 Ryosuke Fukuda
Ryosuke Fukuda Tsukasa Okiyoneda
Tsukasa Okiyoneda
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Pharmacol. , 27 September 2018
Sec. Pharmacology of Ion Channels and Channelopathies
Volume 9 - 2018 | https://doi.org/10.3389/fphar.2018.01100
This article is part of the Research Topic Emerging Therapeutic Approaches for Cystic Fibrosis View all 23 articles
Conformationally defective cystic fibrosis transmembrane conductance regulator (CFTR) including rescued ΔF508-CFTR is rapidly eliminated from the plasma membrane (PM) even in the presence of a CFTR corrector and potentiator, limiting the therapeutic effort of the combination therapy. CFTR elimination from the PM is determined by the conformation-dependent ubiquitination as a part of the peripheral quality control (PQC) mechanism. Recently, the molecular machineries responsible for the CFTR PQC mechanism which includes molecular chaperones and ubiquitination enzymes have been revealed. This review summarizes the molecular mechanism of the CFTR PQC and discusses the possibility that the peripheral ubiquitination mechanism becomes a novel drug target to develop the CFTR stabilizer as a novel class of CFTR modulator.
Cystic fibrosis (CF) is one of the most lethal autosomal-recessive diseases caused by mutation in CFTR (Lopes-Pacheco, 2016). CFTR mutations are classified as I–VII according to their properties (I–protein synthesis defect, II-maturation defect, III-gating defect, IV-conductance defect, V-reduced quantity, VI-reduced PM stability, VII-no mRNA transcription). The most prevalent CF causing mutation, ΔF508, was classically categorized as class II mutation. However, rescued ΔF508 (rΔF508)-CFTR by corrector (e.g., VX-809/lumacaftor) or low temperature culture shows class III and VI phenotypes (Dalemans et al., 1991; Veit et al., 2016a). Although drug targets of the class II or III mutations are well studied, that of the class VI mutation are not because the mechanism of CFTR PM stability regulation are still veiled by numerous undefined molecules involved in CFTR PQC system. In this review, we summarize accumulated findings regarding the CFTR PQC from the molecular and environmental aspects and also discuss the potential of recently identified PQC machineries including endocytic adaptors and ubiquitination enzymes as targets for CFTR stabilizer which anchors the functional channel at the PM and reduces the degradation (Figure 1).
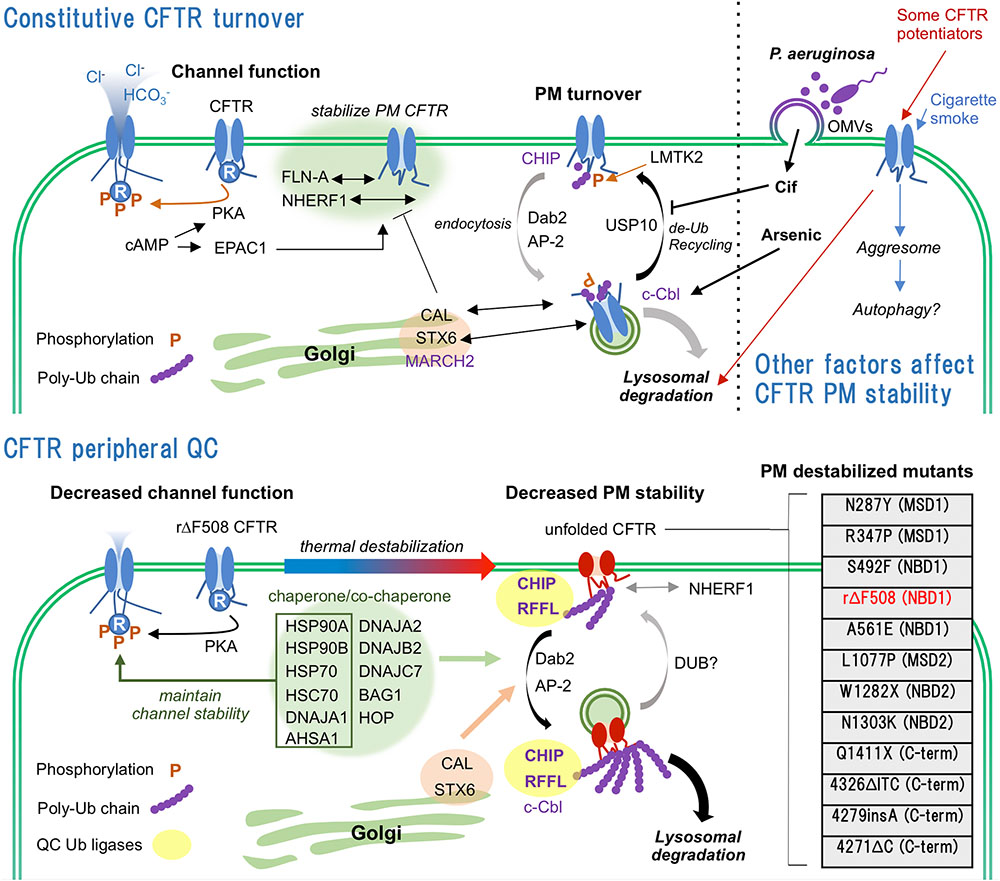
FIGURE 1. Constitutive turnover and PQC of CFTR. (Upper) WT-CFTR is stabilized at the PM by interaction with FLN-A and NHERF1. Internalization of WT-CFTR is regulated by LMTK2-mediated phosphorylation and CHIP-mediated ubiquitination. Predominant endocytosed WT-CFTR is recycled back to the PM by USP10 mediated de-ubiquitination. Golgi localized CAL complex promotes lysosomal sorting of CFTR. CFTR PM stability is deteriorated by infection, CS and CFTR potentiators. (Lower) Unfolded rΔF508-CFTR at the PM is ubiquitinated by PQC Ub ligases CHIP and RFFL, and rapidly internalized probably by CME mediated by DAB2 and AP-2. Chaperone/co-chaperone complex also facilitates the unfolded CFTR internalization while it helps the channel functionality at the PM. Internalized unfolded CFTR could be ubiquitinated at endosomes and targeted to lysosomal degradation. CAL also facilitates the lysosomal degradation of internalized CFTR. PM destabilizing CFTR mutations and their locations are listed (Veit et al., 2016a, Cystic Fibrosis Genetic Consortium Database). N287Y mutation increases CFTR endocytosis without affecting maturation (Silvis et al., 2003). R347P, S492F, rΔF508, A561E, L1077P, and N1303K mutations induce severe maturation defect and PM instability (Van Goor et al., 2014). N287Y and L1077P are localized at intracellular loop 2 (ICL2) of MSD1 and ICL4 of MSD2, respectively. Both mutations are predicted to destabilize the MSD1-NBD2 and MSD2-NBD1 interactions that define CFTR conformational stability.
Nascent wild-type (WT) CFTR is N-glycosylated at the endoplasmic reticulum (ER) during translation and folded by the aid of chaperones such as calnexin (CNX), HSP70 and HSP90 (Amaral, 2004; Kleizen et al., 2005; Okiyoneda et al., 2008; Rosser et al., 2008; Glozman et al., 2009; Kim and Skach, 2012). Properly folded CFTR is then sorted to the Golgi apparatus and processed to complex glycosylation while misfolded CFTR is retained in the ER and consequently degraded by ER-associated degradation (ERAD). The CFTR ERAD is associated with several ER QC processes such as chaperones binding, ubiquitination and retro-translocation from the ER to cytosol (Younger et al., 2006; Turnbull et al., 2007; Morito et al., 2008; Grove et al., 2011; Nery et al., 2011; El Khouri et al., 2013; Matsumura et al., 2013; Ernst et al., 2016; Gong et al., 2016; McClure et al., 2016; Ramachandran et al., 2016; Hou et al., 2018). Properly folded and matured CFTR is trafficked to the PM to function as an ATP-regulated ion-channel (Chen et al., 2000).
The CFTR is internalized by clathrin-mediated endocytosis (CME) and recycled back to the PM. Conformationally defective CFTR produced by genetic mutations (e.g., ΔF508, T70) and/or environmental stresses (e.g., heat) selectively undergoes ubiquitination at the PM by PQC machineries. The ubiquitinated CFTR is rapidly internalized and delivered to lysosome for degradation (Sharma et al., 2004; Okiyoneda et al., 2010). Internalized CFTR could be de-ubiquitinated at endosomes by deubiquitinase (DUB) and recycled back to the PM depending on the conformational states. The class VI mutations render the CFTR unstable at the PM. Additionally, the class I, II and some class III CFTR mutants also show PM instability (Lukacs et al., 1993; Haardt et al., 1999; Silvis et al., 2003; Wang et al., 2014; Veit et al., 2016a). N-glycosylation, especially the core-glycosylation, determines the CFTR PM stability likely by affecting the CFTR conformational stability (Glozman et al., 2009; Cholon et al., 2010). Protein translation kinetics is also a significant factor that modulates proper co-translational folding. Knock down (KD) of ribosomal protein L12 (RPL12) increases ΔF508-CFTR PM expression and stability (Veit et al., 2016b). RPL12 KD might affect protein translation kinetics associated with co-translational protein folding efficiency (Buhr et al., 2016) and thereby improve CFTR thermodynamic stability which also determines the CFTR PM stability (Okiyoneda et al., 2010; Rabeh et al., 2012). Thus, correcting the CFTR structural defects at the ER could improve the PM stability.
The CFTR loss of function induces airway surface liquid (ASL) dysregulation which impairs clearance of infected bacteria and/or fungi, and increases the concentration of other soluble signal mediators such as cytokines, chemokines and growth factors. Pseudomonas aeruginosa (PA) is one of the most common bacteria found in CF respiratory tissue and responsible for lung injury in CF (Koch, 2002; Bhagirath et al., 2016). PA destabilizes PM CFTR by inhibiting endocytic recycling (Swiatecka-Urban et al., 2006). PA secretes CFTR inhibitory factor (Cif) that stabilizes complex formation of ubiquitin (Ub) specific peptidase 10 (USP10) and GTPase activating protein (SH3 domain) binding protein 1 (G3BP1) and inhibits CFTR-USP10 interaction. Cif inhibits internalized CFTR sorting to recycling pathway by suppressing USP10 dependent CFTR de-ubiquitination at endosome, resulting in the lysosomal degradation of WT-CFTR (Bomberger et al., 2011). PA also activates transforming growth factor β1 (TGF-β1) signaling that is an important modifier of lung disease severity in CF (Harris et al., 2011). TGF-β1 inhibits functional PM expression of WT-CFTR and ΔF508-CFTR by reducing mRNA level (Snodgrass et al., 2013; Sun et al., 2014) although its role in the PQC remains unknown.
More than 10 ppb of arsenic induces the WT-CFTR ubiquitination and lysosomal degradation via c-Cbl in CF bronchial epithelial (CFBE) cells (Bomberger et al., 2012). Importantly, the phenotype of arsenic toxicity overlaps with CF patient (Bomberger et al., 2012; Mazumdar et al., 2015). Cadmium (Cd) is a major component of cigarette smoke (CS), and its inhalation is associated with decreased pulmonary function and chronic obstructive pulmonary disease. Cd reduces CFTR PM level, but it remains unknown if it reduces the PM stability (Rennolds et al., 2010).
Cigarette smoke is a major risk factor of chronic obstructive pulmonary disease and interferes with CFTR functionality. Ten minutes of CS exposure transiently suppresses CFTR function, induces internalization and decreases ASL height in human bronchial epithelial (HBE) cells (Clunes et al., 2012). CS promotes CFTR internalization in BHK cells and results in increased insolubility of CFTR and colocalization with vimentin, a filament protein associated with aggresome Ca2+ dependently. This observation suggesting that CS induces PM CFTR destabilization by stimulating internalization and aggregation in addition to suppressing CFTR functionality (Clunes et al., 2012; Rasmussen et al., 2014).
Endocytosis is the critical step of elimination of PM CFTR as a part of PQC and is regulated by several molecules. WT-CFTR is internalized slowly by CME while misfolded rΔF508-CFTR endocytosis is accelerated (Sharma et al., 2004; Swiatecka-Urban et al., 2005; Varga et al., 2008; Okiyoneda et al., 2010). KD of CME adaptor AP-2 μ2 subunit or disabled 2 (DAB2) stabilizes rΔF508-CFTR at the PM by inhibiting endocytosis (Fu et al., 2012, 2015).
CFTR has a postsynaptic density 95, disks large, zonula occludens-1 (PDZ) binding motif at C-terminus and binds with Na+/H+ exchanger regulatory factor (NHERF1) PDZ domain. NHERF1 tethers CFTR with Ezrin and works as a scaffold protein that supports CFTR efficient channel activation and apical PM localization (Favia et al., 2010; Arora et al., 2014; Loureiro et al., 2015). NHERF1 also binds to misfolded ΔF508-CFTR and increases the PM stability by inhibiting carboxy terminus of HSP70-interacting protein (CHIP) Ub ligase interaction (Loureiro et al., 2015). An exchange protein directly activated by cAMP1 (EPAC1) selective activating cAMP analog 007-AM promotes WT-CFTR and NHERF1 interaction and increases CFTR PM stability in CFBE cells by suppressing endocytosis (Lobo et al., 2016). EPAC1 activation can rescue ΔF508-CFTR PM expression, and its effect is further improved with VX-809 combination (Lobo et al., 2016).
The CFTR-associated ligand (CAL) negatively regulates ΔF508-CFTR PM abundance through its PDZ domain (Wolde et al., 2007). CAL inhibition enhances the functional stability of ΔF508-CFTR at the apical PM, implying an attractive therapeutic target for CFTR PM stabilizer (Cushing et al., 2010). However, CAL also interacts with syntaxin 6 (STX6) and Golgi-localized E3-ligase membrane associated RING-CH type finger 2 (MARCH2) and regulates WT-CFTR PM expression (Wolde et al., 2007; Cheng and Guggino, 2013).
Filamin-A (FLN-A) is a membrane tethered actin adaptor protein and interacts with CFTR N-terminus region. S13F mutation of CFTR compromises FLN-A binding and consequently destabilizes the PM CFTR (Thelin et al., 2007). FLN-A binds with both WT and rΔF508-CFTR at similar level, however, its contribution to the CFTR PQC remains unclear.
The CFTR PM stability is regulated by phosphorylation. CFTR is predominantly phosphorylated at the R domain and also at nucleotide binding domain 1 (NBD1) and C-terminus residues by protein kinase A (PKA), protein kinase C (PKC), casein kinase II (CK2) and AMP-activated protein kinase (AMPK) for the channel function (Chappe et al., 2003; Kongsuphol et al., 2009; Luz et al., 2011). CK2 is predicted to regulate CFTR PM stability by phosphorylation at Thr-1471 where NHERF1 could interact (Venerando et al., 2013). Lemur tyrosine kinase 2 (LMTK2) phosphorylates CFTR at Ser-737 (Wang and Brautigan, 2006) and its KD or mutation at CFTR Ser-737 suppresses the endocytosis and increases CFTR PM density and stability (Luz et al., 2014). However, LMKT2 KD only modestly improves the PM function of rΔF508-CFTR (Luz et al., 2014). Spleen tyrosine kinase (SYK) phosphorylates CFTR at Tyr-512 and decreases CFTR PM levels possibly by triggering endocytosis (Luz et al., 2011; Mendes et al., 2011). Mixed-lineage kinase 3 (MLK3) pathway regulates not only ΔF508-CFTR ERQC, but also the PQC by regulating the CFTR proteostasis (Hegde et al., 2015). Inhibition of MLK3 pathway could regulate ΔF508-CFTR folding/degradation switch by impairing interaction with PQC machinery such as HSP70/HSP90 Organizing Protein (HOP) (Hegde et al., 2015).
Molecular chaperones selectively interact with and stabilize unfolded or partially folded protein to acquire a functionally active conformation. Nascent CFTR interacts with a panel of chaperones and co-chaperones including HSC70, HSP70, HSP90, and CNX at the ER (Yang et al., 1993; Pind et al., 1994; Loo et al., 1998; Meacham et al., 1999; Okiyoneda et al., 2004). Even at the post-ER compartments, conformationally defective CFTR such as unfolded rΔF508-CFTR is recognized by chaperone/co-chaperone complex (Okiyoneda et al., 2010). HSC70/HSP90 complex selectively interacts with unfolded rΔF508-CFTR at the post-Golgi and this interaction is crucial for the unfolding dependent ubiquitination (Okiyoneda et al., 2010). KD of HSC70/HSP90 complex (HSP90, HSC70, HOP, AHA1, DNAJB2, DNAJA1, BAG1) increases the rΔF508-CFTR PM stability in HeLa cells (Okiyoneda et al., 2010). The HSC70/HSP90 complex is also essential for maintaining kinetic and thermodynamic stability of rΔF508-CFTR at the PM by reshaping the CFTR conformation during energetic destabilization (Bagdany et al., 2017). This chaperone activity also maintains rΔF508-CFTR channel function at the PM (Bagdany et al., 2017). Thus, modulating the chaperone activity would be a viable target for attenuating the ubiquitination and for stabilizing the CFTR function at the PM.
Ubiquitination determines CFTR elimination not only at the ER, but also from the PM. Ubiquitination is mediated by a sequential action of E1, E2, and E3 enzymes and this modification could be removed by DUB. Specifically, E3 Ub ligase has been proposed to determine the substrate specificity. CHIP is the first identified E3 ligase responsible for the CFTR PQC (Okiyoneda et al., 2010). Consistent with the action at the ER (Meacham et al., 2001), CHIP selectively interacts with unfolded ΔF508-CFTR at the post-Golgi through the HSC70/HSP90 chaperones. CHIP KD reduces the ubiquitination of unfolded ΔF508-CFTR, resulting in the decelerated endocytosis and lysosomal delivery in HeLa cells (Okiyoneda et al., 2010). CHIP KD also stabilizes rΔF508-CFTR at the PM of polarized CFBE cells (Fu et al., 2015).
E3 ligase c-Cbl may play a role in the CFTR peripheral QC, but its contribution could be modest since its KD slightly increases rΔF508-CFTR PM stability in CFBE cells (Cihil et al., 2013; Fu et al., 2015). c-Cbl also binds with WT-CFTR and decreases the PM stability without affecting the ubiquitination, suggesting that c-Cbl could regulate constitutive PM turnover of folded CFTR by inducing endocytosis through its C-terminus adaptor function (Ye et al., 2010).
Nedd4-2 is a member of homologous to the E6-AP carboxyl terminus (HECT) E3 which may regulate the CFTR PM expression. Nedd4-2 KD reduces ΔF508-CFTR ubiquitination at the ER, and increases the PM expression and function in CF pancreatic adenocarcinoma cell 1 (CFPAC1) and IB3-1 cells (Caohuy et al., 2009). Nedd4-2 binds both WT- and ΔF508-CFTR while its role in the WT-CFTR ubiquitination remains controversial (Koeppen et al., 2012). However, Nedd4-2 KD does not stabilize the PM rΔF508-CFTR in CFBE cells, implying its marginal contribution to the CFTR PQC (Koeppen et al., 2012; Fu et al., 2015). Nedd4-2 is unlikely a viable CF drug target because its knock out (KO) induces CF-like lung phenotype by excessive function of epithelial Na+ Channel (ENaC) (Kimura et al., 2011; Rotin and Staub, 2012).
A number of DUBs regulate the CFTR turnover. USP10, a DUB localized at early endosomes, interacts with WT-CFTR and reduces the CFTR poly-ubiquination in CFBE cells. The USP10-mediated deubiquitination enhances the endocytic recycling of WT-CFTR (Bomberger et al., 2009). The role of USP10 in the PM stability of conformationally defective CFTR such as rΔF508-CFTR remains unclear.
Recently, we have discovered RING finger and FYVE like domain containing E3 Ub protein ligase (RFFL) as a novel component of the CFTR PQC machineries by a comprehensive siRNA screen in CFBE cells (Okiyoneda et al., 2018). RFFL selectively recognizes unfolded rΔF508-CFTR through the disordered regions. RFFL promotes K63-linked poly-ubiquitination of the unfolded CFTR in post-Golgi, resulting in accelerated endocytosis and lysosomal degradation. Importantly, RFFL directly interacts with conformationally defective CFTR such as rΔF508-CFTR, but not with folded WT-CFTR at the PM and endosomes. Moreover, the RFFL-mediated ubiquitination is conformation dependent as it selectively ubiquitinates thermally unfolded NBD1. RFFL KD enhances the functional PM expression of rΔF508-CFTR in the presence of VX-809, and this effect is further improved by inhibiting the HSC70-dependent ubiquitination machinery. Thus, RFFL plays an important role in the chaperone-independent CFTR PQC mechanism in HBE cell models.
Pharmacological chaperones affect the CFTR PM stability by direct stabilization. CFTR corrector VX-809 is the first food and drug administration (FDA) approved CFTR corrector in combination with VX-770/ivacaftor (known as Orkambi). VX-809 selectively improves the processing of misfolded CFTR by stabilizing NBD1-membrane spanning domain (MSD) interface but not other misfolded proteins such as human ether-à-go-go-related gene (hERG) mutants (Van Goor et al., 2011; Okiyoneda et al., 2013; Ren et al., 2013; Farinha et al., 2015). VX-809 repairs not only the CFTR folding defect at the ER but also the CFTR PM instability. VX-809 washout prolongs ΔF508-CFTR functional sustainability (Van Goor et al., 2011), suggesting that improvement of the CFTR folding at the ER could increase the thermal stability and proper co- and/or post-translational modifications that renders CFTR more energetic robust conformations even at the PM. VX-809 also promotes ΔF508-CFTR and NHERF1 interaction, that may increase the PM stability (Arora et al., 2014). C3 (CFcor-325/VRT-325) and C4 (Corr-4a) also extend rΔF508-CFTR PM stability in CFBE cells probably by directing binding (Wang et al., 2007; Varga et al., 2008) although their effect could be not specific to the conformationally defective CFTR (Van Goor et al., 2011). Chemical chaperones such as glycerol also increases the rΔF508-CFTR PM stability probably by non-specifically improving the conformational stability (Okiyoneda et al., 2013).
The first FDA approved CFTR potentiator VX-770 improves the gating defect of some CFTR mutants. However, chronic VX-770 treatment destabilizes the PM rΔF508-CFTR in CFBE and ΔF508 homozygous CF patient HBE (CF-HBE) cells (Cholon et al., 2014; Veit et al., 2014). Importantly, chronic VX-770 treatment diminishes the VX-809 therapeutic efficacy by stimulating the elimination of PM rΔF508-CFTR (Cholon et al., 2014; Veit et al., 2014). In addition to VX-770, several CFTR potentiators including P1 (VRT-532) and P2 (PG-01) also decrease the rΔF508-CFTR PM stability (Veit et al., 2014). VX-770 and other potentiators could destabilize a variety of CFTR rare mutants referred to as CFTR2 mutants including E92K and L1077P at the PM (Avramescu et al., 2017). Thus, several CFTR potentiators may decrease the thermal stability of metastable mutant CFTR at the PM by inducing conformational change that positively affects for channel gating but negatively affects stability. High-throughput screening has identified several novel CFTR potentiators such as class A analog 4 (A04) and class P analog 12 (P12) that could not destabilize the PM rΔF508-CFTR (Phuan et al., 2015).
Proteostasis regulating drugs that affect array of proteins regulating CFTR folding and QC also affect the CFTR PM stability. Histone deacetylase (HDAC) inhibitor suberoylanilide hydroxamic acid (SAHA) alters expression of a subset of CF-interacting gene products (e.g., chaperones and DAB2) and sustains PM expression of ΔF508-CFTR in CFBE cells (Hutt et al., 2010). Tissue transglutaminase (TGM2) inhibitor cystamine also stabilizes ΔF508-CFTR at the PM of airway epithelial cells by restoring BECN1 interactome which is sequestrated by CFTR dysfunction (Luciani et al., 2012; Villella et al., 2013). MLK3 pathway inhibitor oxozeaenol has been reported to be effective in correcting the ΔF508-CFTR proteostasis defect in the primary HBE cells (Trzcinska-Daneluti et al., 2012). Oxozeaenol could stabilize ΔF508-CFTR at the PM as MLK3 KD reduces mature ΔF508-CFTR elimination by PQC (Hegde et al., 2015).
HSP70/HSP90 Organizing Protein is adaptor protein which coordinates HSP70 and HSP90 function in protein folding and regulates CFTR maturation and PM stability (Odunuga et al., 2004; Okiyoneda et al., 2010). HOP S-nitrosylation by S-nitrosoglutathione (GSNO) induces HOP degradation and increases ΔF508-CFTR PM expression (Marozkina et al., 2010). Levels of S-nitrosothiols such as GSNO are low in CF airway (Grasemann et al., 1999) and S-nitrosothiol decreases the internalization rate of rΔF508-CFTR in HBE cells (Grasemann et al., 1999, 2000; Zaman et al., 2014). Cavosonstat (N91115) is an orally bioavailable inhibitor of GSNO reductase and restores GSNO levels (Donaldson et al., 2017). Cavosonstat is the first CFTR stabilizer in phase II trials, but it was not beneficial for improvement of lung function in combination with ivacaftor.
CFTR-associated ligand binds CFTR via a PDZ interaction domain and targets CFTR for lysosomal degradation (Cheng et al., 2004). CAL inhibition increases the PM stability of ΔF508-CFTR (Cushing et al., 2010) and cell penetrating CAL inhibiting peptide is established (Qian et al., 2015). CAL inhibitor has been developed as a cell surface CFTR stabilizer in pre-clinical level while its therapeutic efficacy and conformational selectivity remain unclear.
RING finger protein 5 (RNF5/RMA1) is an ER associated E3 Ub ligase that regulates early stage CFTR proteostasis at the ER (Younger et al., 2006). A RNF5 inhibitor Inh-2 identified by homology modeling and virtual ligand screening causes significant rescue of ΔF508-CFTR in immortalized and primary HBE cells from CF patients (Sondo et al., 2018). Intriguingly, Inh-2 modestly increases mature ΔF508-CFTR half-life and this stabilization effect is further improved by VX-809. While the contribution of RNF5 in the CFTR peripheral QC remains unclear, RNF5 inhibitor may be useful to overcome the CFTR instability.
Currently, CHIP and RFFL are the only Ub ligases responsible for the CFTR peripheral QC (Okiyoneda et al., 2010, 2018). Thus, inhibiting their activity could selectively reduce the ubiquitination and elimination of unfolded CFTR from the PM, improving the limited efficacy of CF combination therapy. CHIP binds and regulates a number of substrates via chaperones (Connell et al., 2001). Moreover, inhibiting the CHIP activity induces deleterious effect as the CHIP KO mice result in the abnormal phenotypes including ataxia and pre-mature death1. In contrast, RFFL could bind and regulate a limited number of substrates because of its nature of direct binding to the CFTR through the disordered regions (Okiyoneda et al., 2018). More importantly, inhibiting the RFFL activity seems to have no venomousness since the RFFL KO mice exhibit no abnormal phenotype (Ahmed et al., 2009). Therefore, counteracting RFFL activity may provide a preferable therapeutic approach as a CFTR stabilizer that is a class of drugs that extends the PM resident time of CFTR class VI mutants. Although future studies are needed to validate the impact on ΔF508-CFTR in CF-HBE cells, developing agents selectively inhibiting RFFL-mediated CFTR ubiquitination may help improve the efficacy of CF pharmacological therapy.
Beside the progresses of CF pharmacological therapy, stabilizing the cell surface CFTR remains challenging and is necessary to improve the limited therapeutic efficacy. Recent studies have revealed some of the CFTR PQC mechanism eliminating the functional but conformationally defective CFTR from the PM. Understanding the CFTR PQC mechanism help the development of the CFTR stabilizer, a novel class of CFTR modulator necessary to establish the robust CF pharmacological therapy.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This work was supported by JSPS KAKENHI grant numbers JP25893275, 15H05643, and 15H01192.
TO has a patent pending in Japan for methodology to identify inhibitors of RFFL-mediated CFTR ubiquitination (2017-047626).
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Ahmed, A. U., Moulin, M., Coumailleau, F., Wong, W. W., Miasari, M., Carter, H., et al. (2009). CARP2 deficiency does not alter induction of NF-kappaB by TNFalpha. Curr. Biol. 19, R15–R17. doi: 10.1016/j.cub.2008.11.040
Amaral, M. D. (2004). CFTR and chaperones: processing and degradation. J. Mol. Neurosci. 23, 41–48. doi: 10.1385/JMN:23:1-2:041
Arora, K., Moon, C., Zhang, W., Yarlagadda, S., Penmatsa, H., Ren, A., et al. (2014). Stabilizing rescued surface-localized δf508 CFTR by potentiation of its interaction with Na( + )/H( + ) exchanger regulatory factor 1. Biochemistry 53, 4169–4179. doi: 10.1021/bi401263h
Avramescu, R. G., Kai, Y., Xu, H., Bidaud-Meynard, A., Schnúr, A., Frenkiel, S., et al. (2017). Mutation-specific downregulation of CFTR2 variants by gating potentiators. Hum. Mol. Genet. 26, 4873–4885. doi: 10.1093/hmg/ddx367
Bagdany, M., Veit, G., Fukuda, R., Avramescu, R. G., Okiyoneda, T., Baaklini, I., et al. (2017). Chaperones rescue the energetic landscape of mutant CFTR at single molecule and in cell. Nat. Commun. 8:398. doi: 10.1038/s41467-017-00444-4
Bhagirath, A. Y., Li, Y., Somayajula, D., Dadashi, M., Badr, S., and Duan, K. (2016). Cystic fibrosis lung environment and Pseudomonas aeruginosa infection. BMC Pulm. Med. 16:174. doi: 10.1186/s12890-016-0339-5
Bomberger, J. M., Barnaby, R. L., and Stanton, B. A. (2009). The deubiquitinating enzyme USP10 regulates the post-endocytic sorting of cystic fibrosis transmembrane conductance regulator in airway epithelial cells. J. Biol. Chem. 284, 18778–18789. doi: 10.1074/jbc.M109.001685
Bomberger, J. M., Coutermarsh, B. A., Barnaby, R. L., and Stanton, B. A. (2012). Arsenic promotes ubiquitinylation and lysosomal degradation of cystic fibrosis transmembrane conductance regulator (CFTR) chloride channels in human airway epithelial cells. J. Biol. Chem. 287, 17130–17139. doi: 10.1074/jbc.M111.338855
Bomberger, J. M., Ye, S., Maceachran, D. P., Koeppen, K., Barnaby, R. L., O’Toole, G. A., et al. (2011). A Pseudomonas aeruginosa toxin that hijacks the host ubiquitin proteolytic system. PLoS Pathog. 7:e1001325. doi: 10.1371/journal.ppat.1001325
Buhr, F., Jha, S., Thommen, M., Mittelstaet, J., Kutz, F., Schwalbe, H., et al. (2016). Synonymous codons direct cotranslational folding toward different protein conformations. Mol. Cell. 61, 341–351. doi: 10.1016/j.molcel.2016.01.008
Caohuy, H., Jozwik, C., and Pollard, H. B. (2009). Rescue of DeltaF508-CFTR by the SGK1/Nedd4-2 signaling pathway. J. Biol. Chem. 284, 25241–25253. doi: 10.1074/jbc.M109.035345
Chappe, V., Hinkson, D. A., Zhu, T., Chang, X. B., Riordan, J. R., and Hanrahan, J. W. (2003). Phosphorylation of protein kinase C sites in NBD1 and the R domain control CFTR channel activation by PKA. J. Physiol. 548, 39–52. doi: 10.1113/jphysiol.2002.035790
Chen, E. Y., Bartlett, M. C., and Clarke, D. M. (2000). Cystic fibrosis transmembrane conductance regulator has an altered structure when its maturation is inhibited. Biochemistry 39, 3797–3803. doi: 10.1021/bi992620m
Cheng, J., and Guggino, W. (2013). Ubiquitination and degradation of CFTR by the E3 ubiquitin ligase MARCH2 through its association with adaptor proteins CAL and STX6. PLoS One 8:e68001. doi: 10.1371/journal.pone.0068001
Cheng, J., Wang, H., and Guggino, W. B. (2004). Modulation of mature cystic fibrosis transmembrane regulator protein by the PDZ domain protein CAL. J. Biol. Chem. 279, 1892–1898. doi: 10.1074/jbc.M308640200
Cholon, D. M., O’Neal, W. K., Randell, S. H., Riordan, J. R., and Gentzsch, M. (2010). Modulation of endocytic trafficking and apical stability of CFTR in primary human airway epithelial cultures. Am. J. Physiol. Lung. Cell Mol. Physiol. 298, L304–L314. doi: 10.1152/ajplung.00016.2009
Cholon, D. M., Quinney, N. L., Fulcher, M. L., Esther, C. R., Das, J., Dokholyan, N. V., et al. (2014). Potentiator ivacaftor abrogates pharmacological correction of ΔF508 CFTR in cystic fibrosis. Sci. Transl. Med. 6:246ra96. doi: 10.1126/scitranslmed.3008680
Cihil, K. M., Zimnik, A., and Swiatecka-Urban, A. (2013). c-Cbl reduces stability of rescued ΔF508-CFTR in human airway epithelial cells: implications for cystic fibrosis treatment. Commun. Integr. Biol. 6:e23094. doi: 10.4161/cib.23094
Clunes, L. A., Davies, C. M., Coakley, R. D., Aleksandrov, A. A., Henderson, A. G., Zeman, K. L., et al. (2012). Cigarette smoke exposure induces CFTR internalization and insolubility, leading to airway surface liquid dehydration. FASEB J. 26, 533–545. doi: 10.1096/fj.11-192377
Connell, P., Ballinger, C. A., Jiang, J., Wu, Y., Thompson, L. J., Höhfeld, J., et al. (2001). The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins. Nat. Cell Biol. 3, 93–96. doi: 10.1038/35050618
Cushing, P. R., Vouilleme, L., Pellegrini, M., Boisguerin, P., and Madden, D. R. (2010). A stabilizing influence: CAL PDZ inhibition extends the half-life of ΔF508-CFTR. Angew. Chem. Int. Ed. Engl. 49, 9907–9911. doi: 10.1002/anie.201005585
Dalemans, W., Barbry, P., Champigny, G., Jallat, S., Dott, K., Dreyer, D., et al. (1991). Altered chloride ion channel kinetics associated with the delta F508 cystic fibrosis mutation. Nature 354, 526–528. doi: 10.1038/354526a0
Donaldson, S. H., Solomon, G. M., Zeitlin, P. L., Flume, P. A., Casey, A., McCoy, K., et al. (2017). Pharmacokinetics and safety of cavosonstat (N91115) in healthy and cystic fibrosis adults homozygous for F508DEL-CFTR. J. Cyst. Fibros 16, 371–379. doi: 10.1016/j.jcf.2017.01.009
El Khouri, E., Le Pavec, G., Toledano, M. B., and Delaunay-Moisan, A. (2013). RNF185 is a novel E3 ligase of endoplasmic reticulum-associated degradation (ERAD) that targets cystic fibrosis transmembrane conductance regulator (CFTR). J. Biol. Chem. 288, 31177–31191. doi: 10.1074/jbc.M113.470500
Ernst, W. L., Shome, K., Wu, C. C., Gong, X., Frizzell, R. A., and Aridor, M. (2016). VAMP-associated Proteins (VAP) as receptors that couple cystic fibrosis transmembrane conductance regulator (CFTR). proteostasis with lipid homeostasis. J. Biol. Chem. 291, 5206–5220. doi: 10.1074/jbc.M115.692749
Farinha, C. M., Sousa, M., Canato, S., Schmidt, A., Uliyakina, I., and Amaral, M. D. (2015). Increased efficacy of VX-809 in different cellular systems results from an early stabilization effect of F508del-CFTR. Pharmacol. Res. Perspect. 3:e00152. doi: 10.1002/prp2.152
Favia, M., Guerra, L., Fanelli, T., Cardone, R. A., Monterisi, S., Di Sole, F., et al. (2010). Na + /H + exchanger regulatory factor 1 overexpression-dependent increase of cytoskeleton organization is fundamental in the rescue of F508del cystic fibrosis transmembrane conductance regulator in human airway CFBE41o- cells. Mol. Biol. Cell 21, 73–86. doi: 10.1091/mbc.E09-03-0185
Fu, L., Rab, A., Tang, L., Bebok, Z., Rowe, S. M., Bartoszewski, R., et al. (2015). ΔF508 CFTR surface stability is regulated by DAB2 and CHIP-mediated ubiquitination in post-endocytic compartments. PLoS One 10:e0123131. doi: 10.1371/journal.pone.0123131
Fu, L., Rab, A., Tang, L. P., Rowe, S. M., Bebok, Z., and Collawn, J. F. (2012). Dab2 is a key regulator of endocytosis and post-endocytic trafficking of the cystic fibrosis transmembrane conductance regulator. Biochem. J. 441, 633–643. doi: 10.1042/BJ20111566
Glozman, R., Okiyoneda, T., Mulvihill, C. M., Rini, J. M., Barriere, H., and Lukacs, G. L. (2009). N-glycans are direct determinants of CFTR folding and stability in secretory and endocytic membrane traffic. J. Cell Biol. 184, 847–862. doi: 10.1083/jcb.200808124
Gong, X., Ahner, A., Roldan, A., Lukacs, G. L., Thibodeau, P. H., and Frizzell, R. A. (2016). Non-native conformers of cystic fibrosis transmembrane conductance regulator NBD1 are recognized by Hsp27 and conjugated to SUMO-2 for degradation. J. Biol. Chem. 291, 2004–2017. doi: 10.1074/jbc.M115.685628
Grasemann, H., Gaston, B., Fang, K., Paul, K., and Ratjen, F. (1999). Decreased levels of nitrosothiols in the lower airways of patients with cystic fibrosis and normal pulmonary function. J. Pediatr. 135, 770–772. doi: 10.1016/S0022-3476(99)70101-0
Grasemann, H., Knauer, N., Büscher, R., Hübner, K., Drazen, J. M., and Ratjen, F. (2000). Airway nitric oxide levels in cystic fibrosis patients are related to a polymorphism in the neuronal nitric oxide synthase gene. Am. J. Respir. Crit. Care Med. 162, 2172–2176. doi: 10.1164/ajrccm.162.6.2003106
Grove, D. E., Fan, C. Y., Ren, H. Y., and Cyr, D. M. (2011). The endoplasmic reticulum-associated Hsp40 DNAJB12 and Hsc70 cooperate to facilitate RMA1 E3-dependent degradation of nascent CFTRDeltaF508. Mol. Biol. Cell 22, 301–314. doi: 10.1091/mbc.E10-09-0760
Haardt, M., Benharouga, M., Lechardeur, D., Kartner, N., and Lukacs, G. L. (1999). C-terminal truncations destabilize the cystic fibrosis transmembrane conductance regulator without impairing its biogenesis. A novel class of mutation. J. Biol. Chem. 274, 21873–21877. doi: 10.1074/jbc.274.31.21873
Harris, W. T., Muhlebach, M. S., Oster, R. A., Knowles, M. R., Clancy, J. P., and Noah, T. L. (2011). Plasma TGF-β1 in pediatric cystic fibrosis: potential biomarker of lung disease and response to therapy. Pediatr. Pulmonol. 46, 688–695. doi: 10.1002/ppul.21430
Hegde, R. N., Parashuraman, S., Iorio, F., Ciciriello, F., Capuani, F., Carissimo, A., et al. (2015). Unravelling druggable signalling networks that control F508del-CFTR proteostasis. Elife 4:e10365. doi: 10.7554/eLife.10365
Hou, X., Wei, H., Rajagopalan, C., Jiang, H., Wu, Q., Zaman, K., et al. (2018). Dissection of the Role of VIMP in endoplasmic reticulum-associated degradation of CFTRΔF508. Sci. Rep. 8:4764. doi: 10.1038/s41598-018-23284-8
Hutt, D. M., Herman, D., Rodrigues, A. P., Noel, S., Pilewski, J. M., Matteson, J., et al. (2010). Reduced histone deacetylase 7 activity restores function to misfolded CFTR in cystic fibrosis. Nat. Chem. Biol. 6, 25–33. doi: 10.1038/nchembio.275
Kim, S. J., and Skach, W. R. (2012). Mechanisms of CFTR folding at the endoplasmic reticulum. Front. Pharmacol. 3:201. doi: 10.3389/fphar.2012.00201
Kimura, T., Kawabe, H., Jiang, C., Zhang, W., Xiang, Y. Y., Lu, C., et al. (2011). Deletion of the ubiquitin ligase Nedd4L in lung epithelia causes cystic fibrosis-like disease. Proc. Natl. Acad. Sci. U.S.A. 108, 3216–3221. doi: 10.1073/pnas.1010334108
Kleizen, B., van Vlijmen, T., de Jonge, H. R., and Braakman, I. (2005). Folding of CFTR is predominantly cotranslational. Mol. Cell. 20, 277–287. doi: 10.1016/j.molcel.2005.09.007
Koch, C. (2002). Early infection and progression of cystic fibrosis lung disease. Pediatr. Pulmonol. 34, 232–236. doi: 10.1002/ppul.10135
Koeppen, K., Chapline, C., Sato, J. D., and Stanton, B. A. (2012). Nedd4-2 does not regulate wt-CFTR in human airway epithelial cells. Am. J. Physiol. Lung. Cell Mol. Physiol. 303, L720–L727. doi: 10.1152/ajplung.00409.2011
Kongsuphol, P., Cassidy, D., Hieke, B., Treharne, K. J., Schreiber, R., Mehta, A., et al. (2009). Mechanistic insight into control of CFTR by AMPK. J. Biol. Chem. 284, 5645–5653. doi: 10.1074/jbc.M806780200
Lobo, M. J., Amaral, M. D., Zaccolo, M., and Farinha, C. M. (2016). EPAC1 activation by cAMP stabilizes CFTR at the membrane by promoting its interaction with NHERF1. J. Cell Sci. 129, 2599–2612. doi: 10.1242/jcs.185629
Loo, M. A., Jensen, T. J., Cui, L., Hou, Y., Chang, X. B., and Riordan, J. R. (1998). Perturbation of Hsp90 interaction with nascent CFTR prevents its maturation and accelerates its degradation by the proteasome. EMBO J. 17, 6879–6887. doi: 10.1093/emboj/17.23.6879
Lopes-Pacheco, M. (2016). CFTR modulators: shedding light on precision medicine for cystic fibrosis. Front. Pharmacol. 7:275. doi: 10.3389/fphar.2016.00275
Loureiro, C. A., Matos, A. M., Dias-Alves,Â, Pereira, J. F., Uliyakina, I., Barros, P., et al. (2015). A molecular switch in the scaffold NHERF1 enables misfolded CFTR to evade the peripheral quality control checkpoint. Sci. Signal 8:ra48. doi: 10.1126/scisignal.aaa1580
Luciani, A., Villella, V. R., Esposito, S., Gavina, M., Russo, I., Silano, M., et al. (2012). Targeting autophagy as a novel strategy for facilitating the therapeutic action of potentiators on ΔF508 cystic fibrosis transmembrane conductance regulator. Autophagy 8, 1657–1672. doi: 10.4161/auto.21483
Lukacs, G. L., Chang, X. B., Bear, C., Kartner, N., Mohamed, A., Riordan, J. R., et al. (1993). The delta F508 mutation decreases the stability of cystic fibrosis transmembrane conductance regulator in the plasma membrane. Determination of functional half-lives on transfected cells. J. Biol. Chem. 268, 21592–21598.
Luz, S., Cihil, K. M., Brautigan, D. L., Amaral, M. D., Farinha, C. M., and Swiatecka-Urban, A. (2014). LMTK2-mediated phosphorylation regulates CFTR endocytosis in human airway epithelial cells. J. Biol. Chem. 289, 15080–15093. doi: 10.1074/jbc.M114.563742
Luz, S., Kongsuphol, P., Mendes, A. I., Romeiras, F., Sousa, M., Schreiber, R., et al. (2011). Contribution of casein kinase 2 and spleen tyrosine kinase to CFTR trafficking and protein kinase A-induced activity. Mol. Cell. Biol. 31, 4392–4404. doi: 10.1128/MCB.05517-11
Marozkina, N. V., Yemen, S., Borowitz, M., Liu, L., Plapp, M., Sun, F., et al. (2010). Hsp 70/Hsp 90 organizing protein as a nitrosylation target in cystic fibrosis therapy. Proc. Natl. Acad. Sci. U.S.A. 107, 11393–11398. doi: 10.1073/pnas.0909128107
Matsumura, Y., Sakai, J., and Skach, W. R. (2013). Endoplasmic reticulum protein quality control is determined by cooperative interactions between Hsp/c70 protein and the CHIP E3 ligase. J. Biol. Chem. 288, 31069–31079. doi: 10.1074/jbc.M113.479345
Mazumdar, M., Christiani, D. C., Biswas, S. K., Ibne-Hasan, O. S., Kapur, K., and Hug, C. (2015). Elevated sweat chloride levels due to arsenic toxicity. N. Engl. J. Med. 372, 582–584. doi: 10.1056/NEJMc1413312
McClure, M. L., Barnes, S., Brodsky, J. L., and Sorscher, E. J. (2016). Trafficking and function of the cystic fibrosis transmembrane conductance regulator: a complex network of posttranslational modifications. Am. J. Physiol. Lung. Cell Mol. Physiol. 311, L719–L733. doi: 10.1152/ajplung.00431.2015
Meacham, G. C., Lu, Z., King, S., Sorscher, E., Tousson, A., and Cyr, D. M. (1999). The Hdj-2/Hsc70 chaperone pair facilitates early steps in CFTR biogenesis. EMBO J. 18, 1492–1505. doi: 10.1093/emboj/18.6.1492
Meacham, G. C., Patterson, C., Zhang, W., Younger, J. M., and Cyr, D. M. (2001). The Hsc70 co-chaperone CHIP targets immature CFTR for proteasomal degradation. Nat. Cell Biol. 3, 100–105. doi: 10.1038/35050509
Mendes, A. I., Matos, P., Moniz, S., Luz, S., Amaral, M. D., Farinha, C. M., et al. (2011). Antagonistic regulation of cystic fibrosis transmembrane conductance regulator cell surface expression by protein kinases WNK4 and spleen tyrosine kinase. Mol. Cell. Biol. 31, 4076–4086. doi: 10.1128/MCB.05152-11
Morito, D., Hirao, K., Oda, Y., Hosokawa, N., Tokunaga, F., Cyr, D. M., et al. (2008). Gp78 cooperates with RMA1 in endoplasmic reticulum-associated degradation of CFTRDeltaF508. Mol. Biol. Cell 19, 1328–1336. doi: 10.1091/mbc.E07-06-0601
Nery, F. C., Armata, I. A., Farley, J. E., Cho, J. A., Yaqub, U., Chen, P., et al. (2011). TorsinA participates in endoplasmic reticulum-associated degradation. Nat. Commun. 2:393. doi: 10.1038/ncomms1383
Odunuga, O. O., Longshaw, V. M., and Blatch, G. L. (2004). Hop: more than an Hsp70/Hsp90 adaptor protein. Bioessays 26, 1058–1068. doi: 10.1002/bies.20107
Okiyoneda, T., Barrière, H., Bagdány, M., Rabeh, W. M., Du, K., Höhfeld, J., et al. (2010). Peripheral protein quality control removes unfolded CFTR from the plasma membrane. Science 329, 805–810. doi: 10.1126/science.1191542
Okiyoneda, T., Harada, K., Takeya, M., Yamahira, K., Wada, I., Shuto, T., et al. (2004). Delta F508 CFTR pool in the endoplasmic reticulum is increased by calnexin overexpression. Mol. Biol. Cell 15, 563–574. doi: 10.1091/mbc.e03-06-0379
Okiyoneda, T., Niibori, A., Harada, K., Kohno, T., Michalak, M., Duszyk, M., et al. (2008). Role of calnexin in the ER quality control and productive folding of CFTR; differential effect of calnexin knockout on wild-type and DeltaF508 CFTR. Biochim. Biophys. Acta 1783, 1585–1594. doi: 10.1016/j.bbamcr.2008.04.002
Okiyoneda, T., Veit, G., Dekkers, J. F., Bagdany, M., Soya, N., Xu, H., et al. (2013). Mechanism-based corrector combination restores ΔF508-CFTR folding and function. Nat. Chem. Biol. 9, 444–454. doi: 10.1038/nchembio.1253
Okiyoneda, T., Veit, G., Sakai, R., Aki, M., Fujihara, T., Higashi, M., et al. (2018). Chaperone-independent peripheral quality control of CFTR by RFFL E3 ligase. Dev. Cell 44, 694–708.e7. doi: 10.1016/j.devcel.2018.02.001
Phuan, P. W., Veit, G., Tan, J. A., Finkbeiner, W. E., Lukacs, G. L., and Verkman, A. S. (2015). Potentiators of defective ΔF508-CFTR gating that do not interfere with corrector action. Mol. Pharmacol. 88, 791–799. doi: 10.1124/mol.115.099689
Pind, S., Riordan, J. R., and Williams, D. B. (1994). Participation of the endoplasmic reticulum chaperone calnexin (p88, IP90) in the biogenesis of the cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 269, 12784–12788.
Qian, Z., Xu, X., Amacher, J. F., Madden, D. R., Cormet-Boyaka, E., and Pei, D. (2015). Intracellular delivery of peptidyl ligands by reversible cyclization: discovery of a PDZ domain inhibitor that rescues CFTR activity. Angew. Chem. Int. Ed. Engl. 54, 5874–5878. doi: 10.1002/anie.201411594
Rabeh, W. M., Bossard, F., Xu, H., Okiyoneda, T., Bagdany, M., Mulvihill, C. M., et al. (2012). Correction of both NBD1 energetics and domain interface is required to restore ΔF508 CFTR folding and function. Cell 148, 150–163. doi: 10.1016/j.cell.2011.11.024
Ramachandran, S., Osterhaus, S. R., Parekh, K. R., Jacobi, A. M., Behlke, M. A., and McCray, P. B. (2016). SYVN1, NEDD8, and FBXO2 proteins regulate ΔF508 cystic fibrosis transmembrane conductance regulator (CFTR). Ubiquitin-mediated proteasomal degradation. J. Biol. Chem. 291, 25489–25504. doi: 10.1074/jbc.M116.754283
Rasmussen, J. E., Sheridan, J. T., Polk, W., Davies, C. M., and Tarran, R. (2014). Cigarette smoke-induced Ca2 + release leads to cystic fibrosis transmembrane conductance regulator (CFTR) dysfunction. J. Biol. Chem. 289, 7671–7681. doi: 10.1074/jbc.M113.545137
Ren, H. Y., Grove, D. E., De La Rosa, O., Houck, S. A., Sopha, P., Van Goor, F., et al. (2013). VX-809 corrects folding defects in cystic fibrosis transmembrane conductance regulator protein through action on membrane-spanning domain 1. Mol. Biol. Cell 24, 3016–3024. doi: 10.1091/mbc.E13-05-0240
Rennolds, J., Butler, S., Maloney, K., Boyaka, P. N., Davis, I. C., Knoell, D. L., et al. (2010). Cadmium regulates the expression of the CFTR chloride channel in human airway epithelial cells. Toxicol. Sci. 116, 349–358. doi: 10.1093/toxsci/kfq101
Rosser, M. F., Grove, D. E., Chen, L., and Cyr, D. M. (2008). Assembly and misassembly of cystic fibrosis transmembrane conductance regulator: folding defects caused by deletion of F508 occur before and after the calnexin-dependent association of membrane spanning domain (MSD). 1 and MSD2. Mol. Biol. Cell 19, 4570–4579. doi: 10.1091/mbc.E08-04-0357
Rotin, D., and Staub, O. (2012). Nedd4-2 and the regulation of epithelial sodium transport. Front. Physiol. 3:212. doi: 10.3389/fphys.2012.00212
Sharma, M., Pampinella, F., Nemes, C., Benharouga, M., So, J., Du, K., et al. (2004). Misfolding diverts CFTR from recycling to degradation: quality control at early endosomes. J. Cell Biol. 164, 923–933. doi: 10.1083/jcb.200312018
Silvis, M. R., Picciano, J. A., Bertrand, C., Weixel, K., Bridges, R. J., and Bradbury, N. A. (2003). A mutation in the cystic fibrosis transmembrane conductance regulator generates a novel internalization sequence and enhances endocytic rates. J. Biol. Chem. 278, 11554–11560. doi: 10.1074/jbc.M212843200
Snodgrass, S. M., Cihil, K. M., Cornuet, P. K., Myerburg, M. M., and Swiatecka-Urban, A. (2013). Tgf-β1 inhibits Cftr biogenesis and prevents functional rescue of ΔF508-Cftr in primary differentiated human bronchial epithelial cells. PLoS One 8:e63167. doi: 10.1371/journal.pone.0063167
Sondo, E., Falchi, F., Caci, E., Ferrera, L., Giacomini, E., Pesce, E., et al. (2018). Pharmacological inhibition of the ubiquitin ligase RNF5 rescues F508del-CFTR in cystic fibrosis airway epithelia. Cell Chem. Biol. 25, 891–905.e8. doi: 10.1016/j.chembiol.2018.04.010
Sun, H., Harris, W. T., Kortyka, S., Kotha, K., Ostmann, A. J., Rezayat, A., et al. (2014). Tgf-beta downregulation of distinct chloride channels in cystic fibrosis-affected epithelia. PLoS One 9:e106842. doi: 10.1371/journal.pone.0106842
Swiatecka-Urban, A., Brown, A., Moreau-Marquis, S., Renuka, J., Coutermarsh, B., Barnaby, R., et al. (2005). The short apical membrane half-life of rescued {Delta}F508-cystic fibrosis transmembrane conductance regulator (CFTR).results from accelerated endocytosis of {Delta}F508-CFTR in polarized human airway epithelial cells. J. Biol. Chem. 280, 36762–36772. doi: 10.1074/jbc.M508944200
Swiatecka-Urban, A., Moreau-Marquis, S., Maceachran, D. P., Connolly, J. P., Stanton, C. R., Su, J. R., et al. (2006). Pseudomonas aeruginosa inhibits endocytic recycling of CFTR in polarized human airway epithelial cells. Am. J. Physiol. Cell Physiol. 290, C862–C872. doi: 10.1152/ajpcell.00108.2005
Thelin, W. R., Chen, Y., Gentzsch, M., Kreda, S. M., Sallee, J. L., Scarlett, C. O., et al. (2007). Direct interaction with filamins modulates the stability and plasma membrane expression of CFTR. J. Clin. Invest. 117, 364–374. doi: 10.1172/JCI30376
Trzcinska-Daneluti, A. M., Nguyen, L., Jiang, C., Fladd, C., Uehling, D., Prakesch, M., et al. (2012). Use of kinase inhibitors to correct ΔF508-CFTR function. Mol. Cell. Proteomics 11, 745–757. doi: 10.1074/mcp.M111.016626
Turnbull, E. L., Rosser, M. F., and Cyr, D. M. (2007). The role of the UPS in cystic fibrosis. BMC Biochem. 8(Suppl. 1):S11. doi: 10.1186/1471-2091-8-S1-S11
Van Goor, F., Hadida, S., Grootenhuis, P. D., Burton, B., Stack, J. H., Straley, K. S., et al. (2011). Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl. Acad. Sci. U.S.A. 108, 18843–18848. doi: 10.1073/pnas.1105787108
Van Goor, F., Yu, H., Burton, B., and Hoffman, B. J. (2014). Effect of ivacaftor on CFTR forms with missense mutations associated with defects in protein processing or function. J. Cyst. Fibros 13, 29–36. doi: 10.1016/j.jcf.2013.06.008
Varga, K., Goldstein, R. F., Jurkuvenaite, A., Chen, L., Matalon, S., Sorscher, E. J., et al. (2008). Enhanced cell-surface stability of rescued DeltaF508 cystic fibrosis transmembrane conductance regulator (CFTR) by pharmacological chaperones. Biochem. J. 410, 555–564. doi: 10.1042/BJ20071420
Veit, G., Avramescu, R. G., Chiang, A. N., Houck, S. A., Cai, Z., Peters, K. W., et al. (2016a). From CFTR biology toward combinatorial pharmacotherapy: expanded classification of cystic fibrosis mutations. Mol. Biol. Cell 27, 424–433. doi: 10.1091/mbc.E14-04-0935
Veit, G., Avramescu, R. G., Perdomo, D., Phuan, P. W., Bagdany, M., Apaja, P. M., et al. (2014). Some gating potentiators, including VX-770, diminish ΔF508-CFTR functional expression. Sci. Transl. Med. 6:246ra97. doi: 10.1126/scitranslmed.3008889
Veit, G., Oliver, K., Apaja, P. M., Perdomo, D., Bidaud-Meynard, A., Lin, S. T., et al. (2016b). Ribosomal stalk protein silencing partially corrects the ΔF508-CFTR functional expression defect. PLoS Biol. 14:e1002462. doi: 10.1371/journal.pbio.1002462
Venerando, A., Franchin, C., Cant, N., Cozza, G., Pagano, M. A., Tosoni, K., et al. (2013). Detection of phospho-sites generated by protein kinase CK2 in CFTR: mechanistic aspects of Thr1471 phosphorylation. PLoS One 8:e74232. doi: 10.1371/journal.pone.0074232
Villella, V. R., Esposito, S., Bruscia, E. M., Maiuri, M. C., Raia, V., Kroemer, G., et al. (2013). Targeting the intracellular environment in cystic fibrosis: restoring autophagy as a novel strategy to circumvent the CFTR defect. Front. Pharmacol. 4:1. doi: 10.3389/fphar.2013.00001
Wang, H., and Brautigan, D. L. (2006). Peptide microarray analysis of substrate specificity of the transmembrane Ser/Thr kinase KPI-2 reveals reactivity with cystic fibrosis transmembrane conductance regulator and phosphorylase. Mol. Cell. Proteomics 5, 2124–2130. doi: 10.1074/mcp.M600188-MCP200
Wang, Y., Liu, J., Loizidou, A., Bugeja, L. A., Warner, R., Hawley, B. R., et al. (2014). CFTR potentiators partially restore channel function to A561E-CFTR, a cystic fibrosis mutant with a similar mechanism of dysfunction as F508del-CFTR. Br. J. Pharmacol. 171, 4490–4503. doi: 10.1111/bph.12791
Wang, Y., Loo, T. W., Bartlett, M. C., and Clarke, D. M. (2007). Correctors promote maturation of cystic fibrosis transmembrane conductance regulator (CFTR)-processing mutants by binding to the protein. J. Biol. Chem. 282, 33247–33251. doi: 10.1074/jbc.C700175200
Wolde, M., Fellows, A., Cheng, J., Kivenson, A., Coutermarsh, B., Talebian, L., et al. (2007). Targeting CAL as a negative regulator of DeltaF508-CFTR cell-surface expression: an RNA interference and structure-based mutagenetic approach. J. Biol. Chem. 282, 8099–8109. doi: 10.1074/jbc.M611049200
Yang, Y., Janich, S., Cohn, J. A., and Wilson, J. M. (1993). The common variant of cystic fibrosis transmembrane conductance regulator is recognized by hsp70 and degraded in a pre-Golgi nonlysosomal compartment. Proc. Natl. Acad. Sci. U.S.A. 90, 9480–9484. doi: 10.1073/pnas.90.20.9480
Ye, S., Cihil, K., Stolz, D. B., Pilewski, J. M., Stanton, B. A., and Swiatecka-Urban, A. (2010). c-Cbl facilitates endocytosis and lysosomal degradation of cystic fibrosis transmembrane conductance regulator in human airway epithelial cells. J. Biol. Chem. 285, 27008–27018. doi: 10.1074/jbc.M110.139881
Younger, J. M., Chen, L., Ren, H. Y., Rosser, M. F., Turnbull, E. L., Fan, C. Y., et al. (2006). Sequential quality-control checkpoints triage misfolded cystic fibrosis transmembrane conductance regulator. Cell 126, 571–582. doi: 10.1016/j.cell.2006.06.041
Keywords: peripheral QC, CFTR, stabilizer, ubiquitination, RFFL
Citation: Fukuda R and Okiyoneda T (2018) Peripheral Protein Quality Control as a Novel Drug Target for CFTR Stabilizer. Front. Pharmacol. 9:1100. doi: 10.3389/fphar.2018.01100
Received: 19 July 2018; Accepted: 10 September 2018;
Published: 27 September 2018.
Edited by:
Nicoletta Pedemonte, Istituto Giannina Gaslini (IRCCS), ItalyReviewed by:
Tingwei Mu, Case Western Reserve University, United StatesCopyright © 2018 Fukuda and Okiyoneda. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tsukasa Okiyoneda, dC1va2l5b25lZGFAa3dhbnNlaS5hYy5qcA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.