 Chiara Giacomelli1
Chiara Giacomelli1 Simona Daniele1
Simona Daniele1 Chiara Romei2,3Laura Tavanti4Tommaso Neri2Ilaria Piano1Alessandro Celi2
Chiara Romei2,3Laura Tavanti4Tommaso Neri2Ilaria Piano1Alessandro Celi2 Claudia Martini1*
Claudia Martini1* Maria L. Trincavelli1
Maria L. Trincavelli1- 1Department of Pharmacy, University of Pisa, Pisa, Italy
- 2Department of Surgical, Medical and Molecular Pathology and Critical Care Medicine, University of Pisa, Pisa, Italy
- 3Radiology Unit, University Hospital of Pisa, Pisa, Italy
- 4Pneumology Unit, Cardio-Thoracic Department, University Hospital of Pisa, Pisa, Italy
The epithelial-mesenchymal transition (EMT) is a complex process in which cell phenotype switches from the epithelial to mesenchymal one. The deregulations of this process have been related with the occurrence of different diseases such as lung cancer and fibrosis. In the last decade, several efforts have been devoted in understanding the mechanisms that trigger and sustain this transition process. Adenosine is a purinergic signaling molecule that has been involved in the onset and progression of chronic lung diseases and cancer through the A2B adenosine receptor subtype activation, too. However, the relationship between A2BAR and EMT has not been investigated, yet. Herein, the A2BAR characterization was carried out in human epithelial lung cells. Moreover, the effects of receptor activation on EMT were investigated in the absence and presence of transforming growth factor-beta (TGF-β1), which has been known to promote the transition. The A2BAR activation alone decreased and increased the expression of epithelial markers (E-cadherin) and the mesenchymal one (Vimentin, N-cadherin), respectively, nevertheless a complete EMT was not observed. Surprisingly, the receptor activation counteracted the EMT induced by TGF-β1. Several intracellular pathways regulate the EMT: high levels of cAMP and ERK1/2 phosphorylation has been demonstrated to counteract and promote the transition, respectively. The A2BAR stimulation was able to modulated these two pathways, cAMP/PKA and MAPK/ERK, shifting the fine balance toward activation or inhibition of EMT. In fact, using a selective PKA inhibitor, which blocks the cAMP pathway, the A2BAR-mediated EMT promotion were exacerbated, and conversely the selective inhibition of MAPK/ERK counteracted the receptor-induced transition. These results highlighted the A2BAR as one of the receptors involved in the modulation of EMT process. Nevertheless, its activation is not enough to trigger a complete transition, its ability to affect different intracellular pathways could represent a mechanism at the basis of EMT maintenance/inhibition based on the extracellular microenvironment. Despite further investigations are needed, herein for the first time the A2BAR has been related to the EMT process, and therefore to the different EMT-related pathologies.
Introduction
The epithelial-mesenchymal transition (EMT) is an evolutionarily conserved biochemical process in which cells undergo conversion from an epithelial to a mesenchymal phenotype. The EMT is characterized by the loss of the epithelial cell–cell adhesion molecule CDH1 (E-cadherin), and/or a concomitant gain of mesenchymal markers such as CDH2 (N-cadherin), VIM (Vimentin), and/or αSMA (alpha-smooth muscle actin) (Nieto et al., 2016; Singh et al., 2017). EMT is a Janus-faced process due to its pivotal role in embryogenesis and wound healing and in the development of chronic pathologies, such as fibrosis and cancer (Lamouille et al., 2014).
The EMT process has been widely correlated with lung embryogenesis (Lee et al., 2006) and cancer (Sung et al., 2016; Legras et al., 2017), but it has only recently been linked to chronic human lung and airway diseases such as chronic obstructive pulmonary disease (COPD) and idiopathic pulmonary fibrosis (IPF) (Willis et al., 2006; Sohal and Walters, 2013; Jonsdottir et al., 2015; Jolly et al., 2017). In fact, it has been proposed that alveolar epithelial cells (AECs), undergoing EMT (De Maio et al., 2012; Yamaguchi et al., 2017) or partial EMT (Morbini et al., 2011), are a source of extracellular matrix-producing fibroblasts/myofibroblasts. It has been widely accepted that several soluble factors such as, growth factors [fibroblast growth factor (FGF), epidermal growth factor (EGF)] and inflammatory cytokines (transforming growth factor-beta (TGF-β1), interleukin-6 (Il-6), tumor necrosis factor-alpha (TNF-α), could trigger and promote the EMT (Nieto et al., 2016; Suarez-Carmona et al., 2017). In the lung, attention has been focused on TGF-β1, which has been found to promote AEC differentiation (Yang et al., 2014; Shi et al., 2016) and the aggressiveness of cancer cells (Sakuma, 2017).
Adenosine is a soluble factor involved in physiological processes; however, after tissue injury, its levels rise to micromolar concentration promoting anti-inflammatory action (Ohta and Sitkovsky, 2001). Recently, a correlation between the increase in adenosine levels and the EMT process has been reported (Guillén-Gómez et al., 2012; Lu and Insel, 2014; Gao et al., 2016). Adenosine binds to four specific cell membrane G-protein-coupled receptors (GPCRs) known as adenosine receptors (ARs): A1, A2A, A2B, and A3. In this context, great attention has been focused on the purine receptor involvement in the EMT process. Only a few studies have correlated the EMT and purinergic receptors P2X and P2Y, such as P2X7, P2Y2, P2Y6, and P2Y12 (review in Martínez-Ramírez et al., 2017). Furthermore, some evidence has correlated the adenosine receptor A2A with the transition (Xiao et al., 2013). However, no evidence has been reported on the role of the A2BAR subtype in the trigger or modulation of induced-EMT.
The A2BAR subtype has been recently linked to cancer aggressiveness (Mittal et al., 2016; Sepúlveda et al., 2016) and fibrotic processes of the heart (Ryzhov et al., 2014; Phosri et al., 2017), kidney (Roberts et al., 2014; Wilkinson et al., 2016) and lung (Zhou et al., 2011; Karmouty-Quintana et al., 2012; Karmouty-Quintana et al., 2013). A2BAR couples to Gαs proteins, resulting in the activation of adenylyl cyclase (AC) and an increase in intracellular cyclic AMP (cAMP) levels that subsequently activate protein kinase A (PKA) (Schulte and Fredholm, 2003; Sun and Huang, 2016). However, A2BAR can also coupled to the Gq-PLC pathway and induces the activation of mitogen-activated protein kinase (MAPK) (Schulte and Fredholm, 2003; Sun and Huang, 2016). In fact, A2BAR induces the phosphorylation of ERK1/2 in human umbilical vein endothelial cells (HUVECs) (Fang and Olah, 2007), human mast cell line (MHC-1 cells) (Feoktistov et al., 1999), and human retinal endothelial cells (HRECs) (Grant et al., 2001). The cAMP role in EMT promotion/blockade is debatable. Pattabiraman et al. (2016) reported that a PKA activator could lead to a mesenchymal-epithelial transition as previously reported by Boucher et al. (2005). Furthermore, the increase in the cAMP intracellular levels, induced by forskolin or phosphodiesterase inhibitors, as well as the administration of a PKA activator counteracts the EMT induced by TGF-β1 (Zhang et al., 2006a,b; Nadella et al., 2008; Lambers et al., 2015). By contrast, other evidence has been reported regarding the positive role of cAMP and PKA in the induction of EMT (Shaikh et al., 2012). Conversely, it has been widely accepted that ERK phosphorylation is one of the mechanisms that promotes the EMT program (Singh et al., 2017). In fact, ERK activation is one of the Smad-independent events that is necessary for TGF-β-mediated EMT (Gui et al., 2012; O’Connor and Gomez, 2014).
Therefore, in the present study, the effects of A2BAR stimulation on EMT was investigated in human lung epithelial cells. Particularly, the role of this adenosine receptor subtype in directly modulating the epithelial-mesenchymal markers under physiological or inflammatory conditions (TGF-β1) was analyzed. Furthermore, the intracellular pathways activated by A2BAR, cAMP/PKA, and MAPK/ERK, involved in EMT, were explored.
Materials and Methods
Material and Reagents
Human type II alveolar epithelial cells (A549, American Type Culture Collection, CCL-195), were kindly provided by Dr. R. Danesi, University of Pisa, Pisa, Italy. The chemicals 2-[6-amino-3,5-dicyano-4-[4-(cyclopropylmethoxy) phenyl]pyridin-2-ylsulfanyl] acetamide (BAY 60-6583) and N-(4-acetylphenyl)-2-[4-(2,3,6,7-tetrahydro-2,6-dioxo-1,3-dipropyl-1H-purin-8-yl)phenoxy]-acetamide (MRS1706) were purchased from Tocris Bioscience (Bristol, United Kingdom). The RNeasy minikit was obtained from Qiagen. The Script cDNA synthesis kit was furnished by Bio-Rad s.r.l. Fluocycle II SYBR was from Euroclone (Milan, Italy). TGF-β1 was purchased from Sigma–Aldrich. All other reagents were obtained from standard commercial sources and were of the highest commercially available grade
Cell Culture
Human type II alveolar epithelial cells (A549) were maintained in DMEM-F12 containing 10% FBS, 2 mM L-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin at 37°C in a humidified 5% CO2 atmosphere. For the different experiments, cultures of cells were maintained in serum-free DMEM for 8 h prior to stimulation with cytokines or other reagents.
RNA Extraction and Real-Time RT-PCR Analysis
A549 cells were untreated or treated with DMSO (CTRL) or BAY 60-6583 (100 nM) in the absence or presence of TGF-β1 (10 ng/ml) and MRS 1706 (1 μM) for the indicated time. At the end of treatments, cells were collected, and total RNA was extracted using Rneasy®Mini Kit (Qiagen, Hilden, Germany) as previously reported (Da Pozzo et al., 2014); residual DNA was removed using RNase free DNAse set (#79254, Qiagen, Hilden, Germany). The RNA purity was checked measuring the A260/280 ratio. cDNA synthesis was performed with 400 ng of RNA using i-Script cDNA synthesis kit (BioRad, Hercules, CA, United States) following manufacturer’s instructions. Real-time RT-PCR reactions consisted of 25 μL Fluocycle®II SYBR® (Euroclone, Milan, Italy), 1.0 μL of both 10 μM forward and reverse primers, 2.5 μL cDNA (100 ng), and 20.5 μL of H2O. All reactions were performed for 40 cycles using the following temperature profiles: 98°C for 30 s (initial denaturation); 55°C for 30 s (annealing); and 72°C for 3 s (extension). The primer sequences were listed in Table 1. When possible, the primers used were designed to span intron/exon boundaries and the β-actin was used as the housekeeping gene. The mRNA levels for each sample were normalized against β-actin mRNA levels, and the relative expression was calculated by using a Ct value. PCR specificity was determined by both the melting curve analysis and gel electrophoresis.
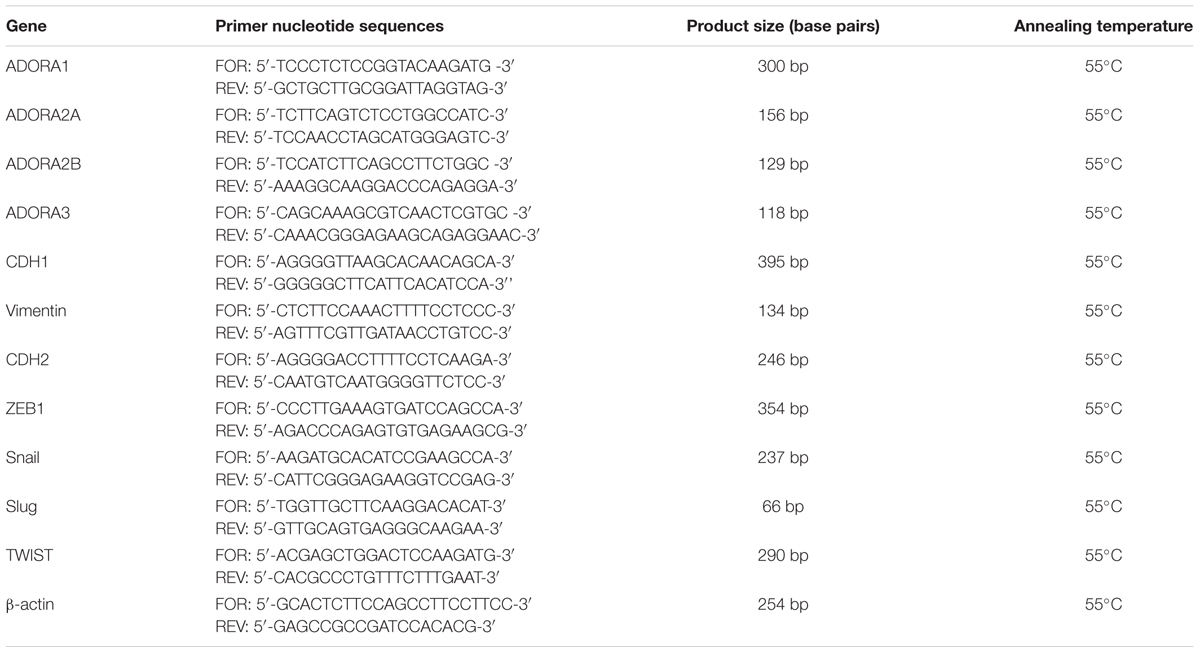
TABLE 1. Primers used for real-time RT-PCR.
Western Blotting Analysis
A549 cells (3.5 × 104 cell/cm2) were maintained untreated or treated with DMSO (CTRL) or BAY 60-6583 (100 nM) in the absence or presence of TGF-β1 (10 ng/ml) and MRS 1706 (1 μM) for 48 h. At the end 200 μl RIPA buffer were added for 60 min at 4°C to lyse the cells. Fifty microgram of total proteins was diluted in Laemmli solution, resolved by SDS-PAGE (7.5%), transferred to PVDF membranes and probed overnight at 4°C with primary anti-A1AR (diluted 1:200, sc-28995; Santa Cruz Biotechnology), anti-A2AAR (diluted 1:200, sc-13937; Santa Cruz Biotechnology), anti-A2BAR (diluted 1:150, sc-28996; Santa Cruz Biotechnology), anti-A3AR (diluted 1:200, sc-13938; Santa Cruz Biotechnology), anti-E-cadherin antibody (diluted 1:200; sc-7870; Santa Cruz Biotechnology), anti-N-cadherin antibody (diluted 1:200; sc-7939; Santa Cruz Biotechnology) anti-Vimentin (diluted 1:1000, #5741; Cell Signaling Technology) or β-actin antibody (diluted 1:1000; MAB1501, Merck KGaA, Darmstadt, Germany). The primary antibody was detected using appropriate secondary antibody. The peroxidase was detected using a chemiluminescent substrate (ECL, Perkin Elmer), and the images were acquired by photographic film or by LAS4010 (GE Health Care Europe, Uppsala, Sweden). Immunoreactive bands were quantified performing a densitometric analysis with Image J Software (version 1.41; Bethesda, MD, United States).
Immunofluorescence Analysis
A549 cells were seeded at 3.5 × 104 cell/cm2 in chamber slide (BD Biosciences, San Jose, CA, United States). After treatment, cells were fixed in 2% paraformaldheyde in 0.1 M phosphate buffer, washed three times with PBS, rinsed, and blocked for 45 min with PBS containing 0.1% Triton-X 100 and 1% BSA. After washing, cells were incubated overnight at 4°C with mouse monoclonal anti-β-actin antibody (diluted 1:500; MAB1501, Merck KGaA, Darmstadt, Germany) or a rabbit polyclonal anti-A2BAR (diluted 1:100, sc-28996; Santa Cruz Biotechnology) primary antibodies diluted in PBS containing 0.03% Triton-X 100 and 1% BSA overnight at 4°C. After washing, to visualize the staining, cells were incubated with Alexa Fluor 488- and Alexa Fluor 568-labeled goat anti-mouse (1:500) or anti-rabbit (1:500) antibodies for 2 h at room temperature. Then slides were covered with Vectashield conjugated with DAPI (Vector Laboratories, Burlingame, CA, United States). Images were obtained with a Nikon Ni-E microscope, using a 20× objective with 1.45 NA and a recommended pinhole size of less than 1.0 mm, and equipped with digital camera Nikon Mod.DS-Ri2. The images were processed with ImageJ software.
Cell Viability Assay
A549 cells were seeded at a density of 3 × 103 cells/well in a black, clear bottom PerkinElmer 96-well CellCarrierTM microplate (#6005550). After 24 h, the cells were starved for 8 h with non-complete medium; then, cells were treated with different concentrations of BAY 60-6583 (0.5 nM–1 μM) or TGF-β1 (5–20 ng/ml) alone or in combination for 48 or 72 h. Following the treatment period, cells were imaged with the EnSightTM multimode plate reader equipped with well-imaging module, and Kaleido Data Acquisition and Analysis Software. Brightfield images were taken before and 48 or 72 h after treatments to allow for a Cell Count comparison between both time points. The cell count was determined using the pre-defined Brightfield Cell Count algorithm provided by the Kaleido software. Cell with a mean area ≥100 μm2 were counted. Data were normalized to the cell count at t0 and expressed as the number of cell per well.
cAMP Quantification
A549 cells were seeded at a density of 3 × 103 cells/well in a black, clear bottom PerkinElmer 96-well CellCarrierTM microplate (#6005550). After 24 h, the cells were starved for 8 h with non-complete medium; then, cells were treated with different concentrations of BAY 60-6583 (0.1 nM–1 μM) in the absence or presence of TGF-β1 (10 ng/ml) in 100 μl of non-complete medium for 48 h. Cells were imaged with the EnSightTM multimode plate reader. The desensitization experiments were performed incubating the cells with BAY 60-6583 (100 nM) in the presence or absence of TGF-β1 (10 ng/ml) for 48 h. At the end the medium was changed and cells were treated with different concentrations of BAY 60-6583 (0.1 nM–1 μM) for 15 min. Then, cells were lysed with 50 μl of 1X lysis buffer (PerkinElmer, #AL003C) supplemented with protease inhibitors and phosphatase inhibitors for 10 min with gentle shaking. Lysates were either tested immediately in AlphaLISA assays or frozen at –80°C for later testing. cAMP levels were quantified using cAMP AlphaLISA kit (PerkinElmer, #AL312) following manufacturer’s instructions. Standard curves for each AlphaLISA immunoassay were performed in the same diluent as the samples being tested (1X lysis buffer with supplements) using the recombinant standards provided in each kit. Curves were plotted with a sigmoidal concentration-response curve with variable slope. Quantitation of protein levels in cellular assays were interpolated off their respective standard curves.
MAPK (Mitogen-Activated Phosphorylation Kinase) Assays
A549 cells were seeded at a density of 3 × 103 cells/well in 96 multi-well plate. After 24 h, the cells were starved for 8 h with non-complete medium; then, cells were treated with BAY 60-6583 (100 nM) in the absence or presence of TGF-β1 (10 ng/ml) and MRS1706 (1 μM) for 5 min, 30 min, 6, 24, and 48 h. In some experiments, before incubation with BAY 60-6583 or TGF-β1, cells were pre-treated for 30 min with PD98059 (1 μM) (MEK, inhibitor). At the end of treatments, cells were fixed with 4% formaldehyde to preserve activation of specific protein modification. Levels of total and phosphorylated extracellular signal-regulated kinases (ERK1/2) were determined by ELISA assays, as previously reported (Giacomelli et al., 2015). Briefly, the cells were washed three times with wash buffer (0.1% Triton X-100 in PBS) and 100 μl of quenching buffer (1% H2O2; 0.1% sodium azide in wash buffer) was added and incubation was protracted for other 20 min. The cells were washed with PBS twice, and then 100 μl of blocking solution (1% BSA; 0.1% Triton X-100 in PBS) was added for 60 min. After blocking, cells were washed three times with wash buffer and the specific primary antibodies (anti-phospho ERK1/2, 1:500, sc-7383 Santa Cruz Biotechnology; anti-ERK1/2, 1:500, #4695 Cell Signaling Technology) were added on at 4°C. Subsequent incubation with secondary HRP-conjugated antibodies and developing solution allowed a colorimetric quantification of total and phosphorylated levels. Blanks were obtained by treating cells in the absence of the primary antibody. The relative number of cells in each well was then determined using Crystal Violet solution. The results were calculated by subtracting the mean background from the values obtained from each test condition; values were normalized to the number of cells in each well, and were expressed as the percentage of untreated cells (basal).
Quantification of E-cad and N-cad Proteins
A549 cells were seeded at a density of 3 × 103 cells/well in a black, clear bottom PerkinElmer 96-well CellCarrierTM microplate (#6005550). After 24 h, the cells were starved for 8 h with non-complete medium; then, cells were treated with different concentrations of BAY 60-6583 (0.1 nM–1 μM) in the absence or presence of TGF-β1 (10 ng/ml) in 100 μl of non-complete medium for 48 h. In some experiments, before incubation with BAY 60-6583 or TGF-β1, cells were pre-treated for 30 min with PD98059 (1 μM, MEK, inhibitor), H89 (100 nM, PKA inhibitor) or 8-Br-cAMP (100 nM–1 μM, cAMP analog & PKA activator). Then, cells were imaged with the EnSightTM multimode plate reader. After imaging, cells were lysed with 50 μl of 1X lysis buffer (PerkinElmer, #AL003C) supplemented with protease inhibitors and phosphatase inhibitors for 10 min with gentle shaking. E-cad were quantified using E-cadherin AlphaLISA kit (PerkinElmer, #AL370), and N-cad were quantified using N-cadherin AlphaLISA kit (PerkinElmer, #AL379) following the manufacturer’s instructions. Standard curves for each AlphaLISA immunoassay were performed in the same diluents as the samples being tested (1X lysis buffer with supplements), using the recombinant standards provided in each kit. Curves were plotted with a sigmoidal concentration-response curve with variable slope. Quantitation of protein levels in cellular assays were interpolated off their respective standard curves.
Statistical Analysis
The Graph-Pad Prism program (GraphPad Software Inc., San Diego, CA, United States) was used for data analysis and graphic presentation. All data are the mean ± SEM of at least three different experiments. Statistical analysis was performed by one-way analysis of variance (ANOVA) with Dunnett post hoc analysis to compare the data to the control, or two-way ANOVA with Bonferroni correction and two-sided tests for multiple comparisons. EC50 values were reported as mean of the values obtained in at least three independent experiments performed in duplicate ± SEM. P ≤ 0.05 was considered statistically significant.
Results
Adenosine Receptor Expression in Human Epithelial Lung Cells and Its Modulation by TGF-β1
The A549 human alveolar epithelial cells have been widely used to study the fibrotic process in the lung and related EMT mechanism (Kim et al., 2007; Ji et al., 2016). Furthermore, these cells were maintained in serum-free medium to increase the epithelial phenotype (Dong et al., 2014).
First, the expression of the AR subtypes in A549 cells was evaluated after incubation in serum-free medium for 48 h (Figure 1A). All the ARs were expressed under this condition, and the A2BAR subtype was the most expressed with a fold change of approximately 200 (Figure 1A).
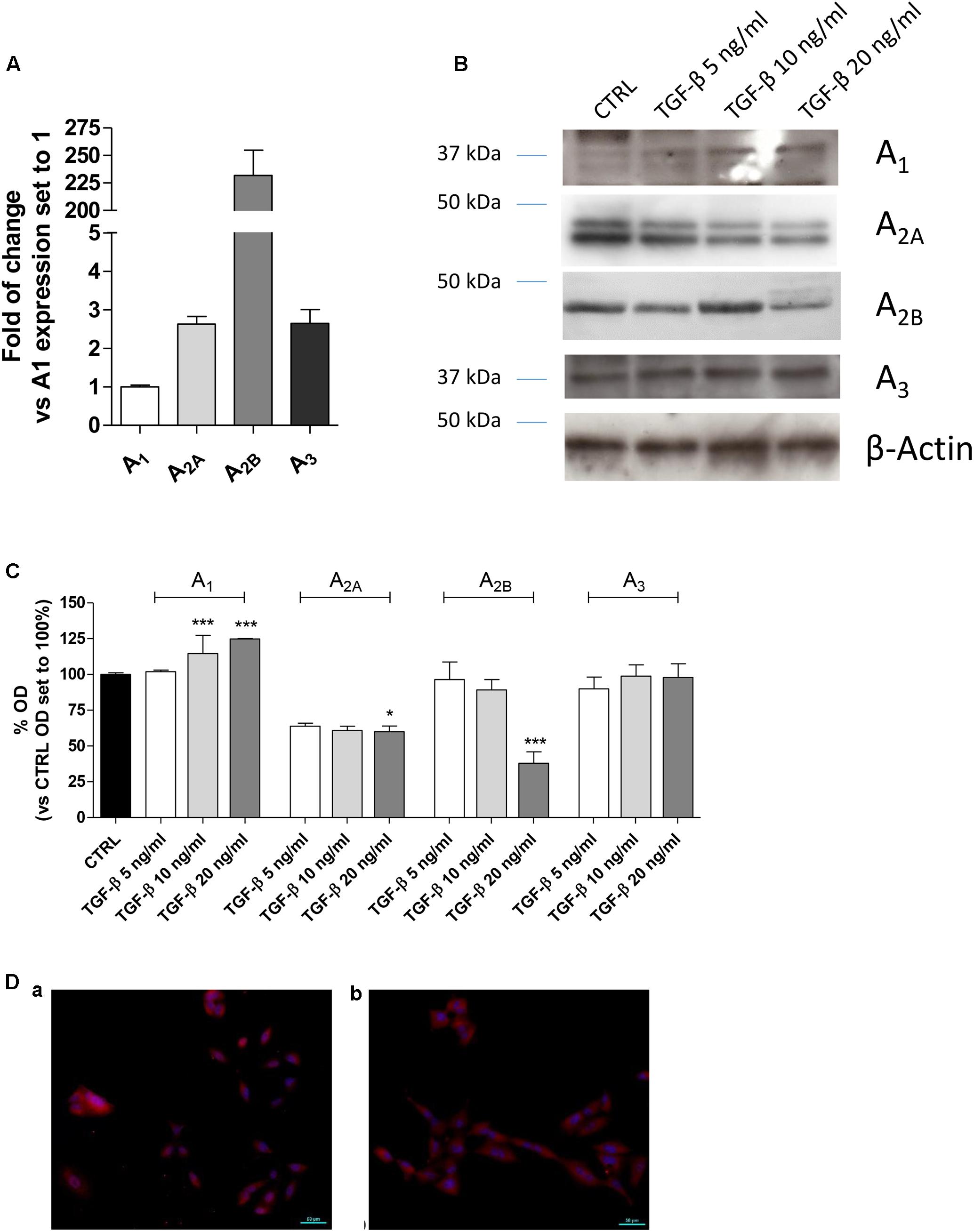
FIGURE 1. Expression of ARs in A549 cells and their modulation in the presence of TGF-β1. (A) A549 cells were maintained in serum-free medium for 48 h. Next, real-time RT-PCR analysis of A1, A2A, A2B, and A3 adenosine receptors was performed. The data were expressed as the fold change vs. A1AR expression, which was set to 1 and are the mean values ± SEM of three different experiments. (B,C) A549 cells were treated with different concentrations of TGF-β1 for 48 h, and the levels of AR subtypes were evaluated by Western blotting. One representative Western blot is presented (C). The bar graph (D) shows the densitometric analysis of the Western blot performed using the ImageJ program. Cells were maintained in serum-free medium for 48 h in the absence (a) or presence (b) of TGF-β1. Next, cells were fixed and stained with anti-β actin and visualized with goat anti-rabbit Alexa Fluor 568 (red). Nuclei were counterstained with DAPI (blue). The data are presented as the means of three different experiments. The significance of the differences was determined by one-way ANOVA, followed by Dunnett’s post hoc test: ∗P ≤ 0.05, ∗∗∗P ≤ 0.001 vs. the CTRL.
TGF-β1 has been reported to affect the expression of several proteins (Zhang, 2017); thus, the effects of cytokine treatment on AR expression were evaluated (Figures 1B–D). Challenging the A549 cells for 48 h with increasing concentrations of TGF-β1 (5–20 ng/ml) modified the expression of the different adenosine subtypes. Particularly, A1AR expression was significantly increased when a high concentration of TGF-β1 was used; conversely, A2A and A3 receptor expression was slightly decreased when increased concentration of the cytokine was applied. Regarding A2BAR expression, low concentrations of TGF-β1 (5 or 10 ng/ml) were not sufficient to modify the receptor expression; by contrast, a higher concentration of the cytokine significantly decreased its expression. In this light, further experiments were performed using a maximum concentration of the cytokine of 10 ng/ml for 48 h to minimize the change in the receptor expression.
TGF-β1 Effects on A2BAR Functionality and Lung Cell Growth
A2BAR stimulation or blockade has been correlated to different levels of cell proliferation depending on the cell type and culture condition (Phosri et al., 2017; Zhou et al., 2017). Thus, the effects of A2BAR stimulation in the absence or presence of TGF-β1 were evaluated in the lung cells (Figure 2). The selective agonist BAY 60-6583 was used to stimulate selectively the A2BAR subtype. The compound could slightly increase the cell proliferation when used at low concentration (50–100 nM) for 48 or 72 h treatment (Figure 2A), conversely, it produced a decrease in proliferation when used at a high concentration (1 μM), with a significant effect after 72 h of treatment (13970 ± 488 n° of cells CTRL, 11038 ± 816 n° of cells BAY; P ≤ 0.05). TGF-β1 alone presented a hormetic concentration-response course: the lower concentration possessed positive effects on cell proliferation that are lost at higher concentrations (Figure 2B). The combined treatment with BAY 60-6583 and TGF-β1 for 48 h did not significantly affect the cell proliferation (Figure 2C). These data confirmed the diverse role of A2BAR in the cell proliferative mechanism. Based on such results, further experiments were then performed using BAY 60-6583 at a concentration (100 nM) not affecting significantly the A549 cell proliferation.
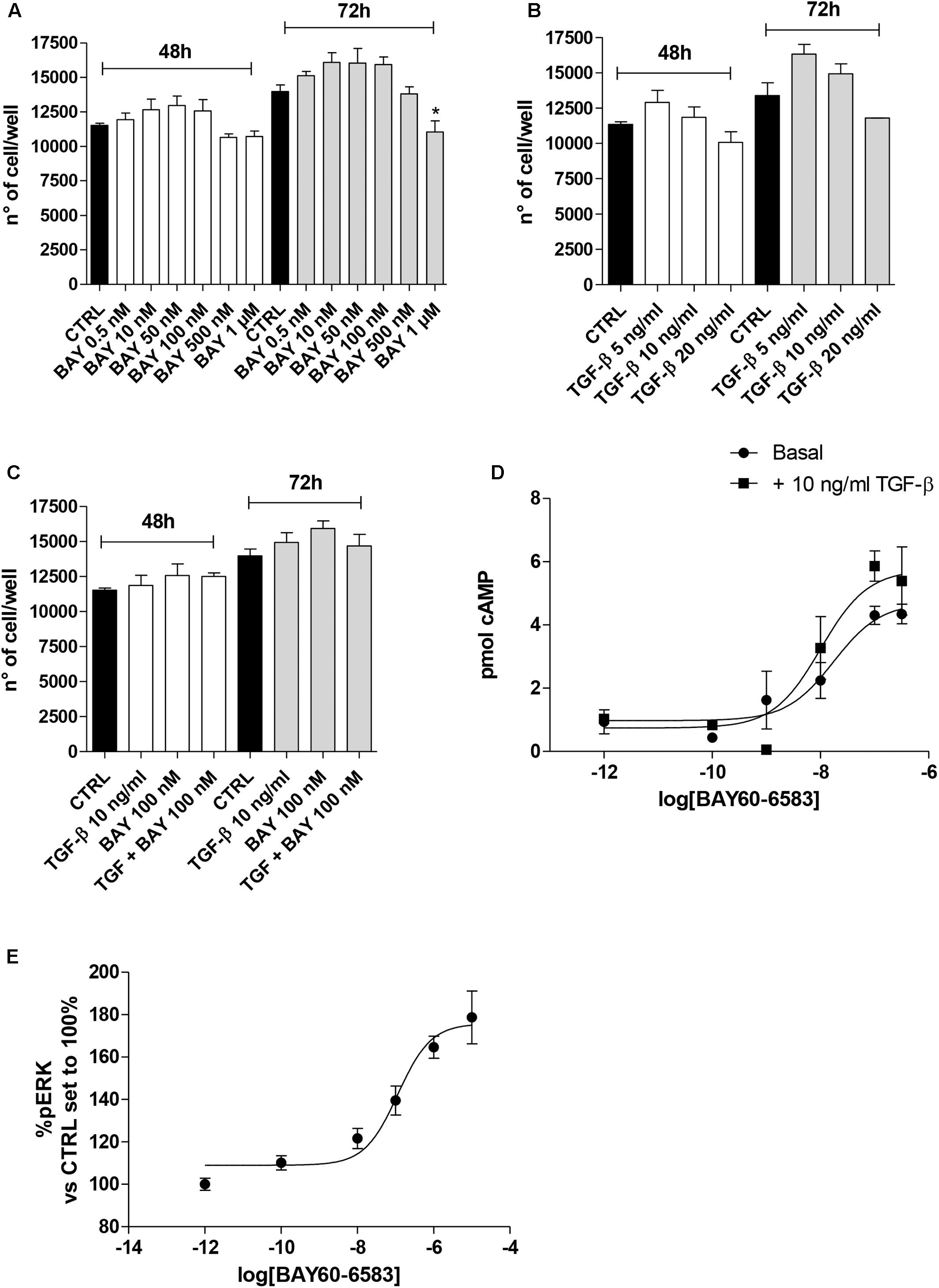
FIGURE 2. Effects of BAY 60-6583 and TGF-β1 on cell proliferation and cAMP accumulation. A549 cells were treated in serum-free medium with different concentrations of BAY 60-6583 (0.5 nM–1 μM) and TGF-β1 (5–20 ng/ml), alone or in combination, for 48 h (A–C). At the end of the treatments, the cell numbers were counted. The data were expressed as cell number per well, and they were presented as the mean values ± SEM of three independent experiments, each performed in duplicate. The significance of the differences was determined by one-way ANOVA, followed by Dunnett’s post hoc test: ∗P ≤ 0.05 vs. the CTRL. (D) A549 cells were maintained in the absence or presence of TGF-β1 (10 ng/ml) for 48 h. Next, cells were challenged with increasing concentrations of BAY 60-6583, and cAMP production was quantified. (E) A549 cells were treated with different concentrations of BAY 60-6583 for 5 min and the levels of pERK were quantified. The data were expressed as pmol of cAMP and are the mean values ± SEM of three independent experiments, each performed in duplicate.
Next, the effect of cytokine treatment on A2BAR functionality was evaluated by measuring the agonist-mediated cAMP production (Figure 2D). The potency of BAY 60-6583 was in the nanomolar range (EC50 = 17.2 ± 2.5 nM), as derived by agonist concentration-response curves, it is a value comparable to that described for the same agonist in transfected cells (Figure 2D) (Trincavelli et al., 2014). The presence of TGF-β1 did not significantly affect the agonist potency (EC50 = 10.0 ± 1.8 nM), even if the maximal effect of BAY in the production of cAMP was slightly increased (Emax = 4.3 pmol – TGF-β1; Emax = 5.8 pmol + TGF-β1, P = 0.0565) even if it was not statistically significant. These results are in accordance with the effect of other cytokines on A2BAR functionality (Daniele et al., 2017); in fact, TNF-α has been demonstrated to increase A2BAR coupling to the Gs protein (Daniele et al., 2017).
In parallel, the BAY-induced ERK phosphorylation was evaluated in order to further assess the A2BAR functionality (Figure 2E). The agonist was able to produce a concentration-dependent increase of pERK with a potency in the sub-micromolar range of (EC50 = 114.8 ± 24.5 nM).
Effects of A2BAR Stimulation on EMT Markers in the Absence or Presence of TGF-β1
Typically, the EMT process is triggered and maintained by the action of different cytokines and extracellular stimuli that, through the activation of specific intracellular pathways, modify the expression of several proteins (Nieto et al., 2016). A549 cells were treated for 48 h with the A2BAR agonist alone or in the presence of TGF-β1 (10 ng/ml), which has been reported to induce EMT (Kawata et al., 2012). First, a morphological analysis of the cells was performed (Figures 3A,B). TGF-β1 treatment induced a change in cell morphology: most of the A549 cells, which normally display an oval shape, showed an elongated shape with a fibroblast-like appearance. Surprisingly, cells challenged with the A2BAR agonist lead to a partial morphological change, with only some cells presenting an elongated shape, suggesting the induction of partial EMT.
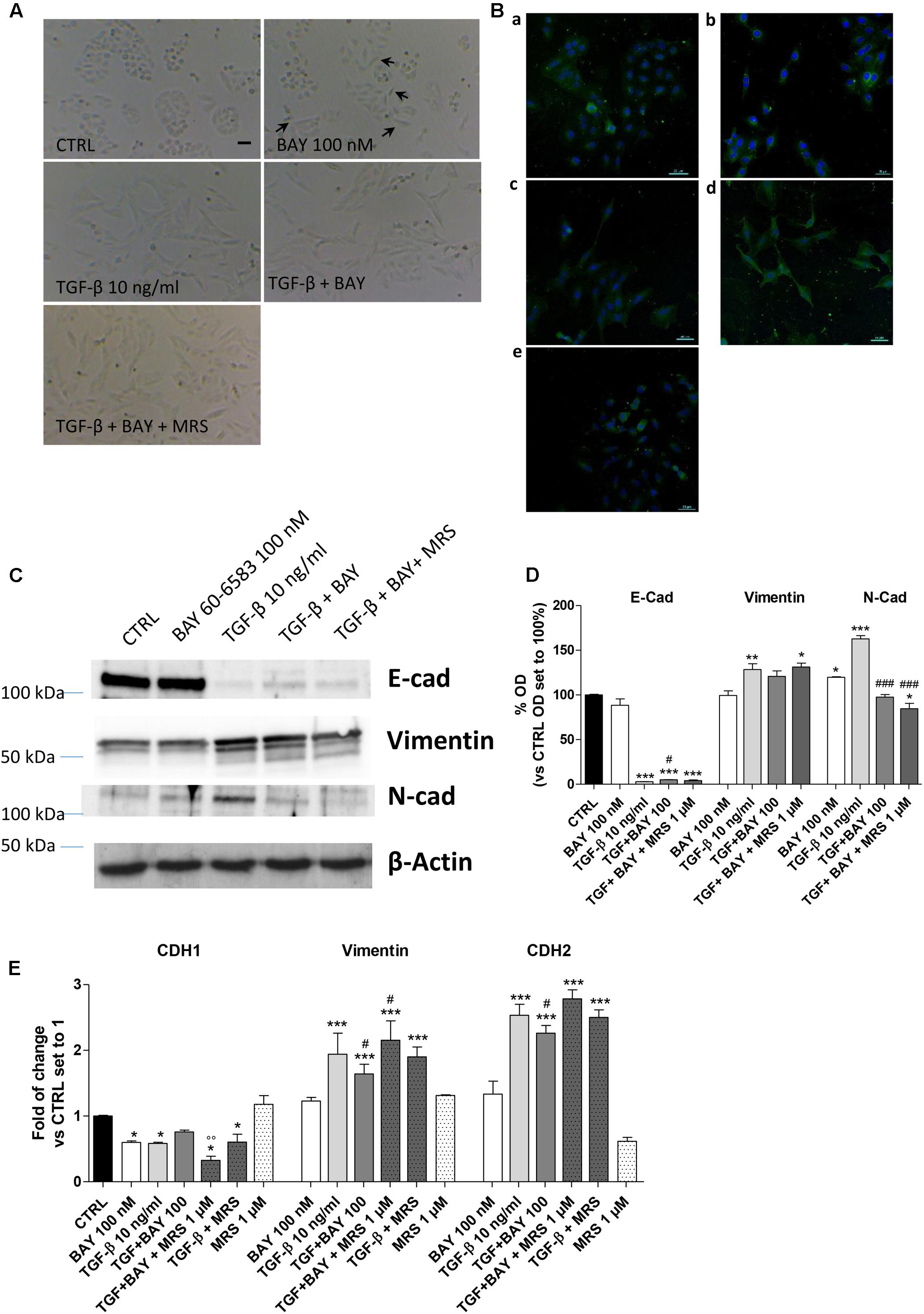
FIGURE 3. A2BAR stimulation affects EMT markers and decreases TGF-β1-induced EMT. A549 cells were treated with BAY 60-6583 (100 nM) in the absence or presence of TGF-β1 (10 ng/ml) for 48 h. When indicated, the A2BAR inverse agonist, MRS 1706 (1 μM), was applied. (A) At the end of the incubation times, representative images were taken. Scale bar = 100 μm. (B) Cells treated as above were fixed and stained with anti-β actin and visualized with goat anti-mouse AlexaFluo 488 (green). Nuclei were counterstained with DAPI (blue). (C,D) A549 cells were treated as described above, and the levels of the EMT markers E-cad, Vimentin and N-cad were evaluated by Western blotting. One representative Western Blot is presented (C). The bar graph (D) shows the densitometric analysis of the Western blot performed using the ImageJ program. The data are presented as the means of three different experiments. (E) Real-time RT-PCR analysis of the same EMT markers were performed. The data were expressed as the fold change vs. the CTRL levels, which were set to 1 and are the mean values ± SEM of three different experiments each performed in duplicate. The significance of the differences was determined by one-way ANOVA, followed by Dunnett’s post hoc test or two-way ANOVA with Bonferroni correction and two-sided tests for multiple comparisons. ∗P ≤ 0.05, ∗∗P ≤ 0.01, ∗∗∗P ≤ 0.001 vs. the CTRL; #P ≤ 0.05, ###P ≤ 0.001 vs. TGF-β1 alone; ∘∘P ≤ 0.01 vs. TGF-β1 + BAY.
The morphological changes, which are characteristic of cells undergoing EMT, are accompanied by a shift in the expression of epithelial genes to a mesenchymal gene repertoire (Nieto et al., 2016). Accordingly, challenging cells with TGF-β1 modified the expression of EMT markers, leading to a significant increase in the expression of the mesenchymal markers, Vimentin and N-cadherin (128.8 ± 6.5, P ≤ 0.01, and 162.7 ± 3.8% P ≤ 0.001 vs. CTRL, respectively; Figures 3C,D) and a concomitant decrease in the expression of the epithelial marker E-cadherin (6.7 ± 2.5% P ≤ 0.001 vs. CTRL; Figures 3C,D). The data were confirmed at the gene expression level by real-time RT-PCR analysis of E-cadherin (CDH1), Vimentin, and N-cadherin (CHD2) expression (Figure 3E).
A2BAR stimulation was able to slightly decrease the expression of E-cad protein (Figures 3C,D) and increase the expression of a mesenchymal marker (N-cad, 119.8 ± 0.6% vs. CTRL; P ≤ 0.05). These effects are in accordance with the effects elicited by BAY on epithelial/mesenchymal gene transcription. Conversely, A2BAR activation in the presence of TGF-β1 produced an opposite effect, significantly counteracting the induced EMT (Figures 3C–E). These effects were almost completely counteracted by concomitant treatment with the A2BAR selective inverse agonist MRS 1706 (1 μM), demonstrating that they were mediated specifically by A2BAR. The treatment with MRS 1706 alone was not able to significantly modify the expression of mesenchymal and epithelial markers in the absence or presence of TGF-β1 demonstrating that the adenosine in the medium was not sufficient to induce a basal A2BAR activation.
Effects of A2BAR Modulation on EMT Major Transcription Factors (EMT-TFs)
The modulation of classical EMT centers on the transcriptional control of different transcription factors: Snail (SNAI1), Slug (SNAI2), ZEB1 and TWIST (Nieto et al., 2016). To gain insight into the mechanisms by which A2BAR affects E-cadherin deficiency alone and in TGF-β1-induced A549 cells, the expression levels of the EMT-TFs were examined using real-time RT-qPCR (Figure 4).
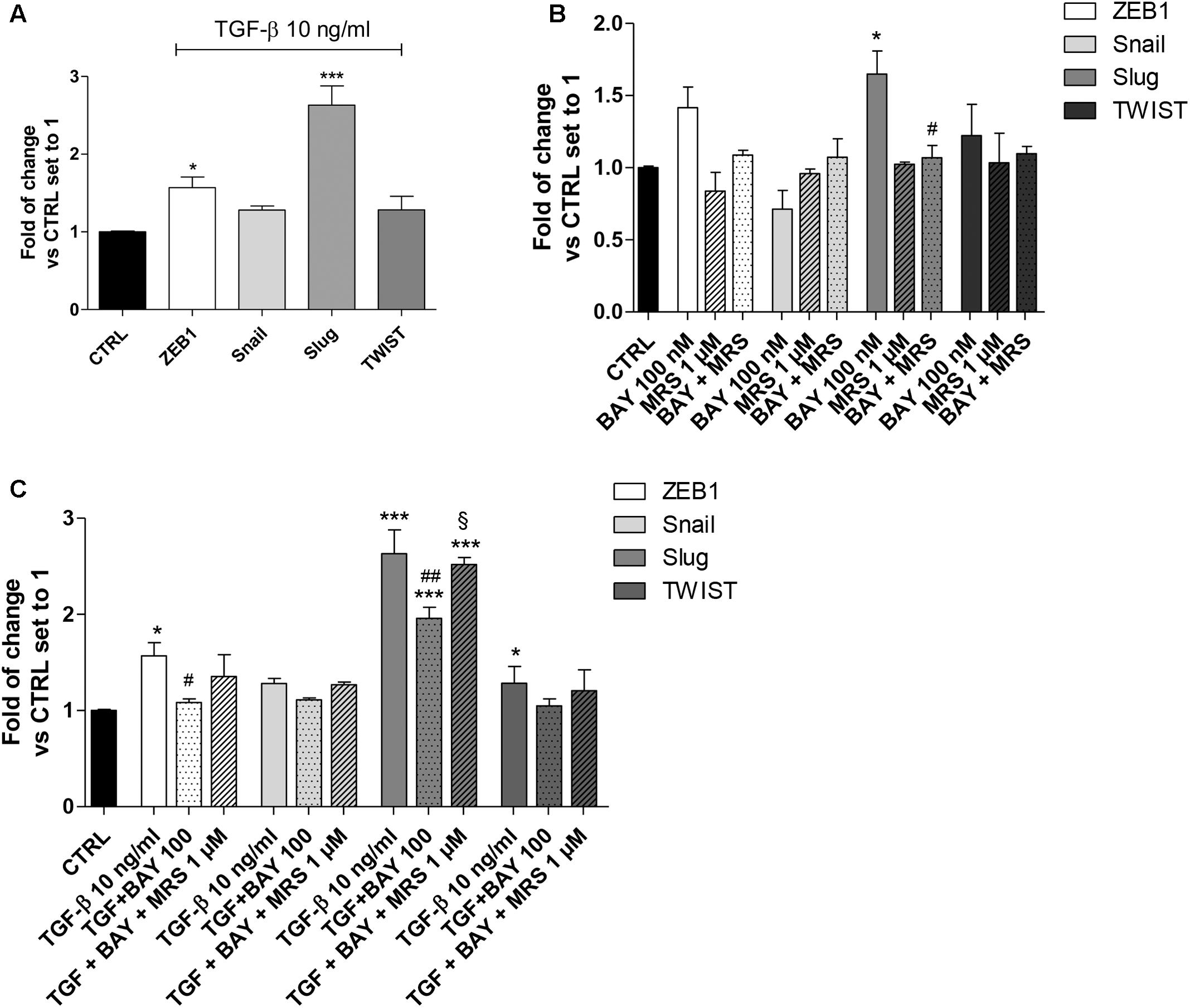
FIGURE 4. A2BAR activation of EMT transcription factors. (A) A549 cells were treated with TGF-β1 (10 ng/ml) for 48 h. At the end of the incubation, real-time RT-PCR analysis of the transcription factors that act as master regulators of EMT (ZEB1, Snail, Slug, TWIST) was performed. (B,C) A549 cells were treated with BAY 60-6583 (100 nM) in the absence or presence of TGF-β1 (10 ng/ml) for 48 h. When indicated, the A2BAR inverse agonist MRS 1706 (1 μM) was applied. At the end of the incubation, real-time RT-PCR analysis of the transcription factors that act as master regulators of EMT (ZEB1, Snail, Slug, TWIST) was performed. The data were expressed as the fold change vs. the CTRL levels, which were set to 1 and are the mean values ± SEM of three different experiments, each performed in duplicate. The significance of the differences was determined by one-way ANOVA, followed by Bonferroni’s post hoc test or two-way ANOVA with Bonferroni correction and two-sided tests for multiple comparisons. ∗P ≤ 0.05, ∗∗∗P ≤ 0.001 vs. the CTRL; #P ≤ 0.05, ##P ≤ 0.01 vs. TGF-β1 alone; § P ≤ 0.05 vs. the TGF-β1 + BAY.
A549 cells expressed Snail and Slug at low levels (Supplementary Figure 1), in accordance with the high expression of E-cad (Figure 2). Treatment with TGF-β1 for 48 h changed the expression pattern of these genes (Figure 4A), with a significant increase in ZEB1 (1.57 ± 0.13-fold; P ≤ 0.05), Slug (2.63 ± 0.25-fold; P ≤ 0.001), in accordance with the literature data (Ji et al., 2016). Next, the effects of A2BAR stimulation (BAY 60-6583) or blockade (MRS 1706) were evaluated (Figure 4B). The MRS alone could not modify the expression of EMT-TFs, highlighting that the endogenous activation of the receptor was not sufficient to modify the related-EMT gene expression. Conversely, A2BAR activation modified the gene repertoire, producing a significant increase in the Slug expression (1.65 ± 0.16-fold; P ≤ 0.05). These data are in accordance with the effects of A2BAR activation on epithelial/mesenchymal markers (Figure 3).
Next, the role of A2BAR stimulation on EMT-TF was evaluated in the presence of TGF-β1 (Figure 4C). As expected, the A2BAR activation counteracted the effects of cytokine treatment, with a significant decrease in ZEB1 and Slug expression. When the A2BAR inverse agonist was applied, the TGF-β1-induced decrease in gene expression was completely counteracted, demonstrating that these effects were mediated by A2BAR.
The Balance of cAMP Production and MAPK/ERK Activation Orchestrates the A2BAR Effects on EMT
A2BAR couples to Gαs proteins, leading to an increase in intracellular cAMP, and to the Gq-PLC pathway, which induces the activation of the MAPK/ERK pathway (Schulte and Fredholm, 2003; Sun and Huang, 2016). Thus, activation of these two intracellular signaling pathways were investigated following A2BAR stimulation, in the absence or presence of TGF-β1 (Figure 5). MRS 1706 alone did not affect the activation of the two pathways, demonstrating that the adenosine levels in our cellular model were not sufficient to activate the receptor. BAY was able to increase the intracellular cAMP concentration (Figure 5A), and significantly enhance ERK1/2 phosphorylation (178.9 ± 10.5% vs. CTRL, P ≤ 0.05; Figure 5B), producing transient activation of these kinases (Figure 5C). The effects evoked by A2BAR stimulation were almost completely reversed by the A2BAR inverse agonist MRS1706, demonstrating the specific involvement of the A2BAR subtype. As expected, TGF-β1 did not produce a significant increase in the cAMP levels (Figure 5A); conversely, it was able to significantly increase ERK1/2 phosphorylation (341.7 ± 16.2% vs. CTRL, P ≤ 0.001; Figure 5B). Both BAY and TGF-β1 induced transient activation of ERK1/2 but with different kinetics; whereas ERK phosphorylation induced by BAY peaked within 30 min and then returned to the basal value within 6 h, TGF-β1 caused a more sustained ERK activation up to 48 h of cell treatment (48 h, 155.2 ± 7.8%, TGF-β1; 85.3 ± 8.8%, BAY; P ≤ 0.05; Figure 5C). These data highlighted a difference in the final reorganization of the intracellular pathways activated by the cytokine and the receptor.
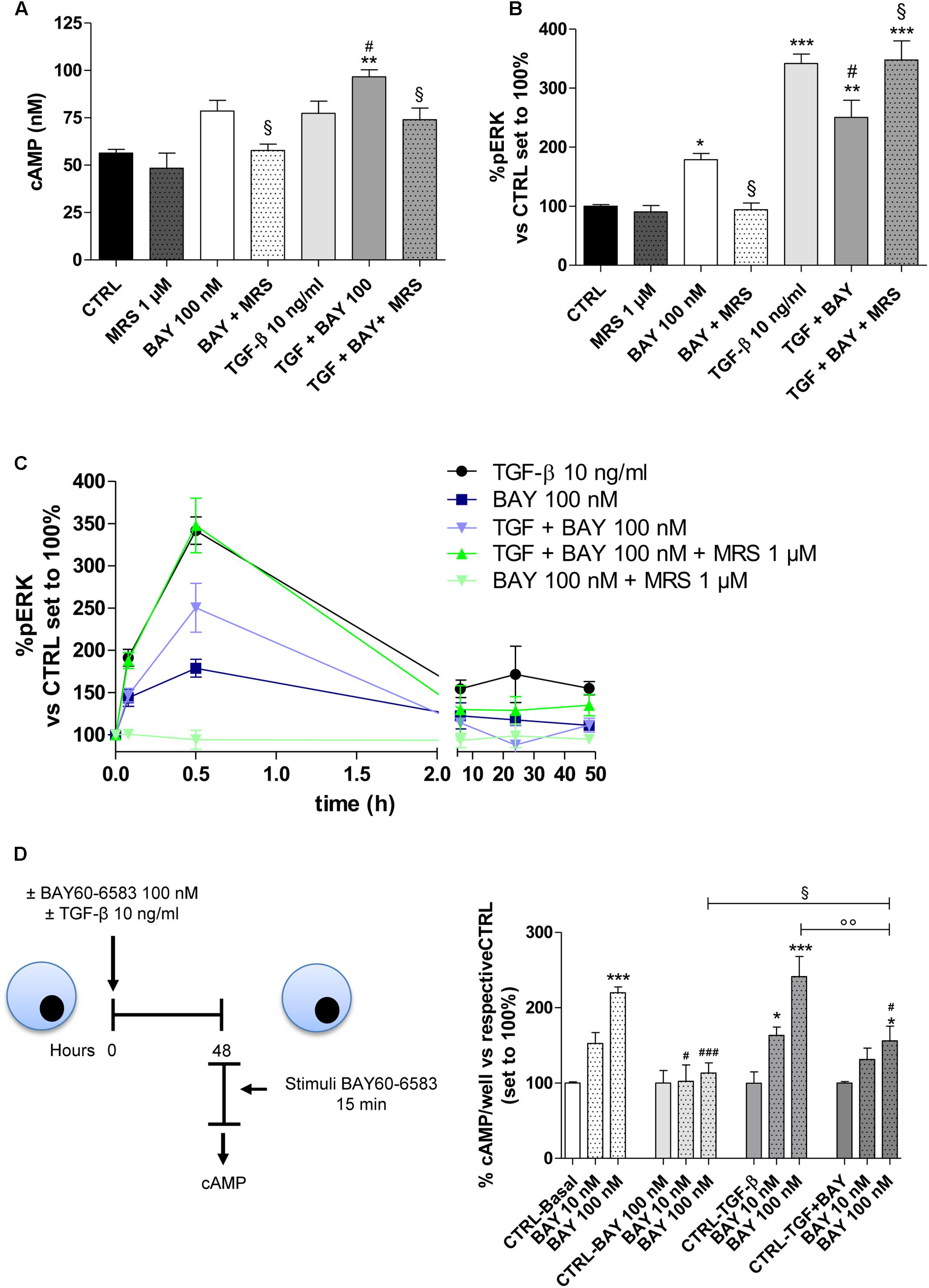
FIGURE 5. Effects of BAY 60-6583 and TGF-β1 on cAMP accumulation and ERK 1/2 phosphorylation. (A) A549 cells were treated with BAY 60-6583 (100 nM) in the absence or presence of TGF-β1 (10 ng/ml) for 48 h. When indicated, the A2BAR inverse agonist, MRS 1706 (1 μM), was applied. At the end of the treatment, the cAMP production was quantified. The data were expressed as cAMP concentrations and were presented as mean values ± SEM of three independent experiments, each performed in duplicate. (B,C) Cells treated as above for different time (30 min-48 h), were fixed, and ERK 1/2 phosphorylation was measured by immunoenzymatic assay. (B) ERK 1/2 phosphorylation after 30 min of treatment. (C) Time-course of ERK 1/2 phosphorylation in A549 cells. The data were expressed as the percentage versus untreated cells (CTRL) set to 100% ± SEM of at least three different experiments performed in duplicate. ∗P ≤ 0.05, ∗∗P ≤ 0.01, ∗∗∗P ≤ 0.001 vs. the CTRL; #P ≤ 0.05 vs. TGF-β1 alone; § P ≤ 0.05 vs. the TGF-β1 + BAY. (D) Cells were treated with BAY 60-6583 (100 nM) in the absence or presence of TGF-β1 (10 ng/ml) for 48 h. After extensive washing, cells were treated for 15 min with different concentrations of BAY60-6583. Next, intracellular cAMP levels were evaluated. The data were expressed as the percentage versus untreated cells (CTRL-Basal) set to 100% ± SEM of at least three different experiments performed in duplicate. ∗P ≤ 0.05, ∗∗∗P ≤ 0.001 vs. the CTRL-Basal; #P ≤ 0.05, ###P ≤ 0.001 vs. respective basal condition. The significance of the differences was determined by one-way ANOVA, followed by Dunnett’s post hoc test or two-way ANOVA with Bonferroni correction and two-sided tests for multiple comparisons.
When A2BAR was activated in the presence of TGF-β1, the receptor activation produced a significant increase in the intracellular cAMP concentration (56.4 ± 1.9 nM, CTRL; 96.6 ± 5.3 nM, TGF-β1 + BAY P ≤ 0.01; Figure 5A). This effect was in accordance with the slight increase in A2BAR functionality that was noticed in the presence of the cytokine (Figure 2D). Regarding pERK1/2, the co-treatment led to a modest decrease in phosphorylation. The BAY activities on cAMP and pERK1/2 were most completely counteracted by cell pre-incubation with the A2BAR inverse agonist MRS1706. Finally, TGF-β1 and BAY did not affect the amount of total ERK (Supplementary Figure 2).
To elucidate if the different activation of cAMP and ERK pathway in the presence of the cytokine, could be related to A2BAR desensitization, the functionality of the receptor was evaluated after 48 h of cell treatment in the absence or presence of BAY and TGF-β1 alone or in combination (Figure 5D). In the basal condition (48 h without treatment), A2BAR maintained its functional response. The cell treatment with BAY (100 nM) for 48 h induced the desensitization of the A2B receptor (219.9 ± 7.7% in Basal condition; 113.3 ± 13.6% in BAY pre-treated cells; P ≤ 0.001). Cytokine alone was not able to significantly interfere on the receptor desensitization. Interestingly, when cells were treated with BAY in the presence of TGF-β1 for 48 h, the desensitization of the receptor was partially impaired (164.2 ± 8.7%; P ≤ 0.01).
cAMP/PKA Regulation of EMT Markers in Human Epithelial Cells
The role of the cAMP pathway in EMT is controversial, depending on the cell type, the nature of the EMT inducers and maximum levels of the intracellular cAMP obtained (Weng et al., 2015). Thus, the effects of different levels of cAMP on EMT markers in our cellular model were investigated using the cAMP analog Br-cAMP (8-bromoadenosine 3′,5′-cyclic monophosphate) as the PKA activator (Figure 6). Br-cAMP (100 nm–1 μM) induced a concentration-dependent increase in the intracellular cAMP levels (Figure 6A). Surprisingly, different concentrations of the activator caused opposite effects on E-cad (Figure 6B) and N-cad (Figure 6C) expression. A high Br-cAMP concentration increased E-cad and decreased N-cad expression. A low Br-cAMP concentration (100 nM) was sufficient to determine a modest decrease of E-cad (1646 ± 56 ng/ml, CTRL; 1540 ± 93 ng/ml, Br-cAMP; Figure 6B) and an increase of N-cad (246 ± 35 ng/ml, CTRL; 270 ± 8 ng/ml, Br-cAMP; Figure 6C) even if these variations were not statistically significant. When TGF-β1 and BAY were used simultaneously, the activator produced higher cAMP levels and a concentration-dependent increase in E-cad and decrease in N-cad.
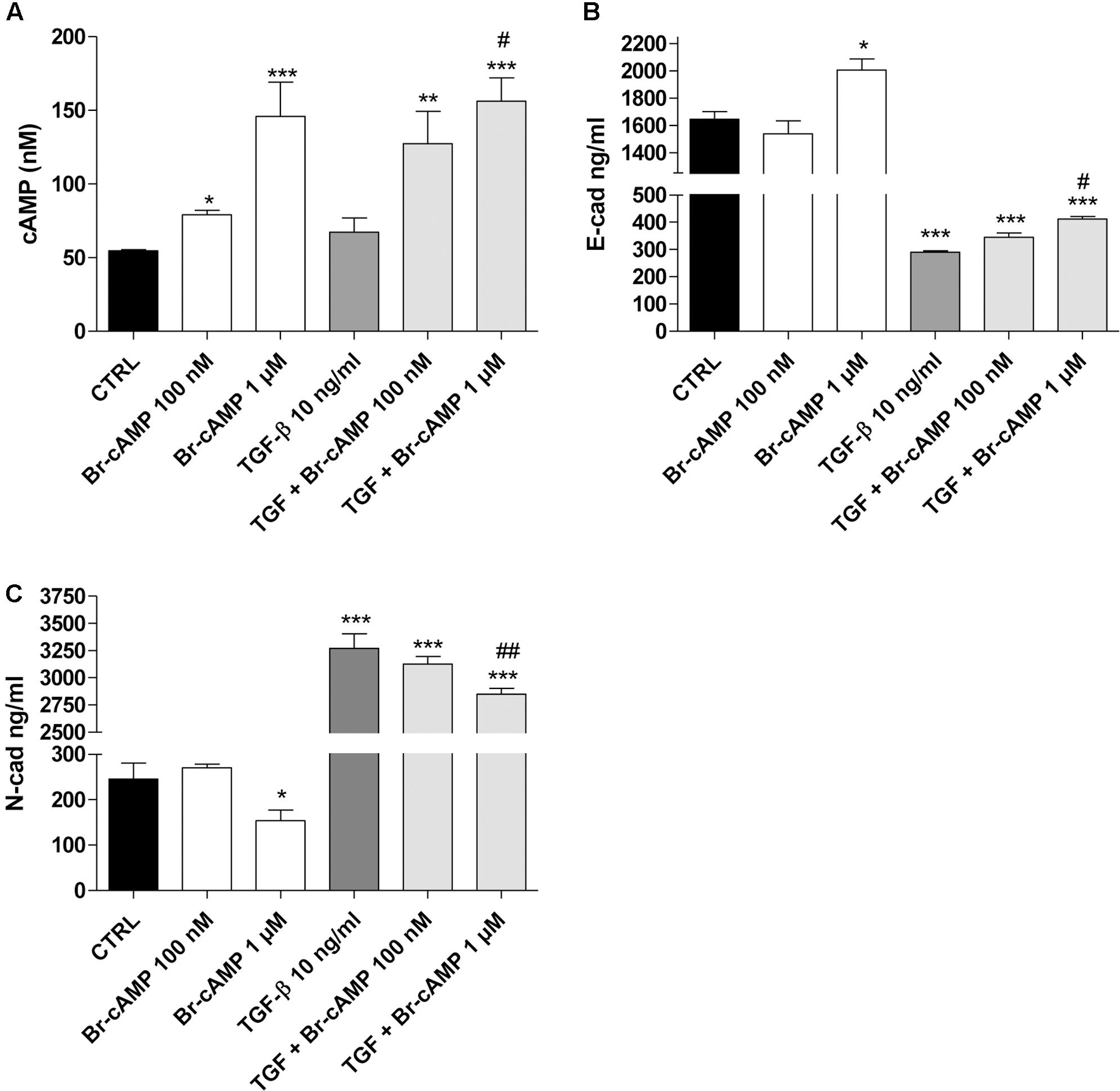
FIGURE 6. Effect of the cAMP pathway on E-cadherin and N-cadherin expression. A549 cells were treated with Br-cAMP (100 nm–1 μM) in the absence or presence of TGF-β1 (10 ng/ml) for 48 h. (A) At the end of the treatment, cAMP production was quantified. The data were expressed as cAMP concentrations and are presented as mean values ± SEM of three independent experiments, each performed in duplicate. Cells treated as above were lysed, and the expression of E-cadherin (B) or N-cadherin (C) was quantified using AlphaLISA kits. The data were expressed as E-cadherin and N-cadherin concentrations (ng/ml) and are presented as mean values ± SEM of three independent experiments, each performed in duplicate. The significance of the differences was determined by two-way ANOVA with Bonferroni correction and two-sided tests for multiple comparisons. ∗P ≤ 0.05, ∗∗P ≤ 0.01, ∗∗∗P ≤ 0.001 vs. the CTRL; #P ≤ 0.05, ##P ≤ 0.01, vs. TGF-β1 alone.
These data demonstrate that, in our cellular model, when the cAMP was increased up to high concentrations, it could negatively affect the EMT process. Conversely, when cAMP levels remained low, it seems to mediate the opposite effects, promoting a slight decrease in epithelial markers and an increase in the mesenchymal markers. This controversial nature of the cAMP analogue on the EMT process is in accordance with the recent data reported by Zuccarini et al. (2017).
The cAMP/PKA-MAPK/ERK Balance in the A2BAR Modulation of EMT Markers and EMT-TFs
Nevertheless, high levels of cAMP counteract EMT, and ERK phosphorylation mediated by MAPK activation has been demonstrated to promote EMT (Singh et al., 2017). Thus, the ability of A2BAR to modify the amounts of both pERK and of cAMP could be a possible mechanism by which the receptor agonist affects epithelial/mesenchymal markers. In this respect, the involvement of A2BAR in the regulation of EMT markers and transcription factors was investigated in the presence of PKA inhibitor (H89, 100 nM) and a mitogen-activated protein kinase inhibitor (PD98059, 1 μM) (Figure 7, Supplementary Figure 3). The BAY-mediated increase in the intracellular cAMP levels was not affected by H89 treatment (Figure 7A); conversely, PD98059 was able to block BAY-mediated ERK phosphorylation (Figure 7B). In this respect, the inhibitors were used to unveil the involvement of the two signaling pathways in A2BAR mediated triggering of EMT.
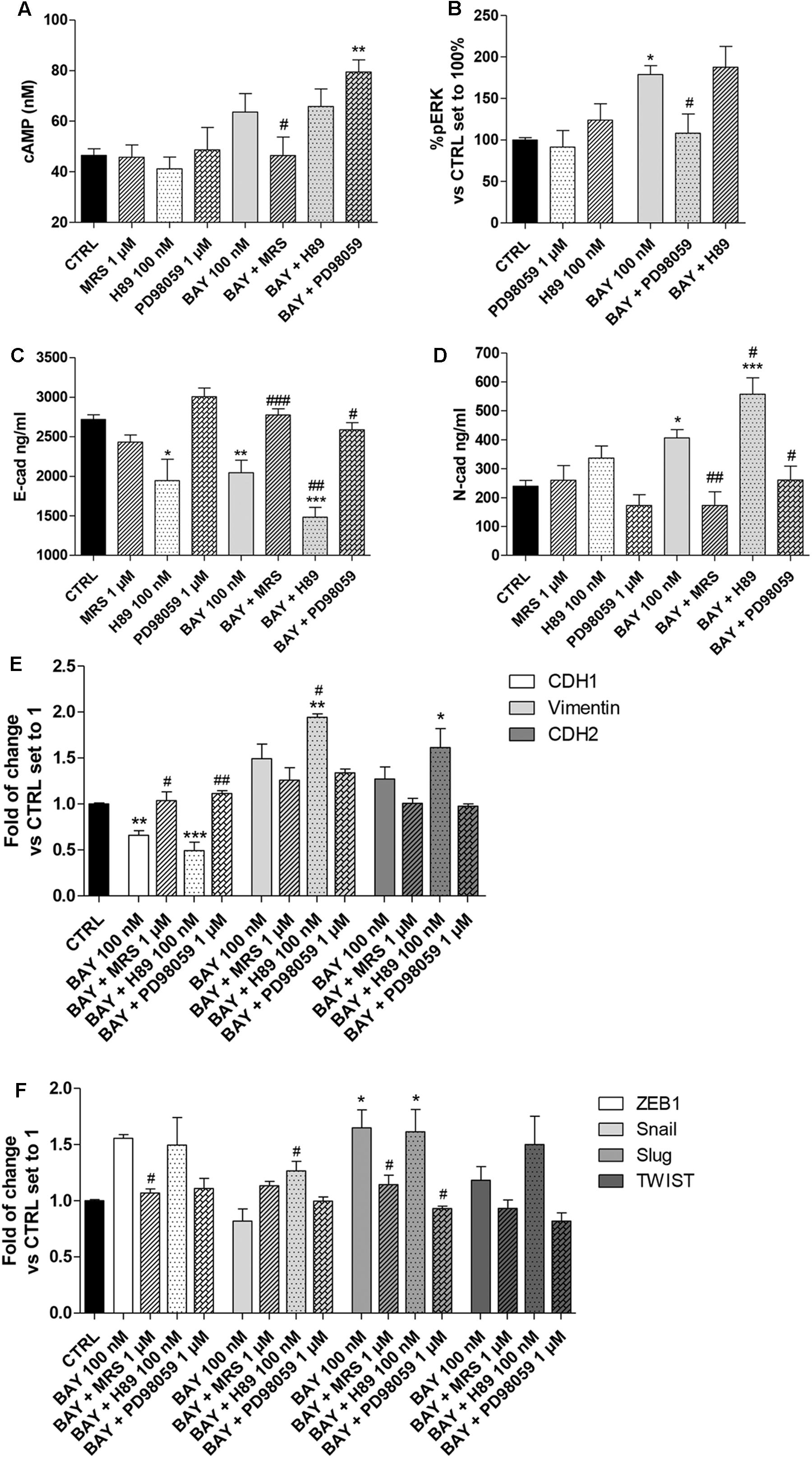
FIGURE 7. Effect of cAMP and ERK1/2 phosphorylation blockade on A2BAR-mediated changes in EMT markers. A549 cells were treated with BAY 60-6583 (100 nM) in the absence or presence of MRS 1706 (1 μM), PKA inhibitor H89 (100 nM) or MEK1/2 inhibitor PD98059 (1 μM) for 48 h. When indicated, the antagonist and inhibitor were applied 30 min before treatment with BAY 60-6583. (A) At the end of the treatment, cAMP production was quantified. The data were expressed as cAMP concentrations and are the mean values ± SEM of three independent experiments, each performed in duplicate. (B) Cells were treated as above and ERK 1/2 phosphorylation was evaluated after 30 min of treatment. The data were expressed as the percentage versus untreated cells (CTRL) set to 100%±SEM of at least three different experiments performed in duplicate. Cells treated as above were lysed, and the expression of E-cadherin (C) or N-cadherin (D) were quantified using AlphaLISA kits. The data were expressed as E-cadherin and N-cadherin concentrations (ng/ml) and are the mean values±SEM of three independent experiments, each performed in duplicate. The significance of the differences was determined by one-way ANOVA, followed by Dunnett’s post hoc test or two-way ANOVA with Bonferroni correction and two-sided tests for multiple comparisons. ∗P ≤ 0.05, ∗∗P ≤ 0.01, ∗∗∗P ≤ 0.001 vs. the CTRL; #P ≤ 0.05, ##P ≤ 0.01, ###P ≤ 0.001 vs. BAY alone. (E,F) Cells were treated as above and real-time RT-PCR analysis of EMT markers (E-cad, N-cad and Vimentin) (E), and EMT transcription factors (ZEB1, Snail, Slug, TWIST) (F) was performed. The data were expressed as the fold change vs. the CTRL levels, which were set to 1 and are the mean values±SEM of three different experiments each performed in duplicate. The significance of the differences was determined by one-way ANOVA, followed by Dunnett’s post hoc test or two-way ANOVA with Bonferroni correction and two-sided tests for multiple comparisons. ∗P ≤ 0.05, ∗∗P ≤ 0.01, ∗∗∗P ≤ 0.001 vs. the CTRL; #P ≤ 0.05, ##P ≤ 0.01 vs. BAY alone.
BAY decreased the protein density of E-cad and increased that of the N-cad one; these effects were lost in the presence of the inverse agonist MRS 1706, in accordance with the abovementioned data (Figure 3). The ability of BAY to decrease the expression of E-cad was augmented by pretreatment with the PKA inhibitor (2718 ± 58 ng/ml, CTRL; 2045 ± 157 ng/ml, BAY; P ≤ 0.01; 1483 ± 122 ng/ml, BAY + H89; P ≤ 0.001 vs. CTRL, P ≤ 0.01 vs BAY, Figure 7C). Similarly, A2BAR mediated increase in N-cad was enhanced when the cAMP/PKA axis was abrogated (239 ± 20 ng/ml, CTRL; 406 ± 29 ng/ml, BAY; P ≤ 0.05; 558 ± 72 ng/ml, BAY + H89; P ≤ 0.001 vs. CTRL, P ≤ 0.05 vs BAY; Figure 7D). These effects highlighted that cAMP, through PKA activation, could partially antagonize A2BAR-mediated EMT induction. When the MEK inhibitor was used, the changes induced by A2BAR stimulation were almost completely counteracted, restoring the basal conditions (E-cad, 2586 ± 90 ng/ml, BAY + PD98059; P ≤ 0.05 vs BAY; N-cad, 260 ± 49 ng/ml, BAY + PD89059; P ≤ 0.05 vs BAY, Figures 7C,D). These results are in accordance with the effects of the inhibitor treatments on A2BAR-mediated alteration of the expression of CDH1, CDH2 and vimentin genes (Figure 7E).
Finally, the modulation of the EMT-TF gene expression by BAY was evaluated in the presence of H89 and PD98059. Similarly, regarding the EMT markers, the PKA inhibitor slightly exacerbated the A2BAR-mediated effects; conversely, the MEK inhibitor counteracted the BAY-mediated increase in gene expression. The PD98059 activity was more pronounced on Slug expression, causing a significant decrease of gene expression (1.65 ± 0.16-fold, BAY; 1.61 ± 0.20-fold, BAY + H89; 0.93 ± 0.09-fold vs. CTRL, BAY + PD98059; P ≤ 0.05). These data are consistent with the reported role of ERK phosphorylation in the induction of Slug expression (Choi et al., 2007; Joannes et al., 2014).
Discussion
Adenosine receptors (ARs) have attracted great attention as possible targets to control the EMT process in pathologies such as fibrosis and cancer (Lu and Insel, 2014). However, among the AR subtypes, no data have been reported that correlate A2BAR activation with EMT in epithelial cells. In this respect, herein, we report for the first time the ability of A2BAR activation to promote EMT or contrast the effects of the extracellular inducer, TGF-β1, in human lung epithelial cells. Furthermore, the ability of the receptor activation to modulate two signaling pathways involved in EMT, cAMP/PKA, and MAPK/ERK, was demonstrated as a possible mechanism explaining the different effects mediated by A2BAR stimulation in different extracellular microenvironment (Figure 8).
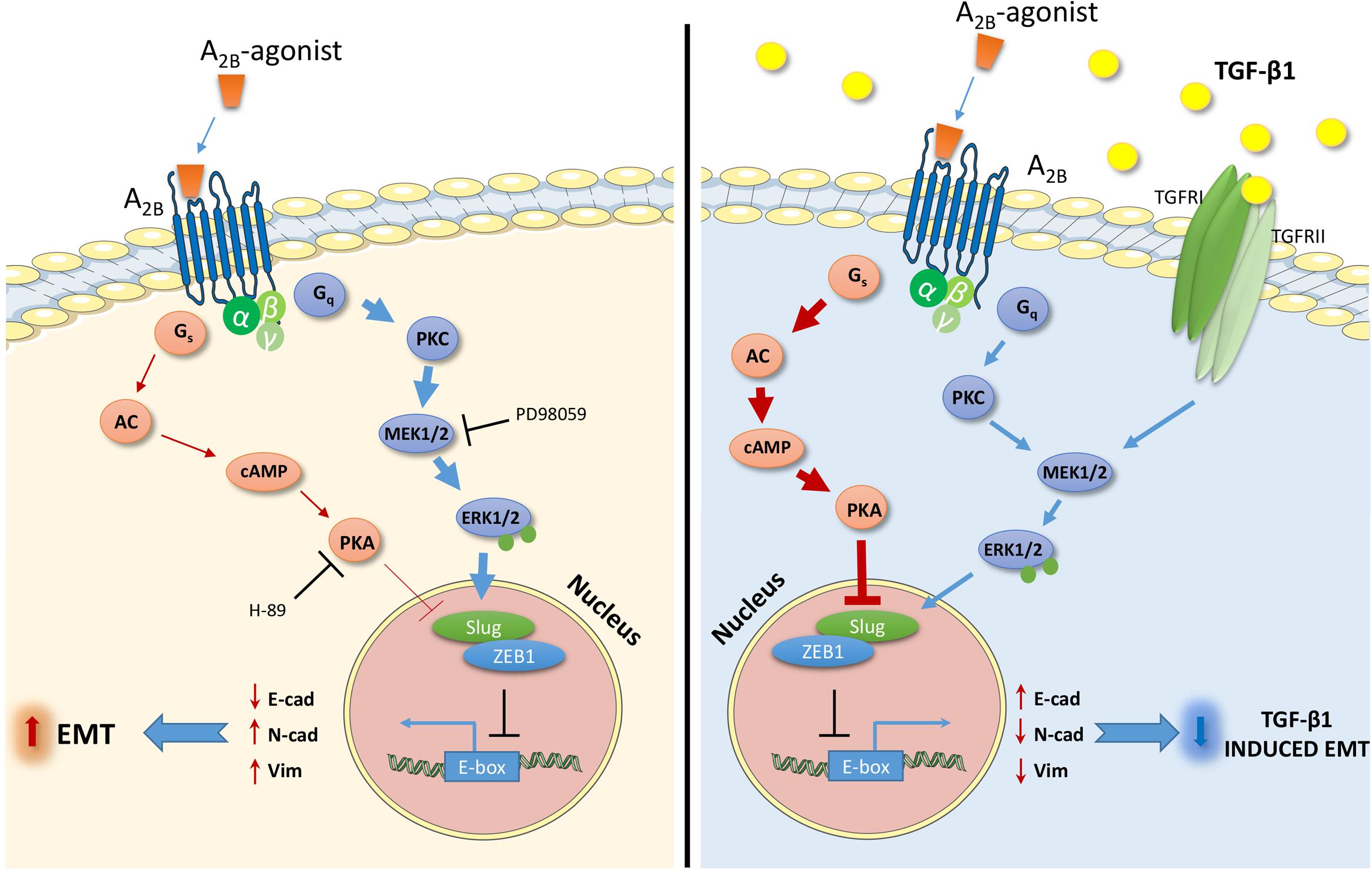
FIGURE 8. Schematic representation of the effects and the intracellular signals activated by the A2BAR stimulation in the absence (left) or presence (right) of TGF-β1. The A2BAR activates cAMP/PKA pathways and ERK1/2 phosphorylation. The extent of the activation changes in the presence of the TGF-β1 cytokine (bold arrow), causing different effects on the expression of transcription factors and markers related to EMT. The receptor activation increases the EMT process (left), and counteracts the TGF-β1-induced EMT (right).
Human carcinoma epithelial lung cells (A549) were maintained under serum deprivation, a condition that has been demonstrated to amplify the epithelial phenotype (Dong et al., 2014). Although all the ARs are expressed in epithelial lung cells, the A2B receptor subtype was demonstrated to be the most represented, as previously reported (Roman et al., 2006). Challenging these cells with TGF-β1 produced a marked change in the phenotype through the simultaneous decrease in epithelial markers (E-cad) and increase in mesenchymal markers (N-cad). In accordance with the literature (Kawata et al., 2012), these data demonstrated the suitability of the model to study the A2BAR involvement in the EMT process.
A2BAR stimulation modified the expression of epithelial/mesenchymal markers with a low but significant amplitude. Surprisingly, the same receptor could counteract the EMT induction mediated by TGF-β1. These opposite effects elicited by A2BAR stimulation in lung cells reflect the well documented debated role of this receptor in the onset and progression of different pathologies. The receptor activation has been reported to protect the lungs and other tissues from inflammation and acute injury (Eckle et al., 2008; Davies et al., 2014; Hoegl et al., 2015). By contrast, other evidence has been reported concerning the A2BAR-mediated pro-fibrotic effects and promotion of cancer development (Karmouty-Quintana et al., 2012; Huerter et al., 2016; Sepúlveda et al., 2016). Furthermore, Zhou et al. (2011) have shown that A2BAR genetic removal slightly affects acute lung injury but reduces lung fibrosis, supporting a pro-fibrotic role for this receptor.
In our study, the A2BAR selective agonist regulated the gene transcription of different EMT-transcription factors (EMT-TFs). EMT is a complex process regulated by the orchestration of several transcription factors. Among these EMT-TFs, ZEB1, Snail and Slug (SNAI2) are the major transcription factors involved in the promotion of the EMT process in A549 cells, as previously reported (Ji et al., 2016). A2BAR stimulation did not affect significantly the expression of Snail and TWIST, other master genes of EMT. However, A2BAR activation modulate the expression of Slug and, to a lesser extent, that of ZEB1, which are commonly considered the major actors in myofibroblast differentiation and fibrogenesis (Lamouille et al., 2014). Furthermore, Slug expression has been correlated to direct repression of E-cadherin expression (Choi et al., 2007). This evidence was in accordance with the increase in E-cadherin induced by A2BAR stimulation.
The EMT-TFs are regulated by the activation of different intracellular pathways, which in turn regulate the balance between the epithelial and mesenchymal markers. ERK1/2, GSK3β, p38, STAT3, and cAMP are only few of all the proteins involved in the promotion/inhibition of EMT (Nieto et al., 2016). In our cellular model, activation of the cAMP/PKA axis promoted opposite effects depending on the levels of intracellular the cAMP. Low concentrations led to a slight promotion or no effects on epithelial/mesenchymal markers; conversely, high levels of cAMP could counteract EMT progression. In accordance with our results, it has been reported that cAMP/PKA activation could exert opposite effects on EMT (Zhang et al., 2006a,b; Nadella et al., 2008; Shaikh et al., 2012; Lambers et al., 2015) that could be ascribed to the different cell models, type of EMT inducer and maximum levels of intracellular cAMP obtained (Weng et al., 2015).
A2BAR activation increases the intracellular cAMP levels and promotes the phosphorylation of ERK 1/2 (Grant et al., 2001; Zhou et al., 2017). In this respect, we hypothesize that the controversial role of A2BAR in promoting EMT or reversing TGF-β1-induced EMT could be ascribed to the different balance of the intracellular pathways activated. The activation of other GPCRs coupled to different G proteins has been reported to elicit opposite effects on EMT. Zhang et al. (2016) have demonstrated that norepinephrine, which activates Gs protein, induces the epithelial-mesenchymal transition in A549 cells; similarly, the P2X7 stimulation, which activates the PI3K/Akt and ERK1/2 signaling pathways, promotes the mesenchymal phenotype in epithelial renal cells (Zuccarini et al., 2017). A2AARs have been reported to modify the epithelial/mesenchymal phenotype in renal cells by downregulating TGF-β1-induced EMT (Zuccarini et al., 2017) in accordance with the effects elicited by A2BAR activation in our cellular model when the cytokine was present. TGF-β1 activates both Smad signaling and non-Smad signaling including the ERK pathway, critically regulating EMT (Xie et al., 2004). A2BAR stimulation could counteract cytokine-induced EMT, reducing the transition progression. These effects were correlated with a robust increase in cAMP production and with a concomitant decrease in ERK phosphorylation. In this respect, we could speculate that the increase in the cAMP, mediated by A2BAR activation, could counteract the cytokine-mediated effects on EMT. This is in accordance with the ability of other Gs coupled receptors (i.e., A2AAR) or cAMP inducers (i.e., forskolin, 8-Br-cAMP) to prevent TGF-β1-mediated EMT (Zhang et al., 2006a,b; Zuccarini et al., 2017). It should also be considered that cAMP/PKA has a negative effect on TGF-β induced pERK 1/2, but only when the cytokine downstream pathways are highly expressed (Weng et al., 2015). This mechanism could explain the reduction in ERK1/2 phosphorylation in the presence of the A2BAR agonist.
The modulation of cAMP production and ERK phosphorylation could be affected by internalization or desensitization processes of the receptor. Several GPCRs employ a variety of signaling mechanisms to exert their functions. The production of second messengers, such as cAMP, is the results of the receptor-G protein coupling. However, other signals produced in conjunction with, or even independent of heterotrimeric G protein could be activated. It is well known that the arrestins, a family of four GPCR-binding proteins originally described for their role in the GPCR desensitization, have been found to interact with Src family kinases and to serve as scaffolds for activation of ERK1/2 and c-Jun N-terminal kinase 3 mitogen-activated protein kinases (Ferguson, 2001; Luttrell, 2005; Lee et al., 2008). Thus, the desensitization could represent a mechanism to maintain the activation of G-protein independent pathways. The activation of the A2BAR for long time produced the desensitization of the receptor. This may be another mechanism through which A2BAR mediate the ERK phosphorylation favoring the EMT traits. Conversely, the TGF-β1 was able to reduce the agonist-induced A2BAR desensitization, maintaining the ability of A2BAR to produce cAMP that negatively affected EMT process. These data could better highlight the controversial role of A2BAR in the EMT. In different pathological status in which adenosine raise to micromolar concentration, the extracellular microenvironment and the localization of the receptor could interfere with the levels of the activated intracellular signaling shifting the balance in favor of or against the EMT.
In our cellular model, A2BAR activation was able to induce partial EMT; however, it was not sufficient to promote a complete epithelial-mesenchymal transformation of the lung cells. The involvement of the cAMP/PKA and MAPK/ERK signaling pathways was demonstrated by the use of selective inhibitors. The EMT traits, in the presence of A2BAR agonist was enhanced by PKA inhibition and decreased by the ERK1/2 phosphorylation blockade. Furthermore, the pretreatment with a PKA inhibitor did not enhance receptor effects on TF expression. Conversely, the presence of the MEK inhibitor completely counteracted the A2B-mediated increase in Slug and ZEB1 gene expression, demonstrating the involvement of MAPK/ERK pathway activation in A2BAR-mediated EMT induction.
Conclusion
For the first time, these results highlight the possibility that A2BAR is one of the numerous receptors that could be involved in EMT regulation. Although the expression of the other adenosine receptor subtypes are low, in physiological condition the involvement of all the AR in the EMT regulation could not be excluded. A2BAR activation is not enough to trigger a complete transition; it can affect the expression of the epithelial and mesenchymal markers promoting EMT traits. These effects are related to the ability of A2BAR to modify the balance between cAMP/PKA and MAPK/ERK activation. The presence of extracellular cytokines that activate the transition could change the balance and levels of these two intracellular pathways, shifting the effects exerted by the receptor stimulation on EMT trigger and maintenance. Thus, the A2BAR seems to act as an EMT regulator rather than a main actor in the EMT induction. Although the EMT is a complex machinery and further investigation are needed to better elucidate the effects of adenosine and the contribution of the other AR subtypes in physiopathological conditions, herein, for the first time, the A2BAR was shown to be related to the EMT process, highlighting its potential role as pharmacological target in EMT-related pathologies.
Author Contributions
CG designed and performed the biological experiments. CG, SD, and MT. analyzed the data and wrote the manuscript. IP performed the immunofluorescence analysis. CR, LT, and TN contributed to the experimental work. MT, AC, and CM designed the study and played a key role as project supervisor. MT and CM coordinated the project. All the authors contributed to and approved the final manuscript.
Funding
This work was financially supported by the Italian Ministry of Education (Project of National Research Interest PRIN 2015, 2015E8EMCM_007).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2018.00054/full#supplementary-material
References
Boucher, M. J., Laprise, P., and Rivard, N. (2005). Cyclic AMP-dependent protein kinase A negatively modulates adherens junction integrity and differentiation of intestinal epithelial cells. J. Cell. Physiol. 202, 178–190. doi: 10.1002/jcp.20104
Choi, J., Park, S. Y., and Joo, C. K. (2007). Transforming growth factor-beta1 represses E-cadherin production via slug expression in lens epithelial cells. Invest. Ophthalmol. Vis. Sci. 48, 2708–2718. doi: 10.1167/iovs.06-0639
Da Pozzo, E., La Pietra, V., Cosimelli, B., Da Settimo, F., Giacomelli, C., Marinelli, L., et al. (2014). p53 functional inhibitors behaving like pifithrin-β counteract the Alzheimer peptide non-β-amyloid component effects in human SH-SY5Y cells. ACS Chem. Neurosci. 5, 390–399. doi: 10.1021/cn4002208
Daniele, S., Natali, L., Giacomelli, C., Campiglia, P., Novellino, E., Martini, C., et al. (2017). Osteogenesis is improved by low tumor necrosis factor alpha concentration through the modulation of Gs-coupled receptor signals. Mol. Cell. Biol. 37:e00442-16. doi: 10.1128/MCB.00442-16
Davies, J., Karmouty-Quintana, H., Le, T. T., Chen, N. Y., Weng, T., Luo, F., et al. (2014). Adenosine promotes vascular barrier function in hyperoxic lung injury. Physiol. Rep. 2:e12155. doi: 10.14814/phy2.12155
De Maio, L., Buckley, S. T., Krishnaveni, M. S., Flodby, P., Dubourd, M., Banfalvi, A., et al. (2012). Ligand-independent transforming growth factor-β type I receptor signalling mediates type I collagen-induced epithelial-mesenchymal transition. J. Pathol. 226, 633–644. doi: 10.1002/path.3016
Dong, S., Khoo, A., Wei, J., Bowser, R. K., Weathington, N. M., Xiao, S., et al. (2014). Serum starvation regulates E-cadherin upregulation via activation of c-Src in non-small-cell lung cancer A549 cells. Am. J. Physiol. Cell Physiol. 307, C893–C899. doi: 10.1152/ajpcell.00132.2014
Eckle, T., Grenz, A., Laucher, S., and Eltzschig, H. K. (2008). A2B adenosine receptor signaling attenuates acute lung injury by enhancing alveolar fluid clearance in mice. J. Clin. Invest. 118, 3301–3315. doi: 10.1172/JCI34203
Fang, Y., and Olah, M. E. (2007). Cyclic AMP-dependent, protein kinase A-independent activation of extracellular signal-regulated kinase 1/2 following adenosine receptor stimulation in human umbilical vein endothelial cells: role of exchange protein activated by cAMP 1 (Epac1). J. Pharmacol. Exp. Ther. 322, 1189–1200. doi: 10.1124/jpet.107.119933
Feoktistov, I., Goldstein, A. E., and Biaggioni, I. (1999). Role of p38 mitogen-activated protein kinase and extracellular signal-regulated protein kinase kinase in adenosine A2B receptor-mediated interleukin-8 production in human mast cells. Mol. Pharmacol. 55, 726–734.
Ferguson, S. S. (2001). Evolving concepts in G protein-coupled receptor endocytosis: the role in receptor desensitization and signaling. Pharmacol. Rev. 53, 1–24.
Gao, Z. W., Wang, H. P., Dong, K., Lin, F., Wang, X., and Zhang, H. Z. (2016). Adenosine inhibits migration, invasion and induces apoptosis of human cervical cancer cells. Neoplasma 63, 201–207. doi: 10.4149/204_150723N407
Giacomelli, C., Trincavelli, M. L., Satriano, C., Hansson,Ö, La Mendola, D., Rizzarelli, E., et al. (2015). ♦Copper (II) ions modulate Angiogenin activity in human endothelial cells. Int. J. Biochem. Cell Biol. 60, 185–196. doi: 10.1016/j.biocel.2015.01.005
Grant, M. B., Davis, M. I., Caballero, S., Feoktistov, I., Biaggioni, I., and Belardinelli, L. (2001). Proliferation, migration, and ERK activation in human retinal endothelial cells through A(2B) adenosine receptor stimulation. Invest. Ophthalmol. Vis. Sci. 42, 2068–2073.
Gui, T., Sun, Y., Shimokado, A., and Muragaki, Y. (2012). The roles of mitogen-activated protein kinase pathways in TGF-β-induced epithelial-mesenchymal transition. J. Signal Transduct. 2012:289243. doi: 10.1155/2012/289243
Guillén-Gómez, E., Pinilla-Macua, I., Pérez-Torras, S., Choi, D. S., Arce, Y., Ballarín, J. A., et al. (2012). New role of the human equilibrative nucleoside transporter 1 (hENT1) in epithelial-to-mesenchymal transition in renal tubular cells. J. Cell Physiol. 227, 1521–1528. doi: 10.1002/jcp.22869
Hoegl, S., Brodsky, K. S., Blackburn, M. R., Karmouty-Quintana, H., Zwissler, B., and Eltzschig, H. K. (2015). Alveolar epithelial A2B adenosine receptors in pulmonary protection during acute lung injury. J. Immunol. 195, 1815–1824. doi: 10.4049/jimmunol.1401957
Huerter, M. E., Sharma, A. K., Zhao, Y., Charles, E. J., Kron, I. L., and Laubach, V. E. (2016). Attenuation of pulmonary ischemia-reperfusion injury by adenosine A2B receptor antagonism. Ann. Thorac. Surg. 102, 385–393. doi: 10.1016/j.athoracsur.2016.02.060
Ji, Y., Dou, Y. N., Zhao, Q. W., Zhang, J. Z., Yang, Y., Wang, T., et al. (2016). Paeoniflorin suppresses TGF-β mediated epithelial-mesenchymal transition in pulmonary fibrosis through a Smad-dependent pathway. Acta Pharmacol. Sin. 37, 794–804. doi: 10.1038/aps.2016.36
Joannes, A., Grelet, S., Duca, L., Gilles, C., Kileztky, C., Dalstein, V., et al. (2014). Fhit regulates EMT targets through an EGFR/Src/ERK/Slug signaling axis in human bronchial cells. Mol. Cancer Res. 12, 775–783. doi: 10.1158/1541-7786.MCR-13-0386-T
Jolly, M. K., Ward, C., Eapen, M. S., Myers, S., Hallgren, O., Levine, H., et al. (2017). Epithelial-mesenchymal transition, a spectrum of states: role in lung development, homeostasis, and disease. Dev. Dyn. doi: 10.1002/dvdy.24541 [Epub ahead of print].
Jonsdottir, H. R., Arason, A. J., Palsson, R., Franzdottir, S. R., Gudbjartsson, T., Isaksson, H. J., et al. (2015). Basal cells of the human airways acquire mesenchymal traits in idiopathic pulmonary fibrosis and in culture. Lab. Invest. 95, 1418–1428. doi: 10.1038/labinvest.2015.114
Karmouty-Quintana, H., Weng, T., Garcia-Morales, L. J., Chen, N. Y., Pedroza, M., Zhong, H., et al. (2013). Adenosine A2B receptor and hyaluronan modulate pulmonary hypertension associated with chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 49, 1038–1047. doi: 10.1165/rcmb.2013-0089OC
Karmouty-Quintana, H., Zhong, H., Acero, L., Weng, T., Melicoff, E., West, J. D., et al. (2012). The A2B adenosine receptor modulates pulmonary hypertension associated with interstitial lung disease. FASEB J. 26, 2546–2557. doi: 10.1096/fj.11-200907
Kawata, M., Koinuma, D., Ogami, T., Umezawa, K., Iwata, C., Watabe, T., et al. (2012). TGF-β-induced epithelial-mesenchymal transition of A549 lung adenocarcinoma cells is enhanced by pro-inflammatory cytokines derived from RAW 264.7 macrophage cells. J. Biochem. 151, 205–216. doi: 10.1093/jb/mvr136
Kim, J. H., Jang, Y. S., Eom, K. S., Hwang, Y. I., Kang, H. R., Jang, S. H., et al. (2007). Transforming growth factor beta1 induces epithelial-to-mesenchymal transition of A549 cells. J. Korean Med. Sci. 22, 898–904. doi: 10.3346/jkms.2007.22.5.898
Lambers, C., Costa, L., Ying, Q., Zhong, J., Lardinois, D., Dekan, G., et al. (2015). Aclidinium bromide combined with formoterol inhibits remodeling parameters in lung epithelial cells through cAMP. Pharmacol. Res. 102, 310–318. doi: 10.1016/j.phrs.2015.09.010
Lamouille, S., Xu, J., and Derynck, R. (2014). Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 15, 178–196. doi: 10.1038/nrm3758
Lee, J. M., Dedhar, S., Kalluri, R., and Thompson, E. W. (2006). The epithelial-mesenchymal transition: new insights in signaling, development, and disease. J. Cell Biol. 172, 973–981. doi: 10.1083/jcb.200601018
Lee, M. H., El-Shewy, H. M., Luttrell, D. K., and Luttrell, L. M. (2008). Role of beta-arrestin-mediated desensitization and signaling in the control of angiotensin AT1a receptor-stimulated transcription. J. Biol. Chem. 283, 2088–2097. doi: 10.1074/jbc.M706892200
Legras, A., Pécuchet, N., Imbeaud, S., Pallier, K., Didelot, A., Roussel, H., et al. (2017). Epithelial-to-mesenchymal transition and MicroRNAs in lung cancer. Cancers 9:E101. doi: 10.3390/cancers9080101
Lu, D., and Insel, P. A. (2014). Cellular mechanisms of tissue fibrosis. 6. Purinergic signaling and response in fibroblasts and tissue fibrosis. Am. J. Physiol. Cell Physiol. 306, C779–C788. doi: 10.1152/ajpcell.00381.2013
Luttrell, L. M. (2005). Composition and function of g protein-coupled receptor signalsomes controlling mitogen-activated protein kinase activity. J. Mol. Neurosci. 26, 253–264. doi: 10.1385/JMN:26:2-3:253
Martínez-Ramírez, A. S., Díaz-Muñoz, M., Butanda-Ochoa, A., and Vázquez-Cuevas, F. G. (2017). Nucleotides and nucleoside signaling in the regulation of the epithelium to mesenchymal transition (EMT). Purinergic Signal. 13, 1–12. doi: 10.1007/s11302-016-9550-3
Mittal, D., Sinha, D., Barkauskas, D., Young, A., Kalimutho, M., Stannard, K., et al. (2016). Adenosine 2B receptor expression on cancer cells promotes metastasis. Cancer Res. 76, 4372–4382. doi: 10.1158/0008-5472.CAN-16-0544
Morbini, P., Inghilleri, S., Campo, I., Oggionni, T., Zorzetto, M., and Luisetti, M. (2011). Incomplete expression of epithelial-mesenchymal transition markers in idiopathic pulmonary fibrosis. Pathol. Res. Pract. 207, 559–567. doi: 10.1016/j.prp.2011.06.006
Nadella, K. S., Jones, G. N., Trimboli, A., Stratakis, C. A., Leone, G., and Kirschner, L. S. (2008). Targeted deletion of Prkar1a reveals a role for protein kinase A in mesenchymal-to-epithelial transition. Cancer Res. 68, 2671–2677. doi: 10.1158/0008-5472.CAN-07-6002
Nieto, M. A., Huang, R. Y., Jackson, R. A., and Thiery, J. P. (2016). EMT: 2016. Cell 166, 21–45. doi: 10.1016/j.cell.2016.06.028
O’Connor, J. W., and Gomez, E. W. (2014). Biomechanics of TGFβ-induced epithelial-mesenchymal transition: implications for fibrosis and cancer. Clin. Transl. Med. 3:23. doi: 10.1186/2001-1326-3-23
Ohta, A., and Sitkovsky, M. (2001). Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature 414, 916–920. doi: 10.1038/414916a
Pattabiraman, D. R., Bierie, B., Kober, K. I., Thiru, P., Krall, J. A., Zill, C., et al. (2016). Activation of PKA leads to mesenchymal-to-epithelial transition and loss of tumor-initiating ability. Science 351:aad3680. doi: 10.1126/science.aad3680
Phosri, S., Arieyawong, A., Bunrukchai, K., Parichatikanond, W., Nishimura, A., Nishida, M., et al. (2017). Stimulation of Adenosine A2B receptor inhibits endothelin-1-induced cardiac fibroblast proliferation and α-smooth muscle actin synthesis through the cAMP/Epac/PI3K/Akt-signaling pathway. Front. Pharmacol. 8:428. doi: 10.3389/fphar.2017.00428
Roberts, V. S., Cowan, P. J., Alexander, S. I., Robson, S. C., and Dwyer, K. M. (2014). The role of adenosine receptors A2A and A2B signaling in renal fibrosis. Kidney Int. 86, 685–692. doi: 10.1038/ki.2014.244
Roman, J., Rivera, H. N., Roser-Page, S., Sitaraman, S. V., and Ritzenthaler, J. D. (2006). Adenosine induces fibronectin expression in lung epithelial cells: implications for airway remodeling. Am. J. Physiol. Lung Cell. Mol. Physiol. 290, L317–L325. doi: 10.1152/ajplung.00118.2005
Ryzhov, S., Sung, B. H., Zhang, Q., Weaver, A., Gumina, R. J., Biaggioni, I., et al. (2014). Role of adenosine A2B receptor signaling in contribution of cardiac mesenchymal stem-like cells to myocardial scar formation. Purinergic Signal. 10, 477–486. doi: 10.1007/s11302-014-9410-y
Sakuma, Y. (2017). Epithelial-to-mesenchymal transition and its role in EGFR-mutant lung adenocarcinoma and idiopathic pulmonary fibrosis. Pathol. Int. 67, 379–388. doi: 10.1111/pin.12553
Schulte, G., and Fredholm, B. B. (2003). Signalling from adenosine receptors to mitogen-activated protein kinases. Cell. Signal. 15, 813–827. doi: 10.1016/S0898-6568(03)00058-5
Sepúlveda, C., Palomo, I., and Fuentes, E. (2016). Role of adenosine A2b receptor overexpression in tumor progression. Life Sci. 166, 92–99. doi: 10.1016/j.lfs.2016.10.008
Shaikh, D., Zhou, Q., Chen, T., Ibe, J. C., Raj, J. U., and Zhou, G. (2012). cAMP-dependent protein kinase is essential for hypoxia-mediated epithelial-mesenchymal transition, migration, and invasion in lung cancer cells. Cell. Signal. 24, 2396–2406. doi: 10.1016/j.cellsig.2012.08.007
Shi, L., Dong, N., Fang, X., and Wang, X. (2016). Regulatory mechanisms of TGF-β1-induced fibrogenesis of human alveolar epithelial cells. J. Cell Mol. Med. 20, 2183–2193. doi: 10.1111/jcmm.12918
Singh, M., Yelle, N., Venugopal, C., and Singh, S. K. (2017). EMT: mechanisms and therapeutic implications. Pharmacol. Ther. doi: 10.1016/j.pharmthera.2017.08.009 [Epub ahead of print].
Sohal, S. S., and Walters, E. H. (2013). Role of epithelial mesenchymal transition (EMT) in chronic obstructive pulmonary disease (COPD). Respir. Res. 14:120. doi: 10.1186/1465-9921-14-120
Suarez-Carmona, M., Lesage, J., Cataldo, D., and Gilles, C. (2017). EMT and inflammation: inseparable actors of cancer progression. Mol. Oncol. 11, 805–823. doi: 10.1002/1878-0261.12095
Sun, Y., and Huang, P. (2016). Adenosine A2B receptor: from cell biology to human diseases. Front. Chem. 4:37. doi: 10.3389/fchem.2016.00037
Sung, W. J., Kim, H., and Park, K. K. (2016). The biological role of epithelial-mesenchymal transition in lung cancer (Review). Oncol. Rep. 36, 1199–1206. doi: 10.3892/or.2016.4964
Trincavelli, M. L., Giacomelli, C., Daniele, S., Taliani, S., Cosimelli, B., Laneri, S., et al. (2014). Allosteric modulators of human A2B adenosine receptor. Biochim. Biophys. Acta 1840, 1194–1203. doi: 10.1016/j.bbagen.2013.12.021
Weng, L., Wang, W., Su, X., Huang, Y., Su, L., Liu, M., et al. (2015). The effect of cAMP-PKA activation on TGF-β1-induced profibrotic signaling. Cell Physiol. Biochem. 36, 1911–1927. doi: 10.1159/000430160
Wilkinson, P. F., Farrell, F. X., Morel, D., Law, W., and Murphy, S. (2016). Adenosine signaling increases proinflammatory and profibrotic mediators through activation of a functional adenosine 2B receptor in renal fibroblasts. Ann. Clin. Lab. Sci. 46, 339–345.
Willis, B. C., duBois, R. M., and Borok, Z. (2006). Epithelial origin of myofibroblasts during fibrosis in the lung. Proc. Am. Thorac. Soc. 3, 377–382. doi: 10.1513/pats.200601-004TK
Xiao, H., Shen, H. Y., Liu, W., Xiong, R. P., Li, P., Meng, G., et al. (2013). Adenosine A2A receptor: a target for regulating renal interstitial fibrosis in obstructive nephropathy. PLOS ONE 8:e60173. doi: 10.1371/journal.pone.0060173
Xie, L., Law, B. K., Chytil, A. M., Brown, K. A., Aakre, M. E., and Moses, H. L. (2004). Activation of the Erk pathway is required for TGF-beta1-induced EMT in vitro. Neoplasia 6, 603–610. doi: 10.1593/neo.04241
Yamaguchi, M., Hirai, S., Tanaka, Y., Sumi, T., Miyajima, M., Mishina, T., et al. (2017). Fibroblastic foci, covered with alveolar epithelia exhibiting epithelial-mesenchymal transition, destroy alveolar septa by disrupting blood flow in idiopathic pulmonary fibrosis. Lab. Invest. 97, 232–242. doi: 10.1038/labinvest.2016.135
Yang, J., Velikoff, M., Canalis, E., Horowitz, J. C., and Kim, K. K. (2014). Activated alveolar epithelial cells initiate fibrosis through autocrine and paracrine secretion of connective tissue growth factor. Am. J. Physiol. Lung Cell. Mol. Physiol. 306, L786–L796. doi: 10.1152/ajplung.00243.2013
Zhang, A., Dong, Z., and Yang, T. (2006a). Prostaglandin D2 inhibits TGF-beta1-induced epithelial-to-mesenchymal transition in MDCK cells. Am. J. Physiol. Renal Physiol. 291, F1332–F1342. doi: 10.1152/ajprenal.00131.2006
Zhang, A., Wang, M. H., Dong, Z., and Yang, T. (2006b). Prostaglandin E2 is a potent inhibitor of epithelial-to-mesenchymal transition: interaction with hepatocyte growth factor. Am. J. Physiol. Renal Physiol. 291, F1323–F1331. doi: 10.1152/ajprenal.00480.2005
Zhang, J., Deng, Y. T., Liu, J., Wang, Y. Q., Yi, T. W., Huang, B. Y., et al. (2016). Norepinephrine induced epithelial-mesenchymal transition in HT-29 and A549 cells in vitro. J. Cancer Res. Clin. Oncol. 142, 423–435. doi: 10.1007/s00432-015-2044-9
Zhang, Y. E. (2017). Non-Smad signaling pathways of the TGF-β family. Cold Spring Harb. Perspect. Biol. 9:a022129. doi: 10.1101/cshperspect.a022129
Zhou, Y., Chu, X., Deng, F., Tong, L., Tong, G., Yi, Y., et al. (2017). The adenosine A2b receptor promotes tumor progression of bladder urothelial carcinoma by enhancing MAPK signaling pathway. Oncotarget 8, 48755–48768. doi: 10.18632/oncotarget.17835
Zhou, Y., Schneider, D. J., Morschl, E., Song, L., Pedroza, M., Karmouty-Quintana, H., et al. (2011). Distinct roles for the A2B adenosine receptor in acute and chronic stages of bleomycin-induced lung injury. J. Immunol. 186, 1097–1106. doi: 10.4049/jimmunol.1002907
Keywords: adenosine A2B receptor, epithelial mesenchymal transition, lung, MAPK-ERK, cAMP-PKA, transforming growth factor-β1
Citation: Giacomelli C, Daniele S, Romei C, Tavanti L, Neri T, Piano I, Celi A, Martini C and Trincavelli ML (2018) The A2B Adenosine Receptor Modulates the Epithelial– Mesenchymal Transition through the Balance of cAMP/PKA and MAPK/ERK Pathway Activation in Human Epithelial Lung Cells. Front. Pharmacol. 9:54. doi: 10.3389/fphar.2018.00054
Received: 04 October 2017; Accepted: 15 January 2018;
Published: 31 January 2018.
Edited by:
Francisco Ciruela, University of Barcelona, SpainCopyright © 2018 Giacomelli, Daniele, Romei, Tavanti, Neri, Piano, Celi, Martini and Trincavelli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Claudia Martini, Y2xhdWRpYS5tYXJ0aW5pQHVuaXBpLml0