
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol. , 17 April 2018
Sec. Ethnopharmacology
Volume 9 - 2018 | https://doi.org/10.3389/fphar.2018.00180
 Yuyang Du1,2†
Yuyang Du1,2† Min Wang1,2†
Min Wang1,2† Xuesong Liu3
Xuesong Liu3 Jingyi Zhang1,2Xudong Xu1,2Huibo Xu4*Guibo Sun1,2*
Jingyi Zhang1,2Xudong Xu1,2Huibo Xu4*Guibo Sun1,2* Xiaobo Sun1,2*
Xiaobo Sun1,2*Araloside C (AsC) is a cardioprotective triterpenoid compound that is mainly isolated from Aralia elata. This study aims to determine the effects of AsC on hypoxia-reoxygenation (H/R)-induced apoptosis in H9c2 cardiomyocytes and its underlying mechanisms. Results demonstrated that pretreatment with AsC (12.5 μM) for 12 h significantly suppressed the H/R injury in H9c2 cardiomyocytes, including improving cell viability, attenuating the LDH leakage and preventing cardiomyocyte apoptosis. AsC also inhibited H/R-induced ER stress by reducing the activation of ER stress pathways (PERK/eIF2α and ATF6), and decreasing the expression of ER stress-related apoptotic proteins (CHOP and caspase-12). Moreover, AsC greatly improved the expression level of HSP90 compared with that in the H/R group. The use of HSP90 inhibitor 17-AAG and HSP90 siRNA blocked the above suppression effect of AsC on ER stress-related apoptosis caused by H/R. Taken together, AsC could reduce H/R-induced apoptosis possibly because it attenuates ER stress-dependent apoptotic pathways by increasing HSP90 expression.
According to the report released by the World Health Organization (2017), ischemic heart disease is the world’s biggest killers in 2015 worldwide, and it has remained the leading causes of death globally in the last 15 years. The most effective therapeutic intervention for reducing acute myocardial ischemic injury and limiting the size of myocardial infarction is timely and effective myocardial reperfusion using either thrombolytic therapy or primary percutaneous coronary intervention. However, myocardial reperfusion may itself contribute to further tissue damage, known as myocardial reperfusion injury (Hausenloy and Yellon, 2013; Binder et al., 2015). Molecular and cellular events underlying myocardial I/R injury are complex; nevertheless, scholars have provided increasing lines of evidence regarding the critical role of ER stress in pathological I/R injury process (Qi et al., 2007; Yu et al., 2016; Zhang et al., 2017).
Proper synthesis and correct folding of proteins in the ER are important for normal heart functions. During myocardial I/R, changes in cellular energy levels, redox state and Ca2+ concentration can disturb the ER protein-folding capacity and cause accumulation of unfolded proteins in the ER lumen; this condition, also called ER stress, activates UPR (Martindale et al., 2006; Gorman et al., 2012). The UPR is mediated through three transmembrane signal proteins: PERK, IRE1 and ATF6, which are inactivated by the ER chaperone GRP78. Once the unfolded proteins accumulating in the ER attract GRP78, the sensors start their signaling cascades (Bertolotti et al., 2000). The initial intent of the UPR is to adapt to the changing environment and re-establish normal ER function. However, under prolonged or excessive stress, the UPR can trigger pro-apoptotic signals, including CHOP, caspase-12 and JNK-dependent pathways (Gorman et al., 2012). Attenuation of ER stress induced apoptosis can protect the heart against I/R injury (Zheng et al., 2015; Gao et al., 2016; Yu et al., 2016), and inhibition of ER stress through certain proteins or signal pathways may represent a novel therapeutic mechanism. For example, modulating SERCA activity by activating of PI3K/Akt signaling may effectively suppress ER stress during I/R injury (Guo et al., 2013). Enhancing cardiac SIRT1 signaling by diallyl trisulfide can suppress PERK/eIF2α/ATF4/CHOP-mediated ER stress level, thereby reducing myocardial apoptosis and eventually preserving cardiac function (Yu et al., 2017). Upregulation of calpain-1 is sufficient to induce ER stress and apoptosis in cardiomyocytes, and inhibition of calpain-1 prevents ER stress and apoptotic cell death in H/R-stimulated cardiomyocytes (Zheng et al., 2015). Therefore, regulating ER stress to attenuate ER stress-induced apoptosis may become the focus of evolving strategies to ameliorate myocardial I/R injury.
HSP90, which accounts for approximately 1% of the total soluble proteins in resting mammalian cells (Latchman, 2001), is an ATP-dependent molecular chaperone responsible for managing protein folding and quality control in the crowded environment of the cell. HSP90 mediates cardioprotective effect by attenuating IRE1 activity in the ER through interacting with HAX-1 (Lam et al., 2013), implying its involvement in regulating UPR. Besides, researches also indicated the protection of HSP90 against myocardial I/R injury. Overexpression of HSP90 could attenuate cell apoptosis (Zhu et al., 2016) and reduce infarct size and myocardial dysfunction (Kupatt et al., 2004). Inhibition of HSP90 function or expression completely suppresses the protection of hypoxic preconditioning (Jiao et al., 2008). Therefore, modulation of HSP90 may be a plausible strategy for regulating the ER stress and alleviate the I/R injury.
Aralia elata (Miq.) Seem, a shrub belonging to the family Araliaceae, is mainly distributed in Northeast China, Far East Russia, Korea, and Japan (Shikov et al., 2016); this plant has been traditionally used in China as a tonic and medicine for the treatment of rheumatoid arthritis, diabetes and hepatitis (Zhang et al., 2013; Shikov et al., 2016). The main pharmacologically active ingredient in A. elata are saponins(Xu et al., 1997). Both in vivo and in vitro studies have shown that the total saponin extracted from A. elata can improve cardiac function (Wang et al., 2014b) and protect against myocardial ischemic (Sun et al., 2006), arrhythmia (Maslov and Guzarova, 2007; Maslov et al., 2009) and diabetic cardiomyopathy (Xi et al., 2009). AsC (Figure 2A) is one of the most abundant triterpenoid compounds isolated from the bark and root of A. elata. Our group previously reported that AsC can improve heart function following I/R injury possibly by binding to the HSP90 protein (Wang et al., 2017a). However, the further cardioprotective properties and whether and how HSP90 is involved in the protective effects of AsC are still unknown.
This study aims to determine: (1) whether AsC exerts protective effects on myocardial hypoxia-reperfusion (H/R) injury; (2) the effect of AsC on H/R-induced ER stress; and (3) the potential role of HSP90 and its modulation of ER stress in the cardioprotective effect of AsC.
Araloside C was synthesized as previously reported (Wang et al., 2017a) at the Institute of Medicinal Plant Development (Beijing, China). Tunicamycin from Streptomyces was purchased from Sigma (St. Louis, MO, United States), and the 4-PBA (CAS.NO. 1821-12-1) was purchased from Sinopharm Chemical Reagent Co., Ltd (Beijing, China). All cell culture materials were from GIBCO (Grand Island, NY, United States). DMSO, MTT and 17-AAG were the products of Sigma Chemical Co. (St. Louis, MO, United States). The kits for determining LDH was obtained from Jiancheng Bioengineering Institute (Nanjing, China). The JC-1 was obtained from Beyotime Biotechnology Inc. (Beijing, China). The Alexa Fluor® 488 annexinV/Dead Cell Apoptosis Kit, HSP90 siRNA and Lipofectamine® RNAiMAX Reagent were acquired from Invitrogen (Carlsbad, CA, United States). The caspase-3 activity kit was purchased from BioVision, Inc. (Mountain View, CA, United States). RIPA lysis buffer, Protease Inhibitor Cocktail, BCA Protein Assay Kit, and Enhanced Chemiluminescence Western Blot Detection Kits were supplied by CWbiotech (Beijing, China). Primary antibodies against PARP, HSP90 and IRE1 were obtained from Abcam (Cambridge, United Kingdom). Primary antibodies against caspase-9, caspase-3, caspase-12, p-eIF2α and eIF2α were obtained from Cell Signaling Technology Inc. (Danvers, MA, United States). All other antibodies were purchased from Santa Cruz Biotechnology (Dallas, TX, United States). All chemical reagents were of at least analytical grade.
Rat embryonic cardiomyoblast-derived H9c2 cardiomyocytes were obtained from the Chinese Academy of Sciences Cell Bank (Shanghai, China) and cultured in high glucose DMEM supplemented with 10% (v/v) fetal bovine serum, 1% penicillin/streptomycin (v/v), and 2 mM L-glutamine. The cells were maintained in a humidified incubator with 95% air and 5% CO2 at 37°C. For all experiments, cells were plated at an appropriate density according to the experimental design and grown for 24 h to reach 70–80% confluence before experimentations began.
The H/R model was built using a modified process(Sun et al., 2013). Briefly, H9c2 cardiomyocytes were incubated at 37°C in an anaerobic glove box (Coy Laboratory, United States), where normal air was replaced by a combination of 5% CO2, 5% H2, and 90% N2, with the high glucose DMEM medium replaced by no-glucose DMEM to mimic ischemia. The cells were cultured under hypoxia for 6 h, and then removed to the regular incubator with the medium replaced by high glucose medium and were maintained for 12 h to mimic reperfusion.
Cultured H9c2 cardiomyocytes were randomly divided into different groups. In the control group, H9c2 cardiomyocytes were incubated in high glucose DMEM under normoxic conditions for equivalent durations. The H/R group was conducted as described in the preceding section. When employing TM to cause cell damage, H9c2 cardiomyocytes were incubated with TM (1 μg/ml) for 24 h. In the AsC- treated group (AsC+ H/R or AsC+ TM), H9c2 cardiomyocytes were treated with AsC (12.5 μM) for 12 h before H/R or TM. In 4-PBA treated group (PBA+ TM), the cells were incubated with 4-PBA (0.5 mM) for 12 h before exposed to TM. The inhibitor-treated groups (AAG+ AsC+ H/R) were processed similar to the AsC+ H/R group, but the cells were incubated with 0.1 μM 17-AAG for 1 h before treated with AsC.
The groups were clustered into three categories for experiment: the first category includes Control, H/R, H/R+ AsC and AsC groups; the second category includes Control, TM, AsC+ TM, and PBA+ TM groups; and the third category includes the Control, H/R, H/R+ AsC and AAG +AsC+ H/R groups.
Cell viability was determined by MTT assay. Briefly, H9c2 cardiomyocytes were plated on 96-well plates at a density of 1 × 104 cells/well. After designated treatment, 20 μL of MTT (5 mg/mL) was added to each well and cells were incubated at 37°C for 4 h. Then, the culture medium with MTT was removed and the colored formazan crystals were dissolved in 150 μL of DMSO. Absorption was measured at 570 nm using a microplate reader (TECAN Infinite M1000, Austria). The viability of H9c2 cells in each well was presented as percentage of control cells.
H9c2 cardiomyocytes were cultured at 2 × 105 cells/well in 6- well plates for 24 h. After H/R treatment with or without AsC pretreatment, the medium was collected to measure LDH release using a LDH assay kits.
The change in mitochondrial membrane potential was detected by JC-1 staining. After treatment, the cells were incubated with JC-1 (2 μM final concentration) at 37°C in the dark for 30 min. Then the cells were washed three times with PBS and observed using fluorescence microscopy (EVOS® FL Color, Life Technologies). For further analysis of the fluorescence intensity, the treated cells were harvested and incubated with JC-1 at 37°C in the dark for 30 min. The fluorescence intensity changes were analyzed using a microplate reader (TECAN Infinite M1000, Austria). The green JC-1 signal was detected at excitation wavelengths of 514 nm and emission wavelengths of 529 nm, the red signal was detected at excitation wavelengths of 585 nm and emission wavelengths of 590 nm.
The percentage of early Apoptosis rate was determined using the Annexin V/PI assay kit according to the manufacturer’s instruction. After treatment, the cells were harvested, washed with cold PBS, and incubated with 5 μL FITC-Annexin V and 1 μL PI working solution (100 μg/mL) in the dark for 15 min at room temperature. Cellular fluorescence was determined using a FACS Calibur flow cytometer (BD Biosciences, CA, United States). The early apoptosis rate was expressed as the ratio of Annexin V-positive/PI-negative cells to total cells.
The activities of caspase-3 in H9c2 cardiomyocytes were measured using a fluorometric assay kit. Fluorescence intensity was detected using a microplate reader at excitation and emission wavelengths of 485 and 535 nm, respectively. The results are described as fold changes compared with the control group.
H9c2 cardiomyocytes were cultured in 6-well plates. When 50% confluence was reached, the cells were transfected with HSP90 siRNA (50 nM) or equivalent concentrations of control siRNA diluting by Lipofectamine RNAiMAX. The cells were transfected for 24h and incubated with 12.5 μM AsC for 12 h, followed with H/R treatment.
Protein expression was analyzed by western blot as described previously (Sun et al., 2013). After treatment, H9c2 cardiomyocytes were harvested and lysed with cell lysis buffer containing 1% protease inhibitor cocktail. The lysate was centrifuged for 20 min at 12,000 g and 4°C to remove insoluble materials. Equal amounts of protein from each sample were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a nitrocellulose membrane (Millipore Corporation, United States). After blocked with 5% (w/v) non-fat milk powder in Tris-buffer that contains 0.05% (v/v) Tween-20 (TBST) at room temperature for at least 2 h, the membranes were incubated overnight at 4°C with appropriate primary antibodies. Then the membrane was washed three times with TBST, incubated with secondary antibody at a 1: 2,000 dilution for 2 h, and washed with TBST again. Finally, the blots were visualized by enhanced chemiluminescence using a BioRad imaging system (Bio-Rad, Hercules, CA, United States). Densitometric analysis of the bands was performed using Gel Pro software (Media Cybernetics, Rockville, MD, United States).
The results were expressed as mean ± SD. Comparisons between groups were performed by Student’s t-test or one-way ANOVA followed by Newman-Keuls multiple comparison test using Prism 5.00 software (GraphPad Software, La Jolla, CA, United States). A p-value less than 0.5 was considered statistically significant. All experiments were repeated at least three times.
We first examined the effects of reoxygenation time on cell viability and expression of representative apoptotic-related proteins. H9c2 cardiomyocytes were exposed to hypoxia for 6 h and reoxygenation for 0, 2, 6, 12, and 24 h. Cell viability was detected by MTT assay, and the percentage of viable cell in each group was calculated relative to control. As shown in Figure 1A, hypoxia for 6 h decreased the cell viability, and this effect was time-dependently promoted by reoxygenation, which was consistent with the literature report (Yu et al., 2016). We then conducted Western Blot analysis to evaluate the expression of proteins related to apoptosis at different reoxygenation times. The expression of cleaved caspase-3 increased after 6h of reoxygenation and decreased after 12 h; meanwhile, the ratio of Bcl-2 to Bax decreased since hypoxia (Figures 1B,C). The expression levels of CHOP and caspase-12, which are associated with ER stress-induced apoptosis signaling pathways, were rapidly induced after reoxygenation and reached the peak at 6 and 12 h, respectively (Figures 1B,C). These results indicated that ER stress was activated after reoxygenation, and apoptosis was initiated thereafter. After reoxygenation for 12 h, ER stress-induced apoptosis weakened. Therefore, hypoxia for 6 h and reoxygenation for 12 h were chosen as optimal conditions for subsequent experiments.
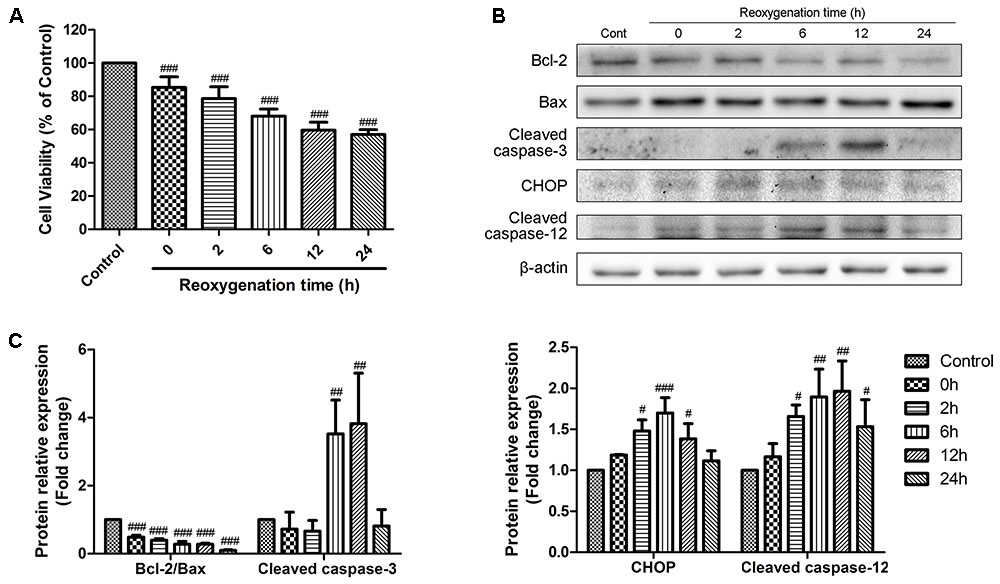
FIGURE 1. Effects of reoxygenation time on H9c2 cardiomyocytes cell viability and apoptotic proteins. H9c2 cardiomyocytes were subjected to 6h of hypoxia and then exposed to different duration (0, 2, 6, 12, 24 h) of reoxygenation. (A) Cell viability was measured by MTT assays. (B,C) The expression of cleaved caspase-3, Bcl-2, Bax, CHOP and cleaved caspase-12 were analyzed by western blotting, with representative bands quantified in the corresponding bar graph. β-actin expression was examined as the protein loading control. The values are expressed as the mean ± SD three independent experiments. #P < 0.05 vs. Control, ##P < 0.01 vs. Control, ###P < 0.001 vs. Control.
Next, we estimated the potential cardioprotective effects of AsC (Figure 2A) on H9c2 cardiomyocytes against H/R injury by using MTT and LDH assays. As shown in Figure 2B, AsC did not demonstrate any cytotoxicity after 12 h of pretreatment with virous concentrations. H9c2 cardiomyocytes exposed to H/R exhibited decreased cell viability compared to control, whereas pretreatment with different concentrations of AsC for 12 h conspicuously increased cell viability in a does-dependent manner (Figure 2C). The total saponins of Aralia elata (AS) (Sun et al., 2006; Wang et al., 2014b) and hydroxysafflor yellow A (HSYA) (Liu et al., 2012; Min and Wei, 2017), which were shown to be protective in myocardial ischemia/reperfusion injury, were used as positive controls here (Supplementary Figure S3). As a biomarker of cell death, LDH leakage was also detected. As shown in Figure 2D, H/R-induced LDH release was significantly decreased when the cells were pre-incubted with AsC (6.25- 12.5 μM) for 12 h.
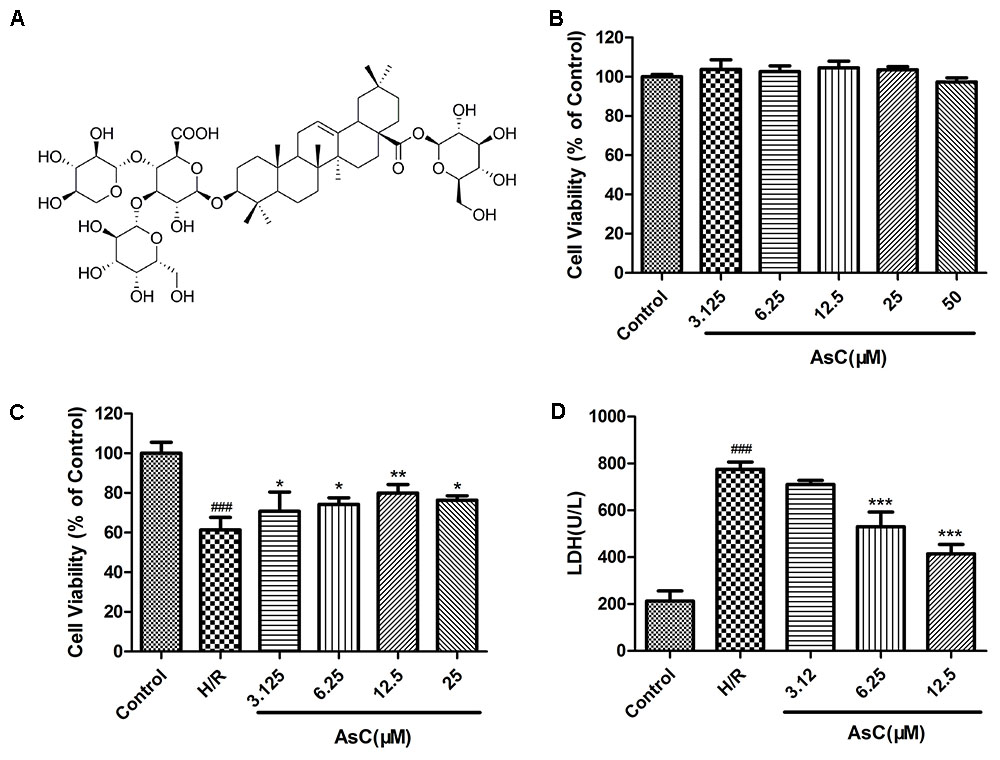
FIGURE 2. Effects of Araloside C on H/R-induced cell injury. (A) The chemical structure of AsC. (B) Cell viability of H9c2 cardiomyocytes incubated with different concentrations of AsC for 12 h. H9c2 cardiomyocytes were incubated with or without AsC for 12 h and then exposed to 6 h of hypoxia and 12 h of reoxygenation. (C) Cell viability was detected by MTT assay. (D) Effects of AsC on H/R-induced LDH leakage. The values are expressed as the mean ± SD three independent experiments. ###P < 0.001 vs. Control; ∗P < 0.05 vs. H/R group, ∗∗P < 0.01 vs. H/R group, ∗∗∗P < 0.001 vs. H/R group.
Mitochondria plays a key role in I/R injury, and the disruption of mitochondrial membrane potential (ΔΨm) is an early event in the apoptotic cascade (Gustafsson and Gottlieb, 2008). Thus, we determined the effect of AsC on ΔΨm by JC-1 staining. The results showed that the H/R-treated group exhibited a significant decrease in the ratio of red/green fluorescence intensity, indicating ΔΨm dissipation; however, this effect was reversed by pretreatment with 12.5 μM AsC for 12 h (Figures 3A,C).
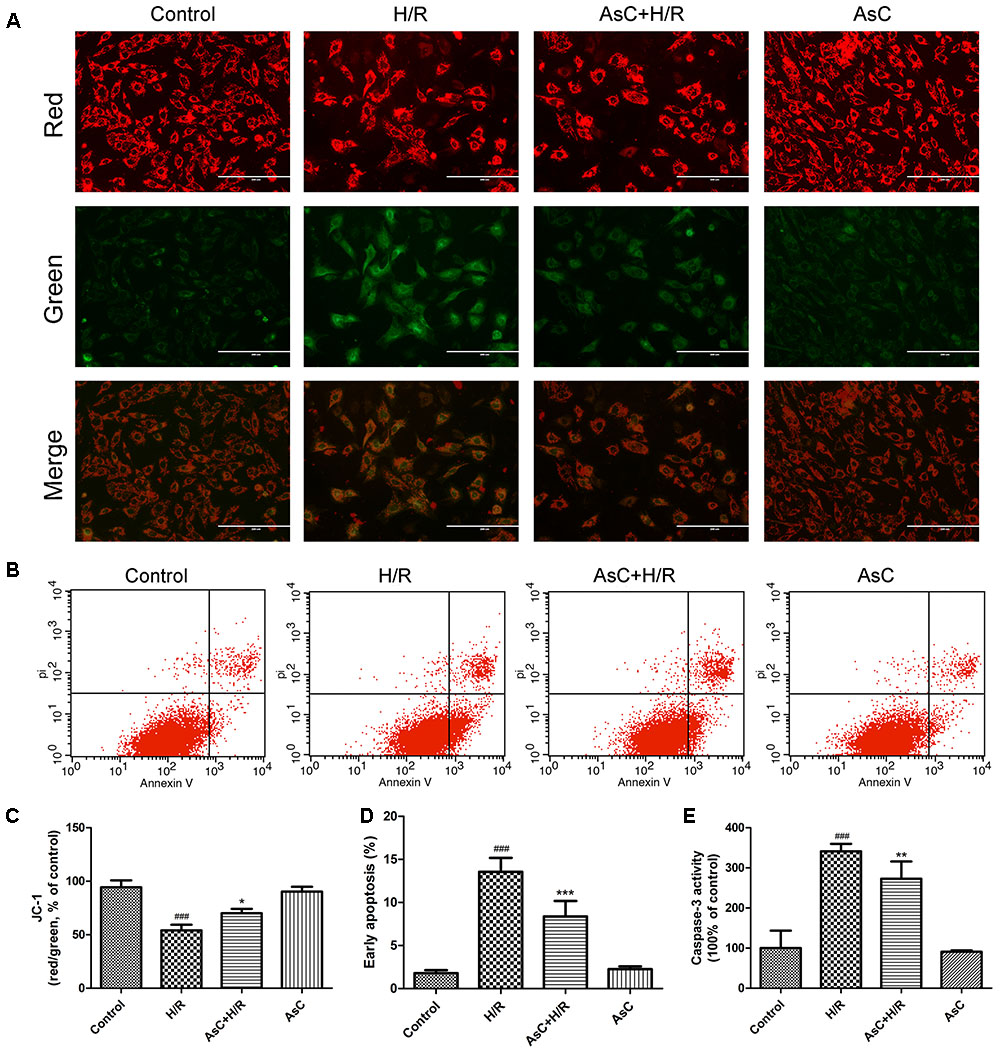
FIGURE 3. Araloside C protected against H/R-induced apoptosis in H9c2 cardiomyocytes. H9c2 cardiomyocytes were incubated with or without AsC for 12 h and then exposed to 6h of hypoxia and 12h of reoxygenation. (A) ΔΨm was assessed by fluorescence microscopy with JC-1 staining. (B) Apoptosis in H9c2 cardiomyocytes was analyzed by flow cytometry. (C) Quantitative analysis of mitochondrial membrane potential (ΔΨm) by plate reader. (D) Quantitative analysis of the percentages of early apoptotic cells. (E) caspase-3 activity was measured using a fluorometric assay. The values are expressed as the mean ± SD three independent experiments. ###P < 0.001 vs. Control; ∗P < 0.05 vs. H/R group, ∗∗P < 0.01 vs. H/R group, ∗∗∗P < 0.001 vs. H/R group.
The Annexin V-FITC/PI staining assay demonstrated that the number of early apoptotic cells significantly increased in H/R-treated H9c2 cardiomyocytes compared with that in the control group, while incubation with 12.5 μM AsC effectively alleviated H/R-induced early apoptosis (Figures 3B,D).
To further confirm the anti-apoptotic effects of AsC, we evaluated the expression of apoptosis-related proteins by western blot. As shown in Figures 4A,B, H/R decreased the ratio of Bcl-2/Bax, increased the expression level of cleaved caspase-9, cleaved caspase-3 and PARP. These changes were reversed by AsC treatment. We also assessed caspase-3 activity by fluorimetric assay. Consistent with the effect on the expression of cleaved caspase-3, AsC pretreatment for 12h remarkably reduced caspase-3 activation induced by H/R (Figure 3E).
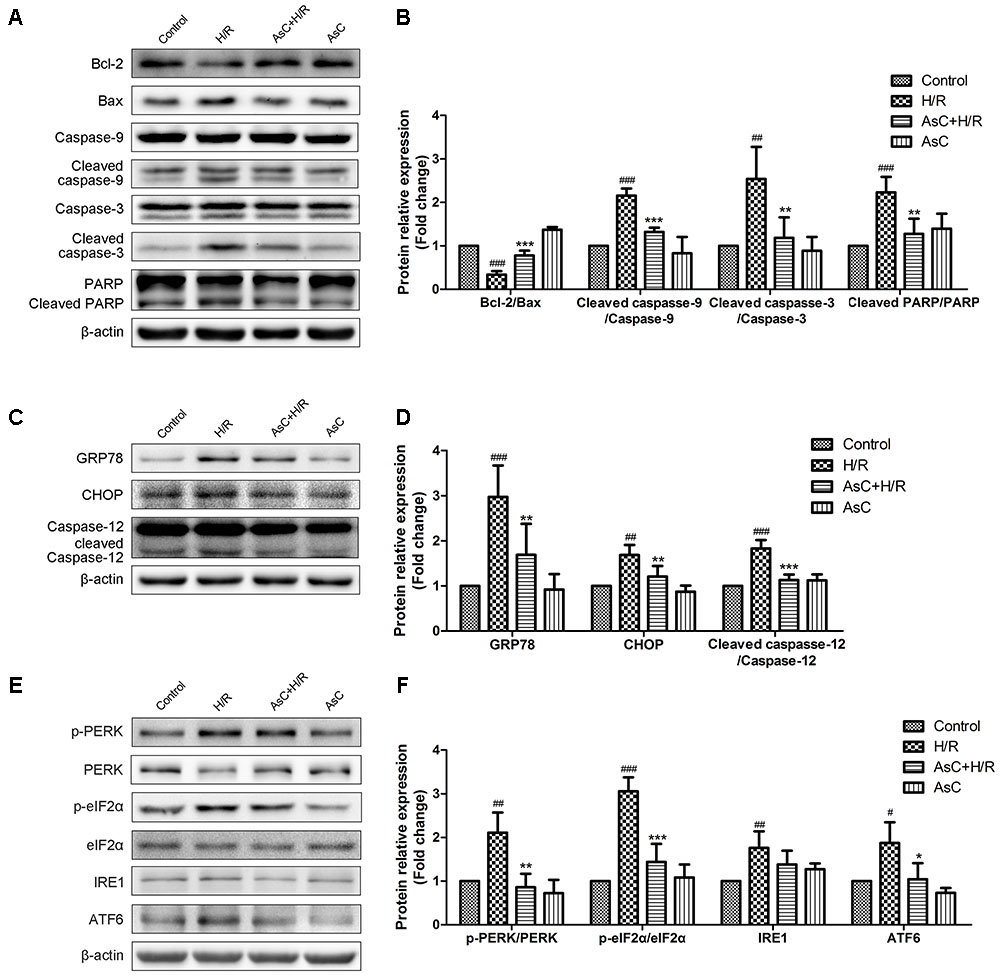
FIGURE 4. Araloside C suppressed H/R-induced apoptosis and ER stress in H9c2 cardiomyocytes. H9c2 cardiomyocytes were incubated with or without AsC for 12 h and then exposed to 6 h of hypoxia and 12 h of reoxygenation. (A,B) The expression of Bcl-2, Bax, caspase-9, caspase-3 and PARP were analyzed by western blotting, with representative bands quantified in the corresponding bar graph. (C,D) The ER stress marker GRP78 and ER stress-related apoptotic proteins (CHOP and caspase-12) were evaluated by western blot analysis. (E,F) The expression level of UPR pathway proteins were detected using western blot analysis and the relative protein expression of p-PERK to PERK, p- eIF2α to eIF2α, IRE1 and ATF6 to β-actin are expressed in the bar graphs. The values are expressed as the mean ± SD three independent experiments. #P < 0.05 vs. Control. ##P < 0.01 vs. Control. ###P < 0.001 vs. Control; ∗P < 0.05 vs. H/R group, ∗∗P < 0.01 vs. H/R group, ∗∗∗P < 0.001 vs. H/R group.
All these results indicated that AsC protected the cardiomyocytes against H/R injury partly by attenuating apoptosis activation.
The ER stress-responsive marker GRP78 and ER stress-dependent apoptotic proteins, namely CHOP and caspasse-12, were evaluated through Western Blot analysis to determine the effect of AsC on H/R-induced ER stress. As shown in Figures 4C,D, pharmacological intervention with 12.5 μM AsC for 12 h significantly suppressed the H/R-induced upregulation of GRP78 and the expression of the pro-apoptotic proteins (CHOP and caspase-12) compared with those in the control.
To further confirm whether or not AsC protected cardiomyocytes by modulating ER stress-dependent apoptosis, we used a typical ER stress inducer tunicamycin (TM) to stimulate ER stress and induce cell damage. We assessed the effect of AsC on TM-induced ER stress by MTT assay and Western Blot analysis with 4-PBA as positive control. As shown in Figure 5, treatment of 1 μg/ml TM for 24 h decreased the cell viability and the ratio of Bcl-2/Bax and promoted the activation of caspase-3, CHOP, caspase-12 and GRP78. Moreover, both AsC and 4-PBA preconditioning greatly reduced TM-induced injury and ER stress-related apoptosis. These results suggested that ER stress participated in H/R-induced injury, and the cardioprotective effect of AsC may partly rely on its suppression on ER stress-related apoptosis.
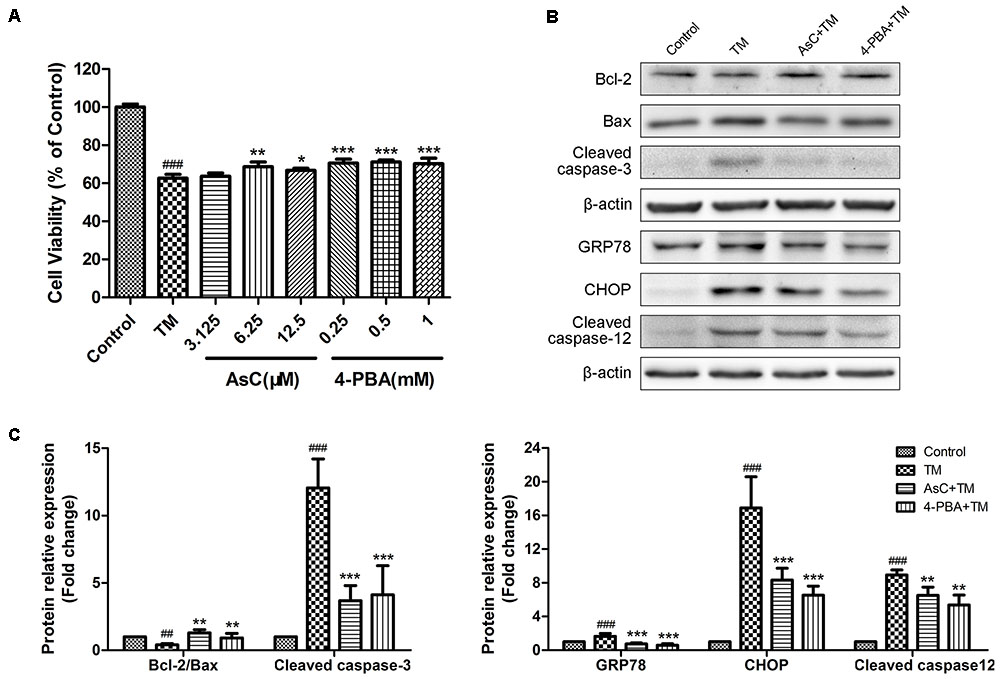
FIGURE 5. Araloside C inhibited TM-induced cell death and ER stress in H9c2 cardiomyocytes. (A) H9c2 cardiomyocytes were incubated with different concentrations of AsC (3.125, 6.25 and 12.5 μM) or 4-PBA (0.25, 0.5, and 1 mM) for 12 h, followed by treatment with TM (1 μg/ml) for another 24 h. Cell viability was detected by MTT assay. (B,C) H9c2 cardiomyocytes pretreated with AsC (12.5 μM) or 4-PBA (1 mM) for 12 h were exposed to TM (1 μg/ml) for 24 h. Representative apoptotic proteins (cleaved caspase-3, Bcl-2 and Bax), as well as ER stress marker GRP78 and ER stress-related apoptotic proteins (CHOP and cleaved caspase-12) were evaluated by western blot analysis. The values are expressed as the mean ± SD three independent experiments. ##P < 0.01 vs. Control. ###P < 0.001 vs. Control; ∗P < 0.05 vs. H/R group, ∗∗P < 0.01 vs. H/R group, ∗∗∗P < 0.001 vs. H/R group.
To investigate the mechanism whereby AsC prevents cell death in response to H/R-induced ER stress, we next evaluated the effects of AsC on the UPR signal in stressed cardiomyocytes. Western blot analysis showed that hypoxia for 6 h and reoxygenation for 12 h activated the three arms of the UPR compared to control. In comparison with H/R treated cells, 12 h pretreatment with 12.5 μM AsC remarkably reduced the phosphorylation of PERK and eIF2α and decreased ATF6 expression, while there was no significant effect on the increased expression of IRE1 (Figures 4E,F).
We first detected the effects of AsC on HSP90 expression levels in H/R-treated H9c2 cardiomyocytes. Consistent with our previous reports (Wang et al., 2017a), AsC treatment significantly inhibited the H/R-induced down-regulation of HSP90 expression (Figures 6A,B).
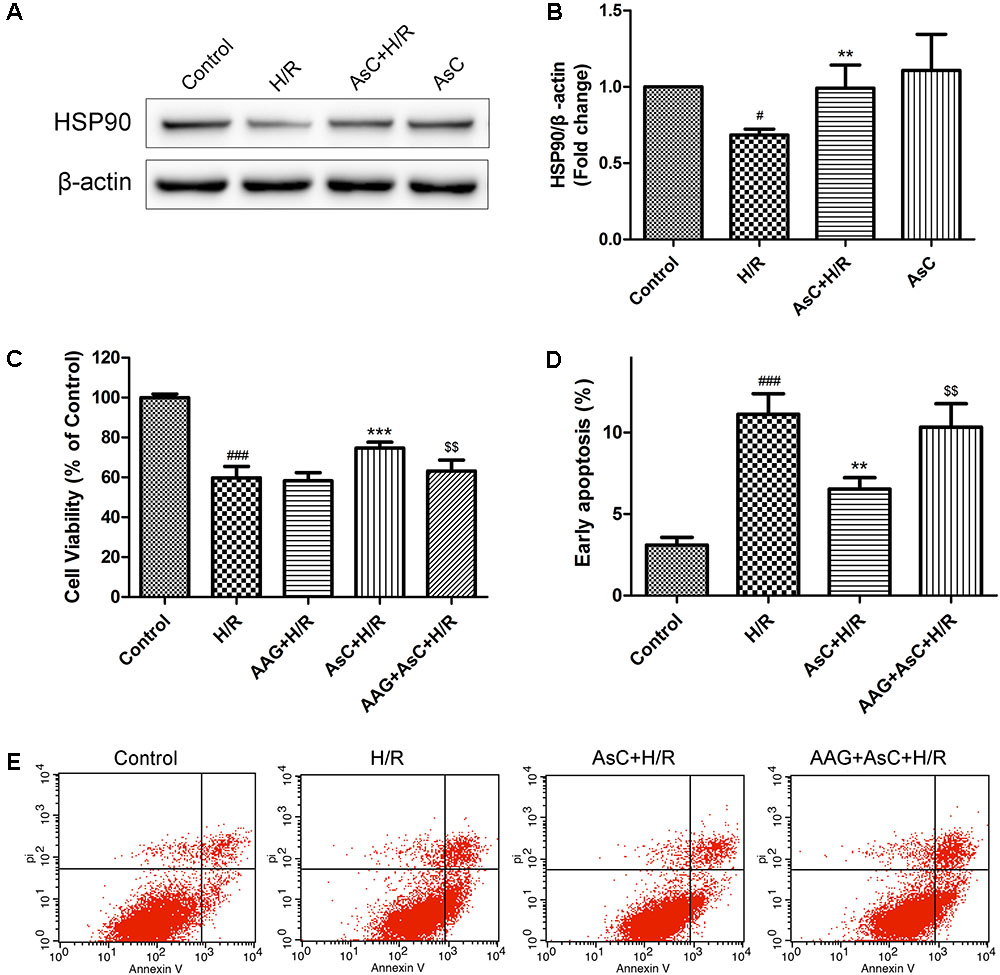
FIGURE 6. Araloside C protected H9c2 cardiomyocytes against H/R-induced cell death by increasing the expression of HSP90. (A,B) H9c2 cardiomyocytes were incubated with or without AsC for 12 h and then exposed to 6h of hypoxia and 12 h of reoxygenation. The expression of HSP90 were analyzed by western blotting. Then 17-AAG was used to inhibited HSP90 in order to explore the involvement of HSP90 in the cardioprotective effect of AsC. H9c2 cardiomyocytes were incubated with 17-AAG (0.1 μM) for 1 h before treated with AsC. (C) Cell viability was detected by MTT assay. (D) Quantitative analysis of the percentages of early apoptotic cells. (E) Apoptosis in H9c2 cardiomyocytes was analyzed by flow cytometry. The values are expressed as the mean ± SD three independent experiments. #P < 0.05 vs. Control, ###P < 0.001 vs. Control; ∗∗P < 0.01 vs. H/R group, ∗∗∗P < 0.001 vs. H/R group; $$P < 0.01 vs. AsC+ H/R.
Then we utilized 17-AAG, a selective inhibitor of HSP90, to further investigate the contribution of the elevated expression of HSP90 to AsC-mediated protection against H/R-induced ER stress and apoptosis. Compared with that in the H/R group, pretreatment with AsC increased the cell viability and the ratio of Bcl-2/Bax, but significantly reduced the rate of early apoptosis and the expression level of cleaved caspase-9 and caspase-3. However, these effects were reversed by pretreatment with 0.1 μM 17-AAG for 1 h (Figures 6C–E, 7A,B).
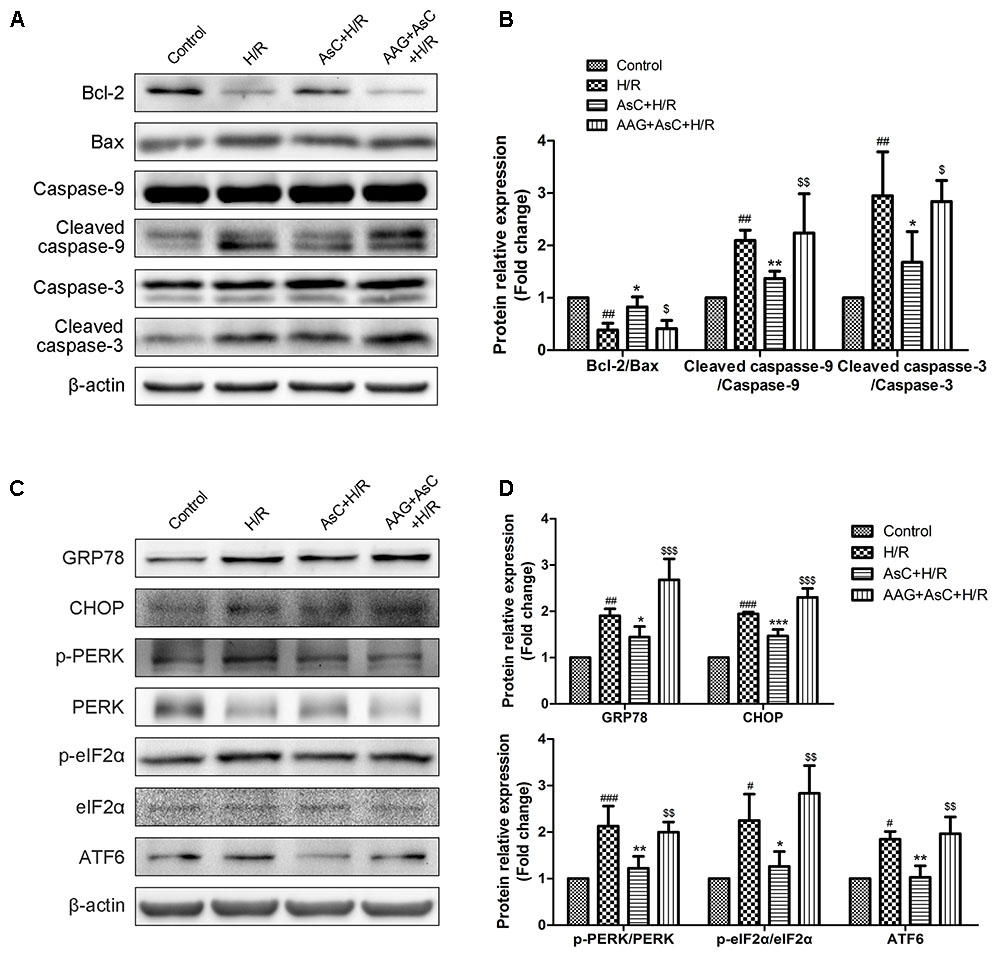
FIGURE 7. Inhibition of HSP90 by 17-AAG reversed the suppressive effect of AsC on H/R-induced ER stress. H9c2 cardiomyocytes were incubated with 17-AAG (0.1 μM) for 1 h and treated with AsC (12.5 μM) for another 12 h, followed by 6 h of hypoxia and 12 h of reoxygenation. (A,B) Expression of Bcl-2, Bax, caspase-9 and caspase-3 were analyzed by western blotting, with representative bands quantified in the corresponding bar graph. (C,D) ER stress-associated proteins (GRP78, CHOP, p-PERK/PERK, p-eIF2α/eIF2α and ATF6) were detected using western blot analysis, with representative bands quantified in the corresponding bar graph. The values are expressed as the mean ± SD three independent experiments. #P < 0.05 vs. Control, ##P < 0.01 vs. Control, ###P < 0.001 vs. Control; ∗P < 0.05 vs. H/R group, ∗∗P < 0.01 vs. H/R group, ∗∗∗P < 0.001 vs. H/R group; $P < 0.05 vs. AsC+ H/R, $$P < 0.01 vs. AsC+ H/R, $$$P < 0.001 vs. AsC+ H/R.
Additionally, the inhibitive effect of AsC on H/R-induced ER stress was abolished when the H9c2 cardiomyocytes were pretreated with 17-AAG, which were demonstrated by the upregulated protein expression level of p-PERK to PERK, p- eIF2α to eIF2α, CHOP and ATF6 to β-actin in 17-AAG pretreated group compared with those in AsC + H/R group (Figures 7C,D). Consistent with the effect of 17-AAG, interfering the expression of HSP90 with siRNA also reversed the suppressive effect of AsC on H/R-induced ER stress apoptosis. Interestingly, transfected with HSP90 siRNA in H9c2 cardiomyocytes removed the inhibition of AsC on the phosphorylation of PERK and eIF2α, but had no influence on the suppressive effect on ATF6 (Figure 8). Therefore, AsC inhibited H/R-induced over-activation of ER stress by increasing the expression level of HSP90.
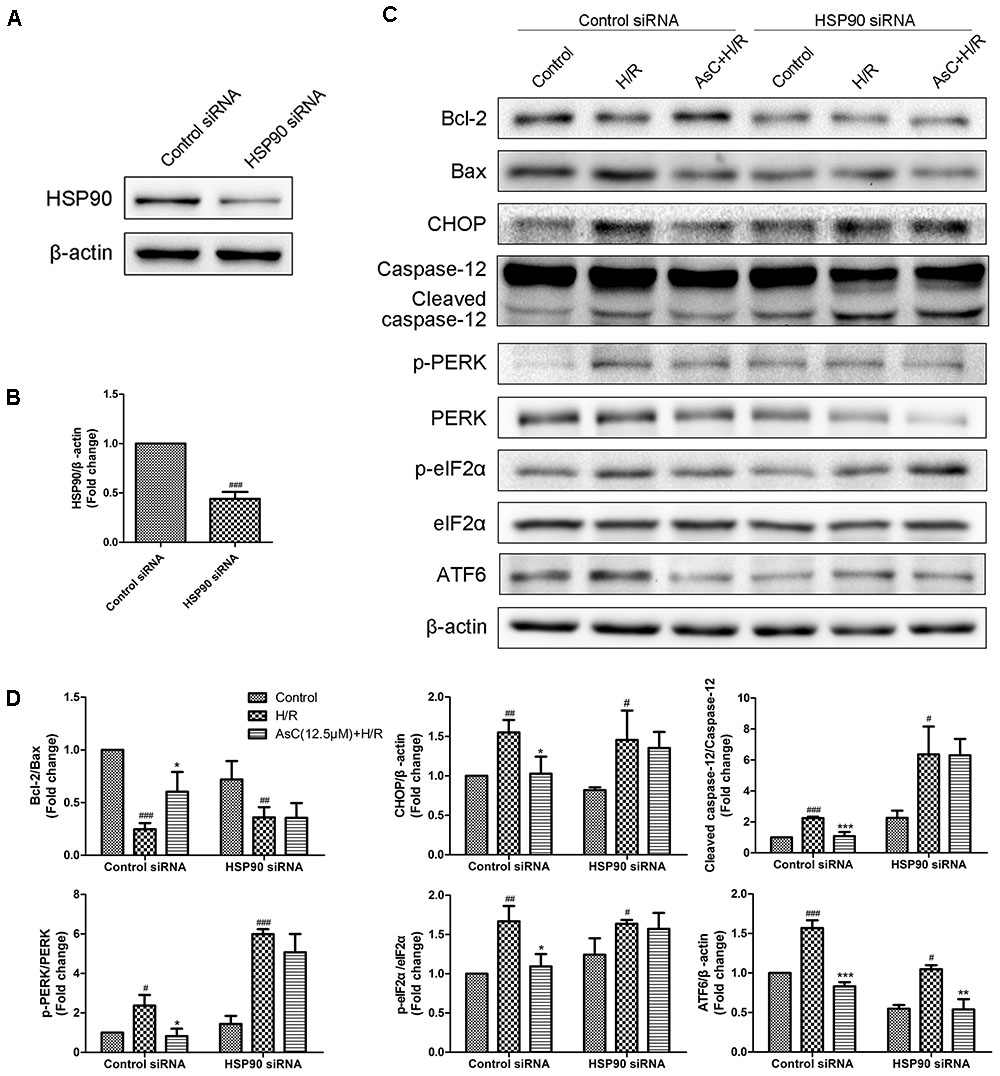
FIGURE 8. Interference of HSP90 by siRNA reversed the suppressive effect of AsC on H/R-induced ER stress. (A,B) H9c2 cardiomyocytes were transfected with control siRNA or HSP90 siRNA for 24h. These cells were then harvested, lysed, and analyzed for HSP90 protein expression levels by western blotting. (C,D) H9c2 cardiomyocytes transfected with control siRNA or HSP90 siRNA were incubated with AsC (12.5 μM) for 12 h and then subjected to 6h of hypoxia and 12 h of reoxygenation. The expression levels of ER stress-assoceated proteins were detected using western blot analysis. The relative protein expression of Bcl-2 to Bax, CHOP, caspase-12, ATF6 to β-actin, p-PERK to PERK and p- eIF2α to eIF2α were expressed in the bar graphs. The values are expressed as the mean ± SD three independent experiments. #P < 0.05 vs. Control, ##P < 0.01 vs. Control, ###P < 0.001 vs. Control; ∗P < 0.05 vs. H/R group, ∗∗P < 0.01 vs. H/R group, ∗∗∗P < 0.001 vs. H/R group.
Ischemic myocardial injury occurs under many clinical conditions, such as heart transplantation, cardiac bypass, and coronary stenting after acute myocardial infarction. However, the concept of the so-called “reperfusion injury” has not been totally accepted until recent years (Zhao et al., 2003; Bopassa, 2012). In the present study, we demonstrated that reperfusion for 2–24 h significantly decreased the cell viability and the expression ratio of Bcl-2/Bax compared with reperfusion for 0 h group, namely, ischemic group; reperfusion for 6–12 h promoted the activation of caspase-3 (Figure 1). These findings are consistent with previous studies (Liu et al., 2015) and confirmed the existence of reperfusion injury. Both necrosis and apoptosis were shown to be involved in H/R-induced cell death (Supplementary Figure S1) (McCully et al., 2004; Koshinuma et al., 2014). However, as shown in Supplementary Figure S2, cells in Annexin V positive and PI negative (AV+/PI-, represented early apoptosis) were markedly higher than that in Annexin V positive and PI positive (AV+/PI+, represented late apoptosis and necrosis) after H/R treatment, suggesting that apoptosis plays a critical role in pathogenesis of I/R injury (Eefting et al., 2004; Liu et al., 2015).
An increasing number of studies have suggested that ER stress plays a crucial role in I/R-induced apoptosis of cardiomyocytes (Lam et al., 2013; Zhang et al., 2017). ER stress sensors initiating the UPR are IRE1, ATF6, and PERK which can inhibit general protein translation through the phosphorylation of eIF2α (Harding et al., 2000). Although the UPR is a defense mechanism directed toward cellular adaptation to alleviate the unfolded protein load, prolonged stress is associated with the activation of apoptotic proteins like CHOP and caspase-12 (Gorman et al., 2012). In the present study, the protein level of the ER stress marker GRP78 was significantly increased and the activation of ATF6, IREα, PERK and eIF2α as well as the downstream apoptosis factors CHOP and caspase-12 were promoted in H9c2 cardiomyocytes subjected to H/R, indicating that ER stress was excessively activated and partly contributed to the cell apoptosis during H/R. However, this H/R-induced processing was remarkably suppressed by AsC pretreatment. Interestingly, AsC had no influence on H/R-induced upregulation of IRE1 (Figures 4E,F), implying that AsC might selectively inhibit the PERK and ATF6 branch in the UPR. Some research found that cell death under ER stress depended on the core mitochondrial apoptosis pathway, which is regulated by the Bcl-2 protein family (Tait and Green, 2010). CHOP, which is positively controlled by the PERK- eIF2α-ATF4 axis, promotes both the transcription of BIM and the downregulation of Bcl-2 expression, contributing to the induction of apoptosis (Li et al., 2014). Consistent with previous reports, our study confirmed that the reversal of ER stress by AsC was accompanied by increased Bcl-2/Bax ratio and reduced apoptotic cardiomyocytes (Figures 4A,B).
To further identify the effect of AsC on ER stress, we treated H9c2 cardiomyocytes with tunicamycin (TM), a well characterized ER stress inducing agent that can initiate ER stress-induced apoptosis by preventing glycosylation and protein folding and by inducing prolonged ER stress in treated cells (Wang et al., 2017b). 4-PBA is a chemical chaperone that improves ER folding capacity and facilitating the trafficking of mutant proteins (Jian et al., 2016a). It shows protective effects against ER stress-induced neuronal and myocardial cell death (Mimori et al., 2012; Jian et al., 2016a,b). Hence, 4-PBA was employed here as positive control. Both AsC and 4-PBA significantly ameliorated H/R-induced cell apoptosis and ER stress (Figure 5), which manifested the direct inhibitory effect of AsC on ER stress.
The total saponins are suggested to be the main pharmacological active ingredients of A. elata and exhibit anti-myocardial ischemic, anti-apoptotic and anti-inflammatory properties (Sun et al., 2006; Wang et al., 2014b; Chen et al., 2015). They also the main pharmacodynamic ingredient of Long Ya Guan Xin Kang Jiao Nang (Chinese name), a Chinese pharmaceutical drug developed for the treatment of angina pectoris (Chen et al., 2015). Our laboratory previously found that the total saponins extracted from A. elata and a saponin monomer can inhibit ER stress-related apoptosis and play protective roles in non-alcoholic steatohepatitis (Luo et al., 2015) and I/R injury (Wang et al., 2014a, 2015), respectively. As one of the major triterpenoid compounds isolated from A. elata, AsC can attenuate H/R induced ER stress-dependent apoptosis. Therefore, these results not only partially suggest the underlying mechanism of the cytoprotective effect of AsC, but also provide theoretical supports for the clinical application and further development of A. elata.
HSP90, a well-characterized, conserved and essential chaperone protein in eukaryotes, is shown to interact with the cytoplasmic parts of PERK (Marcu et al., 2002). Agents that has higher affinity, such as HSP90 inhibitors, may destabilize the activity of PERK by displacing them from HSP90 and subsequently regulate the ER stress (Marcu et al., 2002; Lam et al., 2013). A growing body of evidence implicates that HSP90 plays a critical role in the protective effect of myocardial ischemic preconditioning (Jiao et al., 2008; Amour et al., 2009) and against I/R injury (Kupatt et al., 2004; Zhu et al., 2016). Our oratory previously revealed that AsC was able to bind to the HSP90 protein in vitro in a dose-dependent manner with the KD 29 μM (Wang et al., 2017a).The current results confirmed that AsC significantly attenuated the apoptosis and the down-regulation of HSP90 expression induced by H/R H9c2 cardiomyocytes. By contrast, inhibition of HSP90 by 17-AAG, a well-known inhibitor of HSP90, notably blocked the AsC-induced cardioprotective effect against H/R injury (Figures 6C–E). In present study, we also found that 17-AAG and interference of HSP90 by siRNA remarkably reversed the suppression effect of AsC on H/R-induced UPR and ER stress-dependent apoptosis (Figures 7, 8), indicating that the myocardial protective effect of AsC was likely due to its ability to increase HSP90. It was notable that HSP90 siRNA abolished the inhibition effect of AsC on PERK/eIF2α pathway, while it did not affect that on ATF6. Therefore, AsC probably protected against H/R injury through mediating the interaction between HSP90 and PERK. However, further research is needed to investigate and confirm the interaction of AsC with ER stress signal pathways and HSP90.
The following study limitations should be acknowledged. In this study, we showed that AsC can alleviate H/R-induced ER stress-associated cardiomyocyte apoptosis as it can downregulate the expression of CHOP and inhibit caspase-12 cleavage. Caspase-12 is shown to be a ER resident caspase and activated by ER stress. Mice deficient in caspase-12 are resistant to ER-induced apoptosis (Nakagawa et al., 2000). Terai K and collaborators have demonstrated that knockdown of caspase-12 through small interfering RNA (siRNA) resulted in decreased expression of cleaved PARP following exposure to hypoxia (Terai et al., 2005). By contrast to these studies, other work using caspase-12-/- MEFs observed no resistance to ER stressors such as thapsigargin (Saleh et al., 2006). These results suggest that caspase-12 may not be a predominant apoptotic pathway activated by ER stress in some types of cells and the predominant apoptotic pathway may differ among cell type and differentiation stage (Momoi, 2004). Moreover, it has been reported that caspase-12 is phylogenetically one of the inflammatory group caspases (Martinon and Tschopp, 2004). It is generally recognized as a negative regulator of the inflammatory response because it inhibits the activation of caspase-1 in inflammasome complexes, thereby modulating the production of IL-1b and IL-18 (Hermel and Klapstein, 2011). Murine caspase-12 deficiency confers resistance to sepsis and its presence exerts a direct suppressive effect on caspase-1, resulting in enhanced vulnerability to bacterial infection and septic shock (Saleh et al., 2006). In our research, we didn’t use caspase-12 inhibitor to further focus on the role of caspase-12 in I/R mediated apoptosis, so we cannot rule out the possibility that caspase-12 might be involved in inflammatory response in our experiment and the protective effect of AsC against H/R injury might be partially resulted from its inhibition of inflammatory. Further investigations need to be conducted to determine the role of caspase-12, as well as AsC, on H/R-induced inflammatory.
In summary, our data demonstrated that AsC protected against H9c2 cardiomyocytes against H/R injury, at least in part, by alleviating ER stress-associated apoptosis via increasing the expression of the HSP90 protein (Figure 9). It might provide some references for the development of AsC into cardiovascular drugs.
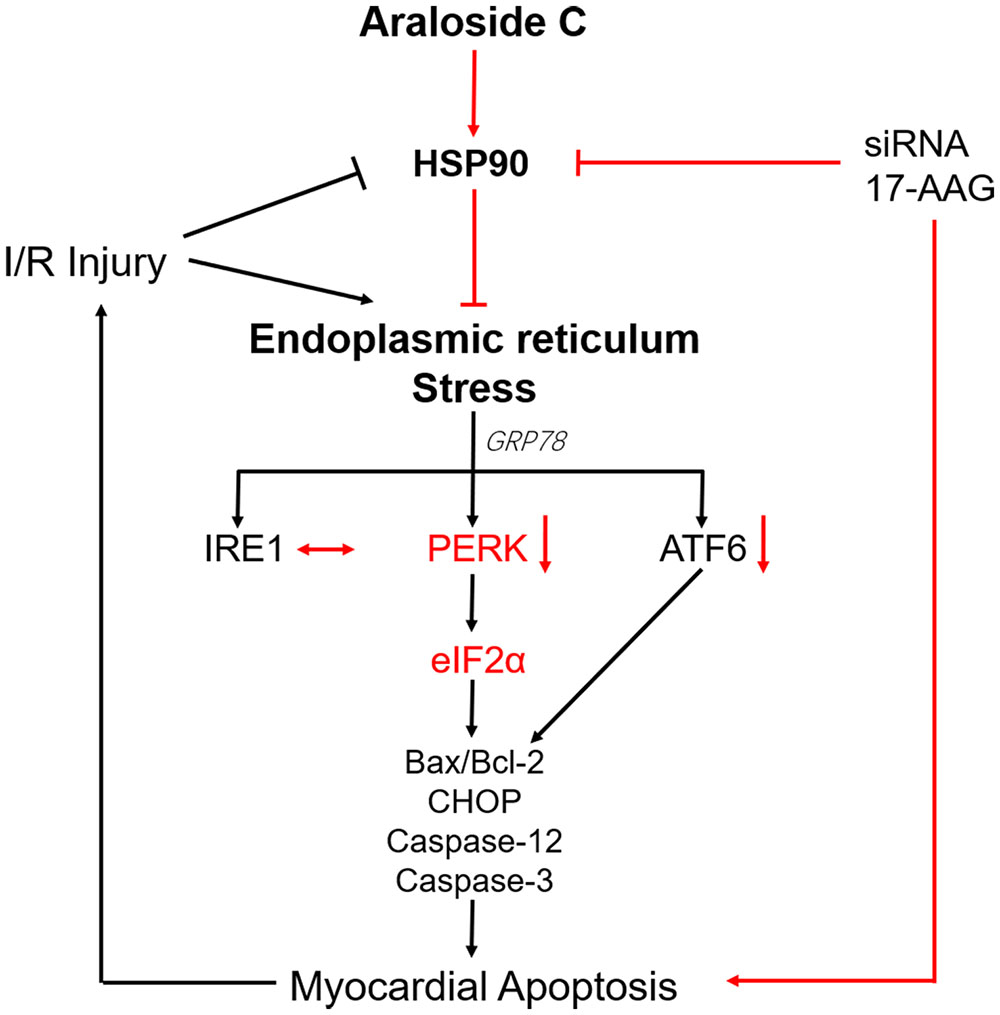
FIGURE 9. Schematic representation of AsC-mediated cardioprotective effect against H/R injury. AsC inhibited ER stress-dependent apoptotic pathways though attenuating PERK/eIF2α signal pathway in ER stress. This effect was mediated by the increase of HSP90 expression. 17-AAG and siRNA abolished the protective effect of AsC against H/R injury.
XS, GS, and HX supervised the project. YD, MW, XL, and JZ designed the detailed experiments, performed the study, and collected and analyzed data. HX and XX contributed reagents and materials. YD and MW wrote or contributed to the writing and revision of the manuscript. All authors commented the study and approved the final manuscript.
This study was supported by the National Natural Science Foundation of China (Grant Nos. 81473380 and 81603333), the Natural Sciences Foundation of Beijing (Grant No. 7152102), the Special Research Project for TCM (Grant No. 201507004), CAMS Innovation Fund for Medical Sciences (CIFMS) (Grant No. 2016-I2M-1-012), and the Program for Innovative Research Team in IMPLAD (Grant No. IT1301).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2018.00180/full#supplementary-material
FIGURE S1 | Effects of zVAD-fmk (zVAD, apoptosis inhibitor) and Necrostatin-1 (Nec-1, necrosis inhibitor) on H/R-induced cell injury. H9c2 cardiomyocytes were treated with different concentrations of zVAD and Nec-1 for 12 h, followed by 6 h of hypoxia and 12 h of reoxygenation. Cell viability was measured by MTT assays. The values are expressed as the mean ± SD four independent experiments. ###P < 0.001 vs. Control; ∗∗∗P < 0.001 vs. H/R group.
FIGURE S2 | Quantitative analysis of the percentages of Annexin V positive and PI negative (AV+/PI-) cells and Annexin V positive and PI positive (AV+/PI+) cells with or without H/R treatment. Annexin V/PI assay kit was used and cellular fluorescence was determined by flow cytometer. The values are expressed as the mean ± SD four independent experiments. ###P < 0.001 vs. Control; ∗∗∗P < 0.001 vs. H/R group in AV+/PI-) cells.
FIGURE S3 | Effects of AsC on H/R-induced cell injury. H9c2 cardiomyocytes were treated with different concentrations of AsC and Nec-1 for 12 h, followed by 6 h of hypoxia and 12 h of reoxygenation. Total saponins of Aralia elata (AS, 12.5 μg/ml) and hydroxysafflor yellow A (HSYA, 3 μM) were used as positive controls. Cell viability was measured by MTT assays. The values are expressed as the mean ± SD four independent experiments. ###P < 0.001 vs. Control; ∗∗P < 0.01 vs. H/R group, ∗∗∗P < 0.001 vs. H/R group.
17-AAG, 17-allylamino-17-demethoxygeldanamycin; 4-PBA, 4-phenylbutyric acid; AsC, Araloside C; ATF6, activating transcription factor 6; CHOP, CCAAT/enhancer binding protein homologous protein; DMSO, Dimethylsulfoxide; eIF2α, eukaryotic translation initiator factor 2α; ER, endoplasmic reticulum; GRP, Glucose Regulated Protein; H/R, hypoxia-reoxygenation; HSP, heat shock protein; I/R, ischemic/reperfusion; IRE1, inositol-requiring enzyme 1; LDH, lactate dehydrogenase; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; PARP, poly ADP-ribose polymerase; PERK, pancreatic ER kinase (PKR)-like ER kinase; PI, propidium iodide; TM, tunicamycin; UPR, unfolded protein respond.
Amour, J., Brzezinska, A. K., Weihrauch, D., Billstrom, A. R., Zielonka, J., Krolikowski, J. G., et al. (2009). Role of heat shock protein 90 and endothelial nitric oxide synthase during early anesthetic and ischemic preconditioning. Anesthesiology 110, 317–325. doi: 10.1097/ALN.0b013e3181942cb4
Bertolotti, A., Zhang, Y., Hendershot, L. M., Harding, H. P., and Ron, D. (2000). Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2, 326–332. doi: 10.1038/35014014
Binder, A., Ali, A., Chawla, R., Aziz, H. A., Abbate, A., and Jovin, I. S. (2015). Myocardial protection from ischemia-reperfusion injury post coronary revascularization. Expert Rev Cardiovasc. Ther. 13, 1045–1057. doi: 10.1586/14779072.2015.1070669
Bopassa, J. C. (2012). Protection of the ischemic myocardium during the reperfusion. Am. J. Cardiovasc. Dis. 2, 223–236.
Chen, R.-C., Wang, J., Yu, Y.-L., Sun, G.-B., and Sun, X.-B. (2015). Protective effect of total saponins of Aralia elata (Miq) Seem on lipopolysaccharide-induced cardiac dysfunction via down-regulation of inflammatory signaling in mice. RSC Adv. 5, 22560–22569. doi: 10.1039/c4ra16353b
Eefting, F., Rensing, B., Wigman, J., Pannekoek, W. J., Liu, W. M., Cramer, M. J., et al. (2004). Role of apoptosis in reperfusion injury. Cardiovasc. Res. 61, 414–426. doi: 10.1016/j.cardiores.2003.12.023
Gao, Y., Jia, P., Shu, W., and Jia, D. (2016). The protective effect of lycopene on hypoxia/reoxygenation-induced endoplasmic reticulum stress in H9C2 cardiomyocytes. Eur. J. Pharmacol. 774, 71–79. doi: 10.1016/j.ejphar.2016.02.005
Gorman, A. M., Healy, S. J., Jager, R., and Samali, A. (2012). Stress management at the ER: regulators of ER stress-induced apoptosis. Pharmacol. Ther. 134, 306–316. doi: 10.1016/j.pharmthera.2012.02.003
Guo, J., Bian, Y., Bai, R., Li, H., Fu, M., and Xiao, C. (2013). Globular adiponectin attenuates myocardial ischemia/reperfusion injury by upregulating endoplasmic reticulum Ca(2)(+)-ATPase activity and inhibiting endoplasmic reticulum stress. J. Cardiovasc. Pharmacol. 62, 143–153. doi: 10.1097/FJC.0b013e31829521af
Gustafsson, A. B., and Gottlieb, R. A. (2008). Heart mitochondria: gates of life and death. Cardiovasc. Res. 77, 334–343. doi: 10.1093/cvr/cvm005
Harding, H. P., Novoa, I., Zhang, Y., Zeng, H., Wek, R., Schapira, M., et al. (2000). Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 6, 1099–1108. doi: 10.1016/S1097-2765(00)00108-8
Hausenloy, D. J., and Yellon, D. M. (2013). Myocardial ischemia-reperfusion injury: a neglected therapeutic target. J. Clin. Invest. 123, 92–100. doi: 10.1172/JCI62874
Hermel, E., and Klapstein, K. D. (2011). A possible mechanism for maintenance of the deleterious allele of human CASPASE-12. Med. Hypotheses 77, 803–806. doi: 10.1016/j.mehy.2011.07.041
Jian, L., Lu, Y., Lu, S., and Lu, C. (2016a). Chemical chaperone 4-phenylbutyric acid protects H9c2 cardiomyocytes from ischemia/reperfusion injury by attenuating endoplasmic reticulum stress-induced apoptosis. Mol. Med. Rep. 13, 4386–4392. doi: 10.3892/mmr.2016.5063
Jian, L., Lu, Y., Lu, S., and Lu, C. (2016b). Chemical chaperone 4-phenylbutyric acid reduces cardiac ischemia/reperfusion injury by alleviating endoplasmic reticulum stress and oxidative stress. Med. Sci. Monit. 22, 5218–5227. doi: 10.12659/msm.898623
Jiao, J. D., Garg, V., Yang, B., and Hu, K. (2008). Novel functional role of heat shock protein 90 in ATP-sensitive K+ channel-mediated hypoxic preconditioning. Cardiovasc. Res. 77, 126–133. doi: 10.1093/cvr/cvm028
Koshinuma, S., Miyamae, M., Kaneda, K., Kotani, J., and Figueredo, V. M. (2014). Combination of necroptosis and apoptosis inhibition enhances cardioprotection against myocardial ischemia-reperfusion injury. J. Anesth. 28, 235–241. doi: 10.1007/s00540-013-1716-3
Kupatt, C., Dessy, C., Hinkel, R., Raake, P., Daneau, G., Bouzin, C., et al. (2004). Heat shock protein 90 transfection reduces ischemia-reperfusion-induced myocardial dysfunction via reciprocal endothelial NO synthase serine 1177 phosphorylation and threonine 495 dephosphorylation. Arterioscler. Thromb. Vasc. Biol. 24, 1435–1441. doi: 10.1161/01.ATV.0000134300.87476.d1
Lam, C. K., Zhao, W., Cai, W., Vafiadaki, E., Florea, S. M., Ren, X., et al. (2013). Novel role of HAX-1 in ischemic injury protection involvement of heat shock protein 90. Circ. Res. 112, 79–89. doi: 10.1161/CIRCRESAHA.112.279935
Latchman, D. S. (2001). Heat shock proteins and cardiac protection. Cardiovasc. Res. 51, 637–646. doi: 10.1016/S0008-6363(01)00354-6
Li, Y., Guo, Y., Tang, J., Jiang, J., and Chen, Z. (2014). New insights into the roles of CHOP-induced apoptosis in ER stress. Acta Biochim. Biophys. Sin. 46, 629–640. doi: 10.1093/abbs/gmu048
Liu, S. X., Zhang, Y., Wang, Y. F., Li, X. C., Xiang, M. X., Bian, C., et al. (2012). Upregulation of heme oxygenase-1 expression by hydroxysafflor yellow A conferring protection from anoxia/reoxygenation-induced apoptosis in H9c2 cardiomyocytes. Int. J. Cardiol. 160, 95–101. doi: 10.1016/j.ijcard.2011.03.033
Liu, X., Zhang, C., Qian, L., Zhang, C., Wu, K., Yang, C., et al. (2015). NF45 inhibits cardiomyocyte apoptosis following myocardial ischemia-reperfusion injury. Pathol. Res. Pract. 211, 955–962. doi: 10.1016/j.prp.2015.09.018
Luo, Y., Dong, X., Yu, Y., Sun, G., and Sun, X. (2015). Total aralosides of Aralia elata (Miq) seem (TASAES) ameliorate nonalcoholic steatohepatitis by modulating IRE1alpha-mediated JNK and NF-kappaB pathways in ApoE-/- mice. J. Ethnopharmacol. 163, 241–250. doi: 10.1016/j.jep.2015.01.017
Marcu, M. G., Doyle, M., Bertolotti, A., Ron, D., Hendershot, L., and Neckers, L. (2002). Heat shock protein 90 modulates the unfolded protein response by stabilizing IRE1. Mol. Cell. Biol. 22, 8506–8513. doi: 10.1128/mcb.22.24.8506-8513.2002
Martindale, J. J., Fernandez, R., Thuerauf, D., Whittaker, R., Gude, N., Sussman, M. A., et al. (2006). Endoplasmic reticulum stress gene induction and protection from ischemia/reperfusion injury in the hearts of transgenic mice with a tamoxifen-regulated form of ATF6. Circ. Res. 98, 1186–1193. doi: 10.1161/01.RES.0000220643.65941.8d
Martinon, F., and Tschopp, J. (2004). Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell 117, 561–574. doi: 10.1016/j.cell.2004.05.004
Maslov, L. N., and Guzarova, N. V. (2007). [Cardioprotective and antiarrhythmic properties of preparations from Leuzea carthamoides, Aralia mandshurica, and Eleutherococcus senticosus]. Eksp. Klin. Farmakol. 70, 48–54.
Maslov, L. N., Lishmanov, Y. B., Arbuzov, A. G., Krylatov, A. V., Budankova, E. V., Konkovskaya, Y. N., et al. (2009). Antiarrhythmic activity of phytoadaptogens in short-term ischemia-reperfusion of the heart and postinfarction cardiosclerosis. Bull. Exp. Biol. Med. 147, 331–334. doi: 10.1007/s10517-009-0502-6
McCully, J. D., Wakiyama, H., Hsieh, Y. J., Jones, M., and Levitsky, S. (2004). Differential contribution of necrosis and apoptosis in myocardial ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 286, H1923–H1935. doi: 10.1152/ajpheart.00935.2003
Mimori, S., Okuma, Y., Kaneko, M., Kawada, K., Hosoi, T., Ozawa, K., et al. (2012). Protective effects of 4-phenylbutyrate derivatives on the neuronal cell death and endoplasmic reticulum stress. Biol. Pharm. Bull. 35, 84–90. doi: 10.1248/bpb.35.84
Min, J., and Wei, C. (2017). Hydroxysafflor yellow A cardioprotection in ischemia-reperfusion (I/R) injury mainly via akt/hexokinase ii independent of ERK/GSK-3β pathway. Biomed. Pharmacother. 87, 419–426. doi: 10.1016/j.biopha.2016.12.113
Momoi, T. (2004). Caspases involved in ER stress-mediated cell death. J. Chem. Neuroanat. 28, 101–105. doi: 10.1016/j.jchemneu.2004.05.008
Nakagawa, T., Zhu, H., Morishima, N., Li, E., Xu, J., Yankner, B. A., et al. (2000). Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 403, 98–103. doi: 10.1038/47513
Qi, X., Vallentin, A., Churchill, E., and Mochly-Rosen, D. (2007). deltaPKC participates in the endoplasmic reticulum stress-induced response in cultured cardiac myocytes and ischemic heart. J. Mol. Cell. Cardiol. 43, 420–428. doi: 10.1016/j.yjmcc.2007.07.061
Saleh, M., Mathison, J. C., Wolinski, M. K., Bensinger, S. J., Fitzgerald, P., Droin, N., et al. (2006). Enhanced bacterial clearance and sepsis resistance in caspase-12-deficient mice. Nature 440, 1064–1068. doi: 10.1038/nature04656
Shikov, A. N., Pozharitskaya, O. N., and Makarov, V. G. (2016). Aralia elata var. mandshurica (Rupr. & Maxim.) J.Wen: an overview of pharmacological studies. Phytomedicine 23, 1409–1421. doi: 10.1016/j.phymed.2016.07.011
Sun, G. B., Xu, H. B., Wen, F. C., Zhang, W., Ding, T., and Sun, X. B. (2006). Protective effects of aralosides on cultured myocardial cells subjected to anoxia/reoxygenation injury. Chin. Pharmacol. Bull. 22, 1092–1095.
Sun, J., Sun, G., Meng, X., Wang, H., Wang, M., Qin, M., et al. (2013). Ginsenoside RK3 prevents hypoxia-reoxygenation induced apoptosis in H9c2 cardiomyocytes via AKT and MAPK pathway. Evid. Based Complement. Alternat. Med. 2013:690190. doi: 10.1155/2013/690190
Tait, S. W., and Green, D. R. (2010). Mitochondria and cell death: outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 11, 621–632. doi: 10.1038/nrm2952
Terai, K., Hiramoto, Y., Masaki, M., Sugiyama, S., Kuroda, T., Hori, M., et al. (2005). AMP-activated protein kinase protects cardiomyocytes against hypoxic injury through attenuation of endoplasmic reticulum stress. Mol. Cell. Biol. 25, 9554–9575. doi: 10.1128/MCB.25.21.9554-9575.2005
Wang, M., Meng, X. B., Yu, Y. L., Sun, G. B., Xu, X. D., Zhang, X. P., et al. (2014a). Elatoside C protects against hypoxia/reoxygenation-induced apoptosis in H9c2 cardiomyocytes through the reduction of endoplasmic reticulum stress partially depending on STAT3 activation. Apoptosis 19, 1727–1735. doi: 10.1007/s10495-014-1039-3
Wang, M., Sun, G. B., Zhang, J. Y., Luo, Y., Yu, Y. L., Xu, X. D., et al. (2015). Elatoside C protects the heart from ischaemia/reperfusion injury through the modulation of oxidative stress and intracellular Ca2+ homeostasis. Int. J. Cardiol. 185, 167–176. doi: 10.1016/j.ijcard.2015.03.140
Wang, M., Tian, Y., Du, Y. Y., Sun, G. B., Xu, X. D., Jiang, H., et al. (2017a). Protective effects of Araloside C against myocardial ischaemia/reperfusion injury: potential involvement of heat shock protein 90. J. Cell Mol. Med. 21, 1870–1880. doi: 10.1111/jcmm.13107
Wang, M., Xu, X., Xu, H., Wen, F., Zhang, X., Sun, H., et al. (2014b). Effect of the total saponins of Aralia elata (Miq) Seem on cardiac contractile function and intracellular calcium cycling regulation. J. Ethnopharmacol. 155, 240–247. doi: 10.1016/j.jep.2014.05.024
Wang, Y., Xuan, L., Cui, X., Wang, Y., Chen, S., Wei, C., et al. (2017b). Ibutilide treatment protects against ER stress induced apoptosis by regulating calumenin expression in tunicamycin treated cardiomyocytes. PLoS One 12:e0173469. doi: 10.1371/journal.pone.0173469
Xi, S., Zhou, G., Zhang, X., Zhang, W., Cai, L., and Zhao, C. (2009). Protective effect of total aralosides of Aralia elata (Miq) Seem (TASAES) against diabetic cardiomyopathy in rats during the early stage, and possible mechanisms. Exp. Mol. Med. 41, 538–547. doi: 10.3858/emm.2009.41.8.059
Xu, X. D., Yang, J. S., and Zhu, Z. Y. (1997). Advances in the studies of saponins from Aralia. Yao Xue Xue Bao 32, 711–720.
Yu, L., Li, S., Tang, X., Li, Z., Zhang, J., Xue, X., et al. (2017). Diallyl trisulfide ameliorates myocardial ischemia-reperfusion injury by reducing oxidative stress and endoplasmic reticulum stress-mediated apoptosis in type 1 diabetic rats: role of SIRT1 activation. Apoptosis 22, 942–954. doi: 10.1007/s10495-017-1378-y
Yu, Y., Sun, G., Luo, Y., Wang, M., Chen, R., Zhang, J., et al. (2016). Cardioprotective effects of Notoginsenoside R1 against ischemia/reperfusion injuries by regulating oxidative stress- and endoplasmic reticulum stress- related signaling pathways. Sci. Rep. 6:21730. doi: 10.1038/srep21730
Zhang, C., Tang, Y., Li, Y., Xie, L., Zhuang, W., Liu, J., et al. (2017). Unfolded protein response plays a critical role in heart damage after myocardial ischemia/reperfusion in rats. PLoS One 12:e0179042. doi: 10.1371/journal.pone.0179042
Zhang, J. X., Tian, Y., Sun, G. B., Sun, X. B., Zhang, X. P., Zhong, X. M., et al. (2013). Research progress in chemcial constituents of saponins from Aralia elata and their pharmacological activities. Chin. Trad. Herb. Drugs 44, 770–779. doi: 10.7501/j.issn.0253-2670.2013.06.026
Zhao, Z. Q., Corvera, J. S., Halkos, M. E., Kerendi, F., Wang, N. P., Guyton, R. A., et al. (2003). Inhibition of myocardial injury by ischemic postconditioning during reperfusion: comparison with ischemic preconditioning. Am. J. Physiol. Heart Circ. Physiol. 285, H579–H588. doi: 10.1152/ajpheart.01064.2002
Zheng, D., Wang, G., Li, S., Fan, G. C., and Peng, T. (2015). Calpain-1 induces endoplasmic reticulum stress in promoting cardiomyocyte apoptosis following hypoxia/reoxygenation. Biochim. Biophys. Acta 1852, 882–892. doi: 10.1016/j.bbadis.2015.01.019
Keywords: Araloside C, hypoxia/reoxygenation injury, endoplasmic reticulum stress, heat shock protein 90, apoptosis
Citation: Du Y, Wang M, Liu X, Zhang J, Xu X, Xu H, Sun G and Sun X (2018) Araloside C Prevents Hypoxia/Reoxygenation-Induced Endoplasmic Reticulum Stress via Increasing Heat Shock Protein 90 in H9c2 Cardiomyocytes. Front. Pharmacol. 9:180. doi: 10.3389/fphar.2018.00180
Received: 08 December 2017; Accepted: 19 February 2018;
Published: 17 April 2018.
Edited by:
Ruiwen Zhang, University of Houston, United StatesReviewed by:
Gavin P. McStay, New York Institute of Technology, United StatesCopyright © 2018 Du, Wang, Liu, Zhang, Xu, Xu, Sun and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaobo Sun, c3VuX3hpYW9ibzE2M0AxNjMuY29t Guibo Sun, c3VuZ3VpYm9AMTI2LmNvbQ== Huibo Xu, eGhiXzY1MDVAMTYzLmNvbQ==
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.