
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol., 24 October 2017
Sec. Experimental Pharmacology and Drug Discovery
Volume 8 - 2017 | https://doi.org/10.3389/fphar.2017.00727
This article is part of the Research TopicPhenotypic Drug Discovery: Models, Methodology and ApplicationsView all 12 articles
 Lei Qiao1
Lei Qiao1 Xue Zhang1,2Minghao Liu1Xiaoling Liu1Mei Dong1
Xue Zhang1,2Minghao Liu1Xiaoling Liu1Mei Dong1 Jing Cheng1Xinyu Zhang1
Jing Cheng1Xinyu Zhang1 Chungang Zhai1Yu Song1Huixia Lu1*
Chungang Zhai1Yu Song1Huixia Lu1* Wenqiang Chen1*
Wenqiang Chen1*Atherosclerosis (AS) is a lipid-driven disease in which macrophage foam cells play a critical role by increasing vascular lipid accumulation and contributing to plaque instability. Ginsenoside Rb1 (Rb1), the most abundant active component of ginseng, has been found potently to promote lipid metabolism and attenuate lipid accumulation. However, the underlying mechanisms remain unclear. In this study, the effects of Rb1 on lipid accumulation and plaque stability were investigated both in vitro and in vivo by using primary peritoneal macrophages isolated from C57BL/6 mice and an AS model in ApoE-/- mice. The results showed that Rb1 reduced lipid accumulation both in macrophage foam cells and atherosclerotic plaques. Rb1 treatment promoted plaque stability by modifying plaque composition via the activation of autophagy both in vitro and in vivo. Transmission electron microscopy further showed an increased accumulation of autophagolysosomes in Rb1-treated macrophage foam cells. However, the modulation of lipid accumulation by Rb1 was attenuated by autophagy blockage using autophagy-related gene 5 (Atg5) small interfering RNA (siRNA) in vitro. In addition, Rb1 notably increased AMPK phosphorylation both in vitro and in vivo, and the AMPK inhibitor compound C abolished the Rb1-induced autophagy in macrophage foam cells. In conclusion, ginsenoside Rb1 reduced lipid accumulation in macrophage foam cells and enhanced atherosclerotic plaque stability by the induction of macrophage autophagy. Our study provides new evidence for the possible use of Rb1 in the prevention and treatment of AS.
Atherosclerosis (AS) is a chronic lipid-driven disease in large and medium arterial walls (Weber and Noels, 2011). Macrophages serve as the major component of atherosclerotic lesions and are critical to the development of AS by engulfing ox-LDLs to form cholesterol-laden foam cells (Moore et al., 2013), contributing to the increase in atherosclerotic plaque complexity and instability. Therefore, decreasing lipid accumulation in foam cells and atherosclerotic plaque is an efficient way to alleviate or reverse the progression of AS.
Autophagy is an evolutionarily conserved process dedicated to the degradation of cytoplasmic materials, such as long-lived proteins and dysfunctional organelles (Levine and Kroemer, 2008). Previous studies have found that autophagy plays a vital role in lipid metabolism (Singh et al., 2009; Dong and Czaja, 2011), through facilitating the hydrolysis of intracellular lipids and promoting cholesterol efflux in macrophage foam cells (Ouimet et al., 2011). Impaired autophagy efflux of macrophages could lead to an ineffective whole-body clearance of accumulated lipids, which underlies the pathogenesis of many important metabolic disorders, including fatty liver, obesity and AS (Ouimet et al., 2011). Increasing evidence has demonstrated that dysfunctional autophagy is greatly associated with the progression of AS (Razani et al., 2012; Li et al., 2016). Incompetent autophagy is also responsible for excessive lipid accumulation in atherosclerotic plaques. Thus, therapeutic strategies to increase autophagic activity may provide a new approach to facilitate lipid metabolism and plaque stability in advanced atherosclerotic plaques.
Ginseng, a traditional herbal medicine, has been widely used in Eastern Asia and the western world for its alleged tonic effect and restorative properties. Previous studies have shown that ginsenoside Rb1, the most abundant active component of ginseng (Washida and Kitanaka, 2003), plays a protective role in many cardiovascular diseases. For instance, Rb1 prevents homocysteine-induced endothelial dysfunction (Lan et al., 2011), and protects against cardiac remodeling (Zheng et al., 2017) and ischemia/reperfusion-induced myocardial injury (Cui et al., 2017). However, the role of Rb1 in AS has not been reported. Considering the ability of Rb1 to promote lipid metabolism and attenuate lipid accumulation (Park et al., 2002; Shen et al., 2013; Yu et al., 2015), we hypothesized that Rb1 may promote atherosclerotic plaque stability through facilitating lipid metabolism in macrophage foam cells.
To test this hypothesis, primary peritoneal macrophages isolated from C57BL/6 mice and an AS model in the ApoE-/- mouse background were used to explore whether ginsenoside Rb1 was cardioprotective by mitigating lipid accumulation in macrophage foam cells and investigate its mechanism.
All animal experimental protocols complied with the Animal Management Rules of the Chinese Ministry of Health (document no. 55, 2001) and conformed to the NIH guidelines (the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health; NIH Publication No. 85-23, revised 1996).
Mouse primary peritoneal macrophages were obtained as described (Ho et al., 2012). Briefly, 3% thioglycollate was intraperitoneally injected into mice. After 3 days, mice were first anesthetized with a lethal dose of isoflurane and then euthanized by cervical dislocation. Peritoneal macrophages were collected by peritoneal lavage using cold PBS. The cell pellets were obtained after centrifugation at 800 rpm for 5 min. The cells were incubated in 1640 RPMI supplemented with 10% FBS and 1% antibiotics for 3 h and washed three times to remove non-adherent cells. Cells were incubated with ox-LDL (100 μg/ml, Peking Union-Biology, China) for 24 h to induce macrophage foam cell formation. Then, cells were treated with different concentrations of ginsenoside Rb1 (Fleton Natural Products Co. Ltd.) or PBS for another 24 h. Cells were also treated with compound C (CC, 20 μM, sc200689, Santa Cruz Biotechnology, United States) for 2 h following the addition of Rb1 for 24 h. Macrophage foam cells were stained with 0.5% oil red O.
Cultured primary peritoneal macrophages were plated on cover slides in six-well plates and incubated with ox-LDLs for 24 h to form macrophage foam cells and different concentrations of ginsenoside Rb1 or PBS for another 24 h. Cells were washed with cold phosphate-buffered saline (PBS) three times and fixed with 4% paraformaldehyde for 30 min. Subsequently, 0.5% oil red O was added to cells for 30 min. Of note, oil red O needs to be filtered to remove impurities. Foam cells were observed under a microscope at ×100 and ×200 magnification.
Protein extraction and western blotting were performed as described before (Ho et al., 2009). Peritoneal macrophages were washed with ice-cold PBS and lysed on ice using RIPA lysis buffer supplemented with complete protease inhibitor cocktail. Equal amounts of protein (20 μg) were separated by 12% SDS-PAGE gel electrophoresis and transferred onto PVDF membranes (Bio-Rad). Membranes were blocked in 5% non-fat dry milk followed by incubation with primary antibodies. Antibodies purchased from Abcam (Cambridge, United Kingdom) were as follows: LC3B (ab48394), SQSTM1/p62 (ab56416) and Atg5 (ab108327). Antibodies obtained from Cell Signaling Technology (Danvers, MA, United States) included those against AMPKα (5831) and p-AMPKα (Thr172) (2535). Secondary antibodies (ZSJB-BIO, China) were horseradish peroxidase (HRP)-conjugated goat anti-rabbit or goat anti-mouse antibodies.
Small interfering RNA (Biology Engineering Corporation, Shanghai, China) was used to block the expression of Atg5. The mRNA sequence of the Atg5 RNAi was 5′-ACCGGAAACTCATGGAATA-3′ (Seo et al., 2011). Mouse primary peritoneal macrophages (1 × 106 cells/ml) were transfected with con siRNA or Atg5 siRNA using Lipofectamine 2000. After 24 h, western blotting was performed to confirm the knockdown of Atg5 protein for each experiment.
Cells were fixed with 4% glutaraldehyde prior to postfixation in 1% osmium tetroxide. Samples were processed and embedded in Epon-Araldite resin. Ultrathin sections were obtained by using an ultramicrotome and counterstained with lead citrate. A Hitachi H-7500 electron microscope was used to observe lipid droplets and autophagosomes.
A total of 30 apoE-/- mice (male, 8 weeks old) were obtained from Charles River Laboratories (Beijing, China), and all mice were kept on a 12-h light/12-h dark cycle. All mice were fed a high-fat diet (0.25% cholesterol and 15% cocoa butter) for 20 weeks. Subsequently, ApoE-/- mice were randomly divided into 2 groups (n = 15 per group) for treatment: the Rb1 group received an intraperitoneal injection with Rb1 (50 mg/kg/d) for 8 weeks and the control group, an equal volume of saline. After 8 weeks of treatment, mice were sacrificed, and the aorta and heart were harvested. Blood samples were collected to measure serum lipid levels.
Mice were sacrificed, and the heart with attached aortic roots was obtained immediately and fixed overnight in 4% formaldehyde. The aortic roots were embedded in optimum cutting temperature (OCT) compound for cryosections, and were cut into serial 6-μm cross-sections for histological staining and immunofluorescence. IHC staining was undertaken using the rabbit monoclonal antibody against smooth muscle actin (ab5694; Abcam, United States) and rat monoclonal antibody against MOMA-2 (MCA519G; AbD, United Kingdom). The sections were incubated with horseradish peroxidase (HRP)-conjugated goat anti-rabbit or goat anti-rat secondary antibodies (ZSJB-BIO, China). The lipid and collagen content of lesions was detected by oil-red O staining and sirius red staining, respectively. The slides were quantitatively analyzed using Image-Pro Plus 6.0 (IPP 6.0, Media Cybernetics, Rockville, MD, United States).
All values are representative of at least three independent experiments and expressed as the means ± SEM by use of GraphPad Prism 5 (La Jolla, CA, United States). Differences among groups were analyzed by one-way ANOVA followed by a Tukey post hoc test, and t-test was used for the two-group comparisons. Statistical significance was defined as P < 0.05.
Rb1 has been reported to be able to reduce liver lipid accumulation (Shen et al., 2013; Yu et al., 2015). Here, we investigated whether Rb1 had an effect on intracellular lipid accumulation in macrophage foam cells. Macrophages were exposed to 100 μg/ml ox-LDLs for 24 h and further incubated in the absence or presence of 10–80 μM Rb1. Oil red O was used to analyze intracellular lipid accumulation. Treatment of macrophages with ox-LDLs for 24 h resulted in foam cell formation which was characterized by heavy lipid loading. Incubation with Rb1 for another 24 h reduced lipid accumulation, with the minimum effect found at a dose of 20 μM (Figures 1A,B). Therefore, Rb1 reduced lipid accumulation of macrophage foam cells in vitro.

FIGURE 1. Rb1 decreased lipid accumulation in macrophage foam cells. Primary peritoneal macrophages from C57BL/6 mice were incubated with ox-LDL (100 μg/ml) for 24 h and further incubated in the absence or presence of different doses of Rb1 for another 24 h. (A,B) oil red O staining was performed to assess lipid accumulation in macrophages.∗p < 0.05, compared to the ox-LDL group, n = 3.
Autophagy has been found to play critical roles in lipid metabolism (Singh et al., 2009; Dong and Czaja, 2011; Ouimet et al., 2011). The LC3II/LC3I ratio and SQSTM1/p62 protein have been considered to be a standard markers for the detection of autophagy (Mizushima and Yoshimori, 2007; Klionsky et al., 2012). The results showed that treatment with ox-LDLs for 24 h decreased the LC3II/LC3I ratio (Figures 2A,B) with a corresponding increase in the SQSTM1/p62 level in peritoneal macrophages (Figures 2A–C), suggesting that autophagy flux was impaired in cholesterol-laden foam cells. The result was consistent with the previous reports (Singh et al., 2009; Koga et al., 2010; Gu et al., 2016) which showed that increased lipid content might impair autophagy flux. Interestingly, Rb1 treatment rescued the autophagy flux impaired by ox-LDLs in macrophage foam cells, which was evidenced by the increased LC3II/LC3I ratio and SQSTM1/p62 degradation (Figures 2A–C).
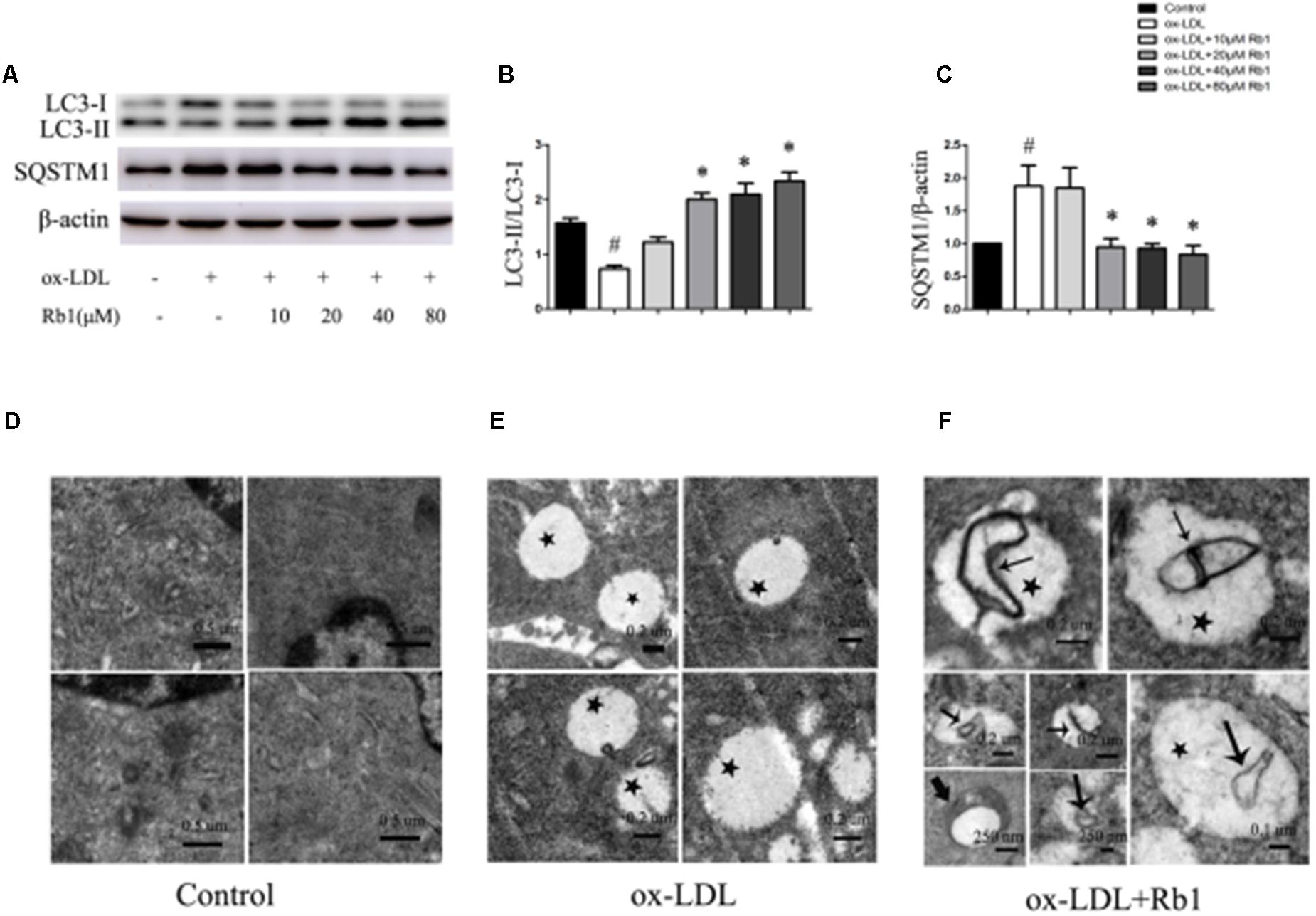
FIGURE 2. Rb1 rescued the impaired autophagy flux in macrophage foam cells. (A) Representative western blot analysis of LC3B and SQSTM1/p62. (B) Quantification of LC3 and (C) SQSTM1/p62 expression relative to the β-actin level in different groups. Electron microscopic analysis of autophagosomes in macrophage foam cells treated with Rb1. Primary peritoneal macrophages were incubated with (D) PBS or (E) ox-LDL (100 μg/ml) for 24 h, and further incubated with (F) Rb1 (20 μM ) for another 24 h. Asterisks indicate lipid droplets (LDs); arrows indicate double-membrane vesicles analogous to autophagosomes (APs) in or around LDs. ∗p < 0.05, compared to the ox-LDL group; #p < 0.05, compared to the control group, n = 3.
To further examine the effect of Rb1 on autophagy in macrophage foam cells, transmission electron microscopy (TEM) was used to explore morphological evidence for autophagy flux. Little lipid droplets (LDs) or autophagosomes (APs) were observed in primary peritoneal macrophages with no treatment (Figure 2D). Treatment with ox-LDL caused lipid accumulation in the form of LDs, which were easily identifiable as round light-density structures without a bilayer lipid membrane (Figure 2E). Rb1 treatment increased the number of double-membrane autophagosomes (APs), which were presented in or around LDs (Figure 2F), suggesting that autophagy occurred on the surface of the LDs. Taken together, our data suggested that Rb1 rescued the impaired autophagy flux in macrophage foam cells.
Because of the demonstrated reduction in lipid accumulation in macrophage foam cells, accompanied by increased expression of autophagy proteins and autophagosomes after Rb1 treatment, we speculated that Rb1 treatment may decrease lipid accumulation by activating autophagy flux in macrophage foam cells. To test this hypothesis, autophagy-related gene 5 (Atg5) small interfering RNA (siRNA) was applied to block autophagic flux in macrophages. Macrophages transfected with Atg5 siRNA showed a decreased level of Atg5 and LC3 II expression (Supplementary Figure S1). In the control siRNA group, Rb1 (20 μM) increased the Atg5 expression and LC3II/LC3I ratio significantly (Figures 3A–C) and reduced the lipid droplet accumulation indicated by oil red O staining (Figures 3D,E). In contrast, in macrophages transfected with Atg5 siRNA, Rb1 failed to alter the levels of autophagy proteins or attenuate intracellular lipid accumulation. These data suggested that Rb1 reduced lipid accumulation of macrophage foam cells by autophagy induction.
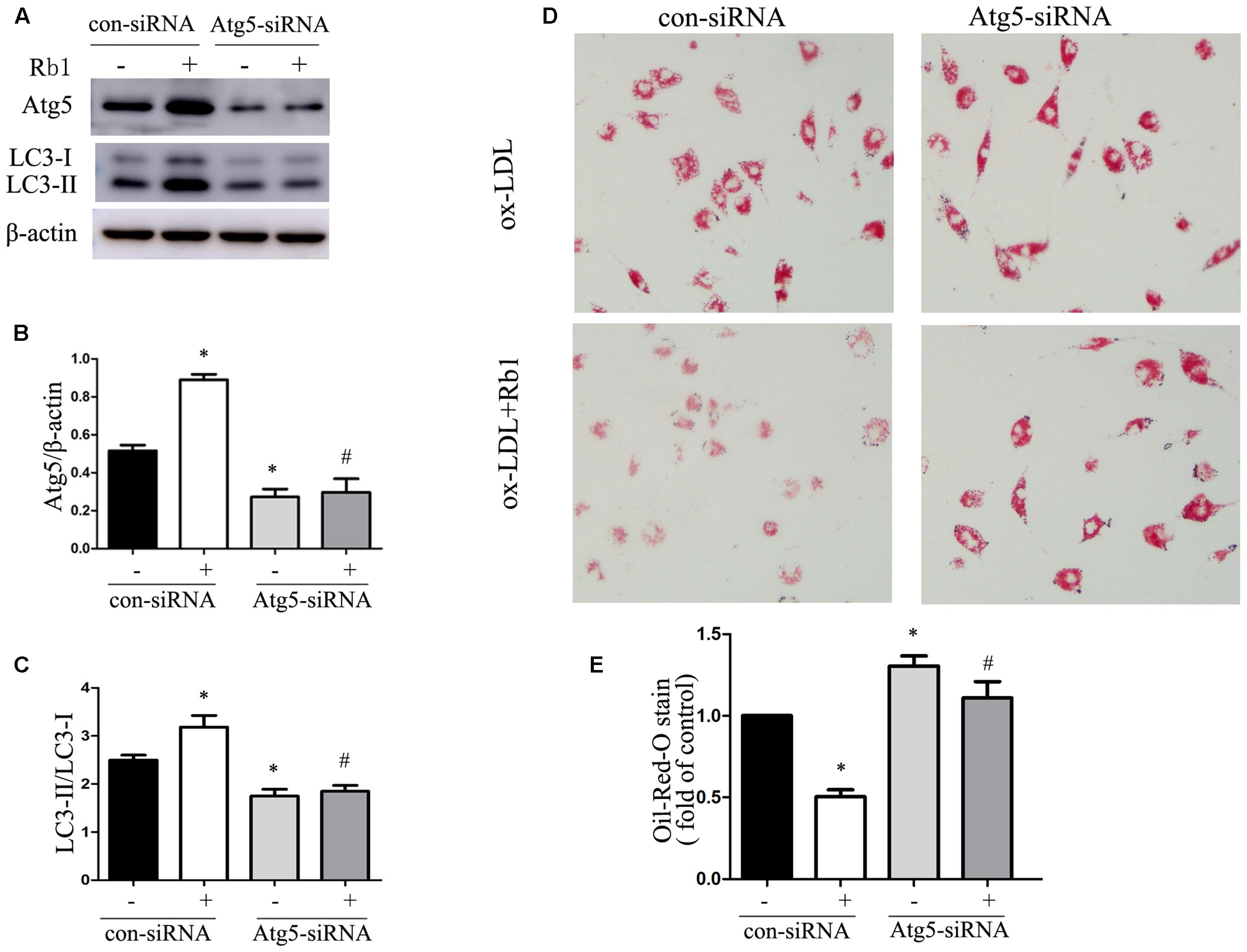
FIGURE 3. Rb1 reduced lipid accumulation of macrophage foam cells by autophagy induction. Primary peritoneal macrophages were transfected with con siRNA or Atg5 siRNA and then treated with ox-LDL (100 μg/ml) in the absence or presence of 20 μM Rb1. (A) Representative western blot analysis of Atg5 and LC3. (B) Quantification of Atg5 and (C) LC3 expression relative to β-actin in macrophages with different treatments. (D) Lipid accumulation in macrophages transfected with Atg5 siRNA with or without Rb1 treatment. (E) Quantitative analysis of lipid accumulation. ∗p < 0.05, compared to con siRNA; #p < 0.05, compared to con siRNA + Rb1, n = 3.
Previous studies have shown that autophagy regulates cholesterol efflux from macrophage foam cells, in which ATP-binding cassette transporter A1 (ABCA1) plays a vital role (Ouimet et al., 2011). As we have proven that Rb1 could promote lipid metabolism in macrophage foam cells via autophagy induction, we indirectly evaluated cholesterol efflux by detecting the expression of ABCA1. Our results showed a significant increase in ABCA1 in macrophage foam cells upon Rb1 treatment, which provides indirect evidence for cholesterol efflux (Supplementary Figure S2).
The results from our research (Figures 1A,B) as well as previous studies (Shen et al., 2013; Yu et al., 2015) showed that Rb1 contributed to reducing lipid accumulation. We next investigated the role of Rb1 in lipid accumulation in vivo in atherosclerotic plaque. Oil red O staining showed that the intraplaque lipid content was significantly decreased in the Rb1 treatment groups, along with decreased macrophage infiltration by histopathological staining (Figures 4A,B). This result was consistent with the in vitro results described above (Figures 1A,B). However, there was no significant difference in serum lipid profiles or blood glucose between the two groups of ApoE-/- mice (Supplementary Table 1). Recent studies have shown that ginsenoside Rb1 plays a protective role in many cardiovascular diseases (Lan et al., 2011; Cui et al., 2017; Zheng et al., 2017); we subsequently investigated the effect of Rb1 on plaque stability. Unstable plaques are characterized by extensive macrophage infiltration and lipid accumulation, as well as a thin cap with less collagen and smooth muscle cell (SMCs) (Dong et al., 2013; Wang et al., 2014). Therefore, the intraplaque content of SMCs and collagen was additionally analyzed by sirius red staining and histopathological staining. The results showed that both of these two beneficial components were markedly increased in the Rb1 treatment groups (Figures 4A,B). Taken together, our data suggested that Rb1 enhanced atherosclerotic plaque stability by modifying the plaque components.
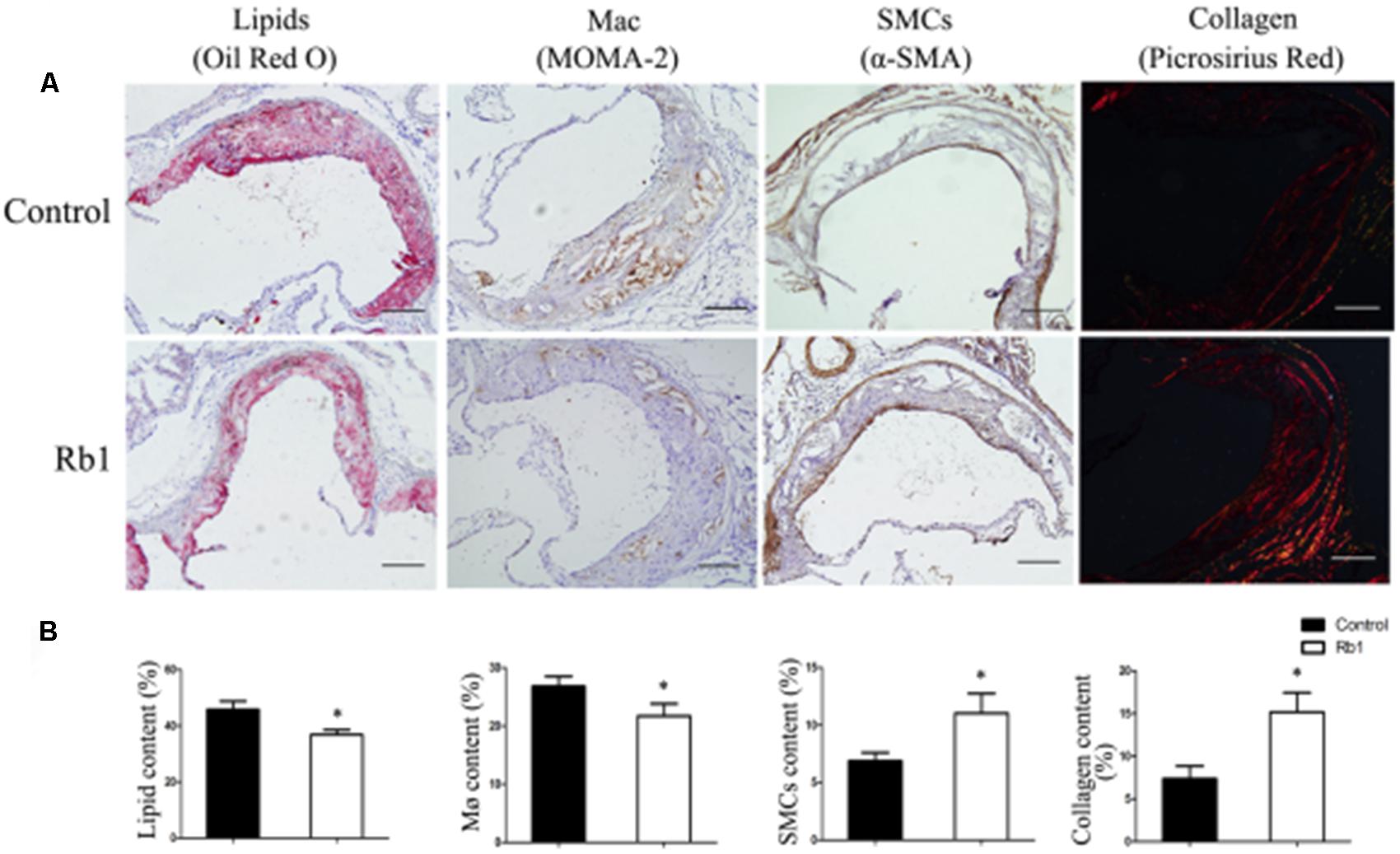
FIGURE 4. Effect of Rb1 treatment on plaque stability of apoE-/- mice. (A) Immunohistochemical staining of oil red O staining (lipids), MOMA-2 (macrophages), α-SMA (smooth muscle cells) and sirius red staining for the quantification of collagen. (B) Area measurement of plaque components. Scale bar: 100 μm. ∗p < 0.05, compared to the control group, n = 6.
Macrophage autophagy plays a protective role in AS (Liao et al., 2012). Advanced AS is accompanied by impaired autophagy flux with decreased Atg5 protein (Li et al., 2016) and increased SQSTM1/p62 protein (Razani et al., 2012). To investigate the role of Rb1 in autophagy activity in atherosclerotic plaques, we prepared aortic lysates from ApoE-/- mice to analyze the protein levels of LC3II and SQSTM1/p62. Not surprisingly, Rb1 treatment promoted the expression of LC3II protein and SQSTM1/p62 degradation in these lysates (Figures 5A,B). These results indicated that Rb1 treatment increased autophagy levels in atherosclerotic plaques, which was possibly responsible for the decrease in the intraplaque lipids content and cardioprotective benefits induced by Rb1.
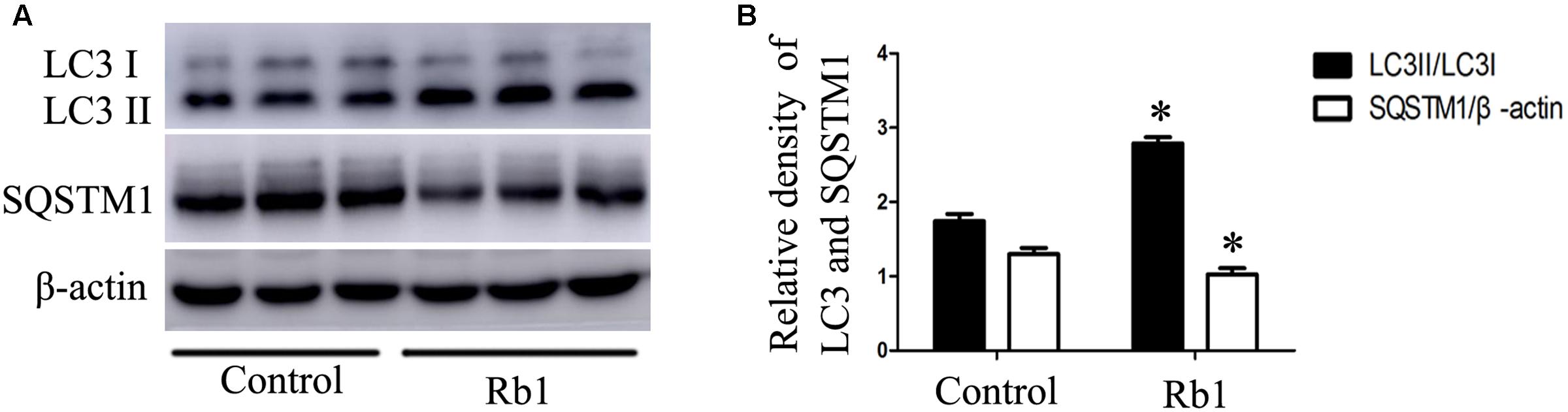
FIGURE 5. Rb1 increased autophagy levels in atherosclerotic plaques in apoE-/- mice. (A) Representative western blot analysis and (B) quantification of LC3-II and SQSTM1/p62 in vivo. ∗p < 0.05, compared to the control group, n = 6.
Previous studies have reported that Rb1 could promote AMPK phosphorylation (Shen et al., 2013; Shen et al., 2015). Therefore, the involvement of AMPK in Rb1-induced autophagy was evaluated in our study. The results showed that Rb1 treatment promoted AMPK phosphorylation both in vivo in atherosclerotic plaques (Figures 6A,B) and in macrophage foam cells (Figures 6C,D). Moreover, compound C, an AMPK inhibitor, abolished the increase in LC3II and SQSTM1/p62 degradation induced by Rb1 in vitro (Figures 6E,F). These data indicated that AMPK played a key role in Rb1-induced autophagy in macrophage foam cells.
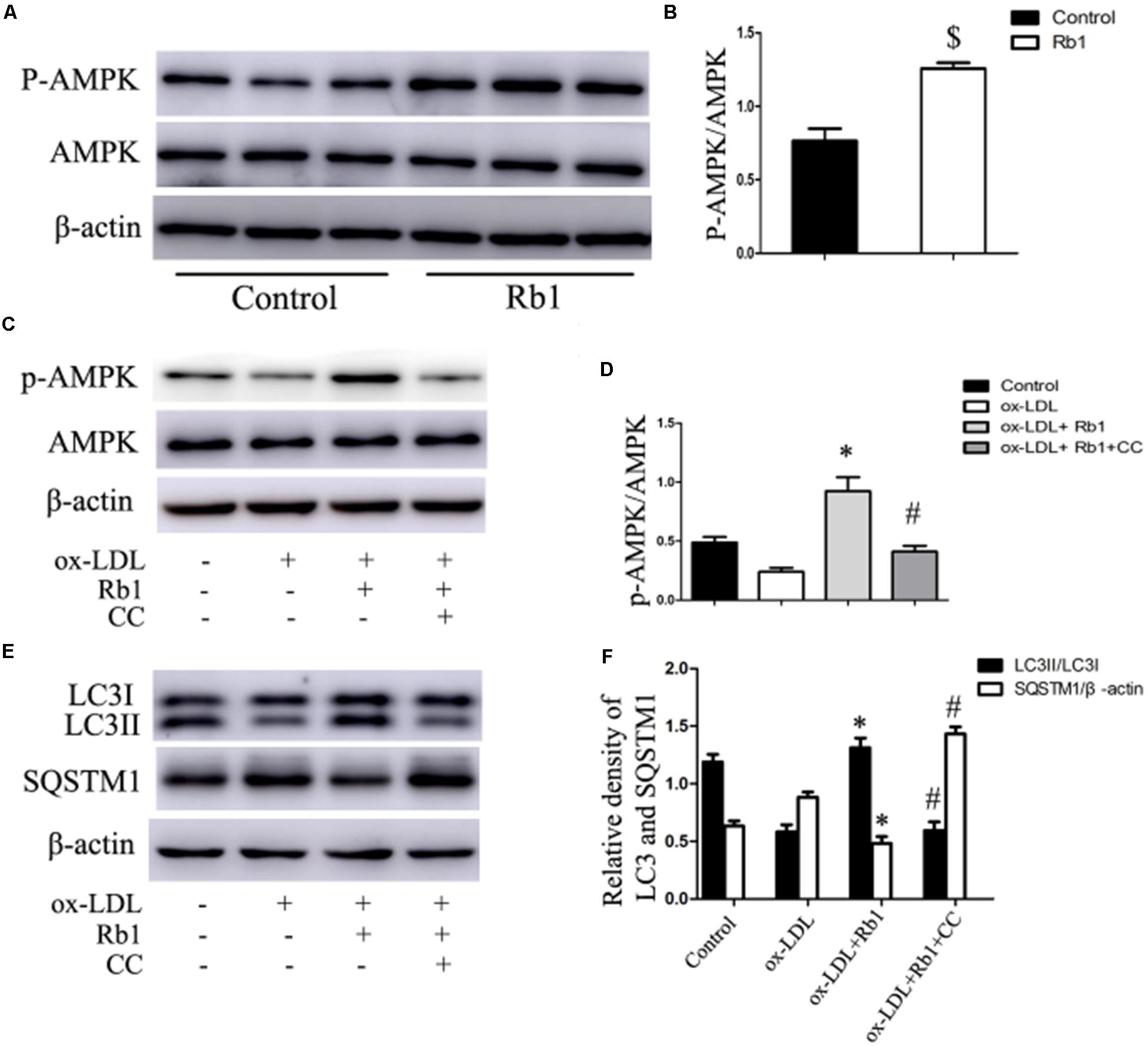
FIGURE 6. Rb1 induced autophagy via promotion of AMPK phosphorylation. (A) Representative western blots and (B) quantification of p-AMPK/AMPK in atherosclerotic plaques of apoE-/- mice treated with Rb1 or saline (control). (C) Representative western blots and (D) quantification of p-AMPK/AMPK in macrophage foam cells treated with Rb1 in the presence or absence of 20 μM compound C (CC). (E) Representative western blots and (F) quantification of LC3-II and SQSTM1/p62 in macrophage foam cells treated with Rb1 in the presence or absence of 20 μM CC. ∗p < 0.05, compared to the ox-LDL group; #p < 0.05, compared to the ox-LDL + Rb1 group; $p < 0.05, compared to the control group, n = 6.
Ginseng is widely used as a general tonic, adaptogen and herbal medicine for many diseases (Dong et al., 2013). Pharmacological effects of ginseng and its constituents have been demonstrated in the CNS and in the endocrine, cardiovascular, and immune systems (Attele et al., 1999). Most pharmacological properties of ginseng are attributed to ginsenosides (Attele et al., 1999). More than 20 ginsenosides have been isolated so far (Gillis, 1997). Ginsenosides exhibit considerable structural variation. Based on chemical structure, there are two major groups: panaxadiols (for example, Rb1, Rb2, Rg3, Rh2 and Rh3) and panaxatriols (for example, Re, Rg1, Rg2 and Rh1) (Attele et al., 1999). Ginsenosides belong to a family of steroids named steroidal saponins, which share structural features with steroid hormones (Kim et al., 1998). Ginsenosides exhibit diverse pharmacological effects. Rb1, Rg1 and Re have been shown to facilitate learning and memory (Takemoto et al., 1984; Salim et al., 1997); Rh2 and Rg3 have been reported to have antineoplastic and immunomodulatory effects (Tode et al., 1993; Mochizuki et al., 1995). Among these ginsenosides, Rb1 is the most abundant one. Rb1 has the structure of a tetracyclic triterpenoid with the molecular formula C54H92O23 and molecular weight of 1,109.26 g/mol (Cho et al., 2004; Xiong et al., 2010). Rb1 shows a wide range of biological activities. For example, Rb1 has a great potential for exerting anti-obesity effects by stimulating c-Fos expression in brain areas involved in energy homeostasis (Xiong et al., 2010); Rb1 also promotes glucose homeostasis by increasing insulin sensitivity (Shen et al., 2015). Recent studies have shown that Rb1 is cardioprotective through endothelial protection (Lan et al., 2011), anti-remodeling of ventricular (Zheng et al., 2017) and protection against IH-induced myocardial injury (Cui et al., 2017). However, the function of Rb1 in AS has not been elucidated.
Atherosclerosis is a chronic lipid-driven disease in the arterial wall. Macrophage foam cells play a crucial role in the development of atherogenesis. Excessive lipid deposition in the arterial wall leads to unbalanced cell apoptosis and inflammatory responses, which contributes to robust increases in atherosclerotic plaque complexity. Thus, removing excess cholesterol from macrophage foam cells in atherosclerotic plaques is necessary. In this study, we observed a reduction in lipid accumulation both in macrophage foam cells and in atherosclerotic plaques upon Rb1 treatment. In addition, Rb1 enhanced atherosclerotic plaque stability by modifying the plaque components to a more stable type. These results showed that Rb1 had potential therapeutic value in AS by decreasing lipid accumulation in macrophage foam cells.
Growing evidence has shown that macrophage autophagy plays a vital role in lipid metabolism and in AS (Singh et al., 2009; Dong and Czaja, 2011; Liao et al., 2012). Autophagy contributes to the hydrolysis of LDs and promotes cholesterol efflux in macrophage foam cells (Dong and Czaja, 2011; Ouimet et al., 2011). Autophagy is active in early atherosclerotic lesions and may alleviate foam cell formation. Autophagic clearance of lipids should be more efficient with increased lipid deposition in advanced atherosclerotic plaques. However, autophagy is increasingly dysfunctional in the progression of AS (Razani et al., 2012; Li et al., 2016). Impaired autophagy efflux results in ineffective clearance of accumulated cholesterol from foam cells. Therefore, pharmacological induction of autophagy in advanced atherosclerotic plaques might be a promising therapeutic approach to promote lipid metabolism and inhibit necrotic core formation.
To investigate whether autophagy was involved in the modulation of lipid content caused by Rb1, western blot and TEM were applied. Our results showed that Rb1 triggered autophagy by increasing the levels of LC3II and the degradation of SQSTM1/p62 and contributed to accumulated number of autophagosomes in macrophage foam cells in vivo and atherosclerotic plaque. Nevertheless, the effect of Rb1 on lipid accumulation was attenuated when autophagy was blocked by using Atg5 siRNA in vitro. Thus, our research showed for the first time that Rb1 reduced lipid accumulation in macrophage foam cells and enhanced atherosclerotic plaque stability by induction of macrophage autophagy.
Then, we further explored the mechanism by which Rb1 restores autophagy flux in macrophage foam cells. AMPK is an evolutionarily conserved sensor of cellular energy receptor and plays a critical role in glucose and lipid metabolism, as well as the induction of autophagy (Kahn et al., 2005; Zhang et al., 2016). Our previous studies have shown that a lower AMPK activation level was associated with coronary atherosclerotic plaque vulnerability by attenuating autophagy in peripheral blood monocytes (Cheng et al., 2015). In addition, Rb1 has also been shown to be able to promote AMPK phosphorylation in rats (Shen et al., 2013, 2015). Therefore, AMPK was studied in our research to explore the potential molecular mechanisms by which Rb1 induced autophagy. The results showed that Rb1 treatment promoted AMPK phosphorylation both in vitro and in vivo in atherosclerotic plaques; the increase in LC3II and SQSTM1/p62 degradation induced by Rb1 was abolished by the AMPK inhibitor compound C in vitro. These data suggested that Rb1 induced autophagy via promotion of AMPK phosphorylation in macrophage foam cells (Figure 7).
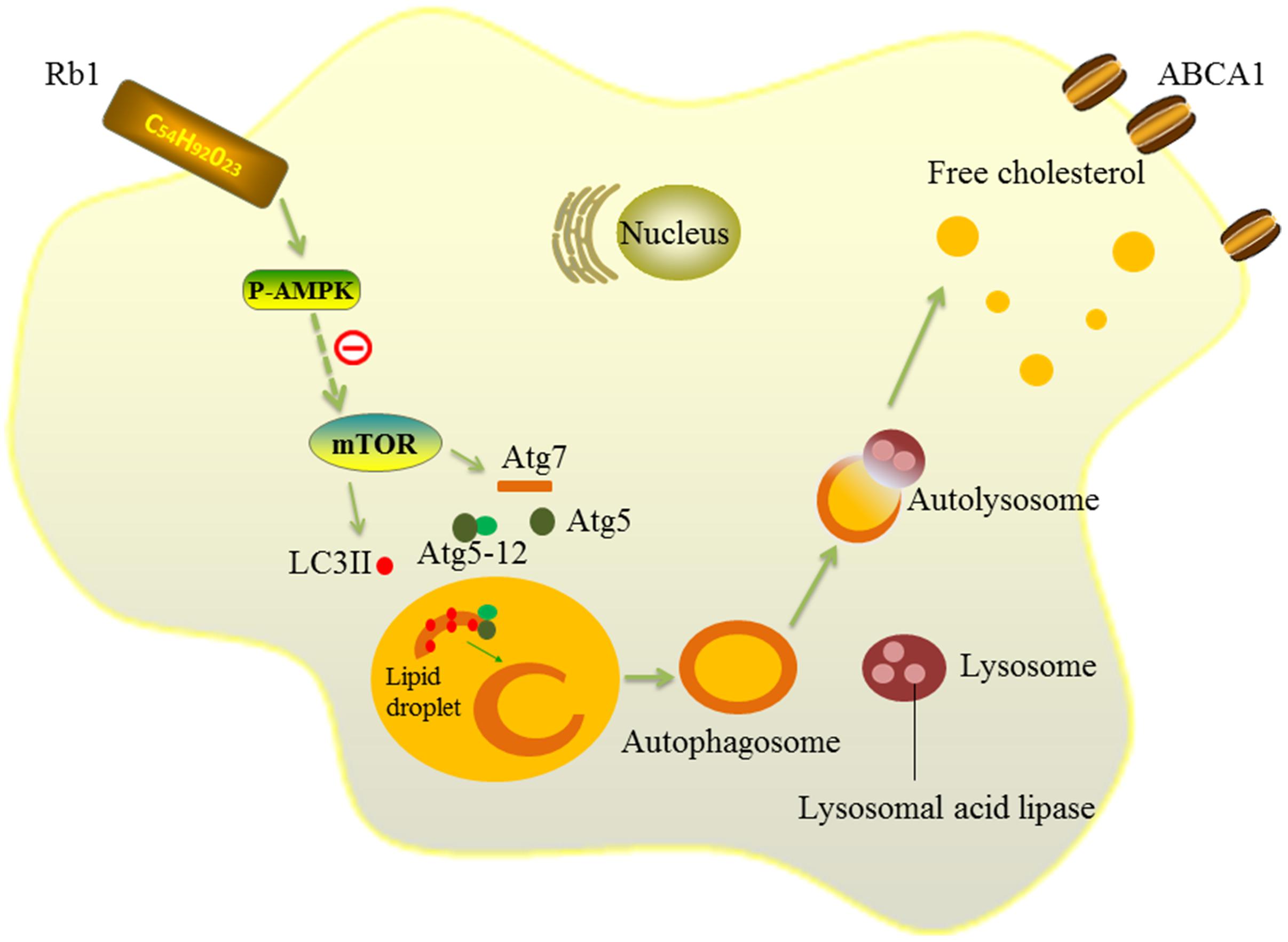
FIGURE 7. Schematic diagram of the effects of Rb1 on autophagy. Rb1 increased AMPK phosphorylation and thus induced autophagy activation in macrophage foam cells. Lipid droplets (LDs) are separated by double-membrane vesicles analogous to autophagosomes and delivered to lysosomes to form an autolysosome. LDs are hydrolyzed by lysosomal acid lipase (LAL) into free cholesterols in the autolysosome. Solid lines ( ) depict the mechanism by which Rb1 improves autophagy and lipid metabolism in macrophage foam cells in our present study. Dotted lines( ) represent a possible signaling molecule (mTOR) that may serve as an upstream regulator of autophagy induced by Rb1, but was not investigated in our study.
All of our findings suggest that ginsenoside Rb1 enhances atherosclerotic plaque stability by improving autophagy and lipid metabolism in macrophage foam cells. Our study supports the possibility that Rb1 might be an optional anti-atherogenic medicine by inducing macrophage autophagy in advanced atherosclerotic plaques.
HL, WC, and LQ conceived and designed experiments; and XZ, ML, XL, and MD conducted the experiments; JC and XZ analyzed the data; CZ and YS made the figures for this manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer CY and handling Editor declared their shared affiliation.
We express our sincere gratitude to everyone who provided technical assistance and participated in this study in the Key Laboratory of Cardiovascular Remodeling and Function Research, Chinese Ministry of Education and Chinese Ministry. This work was supported by the National Natural Science Foundation of China (2016YFC1300403, 81270403, 81400330, and 81300217), the Author of National Excellent Doctoral Dissertation of PR China (201181), China Postdoctoral Science Foundation (2012M521353 and 2014M551914), and the Natural Science Foundation of Shandong Province (2014ZRE27067 and ZR2014CM010). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. We express our sincere gratitude and thanks to Ricky Ngok-shun Wong (Hong Kong Baptist University) for his guidance in shaping this study.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2017.00727/full#supplementary-material
FIGURE S1 | Autophagy was impaired in macrophages by using Atg5 small interfering RNA (siRNA). (A) Representative western blots and (B) quantification of Atg5/β-actin and LC3 II/β-actin in macrophages transfected with con siRNA or Agt5 siRNA. ∗p < 0.05, compared to the con siRNA group, n = 3.
FIGURE S2 | Rb1 treatment promoted the expression of ABCA1 in macrophage foam cell. (A) Representative western blots and (B) quantification of ABCA1/β-actin in macrophage foam cells treated in the presence or absence of 20 μM Rb1. ∗p < 0.05, compared to the control group; #p < 0.05, compared to the ox-LDL group, n = 3.
Attele, A. S., Wu, J. A., and Yuan, C. S. (1999). Ginseng pharmacology: multiple constituents and multiple actions. Biochem. Pharmacol. 58, 1685–1693. doi: 10.1016/S0006-2952(99)00212-9
Cheng, J., Qiao, L., Xu, X., Zhai, C., Zhao, K., Ji, X., et al. (2015). Lower AMP-activated protein kinase level is associated with the vulnerability of coronary atherosclerotic plaques by attenuating the expression of monocyte autophagy. Coron. Artery Dis. 26, 322–327. doi: 10.1097/MCA.0000000000000243
Cho, J., Park, W., Lee, S., Ahn, W., and Lee, Y. (2004). Ginsenoside-Rb1 from Panax ginseng C.A. Meyer activates estrogen receptor-α and -β, independent of ligand binding. J. Clin. Endocrinol. Metab. 89, 3510–3515. doi: 10.1210/jc.2003-031823
Cui, Y. C., Pan, C. S., Yan, L., Li, L., Hu, B. H., Chang, X., et al. (2017). Ginsenoside Rb1 protects against ischemia/reperfusion-induced myocardial injury via energy metabolism regulation mediated by RhoA signaling pathway. Sci. Rep. 7:44579. doi: 10.1038/srep44579
Dong, H., and Czaja, M. J. (2011). Regulation of lipid droplets by autophagy. Trends Endocrinol. Metab. 22, 234–240. doi: 10.1016/j.tem.2011.02.003
Dong, M., Yang, X., Lim, S., Cao, Z., Honek, J., Lu, H., et al. (2013). Cold exposure promotes atherosclerotic plaque growth and instability via UCP1-dependent lipolysis. Cell Metab. 18, 118–129. doi: 10.1016/j.cmet.2013.06.003
Gillis, C. N. (1997). Panax ginseng pharmacology: a nitric oxide link? Biochem. Pharmacol. 54, 1–8. doi: 10.1016/S0006-2952(97)00193-7
Gu, H. F., Li, H. Z., Tang, Y. L., Tang, X. Q., Zheng, X. L., and Liao, D. F. (2016). Nicotinate-curcumin impedes foam cell formation from THP-1 cells through restoring autophagy flux. PLOS ONE 11:e0154820. doi: 10.1371/journal.pone.0154820
Ho, P. C., Lin, Y. W., Tsui, Y. C., Gupta, P., and Wei, L. N. (2009). A negative regulatory pathway of GLUT4 trafficking in adipocyte: new function of RIP140 in the cytoplasm via AS160. Cell Metab. 10, 516–523. doi: 10.1016/j.cmet.2009.09.012
Ho, P. C., Tsui, Y. C., Feng, X., Greaves, D. R., and Wei, L. N. (2012). NF-κB-mediated degradation of the coactivator RIP140 regulates inflammatory responses and contributes to endotoxin tolerance. Nat. Immunol. 13, 379–386. doi: 10.1038/ni.2238
Kahn, B. B., Alquier, T., Carling, D., and Hardie, D. G. (2005). AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 1, 15–25. doi: 10.1016/j.cmet.2004.12.003
Klionsky, D. J., Abdalla, F. C., Abeliovich, H., Abraham, R. T., Acevedo-Arozena, A., Adeli, K., et al. (2012). Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8, 445–544. doi: 10.4161/auto.19496
Kim, Y. S., Kim, D. S., and Kim, S. I. (1998). Ginsenoside Rh2 and Rh3 induce differentiation of HL-60 cells into granulocytes: modulation of protein kinase C isoforms during differentiation by ginsenoside Rh2. Int. J. Biochem. Cell Biol. 30, 327–338. doi: 10.1016/S1357-2725(97)00141-6
Koga, H., Kaushik, S., and Cuervo, A. M. (2010). Altered lipid content inhibits autophagic vesicular fusion. FASEB J. 24, 3052–3065. doi: 10.1096/fj.09-144519
Lan, T. H., Xu, Z. W., Wang, Z., Wu, Y. L., Wu, W. K., and Tan, H. M. (2011). Ginsenoside Rb1 prevents homocysteine-induced endothelial dysfunction via PI3K/Akt activation and PKC inhibition. Biochem. Pharmacol. 82, 148–155. doi: 10.1016/j.bcp.2011.04.001
Levine, B., and Kroemer, G. (2008). Autophagy in the pathogenesis of disease. Cell 132, 27–42. doi: 10.1016/j.cell.2007.12.018
Li, W., Sultana, N., Siraj, N., Ward, L. J., Pawlik, M., Levy, E., et al. (2016). Autophagy dysfunction and regulatory cystatin C in macrophage death of atherosclerosis. J. Cell. Mol. Med. 20, 1664–1672. doi: 10.1111/jcmm.12859
Liao, X., Sluimer, J. C., Wang, Y., Subramanian, M., Brown, K., Pattison, J. S., et al. (2012). Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metab. 15, 545–553. doi: 10.1016/j.cmet.2012.01.022
Mochizuki, M., Yoo, Y. C., Matsuzawa, K., Sato, K., Saiki, I., Tono-oka, S., et al. (1995). Inhibitory effect of tumor metastasis in mice by saponins, ginsenoside-Rb2, 20(R)- and 20(S)-ginsenoside-Rg3, of red ginseng. Biol. Pharm. Bull. 18, 1197–1202. doi: 10.1248/bpb.18.1197
Moore, K. J., Sheedy, F. J., and Fisher, E. A. (2013). Macrophages in atherosclerosis: a dynamic balance. Nat. Rev. Immunol. 13, 709–721. doi: 10.1038/nri3520
Ouimet, M., Franklin, V., Mak, E., Liao, X., Tabas, I., and Marcel, Y. L. (2011). Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metab. 13, 655–667. doi: 10.1016/j.cmet.2011.03.023
Park, K. H., Shin, H. J., Song, Y. B., Hyun, H. C., Cho, H. J., Ham, H. S., et al. (2002). Possible role of ginsenoside Rb1 on regulation of rat liver triglycerides. Biol. Pharm. Bull. 25, 457–460.
Razani, B., Feng, C., Coleman, T., Emanuel, R., Wen, H., Hwang, S., et al. (2012). Autophagy links inflammasomes to atherosclerotic progression. Cell Metab. 15, 534–544. doi: 10.1016/j.cmet.2012.02.011
Salim, K. N., McEwen, B. S., and Chao, H. M. (1997). Ginsenoside Rb1 regulates ChAT, NGF and trkA mRNA expression in the rat brain. Mol. Brain Res. 47, 177–182. doi: 10.1016/S0169-328X(97)00042-9
Seo, G., Kim, S. K., Byun, Y. J., Oh, E., Jeong, S. W., Chae, G. T., et al. (2011). Hydrogen peroxide induces Beclin 1-independent autophagic cell death by suppressing the mTOR pathway via promoting the ubiquitination and degradation of Rheb in GSH-depleted RAW 264.7 cells. Free Radic. Res. 45, 389–399. doi: 10.3109/10715762.2010.535530
Shen, L., Haas, M., Wang, D. Q., May, A., Lo, C. C., Obici, S., et al. (2015). Ginsenoside Rb1 increases insulin sensitivity by activating AMP-activated protein kinase in male rats. Physiol. Rep. 3:e12543. doi: 10.14814/phy2.12543
Shen, L., Xiong, Y., Wang, D. Q., Howles, P., Basford, J. E., Wang, J., et al. (2013). Ginsenoside Rb1 reduces fatty liver by activating AMP-activated protein kinase in obese rats. J. Lipid Res. 54, 1430–1438. doi: 10.1194/jlr.M035907
Singh, R., Kaushik, S., Wang, Y., Xiang, Y., Novak, I., Komatsu, M., et al. (2009). Autophagy regulates lipid metabolism. Nature 458, 1131–1135. doi: 10.1038/nature07976
Takemoto, Y., Ueyama, T., Saito, H., Horio, S., Sanada, S., Shoji, J., et al. (1984). Potentiation of nerve growth factor-mediated nerve fiber production in organ cultures of chicken embryonic ganglia by ginseng saponins: structure–activity relationship. Chem. Pharm. Bull. (Tokyo) 32, 3128–3133. doi: 10.1248/cpb.32.3128
Tode, T., Kikuchi, Y., Kita, T., Hirata, J., Imaizumi, E., and Nagata, I. (1993). Inhibitory effects by oral administration of ginsenoside Rh2 on the growth of human ovarian cancer cells in nude mice. J. Cancer Res. Clin. Oncol. 120, 24–26.
Wang, X. P., Zhang, W., Liu, X. Q., Wang, W. K., Yan, F., Dong, W. Q., et al. (2014). Arginase I enhances atherosclerotic plaque stabilization by inhibiting inflammation and promoting smooth muscle cell proliferation. Eur. Heart J. 35, 911–919. doi: 10.1093/eurheartj/eht329
Washida, D., and Kitanaka, S. (2003). Determination of polyacetylenes and ginsenosides in Panax species using high performance liquid chromatography. Chem. Pharm. Bull. 51, 1314–1317.
Weber, C., and Noels, H. (2011). Atherosclerosis: current pathogenesis and therapeutic options. Nat. Med. 17, 1410–1422. doi: 10.1038/nm.2538
Xiong, Y., Shen, L., Liu, K. J., Tso, P., Xiong, Y., Wang, G., et al. (2010). Antiobesity and antihyperglycemic effects of ginsenoside Rb1 in rats. Diabetes 59, 2505–2512. doi: 10.2337/db10-0315
Yu, X., Ye, L., Zhang, H., Zhao, J., Wang, G., Guo, C., et al. (2015). Ginsenoside Rb1 ameliorates liver fat accumulation by upregulating perilipin expression in adipose tissue of db/db obese mice. J. Ginseng Res. 39, 199–205. doi: 10.1016/j.jgr.2014.11.004
Zhang, D., Wang, W., Sun, X., Xu, D., Wang, C., Zhang, Q., et al. (2016). AMPK regulates autophagy by phosphorylating BECN1 at threonine 388. Autophagy 12, 1447–1459. doi: 10.1080/15548627.2016.1185576
Keywords: ginsenoside Rb1, atherosclerosis, lipid accumulation, macrophage foam cells, autophagy
Citation: Qiao L, Zhang X, Liu M, Liu X, Dong M, Cheng J, Zhang X, Zhai C, Song Y, Lu H and Chen W (2017) Ginsenoside Rb1 Enhances Atherosclerotic Plaque Stability by Improving Autophagy and Lipid Metabolism in Macrophage Foam Cells. Front. Pharmacol. 8:727. doi: 10.3389/fphar.2017.00727
Received: 10 May 2017; Accepted: 27 September 2017;
Published: 24 October 2017.
Edited by:
Satish Kitambi, Karolinska Institute (KI), SwedenReviewed by:
Bashir M. Rezk, Southern University at New Orleans, United StatesCopyright © 2017 Qiao, Zhang, Liu, Liu, Dong, Cheng, Zhang, Zhai, Song, Lu and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huixia Lu, bHVodWl4aWFAc2R1LmVkdS5jbg== Wenqiang Chen, Y2hlbndlbnFpYW5nMzNAc2luYS5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.