 Yu Wang
Yu Wang Lingling Yan1†
Lingling Yan1† Eric Prado
Eric Prado
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol., 20 March 2018
Sec. Respiratory Pharmacology
Volume 9 - 2018 | https://doi.org/10.3389/fphar.2018.00241
Recent advances in epigenetics have made a tremendous impact on our knowledge of biological phenomena and the environmental stressors on complex diseases. Understanding the mechanism of epigenetic reprogramming during the occurrence of pulmonary hypertension (PH) is important for advanced studies and clinical therapy. In this article, we review the discovery of novel epigenetic mechanisms associated with PH including DNA methylation, histone modification, and noncoding RNA interference. In addition, we highlight the role of epigenetic mechanisms in adult PAH resulting from undesirable perinatal environments—Extrauterine growth restriction (EUGR) and Intrauterine growth retardation (IUGR). Lastly, we give a comprehensive summary for the remaining challenges and discuss future methods of epigenetic targeted therapy for pulmonary hypertension.
Pulmonary arterial hypertension (PAH) is clinically defined as an elevation of mean pulmonary artery pressure (mPAP) >25 mmHg at rest (Barst et al., 2004). It is pathologically characterized by proliferation, migration, anti-apoptosis, or phenotype switching of pulmonary arterial endothelial cells (PAEC), pulmonary arterial smooth muscle cells (PASMC), and fibroblasts. The proliferation leads to an “onion-skin” appearance due to the occlusive lesions of small arteries which eventually become completely obstructed from the deposition of extracellular matrix (Edwards, 2004). Progressive remodeling of pulmonary vessels leads to a sustained increase in pulmonary vascular resistance (PVR) and afterload of right ventricle, which ultimately causes irreversible heart failure (Hyduk et al., 2005; Jongmin Kim, 2014). According to the 5th World Symposium of Pulmonary Hypertension held in 2013, PH can be classified into the five following groups: Group 1. Pulmonary arterial hypertension (PAH); Group 2. Pulmonary hypertension due to left heart disease; Group 3. Pulmonary hypertension due to lung diseases and/or hypoxia; Group 4. Chronic thromboembolic pulmonary hypertension (CTEPH); and Group 5. Pulmonary hypertension with unclear multifactorial mechanisms (Simonneau et al., 2013). Multiple etiologies account for PAH, including gene variants, family history, epigenetic changes, levels of sex hormones, risk factors (shear stress, age, drugs), cardiovascular disorders, environmental (hypoxia, virus infection), and nutritional factors (Figure 1; Kim et al., 2011; Tuder et al., 2013). Despite recent achievements in the treatment of PAH, most current therapies provide symptomatic relief for patients rather than cure the underlying disease. One of the challenges that confronts the successful reversing or curing of PAH is the consistent pulmonary hypertension that remains even after being treated maximally (Benza et al., 2012). Thus, there is an urgent need to explore the driving and sustaining molecular pathway of vascular remodeling as well as ventricular maladaptation in PAH for potential targeted therapeutic targets.
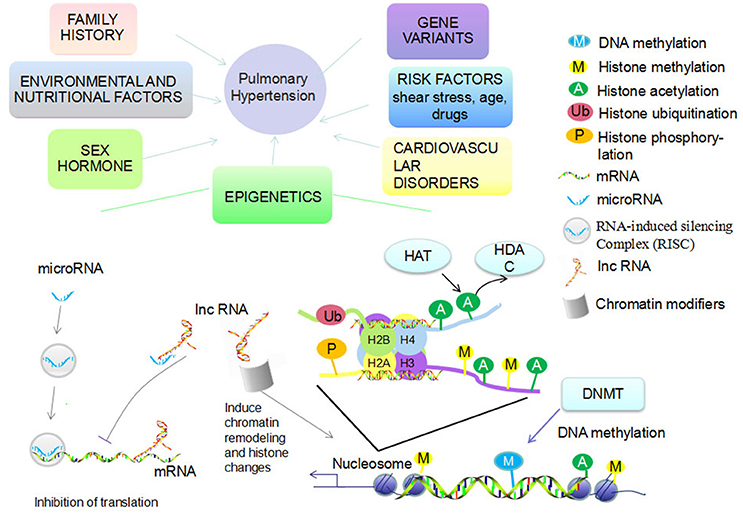
Figure 1. Etiologies account for PAH and epigenetic mechanisms. Multiple etiologies account for PAH, including gene variants, family history, levels of sex hormones, risk factors (shear stress, age, drugs), cardiovascular disorders, environmental (hypoxia, virus infection), and nutritional factors. Epigenetic mechanisms mainly include DNA methylation via DNA methyltransferase (DNMT) and histone modification (methylation, acetylation, ubiquitination, phosphorylation, etc.). Histone acetylation and deacetylation are regulated by histone acetyltransferase (HAT) and histone deacetylases (HDAC). The functional miRNA is incorporated into RNA-induced silencing complex (RISC) to silence gene expression, inhibit translation, or directly promote degradation of target mRNAs. LncRNA recruit chromatin modifiers while inducing chromatin remodeling and histone modifications. LncRNA can then either bind to mRNA to inhibit translation or bind to miRNA to inhibit RISC function.
Epigenetics is the study of inherited changes in gene expression and phenotypic variance caused by mechanisms other than changes in DNA sequence (Saco et al., 2014). There are three main classes of epigenetic information that can be inherited over cell generations: DNA methylation, histone modifications, and non-coding RNA modification (Figure 1; Kim et al., 2011). As an interface between genetics and environmental influences, modification of epigenetic events play an important role in the onset and progression of different human diseases (Herceg, 2016). Thus far, studying epigenetics has enabled researchers to discover the mechanisms of cancer development and has led to FDA approved drugs that reverse epigenetic aberrations for cancer therapy (Manel, 2010). There is increasing evidence that there are important roles of epigenetic regulation in PH, however, the underlying molecular mechanisms remain to be elucidated (Kim et al., 2011). We explore the dysfunctional epigenetic mechanisms associated with PH in this review.
DNA methylation refers to the covalent transfer of methyl groups (-CH3) onto the cytosine (C) pyrimidine ring in dinucleotide CpG (cytosine-guanine) DNA sequences (Bird, 2002). In mammals, it has been widely accepted that DNA methylation predominantly takes place on the fifth carbon of cytosine (5mC). Recently, another form of DNA modification, N6-methyladenine, has been identified in mammalian embryonic stem cells (Wu et al., 2016). DNA methylation is a physiological process that participates in the maintenance of gene activity states (genomic imprinting and silencing), cellular differentiation, and genome defense mechanisms (Paulsen and Fergusonsmith, 2001; Wu and Zhang, 2014). In human genomes, ~60–80% of CpGs within somatic cells are methylated, while the remaining are unmethylated. The unmethylated CpGs gather together to form a CpG-rich sequences known as CpG islands (Smith and Meissner, 2013). It is usually the unregulated hypermethylation of CpG islands that tends to be associated with aberrant transcriptions in cancer cells (Carvalho et al., 2012). This type of modification is set and maintained by several kinds of DNA methyltransferase (DNMTs) such as DNMT1, DNMT 3a, and DNMT 3b. DNA hypermethylation of promoters can frequently result in gene silencing by three of the following mechanisms: directly blocking the transcription factors from binding to specific DNA sequences, forming methyl-CpG binding proteins (MBP) to indirectly disrupt the recruitment of transcription factors, and silencing gene expression by cross-interference with histone modification (Ngo et al., 1995; Jones and Takai, 2001).
Superoxide dismutase-2 (SOD2) is a member of the SOD family, which controls the production of endogenous H2O2. H2O2 has the ability to regulate redox-sensitive targets, such as hypoxia inducible factor-1α (HIF1α) and voltage-gated potassium channel Kv1.5, both of which are important key factors that give rise to the mitochondrial role as a vascular O2 sensor (Sheehan et al., 1993; Eltzschig et al., 2005). SOD2 expression is decreased in PAH patients and fawn hooded rats (FHR). Archer et al. demonstrated that in pulmonary arteries of FHR, the selective hypermethylation of the SOD2 gene is located at both the enhancer region of intron 2 and a promoter region. Increased expression of DNMT1 and DNMT3B was also found. 5-aza-2′-deoxycytidine (Decitabine), a methyltransferase inhibitor, significantly decreased PASMC proliferation by demethylating SOD2, and increasing SOD2 expression (Archer et al., 2010). Recently, a genome-wide DNA methylation profile identified pulmonary endothelial cells genes of from PAH patients. A relationship between ABCA1 downregulation and lipid metabolism was shown which could be relevant to PAH (Hautefort et al., 2017). Reduced levels of global DNA methylation and histone acetylation were found in high-altitude long-term hypoxia induced PH. These epigenetic alterations may lead to PASMC proliferation and vessel remodeling (Yang et al., 2012a). Perros et al. demonstrated a decrease in gene Granulysin (GNLY) demethylation in peripheral blood mononuclear cells and explanted lungs in pulmonary veno-occlusive disease (PVOD) but not in PAH. This is helpful for discriminating these two diseases which share many similarities (Perros et al., 2013).
Histone modification is another key component of cellular regulation and it should always be considered in the topic of epigenetics. Histone modification is implicated in DNA repair processes, chromatin assembly, and genetic imprinting (Polo and Almouzni, 2005; Vidanes et al., 2005; Zhou et al., 2010). The fundamental building block of eukaryotic chromatins is the nucleosome which is comprised of positively charged histones that form an octamer (comprised of H2A, H2B, H3, and H4) with negatively charged DNA wrapped around them (Li, 2002). The N-terminal “tails” of the histones, especially the highly conserved sequences of H3 and H4, can undergo several kinds of post-translational covalent modifications including acetylation, methylation, ubiquitination, phosphorylation, biotinylation, glycosylation (O-GlcNAcylation), ADP-ribosylation, and SUMOylation (Bhaumik et al., 2007; Andreoli and Del, 2014). Histone acetylation enhances DNA transcriptional levels with histone acetyltransferase (HAT) which acts by transferring an acetyl onto the lysine (k) residues of histone proteins. The negatively charged acetyl group neutralizes the positively charged lysine residues, which loosens the binding between DNA and histone proteins. In contrast, histone deacetylation suppresses DNA transcription with histone deacetylases (HDAC) by removing acetyl groups from histones and subsequently tightening the chromatin (Eberharter and Becker, 2002; Verdone et al., 2005). The two processes mentioned above are active in a dynamic relationship depending on the balance between HAT and HDACs (Zentner and Henikoff, 2013). Currently, eight kinds of HAT are classified into two types according to their locations: nuclear or cytoplasmic. Eighteen kinds of HDAC have been discovered which can be classified into four groups (class I, IIa, IIb, III, and IV; Xu et al., 2007). Classification of histone deacetylases is listed in Table 1. Of all these HDACs, HDAC III is nicotinamide adenine dinucleotide (NAD+)-dependent also named as SIRT while the other three classes all have a zinc ion in the catalytic domains, which is required for catalysis (Ropero and Esteller, 2007). Previously, the small-molecule HDAC inhibitors are applied in the preclinical models of PH by chelating zinc in the active sites of HDAC I, II, and IV to increase the histone acetylation and promote the expression of specific genes (Cavasin et al., 2015). An emerging association between aberrant histone acetylation and PAH has been verified though various experiments. For example, there is an increased level of histone H3 and H4 acetylation at the promoter site of the endothelial nitric oxide synthetase (eNOS) gene in pulmonary arterial endothelial cells (PAEC) of persistent pulmonary hypertension of the newborn (PPHN; Xu et al., 2010). SIRT3, a mitochondrial deacetylase, is down regulated in human IPAH PASMC and rats with PAH. Decreased sirt3 suppresses mitochondrial function. It has also been found that sirt3 knockout mice can develop spontaneous PAH (Paulin et al., 2014).

Table 1. Classification of histone deacetylase (HDAC).
Several studies have discovered successful methods to cure PAH by using HDAC inhibitors to decrease arterial pressure. Sodium butyrate (BU), which acts as a histone deacetylase inhibitor, can inhibit PDGF-induced PASMC proliferation and migration (Cantoni et al., 2013). However, nonselective pan-HDAC inhibition with compounds such as TSA will bring about unwanted side-effects in the treatment for PAH and right ventricular (RV) hypertrophy. In Bogaard et al. experiment, treatment using the pulmonary artery banding (PAB) model with TSA did not reverse RV hypertrophy, but instead reduced cardiac output and increased RV dilatation (Bogaard et al., 2011). For this reason, selective HDAC inhibition has been considered to attenuate pulmonary hypertension. HDAC1 and HDAC5 are detected in higher expression in PAH patients and PAH rat models compared with the control group (Zhao et al.). Zhao et al. found that valproic acid (VPA) (an inhibitor to class I HDAC) and suberoylanilide hydroxamic (SAHA) (an inhibitor to classes I, II, and IV HDAC) are able to increase the acetylation of H3, reverse pulmonary hypertension, and reduce right ventricular hypertrophy with no effects to heart rate and systolic pressure (Zhao et al., 2012). Pulmonary adventitial fibroblasts, isolated from chronically hypoxic induced hypertensive calves, showed an accumulation of inflammatory cells and a significantly elevated catalytic activity of HDACs. The epigenetic associations can be illustrated when using class I HDAC inhibitors, which can markedly decrease cytokine/chemokine mRNA expression levels of the inflammatory cells (Courboulin et al., 2011). The class I HDAC inhibitor Apicidin can decrease right ventricular hypertrophy and remodeling of pulmonary arteries in PAEC of neonatal hypoxia induced PAH mice (Yang et al., 2015). The highly selective HDAC inhibitor for HDAC1, 2, 3 (HDACs class I)—MGCD0130 and MS275 were used in the hypoxia-induced PAH rat model to reduce pulmonary arterial pressure. A variety of beneficial effects on the right ventricle were seen though they only modestly reduced right ventricular hypertrophy (Cavasin et al., 2012). In PAH PAECs, binding of class IIa HDACs to Myocyte enhancer factor 2 (MEF2) results in suppression of MEF2 target genes that govern cellular growth and differentiation (Cavasin et al., 2015). Kim et al found that MC1568—pharmacological inhibition of class IIa HDACs, enables the restoration of impaired MEF2 activity and its targets such as miR-424 and miR-503 in PAH PAECs (Kim et al., 2013, 2015). In the monocrotaline (MCT) and Sugen/Hypoxia induced PH models, HDAC6 inhibitor Tubastatin A decreased PAH-PASMC proliferation and reduced mPAP (Boucherat et al., 2017).
The mechanism of HDAC inhibitors is complex. Generally, HDAC inhibitors increase histone acetylation and gene expression. However, HDAC inhibitors can also interact with non-histone substrates, such as transcription factors and coregulators, chaperones, nuclear receptors, nuclear import, signal transduction, DNA repair proteins, and mediators of movement (Marks and Xu, 2009). Therefore, based on these complex interactions, HDAC inhibitors can actually lead to reduced of gene expression. For example, NADPH oxidase 4 (Nox4) is involved in the development of PAH by contributing to reactive oxygen species (ROS) production, altered fibroblast behavior and cellular proliferation (Barman and Fulton, 2017). Nox4 expression is increased in the human PAH, hypoxia-induced, and MCT-induced PH model (Mittal et al., 2007; Barman et al., 2014). In the MCT-induced PH model, HDAC inhibitor VPA reduces right ventricular hypertrophy and rescues pulmonary hypertension by attenuating Nox expression (Chen et al., 2016). Current use of various epigenetic drugs in PAH are summarized in Table 2.
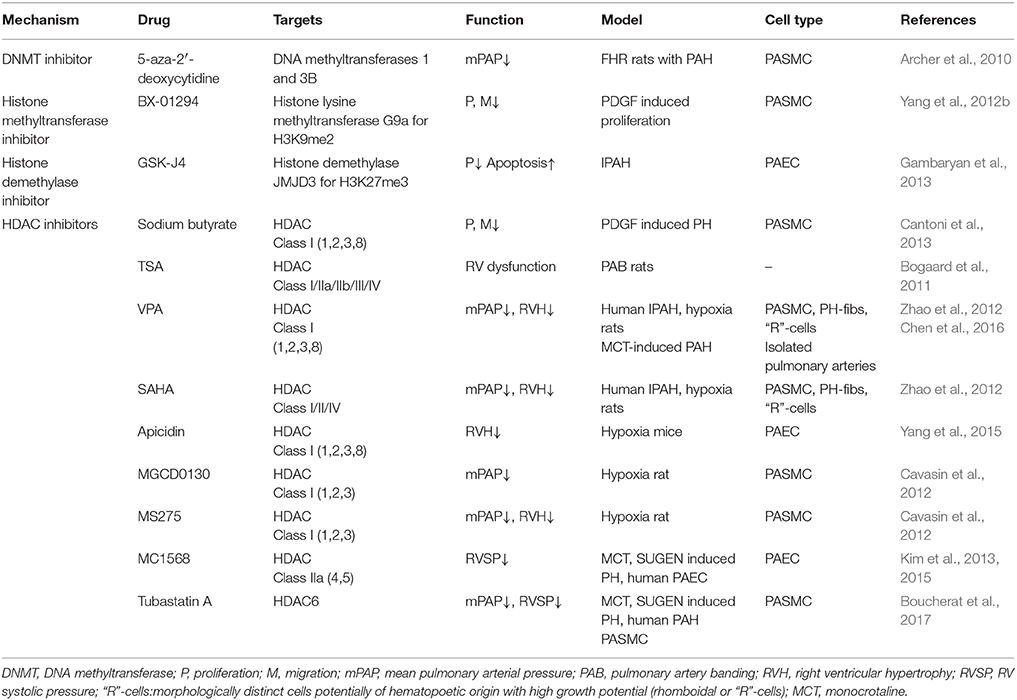
Table 2. Current use of various epigenetic modifiers in PAH.
Similar to DNA methylation, adding one (me1), two (me2), or three (me3) methyl groups into various basic residues of histones, usually targeted on histone H3, is the basic mechanism of histone methylation. On the same nucleosome, there is cross talk for linking DNA and histone methylation (Cheng and Blumenthal, 2010). For instance, methylation of H3K4 has been suggested to protect promoters from de novo DNA methylation in somatic cells to induce gene transcription (Weber et al., 2007). Conversely, methylation at H3K9 is positively correlated with DNA methylation and regarded as a code for transcriptional repression (Nguyen et al., 2002). The gene expression status is mainly determined by the site of a methyl lysine residue on the histone tail and the degree of methylation (me1, me2, or me3). Transcriptional modulator megakaryocytic leukemia 1 (MKL1) could interact with and recruit H3K4 methyltransferase complex in the hypoxia-induced pulmonary hypertension. Endothelial-specific depletion of two key components of the H3K4 methyltransferase complex reduces hypoxia-induced PH (Chen D. et al., 2015). Gambaryan et al. examined the expression of JMJD3, which can specifically demethylate H3K27me3 under the condition of cultured PAEC of PAH. It appears that GSK-J4, a selective JMJD3 inhibitor, can significantly lead to decreased proliferation, increased apoptosis and reduced TNF alpha-induced IL-6 release in a concentration-dependent manner (Gambaryan et al., 2013). Another similar experiment focused on the epigenetic regulatory effects of BX-01294, which is a specific inhibitor for G9a, a key enzyme for H3K9me3. It revealed that BX-01294 can also reduce PDGF-induced proliferation and migration of PASMC of pulmonary hypertensive ovine (Yang et al., 2012b). In the PASMC hypertensive mouse, an increased expression of Enhancer of Zeste Homolog 2 (EZH2), a mammalian histone methyltransferase, was detected. In transfected models, E2H2 can enhance proliferation, migration, and anti-apoptosis of the human PASMCs, compared to the controlled GFP-transfected cells (Aljubran et al., 2012).
“Fetal origins of adult diseases” has gained increased attention in the past few years (Barker et al., 1989; Barker, 2004; Osmond et al., 2011; Szostakwegierek and Szamotulska, 2011). The original model of “the fetal origins of adult diseases” is “the Barker hypothesis.” It suggests that famine exposure during gestation sharply affects children's birth weight and even the susceptibility to diseases in adolescence and adulthood, including type 2 diabetes, impaired glucose tolerance, hypertension, coronary heart disease, metabolic diseases, and so on (Feng et al., 2015). In 2003, the academic community constructed the theory of “the Developmental Origins of Health and Disease” (DOHaD). Based on the theory of DOHaD, the beginning stages of life, including pregnancy, neonatal period, and childhood, are the crucial periods that may increase an individual's sensitivity or risk of developing diseases in adulthood (Barker and Osmond, 1986; Kubota et al., 2015; Dickinson et al., 2016).
Intrauterine growth retardation (IUGR) occurs during unsuitable uterine conditions which result in an average neonatal birth weight in the 10th percentile or 2 standard deviations lower than corresponding gestational age of fetus (Wu et al., 2006). According to a large amount of epidemic and lab research, IUGR is strongly correlated with the formation of adult-onset diseases (Vickers et al., 2000). Fetal tissues initiate some changes in order to adapt themselves to the unsuitable uterine condition. IUGR can lead to epigenetic changes of some related genes, eNOS, and endothelin-1 (ET-1), which make individuals hypersensitive to hypoxia, leading toward pulmonary arterial hypertension (Xu et al., 2013).
Endothelial nitric oxide synthetase (eNOS) catalyses the formation of NO—an endothelium derived relaxing factor which plays a vital role during the regulation of pulmonary arterial pressure. Histone modifications to different sites of eNOS promoter regions can make a difference for its activation or suppression. For instance, H3K9ace and H3K4me3 promote while H3K27me3 and H3K9me3 suppress the transcription of eNOS (Yan et al., 2010). Research based on the human endothelial cells isolated from umbilical veins (hUVEC) from control and IUGR fetuses uncovered the epigenetic mechanism underlying the eNOS changes (Krause et al., 2013). In IUGR-hUVEC, there is a decreased expression of eNOS associated with a hypermethylation of CpG-352 in its promoter. In addition, there is hypomethylation of the hypoxia response element (HRE) that occurs in the eNOS promoter region of IUGR-hUVEC, which is similar to the observation of normal hUVEC cultivated under hypoxic conditions (Casanello et al., 2009). More interestingly, silencing DNMT1 with siRNA against DNMT1 can reverse the eNOS expression and restore the response to hypoxia in hUVEC (Krause et al., 2013).
Endothelin-1 (ET-1), a potent vasoconstrictor peptide, not only fosters the contraction of pulmonary vasculature, but can also stimulate the proliferation, migration, contraction and the deposition of extracellular matrix in vascular smooth muscle cells by activating ET receptors ((Kapakos et al., 2010)). When given a hypoxic stimulus, IUGR rats demonstrated a distinctly higher expression of ET-1 protein. Compared to the normal neonatal rats, the acetylation of histone H3 and H3K9/18 increases in core promoter regions of ET-1 gene in IUGR-hypoxia rats PAEC accompanied with the enriched transcription factor HIF-1α (Figure 2; Xu et al., 2013). Recently discovered, hyperacetylation is observed in the histone H3 in ET-1 promoters of leukocytes from the 1-week IUGR rats with a continued trend 10 weeks after birth (Xu et al., 2016). From this perspective, the epigenetic changes of peripheral leucocytes have a great potential to serve as a biomarker and risk predictor for developing disease (Xu et al., 2016).
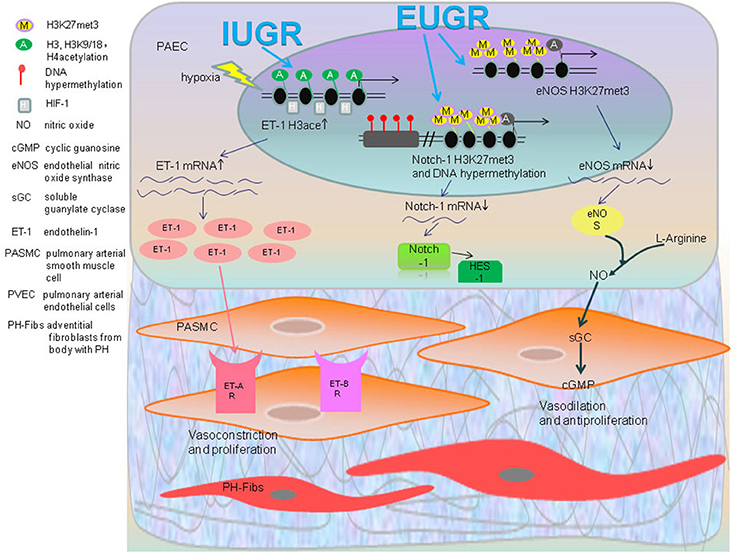
Figure 2. Epigenetic regulatory mechanisms of developmental origin of PAH: IUGR rats become hypersensitive to hypoxia as a result of PH along with an increased expression of ET-1, which is involved in contraction of pulmonary vascular. The acetylation of histone H3, H3K9/18, and H4 of ET-1 gene in IUGR-hypoxia rats PAEC accompanied with enriched transcription factor HIF-1α was found. The increased ET-1 activates ET receptors in PASMC that results in vasoconstriction and proliferation. In the EUGR induced PAH rat model, the levels of H3K27me3 of the eNOS gene were significantly higher. EUGR also caused H3K27me3 and methylation of a CpG site of Notch1 gene. This decreases the expression of eNOS mRNA, Notch1 mRNA and its downstream gene HES1 as a consequence, which impairs anti-proliferation and vasodilation.
Extrauterine growth restriction (EUGR) refers to the measured indicators of growth and development for neonates at the 10th percentile, which is currently inferior to expectations of intrauterine growth rate based on estimated postmenstrual age (Clark et al., 2003). One research study showed both systolic and diastolic blood pressures in adolescents born with EUGR history are dramatically higher than the control group (Ortizespejo and Mercedes, 2013). Another experiment demonstrated that providing IUGR rats with consistent EUGR feeding after birth will have a significantly higher pressure after 5 months (Tare et al., 2012). Hence, these results above indicate that EUGR is an inducer for aberrant functions of blood vessels for the future adult period. Higher pulmonary arterial pressure, increased right heart index, and remolding of pulmonary blood vessels can appear in adulthood of male EUGR rats, which is closely related to the epigenetic change of several key genes associated with PH (Zhang et al., 2014). Decreased expression of the eNOS gene in PAEC of EUGR male rats may arise from the increased presence of histone H3K27me3 and H3K9me3 in the proximal promoter region (Zhang et al., 2014).
Zhang identified that in PAEC of 9-week old EUGR male rats, there is an increased IGF-1 mRNA level, which may have resulted from decreased methylation in PAEC CpG islands as compared to the control groups. In the same experiment, an increased H3K27me3 and H3K9me3 of the PPARγ gene result in a lower expression of its mRNA. This provides the evidence that adult rats with a history of EUGR may undergo remodeling of pulmonary vascular structure and function, making them more susceptible to pulmonary hypertension (Zhang et al., 2014).
Notch1 gene plays an important role for the angiogenesis and repairing of injured vascular endothelial cells caused by ischemia (Limbourg et al., 2005). After forming a complex with NICD and RBP-j, Notch1 binds to the downstream transcription regulatory regions and activates the Notch signaling pathway, mediating the formation, maturation, and remodeling of pulmonary vascularture (Taichman et al., 2002; Gridley, 2007; Kume, 2009). Notch1 can synergistically pair with the VEGF gene to activate PI3K/Akt, which not only promotes the proliferation and migration of PAEC, but also increases the expression of NO to regulate the dilation of vessels by phosphorylation and activation of eNOS (Shiojima and Walsh, 2002; Takeshita et al., 2007). There is a significant reduction in Notch1 along with its downstream gene Hes-1 in the PAEC of EUGR rats of 3w and 9w. This may be related to the increase of histone H3K27me3 in the proximal promoter regions of Notch1 and the DNA hypermethylation of the CpG site in the distal promoter regions (Tang et al., 2015).
In addition to dietary restriction, other adverse environmental conditions, such as smoking, hypoxia, and drugs, are all associated with many diseases in adulthood. Insulin-like growth factor-1(IGF-1) is vital to the growth and metabolism of individuals, especially for the perinatal period. IGF-1 knockout mice suffer devastating embryonic and postnatal growth retardation (Woods et al., 1996). In the aspect of promoting vascular growth, IGF-1 can inhibit cellular apoptosis by phosphorylating apoptosis protein BAD through the IGF-1-IRS-1-PI3K-IGFR-1R/AKT pathway, which has been found to be activated in hypoxia-induced PAH (Madonna et al., 2015). Recent research has discovered that in the PAEC of neonatal hypoxia induced PAH mice, there is aberrant elevated cytosine methylation at the CpG site (position 87857993) in the promoter region of the IGF-1 gene. In addition, the HDAC inhibitor apicidin can diminish right ventricular hypertrophy and remodeling of pulmonary arteries by decreasing the activation of hypoxia-induced IGF-1/PaktS473 signaling pathway (Yang et al., 2015). This study suggests a new relationship between the HDAC and traditional IGF-1 signaling pathway in the regulation of hypoxia-induced PH.
Noncoding RNAs (ncRNAs) are the transcription products of genes that lack the ability to be translated into proteins. Noncoding RNA genes include tRNAs, rRNAs, snoRNAs, microRNAs, siRNAs, snRNAs, exRNAs, piRNAs, and scaRNAs and the long ncRNAs.
MiRNAs are small endogenous noncoding RNA that usually consist of <22 nucleotides. They are transcribed by RNA polymerase II from segments of genomic DNA appearing as primary miRNAs (pri-miRNAs) which then form 60–70 hairpin liked nucleotides known as the precursor miRNAs (pre-miRNAs) with the regulation of Drosha-DGCR8 complex. Then, Exportin-5 assists the GTP-Binding nuclear protein in transporting the pre-miRNA out of the nucleus to form mature miRNAs. The functional miRNA strand is then incorporated into a RNA-induced silencing complex (RISC) to silence gene expression by binding to the 3′-untranslated regions (UTR) of mRNAs, thus inhibiting their translation or directly promoting degradation of the target mRNAs (Lai, 2002; Johar et al., 2015). Comprehensive roles of miRNAs have been confirmed in various cancers, both oncogenes and tumor-suppressors (Volinia et al., 2010). Their potential function in the pathogenesis of cardiovascular diseases and targeted therapies have also become a significant research topic (Sanoudou et al., 2012).
Caruso was the first to report abnormal expressions of miRNA in PAH: downregulation of mir22, mir21, mir30c, and let-7f (Caruso et al., 2010), which created new therapeutic targets for the potential treatment of PAH, of which latter researchers could work with. Paracrine micro-RNA-regulated crosstalk in pulmonary arterial endothelial cells and pulmonary arterial smooth muscle cells has been discovered, for instance miR-17/20a and miR-130/301(Bertero et al., 2014, 2015). It is unclear whether these regulations in the microRNA of endothelial or smooth muscle-based cells have a similar impact on the outcome of PH. However, it is conceivable that these interferences may act on similar progenitors, as endothelial cells can transdifferentiate into smooth muscle cells when stimulated with platelet derived growth factor(PDGF)-BB (Yamashita et al., 2000) or transforming growth factor (TGF)-β (Frid et al., 2002). Furthermore, emerging evidence depicts the concept that miRNAs are affected by regulations from DNA methylation and histone modification, which form an internal regulatory network of epigenetics (Kim et al., 2013, 2015). Alterations of the expressions in different PH models and acting signaling pathways are listed in the Table 3.
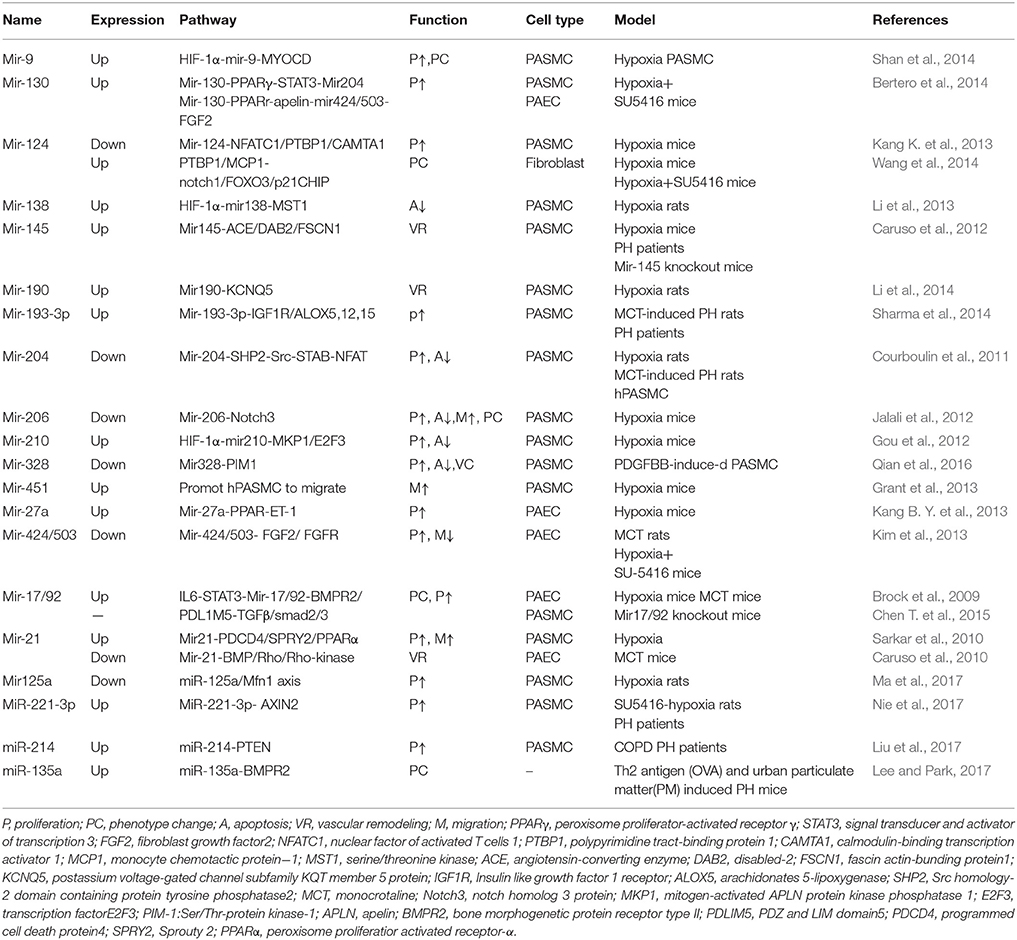
Table 3. MicroRNA mechanisms in PH.
The MiR-17/92 cluster consists of six mature miRNAs (miR-17, miR-18a, miR-19a, miR-19b, miR-20a, miR-92a; Tanzer and Stadler, 2004), which are transcribed simultaneously but have their own respective target mRNAs with different regulatory functions. Down-regulation of bone morphogenetic protein receptor type II (BMPR2) protein, yet not the mRNA in several PAH models (Takahashi et al., 2006; Morty et al., 2007) revealed a posttranscriptional regulatory process mediated by miRNAs exists. Cytokine interleukin(IL)-6 increases expression of miR-17/92 through constant activation of STAT3(signal transducer and activator of transcription 3) binding sites in its promoter region(C13orf25), which inhibits BMPR2 and consequently proliferates the endothelial cells (Brock et al., 2009). MiR-17/92 leads to the expression of contraction proteins of PASMC by suppressing PDZ and LIM domain5(PDLIM5) by binding to its 3′UTR, this protein then induces TGF-β/Smad3 signaling (Chen T. et al., 2015). Pullamsetti et al. reported that miR-17-92 exhibits an over expression in both hypoxia-induced and MCT-induced rat models. An inhibitor of miR-17 can ameliorate disease states in PH (Pullamsetti et al., 2012).
In PH-hPAEC, miR-27a is expressed more with ET-1 by reducing the expression of PPARγ (Kang B. Y. et al., 2013). The micro-130/301 family (miR-130a, miR-130b, miR-301a, and miR-301b) was found to be a master regulator of several subordinate miRNAs. Bertero et al. discovered that in PAECs, miR-130/301-PPARγ contains the same upstream regulatory axis of the APLN-miR-424/503-FGF2 axis that exerts cellular proliferation by repressing apelin and its respective miRNAs (miR-424/503; Kim et al., 2013; Bertero et al., 2014). This miR-130/301-PPARγ is involved in regulating STAT3-miR-204- Scr kinase—NFAT which are substances for the PH-PASMC proliferation and anti-apoptosis (Courboulin et al., 2011). As expressed, the up-regulatory miR130/301 family is of significant importance in the microRNA network of PH, which enables it to become a potential biomarker and antagonistic target for PH therapy (Bertero et al., 2015).
In PASMC isolated from hypoxic rats and patients, miR-124 was down-regulated. The action of miR-124 not only caused a significant decrease in cell proliferation but also maintained a differentiated phenotype by inhibiting its target—nuclear factor of activated T cells (NFAT) signaling pathway (Kang K. et al., 2013; Wang et al., 2014). Hypoxia stimulates the HIF1α induced up regulation of miR-9, leading to the phenotypic switching of PASMC from contractile to proliferative (Shan et al., 2014). MicroRNA21 was found to be down regulated in idiopathic PAH patients' lung tissue and serum as well as MCT-induced PH rats (Caruso et al., 2010). In PASMC isolated from hypoxia induced PH mice, there is an increased expression of miRNA21 which promotes the proliferation and anti-apoptosis of PASMC by down regulating the following target genes: PDCD4, SPRY2, and PPARα (Sarkar et al., 2010). MiR-125a was found to decrease PASMC proliferation during hypoxia by inhibiting Mfn1 expression. MiR-125a agomir had a protective role in mitochondrial dysfunction and abnormal remodeling (Ma et al., 2017). MiR-138 was found to represses serine/threonine kinase Mst1 as a result of activating the Akt signaling pathway, thus functioning as a negative regulator for the apoptosis of PH-PASMC by a mitochondria-mediated caspase-dependent mechanism (Li et al., 2013). Caruso et al. found an elevation of miR-145 expression in PH-PASMC, especially in the lungs of BMPR2-deficient mice and patients with BMPR2 mutations (Caruso et al., 2012). With this current knowledge, the development of a patent for using antisense oligonucleotides (ASOs) against miR-145 has been initiated in chronic hypoxia mouse models, which provides a promising approach for treating the progression of PH (Ogorodnikova and Arenz, 2015). In PH-PASMC, increased miR-190 enhances vasoconstriction by interacting with its target Kcnq5, a subfamily member of voltage-gated K+ channels, which mediates Ca2+ influx (Li et al., 2014). Sharma et al. found that in PAH patients and rodents, miR-193-3p was significantly down regulated. This is due to 4F peptide, an Apolipoprotein A-I (apoA-I) mimetic peptide, which can restore the expression of miR193 by suppressing lipoxygenases (ALOX5,12,15), IGF1R and retinoid X receptor (RXR) α signaling pathway (Sharma et al., 2014). Over-expression of miR-206 in hPASMC can decrease proliferation, increase apoptosis and induce differentiation of α-SMA and calponin by inhibiting Notch3 protein expression (Jalali et al., 2012). MiR-221-3p promotes proliferation of PASMC by targeting AXIN2. This proliferation can be dampened by miR-221-3p inhibitors, AXIN2 over-expression, or β-catenin inhibition (Nie et al., 2017). MiR-210 is an HIF1α-dependent miRNA that can inhibit PASMC apoptosis during hypoxia by directly repressing E2F3 expression (Gou et al., 2012). MiR328 is down regulated in PASMC and functions as an inhibitor of PASMC's proliferation and migration by targeting the Ser/Thr-protein kinase-1 (PIM-1). Based on these findings, measuring serum miR-328 levels is a promising diagnostic tool for CHD-PAH diagnosis (Qian et al., 2016). Over-expression of miR-451 under serum-free conditions only promoted hPASMC to migrate. Temporary inhibition of miR-451 can reduce the development of PAH but genetic deletion of miR-451 had no beneficial effects on PAH (Grant et al., 2013). Increased miR-214 was found to promote hypoxia-induced PH with COPD by targeting PTEN in PASMCs. As such, miR-214 could be a potential circulating biomarker and novel therapeutic target in the treatment of PH (Liu et al., 2017).
Wang et al. demonstrated that in the PH fibroblasts, miR-124 is inhibited with its downstream genes such as Nothch1, PTEN, FOXO3, p21, and p27 by activating polypyrimidine tract-binding protein (PTBP)-1. In addition, there is also increased inflammatory activity associated with miR-204 in PH fibroblasts when searching for its direct target MCP-1—a chemoattractant for macrophages and monocytes, which was shown to have increased levels when transfecting anti-miR-124 (Wang et al., 2014). The study reveals multiple targets (PTBP1 inhibitors, miR-124 agonists) with high therapeutic potential for IPAH patients.
In a PAH model induced by Th2 antigen (OVA) and urban particulate matter (PM), miR-135a was significantly increased, and its target BMPR2 was decreased. MiR-135a could effectively serve as an additional biomarker. A drug that blocks the action of miR-135a could be useful for therapeutic intervention in PAH (Lee and Park, 2017).
Long noncoding RNAs (lncRNAs) are noncoding RNA molecules consisting of greater than >200 nucleotides. Recent evidence points to the involvement of lncRNAs in multiple biological processes including cell proliferation, cell growth, cell differentiation, cell cycle progression, apoptosis, immune response, imprinting, tumorigenesis, pathogenesis of various human diseases (Kogo et al., 2011; Wapinski and Chang, 2011; Ziats and Rennert, 2013; Vencken et al., 2014; Greco et al., 2015; Ling et al., 2015) and other biological processes (Ota et al., 2004; Tupy et al., 2005; Wilusz et al., 2009).
LncRNAs play important roles in epigenetic mechanisms in transcriptional and post-transcriptional regulation by targeting transcription factors, inducing chromatin remodeling, regulating methylation complexes, blocking proximate transcription (Ponting et al., 2009), and RNA based epigenetic regulatory networks through small interfering RNAs (si-RNA; Singh and Prasanth, 2013; Weinberg and Morris, 2013).
Despite the increased knowledge of lncRNAs, the epigenetic mechanisms of lncRNAs involved in PAH remain poorly understood. Recently, some studies have expanded our understanding of lncRNAs in PAH. Chronic thromboembolic pulmonary hypertension (CTEPH) is one of the primary causes of severe pulmonary hypertension. In endothelial tissues from the pulmonary arteries of CTEPH patients, aberrant expression of 185 lncRNAs was observed. The expression levels of lncRNAs, NR_036693, NR_027783, NR_033766, and NR_001284 were significantly altered. The potential role of lncRNAs may involve inflammatory responses, response to endogenous stimulus, antigen processing and presentation (Song et al., 2015). In the hypoxic pulmonary hypertension (HPH) rat model, the expression profile of 362 lncRNAs was significantly altered using a microarray (Wang et al., 2015). Extracellular circulating lncRNAs in human plasma were tested. However, the results showed no changes and very low levels of 84 lncRNAs in plasma of PAH. Therefore, the use of lncRNAs as a potential biomarker for human PAH is limited (Schlosser et al., 2016).
The association between lncRNA's role with miRNAs has also been investigated. Angiotensin II (Ang II) is a small polypeptide hormone involved in hypertension. By using RNA-sequencing, 24 novel lncRNAs were identified in Ang II-induced vascular smooth muscle cells (VSMCs). These 24 novel lncRNAs may play a role in mediating cellular responses of VSMCs to Ang II. Furthermore, Lnc-Ang362 functions as a host transcript for two miRNAs (miR-221 and miR-222) implicated in VSMC proliferation. These results provide novel insights into lncRNAs as potential therapeutic targets in Ang II-associated cardiovascular diseases (Leung et al., 2013).
Pulmonary hypertension (PH) is a progressive and severe vasculopathy characterized by vasoconstriction, inflammation, and remodeling of the pulmonary vessels leading to a sustained increase in pulmonary vascular resistance. Epigenetics not only plays an important role in the normal development of human beings, but is also associated with many human diseases. Researches have made great progress of epigenetic knowledge in PH within the recent years. Studies for the role of epigenetics in PH can help to better understand the pathogenesis of PH. In contrast from permanent changes in DNA sequence that causes disease, many epigenetic changes are reversible. This serves as a great potential for therapeutics interventions that may reverse the phenotype of PH.
Based on the understanding of present epigenetics, the majority of the epigenetic drugs are genetically designed to target DNA methyltransferase and histone deacetylase. All other DNA modified proteins may be potential targets for altering the epigenetic status of cells, such as HAT, Histone methylation enzyme and MeCP2 (methyl CpG binding protein 2; Amir et al., 1999). DNA methylation is set and maintained by several kinds of DNA methyltransferase (DNMTs). Nucleoside DNA methyltransferase inhibitors such as 5-azacytidine and Decitabine have shown antineoplasmic activity. However, they also are severely toxic to cells, requiring careful monitoring of the length of time used for and dose administered. Unfortunately, these drugs are not specific toward the type of tissue acted on—causing many unwanted side effects that may outweigh their benefit. Histone modifications play important roles in the regulation of gene expression. Several animal models using HDAC inhibitors have shown to reduce PH (Bogaard et al., 2011; Zhao et al., 2012; Cantoni et al., 2013). These results provide a theoretical basis of HDAC inhibitors for the treatment of PAH. However, there are some limitations and disadvantages in current epigenetic treatments. Broad spectrum HDAC inhibitors will bring about unwanted cardiac side-effects in the treatment for PAH such as TSA (Bogaard et al., 2011). This undesirable effect from nonspecific targets need to be resolved. Selective isoform drugs and their effect on each organ should be studied further. Different types of histone and HDAC isoforms have different distributions in tissues and diseases. With further understanding of their specific roles and expressions, more target- selective drugs could be designed. For PH, some epigenetic abnormalities are not present throughout the genome, thus providing difficulty when changing the epigenetics of a specific gene for treatment. This remains as a challenging problem for therapy. It is meaningful to study the epigenetic mechanisms of key genes or transcription factors, and further explore the corresponding drugs on this basis. Furthermore, whether epigenetic targets act only on the pulmonary vessels or cause an unexpected decrease in systemic blood pressure at the same time, it is an important problem that requires special attention. These HDAC inhibitors are in the preclinical stage, their clinical trials, safety profile, and effects of drugs still need to be well-confirmed before widespread use.
The concept of adult diseases arising from a developmental origin gives many metabolic diseases and pulmonary diseases a reasonable explanation for how they progress in humans. Pregnancy, the neonatal period and childhood are crucial stages for growth and development. Dietary restriction during pregnancy is an important factor affecting the development of the pulmonary arteries. These findings provide significant advancements of epigenetic mechanisms that take effect during pregnancy and infancy for the risk of developing PH. IUGR rats became hypersensitive to hypoxia as a result of higher right ventricular systolic pressure (RVSP) and expression of ET-1. The acetylated histone H3 of the ET-1 promoter of leucocytes were identified in 1-week IUGR rats (Xu et al., 2016), in which the results of ET-1 serve as a biomarker. Early life has great plasticity and is sensitive to the environment. The changes in the neonatal period are reversible and promote the opportunity to intervene with the disease before birth. With effort, we may be able to establish “preemptive medicine” to prevent the development of “the fetal origins of adult diseases.” Future studies need to be conducted to find out the specific components and time points of dietary restriction in childhood. Multiple signaling pathways associated with endothelial cells have been found, but regulatory mechanism of SMC may be more direct. In addition to dietary restriction, other factors such as alcohol, medications, and caffeine are all need to be investigated for their effects on pulmonary vascular development during the perinatal period.
MiRNA and lncRNA are very promising as diagnostic biomarkers, and their analogs or inhibitors may be novel therapeutic targets in the treatment of PH. More epigenetic mechanisms of microRNA and lncRNA have yet to be discovered, and their relationship with DNA methylation and histone modification may provide enough therapeutic potential to consider further research in epigenetics. Currently, the role of microRNA in pulmonary hypertension is based on known genes or pathways, other genes, transcription factors, and pathways that affect pulmonary arterial function still need to be explored. One miRNA may act on hundreds of genes in different organs, and other gene targets need to be avoided in the use of miRNA for treatment. In addition, mechanisms of these miRNAs are based on different PH animal models or patient cells, on different cells (PAEC, PASMC, PH-Fibs). Whether a miRNA has the same effect on different models or cells need further exploration.
The contribution of epigenetic regulation to pulmonary hypertension is an active but complex field of research. The role taken by the epigenetic regulation in the present clinical management of PH reflects the limitations of our current understanding of the disease to some extent. There is ample room for research and diagnostic translation of the pathobiological studies, aimed at procuring the diagnosis and therapeutic power in the epigenetic regulation of pulmonary hypertension.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by Zhejiang Key Laboratory for Diagnosis and Therapy of Neonatal Diseases, Key Laboratory of Reproductive Genetics (Zhejiang University), Ministry of Education, and by grants from the National Natural Science Foundation of China (No. 81501293 and 81630037).
PH, pulmonary hypertension; EUGR, Extrauterine growth restriction; IUGR, Intrauterine growth retardation; PAH, Pulmonary arterial hypertension; PAEC, pulmonary arterial endothelial cells; PASMC, pulmonary arterial smooth muscle cells; CTEPH, Chronic thromboembolic pulmonary hypertension; DNMTs, DNA methyltransferase; SOD2, Superoxide dismutase-2; HIF1α, hypoxia inducible factor-1α; HAT, histone acetyl transferase; HDAC, histone deacetylases; eNOS, endothelial nitric oxide synthetase; PPHN, persistent pulmonary hypertension of the newborn; DOHaD, Developmental Origins of Health and Disease; ET-1, endothelin-1; hUVEC, human endothelial cells isolated from umbilical veins; IGF-1, Insulin-like growth factor-1; ncRNAs, Noncoding RNAs; BMPR2, bone morphogenetic protein receptor typeII; lncRNAs, Long noncoding RNAs; Ang II, Angiotensin II.
Aljubran, S. A., Ruan Cox, J., Parthasarathy, P. T., Ramanathan, G. K., Rajanbabu, V., Bao, H., et al. (2012). Enhancer of zeste homolog 2 induces pulmonary artery smooth muscle cell proliferation. PLoS ONE 7:e37712. doi: 10.1371/journal.pone.0037712
Amir, R. E., Van den Veyver, I. B., Wan, M., Tran, C. Q., Francke, U., and Zoghbi, H. Y. (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23, 185–188. doi: 10.1038/13810
Andreoli, F., and Del, R. A. (2014). Physicochemical modifications of histones and their impact on epigenomics. Drug Discov. Today 19, 1372–1379. doi: 10.1016/j.drudis.2014.05.005
Archer, S. L., Marsboom, G., Kim, G. H., Zhang, H. J., Toth, P. T., Svensson, E. C., et al. (2010). Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: a basis for excessive cell proliferation and a new therapeutic target. Circulation 121, 2661–2671. doi: 10.1161/CIRCULATIONAHA.109.916098
Barker, D. J. (2004). The developmental origins of adult disease. J. Am. Coll. Nutr. 1(Suppl. 6), 130–141. doi: 10.1080/07315724.2004.10719428
Barker, D. J., and Osmond, C. (1986). Infant mortality, childhood nutrition, and ischaemic heart disease in england and wales. Lancet 1, 1077–1081. doi: 10.1016/S0140-6736(86)91340-1
Barker, D. J., Winter, P. D., Osmond, C., Margetts, B., and Simmonds, S. J. (1989). Weight in infancy and death from ischaemic heart disease. Lancet 2, 577–580. doi: 10.1016/S0140-6736(89)90710-1
Barman, S. A., Chen, F., Su, Y., Dimitropoulou, C., Wang, Y., Catravas, J. D., et al. (2014). NADPH oxidase 4 is expressed in pulmonary artery adventitia and contributes to hypertensive vascular remodeling. Arterioscler. Thromb. Vasc. Biol. 34, 1704–1715. doi: 10.1161/ATVBAHA.114.303848
Barman, S. A., and Fulton, D. (2017). Adventitial fibroblast Nox4 expression and ROS signaling in pulmonary arterial hypertension. Adv. Exp. Med. Biol. 967, 1–11. doi: 10.1007/978-3-319-63245-2_1
Barst, R. J., McGoon, M., Torbicki, A., Sitbon, O., Krowka, M. J., Olschewski, H., et al. (2004). Diagnosis and differential assessment of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 43, S40–S47. doi: 10.1016/j.jacc.2004.02.032
Benza, R. L., Miller, D. P., Barst, R. J., Badesch, D. B., Frost, A. E., and McGoon, M. D. (2012). An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from the REVEAL Registry. Chest 142, 448–456. doi: 10.1378/chest.11-1460
Bertero, T., Cottrill, K., Krauszman, A., Lu, Y., Annis, S., Hale, A., et al. (2015). The microRNA-130/301 family controls vasoconstriction in pulmonary hypertension. J. Biol. Chem. 290, 2069–2085. doi: 10.1074/jbc.M114.617845
Bertero, T., Lu, Y., Annis, S., Hale, A., Bhat, B., Saggar, R., et al. (2014). Systems-level regulation of microRNA networks by miR-130/301 promotes pulmonary hypertension. J. Clin. Invest. 124, 3514–3528. doi: 10.1172/JCI74773
Bhaumik, S., Smith, E., and Shilatifard, A. (2007). Covalent modifications of histones during development and disease pathogenesis. Nat. Struct. Mol. Biol. 14, 1008–1016. doi: 10.1038/nsmb1337
Bird, A. (2002). DNA methylation patterns and epigenetic memory. Genes Dev. 16, 6–21. doi: 10.1101/gad.947102
Bogaard, H. J., Mizuno, S., Hussaini, A. A., Toldo, S., Abbate, A., Kraskauskas, D., et al. (2011). Suppression of histone deacetylases worsens right ventricular dysfunction after pulmonary artery banding in rats. Am. J. Respir. Crit. Care Med. 183, 1402–1410. doi: 10.1164/rccm.201007-1106OC
Boucherat, O., Chabot, S., Paulin, R., Trinh, I., Bourgeois, A., Potus, F., et al. (2017). HDAC6: a novel histone deacetylase implicated in pulmonary arterial hypertension. Sci. Rep. 7:4546. doi: 10.1038/s41598-017-04874-4
Brock, M., Trenkmann, M., Gay, R. E., Michel, B. A., Gay, S., Fischler, M., et al. (2009). Interleukin-6 modulates the expression of the bone morphogenic protein receptor type II through a novel STAT3-microRNA cluster 17/92 pathway. Circ. Res. 104, 1184–1191. doi: 10.1161/CIRCRESAHA.109.197491
Cantoni, S., Galletti, M., Zambelli, F., Valente, S., Ponti, F., Tassinari, R., et al. (2013). Sodium butyrate inhibits platelet-derived growth factor-induced proliferation and migration in pulmonary artery smooth muscle cells through Akt inhibition. FEBS J. 280, 2042–2055. doi: 10.1111/febs.12227
Caruso, P., Dempsie, Y., Stevens, H. C., McDonald, R. A., Long, L., Lu, R., et al. (2012). A role for miR-145 in pulmonary arterial hypertension: evidence from mouse models and patient samples. Circ. Res. 111, 290–300. doi: 10.1161/CIRCRESAHA.112.267591
Caruso, P., MacLean, M. R., Khanin, R., McClure, J., Soon, E., Southgate, M., et al. (2010). Dynamic changes in lung microRNA profiles during the development of pulmonary hypertension due to chronic hypoxia and monocrotaline. Arterioscler. Thromb. Vasc. Biol. 30, 716–723. doi: 10.1161/ATVBAHA.109.202028
Carvalho, D. D. D., Sharma, S., You, J. S., Su, S. F., Taberlay, P. C., Kelly, T. K., et al. (2012). DNA methylation screening identifies driver epigenetic events of cancer cell survival. Cancer Cell 21, 655–667. doi: 10.1016/j.ccr.2012.03.045
Casanello, P., Krause, B., Torres, E., Gallardo, V., González, M., Prieto, C., et al. (2009). Reduced l-arginine transport and nitric oxide synthesis in human umbilical vein endothelial cells from intrauterine growth restriction pregnancies is not further altered by hypoxia. Compar. Stud. Soc. Hist. 30, 625–633. doi: 10.1016/j.placenta.2009.04.010
Cavasin, M. A., Demosdavies, K., Horn, T. R., Walker, L. A., Lemon, D. D., Birdsey, N., et al. (2012). Selective class I histone deacetylase inhibition suppresses hypoxia-induced cardiopulmonary remodeling through an antiproliferative mechanism. Circ. Res. 110, 739–748. doi: 10.1161/CIRCRESAHA.111.258426
Cavasin, M. A., Stenmark, K. R., and McKinsey, T. A. (2015). Emerging roles for histone deacetylases in pulmonary hypertension and right ventricular remodeling (2013 Grover Conference series). Pulm. Circ. 5, 63–72. doi: 10.1086/679700
Chen, D., Yang, Y., Cheng, X., Fang, F., Xu, G., Yuan, Z., et al. (2015). Megakaryocytic leukemia 1 directs a histone H3 lysine 4 methyltransferase complex to regulate hypoxic pulmonary hypertension. Hypertension 65, 821–833. doi: 10.1161/HYPERTENSIONAHA.114.04585
Chen, F., Li, X., Aquadro, E., Haigh, S., Zhou, J., Stepp, D. W., et al. (2016). Inhibition of histone deacetylase reduces transcription of NADPH oxidases and ROS production and ameliorates pulmonary arterial hypertension. Free Radic. Biol. Med. 99, 167–178. doi: 10.1016/j.freeradbiomed.2016.08.003
Chen, T., Zhou, G., Zhou, Q., Tang, H., Ibe, J. C., Cheng, H., et al. (2015). Loss of microRNA-17~92 in smooth muscle cells attenuates experimental pulmonary hypertension via induction of PDZ and LIM domain 5. Am. J. Respir. Crit. Care Med. 191, 678–692. doi: 10.1164/rccm.201405-0941OC
Cheng, X., and Blumenthal, R. M. (2010). Coordinated chromatin control: structural and functional linkage of DNA and histone methylation. Biochemistry 49, 2999–3008. doi: 10.1021/bi100213t
Clark, R. H., Thomas, P., and Peabody, J. (2003). Extrauterine growth restriction remains a serious problem in prematurely born neonates. Pediatrics 111(5 Pt 1), 986–990. doi: 10.1542/peds.111.5.986
Courboulin, A., Paulin, R., Perreault, T., Meloche, J., Paquet, E. R., Biardel, S., et al. (2011). Role for miR-204 in human pulmonary arterial hypertension. J. Exp. Med. 208, 535–548. doi: 10.1084/jem.20101812
Dickinson, H., Moss, T. J., Gatford, K. L., Moritz, K. M., Akison, L., Fullston, T., et al. (2016). A review of fundamental principles for animal models of DOHaD research: an Australian perspective. J. Dev. Orig. Health Dis. 7, 449. doi: 10.1017/S2040174416000477
Eberharter, A., and Becker, P. B. (2002). Histone acetylation: a switch between repressive and permissive chromatin. Second in review series on chromatin dynamics. EMBO Rep. 3, 224–229. doi: 10.1093/embo-reports/kvf053
Edwards, W. D. (2004). Pathology of pulmonary hypertension. Clin. Chest Med. 22, 223–247. doi: 10.1016/j.ccm.2006.11.010
Eltzschig, H. K., Karhausen, J., and Kempf, V. A. (2005). Acute oxygen-sensing mechanisms. N. Engl. J. Med. 353, 2042–2055. doi: 10.1056/NEJMra050002
Feng, A., Wang, L., Chen, X., Liu, X., Li, L., Wang, B., et al. (2015). Developmental Origins of Health and Disease (DOHaD): implications for health and nutritional issues among rural children in China. Biosci. Trends 9:82. doi: 10.5582/bst.2015.01008
Frid, M. G., Kale, V. A., and Stenmark, K. R. (2002). Mature vascular endothelium can give rise to smooth muscle cells via endothelial-mesenchymal transdifferentiation: in vitro analysis. Circ. Res. 90, 1189–1196. doi: 10.1161/01.RES.0000021432.70309.28
Gambaryan, N., Meng, C., Humbert, M., Adcock, I., and Wort, S. (2013). H3K27 histone lysine methylation as potential therapeutic target in pulmonary arterial hypertension. Eur. Respir. J. 42:P5157. Available online at: http://erj.ersjournals.com/content/42/Suppl_57/P5157
Grant, J. S., Morecroft, I., Dempsie, Y., Van Rooij, E., Maclean, M. R., and Baker, A. H. (2013). Transient but not genetic loss of miR-451 attenuates the development of pulmonary arterial hypertension. Pulm. Circ. 3, 840–850. doi: 10.1086/674751
Greco, S., Gorospe, M., and Martelli, F. (2015). Noncoding RNA in age-related cardiovascular diseases. J. Mol. Cell. Cardiol. 83, 142–155. doi: 10.1016/j.yjmcc.2015.01.011
Gridley, T. (2007). Notch signaling in vascular development and physiology. Development 134, 2709–2718. doi: 10.1242/dev.004184
Gou, D., Ramachandran, R., Peng, X., Yao, L., Kang, K., Sarkar, J., et al. (2012). miR-210 has an antiapoptotic effect in pulmonary artery smooth muscle cells during hypoxia. Am. J. Physiol. Lung Cell. Mol. Physiol. 303, 682–691. doi: 10.1152/ajplung.00344.2011
Hautefort, A., Chesne, J., Preussner, J., Pullamsetti, S. S., Tost, J., Looso, M., et al. (2017). Pulmonary endothelial cell DNA methylation signature in pulmonary arterial hypertension. Oncotarget 8, 52995–53016. doi: 10.18632/oncotarget.18031
Herceg, Z. (2016). Epigenetic mechanisms as an interface between the environment and genome. Adv. Exp. Med. Biol. 903, 3–15. doi: 10.1007/978-1-4899-7678-9_1
Hyduk, A., Croft, J. B., Ayala, C., Zheng, K., Zheng, Z. J., and Mensah, G. A. (2005). Pulmonary hypertension surveillance–United States, 1980-2002. MMWR Surveill. Summ. 54, 1–28. Available online at: https://www.cdc.gov/mmwr/preview/mmwrhtml/ss5405a1.htm
Jalali, S., Ramanathan, G. K., Parthasarathy, P. T., Aljubran, S., Galam, L., Yunus, A., et al. (2012). Mir-206 regulates pulmonary artery smooth muscle cell proliferation and differentiation. PLoS ONE 7:e46808. doi: 10.1371/journal.pone.0046808
Johar, D., Siragam, V., Mahood, T. H., and Keijzer, R. (2015). New insights into lung development and diseases: the role of microRNAs. Biochem. Cell Biol. 93, 139–148. doi: 10.1139/bcb-2014-0103
Jones, P. A., and Takai, D. (2001). The role of DNA methylation in mammalian epigenetics. Science 293, 1068–1070. doi: 10.1126/science.1063852
Kang, B. Y., Park, K. K., Green, D. E., Bijli, K. M., Searles, C. D., Sutliff, R. L., et al. (2013). Hypoxia mediates mutual repression between microRNA-27a and PPARγ in the pulmonary vasculature. PLoS ONE 8:e79503. doi: 10.1371/journal.pone.0079503
Kang, K., Peng, X., Zhang, X., Wang, Y., Zhang, L., Gao, L., et al. (2013). MicroRNA-124 suppresses the transactivation of nuclear factor of activated T cells by targeting multiple genes and inhibits the proliferation of pulmonary artery smooth muscle cells. J. Biol. Chem. 288, 25414–25427. doi: 10.1074/jbc.M113.460287
Kapakos, G., Bouallegue, A., Daou, G. B., and Srivastava, A. K. (2010). Modulatory role of nitric oxide/cGMP system in endothelin-1-induced signaling responses in vascular smooth muscle cells. Curr. Cardiol. Rev. 6, 247–254. doi: 10.2174/157340310793566055
Kim, G. H., Ryan, J. J., Marsboom, G., and Archer, S. L. (2011). Epigenetic mechanisms of pulmonary hypertension. Pulm. Circ. 1, 347–356. doi: 10.4103/2045-8932.87300
Kim, J. (2014). Apelin-APJ signaling: a potential therapeutic target for pulmonary arterial hypertension. Mol. Cells 37, 196–201. doi: 10.14348/molcells.2014.2308
Kim, J., Hwangbo, C., Hu, X., Kang, Y., Papangeli, I., Mehrotra, D., et al. (2015). Restoration of impaired endothelial myocyte enhancer factor 2 function rescues pulmonary arterial hypertension. Circulation 131, 190–199. doi: 10.1161/CIRCULATIONAHA.114.013339
Kim, J., Kang, Y., Kojima, Y., Lighthouse, J. K., Hu, X., Aldred, M. A., et al. (2013). An endothelial apelin-FGF link mediated by miR-424 and miR-503 is disrupted in pulmonary arterial hypertension. Nat. Med. 19, 74–82. doi: 10.1038/nm.3040
Kogo, R., Shimamura, T., Mimori, K., Kawahara, K., Imoto, S., Sudo, T., et al. (2011). Long noncoding RNA HOTAIR regulates polycomb-dependent chromatin modification and is associated with poor prognosis in colorectal cancers. Cancer Res. 71, 6320–6326. doi: 10.1158/0008-5472.CAN-11-1021
Krause, B. J., Costello, P. M., Muñozurrutia, E., Lillycrop, K. A., Hanson, M. A., and Casanello, P. (2013). Role of DNA methyltransferase 1 on the altered eNOS expression in human umbilical endothelium from intrauterine growth restricted fetuses. Epigenetics 8, 944–952. doi: 10.4161/epi.25579
Kubota, T., Miyake, K., Hariya, N., and Mochizuki, K. (2015). Understanding the epigenetics of neurodevelopmental disorders and DOHaD. J. Dev. Orig. Health Dis. 6, 1–9. doi: 10.1017/S2040174415000057
Kume, T. (2009). Novel insights into the differential functions of Notch ligands in vascular formation. Vasc. Cell 1, 1–9. doi: 10.1186/2040-2384-1-8
Lai, E. C. (2002). Micro RNAs are complementary to 3′ UTR sequence motifs that mediate negative post-transcriptional regulation. Nat. Genet. 30, 363–364. doi: 10.1038/ng865
Lee, H. W., and Park, S. H. (2017). Elevated microRNA-135a is associated with pulmonary arterial hypertension in experimental mouse model. Oncotarget 8, 35609–35618. doi: 10.18632/oncotarget.16011
Leung, A., Trac, C., Jin, W., Lanting, L., Akbany, A., Sætrom, P., et al. (2013). Novel long noncoding RNAs are regulated by angiotensin II in vascular smooth muscle cells. Circ. Res. 113, 266–278. doi: 10.1161/CIRCRESAHA.112.300849
Li, E. (2002). Chromatin modification and epigenetic reprogramming in mammalian development. Nat. Rev. Genet. 3, 662–673. doi: 10.1038/nrg887
Li, S., Ran, Y., Zhang, D., Chen, J., Li, S., and Zhu, D. (2013). MicroRNA-138 plays a role in hypoxic pulmonary vascular remodelling by targeting Mst1. Biochem. J. 452, 281–291. doi: 10.1042/BJ20120680
Li, S. S., Ran, Y. J., Zhang, D. D., Li, S. Z., and Zhu, D. (2014). MicroRNA-190 regulates hypoxic pulmonary vasoconstriction by targeting a voltage-gated K? channel in arterial smooth muscle cells. J. Cell. Biochem. 115, 1196–1205. doi: 10.1002/jcb.24771
Limbourg, F. P., Takeshita, K., Radtke, F., Bronson, R. T., Chin, M. T., and Liao, J. K. (2005). Essential role of endothelial notch1 in angiogenesis. Circulation 111, 1826–1832. doi: 10.1161/01.CIR.0000160870.93058.DD
Ling, H., Vincent, K., Pichler, M., Fodde, R., Berindanneagoe, I., Slack, F. J., et al. (2015). Junk DNA and the long non-coding RNA twist in cancer genetics. Oncogene 34, 5003–5011. doi: 10.1038/onc.2014.456
Liu, H., Yin, T., Yan, W., Si, R., Wang, B., Chen, M., et al. (2017). Dysregulation of microRNA-214 and PTEN contributes to the pathogenesis of hypoxic pulmonary hypertension. Int. J. Chron. Obstruct. Pulmon. Dis. 12, 1781–1791. doi: 10.2147/COPD.S104627
Ma, C., Zhang, C., Ma, M., Zhang, L., Zhang, L., Zhang, F., et al. (2017). MiR-125a regulates mitochondrial homeostasis through targeting mitofusin 1 to control hypoxic pulmonary vascular remodeling. J. Mol. Med. 95, 977–993. doi: 10.1007/s00109-017-1541-5
Madonna, R., Caterina, R. D., and Geng, Y. J. (2015). Epigenetic regulation of insulin-like growth factor signaling: a novel insight into the pathophysiology of neonatal pulmonary hypertension. Vascul. Pharmacol. 73, 4–7. doi: 10.1016/j.vph.2015.08.002
Marks, P. A., and Xu, W. S. (2009). Histone deacetylase inhibitors: potential in cancer therapy. J. Cell. Biochem. 107, 600–608. doi: 10.1002/jcb.22185
Mittal, M., Roth, M., Konig, P., Hofmann, S., Dony, E., Goyal, P., et al. (2007). Hypoxia-dependent regulation of nonphagocytic NADPH oxidase subunit NOX4 in the pulmonary vasculature. Circ. Res. 101, 258–267. doi: 10.1161/CIRCRESAHA.107.148015
Morty, R. E., Nejman, B., Kwapiszewska, G., Hecker, M., Zakrzewicz, A., Kouri, F. M., et al. (2007). Dysregulated bone morphogenetic protein signaling in monocrotaline-induced pulmonary arterial hypertension. Arterioscler. Thromb. Vasc. Biol. 27, 1072–1078. doi: 10.1161/ATVBAHA.107.141200
Ngo, V. M., Laverriere, J. N., and Gourdji, D. (1995). CpG methylation represses the activity of the rat prolactin promoter in rat GH3 pituitary cell lines. Mol. Cell. Endocrinol. 108, 95–105. doi: 10.1016/0303-7207(94)03462-3
Nguyen, C. T., Weisenberger, D. J., Velicescu, M., Gonzales, F. A., Lin, J. C., Liang, G., et al. (2002). Histone H3-lysine 9 methylation is associated with aberrant gene silencing in cancer cells and is rapidly reversed by 5-aza-2′-deoxycytidine. Cancer Res. 62, 6456–6461.
Nie, X., Chen, Y., Tan, J., Dai, Y., Mao, W., Qin, G., et al. (2017). MicroRNA-221-3p promotes pulmonary artery smooth muscle cells proliferation by targeting AXIN2 during pulmonary arterial hypertension. Vascul. Pharmacol. doi: 10.1016/j.vph.2017.07.002. [Epub ahead of print].
Ogorodnikova, N., and Arenz, C. (2015). MicroRNA-145-targeted drug and its preventive effect on pulmonary arterial hypertension (patent WO2012153135 A1). Expert Opin. Ther. Pat. 25, 1–5. doi: 10.1517/13543776.2015.1025751
Ortizespejo, M., and Mercedes, G. C. (2013). Nutritional assessment in neonatal and prepubertal children with a history of extrauterine growth restriction. Early Hum. Dev. 89, 763–768. doi: 10.1016/j.earlhumdev.2013.06.003
Osmond, C., Barker, D. J., Winter, P. D., Fall, C. H., and Simmonds, S. J. (2011). Early growth and death from cardiovascular disease in women. BMJ 307, 1519–1524. doi: 10.1136/bmj.307.6918.1519
Ota, T., Suzuki, Y., Nishikawa, T., Otsuki, T., Sugiyama, T., Irie, R., et al. (2004). Complete sequencing and characterization of 21,243 full-length human cDNAs. Nat. Genet. 36, 40–45. doi: 10.1038/ng1285
Paulin, R., Dromparis, P., Sutendra, G., Gurtu, V., Zervopoulos, S., Bowers, L., et al. (2014). Sirtuin 3 deficiency is associated with inhibited mitochondrial function and pulmonary arterial hypertension in rodents and humans. Cell Metab. 20, 827–839. doi: 10.1016/j.cmet.2014.08.011
Paulsen, M., and Fergusonsmith, A. C. (2001). DNA methylation in genomic imprinting, development, and disease. J. Pathol. 195, 97–110. doi: 10.1002/path.890
Perros, F., Cohen-Kaminsky, S., Gambaryan, N., Girerd, B., Raymond, N., Klingelschmitt, I., et al. (2013). Cytotoxic cells and granulysin in pulmonary arterial hypertension and pulmonary veno-occlusive disease. Am. J. Respir. Crit. Care Med. 187, 189–196. doi: 10.1164/rccm.201208-1364OC
Polo, S. E., and Almouzni, G. (2005). Histone metabolic pathways and chromatin assembly factors as proliferation markers. Cancer Lett. 220, 1–9. doi: 10.1016/j.canlet.2004.08.024
Ponting, C. P., Oliver, P. L., and Reik, W. (2009). Evolution and functions of long noncoding RNAs. Cell 136, 629–641. doi: 10.1016/j.cell.2009.02.006
Pullamsetti, S. S., Doebele, C., Fischer, A., Savai, R., Kojonazarov, B., Dahal, B. K., et al. (2012). Inhibition of microRNA-17 improves lung and heart function in experimental pulmonary hypertension. Am. J. Respir. Crit. Care Med. 185, 409–419. doi: 10.1164/rccm.201106-1093OC
Qian, Z., Zhang, L., Chen, J., Li, Y., Kang, K., Qu, J., et al. (2016). MiR-328 targeting PIM-1 inhibits proliferation and migration of pulmonary arterial smooth muscle cells in PDGFBB signaling pathway. Oncotarget 7, 54998–55011. doi: 10.18632/oncotarget.10714
Ropero, S., and Esteller, M. (2007). The role of histone deacetylases (HDACs) in human cancer. Mol. Oncol. 1, 19–25. doi: 10.1016/j.molonc.2007.01.001
Saco, T. V., Parthasarathy, P. T., Cho, Y., Lockey, R. F., and Kolliputi, N. (2014). Role of epigenetics in pulmonary hypertension. Am. J. Physiol. Cell Physiol. 306, 1101–1105. doi: 10.1152/ajpcell.00314.2013
Sanoudou, D., Tousoulis, D., and Cokkinos, D. V. (2012). The role of microRNAs in cardiovascular disease. Curr. Med. Chem. 19, 2605–2610. doi: 10.2174/092986712800493048
Sarkar, J., Gou, D., Turaka, P., Viktorova, E., Ramchandran, R., and Raj, J. U. (2010). MicroRNA-21 plays a role in hypoxia-mediated pulmonary artery smooth muscle cell proliferation and migration. Am. J. Physiol. Lung Cell. Mol. Physiol. 299, 861–871. doi: 10.1152/ajplung.00201.2010
Schlosser, K., Hanson, J., Villeneuve, P. J., Dimitroulakos, J., McIntyre, L., Pilote, L., et al. (2016). Assessment of circulating LncRNAs under physiologic and pathologic conditions in humans reveals potential limitations as biomarkers. Sci. Rep. 6:36596. doi: 10.1038/srep36596
Shan, F., Li, J., and Huang, Q. Y. (2014). HIF-1 alpha-induced up-regulation of miR-9 contributes to phenotypic modulation in pulmonary artery smooth muscle cells during hypoxia. J. Cell. Physiol. 229, 1511–1520. doi: 10.1002/jcp.24593
Sharma, S., Umar, S., Potus, F., Iorga, A., Wong, G., Meriwether, D., et al. (2014). Apolipoprotein A-I mimetic peptide 4F rescues pulmonary hypertension by inducing microRNA-193-3p. Circulation 130, 776–785. doi: 10.1161/CIRCULATIONAHA.114.007405
Sheehan, D. W., Giese, E. C., Gugino, S. F., and Russell, J. A. (1993). Characterization and mechanisms of H2O2-induced contractions of pulmonary arteries. Am. J. Physiol. 264(5 Pt 2), H1542–H1547. doi: 10.1152/ajpheart.1993.264.5.H1542
Shiojima, I., and Walsh, K. (2002). Role of Akt signaling in vascular homeostasis and angiogenesis. Circ. Res. 90, 1243–1250. doi: 10.1161/01.RES.0000022200.71892.9F
Simonneau, G., Gatzoulis, M. A., Adatia, I., Celermajer, D., Denton, C., Ghofrani, A., et al. (2013). Updated clinical classification of pulmonary hypertension. J. Am. Coll. Cardiol. 62, D34–D41. doi: 10.1016/j.jacc.2013.10.029
Singh, D. K., and Prasanth, K. V. (2013). Functional insights into the role of nuclear-retained long noncoding RNAs in gene expression control in mammalian cells. Chromosome Res. 21, 695–711. doi: 10.1007/s10577-013-9391-7
Smith, Z. D., and Meissner, A. (2013). DNA methylation: roles in mammalian development. Nat. Rev. Genet. 14, 204–220. doi: 10.1038/nrg3354
Song, G. U., Guanghui, L. I., Zhang, X., Yan, J., Gao, J., Xiangguang, A. N., et al. (2015). Aberrant expression of long noncoding RNAs in chronic thromboembolic pulmonary hypertension. Mol. Med. Rep. 11, 2631–2643. doi: 10.3892/mmr.2014.3102
Szostakwegierek, D., and Szamotulska, K. (2011). Fetal development and risk of cardiovascular diseases and diabetes type 2 in adult life. Med. Wieku Rozwoj. 15, 203–215.
Taichman, D. B., Loomes, K. M., Schachtner, S. K., Guttentag, S., Vu, C., Williams, P., et al. (2002). Notch1 and Jagged1 expression by the developing pulmonary vasculature. Dev. Dyn. 225, 166–175. doi: 10.1002/dvdy.10146
Takahashi, H., Goto, N., Kojima, Y., Tsuda, Y., Morio, Y., Muramatsu, M., et al. (2006). Downregulation of type II bone morphogenetic protein receptor in hypoxic pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 290, 450–458. doi: 10.1152/ajplung.00206.2005
Takeshita, K., Satoh, M., Ii, M., Silver, M., Limbourg, F. P., Mukai, Y., et al. (2007). Critical role of endothelial Notch1 signaling in postnatal angiogenesis. Circ. Res. 100, 70–78. doi: 10.1161/01.RES.0000254788.47304.6e
Tang, L. L., Zhang, L. Y., Lao, L. J., Hu, Q. Y., Gu, W. Z., Fu, L. C., et al. (2015). Epigenetics of Notch1 regulation in pulmonary microvascular rarefaction following extrauterine growth restriction. Respir. Res. 16, 1–10. doi: 10.1186/s12931-015-0226-2
Tanzer, A., and Stadler, P. F. (2004). Molecular evolution of a microRNA cluster. J. Mol. Biol. 339, 327–335. doi: 10.1016/j.jmb.2004.03.065
Tare, M., Parkington, H. C., Bubb, K. J., and Wlodek, M. E. (2012). Uteroplacental insufficiency and lactational environment separately influence arterial stiffness and vascular function in adult male rats. Hypertension 60, 378–386. doi: 10.1161/HYPERTENSIONAHA.112.190876
Tuder, R. M., Archer, S. L., Dorfmüller, P., Erzurum, S. C., Guignabert, C., Michelakis, E., et al. (2013). Relevant issues in the pathology and pathobiology of pulmonary hypertension. J. Am. Coll. Cardiol. 62, 4–12. doi: 10.1016/j.jacc.2013.10.025
Tupy, J. L., Bailey, A. M., Dailey, G., Evans-Holm, M., Siebel, C. W., Misra, S., et al. (2005). Identification of putative noncoding polyadenylated transcripts in Drosophila melanogaster. Proc. Natl. Acad. Sci. U.S.A. 102, 5495–5500. doi: 10.1073/pnas.0501422102
Vencken, S. F., Greene, C. M., and McKiernan, P. J. (2014). Non-coding RNA as lung disease biomarkers. Thorax 70, 501–503. doi: 10.1136/thoraxjnl-2014-206193
Verdone, L., Caserta, M., and Di Mauro, E. (2005). Role of histone acetylation in the control of gene expression. Biochem. Cell Biol. 83, 344–353. doi: 10.1139/o05-041
Vickers, M. H., Breier, B. H., Cutfield, W. S., Hofman, P. L., and Gluckman, P. D. (2000). Fetal origins of hyperphagia, obesity, and hypertension and postnatal amplification by hypercaloric nutrition. Am. J. Physiol. Endocrinol. Metab. 279:E83. doi: 10.1152/ajpendo.2000.279.1.E83
Vidanes, G. M., Bonilla, C. Y., and Toczyski, D. P. (2005). Complicated tails: histone modifications and the DNA damage response. Cell 121, 973–976. doi: 10.1016/j.cell.2005.06.013
Volinia, S., Galasso, M., Costinean, S., Tagliavini, L., Gamberoni, G., Drusco, A., et al. (2010). Reprogramming of miRNA networks in cancer and leukemia. Genome Res. 20, 589–599. doi: 10.1101/gr.098046.109
Wang, D., Zhang, H., Li, M., Frid, M. G., Flockton, A. R., McKeon, B. A., et al. (2014). MicroRNA-124 controls the proliferative, migratory, and inflammatory phenotype of pulmonary vascular fibroblasts. Circ. Res. 114, 67–78. doi: 10.1161/CIRCRESAHA.114.301633
Wang, X., Chao, Y., Xu, X., Dong, L., Hua, S., Hu, Y., et al. (2015). Long noncoding RNA expression profiles of hypoxic pulmonary hypertension rat model. Gene 579, 23–28. doi: 10.1016/j.gene.2015.12.044
Wapinski, O., and Chang, H. Y. (2011). Long noncoding RNAs and human disease. Trends Cell Biol. 21, 354–361. doi: 10.1016/j.tcb.2011.04.001
Weber, M., Hellmann, I., Stadler, M. B., Ramos, L., Pääbo, S., Rebhan, M., et al. (2007). Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat. Genet. 39, 457–466. doi: 10.1038/ng1990
Weinberg, M. S., and Morris, K. V. (2013). Long non-coding RNA targeting and transcriptional de-repression. Nucleic Acid Ther. 23, 9–14. doi: 10.1089/nat.2012.0412
Wilusz, J. E., Sunwoo, H., and Spector, D. L. (2009). Long noncoding RNAs: functional surprises from the RNA world. Genes Dev. 23, 1494–1504. doi: 10.1101/gad.1800909
Woods, K. A., Camachohübner, C., Savage, M. O., and Clark, A. J. L. (1996). Intrauterine growth retardation and postnatal growth failure associated with deletion of the insulin-like growth factor I gene. N. Engl. J. Med. 335, 1363–1367. doi: 10.1056/NEJM199610313351805
Wu, G., Bazer, F. W., Wallace, J. M., and Spencer, T. E. (2006). Board-invited review: intrauterine growth retardation: implications for the animal sciences. J. Anim. Sci. 84, 2316–2337. doi: 10.2527/jas.2006-156
Wu, H., and Zhang, Y. (2014). Reversing DNA methylation: mechanisms, genomics, and biological functions. Cell 156, 45–68. doi: 10.1016/j.cell.2013.12.019
Wu, T. P., Wang, T., Seetin, M. G., Lai, Y., Zhu, S., Lin, K., et al. (2016). DNA methylation on N6-adenine in mammalian embryonic stem cells. Nature 532, 329–333. doi: 10.1038/nature17640
Xu, W. S., Parmigiani, R. B., and Marks, P. A. (2007). Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene 26, 5541–5552. doi: 10.1038/sj.onc.1210620
Xu, X. F., Lv, Y., Gu, W. Z., Tang, L. L., Wei, J. K., Zhang, L. Y., et al. (2013). Epigenetics of hypoxic pulmonary arterial hypertension following intrauterine growth retardation rat: epigenetics in PAH following IUGR. Respir. Res. 14:20. doi: 10.1186/1465-9921-14-20
Xu, X. F., Ma, X. L., Shen, Z., Wu, X. L., Cheng, F., and Du, L. Z. (2010). Epigenetic regulation of the endothelial nitric oxide synthase gene in persistent pulmonary hypertension of the newborn rat. J. Hypertens. 28, 2227–2235. doi: 10.1097/HJH.0b013e32833e08f1
Xu, X. F., Xu, S. S., Fu, L. C., Hu, Q. Y., Lv, Y., and Du, L. Z. (2016). Epigenetic changes in peripheral leucocytes as biomarkers in intrauterine growth retardation rat. Biomed. Rep. 5, 548–552. doi: 10.3892/br.2016.775
Yamashita, J., Itoh, H., Hirashima, M., Ogawa, M., Nishikawa, S., Yurugi, T., et al. (2000). Flk1-positive cells derived from embryonic stem cells serve as vascular progenitors. Nature 408, 92–96. doi: 10.1038/35040568
Yan, M. S., Matouk, C. C., and Marsden, P. A. (2010). Epigenetics of the vascular endothelium. J. Appl. Physiol. 109, 916–926. doi: 10.1152/japplphysiol.00131.2010
Yang, Q., Lu, Z., Ramchandran, R., Longo, L. D., and Raj, J. U. (2012a). Pulmonary artery smooth muscle cell proliferation and migration in fetal lambs acclimatized to high-altitude long-term hypoxia: role of histone acetylation. Am. J. Physiol. Lung Cell. Mol. Physiol. 303, L1001–L1010. doi: 10.1152/ajplung.00092.2012
Yang, Q., Lu, Z., Singh, D., and Raj, J. U. (2012b). BIX-01294 treatment blocks cell proliferation, migration and contractility in ovine foetal pulmonary arterial smooth muscle cells. Cell Prolif. 45, 335–344. doi: 10.1111/j.1365-2184.2012.00828.x
Yang, Q., Sun, M., Ramchandran, R., and Raj, J. U. (2015). IGF-1 signaling in neonatal hypoxia-induced pulmonary hypertension: role of epigenetic regulation. Vascul. Pharmacol. 127, 43–60. doi: 10.1016/j.vph.2015.04.005
Zentner, G. E., and Henikoff, S. (2013). Regulation of nucleosome dynamics by histone modifications. Nat. Struct. Mol. Biol. 20, 259–266. doi: 10.1038/nsmb.2470
Zhang, L., Tang, L., Wei, J., Lao, L., Gu, W., Hu, Q., et al. (2014). Extrauterine growth restriction on pulmonary vascular endothelial dysfunction in adult male rats: the role of epigenetic mechanisms. J. Hypertens. 32, 2188–2198. doi: 10.1097/HJH.0000000000000309
Zhao, L., Chen, C. N., Hajji, N., Oliver, E., Cotroneo, E., Wharton, J., et al. (2012). Histone deacetylation inhibition in pulmonary hypertension: therapeutic potential of valproic acid and suberoylanilide hydroxamic acid. Circulation 126, 455–467. doi: 10.1161/CIRCULATIONAHA.112.103176
Zhou, V. W., Goren, A., and Bernstein, B. E. (2010). Charting histone modifications and the functional organization of mammalian genomes. Nat. Rev. Genet. 12, 7–18. doi: 10.1038/nrg2905
Keywords: epigenetics, pulmonary hypertension, DNA methylation, histone modifications, noncoding RNAs, IUGR, EUGR
Citation: Wang Y, Yan L, Zhang Z, Prado E, Fu L, Xu X and Du L (2018) Epigenetic Regulation and Its Therapeutic Potential in Pulmonary Hypertension. Front. Pharmacol. 9:241. doi: 10.3389/fphar.2018.00241
Received: 20 September 2017; Accepted: 05 March 2018;
Published: 20 March 2018.
Edited by:
Nadia Mores, Università Cattolica del Sacro Cuore, ItalyReviewed by:
Arunava Roy, University of South Florida, United StatesCopyright © 2018 Wang, Yan, Zhang, Prado, Fu, Xu and Du. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lizhong Du, ZHVsaXpob25nQHpqdS5lZHUuY24=
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.