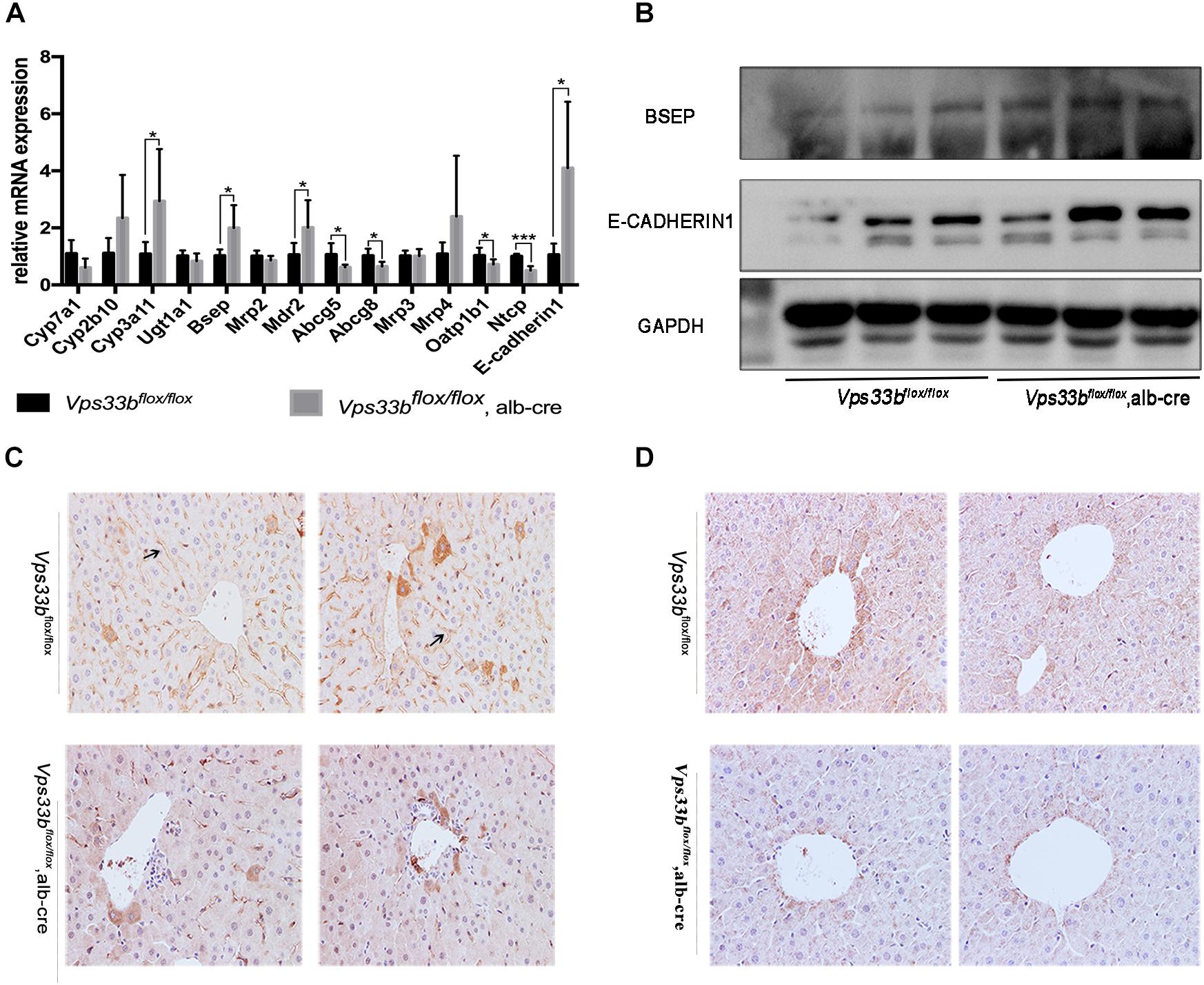
Figure 4. Bile homeostasis-related gene expression levels (A). Western blotting (B) and IHC (C) detection of BSEP, and E-cadherin1 (A,B) and CLAUDIN-1 (D) cell junction protein expression levels.
In addition, mRNA expression of hepatic canalicular membrane transporters (Bsep and Mdr2) were upregulated in Vps33bflox/flox, alb-cre mice, while Abcg5/8 expression was downregulated compared to those in Vps33bflox/flox mice. Another canalicular transporter, Mrp2, was unchanged in Vps33bflox/flox, alb-cre mice. In addition, sinusoidal membrane multidrug resistance protein bile acid efflux transporters (Mrp3 and Mrp4) were not significantly regulated by hepatic Vps33b depletion. However, bile acid reuptake transporters (Oatp1b1 and Ntcp) were significantly downregulated in Vps33bflox/flox, alb-cre mice (Figure 4A).
Vps33b function was related to hepatocyte polarity, which is vital for cell junction formation and specific protein localization. Furthermore, we performed immunohistochemistry staining of the bile acid export pump BSEP and measured the levels of representative cell junction proteins (E-CADHERIN1, CLAUDIN-1). As shown in Figures 4A,B, BSEP mRNA and protein levels were significantly increased in Vps33bflox/flox, alb-cre mice compared to those in Vps33bflox/flox mice. Moreover, BSEP distribution features were subdued in Vps33bflox/flox, alb-cre mice, which were no longer localized along the specific side of hepatocytes. Livers express different kinds of cell tight junction proteins, and we chose E-CADHERIN1 and CLAUDIN-1 for detection. Increased E-CADHERIN1 levels and decreased CLAUDIN-1 levels were observed in Vps33bflox/flox, alb-cre mice compared with those in the Vps33bflox/flox mice (Figures 4B,D). Collectively, these data indicated that Vps33bflox/flox, alb-cre mice displayed disrupted bile acid homeostasis in accordance with an altered targeted-bile acid metabolomics pattern compared to that in Vps33bflox/flox mice. Irregular BSEP localization and altered cell tight junction expression suggested impaired hepatocyte polarity caused by Vps33b hepatic depletion.
Because lipid metabolism is integrally connected to bile acid metabolism, we speculated whether the lipid metabolite pattern was also affected by hepatic Vps33b deletion. Upon UPLC-ESI-HRMS-based lipidomics analysis, we measured serum and liver samples of Vps33bflox/flox, alb-cre mice and Vps33bflox/flox mice to reveal the lipid profiles within specific lipid species.
Principal component analysis of serum and livers showed a clear difference between Vps33bflox/flox and Vps33bflox/flox, alb-cre mice (Figures 5A,E). Liver scatter plots for Vps33bflox/flox mice were in the second quadrants, while scatter plots for Vps33bflox/flox, alb-cre mice were mainly located in the third and fourth quadrants (Figure 5E). To identify the lipids that contribute to the unambiguous separation between Vps33bflox/flox, alb-cre mice and Vps33bflox/flox mice, Lipid Search software was used to identify lipid molecular species based on the accurate mass values of each ion and MS2 pattern. The outstanding matched mass spectrums were shown in Supplementary Figure 5. An OPLS-DA score plot (Figures 5B,F) and s-plot (Figures 5C,G) was performed to screen the specific lipids altered in Vps33bflox/flox, alb-cre mice among all identified lipid molecular species. VIP values > 0.8 and absolute p(corr) value > 0.6 are highlighted in green and red, respectively, in the s-plot (Figures 5D,H). Using the FDR (false discovery rate) test, screened specific lipid species of serum and livers with p-values < 0.05 are shown in the bar graph (Figures 6, 7). It is notable that many lipid species were altered in the serum and liver of hepatic Vps33b-depleted mice according to normalized heatmap pictures (Supplementary Figures 6, 7). TGs were the most abundant lipid species that were altered significantly in serum. Interestingly, regardless of the number of carbons or double bonds in the fatty acid chain, all species of TGs were significantly increased in serum but decreased in the liver of Vps33bflox/flox, alb-cre mice compared with those in the Vps33bflox/flox mice (Figures 6H–J, 7I). SMs were another type of lipid that exhibited a similar altered trend as TGs, in which the number of SM lipids was apparently less than that of TGs (Figures 6B, 7D). In addition, hepatic Vps33b depletion induced an upregulation of ceramides and a downregulation of PE in both serum and liver compared with those of Vps33bflox/flox mice (Figures 6A,E, 7E). Lipids, such as PI, PS and LPC, were significantly increased in the serum of Vps33bflox/flox, alb-cre mice (Figures 6C,D,F). However, some lipids were significantly altered with a different variation trend in Vps33bflox/flox, alb-cre mice, such as PC in serum and PI, PC, and PS in livers (Figures 6G, 7C,F,H). Moreover, PA and CL were increased, and PG was decreased in the livers of Vps33bflox/flox, alb-cre mice, which were not significantly changed in serum compared with those in the Vps33bflox/flox mice.
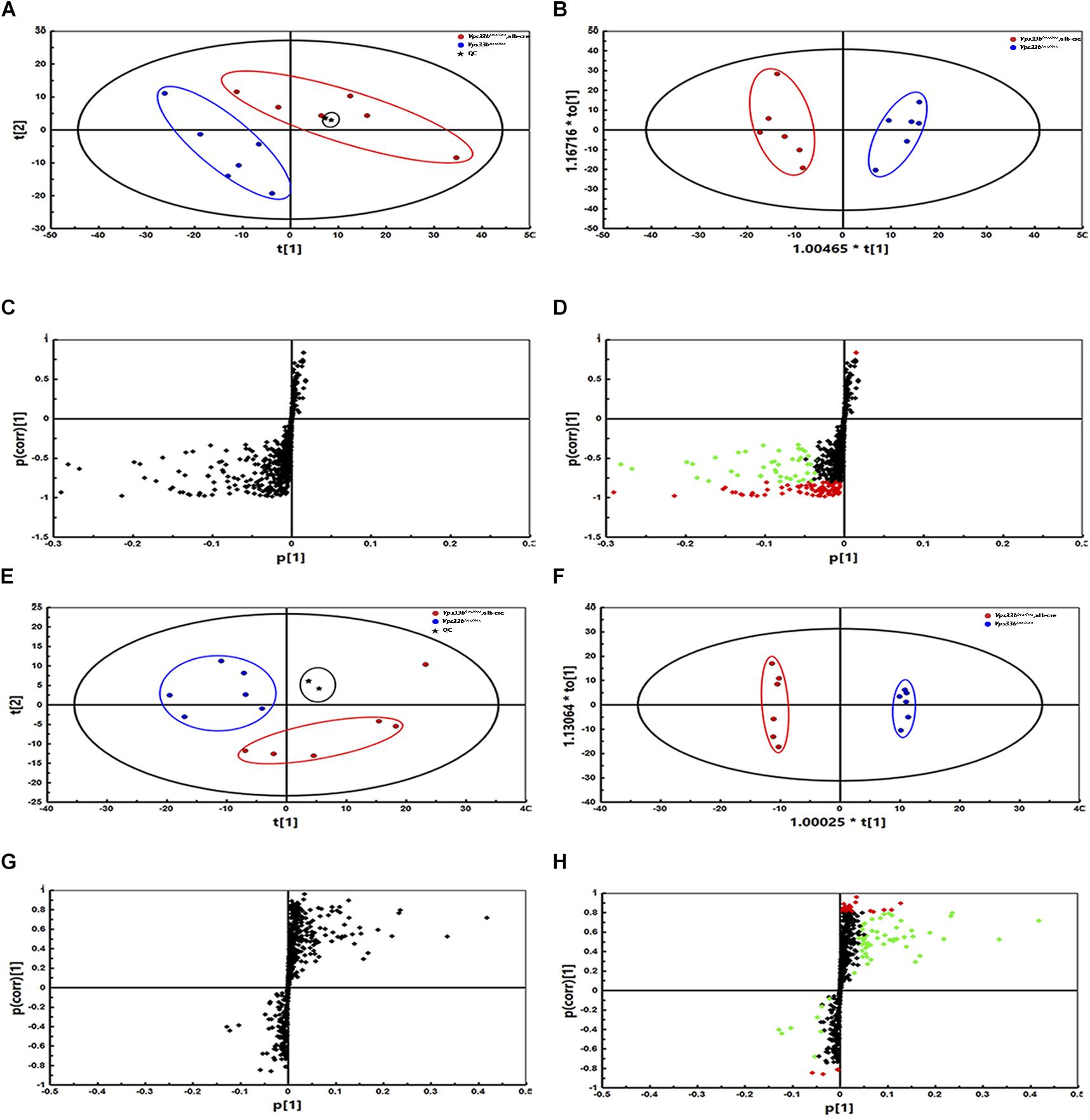
Figure 5. Principal component analysis, OPLS-DA score plot and s-plot of serum and liver lipid profiles in Vps33bflox/flox, alb-cre mice and Vps33bflox/flox mice. The PCA as well as OPLS-DA of serum (A,B) and liver (E,F) show a clear difference between the two groups (blue dots for Vps33bflox/flox mice, red dots for Vps33bflox/flox, alb-cre mice and black dots for QC control). The supervised multivariate analysis shows R2Y = 0.94 and Q2 = 0.877 in serum, while R2Y = 0.99 and Q2 = 0.498 in livers. s-plots (C,D,G,H) highlighted the possible calculated lipid components with p-value less than 0.05. [green dots represent VIP > 0.8, while red dots represent p(corr) < -0.6 or p(corr) > 0.6].
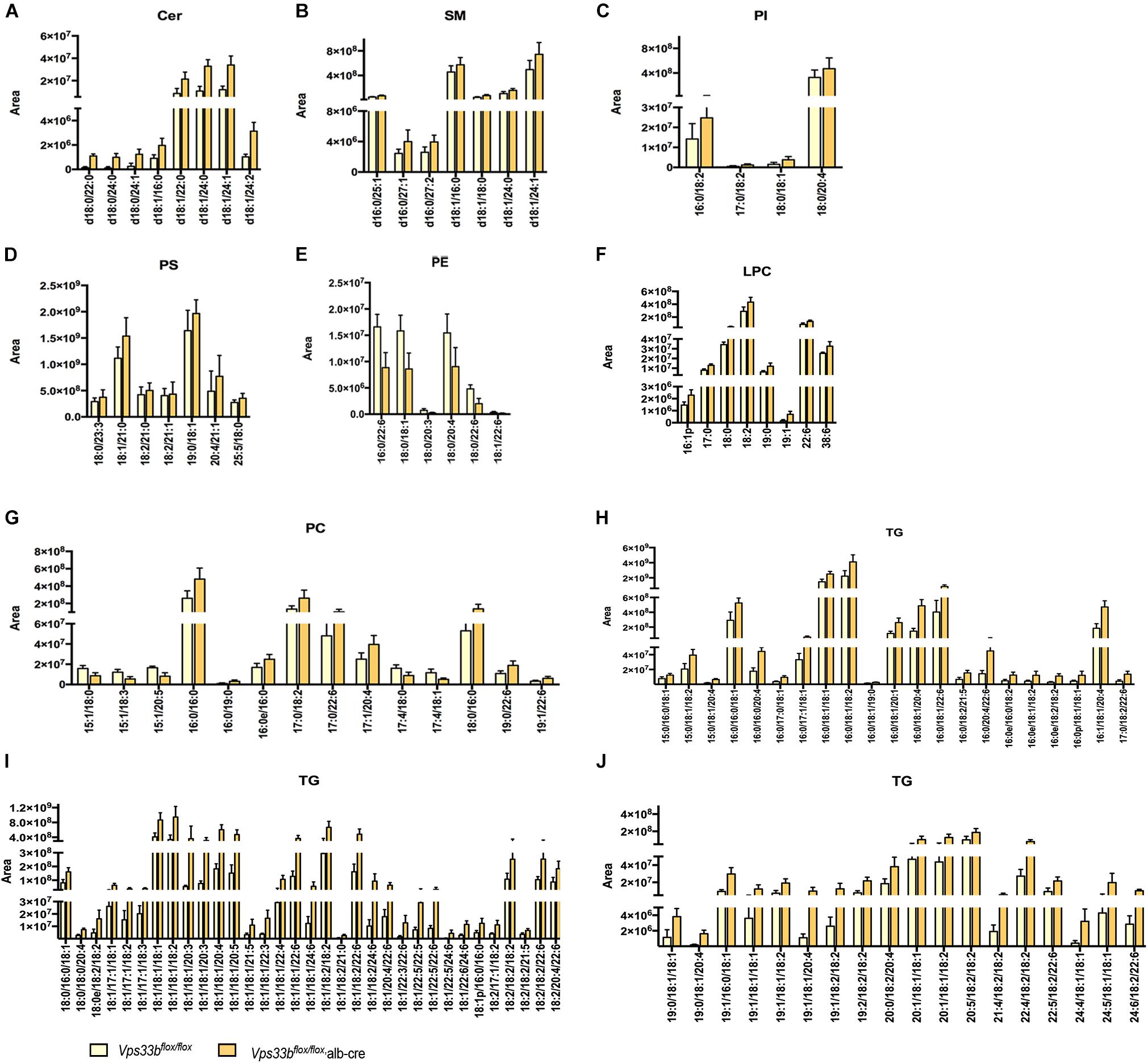
Figure 6. Lipid catalogs and species in serum were significantly different between Vps33bflox/flox, alb-cre and Vps33bflox/flox, mice. [(A) ceramides, (B) SM: sphingomyelin, (C) PI: phosphatidylinositol, (D) PS: phosphatidylserine, (E) PE: phosphatidyl ethanolamine, (F) LPC: lysophosphatidylcholine, (G) PC: phosphatidylcholine, and (H–J) TG: triglyceride].
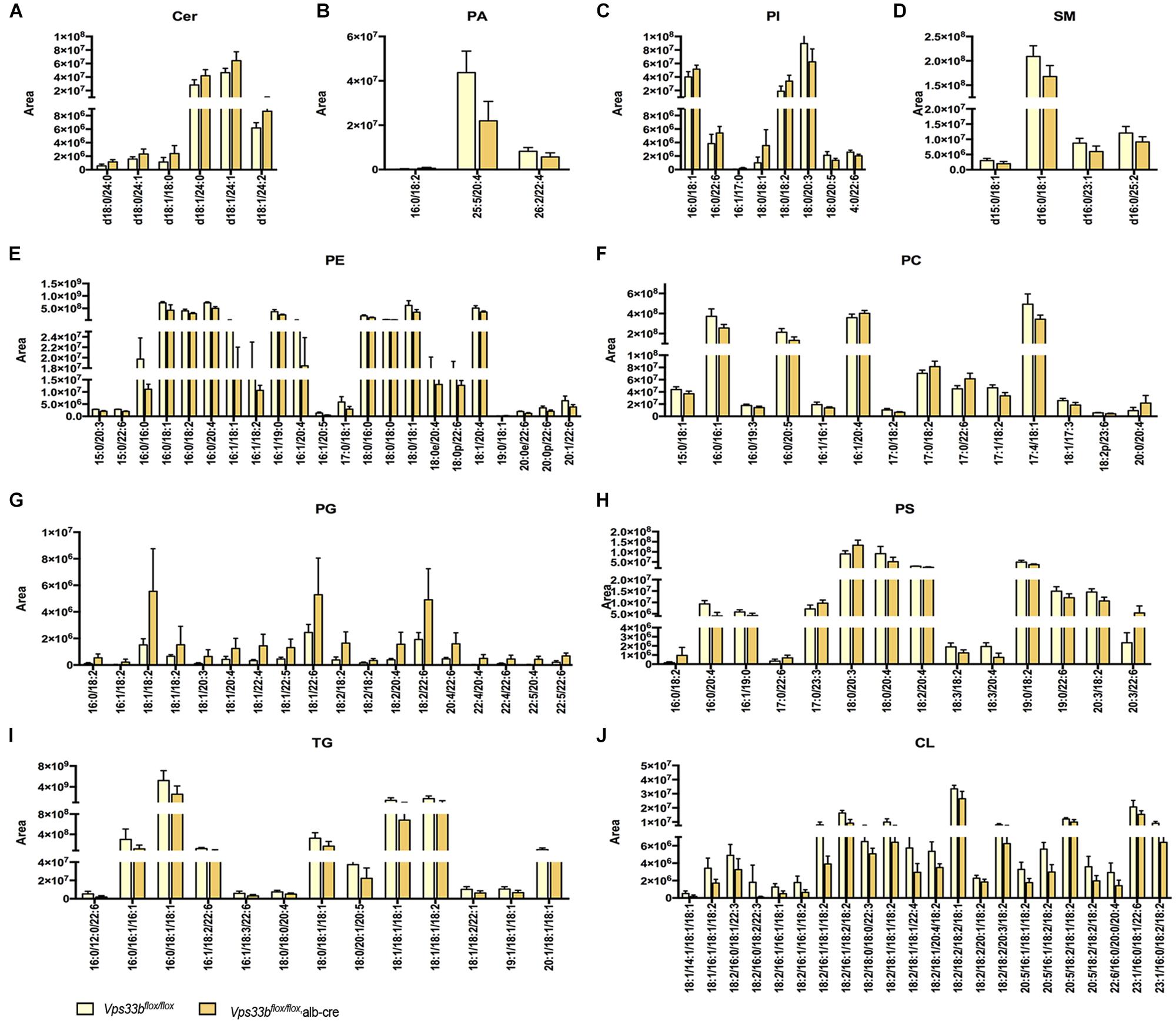
Figure 7. Lipid catalogs and species in the liver were significantly different between Vps33bflox/flox, alb-cre mice and Vps33bflox/flox mice. [(A) ceramides, (B) PA: phosphatidic acid, (C) PI: phosphatidylinositol, (D) SM: sphingomyelin, (E) PE: phosphatidyl ethanolamine, (F) PC: phosphatidylcholine, (G) PG: phosphatidyl glycerol, (H) PS: phosphatidylserine, (I) TG: triglyceride, (J) CL: cardiolipin].
In addition, we detected the expression of lipogenesis-related genes and specific lipid metabolism-related genes. As shown in Figure 8, fatty acid uptake genes were significantly decreased in the livers of Vps33bflox/flox, alb-cre mice compared with those in the Vps33bflox/flox mice, while FA synthesis-related genes displayed an inconsistent change, as Fas and Acc1 increased and Scd1 decreased. TG metabolism-related genes were reduced, especially Pnpla2, in accordance with the decreased level of TG in the livers of Vps33bflox/flox, alb-cre mice. Meanwhile, CL-related genes were decreased as their amount was also lower in the livers of Vps33bflox/flox, alb-cre mice compared with those in the Vps33bflox/flox mice. Lipidomics analysis indicated that hepatic Vps33b regulated the lipid pattern of serum and livers in mice. Without Vps33b expression in the liver, the lipid species were distinct from Vps33bflox/flox mice, which might contribute to the pathology of cholestasis.
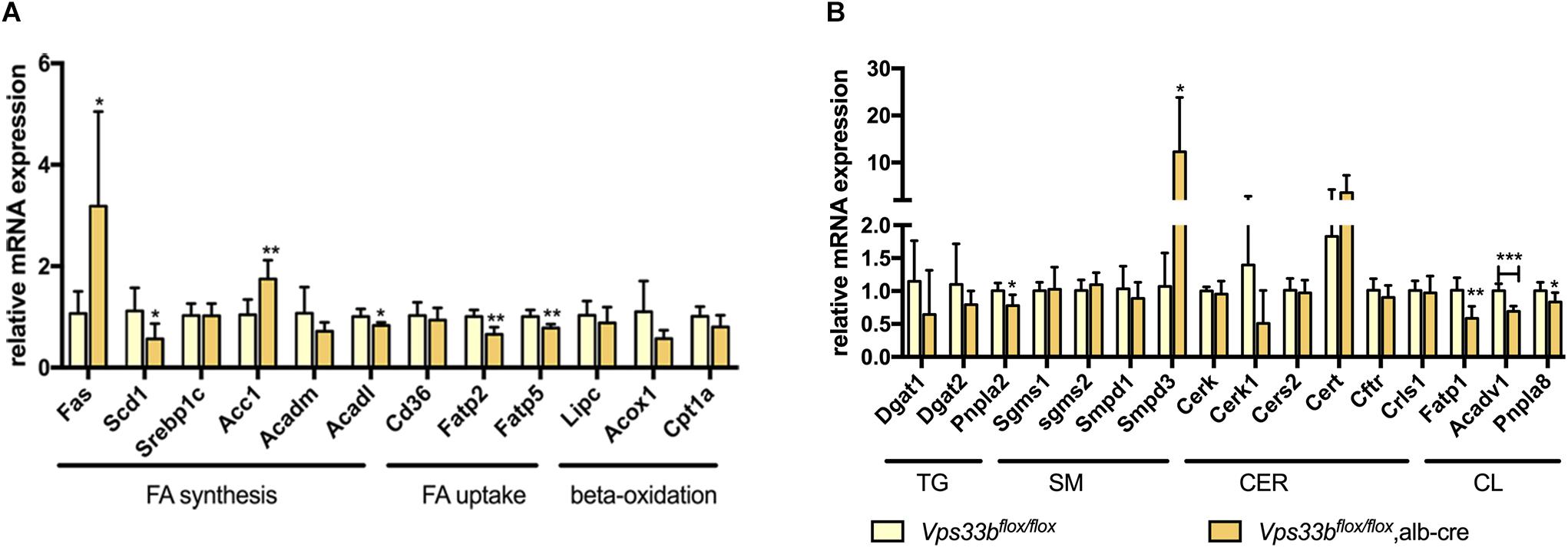
Figure 8. Lipid homeostasis related gene expression levels in the livers of Vps33bflox/flox, alb-cre mice and Vps33bflox/flox mice (A,B). ∗p-value < 0.5, ∗∗p-value < 0.1, and ∗∗∗p-value < 0.001.
ARC syndrome, caused by VPS33B mutations, is a disease involving in multiple organs with a variety of clinical features (Cullinane et al., 2009), and alternative diagnostic approaches should be suggested to replace organ biopsies as a first-line diagnostic test for most children who are suspected to have ARC syndrome (Gissen et al., 2006). Therefore, basic studies have been increasingly performed to determine the tissue-specific function of Vps33b. For example, it has been reported that Vps33b plays an important role in platelet a-granule formation, and megakaryocyte Vps33b-depleted mice were used as an investigation model (Dai et al., 2016). However, few studies have investigated the role of Vps33b in the liver, with the exception of one study, which determined that Vps33b maintains hepatocyte polarity by using hepatic Vps33b-deficient mice (Hanley et al., 2017). Cholestasis is one of the syndrome of ARC, however, the role of hepatic Vps33b deficiency in the bile acids homeostasis and lipid metabolism remains unclear.
In this study, we investigated bile acid deposition and mapped lipid profiles of hepatic Vps33b-depleted mice based on an omics study strategy. We found that hepatic Vps33b depletion caused cholestasis and slight liver injury in mice, which was also commonly observed in ARC patients as previously reported. A targeted bile acid study confirmed disrupted bile acid homeostasis associated with Vps33b deficiency in the liver, and genes involved in bile acid circulation were also transcriptionally regulated. The mis-location of the BSEP apical membrane transporter was evidence of the loss of hepatocyte polarity. Cell junction proteins regulate the injured cell barrier, which might contribute to deteriorating cholestasis (Anderson, 1996). In addition, alterations in lipidomics profiles and the expression of genes involved in lipogenesis revealed a potentially vital role of Vps33b in liver lipid metabolism. Overall, our study characterized metabolic and lipidomic alterations in hepatic Vps33b-depleted mice (Supplementary Figure 8), which provided insights on the function of Vps33b on bile acids and lipid homeostasis.
Hepatic Vps33b knockout mice displayed a higher serum ALP level compared to Vps33bflox/flox mice, which is a marker for liver disease, specifically cholestasis. Additionally, the elevated serum ALT/AST levels indicated slight liver injury, which is similar to clinical ARC syndrome caused by a Vps33b mutation. In the clinic, ARC patients have been characterized by increased bilirubin (Gissen et al., 2006) with normal or lower levels of GGT, mildly elevated ALT/AST levels and substantially increased ALP levels (Malaki et al., 2012; Ilhan et al., 2016). We found that hepatic Vps33b-depleted mice displayed a similar phenotype to ARC patients in accordance with a previous report (Hanley et al., 2017). Therefore, hepatic Vps33b-depleted mice are a valuable experimental model for studying the therapeutic role of Vps33b in ARC syndrome.
Cholestasis is a complicated disease with disrupted bile acid homeostasis that is directly regulated by the synthesis and disposition systems consisting of cytochrome P450 enzymes, bile acid transporters and tight junctions (Gissen and Arias, 2015). We found that several CYP450 enzymes and bile acid transporters were transcriptionally altered in the livers of Vps33bflox/flox, alb-cre mice compared to those in Vps33bflox/flox mice. There is a metabolism loop that stabilizes the bile acid pool. Targeted bile acid metabolomics analysis indicated elevated taurine-conjugated bile acids in serum, which was decreased in the liver and bile of Vps33bflox/flox, alb-cre mice compared with those in Vps33bflox/flox mice. Our results were consistent with previous studies. In a previous study, an increase in TCA levels was observed in the plasma of Vps33bflox/flox, alb-cre mice fed with 0.5% CA chow. Furthermore, it was reported that 0.5% CA-fed Vps33bflox/flox, alb-cre mice showed a slight decrease in TCA levels in bile, but this decrease was not statistically significant (Hanley et al., 2017). Overall, alterations of bile acids among different tissues characterized the pathogenic conditions of cholestasis in hepatic Vps33b-depleted mice. The self-protection function in the liver allows it to attenuate injury under pathogenic conditions, which was observed in our results as the downregulated expression of bile acid synthesis enzyme Cyp7a1 and bile acid reuptake transporters Oatp1b1 and Ntcp. In addition to Bsep, the expression of other bile acid export transporters, Mrp2/3/4, did not change.
A clinical study demonstrated that the VPS33B mutation in ARC patients induced an unusual distribution of canalicular plasma membrane proteins. Liver biopsies of ARC patients revealed a clear disturbance in carcinoembryonic antigen (CEA) localization, which was present only at the canaliculus (Gissen et al., 2004). Furthermore, some BSEPs seemed to be expressed only at the basolateral hepatocyte membrane in the patient’s liver, which is supposed to be sorted to the apical membrane via RAB11A-positive apical recycling endosomes (Cullinane et al., 2010) to export bile acids from hepatocytes to the bile duct. In addition, the localization of another apical membrane transporter of hepatocytes, MRP2, did not change. In our study, the BSEP expression pattern in hepatocytes of hepatic Vps33b-depleted mice was quite different from those of Vps33bflox/flox mice. As an apically localized protein, BSEP might be predominantly distributed in the cytoplasm in the intrahepatic ABC-transporter pool, which is in an inactive state but is trafficked to the apical membrane to function as a bile acid exporting pump (Baier et al., 2006). It is clear that liver BSEP in Vps33bflox/flox mice completed this intracellular trafficking process, which was stained around the apical membrane of hepatocytes and a small bile duct through its 3D structures (de Aguiar Vallim et al., 2013). However, Vps33bflox/flox, alb-cre mice showed more cytoplasmic staining without representative localization in membranes compared to Vps33bflox/flox mice, even though its expression level was elevated in the liver. Overall, we concluded that mislocalization of BSEP contributed to cholestasis caused by hepatic Vps33b deficiency.
Cell junction proteins play an important role in sustaining hepatocyte polarity, which may serve as a belt junction between hepatocytes to prevent bile acid invasion from the bile duct. A study of mIMCD-3 cell polarity clarified that Vps33b deficiency caused structural and functional abnormalities, such as downregulated CLAUDIN-1 and E-CADHERIN1 expression (Cullinane et al., 2010). The absence of CLAUDIN-1, a key regulator of paracellular permeability, leads to a severe autosomal recessive disorder whose phenotype is similar to ARC syndrome (Colegio et al., 2003; Hadj-Rabia et al., 2004). Together with a previous report, CLAUDIN-1 displayed irregular and tortuous distribution in the liver of hepatic Vps33b knockout mice (Hanley et al., 2017), indicating that downregulated expression of CLAUDIN-1 in Vps33bflox/flox, alb-cre mice resulted in abnormalities in hepatocyte polarity. Furthermore, we found a converse change in E-CADHERIN1 expression in Vps33bflox/flox, alb-cre mice, which was supposed to be reduced as characterized by impaired cell polarity. However, the involvement of E-cadherin in regulating liver pathophysiology remains unclear (Gonzalez-Sanchez et al., 2015). As we mentioned previously, Vps33b functions on the apical protein sorting procedure via RAB11A-positive apical recycling endosomes. One possible pathway by which E-CADHERIN1 reaches the cell surface involves the Rab11-positive recycling complex rather than direct movement from the Golgi complex to the plasma membrane (Lock and Stow, 2005). Therefore, the correct localization of E-CADHERIN1, rather than its expression, might also be critical for liver function.
Cholestatic liver disease disturbs lipid absorption and metabolism. Furthermore, biliary secretion of cholesterol and phospholipids via ABCG5/8 and MDR2 in mice plays an important role in lipid homeostasis (Hanley et al., 2017). A previous study reported the mislocalization of ABCG8 in hepatic Vps33b knockout mice. In our study, we found a significant alteration in the expression of these transporters in Vps33bflox/flox, alb-cre mice. Additionally, different serum and liver lipid profiles with altered lipid-related gene expression in the liver were observed in Vps33bflox/flox, alb-cre mice. Serum TG, which is one of the signatures of intrahepatic cholestasis of pregnancy and PFIC in the clinic (Jankowska et al., 2016), showed an increasing trend in Vps33bflox/flox, alb-cre mice, which is consistent with the reduction of mRNA expression of fatty acid uptake genes Fatp2 and Fatp5 and TG synthesis genes.
Sphingomyelins (SMs), which are hydrolyzed by sphingomyelin phosphodiesterases (SMPDs) to form ceramides, act as secondary messengers in many physiological processes including apoptosis. Generally, the SM level is controlled by the balance of SM synthase (SGMS) and SMPD function. SMPD inhibitors have been reported to protect against bile acid-induced primary hepatocyte apoptosis (Qiao et al., 2002). Additionally, LCA-induced intrahepatic cholestasis showed elevated levels of ceramides (Cer) in mouse livers and hepatic expression of Smpd3, which were indicated as key factors that accelerated cholestasis (Matsubara et al., 2011). As we have shown in this study, hepatic Vps33b knockout mice had a disturbed balance of Cer and SM levels in mouse livers, with a significant increase in Smpd3 expression in Vps33bflox/flox, alb-cre mice.
After mapping the lipid profiles in Vps33bflox/flox, alb-cre mice, we found that the lipid components of Vps33b hepatic knockout mice were altered significantly compared to those of Vps33bflox/flox mice, indicating an important role of Vps33b in lipid metabolism. Further study of the relationship between Vps33b and lipid metabolism at the molecular level should be performed.
In summary, we investigated the phenotype of hepatic Vps33b knockout mice, providing additional evidence of the effect of Vps33b on ARC like cholestastic liver injury. In addition, our study demonstrated that Vps33b influences the bile acid homeostasis and lipid metabolism in mice, which is involved in the progression of liver cholestasis, indicating potential therapeutic targets and diagnostic markers of ARC.
All datasets generated for this study are included in the manuscript and/or the Supplementary Files.
KF contributed to the study execution and manuscript preparation. CW supervised the weaning and genotyping of Vps33b hepatic knockout mice. YG, SF, HZ, CL, and LG contributed to the sample extraction for metabolomics studies and data analysis. JS and YJ reviewed the manuscript. HB, MH, and JL supervised the study progress and data analysis, revised the manuscript, and approved the final version of this manuscript for submission.
This study was supported by the National Natural Science Foundation of China (Grants: 81522047, 81573489, 81730103, and 81320108027), the National Key Research and Development Program (Grants: 2017YFE0109900, and 2017YFC0909303), the 111 Project (Grant: B16047), the Natural Science Foundation of Guangdong (Grant: 2017A030311018), and the Guangdong Provincial Key Laboratory of Construction Foundation (Grant: 2017B030314030).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2019.00276/full#supplementary-material
Abdullah, M. A., Al-Hasnan, Z., Okamoto, E., and Abomelha, A. M. (2000). Arthrogryposis, renal dysfunction and cholestasis syndrome. Saudi Med. J. 21, 297–299.
Ackermann, O., Gonzales, E., Keller, M., Guettier, C., Gissen, P., and Jacquemin, E. (2014). Arthrogryposis, renal dysfunction, and cholestasis syndrome caused by VIPAR mutation. J. Pediatr. Gastroenterol. Nutr. 58, e29–e32. doi: 10.1097/MPG.0b013e318298108f
Akbar, M. A., Mandraju, R., Tracy, C., Hu, W., Pasare, C., and Kramer, H. (2016). ARC syndrome-linked Vps33B protein is required for inflammatory endosomal maturation and signal termination. Immunity 45, 267–279. doi: 10.1016/j.immuni.2016.07.010
Anderson, J. M. (1996). Leaky junctions and cholestasis: a tight correlation. Gastroenterology 110, 1662–1665. doi: 10.1053/gast.1996.v110.agast961662
Baier, P. K., Hempel, S., Waldvogel, B., and Baumgartner, U. (2006). Zonation of hepatic bile salt transporters. Dig. Dis. Sci. 51, 587–593. doi: 10.1007/s10620-006-3174-3
Chen, C. H., Lo, R. W., Urban, D., Pluthero, F. G., and Kahr, W. H. (2017). alpha-granule biogenesis: from disease to discovery. Platelets 28, 147–154. doi: 10.1080/09537104.2017.1280599
Chen, P., Li, D., Chen, Y., Sun, J., Fu, K., Guan, L., et al. (2017). p53-mediated regulation of bile acid disposition attenuates cholic acid-induced cholestasis in mice. Br. J. Pharmacol. 174, 4345–4361. doi: 10.1111/bph.14035
Colegio, O. R., Van Itallie, C., Rahner, C., and Anderson, J. M. (2003). Claudin extracellular domains determine paracellular charge selectivity and resistance but not tight junction fibril architecture. Am. J. Physiol. Cell Physiol. 284, C1346–C1354. doi: 10.1152/ajpcell.00547.2002
Cullinane, A. R., Straatman-Iwanowska, A., Seo, J. K., Ko, J. S., Song, K. S., Gizewska, M., et al. (2009). Molecular investigations to improve diagnostic accuracy in patients with ARC syndrome. Hum. Mutat. 30, E330–E337. doi: 10.1002/humu.20900
Cullinane, A. R., Straatman-Iwanowska, A., Zaucker, A., Wakabayashi, Y., Bruce, C. K., Luo, G., et al. (2010). Mutations in VIPAR cause an arthrogryposis, renal dysfunction and cholestasis syndrome phenotype with defects in epithelial polarization. Nat. Genet. 42, 303–312. doi: 10.1038/ng.538
Dai, J., Lu, Y., Wang, C., Chen, X., Fan, X., Gu, H., et al. (2016). Vps33b regulates Vwf-positive vesicular trafficking in megakaryocytes. J. Pathol. 240, 108–119. doi: 10.1002/path.4762
de Aguiar Vallim, T. Q., Tarling, E. J., and Edwards, P. A. (2013). Pleiotropic roles of bile acids in metabolism. Cell Metab. 17, 657–669. doi: 10.1016/j.cmet.2013.03.013
Dehghani, S. M., Bahador, A., Nikeghbalian, S., Salahi, H., Geramizadeh, B., Malekpour, A., et al. (2013). Liver transplant in a case of arthrogryposis-renal tubular dysfunction-cholestasis syndrome with severe intractable pruritus. Exp. Clin. Transplant 11, 290–292. doi: 10.6002/ect.2012.0202
Eastham, K. M., Mckiernan, P. J., Milford, D. V., Ramani, P., Wyllie, J., Van’t Hoff, W., et al. (2001). ARC syndrome: an expanding range of phenotypes. Arch. Dis. Child. 85, 415–420. doi: 10.1136/adc.85.5.415
Gissen, P., and Arias, I. M. (2015). Structural and functional hepatocyte polarity and liver disease. J. Hepatol. 63, 1023–1037. doi: 10.1016/j.jhep.2015.06.015
Gissen, P., Johnson, C. A., Morgan, N. V., Stapelbroek, J. M., Forshew, T., Cooper, W. N., et al. (2004). Mutations in VPS33B, encoding a regulator of SNARE-dependent membrane fusion, cause arthrogryposis-renal dysfunction-cholestasis (ARC) syndrome. Nat. Genet. 36, 400–404. doi: 10.1038/ng1325
Gissen, P., Tee, L., Johnson, C. A., Genin, E., Caliebe, A., Chitayat, D., et al. (2006). Clinical and molecular genetic features of ARC syndrome. Hum. Genet. 120, 396–409. doi: 10.1007/s00439-006-0232-z
Gonzalez-Sanchez, E., Vaquero, J., Fouassier, L., and Chignard, N. (2015). E-cadherin, guardian of liver physiology. Clin. Res. Hepatol. Gastroenterol. 39, 3–6. doi: 10.1016/j.clinre.2014.09.008
Hadj-Rabia, S., Baala, L., Vabres, P., Hamel-Teillac, D., Jacquemin, E., Fabre, M., et al. (2004). Claudin-1 gene mutations in neonatal sclerosing cholangitis associated with ichthyosis: a tight junction disease. Gastroenterology 127, 1386–1390. doi: 10.1053/j.gastro.2004.07.022
Hanley, J., Dhar, D. K., Mazzacuva, F., Fiadeiro, R., Burden, J. J., Lyne, A. M., et al. (2017). Vps33b is crucial for structural and functional hepatocyte polarity. J. Hepatol. 66, 1001–1011. doi: 10.1016/j.jhep.2017.01.001
Hayes, J. A., Kahr, W. H., Lo, B., and Macpherson, B. A. (2004). Liver biopsy complicated by hemorrhage in a patient with ARC syndrome. Paediatr. Anaesth. 14, 960–963. doi: 10.1111/j.1460-9592.2004.01301.x
Horslen, S. P., Quarrell, O. W., and Tanner, M. S. (1994). Liver histology in the arthrogryposis multiplex congenita, renal dysfunction, and cholestasis (ARC)syndrome:report of three new cases and review. J. Med. Genet. 31, 62–64. doi: 10.1136/jmg.31.1.62
Huizing, M., Didier, A., Walenta, J., Anikster, Y., Gahl, W. A., and Kramer, H. (2001). Molecular cloning and characterization of human VPS18, VPS 11, VPS16, and VPS33. Gene 264, 241–247. doi: 10.1016/S0378-1119(01)00333-X
Ilhan, O., Ozer, E. A., Ozdemir, S. A., Akbay, S., Memur, S., Kanar, B., et al. (2016). Arthrogryposis-renal tubular dysfunction-cholestasis syndrome: a cause of neonatal cholestasis. Case report. Arch. Argent Pediatr. 114, e9–e12. doi: 10.5546/aap.2016.eng.e9
Jankowska, I., Czubkowski, P., Wierzbicka, A., Pawlowska, J., Kalicinski, P., and Socha, P. (2016). Influence of partial external biliary diversion on the lipid profile in children with progressive familial intrahepatic cholestasis. J. Pediatr. Gastroenterol. Nutr. 63, 598–602. doi: 10.1097/MPG.0000000000001185
Li, J., Gao, Y., Guan, L., Zhang, H., Sun, J., Gong, X., et al. (2018). Discovery of phosphatidic acid, phosphatidylcholine, and phosphatidylserine as biomarkers for early diagnosis of endometriosis. Front. Physiol. 9:14. doi: 10.3389/fphys.2018.00014
Lobingier, B. T., and Merz, A. J. (2012). Sec1/Munc18 protein Vps33 binds to SNARE domains and the quaternary SNARE complex. Mol. Biol. Cell 23, 4611–4622. doi: 10.1091/mbc.E12-05-0343
Lock, J. G., and Stow, J. L. (2005). Rab11 in recycling endosomes regulates the sorting and basolateral transport of E-cadherin. Mol. Biol. Cell 16, 1744–1755. doi: 10.1091/mbc.e04-10-0867
Malaki, M., Mandana, R., and Ghaffari, S. (2012). ARC syndrome with complex renal problems: nephrocalcinosis, proximal and hyperkalemic distal RTA and nephrogenic diabetes insipidus. Saudi J. Kidney Dis. Transpl. 23, 804–809. doi: 10.4103/1319-2442.98165
Matsubara, T., Tanaka, N., Patterson, A. D., Cho, J. Y., Krausz, K. W., and Gonzalez, F. J. (2011). Lithocholic acid disrupts phospholipid and sphingolipid homeostasis leading to cholestasis in mice. Hepatology 53, 1282–1293. doi: 10.1002/hep.24193
Matyash, V., Liebisch, G., Kurzchalia, T. V., Shevchenko, A., and Schwudke, D. (2008). Lipid extraction by methyl-tert-butyl ether for high-throughput lipidomics. J. Lipid Res. 49, 1137–1146. doi: 10.1194/jlr.D700041-JLR200
Qiao, L., Yacoub, A., Studer, E., Gupta, S., Pei, X. Y., Grant, S., et al. (2002). Inhibition of the MAPK and PI3K pathways enhances UDCA-induced apoptosis in primary rodent hepatocytes. Hepatology 35, 779–789. doi: 10.1053/jhep.2002.32533
Rogerson, C., and Gissen, P. (2018). VPS33B and VIPAR are essential for epidermal lamellar body biogenesis and function. Biochim. Biophys. Acta 1864, 1609–1621. doi: 10.1016/j.bbadis.2018.01.028
Sudhof, T. C., and Rothman, J. E. (2009). Membrane fusion: grappling with SNARE and SM proteins. Science 323, 474–477. doi: 10.1126/science.1161748
Wang, C., Cheng, Y., Zhang, X., Li, N., Zhang, L., Wang, S., et al. (2018). Vacuolar protein sorting 33B Is a tumor suppressor in hepatocarcinogenesis. Hepatology 68, 2239–2253. doi: 10.1002/hep.30077
Zeng, H., Jiang, Y., Chen, P., Fan, X., Li, D., Liu, A., et al. (2017). Schisandrol B protects against cholestatic liver injury through pregnane X receptors. Br. J. Pharmacol. 174, 672–688. doi: 10.1111/bph.13729
Zhang, H., Gao, Y., Sun, J., Fan, S., Yao, X., Ran, X., et al. (2017a). Optimization of lipid extraction and analytical protocols for UHPLC-ESI-HRMS-based lipidomic analysis of adherent mammalian cancer cells. Anal. Bioanal. Chem. 409, 5349–5358. doi: 10.1007/s00216-017-0483-7
Zhang, H., Jiang, Y., Wu, J., Zheng, C., Ran, X., Li, D., et al. (2017b). Metabolic mapping ofSchisandra sphenantheraextract and its active lignans using a metabolomic approach based on ultra high performance liquid chromatography with high-resolution mass spectrometry. J. Separat. Sci. 40, 574–586. doi: 10.1002/jssc.201600954
Keywords: VPS33B, cholestasis, bile acids, metabolomics, lipidomics
Citation: Fu K, Wang C, Gao Y, Fan S, Zhang H, Sun J, Jiang Y, Liu C, Guan L, Liu J, Huang M and Bi H (2019) Metabolomics and Lipidomics Reveal the Effect of Hepatic Vps33b Deficiency on Bile Acids and Lipids Metabolism. Front. Pharmacol. 10:276. doi: 10.3389/fphar.2019.00276
Received: 20 November 2018; Accepted: 04 March 2019;
Published: 22 March 2019.
Edited by:
Houkai Li, Shanghai University of Traditional Chinese Medicine, ChinaCopyright © 2019 Fu, Wang, Gao, Fan, Zhang, Sun, Jiang, Liu, Guan, Liu, Huang and Bi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Junling Liu, bGl1amxAc2hzbXUuZWR1LmNu Huichang Bi, YmloY2hhbmdAbWFpbC5zeXN1LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.