 Gantumur Battogtokh
Gantumur Battogtokh Yong-Yeon Cho
Yong-Yeon Cho Han Chang Kang
Han Chang Kang- Department of Pharmacy, College of Pharmacy, The Catholic University of Korea, Bucheon, South Korea
The mitochondrion is an important intracellular organelle for drug targeting due to its key roles and functions in cellular proliferation and death. In the last few decades, several studies have revealed mitochondrial functions, attracting the focus of many researchers to work in this field over nuclear targeting. Mitochondrial targeting was initiated in 1995 with a triphenylphosphonium-thiobutyl conjugate as an antioxidant agent. The major driving force for mitochondrial targeting in cancer cells is the higher mitochondrial membrane potential compared with that of the cytosol, which allows some molecules to selectively target mitochondria. In this review, we discuss mitochondria-targeting ligand-conjugated anticancer agents and their in vitro and in vivo behaviors. In addition, we describe a mitochondria-targeting nanocarrier system for anticancer drug delivery. As previously reported, several agents have been known to have mitochondrial targeting potential; however, they are not sufficient for direct application for cancer therapy. Thus, many studies have focused on direct conjugation of targeting ligands to therapeutic agents to improve their efficacy. There are many variables for optimal mitochondria-targeted agent development, such as choosing a correct targeting ligand and linker. However, using the nanocarrier system could solve some issues related to solubility and selectivity. Thus, this review focuses on mitochondria-targeting drug conjugates and mitochondria-targeted nanocarrier systems for anticancer agent delivery.
Introduction
Mitochondria play important roles in living cells as an energy source and as a generator of reactive substrates, which are harmful to living cells. Therefore, targeting mitochondria is attracting great attention in medicine. The idea of mitochondrial targeting was posed in 1950 after determining the mitochondrial structure and some molecules with mitochondrial affinity (Palade, 1952, 1964; Ernster and Schatz, 1981). Delocalized lipophilic cations (DLCs), such as triphenylphosphonium, dequalinium, and rhodamine 123, were the first mitochondria-targeting molecules (Ernster and Schatz, 1981; Lampidis et al., 1985; Weiss et al., 1987; Christman et al., 1990; Dairkee and Hackett, 1991). Later, many studies used those ligands in mitochondria-targeted antioxidant and anticancer drug development. The concept of mitochondrial targeting is based on the mitochondrial membrane potential, which differs from the plasma membrane potential. The mitochondrial membrane potential is approximately -180 mV, which is 3- to 5-fold higher than that of the plasma membrane (Zhang and Zhang, 2016b; Zielonka et al., 2017). Thus, positively charged molecules easily accumulate in the mitochondria. However, since mitochondria have a four-layer structure (outer membrane, intermembrane space, inner membrane, and matrix), it is difficult for many molecules to enter the mitochondria. Therefore, lipophilic molecules are used due to their membrane-permeability, allowing the molecules to traverse the mitochondrial membranes by hydrophobic interactions. In several cases, the mitochondria of cancer cells and transformed cells exhibit significantly increased transmembrane potentials compared with normal cells (Dairkee and Hackett, 1991). This biological difference has been utilized as a basis to develop mitochondria-targeting compounds that may be preferentially accumulated within the mitochondria of cancer cells. An alternative option for cancer cell mitochondria targeting over normal cell mitochondria is its ability to generate high reactive oxygen species (ROS) and adenosine triphosphate (ATP) by aerobic glycolysis as compared to that in normal cells by Krebs cycle. Therefore, mitochondria are vulnerable to dysfunction and mitochondrial DNA mutation. Aerobic glycolysis in cancer cells is known as the Warburg effect (Wallace, 2005). Additionally, many anticancer drugs (doxorubicin, cisplatin, paclitaxel, resveratrol, and porphyrin derivatives) are known to be activated in the membrane and the matrix of mitochondria and by mitochondrial nucleases (Wang et al., 2010; Heller et al., 2012). If we can enhance the mitochondrial accumulation of anticancer drugs, the therapeutic effect could be improved, and drug resistance may be resolved. On the other hand, mitochondria targeting anticancer agents are called “mitocans,” which selectively accumulate in mitochondria and damage mitochondrial membrane, leading ROS generation and apoptotic cell death (Neuzil et al., 2013).
There are two well-known approaches for mitochondria drug delivery: direct conjugation of the targeting ligand to drugs and attachment of the targeting ligand to a nanocarrier. In this review, we discuss these approaches and recent achievements. In the case of direct conjugation of the targeting ligand to a drug, many aspects should be considered, including the type of linkage and the type of targeting ligand. There are a number of mitochondria-targeting conjugates, most of which are triphenylphosphonium (TPP)-based conjugates due to TPP’s strong mitochondrial targeting ability (Wang et al., 2017; Zielonka et al., 2017). In the last three decades, nanotechnology has captured great attention in medicine, and some nanoparticles have been investigated for mitochondrial targeting. The first nanomaterials applied for mitochondrial targeting were dequalinium (DQA) micelles (DQAsomes), which exhibit liposome-like self-assembly in aqueous solution (Weissig et al., 1998; Weissig and Torchilin, 2001; D’Souza et al., 2003). Then, other nanomaterials, including polymeric nanoparticles, metallic nanoparticles, and liposomes, were prepared as mitochondrial drug delivery carriers (Zhang and Zhang, 2016a; Wang et al., 2017).
Direct drug-targeting ligand conjugation is simple and easy to control, and the drugs can readily reach the mitochondria; however, the conjugation procedure can diminish the therapeutic effects and solubility of the drugs. In the case of the nanocarrier system, there is no concern regarding a loss of therapeutic effect because the physical interaction and solubility issue would be solved, but optimization has remained a challenge due to the use of many different compositions to prepare the nanocarrier, and mitochondrial delivery may also be delayed due to the nanocarrier features.
Mitochondrial Targeting Ligand-Anticancer Drug Conjugates
Direct conjugation of therapeutic agents to molecules to enhance their physicochemical and biopharmaceutical efficiency results in a prodrug. In most cases, the prodrug is inactive until it is metabolized. The most successful examples are antibody-drug conjugates that can be activated by enzymatic cleavage after reaching the target site (Lambert and Morris, 2017). To produce a successful prodrug, selection of the linkage plays an important role. Generally, the linkages (bonds) are stimulus-responsive, for example, responsive to an enzyme, pH, temperature, or irradiation. Common bonds for prodrug formation are the amide bond, which is main bond in protein and peptide formation in living system and is cleavable by the protease enzyme; the hydrazone bond, which is degradable under acidic conditions; the ester bond, which is easily hydrolyzed in aqueous solution; the disulfide bond, which is cleavable by glutathione; and the ether bond, which is degraded under strong acidic or basic conditions.
In contrast, mitochondria-targeting ligand-anticancer drug conjugates can be active before and after cleavage. Therefore, the bond is not as important as in the prodrug, but it should be considered. A number of direct conjugates have been reported for mitochondrial delivery of anticancer drugs using various targeting moieties, including peptides [mitochondria-penetrating peptide (MPP), mitochondria-targeting sequence (MTS) peptide, and SS peptides] and lipophilic cations (TPP, rhodamine 123, and DQA) with various linkages. The most commonly used anticancer agents for mitochondrial delivery are doxorubicin (Dox), 5-fluorouracil (5-FU), paclitaxel, cisplatin, chlorambucil, porphyrin derivatives, and coumarin because those agents can be activated in mitochondria and exert therapeutic efficacy. In brief, Dox is an anti-cancer drug that intercalates into DNA and inhibits the progression of Topoisomerase II, leading to cell death. In addition, Dox derivatives have been known to interact with mitochondria by the disruption of major mitochondrial functions, damaging membrane-bound proteins and enzymes (Jung and Reszka, 2001). Cisplatin binds to nuclear DNA and induces DNA cross-linking and some studies confirmed that it can interact with mitochondrial DNA as well (Alexeyev et al., 2013). Paclitaxel derivatives are known to depolymerize microtubules and also some studies have shown that they can induce cytochrome c release by activating mitochondrial membrane proteins (D’Souza and Weissig, 2009). Chlorambucil is a DNA alkylating agent, which can alkylate both nuclear and mitochondrial DNA (Mourtada et al., 2013). Many porphyrin derivatives have been known to induce mitochondria functions in the presence of irradiation (Zielonka et al., 2017). Here, we compare various conjugates obtained from a variety of targeting ligands through different linkages, as well as the effect of the linker and targeting moiety for their activity. We summarize some specific properties of the mitochondria-targeted drug-conjugates in Table 1.
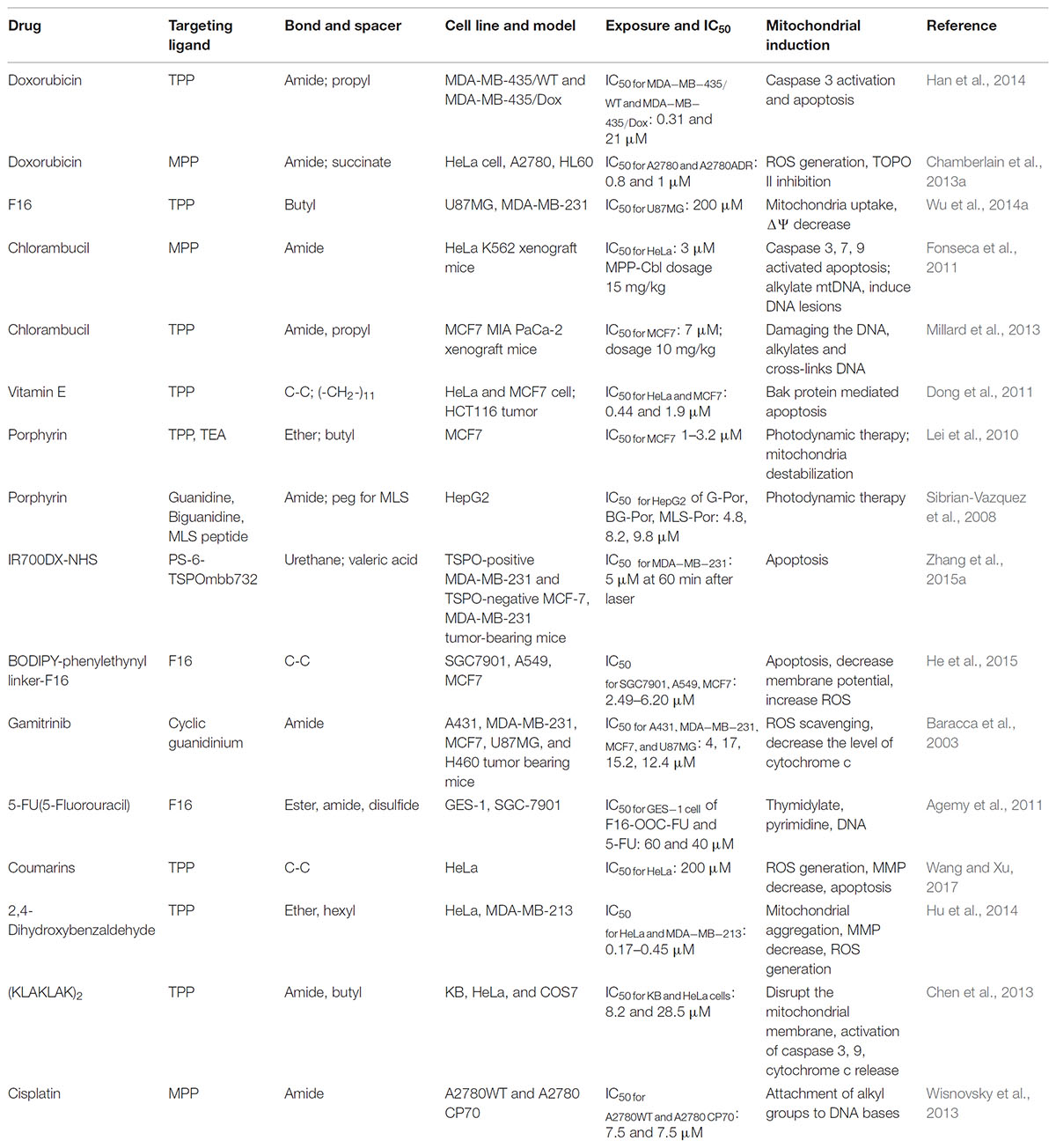
TABLE 1. Some specific characteristics of mitochondria-targeting ligands and anticancer drug conjugates.
Peptide-Anticancer Drug Conjugates
Peptides are favorable molecules for medical application due to their biocompatibility in physiology. As targeting ligands, many peptides have been developed, and the most widely used ones are cell-penetrating peptides (5–30 amino acids) such as TAT (transactivator of transcription, GRKKRRQRRRPQ) (Vives et al., 2008). Most peptides have a positive charge, which promotes cell membrane penetration, and short sequences. For mitochondrial targeting, some peptides have been prepared and successfully applied based on the cell-penetrating peptide sequence. The mitochondria-targeting peptides (mitochondria-penetrating peptide, mitochondria-targeting sequence, SS peptide, and other peptides) were conjugated with various anticancer agents to improve their therapeutic efficacy.
Mitochondria-Penetrating Peptide (MPP)-Drug Conjugates
Kelley’s group developed a series of short peptides for mitochondrial targeting based on cell-penetrating peptides such as TAT (Horton et al., 2008; Yousif et al., 2009a; Jean et al., 2016). Her group introduced the hydrophobic cyclohexyl monomer between arginine molecules to improve its lipophilicity and further explored the mitochondrial targeting ability of sequences of short peptides (Horton et al., 2008; Yousif et al., 2009a). The peptides were attached to several anticancer drugs to assess whether the peptides could improve mitochondrial targeting and enhance the therapeutic effect. Chamberlain et al. (2013a) hypothesized that this approach could prevent drug resistance, which is the most crucial problem associated with current chemotherapy for many cancers.
Among several short peptides (L-cyclohexyl alanine-D-arginine)3 (Fx,r)3 is known to hold promising mitochondria-targeting properties (Horton et al., 2008). Therefore, the peptide has been attached to Dox, cisplatin, and chlorambucil to explore its activity in drug-resistant cells (Fonseca et al., 2011; Chamberlain et al., 2013a; Mourtada et al., 2013; Wisnovsky et al., 2013). The peptide-Dox conjugate (mtDox) was prepared using an amide bond between the free amine group of Dox and carboxyl end of the short peptide (Chamberlain et al., 2013a). For Dox, it is well-known that Dox is pumped out by p-glycoprotein (Pgp) transporter in drug-resistant cells and also that Dox can be accumulated in lysosomes by lysosomal Pgp activity (Seebacher et al., 2016). Nevertheless, mtDox showed high accumulation in mitochondria compared with free Dox (Figure 1). Further studies have been carried out in drug-sensitive and resistant cells. Although mtDox has less cell killing effects than free Dox in drug-sensitive cells, it exhibited strong activity in drug-resistant cells (Figure 1B). The reason for this behavior is that mtDox (1) is not pumped out by p-glycoprotein (Pgp) transporter even as a substrate due to blockage of the free amine group of Dox, which is the reactive part required for Pgp-transporter binding, and (2) is selective for mitochondrial localization. These authors claimed that mtDox intercalated into mtDNA, and it is known that Dox functions by inhibiting DNA topoisomerase II protein and triggers apoptosis (Pommier et al., 2010).
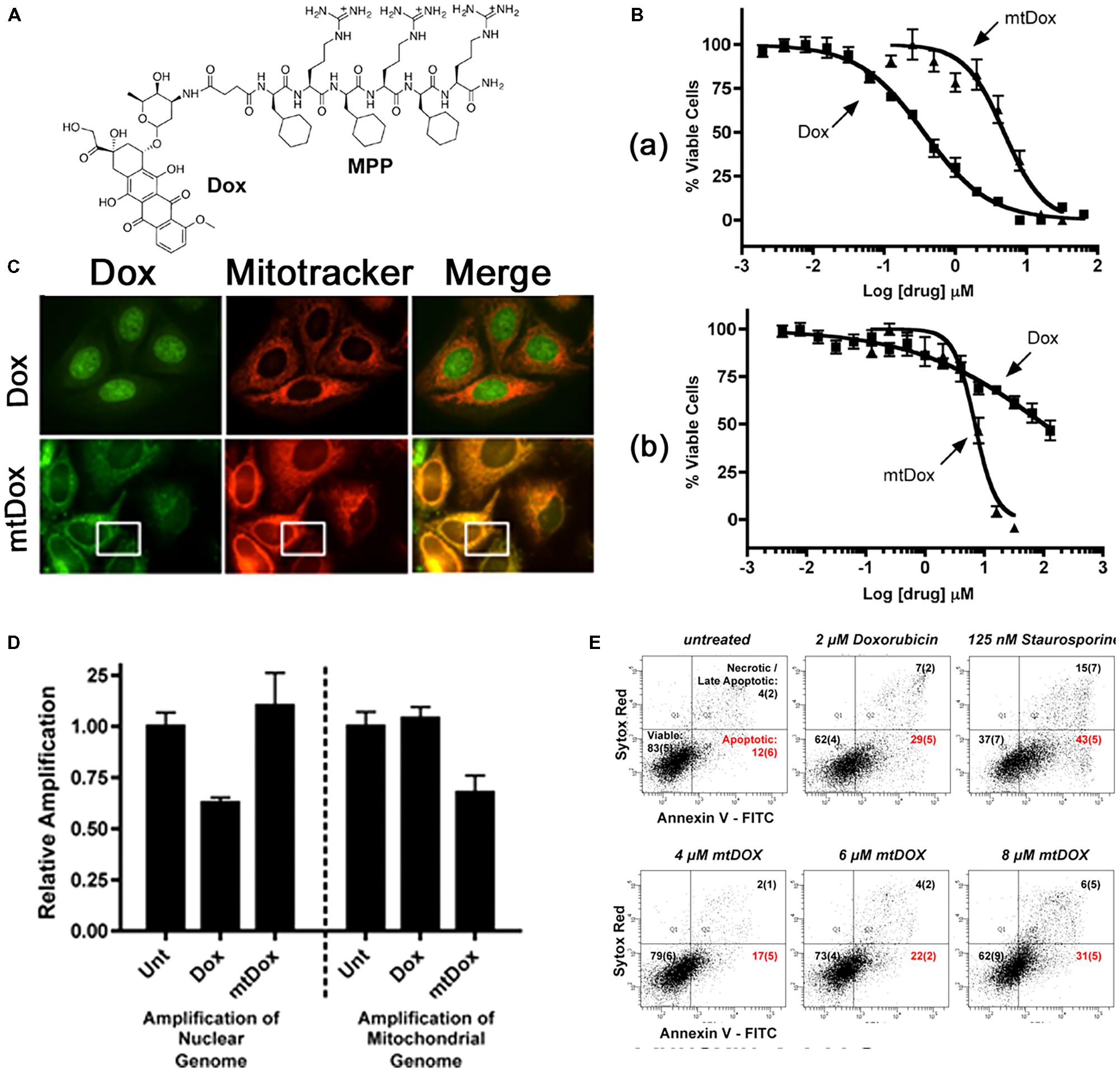
FIGURE 1. (A) Chemical structure of mtDox. (B) Cytotoxicity data. (a) mtDox and Dox are both toxic to sensitive A2780 cells. (b) The toxicity of Dox is highly attenuated in A2780ADR Pgp-expressing cells. (C) Intracellular localization of mtDox by confocal microscopy, Dox and mtDox (green channel) and Mitotracker 633 (red channel). (D) Assessment of DNA strand breaks in cells by measuring the relative amplification of nuclear and mitochondrial DNA segments. (E) Flow cytometry analysis of A2780 Annexin V-FITC (A-FITC)/SYTOX Red (SR) cell staining. Images were adapted with permission from reference (Chamberlain et al., 2013b).
In contrast, this peptide MPP-cisplatin conjugate (mtPt) was prepared to overcome cisplatin resistance in cancer cells because cisplatin, a widely used anticancer drug, is known to be resistant to various cancers (Wisnovsky et al., 2013). The activity of mtPt is quite similar in both sensitive and resistant cells [A2780 wild-type (WT) and cisplatin-resistant A2780 CP70 cell lines]. A subcellular uptake study showed that mtPt localized preferentially in mitochondria, and further study demonstrated that mtPt generated approximately two times higher levels of ROS than its free counterpart, damaging mtDNA and inducing apoptosis. Furthermore, through conjugation of the short peptide to chlorambucil to improve the therapeutic effect of chlorambucil, the MPP-chlorambucil (mtCbl) conjugate was also obtained and confirmed to localize in mitochondria due to the cyclohexylalanines and positive charge of the peptide sequence (Mourtada et al., 2013). Initial results showed that the conjugate was able to induce apoptosis, but a later study demonstrated that mtCbl led to necrotic cell death due to alkylation activity, which was confirmed by activation of caspase 3/7 and increases in protease activity (Mourtada et al., 2013). Among these conjugates, the most potent one is mtCbl, followed by mtDox and mtPt, compared to their counterparts.
Mitochondria-Targeting Sequence (MTS) Peptide-Drug Conjugates
Mitochondria-targeting sequence peptides consist of 20–40 amino acids that are recognized by receptors at the mitochondrial surface. Various translocases of the outer and inner membranes mediate the import and intramitochondrial sorting of MTS-containing peptides, which is driven by ATP or by the mitochondrial transmembrane potential (Yousif et al., 2009a). Multiple MTSs have been successfully applied for the mitochondrial delivery of chemically different cargos, including proteins, catalytically proficient enzymes and nucleic acids (Vestweber and Schatz, 1989; Srivastava and Moraes, 2001; Mukhopadhyay et al., 2005). The major drawbacks of this approach are associated with the large molecular size of MTSs and their solubility and intrinsically poor membrane permeability (Yousif et al., 2009a,b). Lin et al. (2015) conjugated MTS peptide (MLRAALSTARRGPRLSRLL), a well-known natural presequence from mitochondrial-oriented protein aldehyde dehydrogenase (ALDH), to a cell-penetrating peptide (R8) due to its cell internalization ability to enhance cell penetration. Three histidines were introduced into the conjugate to supply a buffering effect upon protonation of the imidazole ring inside endosomes or lysosomes, inducing rupture of endosomal/lysosomal membranes for effective release of the entrapped conjugate into the cytosol. The hydrophobic porphyrin molecule was also conjugated to the MLS peptide (MSVLTPLLLRGLTGSARRLPVPRAKIHSL, isolated from subunit VII of human cytochrome c oxidase), which is encoded in a commercially available expression vector (pShooter) that targets recombinant protein to the mitochondria of mammalian cells, in the presence of a short poly(ethylene glycol) (PEG) spacer (Sibrian-Vazquez et al., 2008). It showed high levels of cell uptake but low mitochondrial uptake. The high cell uptake might be related to the reduced aggregation tendency of the conjugate in aqueous media due to fewer intra- and intermolecular interactions. The conjugate showed less localization in mitochondria than in lysosomes because of the important roles of substituents of the porphyrin periphery in cellular uptake and subcellular distribution mechanisms (Horobin, 2002). In addition, since the conjugate localized extensively to lysosomes, it might exhibit poor endosome escape, resulting in low mitochondrial accumulation.
Delocalized Lipophilic Cation (DLC)-Anticancer Drug Conjugates
Delocalized lipophilic cations are molecules that possess both lipophilicity and a positive charge. Therefore, they can easily pass through phospholipid membranes, plasma membranes, and mitochondrial outer membranes through diffusion. The delocalized positive charge allows the molecules to enter the matrix of mitochondria, which is highly negatively charged with a strong negative membrane potential (-180 mV), through the protein gradient process (Chen, 1988; Torchilin, 2006; Heller et al., 2012). Several DLCs are used as mitochondria-targeting ligands, such as TPP, rhodamine 123, DQA, guanidine, and F16 (Wang et al., 2010; Modica-Napolitano and Weissig, 2015; Zielonka et al., 2017). These mitochondria-targeting ligands can be linked to various anticancer agents through covalent bonds, such as amide, disulfide, ester, ether, and hydrazine bonds (Table 1). We discuss in more detail the conjugates and their specific results in the examples below.
Triphenyl Phosphonium (TPP)-Drug Conjugates
The most common example of a DLC is TPP, which is a positively charged phosphorus atom surrounded by three lipophilic phenyl groups that increase its combination. The TPP moiety has been employed as a probe to explore the mitochondrial membrane potential for more than 40 years (Lampidis et al., 1985). Consequently, its behavior and interaction with mitochondria are well-defined (Zhang and Zhang, 2016a). The relative TPP concentrations inside the negatively charged membrane compartments are increased by one order of magnitude for every 60 mV of negative membrane potential. Given the active transport of salt ions by membrane-bound pumps, the interior part of the plasma membrane is negatively charged compared with the exterior part. As a result, the plasma membrane potential generally ranges between -30 and -60 mV, which is sufficient to promote up to a 10-fold accumulation of TPP inside the cell. Many TPP-anticancer agent conjugates, including TPP-doxorubicin (Han et al., 2014), TPP-chlorambucil (Millard et al., 2013), TPP-F16 (Wu et al., 2014a), TPP-vitamin E (Dong et al., 2011), TPP-therapeutic peptide (Chen et al., 2013), TPP-porphyrin photosensitizers (Lei et al., 2010; Hu et al., 2014; Zhang et al., 2015a), and TPP-coumarin (Wang and Xu, 2017), are known and were described in a recent review (Zielonka et al., 2017). Here, we briefly discuss and compare some specific examples. Han et al. (2014) synthesized TPP-doxorubicin (TPP-Dox) via the reaction between the carboxyl group of the TPP moiety and the amine group of Dox to deliver the drug into mitochondria with the aim of overcoming drug resistance (Figure 2A). A cell uptake study revealed that the TPP-Dox conjugate had lower uptake by MDA-MB-435/WT cells than free Dox, indicating that the TPP moiety affected cell membrane transport. In contrast, in MDA-MB-435/Dox cells, TPP-Dox showed much higher cell uptake than free Dox, supporting the possibility of TPP-Dox by-passage through P-glycoprotein and other pumps overexpressed in a resistant cell line. A subcellular localization study by confocal microscopy revealed that in drug sensitive cells, free Dox was mostly found in the nucleus (Figure 2B), while it was not found in the nucleus in drug-resistant cells. In contrast, in both cell lines, the TPP-Dox conjugate was not localized in the nucleus and accumulated extensively in mitochondria (Figure 2B). A further time-dependent cytotoxicity study showed that TPP-Dox had an approximately fourfold lower IC50 than free Dox in drug-resistant cells after 48 and 72 h of incubation (Figure 2C). These results implied that the TPP-Dox conjugate was able to overcome drug resistance, but it was difficult to apply it in vivo due to its low solubility. Similar studies have been reported using Dox-nitrooxy or 3-phenylsulfonylfuroxan conjugates in a human colon cancer cell line (Riganti et al., 2013).
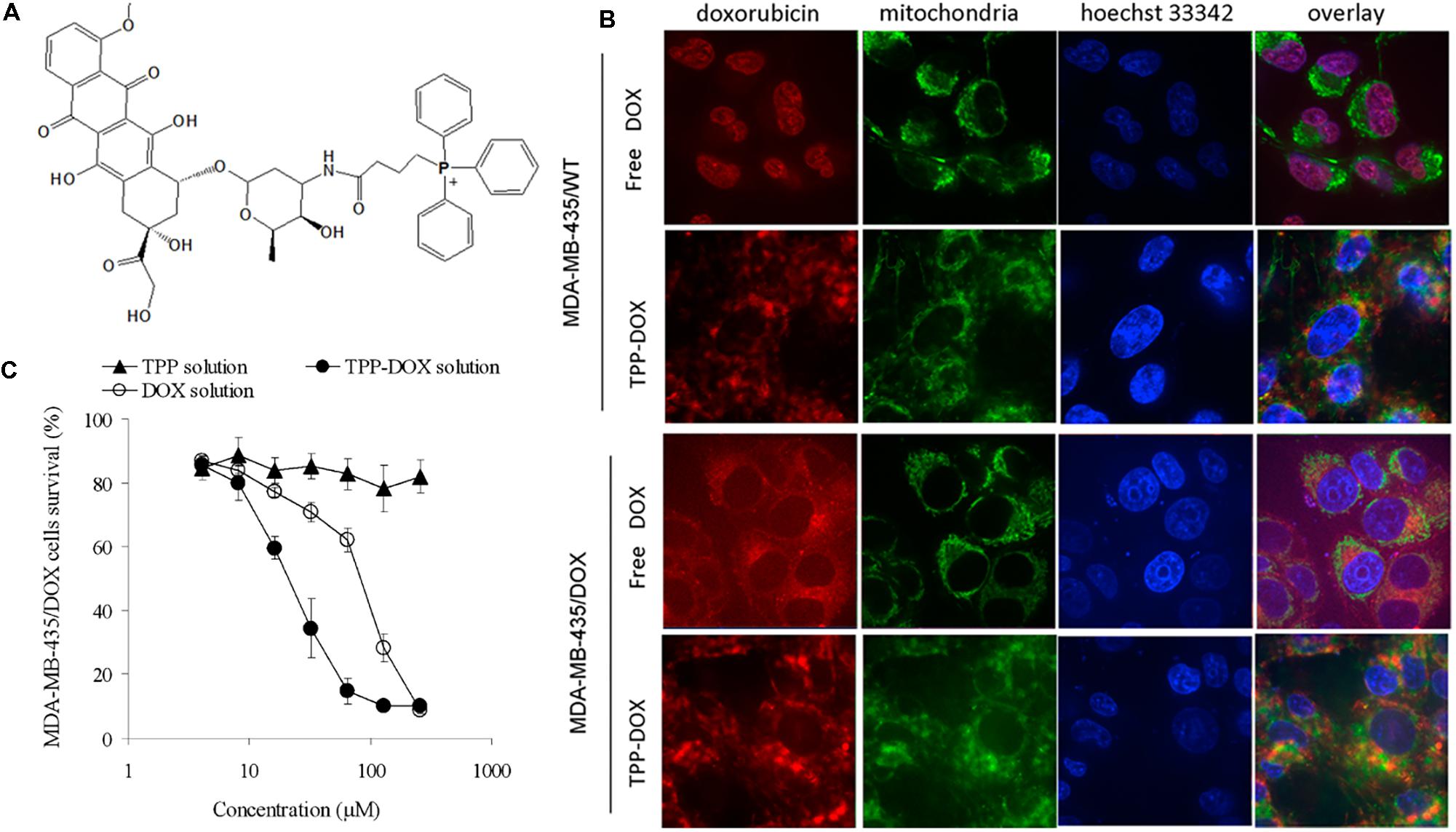
FIGURE 2. (A) Chemical structure of TPP-DOX. (B) Subcellular localization and (C) cytotoxicity of free DOX and TPP-DOX in drug-sensitive and resistant cancer cells; adapted with permission from reference (Han et al., 2014).
Another example is provided by vitamin E (VE) analog conjugates with the TPP molecule (Dong et al., 2011). VE analogs are known to induce cell apoptosis by affecting mitochondrial complex II, where they affect ubiquinone (UbQ) as a natural electron acceptor via the succinate dehydrogenase activity of complex II during the conversion of succinate to fumarate (Neuzil et al., 2001a,b, 2007). As anticancer agents, VE analog α-tocopheryl succinate (α-TOS), vitamin E succinate (VES), and VE were conjugated to the TPP moiety. Among the conjugates, MitoVES, which possesses an 11-carbon linker between tocopheryl succinate and the TPP moiety, strongly induced apoptosis (more than 90%) in various cancer cell lines compared with the other conjugates and its counterpart. The reduced apoptotic activity of VES4TPP was due to blockade of the free carboxyl group, which is essential for the apoptogenic activity of the compound (Dong et al., 2011). In addition, MitoVES did not induce apoptosis in non-malignant cells. The IC50 values of MitoVES in various cancer cell lines were 10- to 30-fold lower than those of the non-targeted counterpart, whereas the IC50 of MitoVES was much greater in non-malignant cells than in malignant cells. Furthermore, flow cytometry analysis showed that MitoVES generated ROS. Greater mitochondrial localization of MitoVES was confirmed by confocal microscopy due to affinity of the positive TPP group to the high negative potential at the matrix face of the mitochondrial inner membrane (MIM). MitoVES is known to induce mitochondria-dependent apoptosis, which involves the transcriptional modulation of bcl-2 family proteins. Furthermore, an antitumor study revealed that MitoVES greatly suppressed tumor growth compared with α-TOS in tumor-bearing mice.
In addition, Millard et al. (2013) compared a TPP-chlorambucil conjugate with other similar cationic moiety-linked chlorambucil conjugates with regard to enhanced therapeutic activity. As mentioned previously, chlorambucil is an anticancer agent that damages DNA. The authors synthesized three different conjugates, TPP-, tributyl phosphonium (TBP)-, and tricyclohexylphosphonium (TCP)-chlorambucil, and one negative control, TPP-dihydroxychlorambucil, and screened the cell cytotoxicity in the MCF7 cancer cell line. The IC50 values of the conjugates were 100, 7, 35, 40, and 80 μM for free chlorambucil, TPP-chlorambucil, TBP-chlorambucil, TCP-chlorambucil, and the negative control, respectively. This result indicated that the presence of aryl rings resulted in a higher potency than the presence of saturated hydrocarbons. Moreover, a cytotoxicity study of free chlorambucil and TPP-chlorambucil was performed in 10 other cancer cell lines, and in all cases, the conjugate had a higher potency, with a sensitivity ratio ranging from 7.7 to 79.9. Next, they found that the conjugate accumulated extensively in mitochondria and caused mitochondrial DNA damage, leading to nitrogen mustard-induced cell death. An in vivo antitumor study revealed that the TPP-chlorambucil conjugate possessed greater tumor suppression efficiency compared with the free counterpart, but it could not completely suppress tumor growth.
Chen et al. (2013) developed a dual-targeted pro-apoptotic peptide (KLAKLAK)2 for delivery into mitochondria and enhanced activity of the peptide. TPP was attached to the peptide via an amide bond, and folic acid was linked to the cell membrane-targeting ligand, enhancing cellular uptake of the peptide via receptor-mediated endocytosis (Figure 3A). The pro-apoptotic peptide is known to disrupt the mitochondrial membrane and induce mitochondria-dependent cell-free apoptosis without toxicity outside of cells (Ellerby et al., 1999; Agemy et al., 2011). To avoid degradation by proteases, D-amino acids were applied for the synthesis. The conjugate (IC50 8.2 μM) showed much stronger cytotoxicity than folate-KLA peptide conjugate alone (IC50 28.5 μM) in KB and HeLa cells (Figure 3B), indicating that TPP targeting assisted cytotoxicity. Further studies showed that the conjugate could activate cytochrome c and caspase 3/9 proteins in cancer cells, leading to apoptotic cell death (Figure 3C). In addition, TEM images of the cells confirmed that mitochondrial disruption occurred after the conjugate treatment (Figure 3D).
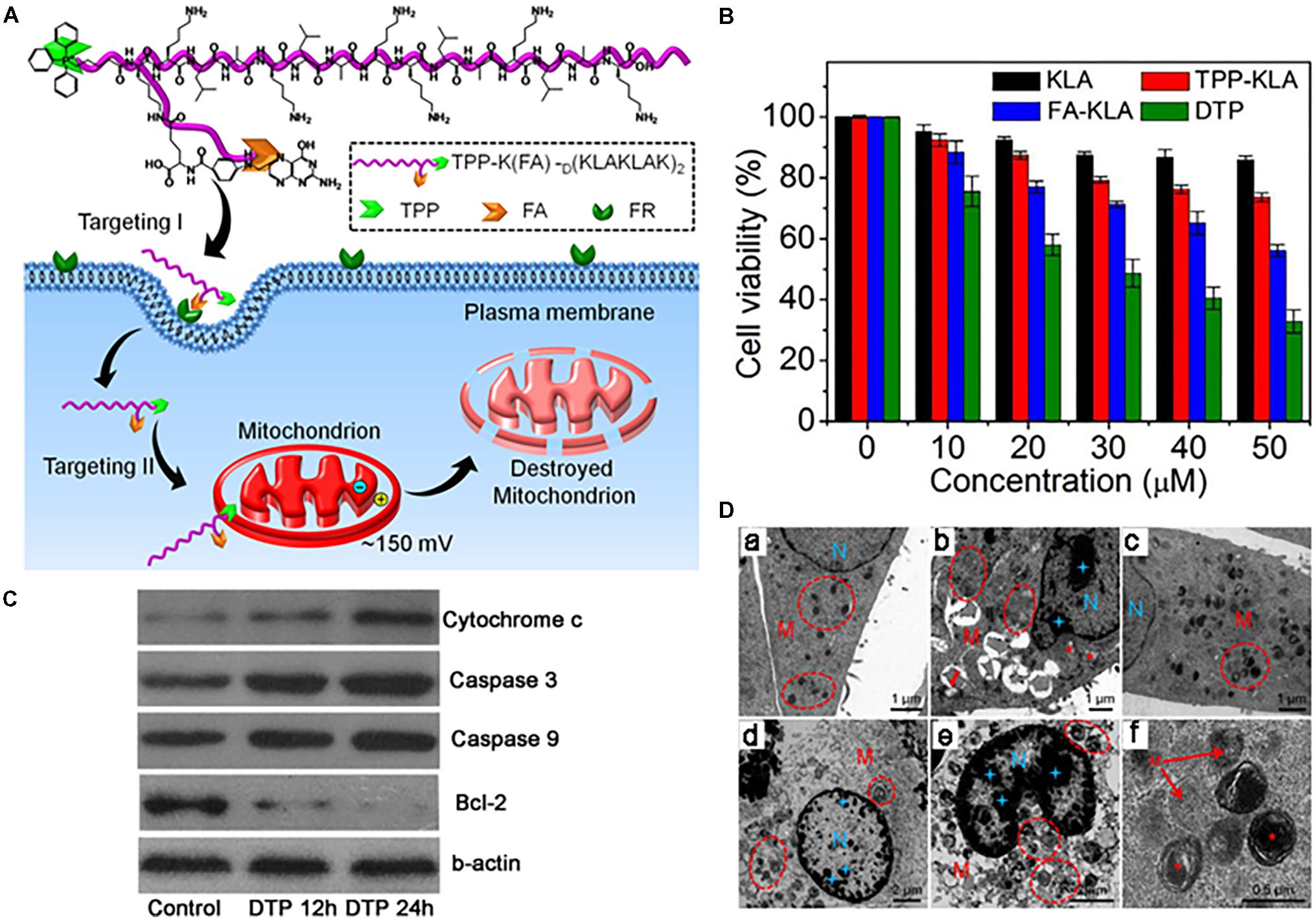
FIGURE 3. (A) Dual-targeting pro-apoptotic peptide (DTP) to selectively target cancer cells and specifically damage mitochondria to lead to programmed cell death. (B) Cell viability of folate receptor (FR) (+) HeLa cells incubated with KLA, TPP-KLA, FA-KLA, and DTP for 48 h. (C) Western blot analysis of apoptosis-related proteins in KB cells incubated with DTP for different times, using β-actin as the control. (D) TEM images of (a) KB cells incubated with no DTP, (b,c) KB cells incubated with DTP (20 mM) for 12 h, and (d) enlarged circled area in (c). (e,f) KB cells incubated with DTP (20 mM) for 24 h. Mitochondria (M) are highlighted by red circles and red pentacles. Nucleus (N) is highlighted by blue doji stars. Images were adapted with permission from reference (Chen et al., 2013).
F16 ((E)-4-(1H-indol-3-ylvinyl)-N-methylpyridinium iodide)-Drug Conjugates
F16 is a delocalized cation and accumulates in the mitochondrial matrix. Initially, Xiang et al. (2013) prepared the F16-5-FU direct conjugate, but it reduced the efficacy of 5-FU. Therefore, they decided to use other linkers that are vulnerable bonds, including the ester bond, amide bond, and disulfide bond. Among the conjugates, F16-OOC-FU showed the strongest toxicity in the SGC-790 (human gastric carcinoma) cell line (Wang et al., 2016). They examined whether this toxicity was associated with cell uptake. The results illustrated that F16-OOC-FU and F16-SS-FU were internalized rapidly by the cells, whereas F16-5FU and F16-NHOC-FU were not internalized well, potentially due to lipophilicity. Enhanced cell uptake of F16-OOC-FU led to greater cytotoxicity, but F16-SS-FU did not affect cytotoxicity due to insufficient glutathione in the cytosol. In addition, the mitochondrial targeting moiety F16 was introduced to boron-dipyrromethene (BODIPY) via a phenylethynyl linker, and the resultant bifunctional mitochondria-targeting anticancer drug conjugate [F16-phenylethynyl-BODIPY conjugate (FPB)] with therapeutic and imaging properties were synthesized. When FPB was applied to cancer and normal cells, it was internalized to a greater extent by cancer cells (e.g., SGC-7901) than by normal cells (e.g., GES-1) (He et al., 2015).
Rhodamine-Drug Conjugate
Rhodamine derivatives are also mitochondria-targeting agents, given their binding affinity to mitochondrial membranes, which subsequently damages the electron transport chain (Baracca et al., 2003). Mechanistically, their mitochondrial accumulation is caused by their lipophilic and cationic properties, which aid in crossing the double mitochondrial membrane and remaining within the negatively charged mitochondrial matrix (Lampidis et al., 1985). Similar to rhodamine 123, rhodamine 19 is a potential mitochondria-targeting moiety (Antonenko et al., 2011). Using rhodamine, several reports have described antioxidant targeting to mitochondria, but very few have addressed anticancer agents. Rhodamine 123 itself has been studied for cancer treatment by conjugation or without conjugation to another molecule (Lampidis et al., 1985; Baracca et al., 2003; Antonenko et al., 2008, 2011). Despite testing in a clinical study for prostate cancer treatment, the dosage (92 mg/m2) was not tolerable, and thus, the efficiency was insufficient.
Guanidine-Drug Conjugates
Guanidinium and biguanidinium, which hold delocalized positive charges, were conjugated to a hydrophobic porphyrin molecule, which is a photosensitizer (PS) and phototoxic agent, to improve mitochondrial accumulation through the high membrane potential across the inner mitochondrial membrane for the treatment of cancer (Figure 4A) (Sibrian-Vazquez et al., 2008). A cellular uptake and subcellular localization study revealed that guanidine-porphyrin (G-Por) as readily taken up by cells and accumulated in mitochondria and the ER compared with biguanidine-porphyrin (BG-Por), which was mainly localized in cell membranes, lysosomes, and mitochondria, associating with their structure and their ability to interact with negatively charged groups in the cell membrane (Figure 4B). The G-Por conjugate showed 1.8-fold higher phototoxicity than the BG-Por conjugate, which may be related to the difference in cellular uptake and localization (Figure 4C). In addition, the guanidine group is a planar structure that acts as a bidentate ligand, but the biguanidine molecule is non-planar due to the requirement to reduce the allylic strain. Partial mitochondrial localization of G-Por and BG-Por could allow the association of guanidine and biguanidine groups, which play important roles in the lysosome/endosome escape of conjugates through membrane rupture, probably by the proton sponge effect, allowing the PS to be distributed to other organelles. Kang et al. (2009) conjugated Gamitrinib, an Hsp90ATPase inhibitor, in human mitochondria to four kinds of cyclic guanidinium. Monomers, dimers, trimers and tetramers of cyclic guanidinium were introduced to Gamitrinib. Gamitrinib-G1, Gamitrinib-G2, Gamitrinib-G3, and Gamitrinib-G4 were synthesized. Among them, Gamitrinib-G4 and Gamitrinib-G3 exhibited increased mitochondrial accumulation and 6- to 10-fold increased anticancer efficacy in cancer cells compared with Gamitrinib-G1 and Gamitrinib-G2 due to different membrane depolarization activities.
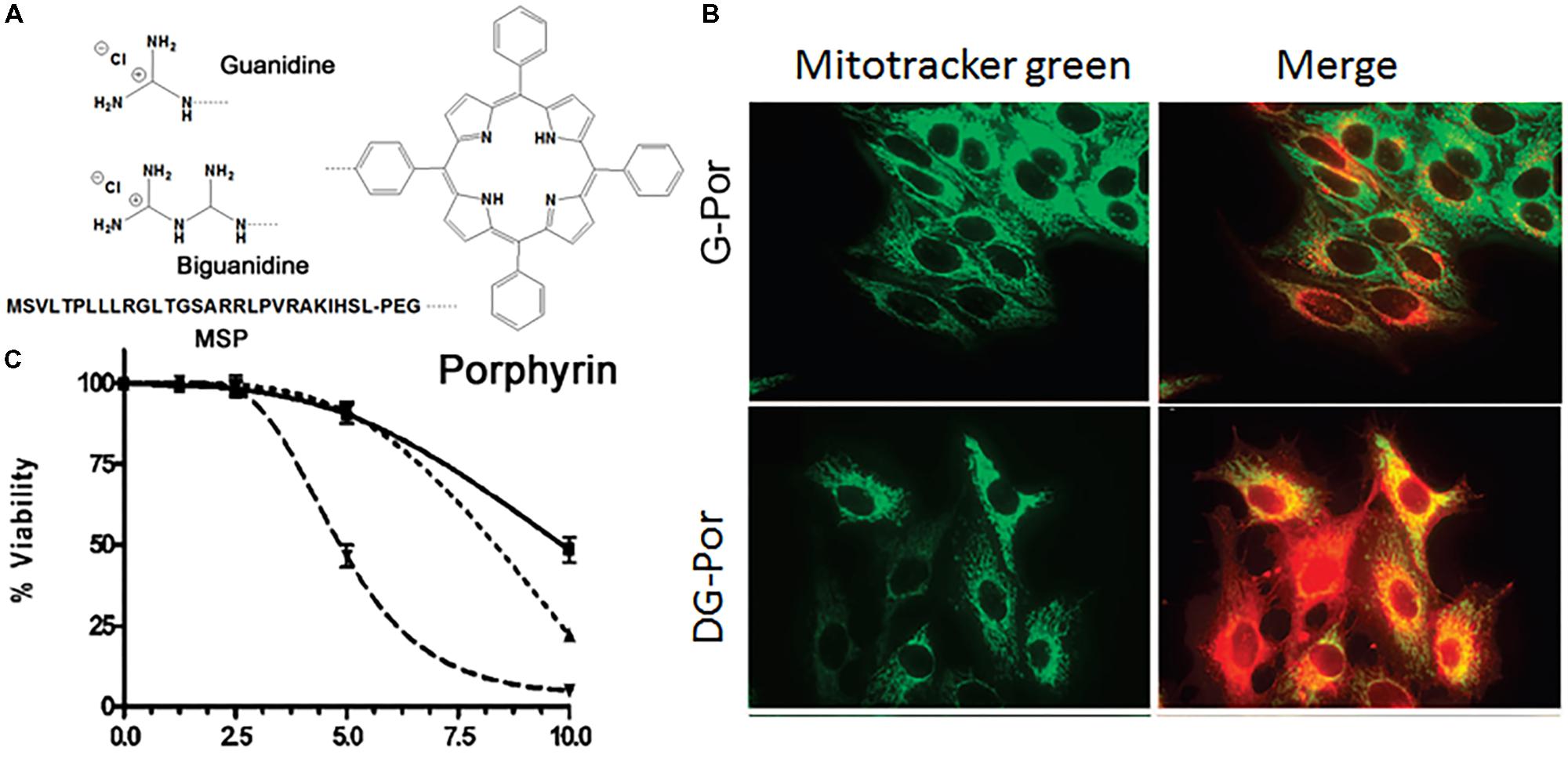
FIGURE 4. (A) Chemical structures of guanidine-porphyrin (G-Por), diguanidine-porphyrin (DG-Por), and peptide-porphyrin conjugates; (B) mitochondrial localization of G-Por and DG-Por in HepG2 cell at 24 h (green for MitoTracker® green and red for porphyrin); (C) phototoxicity of G-Por (dashed line), DG-Por (dotted line), and peptide-Por (continuous line) toward HepG2 cells using a 1 J/cm2 dose of light. Images (B,C) were adapted with permission from reference (Sibrian-Vazquez et al., 2008).
Mitochondria-Targeted Nanocarriers
Although direct conjugation showed promising results for mitochondrial targeting, some challenges persisted in terms of the solubility and clinical application of the therapeutic agents due to poor biocompatibility. Therefore, the use of nanocarriers can enhance the potential of therapeutic agents. Nanotechnology in medicine has captured broad attention over the last three decades due to the large specific surface area, easy functionality, drug-loading capacity, and biocompatibility (Bogart et al., 2014; Wicki et al., 2015). In contrast, disorders such as cancer allow the selective accumulation of nanoparticles in the target area. To date, several kinds of nanocarrier systems, including polymeric nanoparticles, liposomes, micelles, metallic nanoparticles, carbon nanoparticles, and dendrimers, have been developed in medicine, particularly in the drug delivery field. Nanocarrier systems must have some optimal features, such as an average size of 10–200 nm that allows the EPR effect, a neutral surface charge, high biocompatibility, and reduced toxicity. In the case of mitochondrial targeting, the size of nanocarriers should be small as possible. In most cases, a nanoparticle is attached to a targeting ligand to deliver it to mitochondria, but in a few cases, the nanoparticle itself can be the targeting agent to the mitochondria due to its properties. Some physical and biological properties of drug-loaded nanoparticles are summarized in Table 2.
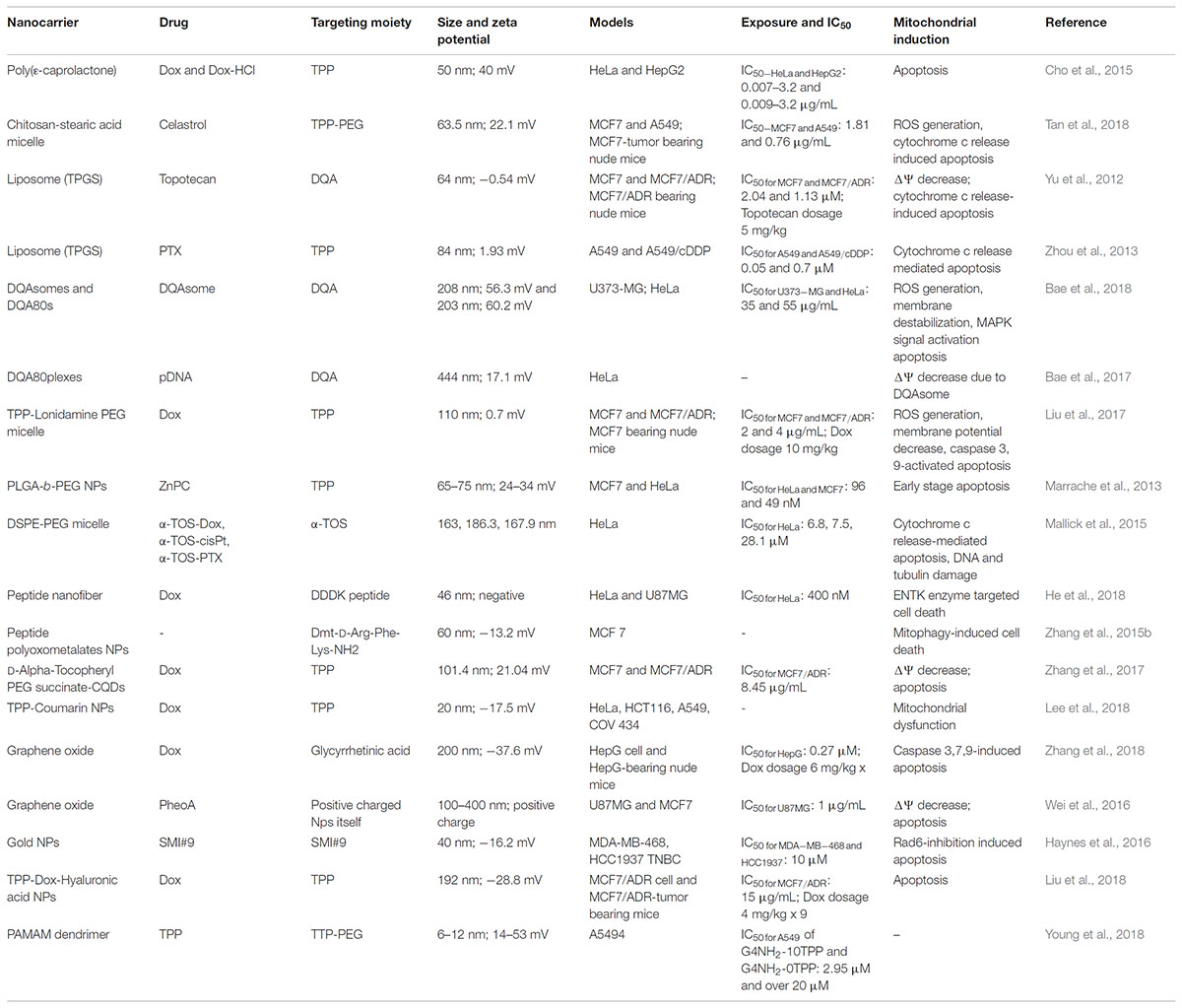
TABLE 2. Some physical and bioactivity characteristics of mitochondria-targeting drug-loaded nanoparticles.
Polymeric Nanoparticles and Micelles
Polymeric nanoparticles are frequently prepared from biodegradable amphiphilic polymers. The advantages of polymeric nanoparticles can be biocompatibility, a small size, high drug loading capacity, good water solubility, low toxicity, and easy modification (Kedar et al., 2010; Oerlemans et al., 2010; Yokoyama, 2010; El-Say and El-Sawy, 2017). Some examples of polymeric nanoparticles for mitochondria-targeted drug delivery are poly(ethylene glycol) (Khatun et al., 2017), poly(𝜀-caprolactone) (PCL) (Cho et al., 2015; Choi et al., 2017), polysaccharides (e.g., chitosan, hyaluronic acid, and dextran) (Kim et al., 2016; Hou et al., 2017; Liu et al., 2018; Tan et al., 2018), poly[(2-hydroxypropyl)-methacrylic acid] (pHPMA) (Sun et al., 2017), poly(lactic-co-glycolic acid) (Marrache and Dhar, 2012; Marrache et al., 2013), and micelles formed from small amphiphilic molecules (He et al., 2018; Lee et al., 2018; Young et al., 2018).
The attachment of mitochondrial targeting ligands to polymeric nanoparticles has been widely explored for the delivery of therapeutic molecules to their active site. The concept of using polymeric nanoparticles for mitochondrial targeting is to deliver therapeutic molecules with reduced toxicity and to improve the biocompatibility of drugs. Polymeric nanoparticles are biocompatible and can carry a large number of cargo materials simultaneously to the target site due to their high loading capacity. Several kinds of polymeric nanoparticles have been applied to mitochondrial targeting, such as PCL with a low molecular weight, chitosan, dendrimers, liposomes, gold nanoparticles, and graphene oxide (Yu et al., 2012; Bielski et al., 2015; Cho et al., 2015; Haynes et al., 2016; Wei et al., 2016; Choi et al., 2017; Tan et al., 2018). Our group developed various TPP-conjugated PCL NPs and demonstrated their mitochondrial targeting ability in cancer cell lines (Cho et al., 2015; Choi et al., 2017; Khatun et al., 2017). We attached the TPP moiety at both ends of low-molecular-weight PCL polymers (0.5, 1.25, and 2 kDa). These polymers self-assemble in an aqueous environment with or without a hydrophobic anticancer drug (Figure 5A). The mean diameters of these NPs are approximately 100 nm, which is an optimal size for a drug carrier. As shown in Figures 5B,C, Dox-loaded PCL NPs preferentially accumulated in the mitochondria of HeLa cells compared to free Dox, resulting in a 3- to 5-fold higher cytotoxicity compared with free Dox. Additionally, the PCL polymeric nanoparticles were able to deliver both hydrophobic and hydrophilic Dox (Dox-HCl) into mitochondria. TPP-related cytotoxicity was observed due to its positive charge. These results support the potential of the polymeric carrier system in mitochondria-targeting drug delivery approaches.
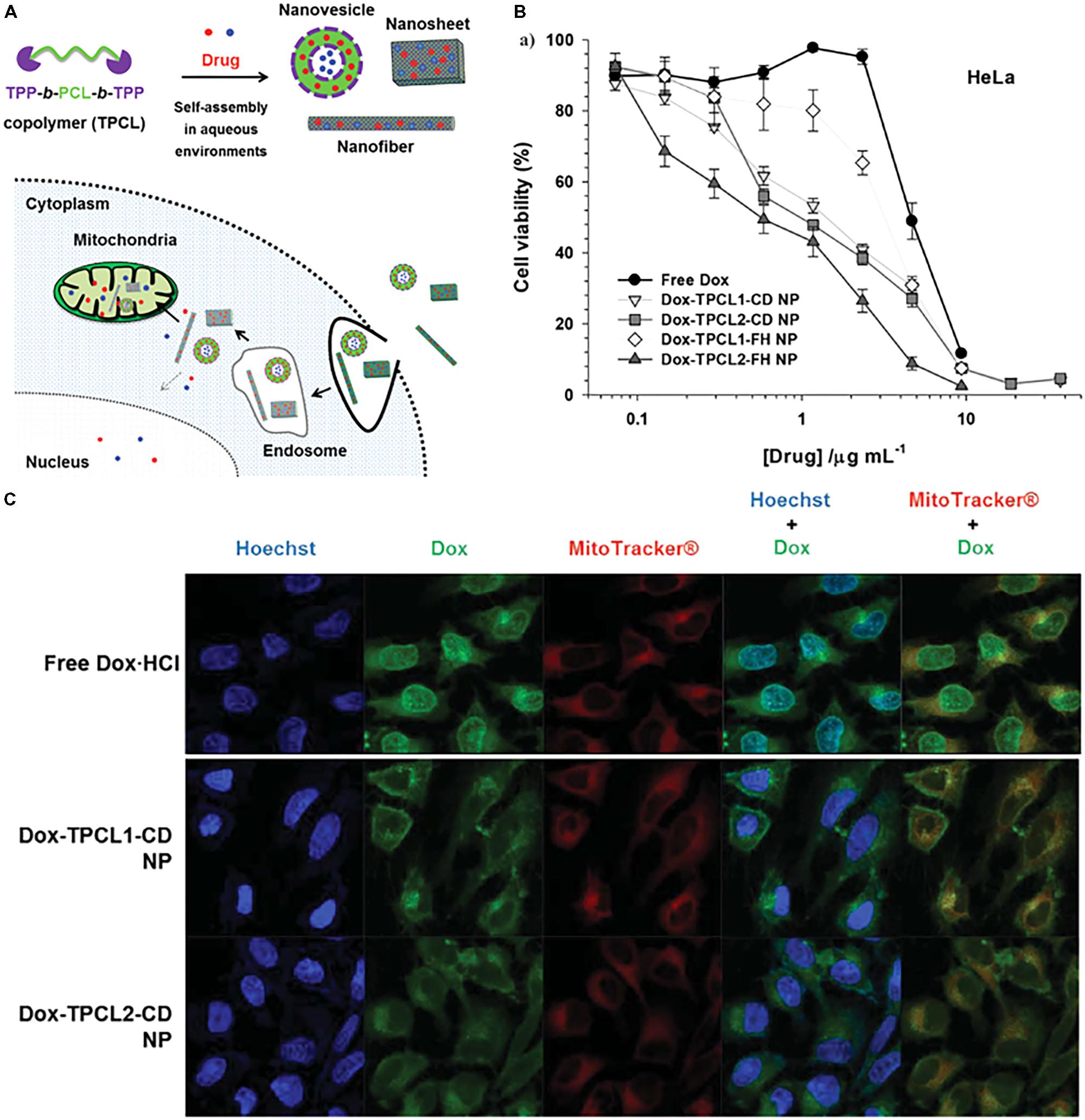
FIGURE 5. (A) Illustrated design concept of drug-loaded TPCL nanoparticles (NPs) in cells. The red solid circle (∙) and blue solid circle (∙) indicate hydrophobic and hydrophilic chemical drugs, respectively. (B) In vitro antitumor effects of Dox-loaded TPCL NPs in HeLa cells. (C) Intracellular distribution of free drugs and drug-loaded TPCL NPs and Dox-TPCL-CD NPs with [Dox] = 1 μg mL-1 in HeLa cells at 4 h post-treatment. Images were adapted with permission from reference (Cho et al., 2015).
Liu et al. (2017) described Dox-loaded TPP-lonidamine self-assembled nanoparticles for mitochondria-targeted cancer treatment). They also attached PEG to the NPs to enhance stability in the blood. The NPs have a size of approximately 110 nm and a neutral surface charge. As claimed by the authors, the NPs showed slow cellular uptake compared with Dox, but they were preferentially localized in mitochondria. Furthermore, the NPs showed greater cytotoxicity in both drug-sensitive and drug-resistant cancer cells compared with the other controls including free Dox. The results confirmed that the NPs generated high ROS levels, decreased the mitochondria membrane potential, and activated caspase 9 and caspase 3 proteins, which induce apoptosis. In addition, the NPs had a 12-fold higher half-life than free Dox in blood and suppressed tumor growth by 2- to 4.5-fold more than in the other groups. Interestingly, the authors claimed that the introduction of PEG to the NPs did not affect their cellular uptake and anticancer efficacy.
Tan et al. (2018) prepared a TPP-attached chitosan-stearic acid conjugate (TPP-CSOSA) loaded with celastrol (Cela), which is a weakly acidic anticancer drug, for drug delivery to mitochondria in a pH-dependent manner. The TPP-CSOSA micelles had a size of 86 nm, and Cela-loading reduced the size to 63 nm. They used a weakly acidic drug, which could easily dissociate in an alkaline pH environment such as the mitochondrial matrix (pH 8). The confocal data showed that FITC-labeled TPP-CSOSA localized in mitochondria in a time-dependent manner. The intracellular localization of NPs was analyzed by TEM imaging and validated their presence in mitochondria. Further studies have confirmed that NPs have greater cytotoxicity in cancer cells than the control groups and generate 3.2-fold higher ROS, which leads to a decrease in membrane potential and depolarization. In vivo studies have revealed that TPP-CSOSA/Cela NPs accumulate to a great extent in tumor tissues and have tumor suppression efficacy. The attachment of TPP to NPs enhanced cell uptake and tumor accumulation, and the use of the weakly acidic drug Cela led to rapid release at a higher pH, resulting in improved pharmacological action. Liu et al. (2018) prepared a hyaluronic acid-Dox-TPP conjugate using a hydrazone linkage to specifically deliver TPP-Dox to mitochondria. The conjugate formed nanoparticles with a size of 192 nm and a negative surface charge. A cell uptake study showed greater mitochondrial accumulation of the NPs in MCF/ADR cells, and further cytotoxicity and antitumor studies confirmed the enhanced efficacy compared with free Dox and TPP-Dox conjugates. Bielski et al. (2015) described a TPP-poly(amidoamine) dendrimer with different numbers of TPP showing different mitochondria-targeting behaviors. The TPP moiety was attached to dendrimers as a direct amide linkage or using the PEG spacer. Both direct conjugation and conjugation with the PEG spacer resulted in enhanced mitochondria-targeting behavior compared with the non-targeted dendrimers. In the case of direct conjugation, there was a direct correlation between mitochondrial targeting and the TPP density in dendrimers, while in the TPP-PEG dendrimer, mitochondrial targeting was not affected. Another example of the use of PEG for mitochondria-targeting NPs was reported to deliver pheophorbide a (PheoA) to mitochondria in the presence of TPCL (Choi et al., 2017). The data showed that the nanoparticle delivered PheoA to mitochondria, but the cell killing effect of free PheoA was not achieved.
Han et al. (2015) linked porphyrin (PpIX), a clinically available photosensitizer, to PEG-proapoptotic peptide (KLAKLAK)2 to obtain a mitochondria-targeted photodynamic agent (Figure 6A). The conjugate was self-assembled and formed nanomicelles with a size of 91 nm. The micelle was found to decrease the mitochondrial membrane potential (Figure 6B), leading to cytochrome c release and apoptotic cell death. Animal studies showed that the micelles accumulated in the tumor area for 8 days (Figure 6C) and strongly suppressed tumor growth.
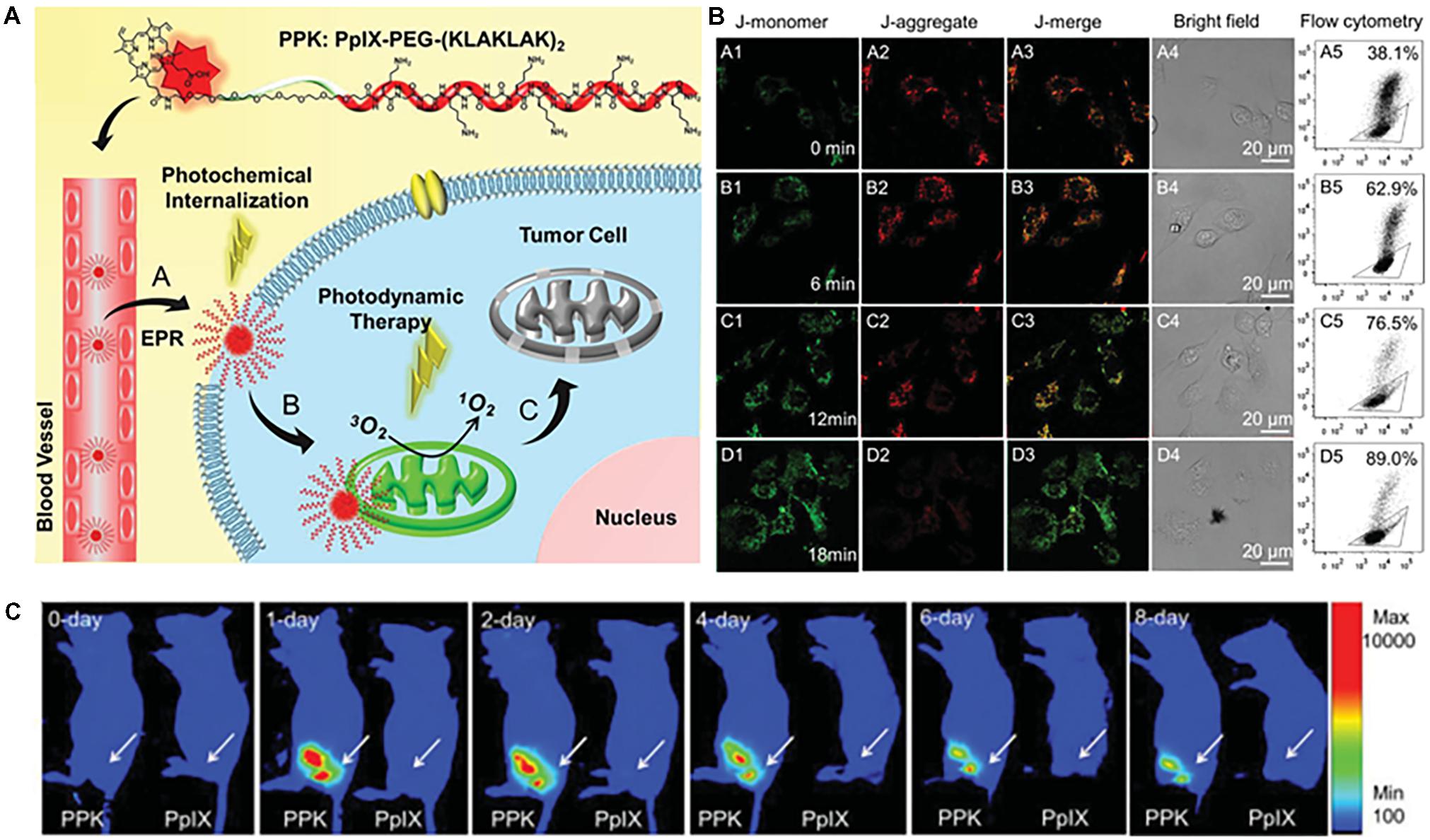
FIGURE 6. (A) Structure of PPK and schematic diagram of the mitochondria-targeted self-delivery process. Passive targeting to tumor cells and the photochemical internalization (PCI) effect enhanced cellular internalization of PPK under short-duration light irradiation. (B) Mitochondria-targeting mediated by (KLAKLAK)2. (C) ROS formation, mitochondrial destruction, and cell death mediated by the synergistic effect between (KLAKLAK)2 and PpIX under long-duration light irradiation. (B) CLSM images and the corresponding flow cytometric analysis of the mitochondrial membrane potential for HeLa cells when the cells were incubated with 40 μg mL-1 of PPK and then exposed to different irradiation durations: (A1–A5) without light irradiation (control); (B1–B5) with 6-min light irradiation; (C1–C5) with 12-min light irradiation; (D1–D5) with 18-min light irradiation. For (A5–D5), the triangle separates populations with high (inside the triangle) and low (outside the triangle) membrane potential. (C) Intratumoral fluorescence distributions of PpIX and PPK on the 10th day after subcutaneous injection. Images were adapted with permission from reference (Han et al., 2015).
DQAsome and Liposomes
DQAsomes were early nanocarriers for mitochondria targeting consisting of a bolaform self-assembly of DQA in aqueous solution due to the presence of a positively charged head group and alkyl chain (Weissig et al., 1998; Weissig and Torchilin, 2001; Weissig, 2015). The DQAsomes were incorporated with pDNA, antioxidant agents, and anticancer agents in the early 2000 (Weissig et al., 1998; Weissig and Torchilin, 2001; D’Souza et al., 2003), but they exhibited a low transfection efficiency and reduced ability to escape from endosomes, raising difficulties in the delivery their cargos to target site (D’Souza et al., 2003). In addition, they showed high cell killing activity in cancer cells compared with normal fibroblasts due to the disruption of mitochondrial transmembrane potential, blockade of ATP synthesis, and ROS generation (Sancho et al., 2007; Pajuelo et al., 2011). Therefore, many studies have focused on DQA-liposomes as a carrier (Yu et al., 2012; Zhou et al., 2013; Bae et al., 2017, 2018). Bae et al. (2017, 2018) developed DQA-liposomes consisting of DQA/DOTAP/DOPE or DQA80s (80% DQA) that can deliver the therapeutic agent to mitochondria and show anticancer effects. The liposomes can escape from lysosomes due to the usage of DOPE, which is a neutral lipid membrane disruption lipid. A cytotoxicity study revealed that DQA80s had higher activity in cancer cells than DQAsomes. A subcellular study showed that DQA80s accumulated to a high extent in mitochondria than simple DQAsomes. The TEM study then confirmed the intracellular localization of DQA80s. Furthermore, DQA80s were incorporated with pDNA and prepared DQAplexes (Bae et al., 2017). The liposome had a size of approximately 400 nm and a positive surface charge.
Drug-encapsulated DQA-based liposomes have been reported to specifically deliver drug to mitochondria (Wang et al., 2011; Yu et al., 2012; Zupancic et al., 2014). Topotecan hydrochloride, a derivative of camptothecin and a topoisomerase I inhibitor, was loaded into DQA and TPGS liposomes (Yu et al., 2012). Mitochondria-targeting topotecan-loaded DQA liposomes showed 2- and 4-fold higher cytotoxicity in MCF7 and MCF7/ADR cells, respectively, than free topotecan. The colocalization study revealed that the liposomes delivered the cargo to mitochondria at levels 21.2-fold higher than the free form and 12.9-fold higher than non-targeted liposomes by flow cytometry. A further study confirmed that liposomes could release cytochrome c following apoptotic cell death. An in vivo biodistribution study showed that Cy7-labeled DQA-liposomes accumulated in tumors, and a further antitumor study in MCF7/ADR tumor-bearing mice demonstrated that the DQA-liposomes strongly suppressed tumor growth compared with the other groups.
Inorganic Nanoparticle Carriers
A number of nanoparticles have been studied for drug delivery, imaging, and therapeutic purposes, such as graphene oxide (GO), gold nanoparticles, silica nanoparticles, quantum dots, and carbon nanospheres, because of their biocompatibility, optimal size, large surface area, easy functionality, and specific physical properties (Wilczewska et al., 2012; Mattoussi and Rotello, 2013). Some mitochondria-targeting nanoparticles that were recently developed include GO (Wei et al., 2016; Zhang et al., 2018), lipid-coated carbon-quantum dots (Zhang et al., 2017), carbon-silica nanoparticles (Wang et al., 2018), polyoxometalates (Zhang et al., 2015b), and gold nanoparticles (Haynes et al., 2016) to deliver the therapeutic agent to mitochondria and overcome drug resistance. NPs have been known to successfully deliver anticancer agents to mitochondria with or without a targeting ligand. Some specific characteristics of nanoparticles are included in Table 2.
Zhang et al. (2018) described glycyrrhetinic acid (GA)-attached GO with Dox as a model drug for mitochondria-targeted cancer treatment (Figure 7A). The authors chose GA for dual targeting to mitochondria and the cell membrane due to its ability to interact with the mitochondrial respiratory chain and binding affinity to protein kinase C (PKC) α, which is overexpressed on some cancer cells (Salvi et al., 2003; Fiore et al., 2004; Stankovich et al., 2006; Wu et al., 2008). In addition, GO is known to have a large specific surface area, high stability, functionality, and an optimal size for drug delivery (Yang et al., 2013; Battogtokh and Ko, 2016). Cell uptake was found to be PKC-dependent via the blockade of GA. Moreover, the NPs were confirmed to be able to reduce the membrane potential and activate caspases 3 and 9, resulting in apoptotic cell death (Figure 7B). The results showed that Dox-loaded GA-GO had twofold high cytotoxicity than free Dox in HepG2 cell, which was confirmed by robust uptake by cells and mitochondria (Figure 7C). The authors found that the NPs accumulated to a high extent in tumor tissue and liver due to the GA-targeting affinity, leading to apoptosis-mediated death (Figure 7D). Another example is the GO-photosensitizer complex, which has been used for mitochondria-targeted cancer therapy (Wei et al., 2016). The authors claimed that the nanoparticle preferentially accumulated in mitochondria compared with free PS due to the strong electronic interactions of the polarized GO with the mitochondria membrane and endosome-escape properties via simple traversal of the lipid membrane (Zhou et al., 2010; Wu et al., 2014b). Further studies have revealed that GO-PS has greater phototoxicity and induces apoptotic cell death.
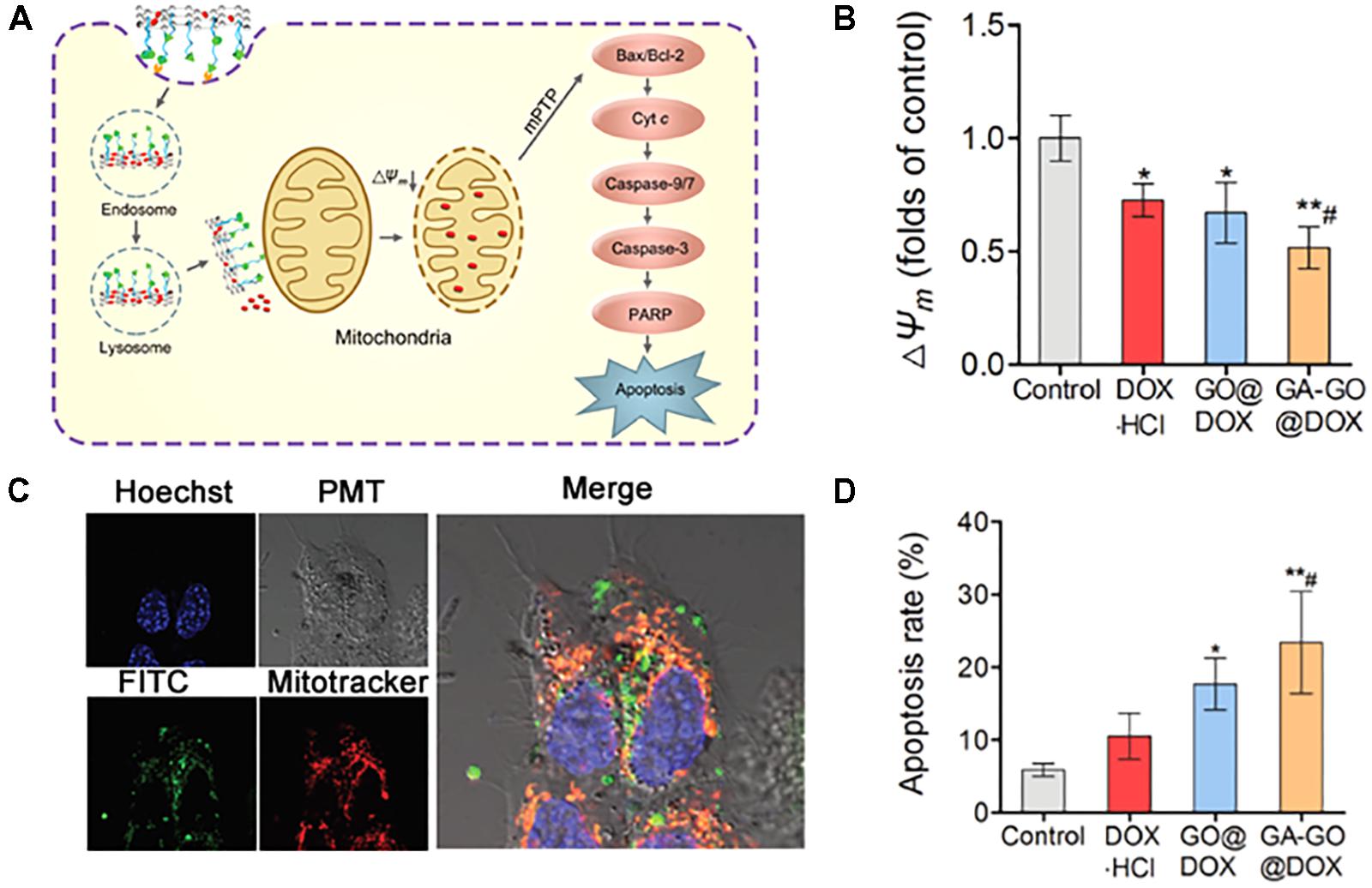
FIGURE 7. (A) Schematic illustration of the proposed mechanism of the MMA pathway of GA-GO@DOX. (B) ΔΨm of HepG2 cells after incubation with DOX⋅HCl, GO@DOX, GA-GO@DOX, and the control at an equivalent DOX dose of 0.5 μg mL-1 for 24 h. (C) CLSM images of the intracellular localization of GA-GO-FITC in HepG2 cells after treatment with GA-GO-FITC (800 μg mL-1) for 6 h. The cells were stained with organelle trackers for 45 min. (D) The apoptosis rate of HepG2 cells after treatment with DOX⋅HCl, GO@DOX, GA-GO@DOX, and the control. Images were adapted with permission from reference (Zhang et al., 2018). Bars represent means ± SD (n = 3); ∗p < 0.05 and ∗∗p < 0.01, compared to control; #p < 0.05, compared to DOX⋅HCl.
Zhang et al. (2017) prepared mitochondria-targeting Dox-loaded carbon quantum dot (CQD) nanoparticles to overcome drug resistance. D-α-Tocopherol polyethylene glycol succinate (TPGS), an inhibitor of Pgp, was included in the nanoparticle to inhibit Pgp expression in drug-resistant cancer cells. CQDs are used as fluorescent probes for bioimaging, biolabeling, and biosensing due to their strong and stable fluorescence and low toxicity (Lim et al., 2015). TPP was conjugated to TPGS and covered the CQDs. The cytotoxicity results revealed that the Dox-loaded CQD nanoparticles had a fivefold lower IC50 value in drug-resistant MCF7 cells compared with free Dox. A further intracellular study revealed preferential mitochondrial localization of the NPs in drug-resistant cells. Mechanistic studies are currently outstanding.
Additionally, Zhang et al. (2015b) developed peptide-polyoxometalate (POM) nanoparticles as mitochondria-targeted anticancer agents. POM nanoparticles have photoluminescence properties, but their rapid quenching effect does not allow cell imaging. As a mitochondria-targeting agent, the positively charged short peptide Dmt-D-Arg-Phe-Lys-NH2 was chosen due to its selective targeting to mitochondria (Zhao et al., 2004). Preferential mitochondrial localization of the nanoparticle was demonstrated by confocal and TEM imaging. The authors claimed that the NPs could induce mitophagy through a Parkin (Parkinson juvenile disease protein 2)-mediated process.
Wang et al. (2018) described pyruvate-based mitochondria-targeting lipid-membrane coated silica-carbon (LSC) hybrid nanoparticles to solve the problem of drug resistance. These nanoparticles could produce ROS through near-infrared (NIR) laser irradiation. During preparation of the nanoparticles, pyruvaldehyde groups were formed with a structure very similar to pyruvate, which can specifically bind to the monocarboxylate transporters on the outer mitochondrial membrane (Pinheiro et al., 2012; Wen et al., 2013). Therefore, pyruvaldehyde could be a mitochondrial targeting moiety for this nanoparticle. Dox was encapsulated in this nanoparticle, and its effects on drug-sensitive and drug-resistant cancer cells were evaluated. Mitochondrial accumulation of the Dox-loaded NPs was demonstrated by confocal microscopy and TEM imaging. Pyruvate-mediated mitochondrial uptake was tested by pre-treatment with pyruvic acid, followed by a 40 to 3% decrease in mitochondrial uptake. A further cytotoxicity study showed that Dox-LSC nanoparticles with laser irradiation had a great killing effect in drug-resistant cancer cells compared with free Dox and LSC NPs. In vivo biodistribution and antitumor studies revealed that LSC NPs preferentially accumulated in tumor tissue and completely suppressed tumor growth. These results were due to the ability of the LSC NPs to target mitochondria and produce ROS under NIR irradiation to oxidize NADH to inhibit the production of ATP, thus reducing the function of efflux pumps and overcoming the drug resistance.
Mitochondria-Targeted Drug Conjugate-Loaded Nanoparticles
In most cases, mitochondria-targeting ligand-drug conjugates are poorly water soluble, and thus they are difficult to apply in preclinical and clinical studies. One solution is to use a carrier system and deliver them to a target site such as a tumor. The conjugates could subsequently be released from the nanocarrier and reach the mitochondria. Several studies, including studies of TPP-docetaxel conjugates and TPP-photosensitizer-loaded albumin nanoparticles (Battogtokh and Ko, 2017; Battogtokh et al., 2018), paclitaxel, cisplatin or Dox-α-TOS conjugate-loaded lipid nanoparticles (Mallick et al., 2015), and DQA-Dox-loaded pH-responsive micelles (Song et al., 2015), have revealed dual-targeting to the tumor area and mitochondria. The directed targeting of nanosized materials could show delayed entry into mitochondria due to their size, but small mitochondria-targeting molecules can easily enter mitochondria. Therefore, the role of nanocarriers could be to deliver small molecules to the tumor area and cellular cytosol.
Butyl carboxyl TPP was conjugated to docetaxel (DTX) via an ester bond and loaded into folate-targeted albumin nanoparticles to enhance the therapeutic activity of DTX (Battogtokh et al., 2018). Albumin was coupled to the cholesteryl anchor to form self-assembled nanoparticles, which is known to have a high drug loading capacity and a size of approximately 120 nm. Thus, the TPP-DTX conjugates were loaded with cholesteryl-albumin and evaluated in vitro and in vivo. The results revealed that the conjugate accumulated in mitochondria and induced apoptosis-mediated cell death. In contrast, TPP-photosensitizer conjugate-loaded albumin nanoparticles were prepared to localize the photosensitizer in mitochondria because they must be localized at the specific target site due to the short half-life of ROS generated from PS through laser irradiation (Battogtokh and Ko, 2017). If the ROS are generated at a site that is distant from the target site, the therapy will fail due to insufficient damage. Some PS are known to accumulate in mitochondria, but they are also localized in other organelles. Therefore, there is a need to selectively deliver PS to mitochondria. TPP-PS conjugate-albumin NPs showed preferential mitochondrial accumulation, mitochondrial membrane destabilization, apoptotic cell death, and enhanced antitumor efficacy in tumor-bearing nude mice.
Song et al. (2015) further described DQA-Dox conjugate-loaded pH-responsive micelles to achieve drug resistance in cancer cells. The authors synthesized DQA-Dox conjugates via an amide linkage and DSPE-hyd-PEG-anisamide (AA) conjugates by the reaction between DSPE (1,2-distearoyl-sn-glycero-3-phosphoethanolamine) and PEG via linkage with the hydrazone bond, with cleavability at a low pH, with the aim of disassembly in lysosomes/endosomes (Figure 8A). To the DSPE-hyd-PEG conjugates was attached AA for active targeting to sigma receptors expressed on cancer cells. The Dox/DQA-Dox was loaded into micelles and assessed in drug-resistant cancer cells. A cytotoxicity study in MDA-MB-231/ADR cells revealed that the Dox/DQA-Dox-loaded nanoparticles had much higher activity (Figure 8B). A subcellular localization study using confocal microscopy showed that the nanoparticles were preferentially localized in mitochondria (Figure 8C). Furthermore, an antitumor study illustrated that nanoparticles with 2 and 10 μmol/kg of Dox had 2- and 5-fold greater tumor suppression effects than Dox (10 μmol/kg) in a Dox-resistant tumor model (Figure 8D). Mallick et al. (2015) directly conjugated α-TOS, a mitochondria-targeting drug, to cisplatin, Dox, or paclitaxel via monocarboxylate, amide, or ester bonds, respectively. The conjugates were then loaded into DSPE-PEG micelles with a size of less than 200 nm. The conjugate-loaded NPs showed much higher cell-killing effects than the free drugs, while their efficacy decreased after loading the conjugates in NPs. These studies revealed that conjugate-loaded nanoparticles localized in mitochondria and damaged the mitochondrial membrane, leading to cytochrome c release and cell apoptosis. The α-TOS-paclitaxel conjugate damaged the tubulin of HeLa cells.
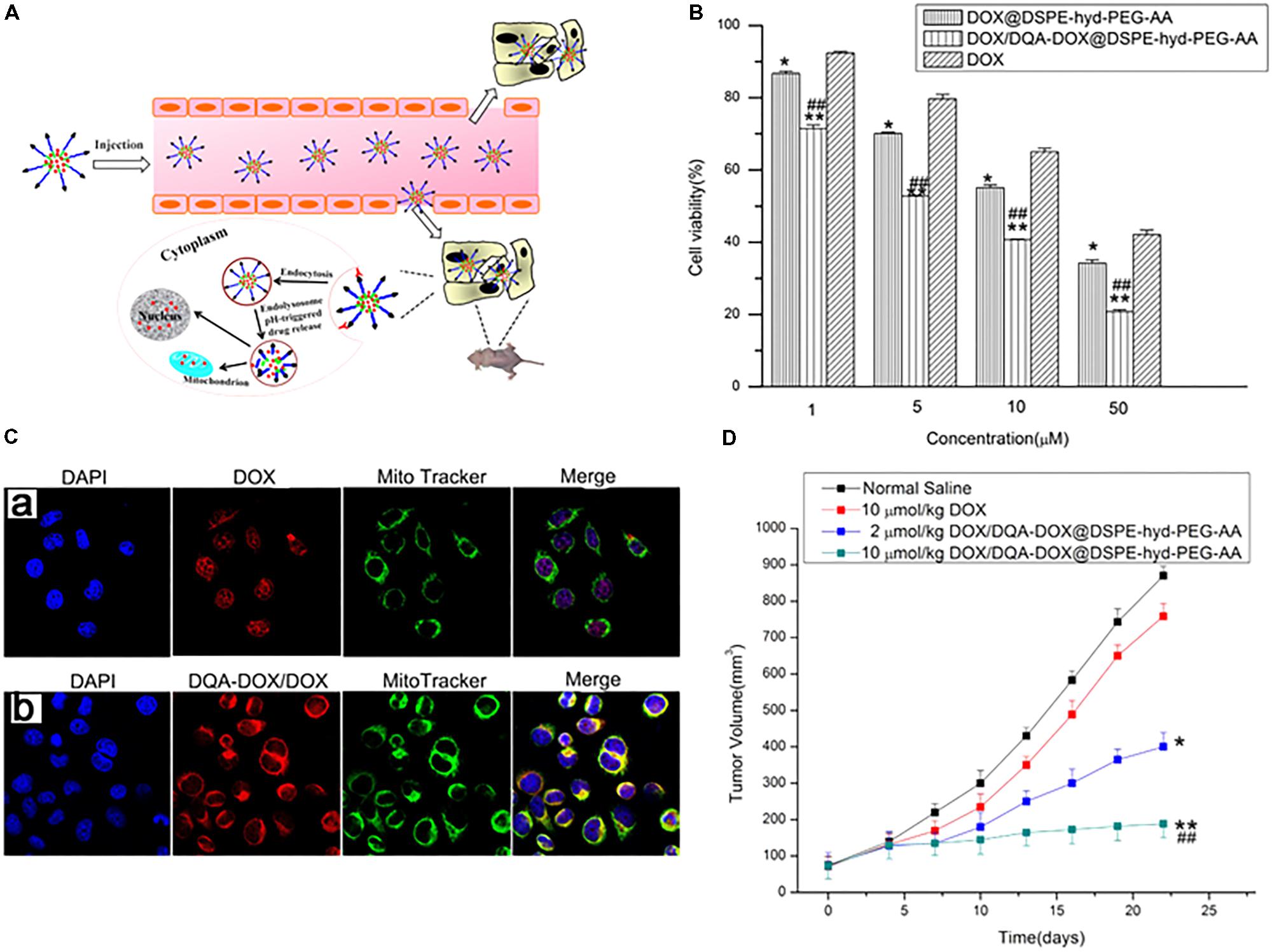
FIGURE 8. (A) Schematic representation of pH-responsive DOX/DQA-DOX@DSPE-hyd-PEG-AA for intracellular doxorubicin delivery in breast cancer. (B) Cytotoxicity of DOX, DOX/DQA-DOX@DSPE-hyd-PEG-AA and DOX@DSPE-hyd-PEG-AA in A549 cells at 48 h. (C) The distribution of DOX in mitochondria after A549 cells were treated with DOX@DSPE-hyd-PEG-AA (a) and DOX/DQA-DOX@DSPE-hyd-PEG-AA (b) at 37°C for 4 h. (D) In vivo antitumor activity of DOX/DQA-DOX@DSPE-hyd-PEG-AA in athymic nude mice xenografted with MDA-MB-231/ADR cells. Images were adapted with permission from reference (Song et al., 2015). Data are presented as mean ± SD, n = 4; ∗p < 0.05 and ∗∗p < 0.01, vs. normal saline; ##p < 0.01, vs. free DOX.
Conclusion
To date, many studies have investigated mitochondria-targeted drug delivery, particularly anticancer drug delivery. Early studies were focused on the direct conjugation of the mitochondria-targeting ligand and anticancer agents. Although direct conjugates were able to solve some issues such as drug-resistance, other challenges such as solubility problems remained, limiting preclinical and clinical applications. In the last decade, many studies have explored mitochondria-targeting carrier development using biomaterials and inorganic nanoparticles to solve the solubility and selectivity issues related to the drugs. Commonly applied mitochondria-targeting ligands are DLCs, particularly TPP and DQA, but those ligands are known to have toxicity problems. Therefore, some recent studies have described the replacement of TPP with other molecules, including short peptides and anionic molecules. Both direct conjugation and nanocarriers are commonly tested in drug-resistant cancer cells because drug resistance is one of the leading limitations of current chemotherapy, and in most cases, the conjugate and nanocarriers provide better results in drug-sensitive cells and animal models. In the future, more detailed preclinical and clinical studies with nanocarrier systems and the development of alternative targeting agents would become the core of the mitochondria-targeting drug delivery field. In addition, we noted that the most frequently used drug for both direct conjugation and nanocarrier for mitochondrial delivery was Dox. As there are several other mitochondria-mediated drugs, those drugs should also be studied.
Author Contributions
GB, Y-YC, JL, HL, and HK collected context information and discussion. GB and HK wrote the manuscript.
Funding
This study was supported by the National Research Foundation of Korea (NRF) funded by the Korean Government (MSIT) (NRF-2017M3A9F5028608 for the Bio & Medical Technology Development Program and NRF-2017R1A4A1015036). Also, the study was supported by BK21PLUS grant of NRF funded by the Korean Government (ME) (22A20130012250).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Agemy, L., Friedmann-Morvinski, D., Kotamraju, V. R., Roth, L., Sugahara, K. N., Girard, O. M., et al. (2011). Targeted nanoparticle enhanced proapoptotic peptide as potential therapy for glioblastoma. Proc. Natl. Acad. Sci. U.S.A. 108, 17450–17455. doi: 10.1073/pnas.1114518108
Alexeyev, M., Shokolenko, I., Wilson, G., and LeDoux, S. (2013). The maintenance of mitochondrial DNA integrity–critical analysis and update. Cold Spring Harb. Perspect. Biol. 5: a012641. doi: 10.1101/cshperspect.a012641
Antonenko, Y. N., Avetisyan, A. V., Bakeeva, L. E., Chernyak, B. V., Chertkov, V. A., Domnina, L. V., et al. (2008). Mitochondria-targeted plastoquinone derivatives as tools to interrupt execution of the aging program. 1. Cationic plastoquinone derivatives: synthesis and in vitro studies. Biochemistry (Mosc) 73, 1273–1287. doi: 10.1134/S0006297908120018
Antonenko, Y. N., Avetisyan, A. V., Cherepanov, D. A., Knorre, D. A., Korshunova, G. A., Markova, O. V., et al. (2011). Derivatives of rhodamine 19 as mild mitochondria-targeted cationic uncouplers. J. Biol. Chem. 286, 17831–17840. doi: 10.1074/jbc.M110.212837
Bae, Y., Jung, M. K., Lee, S., Song, S. J., Mun, J. Y., Green, E. S., et al. (2018). Dequalinium-based functional nanosomes show increased mitochondria targeting and anticancer effect. Eur. J. Pharm. Biopharm. 124, 104–115. doi: 10.1016/j.ejpb.2017.12.013
Bae, Y., Jung, M. K., Song, S. J., Green, E. S., Lee, S., Park, H. S., et al. (2017). Functional nanosome for enhanced mitochondria-targeted gene delivery and expression. Mitochondrion 37, 27–40. doi: 10.1016/j.mito.2017.06.005
Baracca, A., Sgarbi, G., Solaini, G., and Lenaz, G. (2003). Rhodamine 123 as a probe of mitochondrial membrane potential: evaluation of proton flux through F(0) during ATP synthesis. Biochim. Biophys. Acta 1606, 137–146. doi: 10.1016/S0005-2728(03)00110-5
Battogtokh, G., Gotov, O., Kang, J. H., Cho, J., Jeong, T. H., Chimed, G., et al. (2018). Triphenylphosphine-docetaxel conjugate-incorporated albumin nanoparticles for cancer treatment. Nanomedicine (Lond.) 13, 325–338. doi: 10.2217/nnm-2017-0274
Battogtokh, G., and Ko, Y. T. (2016). Graphene oxide-incorporated pH-responsive folate-albumin-photosensitizer nanocomplex as image-guided dual therapeutics. J. Control. Release 234, 10–20. doi: 10.1016/j.jconrel.2016.05.007
Battogtokh, G., and Ko, Y. T. (2017). Mitochondrial-targeted photosensitizer-loaded folate-albumin nanoparticle for photodynamic therapy of cancer. Nanomed. Nanotechnol. Biol. Med. 13, 733–743. doi: 10.1016/j.nano.2016.10.014
Bielski, E. R., Zhong, Q., Brown, M., and da Rocha, S. R. (2015). Effect of the conjugation density of triphenylphosphonium cation on the mitochondrial targeting of poly(amidoamine) dendrimers. Mol. Pharm. 12, 3043–3053. doi: 10.1021/acs.molpharmaceut.5b00320
Bogart, L. K., Pourroy, G., Murphy, C. J., Puntes, V., Pellegrino, T., Rosenblum, D., et al. (2014). Nanoparticles for imaging, sensing, and therapeutic intervention. ACS Nano 8, 3107–3122. doi: 10.1021/nn500962q
Chamberlain, G. R., Tulumello, D. V., and Kelley, S. O. (2013a). Targeted delivery of doxorubicin to mitochondria. ACS Chem. Biol. 8, 1389–1395. doi: 10.1021/cb400095v
Chamberlain, G. R., Tulumello, D. V., and Kelley, S. O. (2013b). Targeted delivery of doxorubicin to mitochondria. ACS Chem. Biol. 8, 1389–1395. doi: 10.1021/cb400095v
Chen, L. B. (1988). Mitochondrial membrane potential in living cells. Annu. Rev. Cell Biol. 4, 155–181. doi: 10.1146/annurev.cb.04.110188.001103
Chen, W. H., Xu, X. D., Luo, G. F., Jia, H. Z., Lei, Q., Cheng, S. X., et al. (2013). Dual-targeting pro-apoptotic peptide for programmed cancer cell death via specific mitochondria damage. Sci. Rep. 3:3468. doi: 10.1038/srep03468
Cho, D. Y., Cho, H., Kwon, K., Yu, M., Lee, E., Huh, K. M., et al. (2015). Triphenylphosphonium-conjugated poly(𝜀-caprolactone)- based self-assembled nanostructures as nanosized drugs and drug delivery carriers for mitochondria-targeting synergistic anticancer drug delivery. Adv. Funct. Mater. 25, 5479–5491. doi: 10.1002/adfm.201501422
Choi, Y. S., Kwon, K., Yoon, K., Huh, K. M., and Kang, H. C. (2017). Photosensitizer-mediated mitochondria-targeting nanosized drug carriers: subcellular targeting, therapeutic, and imaging potentials. Int. J. Pharm. 520, 195–206. doi: 10.1016/j.ijpharm.2017.02.013
Christman, J. E., Miller, D. S., Coward, P., Smith, L. H., and Teng, N. N. (1990). Study of the selective cytotoxic properties of cationic, lipophilic mitochondrial-specific compounds in gynecologic malignancies. Gynecol. Oncol. 39, 72–79. doi: 10.1016/0090-8258(90)90402-7
Dairkee, S. H., and Hackett, A. J. (1991). Differential retention of rhodamine 123 by breast carcinoma and normal human mammary tissue. Breast Cancer Res. Treat. 18, 57–61. doi: 10.1007/BF01975444
Dong, L. F., Jameson, V. J. A., Tilly, D., Prochazka, L., Rohlena, J., Valis, K., et al. (2011). Mitochondrial targeting of alpha-tocopheryl succinate enhances its pro-apoptotic efficacy: a new paradigm for effective cancer therapy. Free Radic. Bio Med. 50, 1546–1555. doi: 10.1016/j.freeradbiomed.2011.02.032
D’Souza, G. G., Rammohan, R., Cheng, S. M., Torchilin, V. P., and Weissig, V. (2003). DQAsome-mediated delivery of plasmid DNA toward mitochondria in living cells. J. Control. Release 92, 189–197. doi: 10.1016/S0168-3659(03)00297-9
D’Souza, G. G., and Weissig, V. (2009). Subcellular targeting: a new frontier for drug-loaded pharmaceutical nanocarriers and the concept of the magic bullet. Expert Opin Drug Deliv. 6, 1135–1148. doi: 10.1517/17425240903236101
Ellerby, H. M., Arap, W., Ellerby, L. M., Kain, R., Andrusiak, R., Rio, G. D., et al. (1999). Anti-cancer activity of targeted pro-apoptotic peptides. Nat. Med. 5, 1032–1038. doi: 10.1038/12469
El-Say, K. M., and El-Sawy, H. S. (2017). Polymeric nanoparticles: promising platform for drug delivery. Int. J. Pharm. 528, 675–691. doi: 10.1016/j.ijpharm.2017.06.052
Ernster, L., and Schatz, G. (1981). Mitochondria: a historical review. J. Cell Biol. 91(3 Pt 2), 227s–255s. doi: 10.1083/jcb.91.3.227s
Fiore, C., Salvi, M., Palermo, M., Sinigaglia, G., Armanini, D., and Toninello, A. (2004). On the mechanism of mitochondrial permeability transition induction by glycyrrhetinic acid. Biochim. Biophys. Acta 1658, 195–201. doi: 10.1016/j.bbabio.2004.05.012
Fonseca, S. B., Pereira, M. P., Mourtada, R., Gronda, M., Horton, K. L., Hurren, R., et al. (2011). Rerouting chlorambucil to mitochondria combats drug deactivation and resistance in cancer cells. Chem. Biol. 18, 445–453. doi: 10.1016/j.chembiol.2011.02.010
Han, K., Lei, Q., Wang, S., Hu, J., Qiu, W., Zhu, J., et al. (2015). Dual-stage-light-guided tumor inhibition by mitochondria targeted photodynamic therapy. Adv. Funct. Mater. 25, 2961–2971. doi: 10.1002/adfm.201500590
Han, M., Vakili, M. R., Soleymani Abyaneh, H., Molavi, O., Lai, R., and Lavasanifar, A. (2014). Mitochondrial delivery of doxorubicin via triphenylphosphine modification for overcoming drug resistance in MDA-MB-435/DOX cells. Mol. Pharm. 11, 2640–2649. doi: 10.1021/mp500038g
Haynes, B., Zhang, Y., Liu, F., Li, J., Petit, S., Kothayer, H., et al. (2016). Gold nanoparticle conjugated Rad6 inhibitor induces cell death in triple negative breast cancer cells by inducing mitochondrial dysfunction and PARP-1 hyperactivation: synthesis and characterization. Nanomedicine 12, 745–757. doi: 10.1016/j.nano.2015.10.010
He, H., Li, D. W., Yang, L. Y., Fu, L., Zhu, X. J., Wong, W. K., et al. (2015). Ael bifunctional mitochondria-targeted anticancer agent with high selectivity for cancer cells. Sci. Rep. 5:13543. doi: 10.1038/srep13543
He, H., Wang, J., Wang, H., Zhou, N., Yang, D., Green, D. R., et al. (2018). Enzymatic cleavage of branched peptides for targeting mitochondria. J. Am. Chem. Soc. 140, 1215–1218. doi: 10.1021/jacs.7b11582
Heller, A., Brockhoff, G., and Goepferich, A. (2012). Targeting drugs to mitochondria. Eur. J. Pharm. Biopharm. 82, 1–18. doi: 10.1016/j.ejpb.2012.05.014
Horobin, R. W. (2002). Biological staining: mechanisms and theory. Biotech. Histochem. 77, 3–13. doi: 10.1080/bih.77.1.3.13
Horton, K. L., Stewart, K. M., Fonseca, S. B., Guo, Q., and Kelley, S. O. (2008). Mitochondria-penetrating peptides. Chem. Biol. 15, 375–382. doi: 10.1016/j.chembiol.2008.03.015
Hou, J., Yu, X., Shen, Y., Shi, Y., Su, C., and Zhao, L. (2017). Triphenyl phosphine-functionalized chitosan nanoparticles enhanced antitumor efficiency through targeted delivery of doxorubicin to mitochondria. Nanoscale Res. Lett. 12:158. doi: 10.1186/s11671-017-1931-1
Hu, Q. L., Gao, M., Feng, G. X., and Liu, B. (2014). Mitochondria-targeted cancer therapy using a light-up probe with aggregation-induced-emission characteristics. Angew. Chem. Int. Edt. 53, 14225–14229. doi: 10.1002/anie.201408897
Jean, S. R., Ahmed, M., Lei, E. K., Wisnovsky, S. P., and Kelley, S. O. (2016). Peptide-mediated delivery of chemical probes and therapeutics to mitochondria. ACC. Chem. Res. 49, 1893–1902. doi: 10.1021/acs.accounts.6b00277
Jung, K., and Reszka, R. (2001). Mitochondria as subcellular targets for clinically useful anthracyclines. Adv. Drug Deliv. Rev. 49, 87–105. doi: 10.1016/S0169-409X(01)00128-4
Kang, B. H., Plescia, J., Song, H. Y., Meli, M., Colombo, G., Beebe, K., et al. (2009). Combinatorial drug design targeting multiple cancer signaling networks controlled by mitochondrial Hsp90. J. Clin. Invest. 119, 454–464. doi: 10.1172/JCI37613
Kedar, U., Phutane, P., Shidhaye, S., and Kadam, V. (2010). Advances in polymeric micelles for drug delivery and tumor targeting. Nanomedicine 6, 714–729. doi: 10.1016/j.nano.2010.05.005
Khatun, Z., Choi, Y. S., Kim, Y. G., Yoon, K., Nurunnabi, M., Li, L., et al. (2017). Bioreducible poly(ethylene glycol)-triphenylphosphonium conjugate as a bioactivable mitochondria-targeting nanocarrier. Biomacromolecules 18, 1074–1085. doi: 10.1021/acs.biomac.6b01324
Kim, Y. K., Zhang, M., Lu, J. J., Xu, F., Chen, B. A., Xing, L., et al. (2016). PK11195-chitosan-graft-polyethylenimine-modified SPION as a mitochondria-targeting gene carrier. J. Drug Target. 24, 457–467. doi: 10.3109/1061186X.2015.1087527
Lambert, J. M., and Morris, C. Q. (2017). Antibody-drug conjugates (ADCs) for personalized treatment of solid tumors: a review. Adv. Ther. 34, 1015–1035. doi: 10.1007/s12325-017-0519-6
Lampidis, T. J., Hasin, Y., Weiss, M. J., and Chen, L. B. (1985). Selective killing of carcinoma cells “in vitro” by lipophilic-cationic compounds: a cellular basis. Biomed. Pharmacother. 39, 220–226.
Lee, J. H., Kim, K. Y., Jin, H., Baek, Y. E., Choi, Y., Jung, S. H., et al. (2018). Self-assembled coumarin nanoparticle in aqueous solution as selective mitochondrial-targeting drug delivery system. ACS Appl. Mater. Interfaces 10, 3380–3391. doi: 10.1021/acsami.7b17711
Lei, W., Xie, J., Hou, Y., Jiang, G., Zhang, H., Wang, P., et al. (2010). Mitochondria-targeting properties and photodynamic activities of porphyrin derivatives bearing cationic pendant. J. Photochem. Photobiol. B 98, 167–171. doi: 10.1016/j.jphotobiol.2009.12.003
Lim, S. Y., Shen, W., and Gao, Z. (2015). Carbon quantum dots and their applications. Chem. Soc. Rev. 44, 362–381. doi: 10.1039/c4cs00269e
Lin, R., Zhang, P., Cheetham, A. G., Walston, J., Abadir, P., and Cui, H. (2015). Dual peptide conjugation strategy for improved cellular uptake and mitochondria targeting. Bioconjug. Chem. 26, 71–77. doi: 10.1021/bc500408p
Liu, H. N., Guo, N. N., Wang, T. T., Guo, W. W., Lin, M. T., Huang-Fu, M. Y., et al. (2018). Mitochondrial targeted doxorubicin-triphenylphosphonium delivered by hyaluronic acid modified and pH responsive nanocarriers to breast tumor: in vitro and in vivo studies. Mol. Pharm. 15, 882–891. doi: 10.1021/acs.molpharmaceut.7b00793
Liu, Y., Zhang, X., Zhou, M., Nan, X., Chen, X., and Zhang, X. (2017). Mitochondrial-targeting lonidamine-doxorubicin nanoparticles for synergistic chemotherapy to conquer drug resistance. ACS Appl. Mater. Interfaces 9, 43498–43507. doi: 10.1021/acsami.7b14577
Mallick, A., More, P., Ghosh, S., Chippalkatti, R., Chopade, B. A., Lahiri, M., et al. (2015). Dual drug conjugated nanoparticle for simultaneous targeting of mitochondria and nucleus in cancer cells. ACS Appl. Mater. Interfaces 7, 7584–7598. doi: 10.1021/am5090226
Marrache, S., and Dhar, S. (2012). Engineering of blended nanoparticle platform for delivery of mitochondria-acting therapeutics. Proc. Natl. Acad. Sci. U.S.A. 109, 16288–16293. doi: 10.1073/pnas.1210096109
Marrache, S., Tundup, S., Harn, D. A., and Dhar, S. (2013). Ex vivo programming of dendritic cells by mitochondria-targeted nanoparticles to produce interferon-gamma for cancer immunotherapy. ACS Nano 7, 7392–7402. doi: 10.1021/nn403158n
Mattoussi, H., and Rotello, V. M. (2013). Inorganic nanoparticles in drug delivery. Adv. Drug Deliv. Rev. 65, 605–606. doi: 10.1016/j.addr.2013.04.012
Millard, M., Gallagher, J. D., Olenyuk, B. Z., and Neamati, N. (2013). A selective mitochondrial-targeted chlorambucil with remarkable cytotoxicity in breast and pancreatic cancers. J. Med. Chem. 56, 9170–9179. doi: 10.1021/jm401243811
Modica-Napolitano, J. S., and Weissig, V. (2015). Treatment strategies that enhance the efficacy and selectivity of mitochondria-targeted anticancer agents. Int. J. Mol. Sci. 16, 17394–17421. doi: 10.3390/ijms160817394
Mourtada, R., Fonseca, S. B., Wisnovsky, S. P., Pereira, M. P., Wang, X., Hurren, R., et al. (2013). Re-directing an alkylating agent to mitochondria alters drug target and cell death mechanism. PLoS One 8:e60253. doi: 10.1371/journal.pone.0060253
Mukhopadhyay, A., Ni, L., Yang, C. S., and Weiner, H. (2005). Bacterial signal peptide recognizes HeLa cell mitochondrial import receptors and functions as a mitochondrial leader sequence. Cell Mol. Life. Sci. 62, 1890–1899. doi: 10.1007/s00018-005-5178-0
Neuzil, J., Dong, L. F., Ramanathapuram, L., Hahn, T., Chladova, M., Wang, X. F., et al. (2007). Vitamin E analogues as ael group of mitocans: anti-cancer agents that act by targeting mitochondria. Mol. Aspects Med. 28, 607–645. doi: 10.1016/j.mam.2007.02.003
Neuzil, J., Dong, L. F., Rohlena, J., Truksa, J., and Ralph, S. J. (2013). Classification of mitocans, anti-cancer drugs acting on mitochondria. Mitochondrion 13, 199–208. doi: 10.1016/j.mito.2012.07.112
Neuzil, J., Weber, T., Gellert, N., and Weber, C. (2001a). Selective cancer cell killing by alpha-tocopheryl succinate. Br. J. Cancer 84, 87–89. doi: 10.1054/bjoc.2000.1559
Neuzil, J., Weber, T., Schroder, A., Lu, M., Ostermann, G., Gellert, N., et al. (2001b). Induction of cancer cell apoptosis by alpha-tocopheryl succinate: molecular pathways and structural requirements. FASEB J. 15, 403–415. doi: 10.1096/fj.00-0251com
Oerlemans, C., Bult, W., Bos, M., Storm, G., Nijsen, J. F., and Hennink, W. E. (2010). Polymeric micelles in anticancer therapy: targeting, imaging and triggered release. Pharm. Res. 27:12. doi: 10.1007/s11095-010-0233-4
Pajuelo, L., Calvino, E., Diez, J. C., Boyano-Adanez Mdel, C., Gil, J., and Sancho, P. (2011). Dequalinium induces apoptosis in peripheral blood mononuclear cells isolated from human chronic lymphocytic leukemia. Invest. New Drugs 29, 1156–1163. doi: 10.1007/s10637-010-9454-y
Palade, G. E. (1952). The fine structure of mitochondria. Anat. Rec. 114, 427–451. doi: 10.1002/ar.1091140304
Palade, G. E. (1964). The organization of living matter. Proc. Natl. Acad. Sci. U.S.A. 52, 613–634. doi: 10.1073/pnas.52.2.613
Pinheiro, C., Longatto-Filho, A., Azevedo-Silva, J., Casal, M., Schmitt, F. C., and Baltazar, F. (2012). Role of monocarboxylate transporters in human cancers: state of the art. J. Bioenerg. Biomembr. 44, 127–139. doi: 10.1007/s10863-012-9428-1
Pommier, Y., Leo, E., Zhang, H., and Marchand, C. (2010). DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem. Biol. 17, 421–433. doi: 10.1016/j.chembiol.2010.04.012
Riganti, C., Rolando, B., Kopecka, J., Campia, I., Chegaev, K., Lazzarato, L., et al. (2013). Mitochondrial-targeting nitrooxy-doxorubicin: a new approach to overcome drug resistance. Mol. Pharm. 10, 161–174. doi: 10.1021/mp300311b
Salvi, M., Fiore, C., Armanini, D., and Toninello, A. (2003). Glycyrrhetinic acid-induced permeability transition in rat liver mitochondria. Biochem. Pharmacol. 66, 2375–2379. doi: 10.1016/j.bcp.2003.08.023
Sancho, P., Galeano, E., Nieto, E., Delgado, M. D., and Garcia-Perez, A. I. (2007). Dequalinium induces cell death in human leukemia cells by early mitochondrial alterations which enhance ROS production. Leuk. Res. 31, 969–978. doi: 10.1016/j.leukres.2006.11.018
Seebacher, N. A., Richardson, D. R., and Jansson, P. J. (2016). A mechanism for overcoming P-glycoprotein-mediated drug resistance:el combination therapy that releases stored doxorubicin from lysosomes via lysosomal permeabilization using Dp44mT or DpC. Cell Death Dis. 7:e2510. doi: 10.1038/cddis.2016.381
Sibrian-Vazquez, M., Nesterova, I. V., Jensen, T. J., and Vicente, M. G. H. (2008). Mitochondria targeting by guanidine-and biguanidine-porphyrin photosensitizers. Bioconjug. Chem. 19, 705–713. doi: 10.1021/bc700393u
Song, Y. F., Liu, D. Z., Cheng, Y., Liu, M., Ye, W. L., Zhang, B. L., et al. (2015). Dual subcellular compartment delivery of doxorubicin to overcome drug resistant and enhance antitumor activity. Sci. Rep. 5:16125. doi: 10.1038/srep16125
Srivastava, S., and Moraes, C. T. (2001). Manipulating mitochondrial DNA heteroplasmy by a mitochondrially targeted restriction endonuclease. Hum. Mol. Genet. 10, 3093–3099. doi: 10.1093/hmg/10.26.3093
Stankovich, S., Dikin, D. A., Dommett, G. H., Kohlhaas, K. M., Zimney, E. J., Stach, E. A., et al. (2006). Graphene-based composite materials. Nature 442, 282–286. doi: 10.1038/nature04969
Sun, W., Li, L., Li, L. J., Yang, Q. Q., Zhang, Z. R., and Huang, Y. (2017). Two birds, one stone: dual targeting of the cancer cell surface and subcellular mitochondria by the galectin-3-binding peptide G3-C12. Acta Pharmacol. Sin. 38, 806–822. doi: 10.1038/aps.2016.137
Tan, Y., Zhu, Y., Zhao, Y., Wen, L., Meng, T., Liu, X., et al. (2018). Mitochondrial alkaline pH-responsive drug release mediated by celastrol loaded glycolipid-like micelles for cancer therapy. Biomaterials 154, 169–181. doi: 10.1016/j.biomaterials.2017.07.036
Torchilin, V. P. (2006). Recent approaches to intracellular delivery of drugs and DNA and organelle targeting. Annu. Rev. Biomed. Eng. 8, 343–375. doi: 10.1146/annurev.bioeng.8.061505.095735
Vestweber, D., and Schatz, G. (1989). DNA-protein conjugates can enter mitochondria via the protein import pathway. Nature 338, 170–172. doi: 10.1038/338170a0
Vives, E., Schmidt, J., and Pelegrin, A. (2008). Cell-penetrating and cell-targeting peptides in drug delivery. Biochim. Biophys. Acta 1786, 126–138. doi: 10.1016/j.bbcan.2008.03.001
Wallace, D. C. (2005). Mitochondria and cancer: warburg addressed. Cold Spring Harb Symp. Quant. Biol. 70, 363–374. doi: 10.1101/sqb.2005.70.035
Wang, F., Ogasawara, M. A., and Huang, P. (2010). Small mitochondria-targeting molecules as anti-cancer agents. Mol. Aspects Med. 31, 75–92. doi: 10.1016/j.mam.2009.12.003
Wang, H., Gao, Z., Liu, X., Agarwal, P., Zhao, S., Conroy, D. W., et al. (2018). Targeted production of reactive oxygen species in mitochondria to overcome cancer drug resistance. Nat. Commun. 9:562. doi: 10.1038/s41467-018-02915-8
Wang, H. A., and Xu, W. Q. (2017). Mito-methyl coumarin, ael mitochondria-targeted drug with great antitumor potential was synthesized. Biochem. Biophys. Res. Commun. 489, 1–7. doi: 10.1016/j.bbrc.2017.05.116
Wang, J., Fan, X. Y., Yang, L., He, H., Huang, R., Jiang, F. L., et al. (2016). Conjugated 5-fluorouracil with mitochondriatargeting lipophilic cation: design, synthesis and biological evaluation. MedChemComm 7, 2016–2019. doi: 10.1039/C6MD00268D
Wang, X. X., Li, Y. B., Yao, H. J., Ju, R. J., Zhang, Y., Li, R. J., et al. (2011). The use of mitochondrial targeting resveratrol liposomes modified with a dequalinium polyethylene glycol-distearoylphosphatidyl ethanolamine conjugate to induce apoptosis in resistant lung cancer cells. Biomaterials 32, 5673–5687. doi: 10.1016/j.biomaterials.2011.04.029
Wang, Z., Guo, W., Kuang, X., Hou, S., and Liu, H. (2017). Nanopreparations for mitochondria targeting drug delivery system: current strategies and future prospective. Asian Pharm. Sci. 12, 498–508. doi: 10.1016/j.ajps.2017.05.006
Wei, Y., Zhou, F., Zhang, D., Chen, Q., and Xing, D. (2016). A graphene oxide based smart drug delivery system for tumor mitochondria-targeting photodynamic therapy. Nanoscale 8, 3530–3538. doi: 10.1039/c5nr07785k
Weiss, M. J., Wong, J. R., Ha, C. S., Bleday, R., Salem, R. R., Steele, G. D., et al. (1987). Dequalinium, a topical antimicrobial agent, displays anticarcinoma activity based on selective mitochondrial accumulation. Proc. Natl. Acad. Sci. U.S.A. 84, 5444–5448. doi: 10.1073/pnas.84.15.5444
Weissig, V. (2015). DQAsomes as the prototype of mitochondria-targeted pharmaceutical nanocarriers: preparation, characterization, and use. Methods Mol. Biol. 1265, 1–11. doi: 10.1007/978-1-4939-2288-8_1
Weissig, V., Lasch, J., Erdos, G., Meyer, H. W., Rowe, T. C., and Hughes, J. (1998). DQAsomes: ael potential drug and gene delivery system made from dequalinium. Pharm. Res. 15, 334–337. doi: 10.1023/A:1011991307631
Weissig, V., and Torchilin, V. P. (2001). Towards mitochondrial gene therapy: DQAsomes as a strategy. J. Drug Target. 9, 1–13. doi: 10.3109/10611860108995628
Wen, S., Zhu, D., and Huang, P. (2013). Targeting cancer cell mitochondria as a therapeutic approach. Future Med. Chem. 5, 53–67. doi: 10.4155/fmc.12.190
Wicki, A., Witzigmann, D., Balasubramanian, V., and Huwyler, J. (2015). Nanomedicine in cancer therapy: challenges, opportunities, and clinical applications. J. Control. Release 200, 138–157. doi: 10.1016/j.jconrel.2014.12.030
Wilczewska, A. Z., Niemirowicz, K., Markiewicz, K. H., and Car, H. (2012). Nanoparticles as drug delivery systems. Pharmacol. Rep. 64, 1020–1037. doi: 10.1016/S1734-1140(12)70901-5
Wisnovsky, S. P., Wilson, J. J., Radford, R. J., Pereira, M. P., Chan, M. R., Laposa, R. R., et al. (2013). Targeting mitochondrial DNA with a platinum-based anticancer agent. Chem. Biol. 20, 1323–1328. doi: 10.1016/j.chembiol.2013.08.010
Wu, S., Cao, Q., Wang, X., Cheng, K., and Cheng, Z. (2014a). Design, synthesis and biological evaluation of mitochondria targeting theranostic agents. Chem. Commun. (Camb) 50, 8919–8922. doi: 10.1039/c4cc03296a
Wu, X. A., Choi, C. H., Zhang, C., Hao, L., and Mirkin, C. A. (2014b). Intracellular fate of spherical nucleic acid nanoparticle conjugates. J. Am. Chem. Soc. 136, 7726–7733. doi: 10.1021/ja503010a
Wu, X., Zhang, L., Gurley, E., Studer, E., Shang, J., Wang, T., et al. (2008). Prevention of free fatty acid-induced hepatic lipotoxicity by 18beta-glycyrrhetinic acid through lysosomal and mitochondrial pathways. Hepatology 47, 1905–1915. doi: 10.1002/hep.22239
Xiang, C., Li, D. W., Qi, Z. D., Jiang, F. L., Ge, Y. S., and Liu, Y. (2013). Synthesis of F16 conjugated with 5-fluorouracil and biophysical investigation of its interaction with bovine serum albumin by a spectroscopic and molecular modeling approach. Luminescence 28, 865–872. doi: 10.1002/bio.2447
Yang, K., Feng, L., Shi, X., and Liu, Z. (2013). Nano-graphene in biomedicine: theranostic applications. Chem. Soc. Rev. 42, 530–547. doi: 10.1039/c2cs35342c
Yokoyama, M. (2010). Polymeric micelles as a new drug carrier system and their required considerations for clinical trials. Expert Opin Drug Deliv. 7, 145–158. doi: 10.1517/17425240903436479
Young, K., Jin, H., Park, J., Ho, S., Ha, J., Park, H., et al. (2018). Mitochondria-targeting self-assembled nanoparticles derived from triphenylphosphonium-conjugated cyanostilbene enable site-specific imaging and anticancer drug delivery. Nano Res. 11:1082. doi: 10.1007/s12274-017-1728-7
Yousif, L. F., Stewart, K. M., Horton, K. L., and Kelley, S. O. (2009a). Mitochondria-penetrating peptides: sequence effects and model cargo transport. Chembiochem 10, 2081–2088. doi: 10.1002/cbic.200900017
Yousif, L. F., Stewart, K. M., and Kelley, S. O. (2009b). Targeting mitochondria with organelle-specific compounds: strategies and applications. Chembiochem 10, 1939–1950. doi: 10.1002/cbic.200900185
Yu, Y., Wang, Z. H., Zhang, L., Yao, H. J., Zhang, Y., Li, R. J., et al. (2012). Mitochondrial targeting topotecan-loaded liposomes for treating drug-resistant breast cancer and inhibiting invasive metastases of melanoma. Biomaterials 33, 1808–1820. doi: 10.1016/j.biomaterials.2011.10.085
Zhang, C., Liu, Z., Zheng, Y., Geng, Y., Han, C., Shi, Y., et al. (2018). Glycyrrhetinic acid functionalized graphene oxide for mitochondria targeting and cancer treatment in vivo. Small 14:1703306. doi: 10.1002/smll.201703306
Zhang, S., Yang, L., Ling, X., Shao, P., Wang, X., Edwards, W. B., et al. (2015a). Tumor mitochondria-targeted photodynamic therapy with a translocator protein (TSPO)-specific photosensitizer. Acta Biomater. 28, 160–170. doi: 10.1016/j.actbio.2015.09.033
Zhang, Z., Zhou, L., Zhou, Y., Liu, J., Xing, X., Zhong, J., et al. (2015b). Mitophagy induced by nanoparticle-peptide conjugates enabling an alternative intracellular trafficking route. Biomaterials 65, 56–65. doi: 10.1016/j.biomaterials.2015.06.029
Zhang, X. Y., and Zhang, P. Y. (2016a). Mitochondria targeting nano agents in cancer therapeutics. Oncol. Lett. 12, 4887–4890. doi: 10.3892/ol.2016.5302
Zhang, X. Y., and Zhang, P. Y. (2016b). Mitochondria targeting nano agents in cancer therapeutics. Oncol. Lett. 12, 4887–4890. doi: 10.3892/ol.2016.5302
Zhang, Y., Zhang, C., Chen, J., Liu, L., Hu, M., Li, J., et al. (2017). Trackable mitochondria-targeting nanomicellar loaded with doxorubicin for overcoming drug resistance. ACS Appl. Mater. Interfaces 9, 25152–25163. doi: 10.1021/acsami.7b07219
Zhao, K., Zhao, G. M., Wu, D., Soong, Y., Birk, A. V., Schiller, P. W., et al. (2004). Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J. Biol. Chem. 279, 34682–34690. doi: 10.1074/jbc.M402999200
Zhou, F., Xing, D., Wu, B., Wu, S., Ou, Z., and Chen, W. R. (2010). New insights of transmembranal mechanism and subcellular localization of noncovalently modified single-walled carbon nanotubes. Nano Lett. 10, 1677–1681. doi: 10.1021/nl100004m
Zhou, J., Zhao, W. Y., Ma, X., Ju, R. J., Li, X. Y., Li, N., et al. (2013). The anticancer efficacy of paclitaxel liposomes modified with mitochondrial targeting conjugate in resistant lung cancer. Biomaterials 34, 3626–3638. doi: 10.1016/j.biomaterials.2013.01.078
Zielonka, J., Joseph, J., Sikora, A., Hardy, M., Ouari, O., Vasquez-Vivar, J., et al. (2017). Mitochondria-targeted triphenylphosphonium-based compounds: syntheses, mechanisms of action, and therapeutic and diagnostic applications. Chem. Rev. 117, 10043–10120. doi: 10.1021/acs.chemrev.7b00042
Keywords: mitochondria-targeting ligand-drug conjugate, mitochondria-targeted nanocarrier system, anticancer agents, triphenylphosphonium, doxorubicin, mitochondria
Citation: Battogtokh G, Cho Y-Y, Lee JY, Lee HS and Kang HC (2018) Mitochondrial-Targeting Anticancer Agent Conjugates and Nanocarrier Systems for Cancer Treatment. Front. Pharmacol. 9:922. doi: 10.3389/fphar.2018.00922
Received: 24 May 2018; Accepted: 26 July 2018;
Published: 17 August 2018.
Edited by:
Alexander S. Sobolev, Lomonosov Moscow State University, RussiaReviewed by:
Nirmalya Chatterjee, Harvard Medical School, United StatesDhanendra Tomar, Temple University, United States
Copyright © 2018 Battogtokh, Cho, Lee, Lee and Kang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Han Chang Kang, aGNrYW5nQGNhdGhvbGljLmFjLmty