 Andrea Grandi1
Andrea Grandi1 Irene Zini1Lisa Flammini1Anna M. Cantoni2Valentina Vivo1Vigilio Ballabeni1
Irene Zini1Lisa Flammini1Anna M. Cantoni2Valentina Vivo1Vigilio Ballabeni1 Elisabetta Barocelli1
Elisabetta Barocelli1 Simona Bertoni1*
Simona Bertoni1*- 1Food and Drug Department, University of Parma, Parma, Italy
- 2Department of Veterinary Sciences, University of Parma, Parma, Italy
The existence of a cholinergic anti-inflammatory pathway negatively modulating the inflammatory and immune responses in various clinical conditions and experimental models has long been postulated. In particular, the protective involvement of the vagus nerve and of nicotinic Ach receptors (nAChRs) has been proposed in intestinal inflammation and repeatedly investigated in DSS- and TNBS-induced colitis. However, the role of α7 nAChRs stimulation is still controversial and the potential contribution of α4β2 nAChRs has never been explored in this experimental condition. Our aims were therefore to pharmacologically investigate the role played by both α7 and α4β2 nAChRs in the modulation of the local and systemic inflammatory responses activated in TNBS-induced colitis in mice and to assess the involvement of the spleen in nicotinic responses. To this end, TNBS-exposed mice were sub-acutely treated with various subcutaneous doses of highly selective agonists (AR-R17779 and TC-2403) and antagonists (methyllycaconitine and dihydro-β-erythroidine) of α7 and α4β2 nAChRs, respectively, or with sulfasalazine 50 mg/kg per os and clinical and inflammatory responses were evaluated by means of biochemical, histological and flow cytometry assays. α4β2 ligands evoked weak and contradictory effects, while α7 nAChR agonist AR-R17779 emerged as the most beneficial treatment, able to attenuate several local markers of colitis severity and to revert the rise in splenic T-cells and in colonic inflammatory cytokines levels induced by haptenization. After splenectomy, AR-R17779 lost its protective effects, demonstrating for the first time that, in TNBS-model of experimental colitis, the anti-inflammatory effect of exogenous α7 nAChR stimulation is strictly spleen-dependent. Our findings showed that the selective α7 nAChRs agonist AR-R17779 exerted beneficial effects in a model of intestinal inflammation characterized by activation of the adaptive immune system and that the spleen is essential to mediate this cholinergic protection.
Introduction
During the last two decades, the existence of a CAP, vagus- and spleen-dependent, has been postulated by several authors (Olofsson et al., 2012; Bonaz et al., 2013; Martelli et al., 2014; Goverse et al., 2016). Apparently pivotal to the anti-inflammatory actions of CAP is the stimulation of α7 nAChR homomeric subtype (Wang et al., 2003). α7 nAChRs are expressed in several regions of the central nervous system and, in the periphery, on neurons, like the adrenergic neurons in the celiac/superior mesenteric ganglia and splenic nerve fibers (Downs et al., 2014), and non-neuronal cells like gut epithelial and glial cells (Costantini et al., 2012), endothelial cells (Saeed et al., 2005) and immune cells, such as macrophages (Wang et al., 2003), T lymphocytes, and dendritic cells (Kawashima et al., 2012). In particular, their activation by nicotine on immune cells can incite an intracellular cascade via Jak2/STAT3 signaling pathway leading to the inhibition of NF-κB transcription factor and, consequently, to the negative regulation of pro-inflammatory cytokines secretion (De Jonge et al., 2005). Accordingly, the anti-inflammatory properties of various selective α7 nAChR agonists have been demonstrated in a number of models of acute inflammation in vivo, such as acute lung injury (Pinheiro et al., 2017), cardiopulmonary bypass (Ge et al., 2016), burn injury (Kashiwagi et al., 2017), mesenteric and renal ischemia/reperfusion (Li et al., 2013; He et al., 2016), sepsis (Nullens et al., 2016) and post-operative ileus (The et al., 2007). More controversial are the effects produced by α7 nAChR agonists on subacute/chronic intestinal inflammation: in dextran sulfate sodium (DSS)-induced colitis, a model mainly dependent on the activation of innate immune responses, alternatively a protective (Salaga et al., 2016), anti-hyperalgesic, but not anti-inflammatory (Costa et al., 2012), or even detrimental (Snoek et al., 2010) role has been evidenced for α7 nAChRs stimulation, while in TNBS-induced colitis, where adaptive immune responses prevail (Kiesler et al., 2015), α7 nAChR agonists seem to exert a protective activity only when acting as partial agonists (Snoek et al., 2010; Salaga et al., 2016).
More recently, a potential counter-inflammatory action has emerged also for α4β2 nAChRs. Heteromeric α4β2 nAChRs, the predominant high-affinity binding sites for nicotine in the central nervous system, are well known to mediate the reinforcing, rewarding and dependence-inducing properties of nicotine (De Biasi and Dani, 2011) and its anti-nociceptive actions (Hurst et al., 2013). In the periphery, α4β2 nAChRs have been identified also on non-neuronal cells, like peritoneal, intestinal (van der Zanden et al., 2009) and alveolar macrophages (Matsunaga et al., 2001): their stimulation may inhibit the release of pro-inflammatory cytokines production through NF-κB inhibition in transfected human cells (Hosur and Loring, 2011) and promote the phagocytic activity of isolated intestinal and peritoneal murine macrophages (van der Zanden et al., 2009). However, no information is available up to now about the ability of α4β2 nAChRs ligands to modulate intestinal inflammation in vivo, as in colitis, where macrophages purportedly play a pivotal role (Mahida et al., 1989).
Starting from these premises, the main aim of the present study was to pharmacologically investigate the role of α7 and α4β2 nAChRs subtypes in the modulation of the local and systemic inflammatory responses in a murine model of colitis characterized by the pronounced activation of the adaptive immune system. To this purpose, we evaluated the effects produced by highly selective agonists (AR-R17779 and TC-2403) (Bencherif et al., 1996; Mullen et al., 2000) and antagonists (MLA and DBE) (Ward et al., 1990; Niimi et al., 2013) of α7 and α4β2 nAChRs, respectively, on the clinical and inflammatory markers increased in mice by TNBS colitis. The α7 nAChR agonist AR-R17779 emerged as the most effective treatment, able to attenuate colitis severity and to revert the rise in splenic T-cells populations and in colonic inflammatory cytokines levels induced by haptenization.
In view of the prominent role played by both the spleen and α7 nAChR signaling in mediating the CAP, we asked whether the protection afforded by AR-R17779 against TNBS-induced colitis could be maintained also after SPX and we demonstrated that, in our model of experimental colitis, the presence of the spleen is essential to the anti-inflammatory effect of exogenous α7 nAChR stimulation.
Materials and Methods
Animals
All animal experiments were performed according to the guidelines for the use and care of laboratory animals and they were authorized by the local Animal Care Committee “Organismo Preposto al Benessere degli Animali” and by Italian Ministry of Health, “Ministero della Salute” (DL 26/2014). Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; Curtis et al., 2015). All appropriate measures were taken to minimize pain or discomfort of animals. Female CD1 Swiss mice (7–12 weeks old) (Charles River Laboratories, Calco, Italy), weighing 25–30 g, were housed five per cage and maintained under standard conditions at our animal facility (12:12 h light–dark cycle, water and food ad libitum, 22–24°C). Intragastric gavage administration was performed using suitable gavage needles (22 gauge, length 3 cm, ball diameter 1.5 mm). All the experimental procedures (induction of colitis, SPX) and euthanasia by CO2 inhalation were performed between 9 a.m. and 12 a.m.
Induction of Colitis
Six days before intrarectal (i.r.) instillation, animals were subjected to skin sensitization through cutaneous application of 50 μL of a 10% (w v-1) TNBS solution in 50% ethanol. After 20 h fasting with free access to water containing 5% glucose, colitis was induced by i.r. instillation of the same volume and concentration of TNBS applied during skin sensitization (Waldner and Neurath, 2009). TNBS instillation was performed using a PE50 catheter, positioned 4 cm from the anus, in anesthetized mice (isoflurane 2%) kept in the head-down position for 3 min to avoid the leakage of intracolonic instillate. The TNBS-induced colitis model with previous skin sensitization was chosen to have a model of colitis characterized by the prominent activation of the adaptive immune system (Te Velde et al., 2006).
Splenectomy
In a second series of experiments, chemical colitis was induced in mice 15 days after SPX. Mice, fasted for 16 h, were anesthetized by intraperitoneal injection of pentobarbital 60 mg⋅kg-1. The spleen was removed after laparotomy and ligation of blood vessels and, after the surgical intervention, mice were monitored daily to examine their health state and scar cicatrisation.
Experimental Design
Pharmacological treatments started 8 h after TNBS enema (day 1) and were applied once (sulfasalazine) or twice daily (b.i.d.), 8 h apart, during the following 2 days by a researcher not involved in biochemical, histological assays or daily monitoring of Disease Activity Index. All the pharmacological treatments were subcutaneously (s.c.) administered except for sulfasalazine, per os (p.o.) applied. Animals were euthanized by CO2 inhalation 3 days after TNBS or saline instillation (day 4).
Mice were assigned through block randomization to the normal group (N; n = 24), i.r. inoculated with 50 μL 0.9% NaCl (saline solution) and administered s.c. 10 mL kg-1 saline (b.i.d.) or to the following experimental groups of colitic mice: saline (CTR, 10 mL kg-1; n = 28), sulfasalazine 50 mg kg-1 die-1 p.o. (S; n = 7), TC-2403 2 mg kg-1 (n = 7) and 5 mg kg-1 (n = 7), DBE 0.5 mg kg-1 (n = 7) and 1.5 mg kg-1 (n = 7), AR-R17779 (A) 0.5 mg kg-1 (n = 7), 1.5 mg kg-1 (n = 16), 5 mg kg-1 (n = 7), MLA 0.1 mg kg-1 (n = 7), 0.5 mg kg-1 (n = 7), 1 mg kg-1 (n = 7). The dosage of each pharmacological treatment was chosen on the basis of the literature (Karimi et al., 2010; Snoek et al., 2010; Joh and Kim, 2011; Hijioka et al., 2012; Niimi et al., 2013). The study was performed using experimental blocks composed by 10 or 12 mice that were randomly assigned to five or six groups of treatment (N and CTR were present in each experimental block), each one encompassing two animals.
Splenectomy mice were assigned through block randomization to the following experimental groups: SPX/N (saline i.r., 10 mL kg-1 saline; n = 15), SPX/C (TNBS i.r., 10 mL kg-1 saline; n = 15) and SPX/A (TNBS i.r., AR-R17779 1.5 mg kg-1; n = 15). SPX study was performed through three experimental blocks composed by 15 mice that were randomly assigned to three groups of treatment, each one comprising five animals.
A group size of n = 7 was determined according to “a priori power analysis,” by setting α = 0.05, 1-β = 0.8 and effect size = 0.6 assuming as primary endpoint the reduction of colonic MPO levels. Due to seasonal variability, N and CTR mice were repeated periodically all over the study in order to verify the attainment of a constant degree of colitis severity with respect to physiological conditions, thus explaining the bigger size of N and CTR experimental groups with respect to the other groups. As regards A1.5, SPX/N, SPX/C, and SPX/A groups, a higher number of mice was necessary to perform the histological and biochemical assays described. Accordingly, each group of animals was randomly subdivided in two subgroups: colons excised from each subset was reserved either for histological analysis or for MPO activity determination and for cytokines assays. In this latter case, the colon was sometimes longitudinally split in two halves, each one dedicated to MPO activity assay or to cytokines determination.
Evaluation of Inflammatory Responses
Body weight, stools consistency and rectal bleeding were examined and registered daily throughout the experimentation by unaware observers, in order to assess the DAI. Immediately after euthanasia the macroscopic damage of colonic mucosa was assessed as MS. The wet weight and the length of each colon were measured and the weight/length ratio was considered as disease-related intestinal wall thickening (Bischoff et al., 2009). Colon, lungs, spleen and MLNs were collected for subsequent microscopic, biochemical or flow cytometry analyses.
Disease Activity Index
Disease Activity Index is a parameter that estimates the severity of the disease; it is based on the daily assignment of a total score, according to Cooper’s modified method (Cooper et al., 1993), on the basis of body weight loss, stool consistency and rectal bleeding. The scores were attributed blindly by two investigators and were quantified as follows: stool consistency: 0 (normal), 1 (soft), 2 (liquid); body weight loss: 0 (<5%), 1 (5–10%), 2 (10–15%), 3 (15–20%), 4 (20–25%), 5 (>25%) and rectal bleeding: 0 (absent), 1 (present).
Colon Macroscopic Damage Score (MS)
After euthanasia, the colon was explanted, opened longitudinally, flushed with saline solution and MS was immediately evaluated through inspection of the mucosa, executed by two investigators unaware of the treatments applied. MS was determined according to previously published criteria (Rapalli et al., 2016), as the sum of scores (max = 12) attributed as follows: presence of strictures and hypertrophic zones (0, absent; 1, one stricture; 2, two strictures; 3, more than two strictures); mucus (0, absent; 1, present); adhesion areas between the colon and other intra-abdominal organs (0, absent; 1, one adhesion area; 2, two adhesion areas; 3, more than two adhesion areas); intraluminal hemorrhage (0, absent; 1, present); erythema (0, absent; 1, presence of a crimsoned area < 1 cm2; 2, presence of a crimsoned area > 1 cm2); ulcerations and necrotic areas (0, absent; 1, presence of a necrotic area < 1 cm2; 2, presence of a necrotic area > 1 cm2).
Colonic Length and Thickness
To evaluate muscular contraction and deposition of fibrotic material induced by a prolonged inflammatory state, the length of the colon and its weight were measured, while weight/length ratio was calculated to estimate colon thickness, according to a previously published method (Bischoff et al., 2009).
Colonic and Pulmonary Myeloperoxidase Activity Assay
Myeloperoxidase activity, marker of granulocytic infiltration within a tissue, was determined according to Krawisz’s modified method (Krawisz et al., 1984). After being weighed, each colonic or lung sample was homogenized in ice-cold potassium phosphate buffer (100 mmolL-1, pH 7.4) containing 1 μg mL-1 aprotinin and centrifuged for 20 min at 12500 RCF at 4°C. Pellets were re-homogenized in five volumes of ice-cold potassium phosphate buffer (50 mmolL-1, pH 6) containing 0.5% hexadecyltrimethylammonium bromide (HTAB) and 1 μg mL-1 aprotinin. Samples were then subjected to three cycles of freezing and thawing and centrifuged for 30 min at 15500 RCF at 4°C. One hundred microliters of the supernatant was then allowed to react with 900 μL of a buffer solution containing 0.167 mg mL-1 o-dianisidine and 0.0005% H2O2. Each assay was performed in duplicate and the rate of change in absorbance was measured spectrophotometrically at 470 nm (Jenway, mod. 6300, Dunmow, Essex, England). The sensitivity of the assay was 10 mU mL-1, 1 unit of MPO being defined as the quantity of enzyme degrading 1 μmol of peroxide per minute at 25°C. Data were normalized with edema values [(wet weight – dry weight) dry weight-1] (Moore-Olufemi et al., 2005) and expressed as U g-1 of dry weight tissue.
Flow Cytometry Assays
Isolation of Splenocytes
Spleen, explanted immediately after euthanasia, was mechanically dispersed through a 100 μm cell-strainer and washed with PBS containing 0.6 mmolL-1 EDTA (PBS-EDTA). The cellular suspension was then centrifuged at 1500 RCF for 10 min at 4°C, the pellet re-suspended in PBS-EDTA and incubated with 2 mL of NH4Cl lysis buffer (0.15 molL-1 NH4Cl, 1 mmolL-1 KHCO3, 0.1 mmolL-1 EDTA in distilled water) for 5 min, in the dark, to induce the lysis of erythrocytes. Afterward, samples were centrifuged at 1500 RCF for 10 min at 4°C, the pellet was washed with PBS-EDTA and re-suspended in 5 mL cell staining buffer (PBS containing 0.5% FCS and 0.1% sodium azide). The obtained cellular suspension was stained with fluorescent antibodies (Kruisbeek, 2001).
Isolation of Mesenteric Lymph Nodes
The lymphoid tissue located in the middle of proximal colon’s mesentery was explanted immediately after euthanasia and flushed with PBS, then MLNs were isolated from adherent adipose and vascular tissue, mechanically dispersed through a 100 μm cell-strainer and washed with Hank’s Balanced Salt Solution (HBSS) containing 5% FCS. The cellular suspension was centrifuged at 1500 RCF for 10 min at 4°C, the pellet washed with HBSS containing 5% FCS and re-suspended in 3 mL cell staining buffer. The obtained cellular suspension was subjected to staining with fluorescent antibodies (Lopez, 2007).
Immunofluorescent Staining
Before the incubation with fluorescent antibodies, the cellular suspension of splenocytes or MLN was incubated with IgG1-Fc (1 μg 10-6 cells) for 10 min in the dark at 4°C, in order to block non-specific binding sites for antibodies. The following antibodies were used: Phycoerythrin-Cyanine 5 (PE-Cy5) conjugated anti-mouse CD3ε (0.25 μg 10-6 cells), Fluorescein Isothiocyanate (FITC) anti-mouse CD4 (0.25 μg 10-6 cells), PE anti-mouse CD8a (0.25 μg 10-6 cells), FITC anti-mouse F4/80 (0.25 μg 10-6 cells), Peridinin Chlorophyll Proteins-Cyanine5.5 (PerCP-Cy5.5) anti-mouse CD25 (1 μg 10-6 cells) and PE anti-mouse FoxP3 (1 μg 10-6 cells). Cells were incubated with antibodies for 1 h in the dark at 4°C, washed with PBS to remove excessive antibody and suspended in cell staining buffer to perform flow cytometry analysis. Because of the intracellular localization of FoxP3, staining with PE anti-mouse FoxP3 was preceded by cells fixation and permeabilization: after staining with cell surface markers, cells were fixed with FOXP3 Fix/Perm Buffer and permeabilized with PBS containing 0.2% Tween 20. Cells were then incubated with PE anti-mouse FoxP3 for 30 min in the dark, at room temperature, washed with PBS to remove excessive antibody and suspended in cell staining buffer (Holmes et al., 2001).
The viability of the cellular suspension was assessed through propidium iodide (PI) staining, a membrane impermeable fluorescent dye, excluded by viable cells, that binds to DNA emitting red fluorescence, thus resulting as a suitable marker for dead cells. Cells were incubated with 10 μg mL-1 PI for 1 min in the dark, at room temperature, and immediately subjected to flow cytometry analysis. Only PI- cells were included in the analysis.
Samples were analyzed using Guava easyCyteTM and InCyteTM software (Merck Millipore, Darmstadt, Germany). The various spleen and MLN cell populations were defined as follows: lymphocytes gated in the Forward Scatter (FSC)-Side Scatter (SSC) plot as FSC low: SSC low; T lymphocytes (CD3+); CD4+ T lymphocytes (CD3+CD4+CD8-); CD8+ T lymphocytes (CD3+CD8+CD4-); CD4+ Tregs (CD4+CD25+FoxP3+); macrophages (CD3-F4/80+). Percentages were reported to the total number of splenocytes or MLN cells of each mouse to calculate the number of cells per population.
Colonic Cytokines Levels
After euthanasia, colon tissues were homogenized for 1 min in tissue lysis buffer containing protease inhibitor cocktail (aprotinin 1 μg mL-1 and leupeptin 1 μg mL-1). Samples were then centrifuged for 30 min at 14000 RCF and the supernatant was collected. Total protein concentration was quantified using Pierce BCA protein assay kit (Thermo Fisher Scientific Inc., Waltham, MA, United States). IL-1β, IL-6, IFNγ, and IL-10 levels were then determined in duplicate in 100 μL samples, using commercially available ELISA kits (Mouse IL-10 ELISA kit, Abcam BiochemicalsTM, Cambridge, United Kingdom; Mouse IL-1β ELISA kit; Mouse IFNγ ELISA kit; Mouse IL-6 ELISA kit, RayBioTM, Norcross, GA, United States) according to the manufacturer’s protocol. The absorbances of the samples were measured spectrophotometrically at 450 nm (TECAN SunriseTM powered by MagellanTM data analysis software, Mannedorf, Switzerland). The assays sensitivity were, respectively, 5 pg mL-1 (IL-1β), 2 pg mL-1 (IL-6), 10 pg mL-1 (IFNγ), and 30 pg mL-1 (IL-10). Results were expressed as pg mg-1 protein.
Colonic Histology
Colonic samples were harvested from N, CTR, A1.5, SPX/N animals and from colitic splenectomized mice, either administered with saline (SPX/C) or with AR-R17779 1.5 mg/kg (SPX/A), immersion-fixed in 10% neutral buffered formalin overnight, dehydrated and embedded in paraffin. For each sample, at least five transverse 5-μm sections were cut in the distal colon, stained with hematoxylin–eosin and blindly examined in a light microscope (Nikon Eclipse E800). The histological damage was quantified according to Rapalli et al. (2016) by an unaware investigator: the grade of mucosal destruction (0, normal; 1, mild; 2, moderate; 3, severe) and the degree of leukocytes infiltration in the lamina propria and submucosa (0, absent; 1, mild; 2, pronounced) were scored (maximum score: 7). Since histological injury produced by TNBS instillation was localized almost exclusively in the distal portion of the colon, the average value of histological score was determined only from sections of distal colon, pooled with those determined for the other animals of the same experimental group and the mean value ± SEM was calculated.
Statistical Analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). All data, each one representing the outcome of a single animal, were presented as means ± SEM. Comparisons among experimental groups were made using analysis of variance (one-way or two-way ANOVA) followed by Bonferroni’s post-test when P < 0.05, chosen as level of statistical significance, was achieved. All analyses were performed using Prism 4 software (GraphPad Software Inc., San Diego, CA, United States).
Materials
Sulfasalazine, MLA, TNBS, ethanol, HTAB and 30% hydrogen peroxide were purchased from Sigma–AldrichTM (St. Louis, MO, United States). AR-R17779 and TC-2403 were purchased from Abcam BiochemicalsTM (Cambridge, United Kingdom), while DBE was purchased from Tocris BioscienceTM (Bristol, United Kingdom). Drugs were dissolved in saline solution the day of the experiment. Fluorescent antibodies used for flow cytometry (FITC anti-mouse CD4, PE anti-mouse CD8, PE anti-mouse FOXP3, PerCP/Cy5.5 anti-mouse CD25), PI and FOXP3 Fix/Perm Buffer were purchased from BioLegendTM (San Diego, CA, United States), PE-Cy5 anti-mouse CD3 from Affymetrix eBioscienceTM (San Diego, CA, United States) and IgG1-Fc from MilliporeTM (Merck, Darmstadt, Germany).
Results
TNBS Triggered a Severe Inflammatory Response Controlled by Sulfasalazine
Animals subjected to hapten-induced colitis showed a marked decrease of body weight and reduction of stools’ consistency compared with normal mice, as indicated by DAI increase from day 1 to day 4 (Figure 1A). Locally, TNBS instillation provoked a strong shortening and thickening of the colon and a substantial increase of macroscopic damage score, due to the appearance of ulcerations, luminal hemorrhages and erythemas (Figures 1B–D). Accordingly, histological observation revealed extensive mucosal necrosis and abundant granulocytes infiltration both in the lamina propria and in the submucosa in TNBS-exposed mice (Figure 2B; histological score: 6.5 ± 0.3 in Supplementary Figure 1A) compared with normal mice (Figure 2A; histological score: 1.0 ± 0.6 in Supplementary Figure 1A). Finally, upon instillation of the haptenating agent we observed a consistent granulocyte infiltration both in the colon and within the lungs (Figures 1E,F).
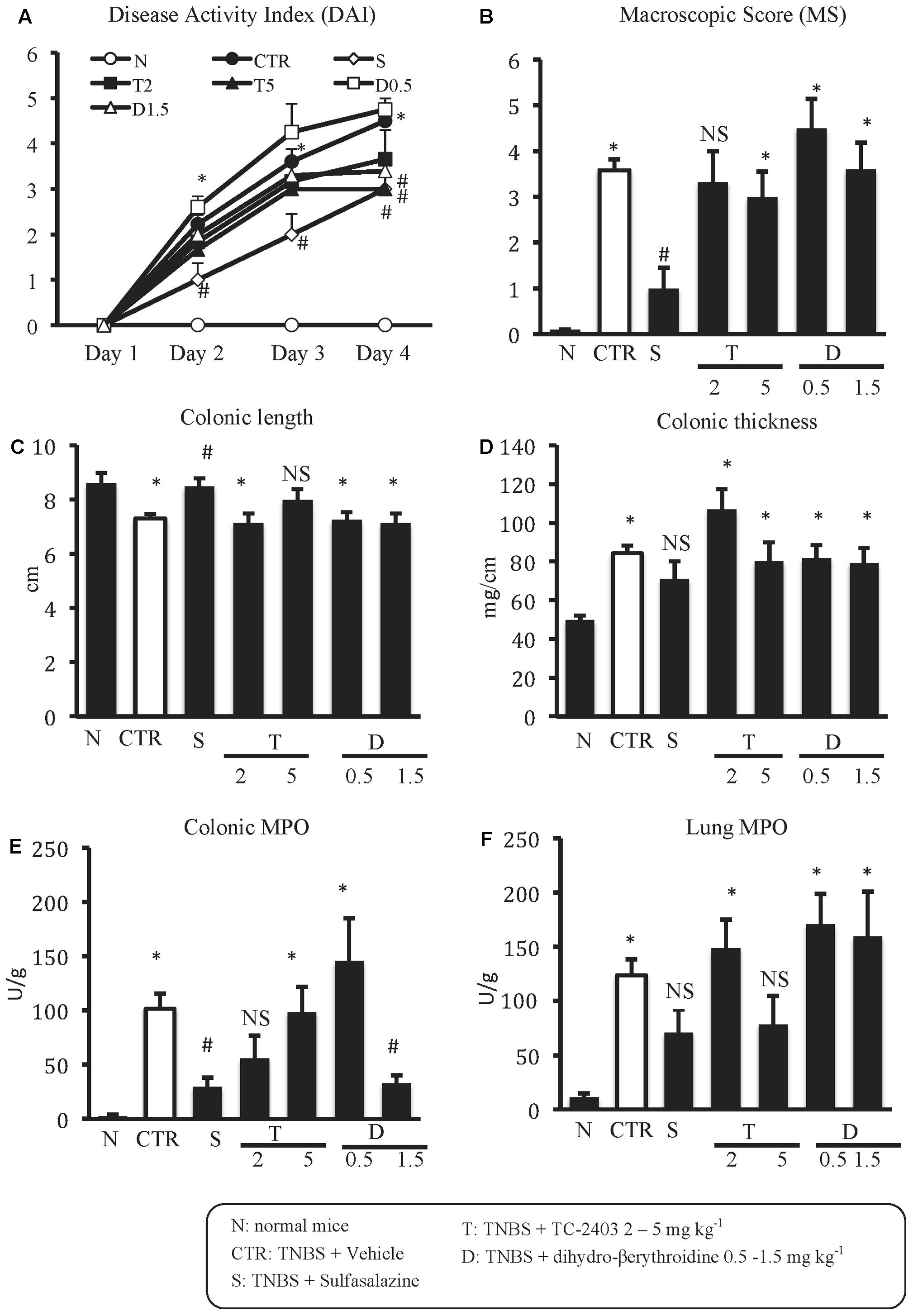
FIGURE 1. Effects of sulfasalazine and α4β2 nAChRs ligands on TNBS-induced inflammatory responses. Disease Activity Index (A), macroscopic score (B), colonic length (C), colonic thickness (D), colonic MPO (E) and lung MPO (F) activity assessed in vehicle-treated normal mice (N) and in TNBS-treated mice administered with vehicle (CTR – white bar), sulfasalazine 50 mg/kg (S), TC-2403 (T) 2 and 5 mg/kg, DBE (D) 0.5 and 1.5 mg/kg (D1.5) (n = 7–12 independent values per group). ∗P < 0.05 vs. N mice; #P < 0.05 vs. CTR mice; NS, not significant vs. N and CTR mice; one-way ANOVA followed by Bonferroni’s post-test.
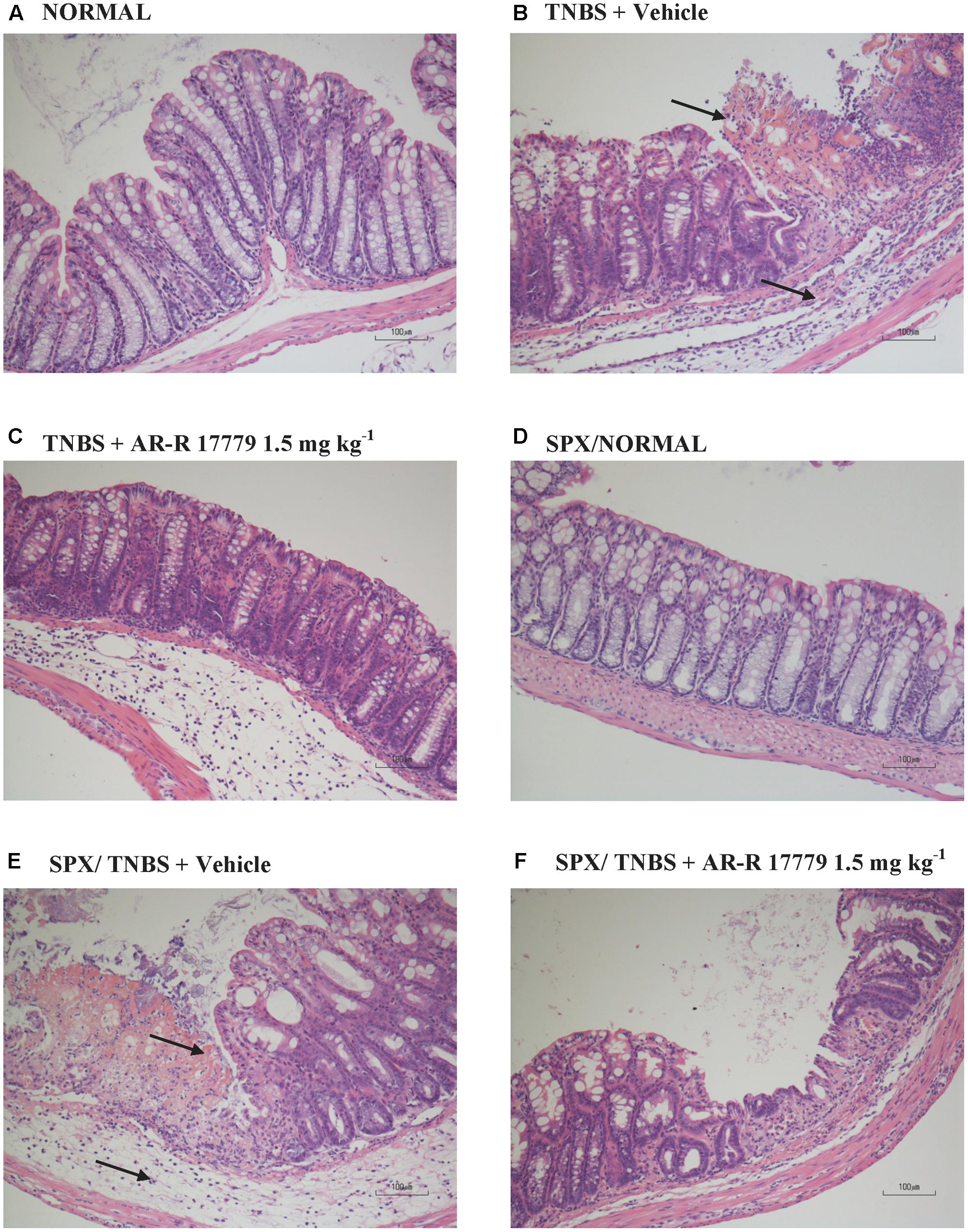
FIGURE 2. Histology. Representative hematoxylin–eosin stained sections of colonic specimens harvested from vehicle-treated normal mice, non-operated (A) or splenectomized (D), and from TNBS-treated mice administered with vehicle, non-operated (B) or splenectomized (E), or with AR-R17779 1.5 mg/kg, non-operated (C) or splenectomized (F). TNBS colonic instillation caused mucosal necrosis, neutrophils infiltration and submucosal oedema (indicated by arrows) in vehicle-treated animals (B,E), attenuated by AR-R17779 treatment in non-operated mice (C) but not in splenectomized animals (F).
As expected, treatment with the anti-inflammatory agent sulfasalazine was able to dampen the local and systemic inflammatory responses induced by TNBS, improving mice health conditions by counteracting both the weight loss and diarrhea over the whole treatment period (Figure 1A), decreasing colon shortening and mucosal damage and mitigating colonic neutrophil infiltration (Figures 1B,C,E).
α4β2 nAChRs Agents Evoked Weak Effects on Experimental Colitis
The repeated administration of α4β2 nAChRs agents in colitic mice elicited weak effects on most of the analyzed inflammatory parameters. Only DAI was significantly reduced on day 4 by both the agonist TC-2403 and the antagonist DBE, when administered at the highest tested dose (Figure 1A); at the same dose, DBE was able to attenuate also colonic MPO activity (Figure 1E).
α7 nAChRs Agonist Protects against Experimental Colitis
Among the three different tested doses, the selective α7 nAChRs agonist AR-R17779 evoked the most prominent effects when administered at 1.5 mg kg-1. In fact, DAI had been ameliorated since day 3, reflecting an attenuation of both body weight loss and diarrhea (Figure 3A). As regards TNBS-induced local damage, treatment with AR-R17779 1.5 mg kg-1 decreased MS, counteracted colonic neutrophil infiltration (Figures 3B,E) and weakly mitigated mucosal destruction (Figure 2C; histological score: 4.0 ± 0.9 in Supplementary Figure 1A).
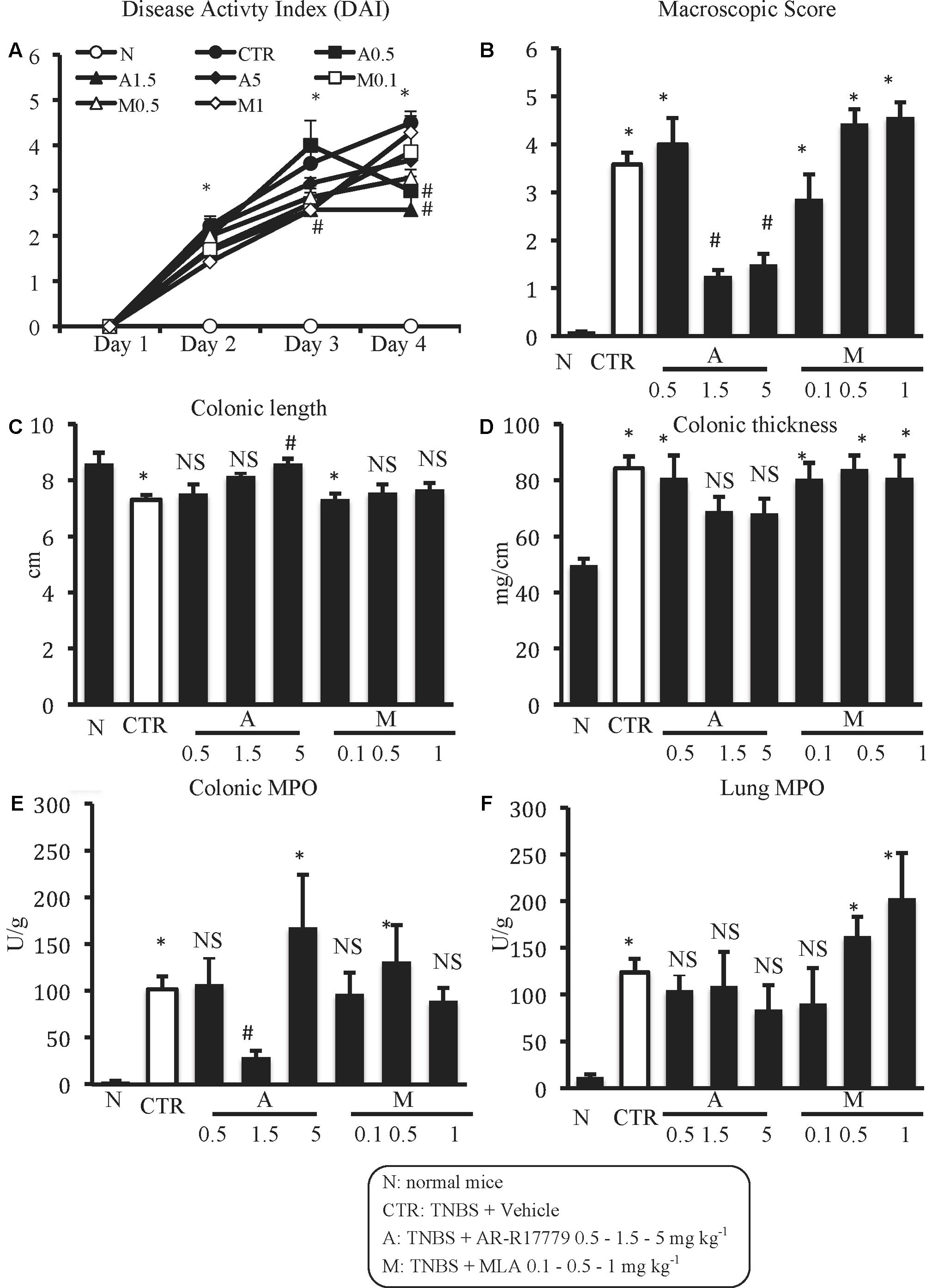
FIGURE 3. Effects of α7 nAChRs ligands on TNBS-induced inflammatory responses. Disease Activity Index (A), macroscopic score (B), colonic length (C), colonic thickness (D), colonic MPO (E), and lung MPO (F) activity assessed in vehicle-treated normal mice (N) and in TNBS-treated mice administered with vehicle (CTR – white bar), AR-R17779 (A) 0.5, 1.5, and 5 mg/kg, MLA (M) 0.1, 0.5 and 1 mg/kg (n = 7–12 independent values per group). N and CTR values were the same already represented in Figure 1. ∗P < 0.05 vs. N mice; #P < 0.05 vs. CTR mice; NS: not significant vs. N and CTR mice; one-way ANOVA followed by Bonferroni’s post-test.
The local beneficial effects of AR-R17779 1.5 mg kg-1 were confirmed by the evaluation of inflammatory cytokines levels in the colon. TNBS exposure produced a remarkable increase of IL-1β and IL-6 levels compared with N mice, whilst moderate, not significant, increase of IFN-γ and reduction of IL-10 concentrations were observed. Treatment with α7 agonist slightly reduced colonic IL-1β and remarkably lowered IL-6 levels, but did not significantly modify either IFN-γ or IL-10 concentrations (Table 1).
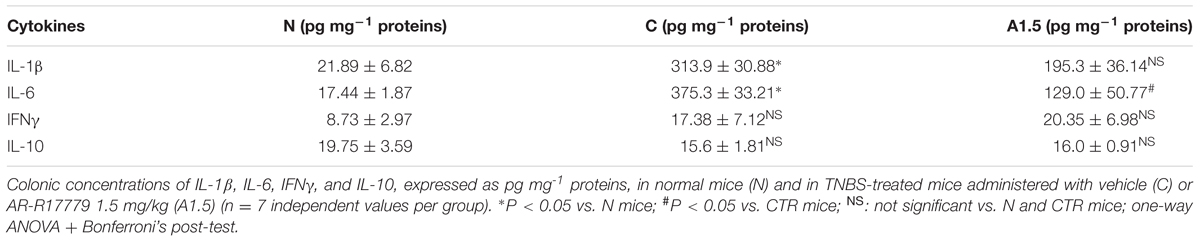
TABLE 1. Effects of AR-R17779 on cytokines levels in TNBS-induced colitis.
AR-R17779 appeared ineffective when tested at 0.5 mg kg-1 and exhibited only sporadic protective effects at 5 mg kg-1. Indeed, treatment with AR-R17779 5 mg kg-1 attenuated MS and colonic shortening and thickening (Figures 3B–D) while no remarkable effects were produced on the other inflammatory parameters with respect to CTR mice.
At contrast with exogenous α7 nAChRs stimulation, treatment with α7 antagonist MLA did not substantially affect the inflammatory parameters taken into consideration, except for MS and pulmonary MPO, which deteriorated, albeit not significantly, in mice administered with MLA 0.5 and 1 mg kg-1 with respect to vehicle-treated mice (Figures 3B,F).
AR-R17779 Attenuated TNBS-Induced Changes on Splenocytes Subpopulations
In order to investigate the mechanism of action of AR-R17779, we performed the phenotypic characterization of splenocytes and MLN lymphocytes in N, CTR and A1.5 animals. TNBS instillation remarkably augmented the number of splenic T lymphocytes, both CD8+ and CD4+ and slightly reduced macrophages count: such changes were mitigated by treatment with α7 agonist (Figure 4). As regards MLN, macrophages could not be identified, while a moderate reduction in the number of lymphocytes, in particular of CD4+ T cells, was elicited by colitis (Table 2). The administration of AR-R17779 did not prevent TNBS-induced MLN depletion, while further diminishing the number of MLN FoxP3+ Tregs (Table 2).
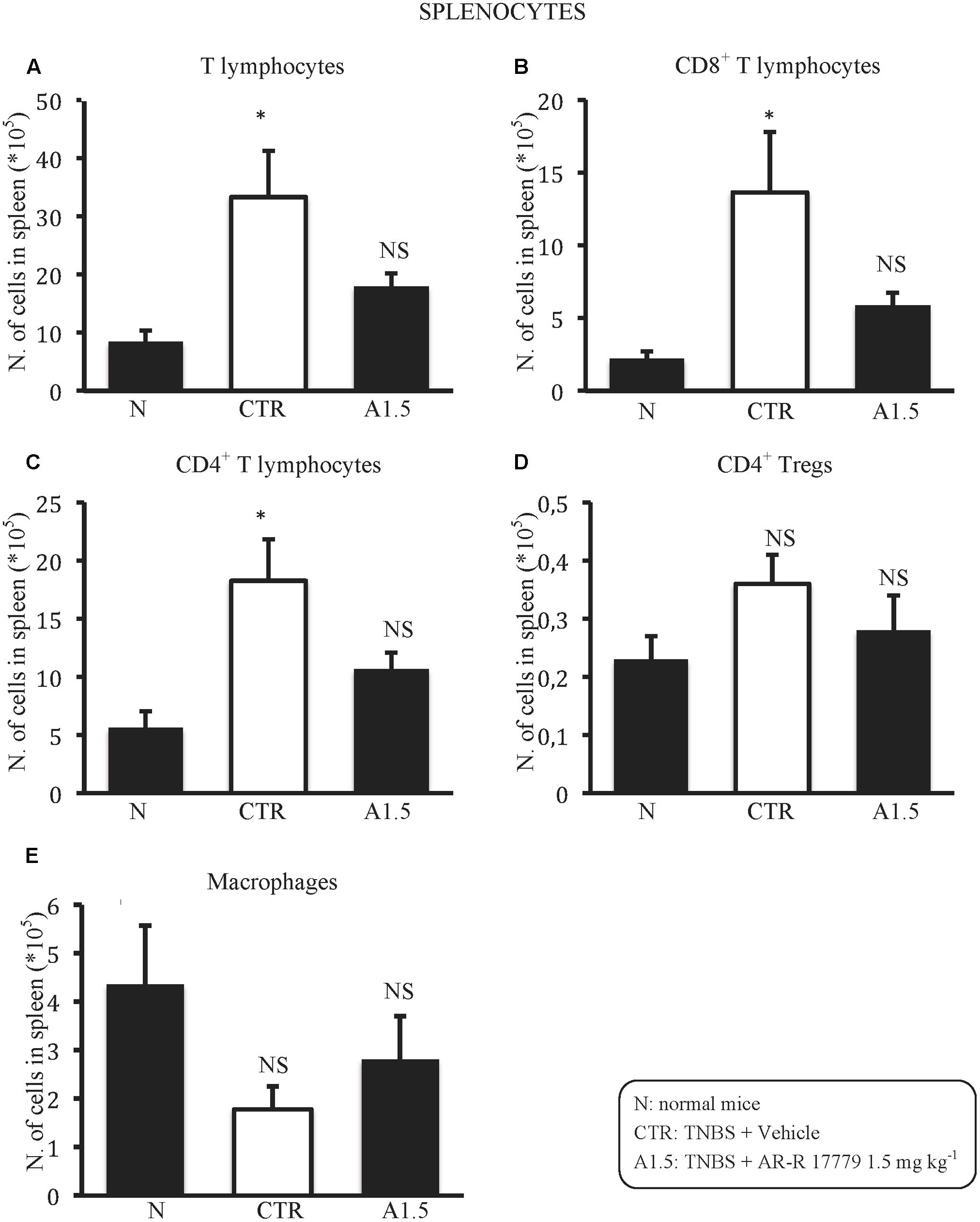
FIGURE 4. AR-R17779 attenuated TNBS-induced changes in splenocytes subpopulations. Number of T lymphocytes (A), CD8+ (B), CD4+ (C), CD4+ Tregs (D) lymphocytes and macrophages (E) in the spleen excised from vehicle-treated normal mice (N) and TNBS-treated mice administered with vehicle (CTR – white bar) or AR-R17779 1.5 mg/kg (A1.5) (n = 7–10 independent values per group). ∗P < 0.05 vs. N mice; NS: not significant vs. N and CTR mice; one-way ANOVA followed by Bonferroni’s post-test.
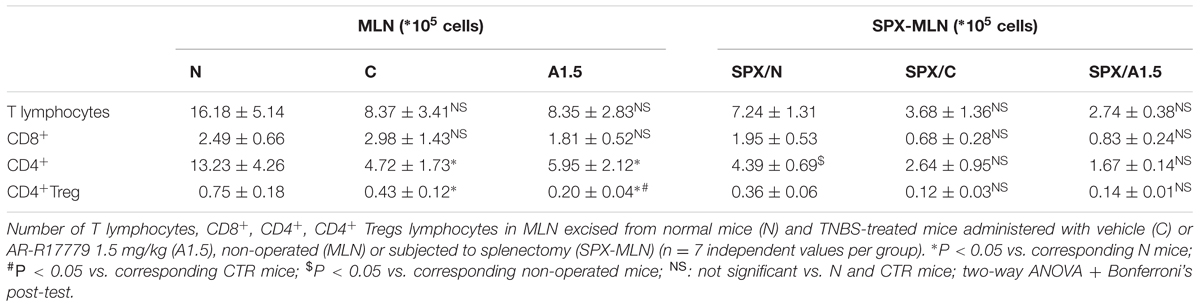
TABLE 2. Effects of AR-R17779 on TNBS-induced changes in MLN subpopulations.
Splenectomy Abolished the Protective Effects of AR-R17779
In SPX mice, colitis severity was similar to that induced in non-operated animals as regards both local and systemic effects, while AR-R17779 lost its protective properties in terms of DAI and colonic granulocytic infiltration, even worsening, instead of diminishing, MS (Figure 5). As regards histological injury, whose score is represented in Supplementary Figure 1B, mucosal damage in vehicle-treated colitic mice (Figure 2E) was increased compared to SPX/N mice (Figure 2D) but was not prevented by AR-R17779 treatment (Figure 2F).
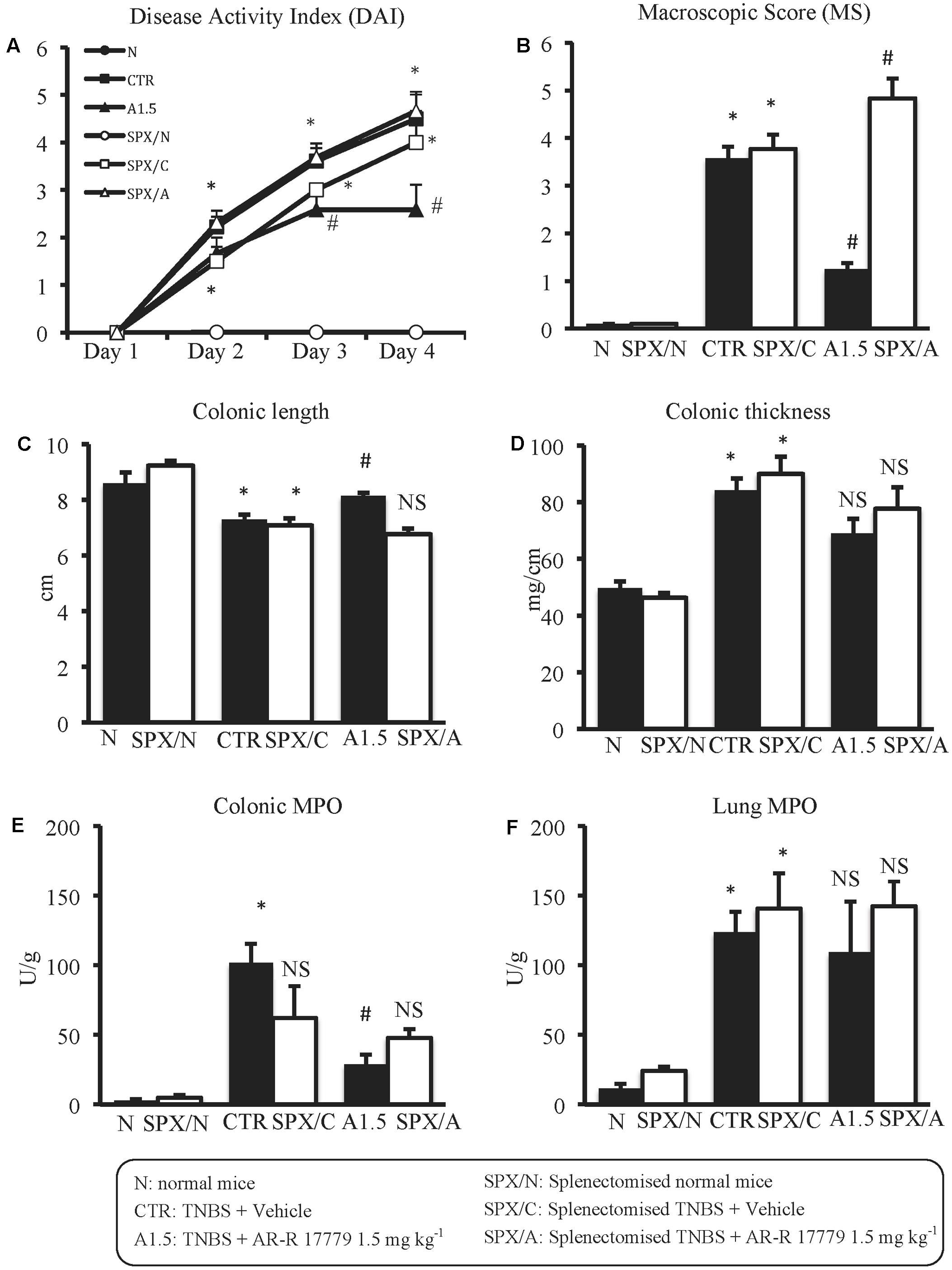
FIGURE 5. Splenectomy abolished the protective effects of AR-R17779 on TNBS-induced colitis. Disease Activity Index (A), macroscopic score (B), colonic length (C), colonic thickness (D), colonic MPO (E) and lung MPO (F) activity assessed in vehicle-treated normal mice (N; SPX/N) and in TNBS-treated mice administered with vehicle (CTR; SPX/C) or AR-R17779 1.5 mg/kg (A1.5; SPX/A), non-operated (black bars) or subjected to SPX (white bars) (n = 10–13 independent values per group). A1.5 values were the same already represented in Figure 3. ∗P < 0.05 vs. N mice; #P < 0.05 vs. CTR mice; NS: not significant vs. N and CTR mice; two-way ANOVA followed by Bonferroni’s post-test.
Regarding cytokines levels, in SPX mice TNBS produced the same changes evoked in non-operated animals; in particular, SPX/C mice exhibited a marked rise in IL-1β and IL-6, a weak increment of IFNγ and a feeble decrease of IL-10 colonic concentrations. Interestingly, after SPX, the treatment with α7 agonist lost completely its protective effects against colitis, being unable to lower IL-1β concentrations and exacerbating TNBS-induced increase of IL-6 levels, while leaving unmodified IFNγ and IL-10 colonic content with respect to SPX/C mice (Figure 6).
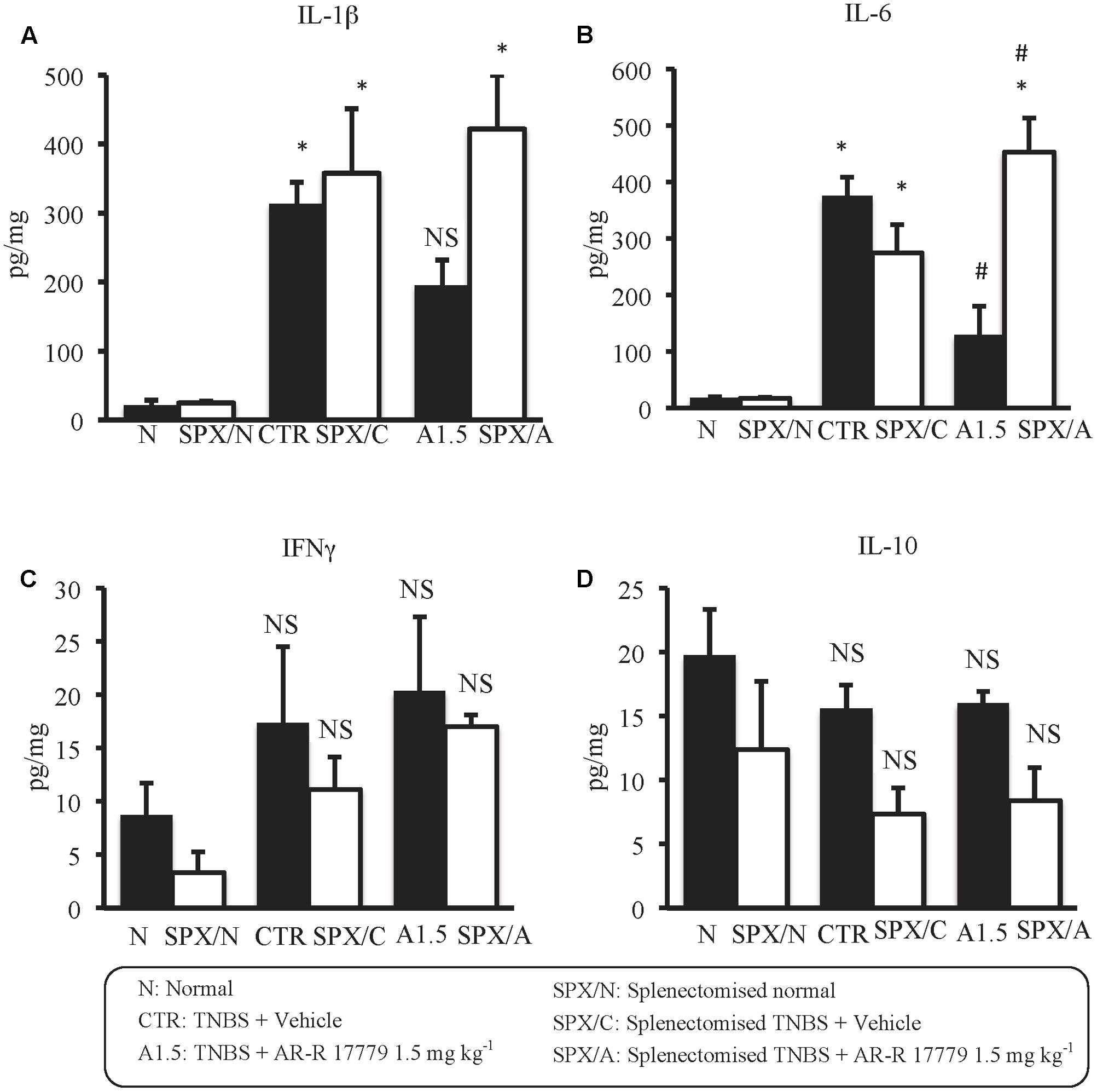
FIGURE 6. Splenectomy abolished the effects of AR-R17779 on cytokines levels in TNBS-induced colitis. Colonic concentrations of IL-1β (A), IL-6 (B), IFNγ (C), and IL-10 (D) in vehicle-treated normal mice (N; SPX/N) and in TNBS-treated mice administered with vehicle (CTR; SPX/C) or AR-R17779 1.5 mg/kg (A1.5; SPX/A), non-operated (black bars) or subjected to SPX (white bars) (n = 7 independent values per group). ∗P < 0.05 vs. N mice; #P < 0.05 vs. CTR mice; NS: not significant vs. N and CTR mice; two-way ANOVA followed by Bonferroni’s post-test.
With respect to MLN T cells subpopulations, SPX slightly decreased T cells number in all the experimental groups, a reduction particularly evident for CD4+ lymphocytes. Administration of α7 agonist was unable to counteract the further reduction in MLN T cells count induced by colitis (Table 2).
Discussion
The results collected in the present study indicate that the exogenous stimulation of α7 nAChRs by AR-R17779 exerted a beneficial effect in mice subjected to TNBS-induced colitis: this protection is spleen-dependent and it apparently involves the regulation of splenic T lymphocytes responses.
Intrarectal instillation of TNBS after skin sensitization produced in mice severe colitis, characterized by progressive deterioration of their clinical conditions with consistent weight loss and diarrhea. Colonic mucosal damage, already evident at the macroscopic observation and confirmed by histological examination, was accompanied by shortening and thickening of the intestinal wall and by the increase in IL-1β and IL-6 levels; a strong neutrophil infiltration was moreover revealed both locally and remotely in the lungs. This intense inflammatory response was mirrored by the remarkable rise in the number of T lymphocytes in the spleen and by their reduction in MLN, changes supporting the strong involvement of adaptive immune responses in our model of experimental colitis. As expected, the anti-inflammatory agent sulfasalazine, conventional therapeutic agent in human IBD (Talley et al., 2011), here employed as positive control, was able to efficiently counteract most of the TNBS-induced alterations, confirming the predictive power of the adopted experimental model.
As regards the effects produced by ligands of α4β2 nAChRs, the interpretation of the data obtained is not immediately straightforward. Indeed, both the agonist TC-2403 and the antagonist dihydro-β erythroidine generally evoked feeble effects and neither of them was able to remarkably modify the severity of TNBS-induced colitis. The only parameter significantly improved was represented by animals’ clinical conditions, surprisingly ameliorated by both ligands. If, in the case of the agonist, its antinociceptive properties may come into play (Rowley et al., 2008), the beneficial action of DBE is less comprehensible: to this end, we should recall the ability of ligands, either agonists or antagonists, to desensitize and/or up-regulate nAChRs following repeated administration (Hurst et al., 2013), leading possibly to overlapping actions. As a result, no firm conclusions can be drawn on the role played by α4β2 nAChRs in our model of intestinal inflammation: it is likely that the involvement of innate immune cells like macrophages, sensitive to α4β2 nAChRs modulation (van der Zanden et al., 2009), is overridden by that of adaptive immune components, whose response to α4β2 nAChRs ligands is yet to be clarified.
With respect to the effects produced by α7 nAChRs ligands, we could notice that repeated administration of α7 antagonist MLA did not significantly modify the local and systemic inflammatory responses evoked in the model we adopted of TNBS-induced colitis. Our findings seem therefore to indicate the absence of an endogenous α7 nAChRs-mediated counter-inflammatory cholinergic tone, similarly to what documented by Di Giovangiulio et al. (2016), who demonstrated that the severity of DSS-induced and T-cell transfer colitis was not increased in α7 nAChR-/- mice.
Contrarily to MLA, α7 nAChRs full agonist AR-R17779 was as effective as sulfasalazine in improving mice clinical conditions, in decreasing colonic macroscopic damage and pro-inflammatory cytokines levels and in preventing local neutrophil infiltration, demonstrating the remarkable protection afforded by exogenous stimulation of this receptor subtype against TNBS-induced colitis. Our results represent therefore a further clue in support of the anti-inflammatory action of α7 nAChRs activation and of the central role played by this receptor subtype in the CAP. Interestingly, AR-R17779 displayed a bell-shaped dose-response curve, exhibiting the greatest efficacy at the intermediate dose of 1.5 mg/kg s.c., thus confirming the tendency of α7 nAChRs to undergo desensitization in a dose-dependent way (Hurst et al., 2013). The findings collected are in line with those presented by Salaga et al. (2016), who demonstrated the protective effects of α7 nAChR partial agonist encenicline against TNBS-induced colitis, supposedly through the modulation of immune cells responses. As regards the investigation by Snoek et al. (2010) apparently denoting a lack of effect of GSK1345038A in the same experimental model, the comparison with our data could be misleading and not relevant, given the different conditions adopted (higher dose and double exposure to TNBS in our model) and the different inflammatory parameters examined by the two studies.
The beneficial effects of AR-R17779 were accompanied by its ability to attenuate the changes caused by TNBS exposure on splenic T lymphocytes, while colitis-induced MLN changes were not antagonized: this observation prompted us to speculate that the protection by the α7 nAChR agonist might be mediated by its immune suppressant effects on splenic T lymphocytes and by its efficacy in preventing their expansion and proliferation. Indeed, the vagal nicotinic down-regulation of splenic CD4+ T cells functions (Karimi et al., 2010) and the proliferative effects of α7 nAChRs antagonists on T cells (De Rosa et al., 2009) would point toward this mechanism of action.
Following SPX, colitis severity was not modified: in operated mice, the local and systemic inflammatory indices induced by haptenization were comparable to those of non-operated animals. Intriguingly, removal of the spleen provoked a proportional decrease in the count of MLN T lymphocytes in normal and colitic mice with respect to non-operated animals, suggesting that MLN T lymphocytes derive partly from spleen trafficking. This observation confirmed us also about the fact that changes in the number of MLN T lymphocytes do not reflect changes in the severity of colonic inflammatory responses. Notably, AR-R17779 lost completely its beneficial effects in colitic splenectomized mice, MS being even worsened by α7 nAChRs agonist. The spleen-dependence of the protective effect of nicotine has been already demonstrated in a model of lethal polymicrobial sepsis, where the activation of the innate immune system was considered pivotal to the development of the inflammatory process (Huston et al., 2006), but the involvement of the spleen in the modulation of adaptive immune responses by selective exogenous stimulation of α7 nAChRs is a novel finding.
Our results show now, for the first time, that AR-R17779 exerts beneficial effects in a model of intestinal inflammation characterized by activation of the adaptive immune system and that the spleen appears essential to the cholinergic protection. Starting from these considerations, intriguing results could arise also from the investigation of AR-R17779 effects in another model of hapten-induced colitis, such as oxazolone colitis, where the protective effect of nicotine has already been demonstrated (Galitovskiy et al., 2011) and where natural killer T cells dictate the Th2-associated cytokine pattern with respect to the Th1 profile typical of TNBS-induced colitis (Kiesler et al., 2015). On the whole, these studies will help to clarify the relationship between α7 nAChRs-mediated protection and splenic T cells functions and its possible implications in view of a therapeutic strategy against chronic inflammatory disorders based on the utilization of selective α7 nAChRs agonists.
Author Contributions
AG, IZ, LF, and VV performed the experiments; AC carried out the histological analysis; VB analyzed the data; EB, SB, and AG designed the research, analyzed the data and wrote the paper; all the authors critically revised the manuscript, finally approved it and agreed to be accountable for all aspects of the work.
Funding
This work was supported by a local grant of University of Parma (FIL 2015).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Preliminary data were presented at Digestive Disease Week 2016, 21–24 May 2016, San Diego, CA, United States. The authors wish to thank Dr. Giuseppe Domenichini for his skillful technical assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2017.00809/full#supplementary-material
Abbreviations
CAP, cholinergic anti-inflammatory pathway; CTR, control; DAI, Disease Activity Index; DBE, dihydro-β-erythroidine; FCS, fetal calf serum; MLA, methyllycaconitine; MLN, mesenteric lymph nodes; MPO, myeloperoxidase; MS, macroscopic score; N, normal; nAChR, nicotinic Ach receptor; RCF, relative centrifugal force; SPX, splenectomy; TNBS, 2,4,6-trinitrobenzene sulfonic acid.
References
Bencherif, M., Lovette, M., Fowler, K., Arrington, S., Reeves, L., Caldwell, W., et al. (1996). RJR-2403: a nicotinic agonist with CNS selectivity I. In vitro characterization. J. Pharmacol. Exp. Ther. 279, 1422–1429.
Bischoff, S. C., Mailer, R., Pabst, O., Weier, G., Sedlik, W., Li, Z., et al. (2009). Role of serotonin in intestinal inflammation: knock out of serotonin reuptake transporter exacerbates 2,4,6-trinitrobenzene sulfonic acid colitis in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 296, G685–G695. doi: 10.1152/ajpgi.90685.2008
Bonaz, B., Picq, C., Sinniger, V., Mayol, J. F., and Clarençon, D. (2013). Vagus nerve stimulation: from epilepsy to the cholinergic anti-inflammatory pathway. Neurogastroenterol. Motil. 25, 208–221. doi: 10.1111/nmo.12076
Cooper, H. S., Murthy, S. N., Shah, R. S., and Sedergran, D. J. (1993). Clinico-pathological study of dextran sulfate sodium experimental murine colitis. Lab. Invest. 69, 238–249.
Costa, R., Motta, E. M., Manjavachi, M. N., Cola, M., and Calixto, J. B. (2012). Activation of the alpha-7 nicotinic acetylcholine receptor (α7 nAchR) reverses referred mechanical hyperalgesia induced by colonic inflammation in mice. Neuropharmacology 63, 798–805. doi: 10.1016/j.neuropharm.2012.06.004
Costantini, T. W., Krzyzaniak, M., Cheadle, G. A., Putnam, J. G., Hageny, A. M., Lopez, N., et al. (2012). Targeting a7 nicotinic acetylcholine receptor in the enteric nervous system: a cholinergic agonist prevents gut barrier failure after severe burn injury. Am. J. Pathol. 181, 478–486. doi: 10.1016/j.ajpath.2012.04.005
Curtis, M. J., Bond, R. A., Spina, D., Ahluwalia, A., Alexander, S. P. A., Giembycz, M. A., et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br. J. Pharmacol. 172, 3461–3471. doi: 10.1111/bph.12856
De Biasi, M., and Dani, J. A. (2011). Reward, addiction, withdrawal to nicotine. Annu. Rev. Sci. 34, 105–130. doi: 10.1146/annurev-neuro-061010-113734
De Jonge, W. J., Van Der Zanden, E. P., The, F. O., Bijlsma, M. F., Van Westerloo, D. J., Bennink, R. J., et al. (2005). Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2/STAT3 signaling pathway. Nat. Immunol. 6, 844–851. doi: 10.1038/ni1229
De Rosa, M. J., Dionisio, L., Agriello, E., Bouzat, C., and Esandi, M. C. (2009). Alpha 7 nicotinic acetylcholine receptor modulates lymphocyte activation. Life Sci. 85, 444–449. doi: 10.1016/j.lfs.2009.07.010
Di Giovangiulio, M., Bosman, G., Meroni, E., Stakenborg, N., Florens, M., Farro, G., et al. (2016). Vagotomy affects the development of oral tolerance and increases susceptibility to develop colitis independently of α7 nicotinic receptor. Mol. Med. 22, 464–476. doi: 10.2119/molmed.2016.00062
Downs, A. M., Bond, C. E., and Hoover, D. B. (2014). Localization of α7 nicotinic acetylcholine receptor mRNA, and protein within the cholinergic anti-inflammatory pathway. Neuroscience 266, 178–185. doi: 10.1016/j.neuroscience.2014.02.011
Galitovskiy, V., Qian, J., Chernyavsky, A. I., Marchenko, S., Gindi, V., Edwards, R. A., et al. (2011). Cytokine-induced alterations of α7 nicotinic receptor in colonic CD4 T cells mediate dichotomous response to nicotine in murine models of Th1/Th17- versus Th2-mediated colitis. J. Immunol. 187, 2677–2687. doi: 10.4049/jimmunol.1002711
Ge, J., Tian, J., Yang, H., Hou, L., Wang, Z., He, Z., et al. (2016). Alpha7 nicotine acetylcholine receptor agonist PNU-282987 attenuates acute lung injury in a cardiopulmonary bypass model in rats. Shock 47, 474–479. doi: 10.1097/SHK.0000000000000744
Goverse, G., Stakenborg, M., and Matteoli, G. (2016). The intestinal cholinergic anti-inflammatory pathway. J. Physiol. 594, 5771–5780. doi: 10.1113/JP271537
He, Y., Ye, Z. Q., Li, X., Zhu, G. S., Liu, Y., Yao, W. F., et al. (2016). Alpha7 nicotinic acetylcholine receptor activation attenuated intestine-derived acute lung injury. J. Surg. Res. 201, 258–265. doi: 10.1016/j.jss.2015.10.046
Hijioka, M., Matsushita, H., Ishibashi, H., Hisatsune, A., Isohama, Y., and Katsuki, H. (2012). α7 nicotinic acetylcholine receptor agonist attenuates neuropathological changes associated with intracerebral hemorrhage in mice. Neuroscience 222, 10–19. doi: 10.1016/j.neuroscience.2012.07.024
Holmes, K., Lantz, L. M., Fowlkes, B. J., Schmid, I., and Giorgi, J. V. (2001). Preparation of cells and reagents for flow cytometry. Curr. Protoc. Immunol. 44, 5.3.1–5.3.24. doi: 10.1002/0471142735.im0503s44
Hosur, V., and Loring, R. H. (2011). α4β2 nicotinic receptors partially mediate anti-inflammatory effects through Janus kinase 2-signal transducer and activator of transcription 3 but not calcium or cAMP signaling. Mol. Pharmacol. 79, 167–174. doi: 10.1124/mol.110.066381
Hurst, R., Rollema, H., and Bertrand, D. (2013). Nicotinic acetylcholine receptors: from basic science to therapeutics. Pharmacol. Ther. 137, 22–54. doi: 10.1016/j.pharmthera.2012.08.012
Huston, J. M., Ochani, M., Rosas-Ballina, M., Liao, H., Ochani, K., Pavlov, V. A., et al. (2006). Splenectomy inactivates the cholinergic antiinflammatory pathway during lethal endotoxemia and polymicrobial sepsis. J. Exp. Med. 203, 1623–1628. doi: 10.1084/jem.20052362
Joh, E. H., and Kim, D. H. (2011). Kalopanaxsaponin A ameliorates experimental colitis in mice by inhibiting IRAK-1 activation in the NF-KB and MAPK pathways. Br. J. Pharmacol. 162, 1731–1742. doi: 10.1111/j.1476-5381.2010.01195.x
Karimi, K., Bienenstock, J., Wang, L., and Forsythe, P. (2010). The vagus nerve modulates CD4+ T cell activity. Brain Behav. Immun. 24, 316–323. doi: 10.1016/j.bbi.2009.10.016
Kashiwagi, S., Khan, M. A., Yasuhara, S., Goto, T., Kem, W. R., Tompkins, R. G., et al. (2017). Prevention of burn-induced inflammatory responses and muscle wasting by GTS-21, a specific agonist for α7 nicotinic acetylcholine receptors. Shock 47, 61–69. doi: 10.1097/SHK.0000000000000729
Kawashima, K., Fujii, T., Moriwaki, Y., Misawa, H., and Horiguchi, K. (2012). Reconciling neuronally and nonneuronally derived acetylcholine in the regulation of immune function. Ann. N. Y. Acad. Sci. 1261, 7–17. doi: 10.1111/j.1749-6632.2012.06516.x
Kiesler, P., Fuss, I. J., and Strober, W. (2015). Experimental models of inflammatory bowel disease. Cell. Mol. Gastroenterol. Hepatol. 1, 154–170. doi: 10.1016/j.jcmgh.2015.01.006
Kilkenny, C., Browne, W. J., Cuthill, I. C., Emerson, M., and Altman, D. G. (2010). Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLOS Biol. 8:e1000412. doi: 10.1371/journal.pbio.1000412
Krawisz, J. E., Sharon, P., and Stenson, W. F. (1984). Quantitative assay for acute intestinal inflammation based on myeloperoxidase activity. Assessment of inflammation in rat and hamster models. Gastroenterology 87, 1344–1350.
Kruisbeek, A. M. (2001). Isolation of mouse mononuclear cells. Curr. Protoc. Immunol. 39, 3.1.1–3.1.5. doi: 10.1002/0471142735.im0301s39
Li, F., Chen, Z., Pan, Q., Fu, S., Lin, F., Ren, H., et al. (2013). The protective effect of PNU-282987, a selective a7 nicotinic acetylcholine receptor agonist, on the hepatic ischemia-reperfusion injury is associated with the inhibition of high-mobility group box 1 protein expression and nuclear factor kB activation in mice. Shock 39, 197–203.
Lopez, M. C. (2007). Fluorescence microscopy and flow cytometric analysis of Peyer’s patches and intestinal immune cells. Curr. Protoc. Immunol. 33, 18.13.1–18.13.20. doi: 10.1002/0471140856.tx1813s33
Mahida, Y. R., Wu, K., and Jewell, D. P. (1989). Enhanced production of interleukin 1-beta by mononuclear cells isolated from mucosa with active ulcerative colitis of Crohn’s disease. Gut 30, 835–838. doi: 10.1136/gut.30.6.835
Martelli, D., McKinley, M. J., and McAllen, R. M. (2014). The cholinergic anti-inflammatory pathway: a critical review. Auton. Neurosci. 182, 65–69. doi: 10.1016/j.autneu.2013.12.007
Matsunaga, K., Klein, T. W., Friedman, H., and Yamamoto, Y. (2001). Involvement of nicotinic acetylcholine receptors in suppression of antimicrobial activity and cytokine responses of alveolar macrophages to Legionella pneumophila infection by nicotine. J. Immunol. 167, 6518–6524. doi: 10.4049/jimmunol.167.11.6518
Moore-Olufemi, S. D., Kozar, R. A., Moore, F. A., Sato, N., Hassoun, H. T., Cox, C. S. Jr., et al. (2005). Ischemic preconditioning protects against gut dysfunction and mucosal injury after ischemia/reperfusion injury. Shock 23, 258–263.
Mullen, G., Napier, J., Balestra, M., DeCory, T., Hale, G., Macor, J., et al. (2000). (-)-Spiro[1-azabicyclo[2.2.2] octane-3,5’-oxazolidin-2’-one], a conformationally restricted analogue of acetylcholine, is a highly selective full agonist at the alpha 7 nicotinic acetylcholine receptor. J. Med. Chem. 43, 4045–4050. doi: 10.1021/jm000249r
Niimi, K., Nishioka, C., Miyamoto, T., Takahashi, E., Miyoshi, I., Itakura, C., et al. (2013). Improvement of spontaneous alternation behavior deficit by activation of α4β2 nicotinic acetylcholine receptor signaling in the ganglioside GM3-deficient mice. Biomed. Res. 34, 189–195. doi: 10.2220/biomedres.34.189
Nullens, S., Staessens, M., Peleman, C., Schrijvers, D. M., Malhotra-Kumar, S., Francque, S., et al. (2016). Effect of GTS-21, an alpha7 nicotinic acetylcholine receptor agonist, on CLP-induced inflammatory, gastrointestinal motility, and colonic permeability changes in mice. Shock 45, 450–459. doi: 10.1097/SHK.0000000000000519
Olofsson, P. S., Rosas-Ballina, M., Levine, Y. A., and Tracey, K. J. (2012). Rethinking inflammation: neural circuits in the regulation of immunity. Immunol. Rev. 248, 188–204. doi: 10.1111/j.1600-065X.2012.01138.x
Pinheiro, N. M., Santana, F. P., Almeida, R. R., Guerreiro, M., Martins, M. A., Caperuto, L. C., et al. (2017). Acute lung injury is reduced by the α7 nAChR agonist PNU-282987 through changes in the macrophage profile. FASEB J. 31, 320–332. doi: 10.1096/fj.201600431R
Rapalli, A., Bertoni, S., Arcaro, V., Saccani, F., Grandi, A., Vivo, V., et al. (2016). Dual role of endogenous serotonin in 2,4,6-Trinitrobenzene Sulfonic acid-induced colitis. Front. Pharmacol. 7:68. doi: 10.3389/fphar.2016.00068
Rowley, T. J., Payappilly, J., Lu, J., and Flood, P. (2008). The antinociceptive response to nicotinic agonists in a mouse model of postoperative pain. Anesth. Analg. 107, 1052–1057. doi: 10.1213/ane.0b013e318165e0c0
Saeed, R. W., Varma, S., Peng-Nemeroff, T., Sherry, B., Balakhaneh, D., Huston, J., et al. (2005). Cholinergic stimulation blocks endothelial cell activation and leukocyte recruitment during inflammation. J. Exp. Med. 201, 1113–1123. doi: 10.1084/jem.20040463
Salaga, M., Blomster, L. V., Piechota-Polanczyk, M., Zielinska, M., Jacenik, D., Cygankiewicz, A. I., et al. (2016). Encenicline, an α7 nicotinic acetylcholine receptor partial agonist, reduces immune cell infiltration in the colon and improves experimental colitis in mice. J. Pharmacol. Exp. Ther. 356, 157–169. doi: 10.1124/jpet.115.228205
Snoek, S. A., Verstege, M. I., Van der Zanden, E. P., Deeks, N., Bulmer, D. C., Skynner, M., et al. (2010). Selective α7 nicotinic acetylcholine receptor agonists worsen disease in experimental colitis. Br. J. Pharmacol. 160, 322–333. doi: 10.1111/j.1476-5381.2010.00699.x
Talley, N. J., Abreu, M. T., Achkar, J. P., Bernstein, C. N., Dubinsky, M. C., Hanauer, S. B., et al. (2011). An evidence-based systematic review on medical therapies for inflammatory bowel disease. Am. J. Gastroenterol. 106, S2–S25. doi: 10.1038/ajg.2011.58
Te Velde, A. A., Verstege, M. I., and Hommes, D. W. (2006). Critical appraisal of the current practice in murine TNBS-induced colitis. Inflamm. Bowel Dis. 12, 995–999. doi: 10.1097/01.mib.0000227817.54969.5e
The, F. O., Boeckxstaens, G. E., Snoek, S. A., Cash, J. A., Bennink, R., and Larosa, G. J. (2007). Activation of the cholinergic anti-inflammatory pathway ameliorates postoperative ileus in mice. Gastroenterology 133, 1219–1228. doi: 10.1053/j.gastro.2007.07.022
van der Zanden, E. P., Snoek, S. A., Heinsbroek, S. E., Stanisor, O. I., Verseijden, C., Boeckxstaens, G. E., et al. (2009). Vagus nerve activity augments intestinal macrophage phagocytosis via nicotinic acetylcholine receptor alpha4beta2. Gastroenterology 13, 1029–1039. doi: 10.1053/j.gastro.2009.04.057
Waldner, M. J., and Neurath, M. F. (2009). Chemically induced mouse models of colitis. Curr. Protoc. Pharmacol. 46, 5.55.1–5.55.15. doi: 10.1002/0471141755.ph0555s46
Wang, H., Yu, M., Ochani, M., Amella, C. A., Tanovic, M., Susarla, S., et al. (2003). Nicotinic acetylcholine receptor α7 subunit is an essential regulator of inflammation. Nature 421, 384–388. doi: 10.1038/nature01339
Keywords: intestine, inflammation, α7 nicotinic agonist, α4β2 nicotinic receptors, sulfasalazine, lymph nodes
Citation: Grandi A, Zini I, Flammini L, Cantoni AM, Vivo V, Ballabeni V, Barocelli E and Bertoni S (2017) α7 Nicotinic Agonist AR-R17779 Protects Mice against 2,4,6-Trinitrobenzene Sulfonic Acid-Induced Colitis in a Spleen-Dependent Way. Front. Pharmacol. 8:809. doi: 10.3389/fphar.2017.00809
Received: 24 July 2017; Accepted: 26 October 2017;
Published: 08 November 2017.
Edited by:
Gabriella Aviello, University of Aberdeen, United KingdomReviewed by:
Ashish Kumar Singh, University College Dublin, IrelandL. Ashley Blackshaw, Queen Mary University of London, United Kingdom
Huaxu Zhu, Nanjing University of Chinese Medicine, China
Copyright © 2017 Grandi, Zini, Flammini, Cantoni, Vivo, Ballabeni, Barocelli and Bertoni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Simona Bertoni, simona.bertoni@unipr.it