 Yiting Wang
Yiting Wang Huanbin Wang
Huanbin Wang Jie Xu
Jie Xu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 22 May 2018
Sec. Pharmacology of Anti-Cancer Drugs
Volume 9 - 2018 | https://doi.org/10.3389/fphar.2018.00536
This article is part of the Research Topic Targeting the PD-1/PD-L1 Cancer Immune Evasion Axis: Challenges and Emerging Strategies, Volume I View all 18 articles
Immune checkpoint blockade therapies (ICBTs) targeting programmed cell death 1 (PD-1) and its ligand programmed death ligand-1 (PD-L1/B7-H1/CD274) have exhibited momentous clinical benefits and durable responses in multiple tumor types. However, primary resistance is found in considerable number of cancer patients, and most responders eventually develop acquired resistance to ICBT. To tackle these challenges, it is essential to understand how PD-L1 is controlled by cancer cells to evade immune surveillance. Recent research has shed new light into the mechanisms of PD-L1 regulation at genetic, epigenetic, transcriptional, translational, and posttranslational levels. In this work, we systematically discuss the mechanisms that control the gene amplification, epigenetic alteration, transcription, subcellular transportation and posttranscriptional modification of PD-L1 in cancer cells. We further categorize posttranscriptional PD-L1 regulations by the molecular modification of PD-L1, including glycosylation, phosphorylation, ubiquitination, deubiquitination, and lysosomal degradation. These findings may provide new routes for targeting tumor immune escape and catalyze the development of small molecular inhibitors of PD-L1 in addition to existing antibody drugs.
Over the past decades, a novel therapy that utilizes human immune system to treat cancer is increasingly popular, which is known as cancer immunotherapy (Yang, 2015). The immunosuppressive microenvironment of tumor is one of the six distinct biological properties that enable tumor growth and metastasis (Hanahan and Weinberg, 2011). Human tumors typically harbor genomic instability, which induce somatic mutations (Hanahan and Weinberg, 2011). Accumulation of mutations may facilitate tumor growth and metastasis, while some non-synonymous mutations, leading to replacement of amino acid residual, create new T cell epitopes (neoepitopes), offering opportunities for immune system to recognize and eliminate cancer cells (Matsushita et al., 2012; Rooney et al., 2015). It has been reported that the number of non-synonymous mutations, defined as mutational load, is closely related with the efficacy of immunotherapy (Danilova et al., 2016). However, cancer cells collaborate with immune cells to dodge the immune destruction, and the anti-cancer pathway is intervened in this microenvironment (Blank et al., 2016; Sukari et al., 2016). The depressed immunology of T cells, if appropriately empowered, may be an efficient and powerful weapon against cancer. Specifically, active vaccination, adoptive cell transfer therapy and immune checkpoint blockade are the three major approaches that could turn on T cell-based anti-cancer immune reaction. In recent years, immune checkpoint blockade therapy (ICBT) has exhibited momentous clinical benefits, placing tumor immunotherapy under the spotlight (Sukari et al., 2016). PD-L1, a type I transmembrane protein with an extracellular N-terminal domain, inhibits the immune response through interaction with receptor PD-1 expressed on T cells (Horita et al., 2017). Under physiological conditions, PD-L1 is expressed in a wide range of cell types and tissues and shown to be overexpressed with immune activation, such as inflammations (Ritprajak and Azuma, 2015). The PD-L1/PD-1 axis maintains the balance between tolerance and autoimmunity and thus deficiency or excess function of it can lead to a variety of disease. Many auto-immune diseases have been found to be associated with PD-L1/PD-1 disruption including arthritis and lupus (Zamani et al., 2016). PD-L1 expression has been found positive in 5–40% tumor cells (Xie et al., 2016; Xiang et al., 2018), helping them to dodge the immune elimination through interaction of PD-L1 on the surface of cancer cells with PD-1 on T cells (Topalian et al., 2015). Thus, blockade of PD-L1/PD-1 axis assists the recognition and elimination of cancer cells. PD-L1 expression on tumor cells has been reasonably detected as a biomarker of ICBT (Ma et al., 2016). Further investigation revealed that the inducible but not continuous expression of PD-L1 is associated with activated CD8+ T cells in hepatocellular carcinoma (Xie et al., 2016), although the expression of PD-L1 is not independently prognostic (Wang X. et al., 2016; Xie et al., 2016).
The binding of immune checkpoint inhibitors and optimal targets is the core idea of ICBT. By inhibiting the immune-suppressive pathways, ICBT allows the clearance of cancer cells by the immune system (Topalian et al., 2015). Several immune checkpoints are discovered to be optimal targets for immune blockade, including the cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed cell-death protein 1 (PD-1)/programmed cell-death 1 ligand 1 (PD-L1) pathways. Drugs targeting these two pathways have nourished recently and many of them have been approved by FDA. Drugs that target PD-1 like Pembrolizumab (Keytruda) and Nivolumab (Opdivo) were approved in 2014. Some PD-L1 inhibitors were also approved including Atezolizumab (Tecentriq) (2016), Avelumab (Bavencio) (2017) and Durvalumab (Imfinzi) (2017). Ipilimumab (Yervoy) is a monoclonal antibody targeting CTLA-4 that gained approval in 2011. Information comes from the official website of United States Food and Drug Administration. Notably, inhibitors targeting PD-1 or PD-L1 have been found to be especially advantageous in the treatment of many kind of cancer, including non-small cell lung carcinoma (NSCLC) (Wang C. et al., 2016), renal cell carcinoma (RCC), bladder cancer, breast cancer (Hu et al., 2017), melanoma (Luke et al., 2017) and Hodgkin’s lymphoma (Allen and Gordon, 2016). The landscape of cancer therapy is evolving with deeper and wider acknowledgment of Immunotherapy with PD-1 or PD-L1 blockade (Pardoll, 2012).
Despite of the promising laboratory results and many positive clinical applications, there seems to be a discount on its overall clinical benefits due to intrinsic and/or acquired resistance to this therapy (Sharma et al., 2017). In certain cancer patients, the significant clinical response and enduring tumor retardation achieved by ICBT have improved patient progress-free survival (PFS) and overall survival (OS). However, the efficacy rate and profits of usage in general patients remain at a modest level, impeding the widespread application of ICBT (Pardoll, 2012). The tumor immunogenicity is a multi-level and delicately modulated process. Therefore, accumulation of mutations may lead to dysregulation of immunogenicity and create an immunosuppressive microenvironment, causing intrinsic resistance to ICBT (Zhao and Subramanian, 2017). Among them is the insufficiency of T cell infiltration (Spranger et al., 2016; Tang et al., 2016). On the other hand, after the significant retardation and durable response of tumor when initially treated with anti-PD-1 therapy, relapses in the long term were observed even after continuous therapy (Zaretsky et al., 2016). The acquired resistance to ICBT in melanoma was reported to be associated to antigen presentation deficiency, in which the interferon signal pathway was involved (Zaretsky et al., 2016). Alternative checkpoints were discovered to be adaptively upregulated after PD-L1 targeting treatment (Koyama et al., 2016). Moreover, PD-L1 upregulation after chemotherapy and nivolumab treatment was reported as a potential cause of acquired resistance (Haratake et al., 2017). In these tumors, immune evasion involves PD-L1/PD-1 interaction, which is the reason why the therapy initially worked. But the aftereffect of increased PD-L1 may have partially restored PD-L1/PD-1 function by providing more PD-L1 sites that were not neutralized by injected antibodies. Nonetheless, not enough investigations have been done to clarify the adaptive upregulation of PD-L1. In this scenario, understanding the mechanisms of PD-L1 regulation in cancer cells would certainly benefit the development of more effective and durable ICBTs.
While the PD-1/PD-L1 pathway has been proven both theoretically and clinically a mature and efficient target for immunotherapy, it is of urgent need to develop more effective approaches to target PD-L1. Firstly, many disadvantages of PD-L1 targeted antibodies are unneglectable. The relatively large size of Mono-antibodies (MAbs) may prohibit its penetration into the complex tumor microenvironment, and thus limiting the therapeutic efficacy (Lee and Tannock, 2010). It is crucial to develop new drugs with smaller sizes and to improve the specificity of tumor PD-L1 targeting, even though existing drugs and research are flourishing (Tan et al., 2016).
Secondly, the primary and acquired resistance to ICBT in many tumors highlights a crucial requirement for developing alternative PD-L1/PD-1-targeting approaches. Several cancer mutations have been suggested to be the cause of PD-L1 suppression and therefore primary resistance to PD-L1 blockade drugs. Inactivation mutations of JAK1/2 is an example (Shin et al., 2017). Thirdly, As a protector of host tissue and regulator of inflammation, PD-1/PD-L1 is located not only on tumor cells but also on normal cells, including anti-tumor T cells and tumor associated macrophages (Tan et al., 2016; Horita et al., 2017). The blockage of physiological PD-1/PD-L1 functions inevitably brings about unfavored results- the depletion of cells which are meant to be activated and functioning. Lastly, the activation of oncogenic pathways, including RAS/RAF/MAPK and PI3K signaling, combined with the complexity of tumor microenvironment, may desensitize anti-tumor immunity (Zhao and Subramanian, 2018). The main components of tumor microenvironment, including infiltrated T cells (Tang et al., 2016), metabolites (will be further discussed) and oxidative stress (Maj et al., 2017), have been reported to be disruptors of anti-tumor immunity. Our understanding on the mechanisms of ICBT resistance and PD-L1 regulation remains rather limited, proposing an urgency to decode the multifaceted roles and complex control of PD-L1 in cancer.
The enthusiastic devotion from both clinical and biological investigators have brought the PD-1/PD-L1 biology into a new era in cancer research. Translational studies targeting the PD-1/PD-L1 pathway have boosted dramatically in recent years. Some progresses in the research of PD-L1 expression in cancer, especially at transcriptional and epigenetic levels, have been forged into a regulatory model for unified explanation (Chen et al., 2016). However, more recent findings that shed light into the multifaceted control of PD-L1 as a membranous protein has not been systematically discussed. In this review, we will summarize the exciting progresses in PD-L1 research in a more comprehensive manner, aiming to facilitate future basic and translational studies in the field of cancer immunotherapy.
Enhanced PD-L1 expression was detected in a wide range of cancers but the prognostic and predictive value of it is controversial (Wang X. et al., 2016). It’s also a sign of efficacy of ICBT targeting PD-1/PD-L1 (Chen et al., 2016), as reported in B-cell lymphomas (Wang X. et al., 2016), breast cancer (Mittendorf et al., 2014), small-cell lung cancer (George et al., 2017) and pancreatic cancer (Wang et al., 2010). Given that many oncogenes are upregulated by gained copy number alterations (CNAs), efforts have been made to clarify the relationship between PD-L1 expression and CNA. As the main form of CNA, PD-L1 copy number amplification directly leads to PD-L1 mRNA upregulation. Tumors harboring PD-L1 amplification presents significantly higher load of mutation, comparing to non-amplified subjects (Budczies et al., 2016). Increased copy number of chromosome 9p24, predominant amplification of focal gene CD274 (which resides on chromosome 9p24.1, as shown in Figure 1), together with abundant PD-L1 expression were observed in a subset of small-cell lung cancer (SCLC) (George et al., 2017). The Janus kinase 2 (JAK2) amplification was documented to be simultaneously activated with 9p24.1 chromosome copy number amplification and upregulated PD-L1 expression in primary cancers (Figure 2), suggesting a possible transactivation between JAK2 and PD-L1 genes (Green et al., 2010; Ikeda et al., 2016; Clave et al., 2018). What’s more, PD-L1/PD-L2 alterations were defined as a feature of Classical Hodgkin lymphomas (cHLs). Specifically, amplification of 9p24.1 was reported to be associated with patients’ advanced stage disease and poor prognosis in cHL and in Epstein-Barr virus-associated gastric cancer (EBVaGC) (Roemer et al., 2016; Saito et al., 2017). These findings collectively suggest that CD274 gene amplification is a crucial factor that drives PD-L1 expression in cancer, and thus targeting PD-L1 at genetic level may be a rationalized strategy in PD-L1 positive tumors. Considering the rapid development of gene therapies, such prospect won’t be infeasible.
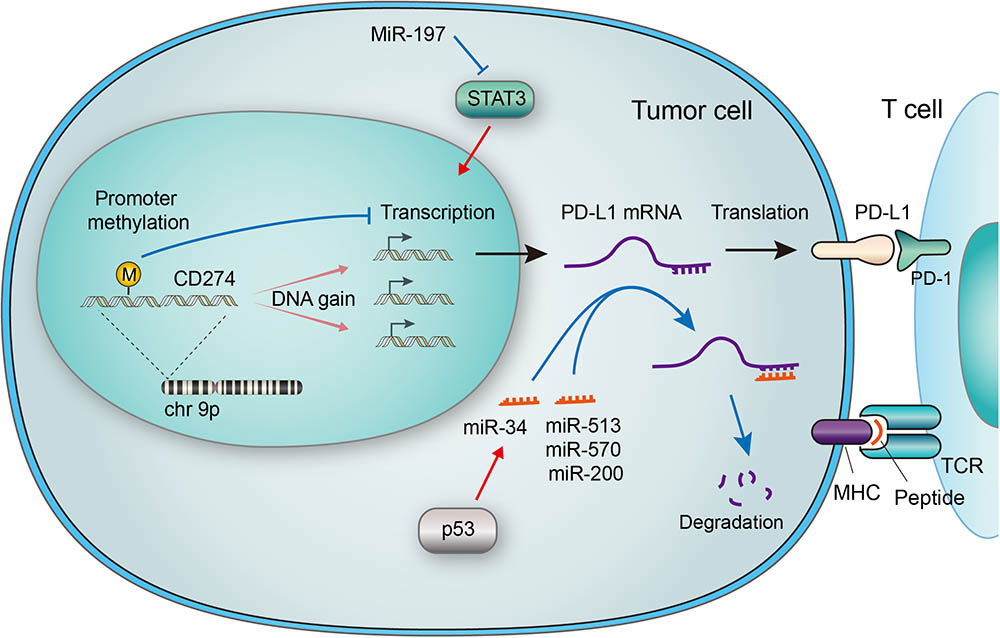
FIGURE 1. Genetic and epigenetic regulations of PD-L1 in tumor cells. The CD274 gene encoding PD-L1 is located on chromosome 9p, which is amplified in a subset of cancers. Increased gene copy number leads to upregulation of mRNA expression, while methylation of the gene promoter suppresses its transcription. Micro RNAs (miRNAs) may regulate PD-L1 expression by suppressing Stat3, which transactivates PD-L1. MiRNAs may also bind to the 3′ UTR of PD-L1 mRNA, leading to its degradation. The p53 tumor suppressor has been reported to downregulate PD-L1 through miR-34, a miRNA that binds to the 3′ UTR of PD-L1 mRNA. The PD-L1/PD-1 interaction and MHC-antigen/TCR interaction collaboratively define an inhibitory output of the immune checkpoint.
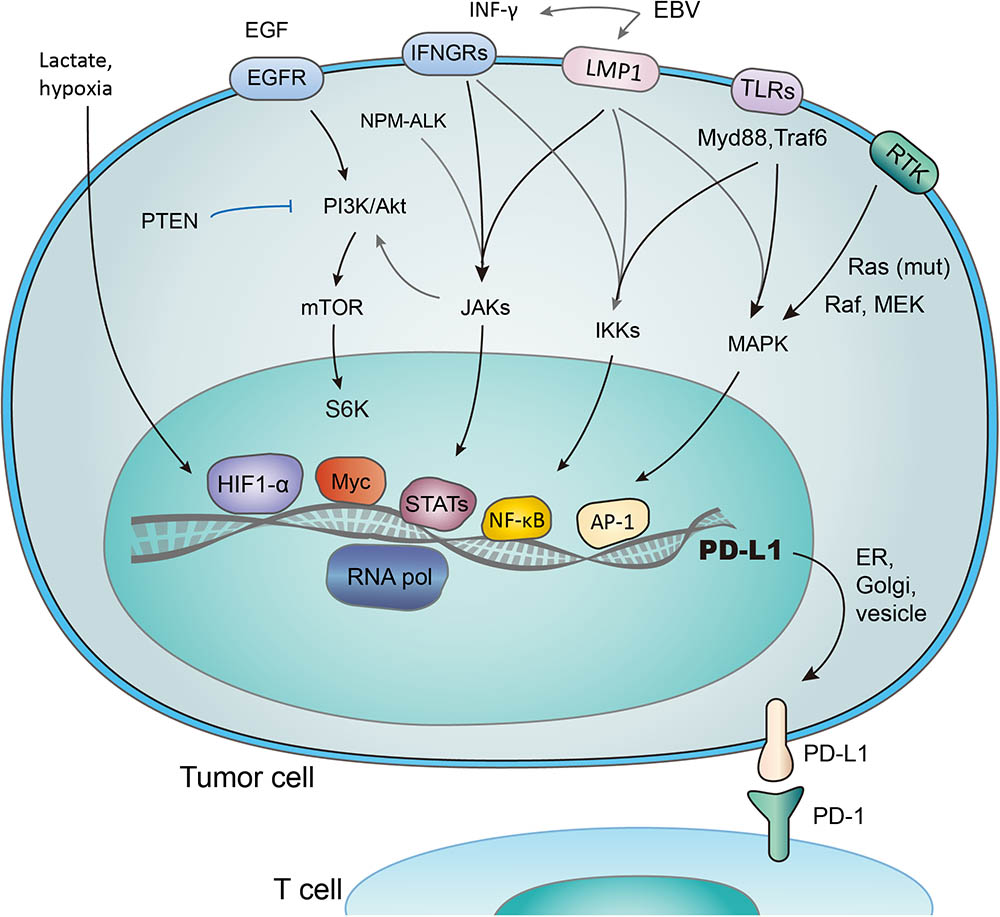
FIGURE 2. Transcriptional activation of PD-L1 in response to different signaling pathways. PD-L1 is transcribed in response to the activation of multiple signaling pathways, and transcription factors (TFs) such as HIF1-α, Myc, Stats, NF- κB, and AP-1 have been reported to bind and transactivate PD-L1. These TFs are controlled by the interconnected pathways involving EGF/PI3K/AKT/MTOR (suppressed by PTEN), RTK/Ras/Raf/MEK/ERK, IFN-γ/JAKs (also induced by mutant NPM-ALK gene and EBV-activated LMP1), TLRs/Myd88/Traf6/IKKs, and lactate-enriched microenvironment.
Structural variations may also be responsible for elevated transcription of PD-L1 (Kataoka et al., 2016). For example, truncation of its 3′UTR was reported to be associated with aberrant PD-L1 expression in multiple cancers (Kataoka et al., 2016).
Epigenetic regulation was revealed to be involved in PD-L1 expression in cancer cells. Micro RNAs (miRNAs), defined as 22–24 nucleotides non-coding single-stranded RNAs, have been implicated in the regulation of PD-L1 expression (Wang Q. et al., 2017). The binding of some miRNAs to the PD-L1 mRNA causes the latter one to degrade and thus PD-L1expression is suppressed. Specifically, the abundance of miR-513, miR-570, miR-34a, and miR-200 were reported to have an inverse correlation with PD-L1 expression (Chen, 2009; Chen et al., 2014; Wang et al., 2015), as described in Figure 1. Among them is miR-513 which inhibits PD-L1 protein translation by binding to 3′ untranslated regions (UTRs) of PD-L1 RNA as complement (Chen, 2009). Supportively, IFN-γ-induced PD-L1 expression was diminished by introducing miR-513 into Jurkat cells, while anti-miR-513 enhanced PD-L1 expression in cholangiocytes (Gong et al., 2009; Jardim et al., 2009). Similar function was found with miR-570. Research has shown that mutation of the PD-L1 3′ UTR which disrupts the association with miR-570, correlated with overexpression of PD-L1 (Wang et al., 2013). P53 was reported to regulate PD-L1 through miR-34 (Cortez et al., 2016). In the case of miR-200, the process of epithelial-to-mesenchymal transition (EMT) is found to be mediated by the regulation of PD-L1 expression by miR-200 (Chen et al., 2014). Moreover, MiR-197 was reported to repress STAT3, a regulator of PD-L1, to decrease PD-L1 expression (Fujita et al., 2015), as demonstrated in Figure 1. Other miRs reported to regulate PD-L1 includes miR-424 (Xu et al., 2016), miR-138 (Zhao et al., 2016), miR-17 (Audrito et al., 2017) and cluster miR-25-93-106b (Cioffi et al., 2017). Most recently, a mechanism that stabilizes PD-L1 mRNA was reported through modulation of the AU-rich element-binding protein tristetraprolin (TTP) (Coelho et al., 2017).
Recent studies have also focused on the promoter methylation of PD-L1 (mPD-L1), which was suggested to be a biomarker for prediction of response to PD-1/PD-L1 targeted ICBT. Significant inverse correlations between mPD-L1 and patient age was reported. The correlation between mPD-L1 and PD-L1 mRNA expression shares similar pattern, indicating a potential interaction between patient age and methylation of PD-L1 gene and that promoter methylation suppresses PD-L1 expression in colorectal cancer (CRC) (Goltz et al., 2017). Correlation between PD-L1 promoter methylation and clinical outcomes was also revealed in other cancers including NSCLC (Wrangle et al., 2013) and prostate cancer (Gevensleben et al., 2016). Moreover, in patients treated with PD-1/PD-L1 targeting drugs, enhanced mPD-L1 is associated with worse overall survival and recurrence-free survival. Epigenetic therapy has also been suggested to sensitize tumor response to PD-L1 targeting drugs (Wrangle et al., 2013). Interestingly, results proved no meaningful correlation between PD-L1 mRNA expression and patients’ outcome. (Goltz et al., 2017)
Several transcriptional factors have been found to control PD-L1 transcriptional activation (Figure 2). As an example, PTEN represses PD-L1 transcription and expression in breast cancer cells, suggesting a new tumor suppressive function of PTEN. In addition, PD-L1 expression decreased after inhibition of phosphoinositide 3-kinase (PI3K) pathway using the AKT inhibitors, further emphasizing the role of PTEN and PI3K signaling in PD-L1 regulation (Mittendorf et al., 2014). Transcription activity, demonstrated by the level of PD-L1 mRNA expression, was promoted through JAK2/STAT1 pathway, as was shown in pancreatic cancer cells treated with anticancer agents (5-fluorouracil, gemcitabine, or paclitaxel) (Wang et al., 2010). Notably, when treated with chemotherapeutic drugs, the MAPK pathway was also reported to upregulate PD-L1 in cancer cells (Chen et al., 2016). While distinct signaling pathways share the ability to control PD-L1 expression by regulating its transcription, the exact mechanisms involved may vary considerably (Chen et al., 2016).
Hypoxia inducible factor 1α (HIF-1α) is a major cancer driver (Ortmann et al., 2014) and a potential therapeutic target (Brown and Wilson, 2004; Vaupel and Mayer, 2007; Wilson and Hay, 2011). The binding of HIF-1α to PD-L1 promoter, a hypoxia response element (HRE), stimulates the transcription of PD-L1 (Noman and Chouaib, 2014). Research has revealed the co-existence of HIF-1α overexpression, increased PD-L1 level, and repression of T-cell function (Noman et al., 2014; Pollizzi and Powell, 2014; Shehade et al., 2014). It was also reported that PD-L1 works predominantly in lactate-enriched tumor microenvironments (Feng et al., 2017). Meanwhile, T cell autophagy is induced in a microenvironment lack of amino acids tryptophan and arginine as well as glucose. In this nutrients-deprived situation, glucose metabolism shrinks while the lactate accumulates, creating an optimal environment for PD-1/PD-L1 interaction and resistance to cancer therapies consequently (Robainas et al., 2017). In other words, Lactate, as a major metabolite under hypoxia condition, may protect tumor cells from cytotoxic T-cell targeting. Accordingly, tumor cell metabolic reprograming was found to correlate with immune suppression (Feng et al., 2017). Taken together, it is suggested that hypoxic environments, which induce activation of HIF-1α and accumulation of lactate (Koukourakis et al., 2005; Marchiq and Pouyssegur, 2016; Ban et al., 2017), contribute to evasion of tumor cells from immune system. The transactivation of PD-L1 by HIF-1 represents a crucial step in the above-mentioned process, and may be a promising target to combat the immune suppression of tumor cells.
STAT3 is another important transcriptional factor that upregulates PD-L1 expression by binding to PD-L1 promoter. Mutations of oncogene chimeric nucleophosmin/anaplastic lymphoma kinase (ALK) have been found to upregulate PD-L1 expression, and this effect could be abolished by silencing STAT3 (Marzec et al., 2008). Furthermore, Latent membrane protein-1 (LMP1) of Epstein-Barr virus was found to increase both PD-L1 expression and STAT3 phosphorylation (p-STAT3) (Fang et al., 2014) (Figure 2). Consistently, the JAK3 inhibitor CP-690550 blocked the above process through suppressing p-STAT3 (Marzec et al., 2008). NF-κB, as a transcriptional factor mediating inflammation-associated tumorigenesis, has been reported to boost PD-L1 expression. However, the exact mechanisms remain unclear. NF-κB is required for LMP1-induced PD-L1 expression, which is evidenced by decreased PD-L1 induction caused by NF-κB inhibitors (Marzec et al., 2008). Notably, the NF-κB inhibitor abolished INF-induced PD-L1 expression, while MAPK, PI3K and STAT3 inhibitors did not. Thus NF-κB also seems to be involved in INF-γ-induced PD-L1 expression (Gowrishankar et al., 2015).
N-glycosylation is a crucial protein modification that determines protein structure and function, especially the function of membrane proteins. By altering protein conformation, glycosylation may modulate protein activities and protein–protein interactions, such as those between ligands and receptors (Ohtsubo and Marth, 2006). In Western Blot assays, the majority of PD-L1 is detected at 45 kDa representing the glycosylated species, while the non-glycosylated form is detected at 33 kDa. By bioinformatics prediction, mass spectrometry and mutagenesis, PD-L1 was found to be exclusively N-glycosylated at N35, N192, N200, and N219 (Li et al., 2016).
The PD-L1 molecule containing N192, N200, and N219 residues forms a region that is the prerequisite for PD-L1 binding to GSK3β, and N-glycosylation on these sites buries the necessary residues and disrupts the interaction between PD-L1 and GSK3β. Glycogen synthase kinase 3beta (GSK3β), a serine/threonine protein kinase, was originally identified as a regulator of glycogen metabolism (Doble and Woodgett, 2003). When bound to non-glycosylated PD-L1, GSK3β leads to phosphorylation and consequent ubiquitination of PD-L1 (Li et al., 2016) (Figure 3). In addition, it was further elucidated that inactivation of GSK3β by activating EGFR enhanced PD-L1 expression by preventing it from being ubiquitinated (Li et al., 2016). Significantly, a small molecular inhibitor of glycosylation, tunicamycin, was found to efficiently decrease PD-L1 expression in cancer cells (Li et al., 2016). Latest results have provided evidence that targeting glycosylated PD-L1 promotes PD-L1 internalization and degradation, leading to eradication of triple-negative breast cancer cells (Li et al., 2018).
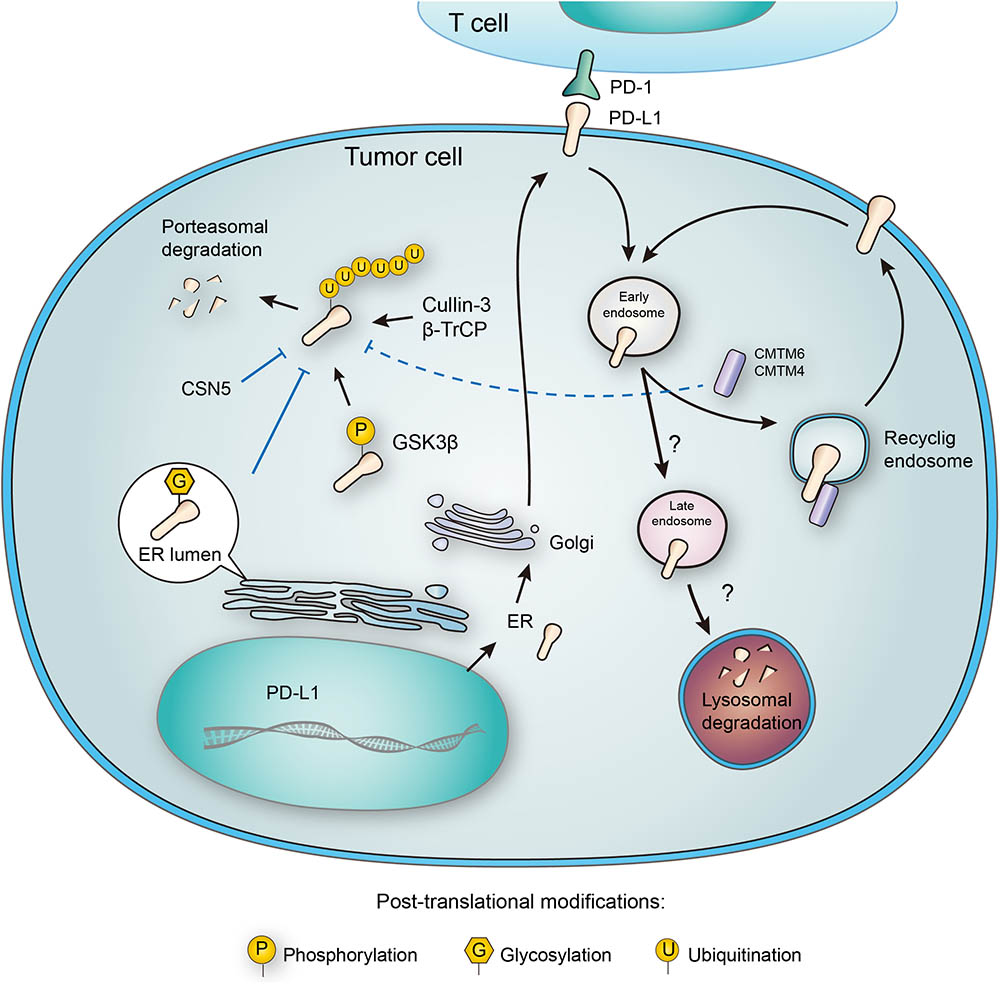
FIGURE 3. Post-translational modifications and subcellular transportation of PD-L1. As a membrane protein, PD-L1 is extensively modified after its translation. N-glycosylation of PD-L1 extracellular domain occurs in the lumen of endoplasmic reticulum (ER), and this modification facilitates the interaction of PD-L1 with lipid membrane. Glycosylation also inhibits phosphorylation by GSK3β, and thereby blocking the ubiquitination by β-TrCP. Deubiquitination by CSN5 also protects PD-L1 from proteasomal degradation. In addition, PD-L1 may also be destructed in lysosome, and this process relies on a series of subcellular transportations from cell membrane to early endosome, late endosome, and finally to lysosome. However, CMTM6 has been found to promote PD-L1 transportation to recycling endosome, causing decreased distribution to late endosome and lysosome. Interestingly, CMTM6 and its homolog CMTM4 may also stabilize PD-L1 by suppressing its ubiquitination.
Phosphorylation involves in a widespread of regulatory mechanisms in cellular signaling, and may affect the conformation, activity, and interactions of proteins. Although one protein may contain multiple phosphorylation sites, the phosphorylation of PD-L1 has been sparsely reported. As mentioned above in the glycosylation part, GSK3β is a multifunctional switch that mediates the direct phosphorylation of a wide range of substrates, including e IF2B, cyclin D1, c-Jun, c-myc, NFAT, MCl-1, and Snail (McCubrey et al., 2014). It also contributes to the phosphorylation of PD-L1 through an evolutionarily conserved GSK3β phosphorylation motif on PD-L1 (Li et al., 2016) (Figure 3). Furthermore, the phosphorylation mediated by GSK3β has been found to initiate the interaction with E3 ligase, which targets proteins to proteasomal degradation (Zhou et al., 2004; Ding et al., 2007; Wang et al., 2018).
Meanwhile, it was reported that treatment of the epidermal growth factor (EGF) would induce tyrosine phosphorylation, together with acetylation and ubiquitination of PD-L1 (Horita et al., 2017). These provide evidential hypothesis for the effects of Gefitinib, an inhibitor of EGFR, in promoting the immune response against breast cancer. Gefitinib was found to cut down on PD-L1 expression and limit its oncogenic potential, therefore promoting T cell immunity. These findings suggest that targeting EGFR by Gefitinib not only suppresses MAPK-dependent tumor proliferation, but also blocks PD-L1-dependent immune suppression (Li et al., 2016). Based on the predicted isoelectric points corresponding to different modifications, the PhosphoSite database has listed potential phosphorylation sites of PD-L1 (basal Isoelectric point = 6.76) (PhosphoSite Plus Protein Page: Pd-L1 Human, 2018). However, no systematic experimental characterization of PD-L1 phosphorylation has been carried out. It also deserves in-depth study how PD-L1 phosphorylation varies and fluctuates in response to distinct microenvironments, therapeutic stresses and interaction with its partner proteins.
Ubiquitination-dependent proteasomal degradation controls the metabolism of many proteins, including membrane proteins like PD-L1 (Zhou et al., 2014). As mentioned above, the EGF treatment may induce tyrosine phosphorylation, acetylation, and ubiquitination of PD-L1 (Horita et al., 2017). The increased PD-L1 mono- and multi-ubiquitination induced by EGF were blocked by gefitinib treatment. Recent study further revealed that ubiquitin E3 is involved in PD-L1 downregulation in EGFR wild-type NSCLC (Wang et al., 2018). In a recent study, cyclin D-CDK4 kinase was reported to destabilize PD-L1 via cullin 3-SPOP, which was proved to be involved in Pd-L1 ubiquitination (Zhang et al., 2018). Surprisingly, the EGF-stimulated PD-L1 mono-ubiquitination not only coexisted with PD-L1 overexpression, but also seemed to occur ahead of its upregulation (Akbay et al., 2013; Chen et al., 2015; Li et al., 2016; Horita et al., 2017). Inhibition of the ubiquitin E1 by blocking its activating enzyme decreased PD-L1 mono- and multi-ubiquitination and total PD-L1 protein expression at the same time, suggesting a possible causal relationship between ubiquitination and overexpression of PD-L1 (Horita et al., 2017).
CMTM6, a type-3 transmembrane protein was recently identified as a positive regulator of PD-L1. Decrease of CMTM6 expression downregulated PD-L1 protein level in a wide range of human tumor cells and in primary human dendritic cells. Apart from CMTM6, its closest family member, CMTM4, was confirmed to share similar function (Figure 3). Of note, the enhancement of PD-L1 protein pool stimulated by CMTM6 was not associated with any variation in PD-L1 transcription. Instead, CMTM6 was found to interact with PD-L1 on cell surface, interfering its ubiquitination to prolong its half-life. It was also functionally confirmed that by enhancing PD-L1 protein pool, CMTM6 improves the evasion ability of PD-L1positive tumor cells to immune elimination (Mezzadra et al., 2017).
On the contrary to ubiquitination, deubiquitination of PD-L1 stabilizes the protein from degradation. The deubiquitination and stabilization of PD-L1 significantly affect the inflammatory response or so-called ‘inflammation-mediated anti-tumor immunity’ (Lim et al., 2016). Recently, COP9 signalosome 5 (CSN5) was identified as a crucial protein that promotes the deubiquitination of PD-L1 (Lim et al., 2016) (Figure 3). It was reported that tumor necrosis factor alpha (TNF-α), as one of the major inflammatory cytokines secreted by macrophages, plays an important role in maintaining cancer cell evasion from immune system. Mechanistically, TNF-α may activate NF-κB and induce CSN5 expression, leading to PD-L1 stabilization. Consistently, CSN5 has been found to be indispensable for TNF-α-mediated PD-L1 stabilization because of its function in deubiquitinating PD-L1 (Lim et al., 2016). With potential translational significance, the authors found that destabilization of PD-L1 by curcumin, an inhibitor for CSN5, may benefit immunotherapy.
PD-L1 functions on the membrane surface, but it may also translocate into the cytoplasm. Many membrane proteins are shuttled between the recycling endosomes and cell surface, and PD-L1 has been tracked in recycling endosomes (Grant and Donaldson, 2009). Furthermore, inhibition of endocytic recycling by primaquine caused vast depletion of membrane PD-L1 protein level in wild-type cells. These results suggest that: first, a large proportion of membrane PD-L1 undergoes metabolism and internalization continuously; second, the dynamic recycling and releasing of PD-L1 maintains the amount of PD-L1 located on cell membrane (Burr et al., 2017). Notably, CMTM6, recognized as a PD-L1 regulator, is predominantly identified in recycling endosomes together with TFRC and RAB11, factors that define the endocytic recycling compartment. What’s more, CMTM6 co-localizes with PD-L1 both on the plasma membrane and in recycling endosomes, so that CMTM6 functions as a protector of PD-L1 that prevents it from being targeted for lysosome-mediated degradation and increases its protein pool (Figure 3).
Interestingly, membrane and cytoplasmic PD-L1 expression is more significant in macrophage cells than in cancer cells (Gong et al., 2017). Studies have been done to test PD-L1 molecule in peripheral blood mononuclear cells (PBMC) and surprisingly revealed a novel human PD-L1 splice variant in activated PBMC. Further studies compared the conventional isoform with the novel isoform and found distinct localization patterns between both proteins. Specifically, the conventional isoform is predominantly expressed on the plasma surface, while the novel isoform is distributed mainly on intracellular membrane. The alternative splicing of PD-L1 may be a posttranscriptional regulator that modulates PD-L1 expression as well as its function in determining the outcome of specific immune responses in the peripheral tissues (He et al., 2005).
In addition to its cellular distribution, PD-L1 has also been detected outside the cells, proposing its potential role as a semi-invasive biomarker. An A/C polymorphism at position 8923 was detected together with increased level of plasma soluble PD-L1 (sPD-L1) in NSCLC patients, especially those with adenocarcinoma (Cheng et al., 2015). Investigation is now undergoing to define the value of plasma PD-L1 protein levels as a predictive biomarker of prognosis in NSCLC and also as a reliable companion diagnostics for individualized treatment with ICBT (Zhu and Lang, 2017).
Unlike cytosolic proteins, many membrane proteins are mainly degraded through the lysosomal pathway. As mentioned in the ubiquitination part, CMTM6 reduces PD-L1 ubiquitination and increases its stability (Mezzadra et al., 2017). Interestingly, different opinion presents another explanation about the stabilization of membrane PD-L1 by CMTM6. In addition to its expression at the plasma membrane, CMTM6 is predominantly identified in recycling endosomes (Zhang et al., 2018). Although CMTM6 is not required for PD-L1 maturation, it functions in protecting PD-L1 from lysosome-mediated degradation (Burr et al., 2017). Thus, CMTM6 depletion, via the reduction of PD-L1, significantly alleviates the suppression of tumor-specific T cell activity in vitro and in vivo (Burr et al., 2017). Although there is no doubt that CMTM6 suppresses PD-L1 degradation, the effect still seems to be indirect, requiring the competitive transportation to the recycling endosome. It remains unclear which protein may directly interact with CMTM6 and transport it to lysosome for degradation (Figure 3). Future efforts to clarify this crucial node would benefit the development of alternative PD-L1-targeting approaches.
Some mutations of PD-L1 gene may impede the protein level of PD-1/PD-L1 but others may cause disturbance on protein folding, and therefore disrupt the interaction of PD-1 and PD-L1. PD-1 and PD-L1 bind through the conserved front and side of their Ig variable (Ig V) domains, representing the structural basis for the design of intervention molecules. By locating the loops at the ends of the IgV domains on the same side of the PD-1/PD-L1 complex, a surface is formed, being similar to the antigen-binding surface of antibodies and T-cell receptors (Zak et al., 2017). Several residues have been identified to play important roles in folding and forming the PD-1/PD-L1 interface (Lin et al., 2008). The immune receptor-like loops provide a new surface for further study and potentially the design of molecules that would affect PD-1/PD-L1 binding and thereby regulate the immune system. Multiple peptides and small-molecular compounds have been evaluated in preclinical models, in order to develop novel PD-1/PD-L1 inhibitors (Zak et al., 2017).
In addition to directly block the interaction between PD-1 and PD-L1, methods have also been developed to inhibit the dimerization of PD-L1, and hence the PD-1/PD-L1 interaction. Particularly, this effect could be achieved by small molecular compounds such as BMS-202 and BMS-8, with considerable translational significance (Zak et al., 2017). Since small molecules behold advantages in terms of production scale, quality standardization, pharmacological kinetics and tissue distribution, it is of enormous interest to discover small molecular drugs targeting the PD-L1/PD-1 axis (Lin et al., 2008). Despite the structural insights provided by recent crystallographic research, it is still unclear how the reported PTMs, e.g., glycosylation, phosphorylation, ubiquitination, etc., may affect the conformation and molecular interactions of PD-L1/PD-1. Understanding these detailed processes would also improve the confidence of structure-based drug design targeting this crucial immune suppression signaling pathway.
PD-L1-targeted ICBT is a promising breakthrough in the field of cancer immunotherapy, but primary and acquired resistances have presented enormous challenges in this fast-evolving area (Pardoll, 2012; Spranger et al., 2016; Zaretsky et al., 2016; Sharma et al., 2017; Zhao and Subramanian, 2017). It has been suggested that the post-treatment positive conversion of PD-L1 expression may be a cause of resistance (Haratake et al., 2017). The regulatory pathways of PD-L1 are of meaningful potential to be translated into therapeutic approaches for tackling the resistance to ICBT (Lee and Tannock, 2010; Tan et al., 2016; Tang et al., 2016; Maj et al., 2017; Shin et al., 2017; Zhao and Subramanian, 2018). The significant PD-L1 overexpression found in multiple cancer types may be an output of interconnected regulatory network, which involves molecular alterations at genetic, epigenetic, transcriptional, translational, post-translational, and structural levels. In fact, several key regulators of PD-L1 have long been established as cancer-related genes, such as JAK2 (Green et al., 2010; Budczies et al., 2016; Ikeda et al., 2016; Clave et al., 2018), PTEN, MAPK, PI3K, HIF-1α, STAT3 (Marzec et al., 2008; Gowrishankar et al., 2015; Chen et al., 2016), TNFα, NF-κB (Gowrishankar et al., 2015), and INF-γ, etc. Existing small molecular compounds targeting these genes/pathways may be repurposed for modulating PD-L1, thus providing readily tools to improve T cell-dependent anticancer immunity. Likewise, the discovery of key post-transcriptional modifications (PTMs) that control PD-L1 stability such as glycosylation, phosphorylation, and ubiquitination also provide alternative strategies for targeting PD-L1 (Zhou et al., 2004; Ding et al., 2007; Li et al., 2016; Lim et al., 2016; Horita et al., 2017). It is worthy to further analyze the function of curcumin (CSN5 inhibitor) and tunicamycin (glycolysis inhibitor) in suppressing PD-1/PD-L1 signaling in vivo and in preclinical models. The inhibitors o In addition, the connection between cancer metabolism and resistance to immunotherapy suggests potential benefit for combined targeting of tumor glycolysis and PD-1/PD-L1 axis (Koukourakis et al., 2005; Vaupel and Mayer, 2007; Wilson and Hay, 2011; Shehade et al., 2014; Marchiq and Pouyssegur, 2016; Feng et al., 2017). Apart from controlling the abundance of PD-L1 in cells, the mechanisms underlying PD-L1 transportation and structural modulation may also provide novel strategies to optimize the blockage of PD-L1 (van Weert et al., 2000; He et al., 2005; Lin et al., 2008; Cheng et al., 2015). With the multifaceted regulation of PD-L1 being revealed, it would be more feasible to develop complementary therapies to sustain the response once cancer cells acquire resistance to the initial treatment.
The prosperity and challenges of immunotherapies targeting the PD-1/PD-L1 axis warrant increasing attentions by biological and pharmaceutical scientists. In our opinion, several research directions would be especially beneficial to a sustained improvement of ICBT.
Firstly, the regulation of PD-L1 should be further clarified in more specified conditions, considering the variations in tumor regions and developmental stages. It has been suggested that PD-L1 expression may differ considerably on the tumor boundary. Cells located here have higher accessibility where immune cells encounter the tumor cells. Thus, tissue sampling by traditional methods may not robustly capture such alterations and result in low fidelity in different assays such as Western Blot, qPCR and microarray tests. On the other hand, hypoxia-related induction of PD-L1 is more likely to occur in the center of solid tumors where oxygen is less accessible. Moreover, our recent study found that PD-L1 is significantly upregulated in metastatic CRCs while compared to primary tumors (Wang H.B. et al., 2017). Thus, the regulation of PD-L1 during metastasis and its corresponding biomarker significance should be considered differentially from those in the primary tumors. To investigate the regulation of PD-L1 in tumors, it is essential to precisely mark the region and stage (e.g., primary vs. metastatic, pre-treatment vs. post-treatment, etc.) of a particular patient, because these variations are associated with the indicated mechanisms.
Secondly, the link between PD-L1 expression and cancer subtyping has been investigated based on genomic and transcriptomic characterizations of tumors. In many tumors, the microsatellite instability (MSI) subtype is linked to PD-L1 positivity and considered as a key factor indicating the suitability for checkpoint blockade therapy (Xiao and Freeman, 2015; Dudley et al., 2016). Even though, more comprehensive understanding on the implications of PD-L1 in cancer subtyping should also be founded by insights into the epigenetic and metabolic reprograming of cancer cells. As described previously, epigenetic and metabolic alterations in tumors are emerging as crucial factors affecting the abundance of PD-L1. In a translational perspective, significant and functional alterations at these facets may also present novel biomarkers and intervention opportunities.
Thirdly, it deserves tremendous efforts to clarify the overlaps and differences between PD-L1 and its homolog PD-L2 in their functions and regulations in various tumors. Although PD-L2 was initially considered to be mainly expressed in immune cells, recent studies have revealed its positive expression in different tumor cells with potential prognostic significance. As an example, we found that PD-L2 is expressed in a considerable subset of CRC cells, with independent association with poor patient survival (Wang H. et al., 2017). It is thus of interest to clarify the relative importance of PD-L1 and PD-L2 in a specific tumor type. Will one protein compensate the function of the other, or be upregulated when its homolog is blocked in immunotherapy? Which ligand of PD-1 may play a predominant role in suppressing T-cell immunity in a given cancer type of patient, and should this be considered when optimizing the strategy for immunotherapy? These questions should be addressed, in order to understand and improve the effectiveness and sustainability of ICBT.
Finally, the structure-based drug design targeting PD-L1 may not be limited in the binding surface to PD-1 or the site mediating its dimerization. If allosteric control of PD-L1 activity could be identified, additional approaches targeting PD-L1 would be feasible. Moreover, the protein interactions between PD-L1 and its reported regulators (e.g., CSN5, CMTM6, etc.) could be characterized in and enough resolution, rational design of blocking peptides or compounds may also be developed. In other words, basic research about the structural dynamics and detailed interaction sites of PD-L1 may provide additional resources for the development of de novo PD-L1 targeting approaches.
Immune checkpoint blockade therapy represents a breakthrough in cancer treatment, but the primary and acquired resistance to immunotherapy warrant further efforts to understand the multifaceted regulation of PD-L1 in cancer. As a cell surface protein that responds to microenvironment stimuli, PD-L1 reacts promptly to balance the outside stresses and inside requirements of cells, representing a key node in the cancer signaling network. In this scenario, the effective and sustained targeting of PD-L1 has to take the complexity of its regulation into account. Identification of the exact causes of PD-L1 upregulation and responsive functional compensations in a broader range of molecular events would improve the targeting specificity and efficiency. A chasm is yet to be crossed by obtaining small molecular inhibitors of PD-L1 in addition to antibody drugs, to improve the cancer distribution and metabolic kinetics of immunotherapeutic medicines. Current approaches for targeting PD-L1 could also affect its normal functions in immune cells, with expected unwanted effects. In these scenarios, targeting PD-L1 effectively and specifically in cancer cells remains a Gordian knot.
YW and JX wrote the manuscript. HW, HY, CL, and J-YF contributed to revisions of the manuscript.
This project was supported by grants from the National Key Research & Development (R&D) Plan (2016YFC0906000 and 2016YFC0906002); National Natural Science Foundation of China (81572326, 81322036, 81272383, 81602518, 81502015, 81572303, 81530072, 81421001, and 81320108024); Top-Notch Young Talents Program of China (ZTZ2015-48); Shanghai Municipal Education Commission-Gaofeng Clinical Medicine Grant Support (20152514); “Shu Guang” project supported by Shanghai Municipal Education Commission and Shanghai Education Development Foundation (15SG16); Tang Scholar (SJTU-JX); and National Key Technology Support Program (2015BAI13B07).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Akbay, E. A., Koyama, S., Carretero, J., Altabef, A., Tchaicha, J. H., Christensen, C. L., et al. (2013). Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 3, 1355–1363. doi: 10.1158/2159-8290.CD-13-0310
Allen, P. B., and Gordon, L. I. (2016). PD-1 blockade in Hodgkin’s lymphoma: learning new tricks from an old teacher. Expert Rev. Hematol. 9, 939–949. doi: 10.1080/17474086.2016.1235970
Audrito, V., Serra, S., Stingi, A., Orso, F., Gaudino, F., Bologna, C., et al. (2017). PD-L1 up-regulation in melanoma increases disease aggressiveness and is mediated through miR-17-5p. Oncotarget 8, 15894–15911. doi: 10.18632/oncotarget.15213
Ban, H. S., Kim, B. K., Lee, H., Kim, H. M., Harmalkar, D., Nam, M., et al. (2017). The novel hypoxia-inducible factor-1alpha inhibitor IDF-11774 regulates cancer metabolism, thereby suppressing tumor growth. Cell Death Dis. 8:e2843. doi: 10.1038/cddis.2017.235
Blank, C. U., Haanen, J. B., Ribas, A., and Schumacher, T. N. (2016). CANCER IMMUNOLOGY. The “cancer immunogram”. Science 352, 658–660. doi: 10.1126/science.aaf2834
Brown, J. M., and Wilson, W. R. (2004). Exploiting tumour hypoxia in cancer treatment. Nat. Rev. Cancer 4, 437–447. doi: 10.1038/nrc1367
Budczies, J., Bockmayr, M., Denkert, C., Klauschen, F., Groschel, S., Darb-Esfahani, S., et al. (2016). Pan-cancer analysis of copy number changes in programmed death-ligand 1 (PD-L1, CD274) - associations with gene expression, mutational load, and survival. Genes Chromosomes Cancer 55, 626–639. doi: 10.1002/gcc.22365
Burr, M. L., Sparbier, C. E., Chan, Y. C., Williamson, J. C., Woods, K., Beavis, P. A., et al. (2017). CMTM6 maintains the expression of PD-L1 and regulates anti-tumour immunity. Nature 549, 101–105. doi: 10.1038/nature23643
Chen, J., Jiang, C. C., Jin, L., and Zhang, X. D. (2016). Regulation of PD-L1: a novel role of pro-survival signalling in cancer. Ann. Oncol. 27, 409–416. doi: 10.1093/annonc/mdv615
Chen, L., Gibbons, D. L., Goswami, S., Cortez, M. A., Ahn, Y. H., Byers, L. A., et al. (2014). Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat. Commun. 5:5241. doi: 10.1038/ncomms6241
Chen, N., Fang, W., Zhan, J., Hong, S., Tang, Y., Kang, S., et al. (2015). Upregulation of PD-L1 by EGFR Activation mediates the immune escape in EGFR-Driven NSCLC: implication for optional immune targeted therapy for NSCLC patients with EGFR mutation. J. Thorac. Oncol. 10, 910–923. doi: 10.1097/JTO.0000000000000500
Chen, X. M. (2009). MicroRNA signatures in liver diseases. World J. Gastroenterol. 15, 1665–1672. doi: 10.3748/wjg.15.1665
Cheng, S., Zheng, J., Zhu, J., Xie, C., Zhang, X., Han, X., et al. (2015). PD-L1 gene polymorphism and high level of plasma soluble PD-L1 protein may be associated with non-small cell lung cancer. Int. J. Biol. Markers 30, e364–e368. doi: 10.5301/jbm.5000170
Cioffi, M., Trabulo, S. M., Vallespinos, M., Raj, D., Kheir, T. B., Lin, M. L., et al. (2017). The miR-25-93-106b cluster regulates tumor metastasis and immune evasion via modulation of CXCL12 and PD-L1. Oncotarget 8, 21609–21625. doi: 10.18632/oncotarget.15450
Clave, S., Pijuan, L., Casadevall, D., Taus, A., Gimeno, J., Hernandez-Llodra, S., et al. (2018). CD274 (PDL1) and JAK2 genomic amplifications in pulmonary squamous-cell and adenocarcinoma patients. Histopathology 72, 259–269. doi: 10.1111/his.13339
Coelho, M. A., de Carne Trecesson, S., Rana, S., Zecchin, D., Moore, C., Molina-Arcas, M., et al. (2017). Oncogenic RAS signaling promotes tumor immunoresistance by stabilizing PD-L1 mRNA. Immunity 47, 1083.e6–1099.e6. doi: 10.1016/j.immuni.2017.11.016
Cortez, M. A., Ivan, C., Valdecanas, D., Wang, X., Peltier, H. J., Ye, Y., et al. (2016). PDL1 Regulation by p53 via miR-34. J. Natl. Cancer Inst. 108:djv303. doi: 10.1093/jnci/djv303
Danilova, L., Wang, H., Sunshine, J., Kaunitz, G. J., Cottrell, T. R., Xu, H., et al. (2016). Association of PD-1/PD-L axis expression with cytolytic activity, mutational load, and prognosis in melanoma and other solid tumors. Proc. Natl. Acad. Sci. U.S.A. 113, E7769–E7777. doi: 10.1073/pnas.1607836113
Ding, Q., He, X., Hsu, J. M., Xia, W., Chen, C. T., Li, L. Y., et al. (2007). Degradation of Mcl-1 by beta-TrCP mediates glycogen synthase kinase 3-induced tumor suppression and chemosensitization. Mol. Cell. Biol. 27, 4006–4017. doi: 10.1128/MCB.00620-06
Doble, B. W., and Woodgett, J. R. (2003). GSK-3: tricks of the trade for a multi-tasking kinase. J. Cell Sci. 116(Pt 7), 1175–1186. doi: 10.1242/jcs.00384
Dudley, J. C., Lin, M. T., Le, D. T., and Eshleman, J. R. (2016). Microsatellite instability as a biomarker for PD-1 blockade. Clin. Cancer Res. 22, 813–820. doi: 10.1158/1078-0432.CCR-15-1678
Fang, W., Zhang, J., Hong, S., Zhan, J., Chen, N., Qin, T., et al. (2014). EBV-driven LMP1 and IFN-gamma up-regulate PD-L1 in nasopharyngeal carcinoma: implications for oncotargeted therapy. Oncotarget 5, 12189–12202. doi: 10.18632/oncotarget.2608
Feng, J., Yang, H., Zhang, Y., Wei, H., Zhu, Z., Zhu, B., et al. (2017). Tumor cell-derived lactate induces TAZ-dependent upregulation of PD-L1 through GPR81 in human lung cancer cells. Oncogene 36, 5829–5839. doi: 10.1038/onc.2017.188
Fujita, Y., Yagishita, S., Hagiwara, K., Yoshioka, Y., Kosaka, N., Takeshita, F., et al. (2015). The clinical relevance of the miR-197/CKS1B/STAT3-mediated PD-L1 network in chemoresistant non-small-cell lung cancer. Mol. Ther. 23, 717–727. doi: 10.1038/mt.2015.10
George, J., Saito, M., Tsuta, K., Iwakawa, R., Shiraishi, K., Scheel, A. H., et al. (2017). Genomic Amplification of CD274 (PD-L1) in small-cell lung cancer. Clin. Cancer Res. 23, 1220–1226. doi: 10.1158/1078-0432.CCR-16-1069
Gevensleben, H., Holmes, E. E., Goltz, D., Dietrich, J., Sailer, V., Ellinger, J., et al. (2016). PD-L1 promoter methylation is a prognostic biomarker for biochemical recurrence-free survival in prostate cancer patients following radical prostatectomy. Oncotarget 7, 79943–79955. doi: 10.18632/oncotarget.13161
Goltz, D., Gevensleben, H., Dietrich, J., and Dietrich, D. (2017). PD-L1 (CD274) promoter methylation predicts survival in colorectal cancer patients. Oncoimmunology 6:e1257454. doi: 10.1080/2162402X.2016.1257454
Gong, A. Y., Zhou, R., Hu, G., Li, X., Splinter, P. L., O’Hara, S. P., et al. (2009). MicroRNA-513 regulates B7-H1 translation and is involved in IFN-gamma-induced B7-H1 expression in cholangiocytes. J. Immunol. 182, 1325–1333. doi: 10.4049/jimmunol.182.3.1325
Gong, Y., Zhang, X., Chen, R., Wei, Y., Zou, Z., and Chen, X. (2017). Cytoplasmic expression of C-MYC protein is associated with risk stratification of mantle cell lymphoma. PeerJ 5:e3457. doi: 10.7717/peerj.3457
Gowrishankar, K., Gunatilake, D., Gallagher, S. J., Tiffen, J., Rizos, H., and Hersey, P. (2015). Inducible but not constitutive expression of PD-L1 in human melanoma cells is dependent on activation of NF-kappaB. PLoS One 10:e0123410. doi: 10.1371/journal.pone.0123410
Grant, B. D., and Donaldson, J. G. (2009). Pathways and mechanisms of endocytic recycling. Nat. Rev. Mol. Cell Biol. 10, 597–608. doi: 10.1038/nrm2755
Green, M. R., Monti, S., Rodig, S. J., Juszczynski, P., Currie, T., O’Donnell, E., et al. (2010). Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood 116, 3268–3277. doi: 10.1182/blood-2010-05-282780
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. doi: 10.1016/j.cell.2011.02.013
Haratake, N., Toyokawa, G., Tagawa, T., Kozuma, Y., Matsubara, T., Takamori, S., et al. (2017). Positive conversion of PD-L1 expression after treatments with chemotherapy and Nivolumab. Anticancer Res. 37, 5713–5717. doi: 10.21873/anticanres.12009
He, X. H., Xu, L. H., and Liu, Y. (2005). Identification of a novel splice variant of human PD-L1 mRNA encoding an isoform-lacking Igv-like domain. Acta Pharmacol. Sin. 26, 462–468. doi: 10.1111/j.1745-7254.2005.00086.x
Horita, H., Law, A., Hong, S., and Middleton, K. (2017). Identifying regulatory posttranslational modifications of PD-L1: a focus on monoubiquitinaton. Neoplasia 19, 346–353. doi: 10.1016/j.neo.2017.02.006
Hu, Z. I., Ho, A. Y., and McArthur, H. L. (2017). Combined radiation therapy and immune checkpoint blockade therapy for breast cancer. Int. J. Radiat. Oncol. Biol. Phys. 99, 153–164. doi: 10.1016/j.ijrobp.2017.05.029
Ikeda, S., Okamoto, T., Okano, S., Umemoto, Y., Tagawa, T., Morodomi, Y., et al. (2016). PD-L1 Is upregulated by simultaneous amplification of the PD-L1 and JAK2 genes in non-small cell lung cancer. J. Thorac. Oncol. 11, 62–71. doi: 10.1016/j.jtho.2015.09.010
Jardim, M. J., Fry, R. C., Jaspers, I., Dailey, L., and Diaz-Sanchez, D. (2009). Disruption of microRNA expression in human airway cells by diesel exhaust particles is linked to tumorigenesis-associated pathways. Environ. Health Perspect. 117, 1745–1751. doi: 10.1289/ehp.0900756
Kataoka, K., Shiraishi, Y., Takeda, Y., Sakata, S., Matsumoto, M., Nagano, S., et al. (2016). Aberrant PD-L1 expression through 3’-UTR disruption in multiple cancers. Nature 534, 402–406. doi: 10.1038/nature18294
Koukourakis, M. I., Giatromanolaki, A., Simopoulos, C., Polychronidis, A., and Sivridis, E. (2005). Lactate dehydrogenase 5 (LDH5) relates to up-regulated hypoxia inducible factor pathway and metastasis in colorectal cancer. Clin. Exp. Metastasis 22, 25–30. doi: 10.1007/s10585-005-2343-7
Koyama, S., Akbay, E. A., Li, Y. Y., Herter-Sprie, G. S., Buczkowski, K. A., Richards, W. G., et al. (2016). Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 7:10501. doi: 10.1038/ncomms10501
Lee, C. M., and Tannock, I. F. (2010). The distribution of the therapeutic monoclonal antibodies cetuximab and trastuzumab within solid tumors. BMC Cancer 10:255. doi: 10.1186/1471-2407-10-255
Li, C. W., Lim, S. O., Chung, E. M., Kim, Y. S., Park, A. H., Yao, J., et al. (2018). Eradication of triple-negative breast cancer cells by targeting glycosylated PD-L1. Cancer Cell 33, 187.e10–201.e10. doi: 10.1016/j.ccell.2018.01.009
Li, C. W., Lim, S. O., Xia, W., Lee, H. H., Chan, L. C., Kuo, C. W., et al. (2016). Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat. Commun. 7:12632. doi: 10.1038/ncomms12632
Lim, S. O., Li, C. W., Xia, W., Cha, J. H., Chan, L. C., Wu, Y., et al. (2016). Deubiquitination and stabilization of PD-L1 by CSN5. Cancer Cell 30, 925–939. doi: 10.1016/j.ccell.2016.10.010
Lin, D. Y., Tanaka, Y., Iwasaki, M., Gittis, A. G., Su, H. P., Mikami, B., et al. (2008). The PD-1/PD-L1 complex resembles the antigen-binding Fv domains of antibodies and T cell receptors. Proc. Natl. Acad. Sci. U.S.A. 105, 3011–3016. doi: 10.1073/pnas.0712278105
Luke, J. J., Flaherty, K. T., Ribas, A., and Long, G. V. (2017). Targeted agents and immunotherapies: optimizing outcomes in melanoma. Nat. Rev. Clin. Oncol. 14, 463–482. doi: 10.1038/nrclinonc.2017.43
Ma, W., Gilligan, B. M., Yuan, J., and Li, T. (2016). Current status and perspectives in translational biomarker research for PD-1/PD-L1 immune checkpoint blockade therapy. J. Hematol. Oncol. 9:47. doi: 10.1186/s13045-016-0277-y
Maj, T., Wang, W., Crespo, J., Zhang, H., Wang, W., Wei, S., et al. (2017). Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat. Immunol. 18, 1332–1341. doi: 10.1038/ni.3868
Marchiq, I., and Pouyssegur, J. (2016). Hypoxia, cancer metabolism and the therapeutic benefit of targeting lactate/H(+) symporters. J. Mol. Med. 94, 155–171. doi: 10.1007/s00109-015-1307-x
Marzec, M., Zhang, Q., Goradia, A., Raghunath, P. N., Liu, X., Paessler, M., et al. (2008). Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1. B7-H1). Proc. Natl. Acad. Sci. U.S.A. 105, 20852–20857. doi: 10.1073/pnas.0810958105
Matsushita, H., Vesely, M. D., Koboldt, D. C., Rickert, C. G., Uppaluri, R., Magrini, V. J., et al. (2012). Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 482, 400–404. doi: 10.1038/nature10755
McCubrey, J. A., Steelman, L. S., Bertrand, F. E., Davis, N. M., Sokolosky, M., Abrams, S. L., et al. (2014). GSK-3 as potential target for therapeutic intervention in cancer. Oncotarget 5, 2881–2911. doi: 10.18632/oncotarget.2037
Mezzadra, R., Sun, C., Jae, L. T., Gomez-Eerland, R., de Vries, E., Wu, W., et al. (2017). Identification of CMTM6 and CMTM4 as PD-L1 protein regulators. Nature 549, 106–110. doi: 10.1038/nature23669
Mittendorf, E. A., Philips, A. V., Meric-Bernstam, F., Qiao, N., Wu, Y., Harrington, S., et al. (2014). PD-L1 expression in triple-negative breast cancer. Cancer Immunol. Res. 2, 361–370. doi: 10.1158/2326-6066.CIR-13-0127
Noman, M. Z., and Chouaib, S. (2014). Targeting hypoxia at the forefront of anticancer immune responses. Oncoimmunology 3:e954463. doi: 10.4161/21624011.2014.954463
Noman, M. Z., Desantis, G., Janji, B., Hasmim, M., Karray, S., Dessen, P., et al. (2014). PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 211, 781–790. doi: 10.1084/jem.20131916
Ohtsubo, K., and Marth, J. D. (2006). Glycosylation in cellular mechanisms of health and disease. Cell 126, 855–867. doi: 10.1016/j.cell.2006.08.019
Ortmann, B., Druker, J., and Rocha, S. (2014). Cell cycle progression in response to oxygen levels. Cell Mol. Life Sci. 71, 3569–3582. doi: 10.1007/s00018-014-1645-9
Pardoll, D. M. (2012). The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 12, 252–264. doi: 10.1038/nrc3239
PhosphoSite Plus Protein Page: Pd-L1 Human (2018). PhosphoSite Plus Protein Page: PD-L1 Human. Available at: https://www.phosphosite.org/proteinAction.action?id=19198&showAllSites=true
Pollizzi, K. N., and Powell, J. D. (2014). Integrating canonical and metabolic signalling programmes in the regulation of T cell responses. Nat. Rev. Immunol. 14, 435–446. doi: 10.1038/nri3701
Ritprajak, P., and Azuma, M. (2015). Intrinsic and extrinsic control of expression of the immunoregulatory molecule PD-L1 in epithelial cells and squamous cell carcinoma. Oral Oncol. 51, 221–228. doi: 10.1016/j.oraloncology.2014.11.014
Robainas, M., Otano, R., Bueno, S., and Ait-Oudhia, S. (2017). Understanding the role of PD-L1/PD1 pathway blockade and autophagy in cancer therapy. Onco Targets Ther. 10, 1803–1807. doi: 10.2147/OTT.S132508
Roemer, M. G., Advani, R. H., Ligon, A. H., Natkunam, Y., Redd, R. A., Homer, H., et al. (2016). PD-L1 and PD-L2 genetic alterations define classical hodgkin lymphoma and predict outcome. J. Clin. Oncol. 34, 2690–2697. doi: 10.1200/JCO.2016.66.4482
Rooney, M. S., Shukla, S. A., Wu, C. J., Getz, G., and Hacohen, N. (2015). Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 160, 48–61. doi: 10.1016/j.cell.2014.12.033
Saito, R., Abe, H., Kunita, A., Yamashita, H., Seto, Y., and Fukayama, M. (2017). Overexpression and gene amplification of PD-L1 in cancer cells and PD-L1(+) immune cells in Epstein-Barr virus-associated gastric cancer: the prognostic implications. Mod. Pathol. 30, 427–439. doi: 10.1038/modpathol.2016.202
Sharma, P., Hu-Lieskovan, S., Wargo, J. A., and Ribas, A. (2017). Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 168, 707–723. doi: 10.1016/j.cell.2017.01.017
Shehade, H., Oldenhove, G., and Moser, M. (2014). Hypoxia in the intestine or solid tumors: a beneficial or deleterious alarm signal? Eur. J. Immunol. 44, 2550–2557. doi: 10.1002/eji.201444719
Shin, D. S., Zaretsky, J. M., Escuin-Ordinas, H., Garcia-Diaz, A., Hu-Lieskovan, S., Kalbasi, A., et al. (2017). Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov. 7, 188–201. doi: 10.1158/2159-8290.CD-16-1223
Spranger, S., Sivan, A., Corrales, L., and Gajewski, T. F. (2016). Tumor and host factors controlling antitumor immunity and efficacy of cancer immunotherapy. Adv. Immunol. 130, 75–93. doi: 10.1016/bs.ai.2015.12.003
Sukari, A., Nagasaka, M., Al-Hadidi, A., and Lum, L. G. (2016). Cancer immunology and immunotherapy. Anticancer Res. 36, 5593–5606. doi: 10.21873/anticanres.11144
Tan, S., Zhang, C. W., and Gao, G. F. (2016). Seeing is believing: anti-PD-1/PD-L1 monoclonal antibodies in action for checkpoint blockade tumor immunotherapy. Signal Transduct. Target Ther. 1:16029. doi: 10.1038/sigtrans.2016.29
Tang, H., Wang, Y., Chlewicki, L. K., Zhang, Y., Guo, J., Liang, W., et al. (2016). Facilitating T cell infiltration in tumor microenvironment overcomes resistance to PD-L1 blockade. Cancer Cell 29, 289–296. doi: 10.1016/j.ccell.2016.08.011
Topalian, S. L., Drake, C. G., and Pardoll, D. M. (2015). Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell 27, 450–461. doi: 10.1016/j.ccell.2015.03.001
van Weert, A. W., Geuze, H. J., Groothuis, B., and Stoorvogel, W. (2000). Primaquine interferes with membrane recycling from endosomes to the plasma membrane through a direct interaction with endosomes which does not involve neutralisation of endosomal pH nor osmotic swelling of endosomes. Eur. J. Cell Biol. 79, 394–399. doi: 10.1078/0171-9335-00062
Vaupel, P., and Mayer, A. (2007). Hypoxia in cancer: significance and impact on clinical outcome. Cancer Metastasis Rev. 26, 225–239. doi: 10.1007/s10555-007-9055-1
Wang, C., Yu, X., and Wang, W. (2016). A meta-analysis of efficacy and safety of antibodies targeting PD-1/PD-L1 in treatment of advanced nonsmall cell lung cancer. Medicine 95:e5539. doi: 10.1097/MD.0000000000005539
Wang, H., Yao, H., Li, C., Liang, L., Zhang, Y., Shi, H., et al. (2017). PD-L2 expression in colorectal cancer: independent prognostic effect and targetability by deglycosylation. Oncoimmunology 6:e1327494. doi: 10.1080/2162402X.2017.1327494
Wang, H. B., Yao, H., Li, C. S., Liang, L. X., Zhang, Y., Chen, Y. X., et al. (2017). Rise of PD-L1 expression during metastasis of colorectal cancer: implications for immunotherapy. J. Dig. Dis. 18, 574–581. doi: 10.1111/1751-2980.12538
Wang, L., Ma, Q., Chen, X., Guo, K., Li, J., and Zhang, M. (2010). Clinical significance of B7-H1 and B7-1 expressions in pancreatic carcinoma. World J. Surg. 34, 1059–1065. doi: 10.1007/s00268-010-0448-x
Wang, Q., Lin, W., Tang, X., Li, S., Guo, L., Lin, Y., et al. (2017). The roles of microRNAs in regulating the expression of PD-1/PD-L1 immune checkpoint. Int. J. Mol. Sci. 18:E2540. doi: 10.3390/ijms18122540
Wang, S., Xu, L., Che, X., Li, C., Xu, L., Hou, K., et al. (2018). E3 ubiquitin ligases Cbl-b and c-Cbl downregulate PD-L1 in EGFR wild-type non-small cell lung cancer. FEBS Lett. 592, 621–630. doi: 10.1002/1873-3468.12985
Wang, W., Li, F., Mao, Y., Zhou, H., Sun, J., Li, R., et al. (2013). A miR-570 binding site polymorphism in the B7-H1 gene is associated with the risk of gastric adenocarcinoma. Hum. Genet. 132, 641–648. doi: 10.1007/s00439-013-1275-6
Wang, X., Li, J., Dong, K., Lin, F., Long, M., Ouyang, Y., et al. (2015). Tumor suppressor miR-34a targets PD-L1 and functions as a potential immunotherapeutic target in acute myeloid leukemia. Cell. Signal. 27, 443–452. doi: 10.1016/j.cellsig.2014.12.003
Wang, X., Teng, F., Kong, L., and Yu, J. (2016). PD-L1 expression in human cancers and its association with clinical outcomes. Onco Targets Ther. 9, 5023–5039. doi: 10.2147/OTT.S105862
Wilson, W. R., and Hay, M. P. (2011). Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 11, 393–410. doi: 10.1038/nrc3064
Wrangle, J., Wang, W., Koch, A., Easwaran, H., Mohammad, H. P., Vendetti, F., et al. (2013). Alterations of immune response of non-small cell lung cancer with azacytidine. Oncotarget 4, 2067–2079. doi: 10.18632/oncotarget.1542
Xiang, X., Yu, P. C., Long, D., Liao, X. L., Zhang, S., You, X. M., et al. (2018). Prognostic value of PD -L1 expression in patients with primary solid tumors. Oncotarget 9, 5058–5072. doi: 10.18632/oncotarget.23580
Xiao, Y., and Freeman, G. J. (2015). The microsatellite instable subset of colorectal cancer is a particularly good candidate for checkpoint blockade immunotherapy. Cancer Discov. 5, 16–18. doi: 10.1158/2159-8290.CD-14-1397
Xie, Q. K., Zhao, Y. J., Pan, T., Lyu, N., Mu, L. W., Li, S. L., et al. (2016). Programmed death ligand 1 as an indicator of pre-existing adaptive immune responses in human hepatocellular carcinoma. Oncoimmunology 5:e1181252. doi: 10.1080/2162402X.2016.1181252
Xu, S., Tao, Z., Hai, B., Liang, H., Shi, Y., Wang, T., et al. (2016). miR-424(322) reverses chemoresistance via T-cell immune response activation by blocking the PD-L1 immune checkpoint. Nat. Commun. 7:11406. doi: 10.1038/ncomms11406
Yang, Y. (2015). Cancer immunotherapy: harnessing the immune system to battle cancer. J. Clin. Invest. 125, 3335–3337. doi: 10.1172/JCI83871
Zak, K. M., Grudnik, P., Magiera, K., Domling, A., Dubin, G., and Holak, T. A. (2017). Structural biology of the immune checkpoint receptor PD-1 and its ligands PD-L1/PD-L2. Structure 25, 1163–1174. doi: 10.1016/j.str.2017.06.011
Zamani, M. R., Aslani, S., Salmaninejad, A., Javan, M. R., and Rezaei, N. (2016). PD-1/PD-L and autoimmunity: a growing relationship. Cell Immunol. 310, 27–41. doi: 10.1016/j.cellimm.2016.09.009
Zaretsky, J. M., Garcia-Diaz, A., Shin, D. S., Escuin-Ordinas, H., Hugo, W., Hu-Lieskovan, S., et al. (2016). Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med. 375, 819–829. doi: 10.1056/NEJMoa1604958
Zhang, J., Bu, X., Wang, H., Zhu, Y., Geng, Y., Nihira, N. T., et al. (2018). Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature 553, 91–95. doi: 10.1038/nature25015
Zhao, L., Yu, H., Yi, S., Peng, X., Su, P., Xiao, Z., et al. (2016). The tumor suppressor miR-138-5p targets PD-L1 in colorectal cancer. Oncotarget 7, 45370–45384. doi: 10.18632/oncotarget.9659
Zhao, X., and Subramanian, S. (2017). Intrinsic resistance of solid tumors to immune checkpoint blockade therapy. Cancer Res. 77, 817–822. doi: 10.1158/0008-5472.CAN-16-2379
Zhao, X., and Subramanian, S. (2018). Oncogenic pathways that affect antitumor immune response and immune checkpoint blockade therapy. Pharmacol. Ther. 181, 76–84. doi: 10.1016/j.pharmthera.2017.07.004
Zhou, B. P., Deng, J., Xia, W., Xu, J., Li, Y. M., Gunduz, M., et al. (2004). Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat. Cell Biol. 6, 931–940. doi: 10.1038/ncb1173
Zhou, M. J., Chen, F. Z., and Chen, H. C. (2014). Ubiquitination involved enzymes and cancer. Med. Oncol. 31:93. doi: 10.1007/s12032-014-0093-6
Keywords: PD-L1, immunotherapy, gene expression, post-translational modification, small molecular inhibitors
Citation: Wang Y, Wang H, Yao H, Li C, Fang J-Y and Xu J (2018) Regulation of PD-L1: Emerging Routes for Targeting Tumor Immune Evasion. Front. Pharmacol. 9:536. doi: 10.3389/fphar.2018.00536
Received: 09 March 2018; Accepted: 03 May 2018;
Published: 22 May 2018.
Edited by:
Ruggero De Maria, Università Cattolica del Sacro Cuore, ItalyReviewed by:
Concetta Quintarelli, Bambino Gesù Ospedale Pediatrico (IRCCS), ItalyCopyright © 2018 Wang, Wang, Yao, Li, Fang and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jie Xu, amlleHVAc2p0dS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.