 Da Zhang
Da Zhang Junbao Du
Junbao Du Chaoshu Tang2,3
Chaoshu Tang2,3 Yaqian Huang
Yaqian Huang Hongfang Jin
Hongfang Jin- 1Department of Pediatrics, Peking University First Hospital, Beijing, China
- 2Key Laboratory of Molecular Cardiology, Ministry of Education, Beijing, China
- 3Department of Physiology and Pathophysiology, Peking University Health Science Center, Beijing, China
At appropriate concentrations, hydrogen sulfide, a well-known gasotransmitter, plays important roles in both physiology and pathophysiology. Increasing evidence suggests that modifying thiol groups of specific cysteines in target proteins via sulfhydration or persulfidation is one of the important mechanisms responsible for the biological functions of hydrogen sulfide. A variety of key proteins of different cellular pathways in mammals have been reported to be sulfhydrated by hydrogen sulfide to participate and regulate the processes of cell survival/death, cell differentiation, cell proliferation/hypertrophy, cellular metabolism, mitochondrial bioenergetics/biogenesis, endoplasmic reticulum stress, vasorelaxtion, inflammation, oxidative stress, etc. Moreover, S-sulfhydration also exerts many biological functions through the cross-talk with other post-translational modifications including phosphorylation, S-nitrosylation and tyrosine nitration. This review summarizes recent studies of hydrogen sulfide-induced sulfhydration as a posttranslational modification, an important biological function of hydrogen sulfide, and sulfhydrated proteins are introduced. Additionally, we discuss the main methods of detecting sulfhydration of proteins.
Introduction
Hydrogen sulfide, a “superstar” gasotransmitter in the gaseous signal molecule family, has been found involved in various physiologic and pathophysiologic processes since the end of the last century. The role of H2S in the nervous, cardiovascular, digestive, and respiratory systems was examined, and the existence of endogenous H2S was verified. To understand how endogenous H2S regulates various cellular processes, researchers identified that H2S was involved in a post-translational modification, called S-sulfhydration, of a large number of proteins (Mustafa et al., 2009; Paul and Snyder, 2012). A variety of key proteins of different cellular pathways in mammals are sulfhydrated by H2S to regulate and affect the processes of cell survival/death, cell differentiation, cell proliferation/hypertrophy, cellular metabolism, mitochondrial bioenergetics/biogenesis, ER stress, vasorelaxtion, inflammation, oxidative stress, for example.
This review summarizes recent studies of H2S-induced sulfhydration as a post-translational modification that plays vital roles in diverse physiologic and pathophysiologic processes. Additionally, we discuss methods to detect sulfhydration of proteins.
Properties of H2S and S-Sulfhydration, and Formation Process of S-Sulfhydration
H2S has some properties different from other gasotransmitters. The most typical difference is its dissociation ability. Its pKa is 6.77; under normal conditions, such as aqueous solutions at pH 7.4, over three quarters of H2S are dissociated to HS-anion and only 20% not dissociated, even though the concentration of S2- is critically low. The H2S pool is believed to consist of H2S, HS- and S2-. Protein persulfidation, or protein S-sulfhydration is regarded as one of the important molecular mechanisms by which H2S plays various biological effects. More accurately, this difference is mainly reflected in the modification of cysteine residues from the -SH to -SSH group. The -SH and -SSH groups differ significantly in properties. As compared with corresponding thiols (-SH), hydropersulfides (-SSH) have a stronger nucleophilic ability, for greater chemical reactivity. When pH is under physiological conditions, because of the lower pKa, hydropersulfides exhibit stronger acidity and will become more active hydrogen donors then thiols (Paul and Snyder, 2012). Another significant difference is the bond dissociation energy of S-H in RSSH or RSH: the former is 70 kcal/mol and the latter is 92 kcal/mol (Benson, 1978). Therefore, perthiyl radicals (RSS •) are more stable than thiyl radicals (RS •) which can be a very efficient antioxidant stress factor.
In recent years, the interests of people on the biological function of sulfhydrated protein has been growing ceaselessly, but the number of the research which addressed on mechanism for the formation process of sulfhydrated proteins is still small. Here we showed some main formation processes of S-sulfhydrated modification which is believed to occur possibly in the following cases: (1) although protein thiols do not react with H2S directly, it can react with sulfenic acids; (2) H2S can react with S-nitrosated cysteines leading to the formation of HSNO or nitroxyl (HNO); (3) H2S can react with cysteine disulfides (-S-S) for sulfhydration formation; (4) reaction between oxidized sulfide species such as polysulfides and cysteine thiols; (5) persulfides play as carriers and engage in “trans-S-sulfhydration” reaction (Figure 1).
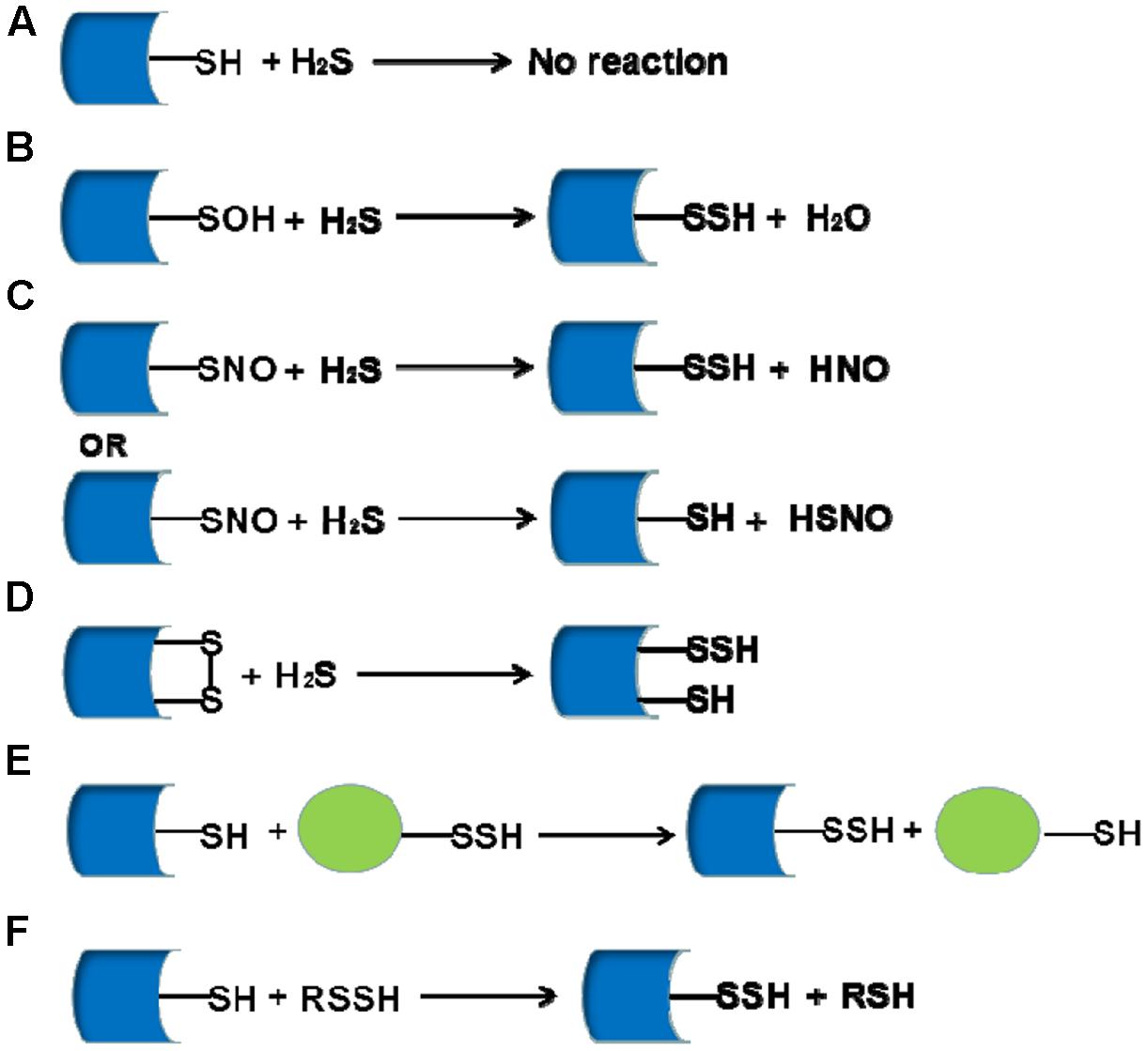
FIGURE 1. The mainly proposed formation processes for sulfhydrated proteins. (A) H2S can not react with protein thiols directly; (B) H2S can react with sulfenic acids for sulfhydration formation; (C) H2S can react with S-nitrosated for sulfhydration formation depending on the environment; (D) H2S reacts with cysteine disulfides (-S-S) for sulfhydration formation; (E,F) Persulfides can be a carrier of sulfhydration and act as the processes of displacement reaction.
Sulfhydration Mediates H2S-Induced Biological Function
S-sulfhydrated modification as a new post-translational modification is involved in many physiological and pathological processes. After S-sulfhydrated, proteins would change their original function, serving as important switchers or regulators. We summarize some literatures on S-sulfhydrated modification targets in recent years and elucidate the important biological function of sulfhydration modification in many physiological and pathophysiological processes (Figure 2 and Table 1).
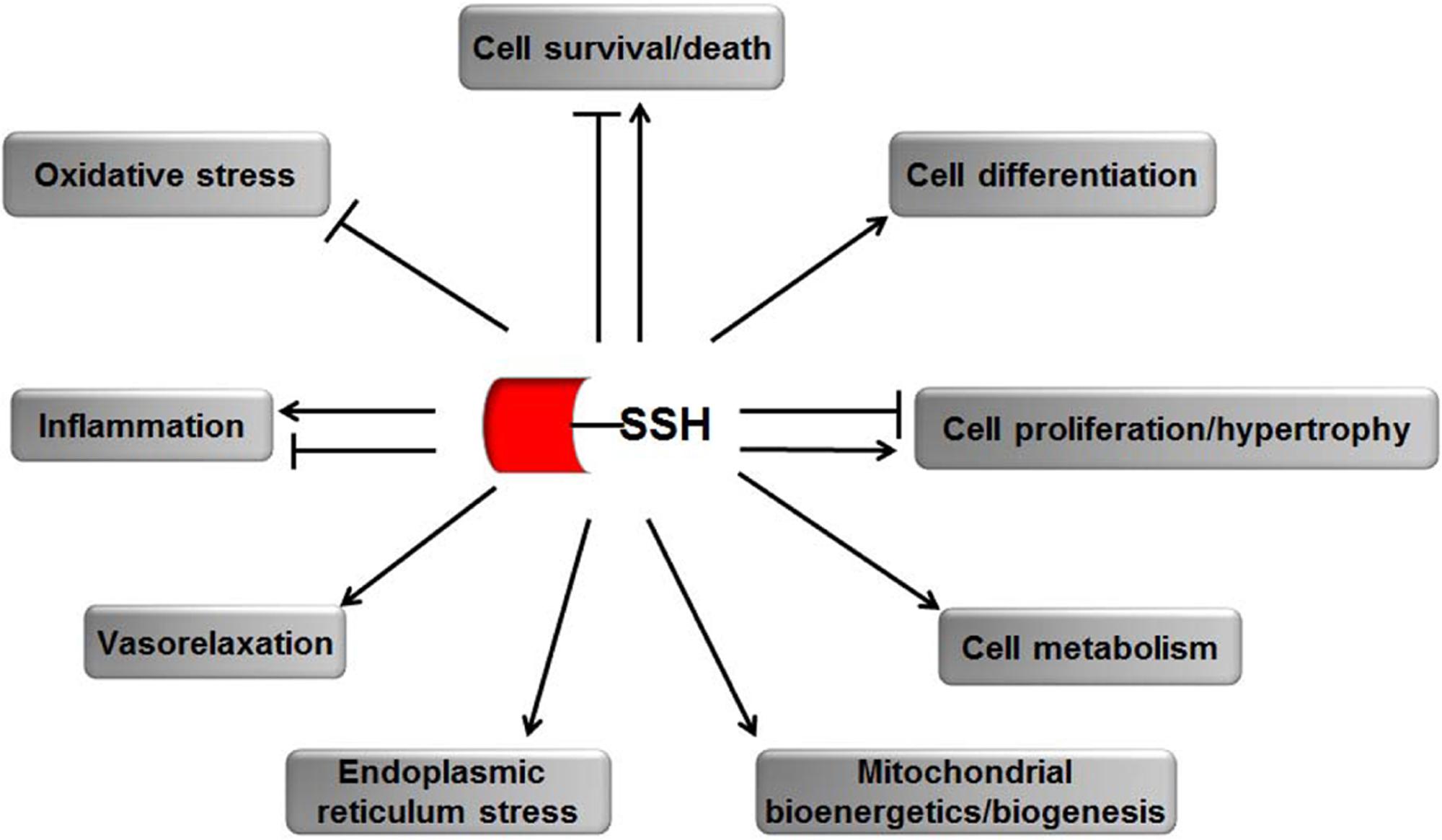
FIGURE 2. The biological functions of S-sulfhydration by H2S. → means stimulating effect, whereas ⊣ means inhibitory effect.
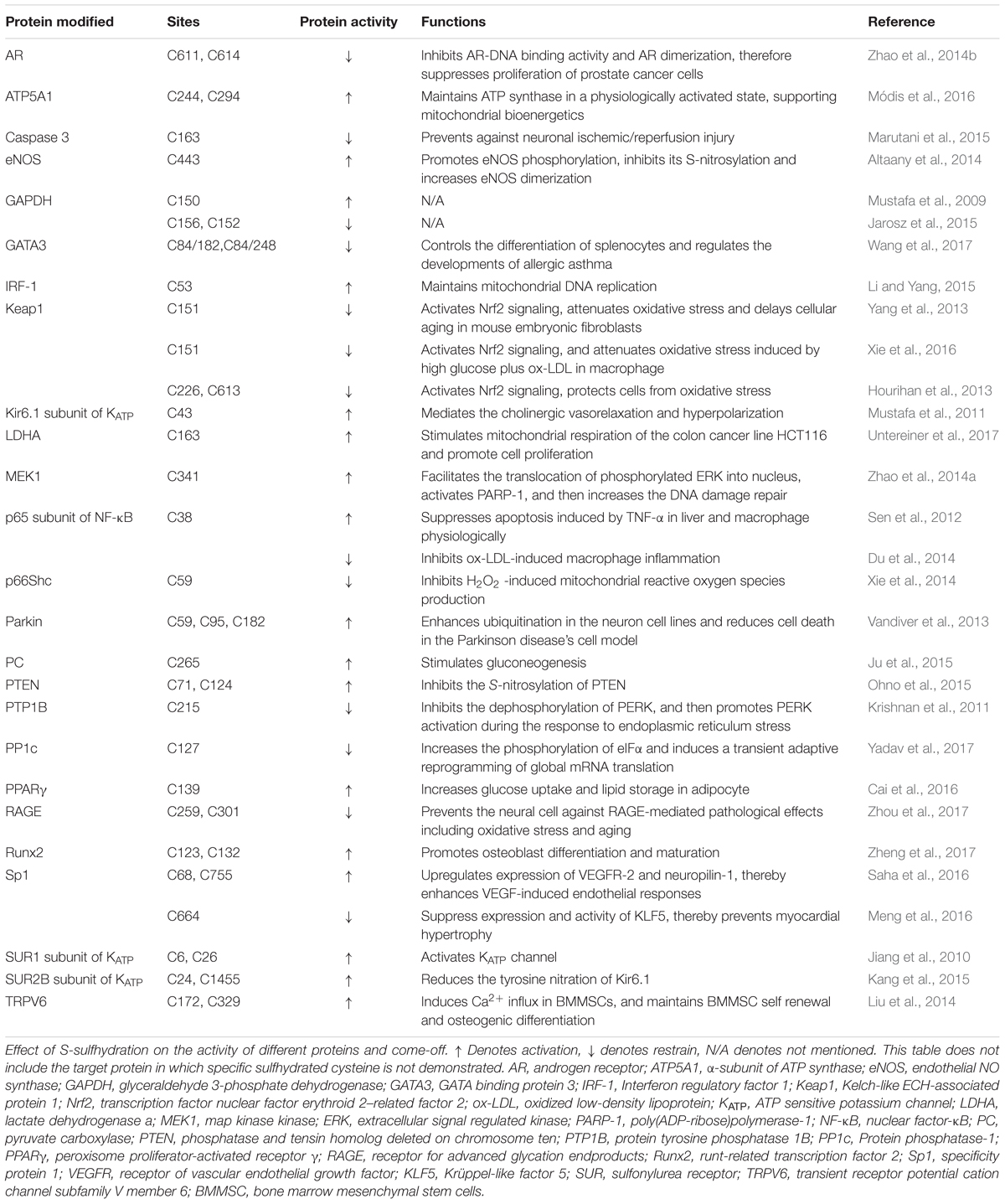
TABLE 1. Examples of sulfhydrated proteins listed in alphabetical order.
S-Sulfhydration and Cell Survival/Death
Apoptosis or programmed cell death is a physiological process that is highly regulated by cells or tissues themselves for various biological processes. GAPDH is among the first proteins found to be modified by S-sulfhydration in the history of post-translational modification by H2S (Mustafa et al., 2009). It plays a significant role in regulating both cell survival and apoptotic death (Colell et al., 2009; Nicholls et al., 2012). GAPDH is an important redox-sensitive protein, the activity of which is largely affected by its highly reactive cysteine residue (Cys). The change in cysteine thiol helps GAPDH translate to the nucleus, where it promotes the degradation of nucleoproteins, inducing cell apoptosis (Hara et al., 2005). Mustafa et al. (2009) showed that H2S could uniquely S-sulfhydrate GAPDH at Cys150 under physiological conditions, thereby enhancing GAPDH catalytic activity. The results in vivo also showed that GAPDH activity was reduced by 25–30% in CSE-/- mice compared with the wild type mice (Mustafa et al., 2009). Another group also confirmed that GAPDH could be S-sulfhydrated by endogenous H2S (Zhang et al., 2014). On the contrary, Jarosz et al. (2015) discovered that polysulfides inactivated the reduced purified GAPDH by 42% through S-sulfhydration on the Cys156. Moreover, polysulfides further decreased the activity of C156S mutant GAPDH via S-sulfhydration on the Cys 152, suggesting S-sulfhydration on the Cys 156 and Cys 152 inactivated GAPDH (Jarosz et al., 2015). Thus, modification of S-sulfydration may regulate GAPDH function, which controls cell apoptosis.
The DNA damage repair is an important response to maintain genomic stability, which is the basis for normal cell development and functions. Poly(ADP-ribose)ation mediated by PARPs is one of the important cellular responses to DNA damage (Audebert et al., 2004). Zhao et al. (2014a) found that H2S activated PARP1 and prevented DNA damage in endothelial cells and fibroblast. The protective effect of H2S involved ERK phosphorylation and nuclear translocation followed S-sulfhydrating map kinase kinase 1 (MEK1) at cysteine 341 (Zhao et al., 2014a). Mutation of cysteine 341 in MEK1 blocked the H2S-induced PARP1 activation, which further supported the major role of H2S S-sulfhydration in the DNA damage repair.
S-sulfhydrations of nuclear factor-κB (NF-κB) p65, parkin and caspase 3 were also involved in the anti-apoptotic/pro-survival effects of H2S. Sen et al. (2012) demonstrated that H2S could modify NF-κB p65 at Cys 38 thiol, enhance the binding of sulfhydrated p65 to its co-activator ribosomal protein S3, and promote the transcription of anti-apoptotic genes such as Bcl-XL and cIAP2. In cystathionine γ- lyase knock out (CSE-/-) mice, anti-apoptotic function and S-sulfhydration of NF-κB was significantly abolished, which strengthened the pro-survival role of H2S (Sen et al., 2012). Parkin is an E3 ubiquitin ligase which participated in the regulation of protein degradation and exerted an important neuroprotective effect. Vandiver et al. (2013) revealed that H2S enhanced parkin activity via sulfhydration on the Cys59, Cys95 and Cys182 sites, and then prevented cell death in the cellular models of PD. Furthermore, a marked decrease in the parkin sulfhydration in PD brain was observed, suggesting that sulfhydration of parkin is essential for neuron survival (Vandiver et al., 2013). Marutani et al. (2015) found that thiosulfate, an oxidation product of H2S, directly inhibited caspase 3 activity through sulfhydration at Cys163, decreased neuronal cell apoptosis, and therefore prevented against neuronal ischemic/reperfusion injury.
S-Sulfhydration and Cell Differentiation
Differentiation from multipotent stem cell to terminal tissue-specific cell is important for the physiological development and pathophysiological tissue repair. In the previous studies, H2S was reported to regulate the differentiation of BMMSCs, periodontal ligament stem cells, neural stem cells, osteoclast and osteoblast (Wang et al., 2013; Gambari et al., 2014; Liu et al., 2014; Su et al., 2015; Zheng et al., 2017). The mechanisms involved the control of Ca2+ transient receptor potential cation channels, PKC/ERK-mediated Wnt/β-catenin signaling, ERK, Nrf2, Akt and Runx2 pathways by H2S. Liu et al found that H2S could sulfhydrate TRPV6 at Cys172 and Cys329 in the BMMSC, induce Ca2+ influx in BMMSCs, and maintain BMMSC self renewal and osteogenic differentiation. Simultaneously, sulfhydration of TRPV3 at Cys131 and sulfhydration of TRPM4 at Cys168 might be also involved in the regulatory effect of H2S on the osteogenic differentiation (Liu et al., 2014). Zheng et al. (2017) demonstrated that H2S promoted osteoblast differentiation and maturation through sulfhyating Runx2 at Cys 123 and Cys 132, which caused transactivation of Runx2.
In addition to the differentiation of stem cells, the differentiation of naïve immune cells is also important for the body homeostasis. GATA-3 is a transcription factor which controls the differentiation of naïve immune cells, demonstrated by inducing Th0 cell differentiation toward Th2 cell subtype, and promotes type-2 immune response. Wang et al found that H2S inhibited the transcriptional activity of GATA-3 through sulfhydrating its Cys84/182 or Cys84/248 sites, promoted the splenocyte differentiation of protective type-1 cytokine-generating cells and suppressed their differentiation toward type-2 cytokine-generating cells. Therefore, age-dependent endogenous H2S generation was correlated with the development of airway inflammation in the allergic asthma (Wang et al., 2017).
S-Sulfhydration and Cell Proliferation/Hypertrophy
Excessive proliferation is the main feature of neoplastic disease. Modulatory effect of H2S on the tumor cell proliferation is different depending on tumor types. Zhao et al. (2014b) found that CSE expression was decreased in both LNCaP-B and prostate cancer tissues. H2S inhibited cell proliferation of both LNCaP and LNCaP-B. Moreover, forced expression of CSE restored the sensitivity of LNCaP-B cells to androgen antagonists. Mechanistically, they demonstrated that AR mediated the abovementioned effect of H2S evidenced by the facts that H2S sulfhydrated AR at Cys 611 and 614, destroyed the functional zinc finger structure in the AR, then inhibited the transcriptional activity of AR, and accordingly suppressed the proliferation of prostate cancer (Zhao et al., 2014b). On the contrary, Untereiner et al. (2017) revealed that endogenous H2S production and expression of its generating enzyme CBS in colon cancer cells were upregulated. H2S promoted the proliferation of colon cancer cell HCT116 via sulfhydrating LDHA at Cys163 to enhance its activity.
Inadequate proliferation of endothelial cell is one of the vital characterizations of endothelial dysfunction, which resulted in vascular injury diseases. Saha et al. (2016) found that cystathionine β-synthase (CBS)-derived H2S maintained the VEGF-dependent cellular response including VEGF-dependent proliferation resulting from increased VEGFR-2 and neuropilin-1 expression in endothelial cell, mediated by the S-sulfhydration of the transcription factor Sp1 at residues Cys68 and Cys755 and therefore enhanced the transcriptional activity of Sp1 (Saha et al., 2016). This study confirmed the deficiency of CBS/H2S-mediated protein S-sulfhydration in the development of vascular dysfunction.
Myocardial hypertrophy is a major adaptive response of cardiomyocyte when it meets various stimulators. The KLF5 was an important signaling contributed to the development of cardiac hypertrophy induced by angiotensin II (Shindo et al., 2002). Meng et al. (2016) found that H2S donor GYY4137 decreased KLF5 promoter activity, reduced KLF5 mRNA expression, inhibited transcriptional activity of KLF5, and therefore prevented cardiomyocyte hypertrophy in vitro and in vivo. The above effects of H2S were mediated by its sulfhydration of Sp1 at Cys664 to block the binding of Sp1 to the KLF5 promoter (Meng et al., 2016).
S-Sulfhydration and Cellular Metabolism
More and more studies confirmed that H2S acted as an important regulator of lipid and glucose metabolism. It was reported that H2S modulated the adipogenesis, lipolysis, apolipoprotein biosynthesis, glucose utilization, glucogenesis, and insulin resistance, etc. The impairment of endogenous H2S generation and function was the important pathogenesis of dyslipidemia and/or hyperglycemia-related diseases. Cai et al found that H2S promoted the triglyceride accumulation in adipocyte differentiation, increased the adipocyte number in mice fed with a high-fat diet for 4 weeks and alleviated insulin resistance of adipose tissues but did not increase the obesity of mice fed with high-fat diet for 13 weeks simultaneously. The mechanism by which H2S changed glucose into triglyceride storage in adipocytes was associated with the facts that H2S induced S-sulfhydration of PPARγ at Cys 139, increased its nuclear translocation and DNA binding activity, and promoted adipogenesis gene expression (Cai et al., 2016). Ju et al. (2015) explored the role of S-sulfhydration modified by CSE-derived H2S in the regulation of glucogenesis. The data showed that H2S donor or overexpression of CSE induced PC sulfhydration, enhanced PC activity, therefore promoted glucose production in liver cells. Furthermore, site-directed mutation at Cys 265 blocked H2S-induced PC sulfhydration and activity (Ju et al., 2015). Additionally, sulfhydration of peroxisome proliferator-activated receptor-γ coactivator-1α, fructose-1,6-bisphosphatase and glucose-6-phosphatase was also involved in the regulation of hepatic glucose production (Untereiner et al., 2016b).
S-Sulfhydration and Mitochondrial Bioenergetics/Biogenesis
The steady state of mitochondria is very important during a cell life and is often metobolism-related, including ATP synthesis and processes that regulate cell growth and death. In the last few years, increasing evidence showed that H2S could stimulate mitochondrial bioenergetics and act as a mitochondrial protectant (Jornayvaz and Shulman, 2010; Bartosz et al., 2014). PPARγ coactivator-related protein (PPRC) has positive effect in maintaining the stability of cell energy metabolism and normal cell viability. PPRC could be S-sulfhydrated by H2S, and the level was lower in untreated CSE–knockout hepatocytes, which regulated cell energy homeostasis under physiological conditions as well as mitochondrial bioenergetics (Untereiner et al., 2016a). H2S can also induce a S-sulfydration of α subunit of ATP synthase (ATP5A1) at Cys244 and Cys294, which maintains ATP synthase activation under physiological condition, thereby supporting mitochondrial bioenergetics (Módis et al., 2016). Li and colleagues confirmed the role of H2S in maintaining mitochondrial DNA replication and mitochondrial marker gene expression. They revealed that H2S sulfhydrated IRF-1 at Cys 53, enhanced its binding with the Dnmt3a promoter, reduced Dnmt3a expression, and induced mitochondrial transcription factor A promoter demethylation and therefore promoted mitochondrial DNA replication (Li and Yang, 2015).
S-Sulfhydration and Endoplasmic Reticulum Stress (ERS)
Endoplasmic reticulum is composed of a membrane in eukaryotic cells and an important organelle for protein synthesis, folding and secretion. External or internal environment changes will lead to ERS. The PTP family is widely recognized as a group of fundamental enzymes that control various biological processes, such as cell–cell communication, cell growth, division and differentiation (Sato et al., 1998). PTP-1B is a vital member of the PTPs protein family; it locates in the cytoplasmic face of ER and plays a key role in ER signaling (Bellomo et al., 2016). PTP-1B loses its enzymatic activity when H2S S-sulfhydrates its active-site Cys215 residue both in vivo and in vitro, thereby promoting the activity of protein kinase RNA-like ER kinase and restoration of ER homeostasis during the response to ERS (Krishnan et al., 2011). Phosphorylation of eIF2α, resulting in inhibition of global protein synthesis, is one of the key biochemical steps for ERS. Yadav et al found that H2S could inhibit PP1c via sulfhydration at Cys127, block the dephosphylation of eIF2α and therefore regulate the ERS (Yadav et al., 2017). Some new pathways by which H2S controls ERS have been recently disclosed, including Akt-heat shock protein 90 pathway (Xie et al., 2012), brain-derived neurotrophic factor-TrkB pathway (Wei et al., 2014), silent mating type information regulator 2 homolog 1 (Li et al., 2014), and Src pathway (Ying et al., 2016), etc. However, most of these studies focused on turnon/off of the protein but not the S-sulfhydrated protein, nor the specific cysteine affected by H2S stimulation. Therefore, further studies are needed to elaborate the mechanism by which H2S inhibits ERS.
S-Sulfhydration and Vasorelaxtion
As one of the important biological functions induced by H2S, vasorelaxation of H2S and its mechanisms have been extensively studied (Hosoki et al., 1997). A series of target proteins including ion channels and second messengers were found to be involved in the control of vessel tone by H2S. Since S-sulfhydration was demonstrated, the molecular mechanisms responsible for H2S-induced vasodilation were understood significantly. KATP are composed of pore-forming subunits and regulatory subunits, including Kir6.x (Kir6.1 or Kir6.2), and SURx (SUR1, SUR2A or SUR2B), which mediated the H2S-induced vasorelaxation in aorta and mesenteric artery (Zhao et al., 2001; Cheng et al., 2004). Jiang et al. (2010) found that Cys6 and Cys26 in the extracellular loop of rat vascular SUR1(rvSUR1) were target of H2S-induced S-sulfhydration. H2S opened the KATP channel to exert a vasorelaxation via S-sulfhydration of KATP channel (Jiang et al., 2010), while a research by Mustafa and colleagues revealed that H2S induced hyperpolarization in endothelial cells mediated by the opening of Kir 6.1 subunit of KATP channel via its sulfhydration at Cys43 (Mustafa et al., 2011). Additionally, S-sulfhydration of endothelial intermediate conductance potassium channel, small conductance potassium channel and TRPV4 might be in part due to vascular relaxation induced by H2S (Mustafa et al., 2011; Naik et al., 2016). Sun et al found that H2S could increase intracellular cGMP level via sulfhydrate phosphodiesterase 5A to inhibit the cGMP degradation (Sun et al., 2017). Moreover, Yu et al. (2017) demonstrated that S-sulfhydration of TRPV1 by CBS-derived H2S in carotid sinus facilitated carotid sinus baroreceptor sensitivity to participated the control of blood pressure.
S-Sulfhydration and Inflammation
The relationship between H2S and inflammation is complex. The anti-inflammatory effect of H2S was reported in carrageenan-induced paw edema (Zanardo et al., 2006), colitis (Fiorucci et al., 2007; Wallace et al., 2009), synovitis (Ekundi-Valentim et al., 2010), monoarthritis (Andruski et al., 2008), atherosclerosis (Wang et al., 2009), ischemia-reperfusion injury (Zuidema et al., 2010), cigarette smoke-induced pulmonary injury (Chen et al., 2011; Han et al., 2011) and diabetic wound healing (Zhao et al., 2017), etc. NF-κB signaling is widely known as an important pathway in regulating inflammatory response. Du et al. (2014) found that H2S inhibited macrophage inflammation induced by oxidized low-density lipoprotein via the sulfhydration of NF-κB p65 at Cys38, thereby restraining NF-κB p65 phosphorylation, nuclear translocation, DNA binding activity and the recruitment to monocyte chemotactic protein-1 promoter. In an experimental model of colitis, endogenous H2S synthesis was upregulated and played a protective role due to the activation of KATP via the S-sulfhydration of its subunit SUR2B (Wallace et al., 2009; Gade et al., 2013). In addition to the abovementioned target proteins, there are many other proteins or pathways involved in the anti-inflammatory effect of H2S. However, whether H2S sulfhydrates those proteins to inhibit the inflammatory response remains unclear.
On the other hand, Bhatia et al. (2005) demonstrated that the treatment with DL-propargylglycine, a CSE inhibitor, significantly reduced the severity of pancreatitis and lung injury induced by caerulein. Similarly, in caecal-ligation and puncture-induced sepsis mice model, CSE gene deletion alleviated the liver and lung injury and reduced inflammation along with the activation of ERK1/2 and NF-κB pathway (Gaddam et al., 2016). Those results suggested that H2S played a role as pro-inflammatory cytokines. Whether H2S is an anti-inflammatory or pro-inflammatory agent is controversial (Whiteman and Winyard, 2011). Therefore, more in-depth studies are needed for broader conclusive answers to elaborate the relationship between H2S and inflammation (Wallace et al., 2012).
S-Sulfhydration and Oxidative Stress
Numerous experimental results show that the oxidative stress sensor protein Keap1 and Nrf2 are closely related to the oxidative stress injury and the antioxidant response (Uesugi et al., 2017; Wasik et al., 2017). Previous studies suggested that H2S played an important role in protecting against oxidative stress by enhancing Nrf2 nuclear translocation and initiating antioxidant response in ischemia-reperfusion injury (Calvert et al., 2009; Guo et al., 2014; Shimada et al., 2015), diabetes-accelerated atherosclerosis (Xie et al., 2016) and high salt-induced renal injury (Huang et al., 2016). Moreover, Nrf2 activation mediated the inhibitory effect of H2S on the oxidative stress-induced cell senescence (Yang et al., 2013). Regarding the molecular mechanism by which H2S activated Nrf2-initiating antioxidant response, Yang et al. (2013) and Xie et al. (2016) elucidated that NaHS could S-sulfhydrate Keap1 at Cys151, which promoted the dissociation of Nrf2 from Keap1, while Hourihan et al. (2013) found that H2S inactivated Keap1 through sulfhydrating Keap1 at Cys226/613 site.
P66Shc, an upstream activator of mitochondrial redox signaling, plays a pivotal role in the regulation of intercellular redox homeostasis. Phosphorylation of p66Shc at Ser36 was regarded as a key step to fire the reactive oxidative species production. Xie et al. (2014) discovered that H2S could sulfhydrate p66Shc at Cys59 to inhibit p66Shc phosphorylation, reduce its translocation to mitochondria, block the mitochondrial reactive oxidative species production, and thereby protect neuronal cells against oxidative stress-induced injury. Activation of the RAGE is the key element in the development of the chronic oxidative stress-induced cytotoxicity. Zhou et al found that the treatment of NaHS reduced H2O2 –induced RAGE dimerization, shortened the half-life of RAGE, decreased the plasma membrane abundance of RAGE and therefore prevented neuron SH-SY5Y cells from cytotoxicity. Mechanistically, cys259 and cys310, which mediated the formation of intermolecular disulfide bond in the RAGE, were verified to be the direct target sites of H2S S-sulfhydration (Zhou et al., 2017). Those abovementioned studies are important for better determining the mechanism by which H2S exerts the protective role in the oxidative stress-induced diseases.
S-Sulfhydration and Other Post-translational Modification
The relationship of S-sulfhydration and phosphorylation: Xie et al. (2014) and Du et al. (2014) found that H2S-induced S-sulfhydration could inhibit phosphorylation of p66Shc and NF-κB p65, and decreased their activity. On the contrary, Altaany et al. (2014) found that H2S enhanced eNOS activity by promoting phosphylation of eNOS, which resulted from H2S-induced S-sulfhydation of eNOS at Cys 443.
The relationship of S-sulfhydration and S-nitrosylation: For a long period, researches have established that nitric oxide (NO) can act as an important regulator in diverse cell signaling pathway via S-nitrosylation happened at cysteine residue of target protein (Jaffrey et al., 2001). Under basal conditions, 10–25% of proteins in liver total proteins were S-sulfhydrated, while 1–2% of proteins were S-nitrosylated (Jaffrey et al., 2001; Mustafa et al., 2009). They have some similar chemical properties. For example, it was proposed that the two modifications preferentially occured at low pKa Cys residues of the protein (Lu et al., 2013). Many proteins have been confirmed to be controlled by both S-nitrosylation and S-sulfhydration such as GAPDH, parkin, eNOS, PPARγ, PTP1B, PTEN, p65, SUB2B and etc (Chung et al., 2004; Hara et al., 2005; Chen et al., 2008; Mustafa et al., 2009; Vandiver et al., 2013; Altaany et al., 2014; Cao et al., 2015; Ohno et al., 2015; Cai et al., 2016). In most cases, S-sulfhydration and S-nitrosylation exert opposite effects. For instance, the glycolytic activity of GAPDH is inhibited by S-nitrosylation (Hara et al., 2005), whereas S-sulfhydration increases its activity about sevenfold (Mustafa et al., 2009). Parkin activity is decreased when S-nitrosylated (Chung et al., 2004) but increased when S-sulfhydrated (Vandiver et al., 2013). S-nitrosylation of PPARγ at Cys139 inhibits PPARγ transcription activity (Cao et al., 2015), but S-sulfhydration of PPARγ at the same residue enhances its activity (Cai et al., 2016). Similarly, S-nitrosylation of PTP1B at Cys 215 prevents it from H2O2-induced inactivation (Chen et al., 2008), but S-sulfhydration of PPARγ at the same residue inhibits its activity (Krishnan et al., 2011). Furthermore, H2S-induced S-sulfhydration could directly inhibit S-nitrosylation of eNOS to prevent eNOS from inactivation (Altaany et al., 2014). Ohno and colleagues found that S-sulfhydration of PTEN at Cys71 and Cys124 by CBS/H2S could prevent its S-nitrosylation induced by NO and restore NO-caused PTEN inactivation under physiological conditions (Ohno et al., 2015). However, Sun et al demonstrated that an increased S-nitrosylation level contributed to the additive myocardial postconditioning protection with H2S donor plus NO donor (Sun et al., 2016). Therefore, the interaction between S-nitrosylation and S-sulfhydration might be one kind of complicate communications between H2S-excited signaling and NO-induced signaling.
The relationship of S-sulfhydration and tyrosine nitration: The tyrosine nitration is a posttranslation modification by peroxynitrite and other reactive nitrogen species which happened at free tyrosine or protein tyrosine residues. Tyrosine nitration is regarded to partly mediate the cytotoxicity of reactive nitrogen species (Franco and Estévez, 2014). Kang et al. (2015) found that S-sulfhydration of SUR2B subunit of KATP channel at Cys 24 and Cys 2455 residues caused by H2S donor NaHS could prevent tyrosine nitration of Kir 6.1, another subunit of KATP channel, and KATP inactivation induced by peroxynitrite. Also, NaHS could decrease the calcium channel nitration and prevent the inhibitory effect of peroxynitrite in CaCl2-induced isolated mouse ileum contraction (Kang et al., 2015). The study demonstrates a new mechanism responsible for cytoprotective effect of H2S in reactive nitrogen species-induced injury and disease.
Methods For S-Sulfhydrated Protein Detection
Establishing a detection method of protein S-sulfhydration has remained challenging for a long period. Scientists have investigated methods of detection to distinguish the persulfide group and free thiols. We here summarize the S-sulfhydration detection methods and discuss their advantages and the possible limitations in the experimental process (Figure 3).
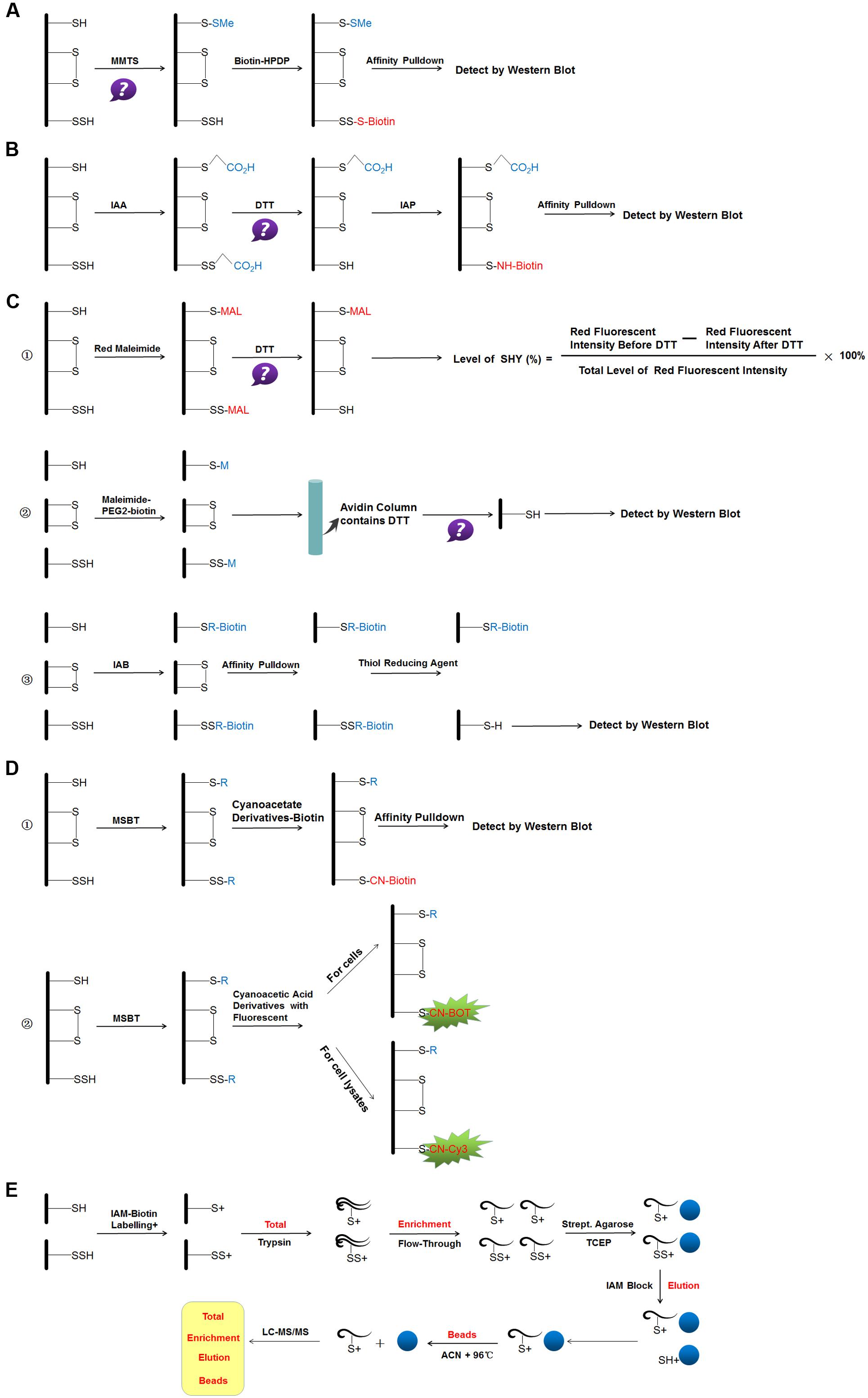
FIGURE 3. Reaction schemes of different methods for S-sulfhydraion detection. (A) biotin-switch assay; (B) cysteinyl labeling assay; (C) ➀ the maleimide assay, ➁ improved method of maleimide assay (Biotin-Thiol Assay) and ➂ protein persulfide detection protocol (ProPerDP); (D) ➀ tag-switch assay and ➁ new tag-switch assay; (E) mass spectrometry assay. MMTS: S-methyl methanethiosulfonate, Biotin-HPDP: N-[6-(biotinamido)hexyl]-3′-(2′-pyridyldithio) propinamide, IAA, Iodoacetic acid; DTT, dithiothreitol; IAP, Iodoacetamide-linked biotin; IAB, Iodoacetyl-PEG2-Biotin; MSBT, methylsulfonyl benzothiazole; CN-BOT, Cyanoacetic acid derivatives with the fluorescent BODIPY moiety; CN-Cy3, Cyanoacetic acid derivatives with the fluorescent Cy3-dye; IAM-Biotin, Iodoacetyl-PEG2-Biotin; TCEP, Tris(2-carboxyethyl)phosphine; IAM, Iodoacetamide; ACN, Acetonitrile; LC-MS/MS, Liquid chromatography and mass spectrometry.
Biotin-Switch Assay
The original method of S-sulfhydrated protein detection, named biotin-switch assay, was described by Mustafa et al. (2009). The authors simplified the original method of detecting nitrosylation for the specific cysteine thiol modification. In the first step, the thiol-blocking reagent MMTS is used to react with the -SH group. In the subsequent step, MMTS was removed by acetone, and persulfides were labeled with biotin-HPDP in dimethyl sulfoxide. Then, biotinylated proteins were pulled down by streptavidin-agarose beads, and then were washed with HENS buffer. For the last step, the biotinylated proteins were eluted by SDS-PAGE sample buffer and examined by western blot analysis. Mustafa and colleagues suggested that up to 25% proteins, especially in liver were sulfhydrated under basal conditions (Mustafa et al., 2009). S-sulfhydration is substantially more prevalent than nitrosylation and represents a previously unappreciated, major post-translational modification. Although this method is the first to identify S-sulfhydrated proteins, it has limitations. MMTS has been widely used in S-nitrosylation detection. The method for S-sulfhydrated group detection is based on the fact that the S-sulfhydrated protein does not react with MMTS as the prerequisite. However, in 2013, a study found that the -SH group could react with MMTS directly, which suggested that the biotin-switch assay was based on an incorrect chemical premise (Pan and Carroll, 2013). The authors explained this phenomenon from the perspective of two mechanisms. First, MMTS may not block and alkylate all of the free thiols; the unblocked free thiols will then react with the pyridyldisulfide biotin reagent. Second, in the presence of a large number of free sulfhydryl groups unblocked in the first step, biotin labeling may be achieved by stepwise thiol–disulfide exchange. Hence, all the different situations that may result in the false-positive results of this method can be considered as not all the free sulfhydryl groups being completely blocked during the MMTS labeling step. Although this method has been in doubt and some other new methods were found later, many research teams also evaluated this method as sensitive and selective. New proteins that can be S-sulfhydrated were found by this original method (Módis et al., 2016; Li et al., 2016).
Cysteinyl Labeling Assay
In 2011, a new method based on a completely opposite principle of chemistry was used. The authors proposed that a new kind of thiol-blocking reagent, named IAA, would react with both S-sulfhydrated protein and free thiols (Krishnan et al., 2011). In the first step, desalting columns were pre-processed by IAA-free lysis buffer and then cell lysates were slowly passed through it. In the next step, DTT was applied to IAA-cleared lysates. During this phase, the authors proposed that the alkylated Cys residue, whether persulfide or another reversibly oxidized form, would reduce back to the thiolate state. So, at a last step, IAP was used to label the particular cysteine. The objection to this method is that it cannot distinguish persulfides from intramolecular, intermolecular and S-nitrosothiols, which will also be cleaved by DTT.
The Maleimide Assay, Biotin-Thiol-Assay and Protein Persulfide Detection Protocol
Sen et al. (2012) suggested a third method based on the chemical characteristics that N-ethyl maleimide (thiol-blocking reagent) would block both free thiol and persulfide. Cy5-conjugated maleimide was used in the first step and DTT in the second. The principle of this method is that the fluorescence signal decreases when the sample contains persulfides, and the decreased ratio of the fluorescence signal is the quantitative index (Sen et al., 2012). The limitation of this method is that it cannot be used widely for proteomic analysis.
A study in 2015 improved this method. The investigators made some changes to the experiment, named BTA (Gao et al., 2015). The authors used NM-Biotin to alkylate both cysteine residues or sulfhydrated cysteine in the first step. In the subsequent step, the avidin column was purified and eluted with DTT for cleaving the disulfide bonds; however, the biotin tag was still left bound to the column. The eluate from the column is further examined by western blot analysis. Furthermore, Dóka and colleagues described another new method named ProPerDP in early 2016. In the first step, IAB was used to alkylate both thiol and persulfide functional groups, but IAB would not discriminate and react with oxidized Cys residues in the original sample. In the second step, steptavidin-coated magnetic beads were used to pull down with alkylated proteins, with oxidized Cys residues maintained in the supernatant. In the last step, reducing buffer was used to re-suspend purified beads for cleaving the original persulfides as thiols selectively from steptavidin-coated magnetic beads, for the next step of determining persulfide proteins (Dóka et al., 2016). The abovementioned two methods still have limitations. First, some proteins contain both sulfhydryl groups of the -SSH group and other non-persulfidated Cys residues. Therefore the protein that contains the -SSH group cannot cleave off from streptavidin beads in the last step, which would lead to false-negative signals. Second, the structure of disulfide bonds in intermolecular protein may result in false-positive persulfidated extra Cys residues on the polypeptide chains, which may also lead to false-positive signals. Third, to overcome the above-mentioned problem, a possible approach is to digest the alkylated protein before the pulldown step, because ensuring that the disulfide and free Cys moieties or persulfidated Cys would maintain the same form and in the same peptide is difficult.
Tag-Switch Assay
Zhang et al. (2014) proposed a new method to detect protein S-sulfhydrated that is based on the different physical and chemical properties between -SH and -SSH groups, named Tag-switch Assay (Zhang et al., 2014). In the first step, a thiol-blocking reagent reacts with both free thiol and persulfide. The authors proposed that, compared with common disulfides in proteins, the disulfide bond in persulfide adducts might have a stronger reactivity to nucleophilicity. Then, a new tag-switching reagent was used to label persulfide protein only. This method has been accepted by some research teams (Park et al., 2015; Zhou et al., 2017). The main challenge for this new method is to ensure that the disulfide linkage in the -SSH group can express a specific and suitable nucleophile and then distinguish persulfide from free thiols. Treating cell lysates with dimedone was suggested to avoid any potential cross reaction (Park et al., 2015).
Wedmann et al. (2016) proposed to improve the original tag-switch method. The biotinylated cyanoacetic acid tag was used for the assay in the original tag-switch method, which requires western blot and unique antibodies for analyzing the final results. To increase the sensitivity, the authors synthesized two new kinds of cyanoacetic acid derivatives with the fluorescent BODIPY moiety (CN-BOT) for labeling cells and Cy3-dye (CN-Cy3) for labeling cell lysates. The improved tag-switch method for persulfide detection provides a new starting point for future researches to elucidate the actual mechanisms for H2S signaling pathway.
However, soon, some other chemists described the deficiencies of the tag-switch method. First, MSBT cannot penetrate the cell membranes, which poses an obstacle for the detection of persulfide in living cells. Besides that, in the “switching” step, methyl cyanoacetate was proposed to uniquely cut off MSBT-labeled dialkyl disulfides among the protein disulfide moieties. Nevertheless, selectivity was specific: it was only assessed on glutathionylated BSA and on the N-tert-butyloxycarbonyl derivatized cystine (Nagy, 2013). Although the tag-switch method for persulfide detection is adopted by increasing numbers of research teams, as a new methodology, it also needs more trials to verify the true reliability and for comparison with previous methods.
Mass Spectrometry Assay
Most recently, Longen et al. (2016) described details of mass spectrometry-based workflow for determining S-sulhydrated proteins and their sites. The proteins collected from cells were precipitated with trichloroacetic acid. The protein thiols and persulfides were labeled with IAMBio. After digestion of the proteins (Total), single peptides containing labeled persulfides or cysteines were concentrated and separated from non-cysteine peptides (flow through) through using streptavidin agarose beads. After several washes, persulfide-containing peptides were eluted by using TCEP with no effect on thiol-containing peptides. The subsequent accessible cysteines were labeled with iodoacetamide (Elution). As a control, labeled thiol peptides remaining on the beads were eluted by using 10 mM TCEP and 80% acetonitrile (Beads). Samples of the total, flow-through, elution and bead fraction were subjected to liquid chromatography and mass spectrometry and the peptides were identified by using PEAKS 7.0 proteomics software (Longen et al., 2016).
The Controversy of Sulfurated Modification
Lately, investigators have discovered that a substance called polysulfide has cytoprotective effects also through mechanisms involving sulfurated modification of target proteins. The oxidation state of sulfur atom in thiol and H2S is -2. Atoms do not react with each other under the same oxidative state; therefore, H2S cannot sulfurate cysteine residues theoretically. The internal sulfur of H2Sn is 0. Therefore, it reacts with thiol readily (Kimura, 2015). However, cysteine residues are oxidized to two main forms —cysteine sulfenic acid or cysteine S-nitrosothiol — under oxidative conditions. H2S shows a stronger ability to sulfurate these oxidized thiols than H2Sn (Kabil and Banerjee, 2014; Kimura, 2015). Therefore, when we verify target proteins that are thiolated, we should further verify and distinguish whether the sulfurated modification is due to H2S or H2Sn. However, sulfurated modification, whether due to H2S or H2Sn, is now well established. At present, the focus of the debate may lie in the specific process of -SSH, but it does not affect consensus on the important role of -SSH in various pathological and physiological processes. The balance between H2S and H2Sn plays a key role in controlling cellular metabolism.
Conclusion and Perspectives
With increasing studies concerning the effect of H2S on phenotype in physiological and pathological processes, the mechanism by which H2S functions in different signaling pathways via S-sulfhydration has gradually been recognized. S-sulfhydration is a new post-translational modification of proteins by H2S. From the beginning of this century, different research groups globally have found that many proteins can be modified by H2S via S-sulfhydration, but there are still a considerable amount of results did not link to the sulfhydration. Furthermore, we still need to elucidate the sites of S-sulfhydration that H2S acts on. Though some new views point out that polysulfides exert a cytoprotective effect, a lot of studies on the biological function of H2S remain. It is gratifying that a large number of laboratory experiments and clinical trials have revealed H2S to have a positive effect on the regulation of physiological and pathological processes and the inhibition of the disease progression. With better understanding of more proteins to be post-modified by H2S via S-sulfhydration, the biological protective effect of H2S will be well recognized.
Author Contributions
DZ, JD, and HJ provided the overall concept and framework of the manuscript. DZ and YH researched and identified appropriate articles. DZ participated in writing the manuscript. JD, HJ and CT revised the manuscript. All authors approved the final version of the manuscript.
Funding
This work was supported by National Natural Science Foundation of China (81370154, 81622004 and 81670395), Major Basic Research Project of China (2013CB933801) and National Program for support of Top-notch Young Professionals.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
AR, androgen receptor; ATP, adenosine triphosphate; ATP5A1, α subunit of ATP synthase; BMMSCs, bone marrow mesenchymal stem cells; biotin-HPDP, N6-(biotinamido)hexyl]-3′-(2′-pyridyldithio) propionamide; BSA, bovine serum albumin; BTA, biotin-thiol-assay; CBS, cystathionine β-synthase; CSE, cystathionine γ-lyase; Cys, cysteine; DTT, dithiothreitol; eNOS, endothelial nitric oxide synthase; ER, endoplasmic reticulum; ERK, extracellular signal-regulated kinase; GAPDH, Glyceraldehyde 3-phosphate dehydrogenase; GATA-3, GATA binding protein 3; HNO, nitroxyl; H2S, hydrogen sulfide; HSNO, thionitrous acid; KATP, ATP sensitive potassium channels; Keap1, Kelch-like ECH-associated protein 1; KLF5, Krüppel-like factor 5; IAA, iodoacetic acid; IAB, biotin-tagged alkylating agent; IAMBio, iodoacetyl-PEG2-Biotin; IAP, iodoacetamide-linked biotin probe; IRF-1, interferon regulatory factor-1; LDHA, lactate dehydrogenase A; LNCaP, androgen-dependent prostate cancer cells; LNCaP-B, antiandrogen-resistant prostate cancer cells; MEK, map kinase kinase; MMTS, S-methyl methanethiosulfonate; MSBT, methylsulfonyl benzothiazole; NaHS, sodium hydrosulfide; NF-κB, nuclear factor-κB; NM-Biotin, maleimide-PEG2-biotin; NO, nitric oxide; Nrf2, nuclear factor erythroid 2–related factor 2; PC, pyruvate carboxylase; PD, Parkinson’s disease; PARP, poly(ADP-ribose)ation polymerases; PP1c, protein phosphatase-1c; PPARγ, peroxisome proliferator activated receptor γ; PPRC, proliferator-activated receptor-γ coactivator-related protein; ProPerDP, protein persulfide detection protocol; PTEN, phosphatase and tensin homolog deleted on chromosome ten; PTP, protein tyrosine phosphatase; RAGE, receptor for advanced glycation endproducts; Runx2, Runt-related transcription factor 2; Sp1, specificity protein 1; TRPV6, transient receptor potential cation channel subfamily V member 6; TCEP, tris(2-carboxyethyl)phosphine; VEGF, vascular endothelial growth factor; VEGFR, VEGF receptor.
References
Altaany, Z., Ju, Y., Yang, G., and Wang, R. (2014). The coordination of S-sulfhydration, S-nitrosylation, and phosphorylation of endothelial nitric oxide synthase by hydrogen sulfide. Sci. Signal. 7, ra87. doi: 10.1126/scisignal.2005478
Andruski, B., McCafferty, D. M., Ignacy, T., Millen, B., and McDougall, J. J. (2008). Leukocyte trafficking and pain behavioral responses to a hydrogen sulfide donor in acute monoarthritis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 3, 814–820. doi: 10.1152/ajpregu.90524.2008
Audebert, M., Salles, B., and Calsou, P. (2004). Involvement of poly(ADP-ribose) polymerase-1 and XRCC1/DNA ligase III in an alternative route for DNA double-strand breaks rejoining. J. Biol. Chem. 279, 55117–55126. doi: 10.1074/jbc.M404524200
Bartosz, S., Katalin, M., Kazunori, Y., Ciro, C., Sophie, L. T., Mark, E. W., et al. (2014). AP39 [10-oxo-10-(4-(3-thioxo-3H-1,2-dithiol-5yl)phenoxy)decyl) triphenylphosphonium bromide], a mitochondrially targeted hydrogen sulfide donor, stimulates cellular bioenergetics, exerts cytoprotective effects and protects against the loss of mitochondrial DNA integrity in oxidatively stressed endothelial cells in vitro. Nitric Oxide 41, 120–130. doi: 10.1016/j.niox.2014.04.008
Bellomo, E., Birla-Singh, K., Massarotti, A., Hogstrand, C., and Maret, W. (2016). The metal face of protein tyrosine phosphatase 1B. Coord. Chem. Rev. 32, 70–83. doi: 10.1016/j.ccr.2016.07.002
Benson, S. W. (1978). Thermochemistry and kinetics of sulfur-containing molecules and radicals. Chem. Rev. 78, 23–35. doi: 10.1021/cr60311a003
Bhatia, M., Wong, F. L., Fu, D., Lau, H. Y., Moochhala, S. M., and Moore, P. K. (2005). Role of hydrogen sulfide in acute pancreatitis and associated lung injury. FASEB J. 6, 623–625. doi: 10.1096/fj.04-3023fje
Cai, J., Shi, X., Wang, H., Fan, J., Feng, Y., Lin, X., et al. (2016). Cystathionine γ lyase-hydrogen sulfide increases peroxisome proliferator-activated receptor γ activity by sulfhydration at C139 site thereby promoting glucose uptake and lipid storage in adipocytes. Biochim. Biophys. Acta 1861, 419–429. doi: 10.1016/j.bbalip.2016.03.001
Calvert, J. W., Jha, S., Gundewar, S., Elrod, J. W., Ramachandran, A., Pattillo, C. B., et al. (2009). Hydrogen sulfide mediates cardioprotection through Nrf2 signaling. Circ. Res. 105, 365–374. doi: 10.1161/CIRCRESAHA.109.199919
Cao, Y., Gomes, S. A., Rangel, E. B., Paulino, E. C., Fonseca, T. L., Li, J., et al. (2015). S-nitrosoglutathione reductase-dependent PPARγ denitrosylation participates in MSC-derived adipogenesis and osteogenesis. J. Clin. Invest. 125, 1679–1691. doi: 10.1172/JCI73780
Chen, Y. H., Wang, P. P., Wang, X. M., He, Y. J., Yao, W. Z., Qi, Y. F., et al. (2011). Involvement of endogenous hydrogen sulfide in cigarette smoke-induced changes in airway responsiveness and inflammation of rat lung. Cytokine 3, 334–341. doi: 10.1016/j.cyto.2010.12.006
Chen, Y. Y., Chu, H. M., Pan, K. T., Teng, C. H., Wang, D. L., Wang, A. H., et al. (2008). Cysteine S-nitrosylation protects protein-tyrosine phosphatase 1B against oxidation-induced permanent inactivation. J. Biol. Chem. 283, 35265–35272. doi: 10.1074/jbc.M805287200
Cheng, Y., Ndisang, J. F., Tang, G., Cao, K., and Wang, R. (2004). Hydrogen sulfide-induced relaxation of resistance mesenteric artery beds of rats. Am. J. Physiol. Heart Circ. Physiol. 287, H2316–H2323. doi: 10.1152/ajpheart.00331.2004
Chung, K. K., Thomas, B., Li, X., Pletnikova, O., Troncoso, J. C., Marsh, L., et al. (2004). S-Nitrosylation of parkin regulates ubiquitination and compromises parkin’s protective function. Science 304, 1328–1331. doi: 10.1126/science.1093891
Colell, A., Green, D. R., and Ricci, J. E. (2009). Novel roles for GAPDH in cell death and carcinogenesis. Cell Death Differ. 12, 1573–1581. doi: 10.1038/cdd.2009.137
Dóka,É., Pader, I., Bíró, A., Johansson, K., Cheng, Q., Ballagó, K., et al. (2016). A novel persulfide detection method reveals protein persulfide- and polysulfide-reducing functions of thioredoxin and glutathione systems. Sci. Adv. 2:e1500968. doi: 10.1126/sciadv.1500968
Du, J., Huang, Y., Yan, H., Zhang, Q., Zhao, M., Zhu, M., et al. (2014). Hydrogen sulfide suppresses oxidized low-density lipoprotein (ox-LDL)-stimulated monocyte chemoattractant protein 1 generation from macrophages via the nuclear factor κB (NF-κB) pathway. J. Biol. Chem. 289, 9741–9753. doi: 10.1074/jbc.M113.517995
Ekundi-Valentim, E., Santos, K. T., Camargo, E. A., Denadai-Souza, A., Teixeira, S. A., Zanoni, C. I., et al. (2010). Differing effects of exogenous and endogenous hydrogen sulphide in carrageenan-induced knee joint synovitis in the rat. Br. J. Pharmacol. 7, 1463–1474. doi: 10.1111/j.1476-5381.2010.00640.x
Fiorucci, S., Orlandi, S., Mencarelli, A., Caliendo, G., Santagada, V., Distrutti, E., et al. (2007). Enhanced activity of a hydrogen sulphide-releasing derivative of mesalamine (ATB-429) in a mouse model of colitis. Br. J. Pharmacol. 8, 996–1002. doi: 10.1038/sj.bjp.0707193
Franco, M. C., and Estévez, A. G. (2014). Tyrosine nitration as mediator of cell death. Cell. Mol. Life Sci. 71, 3939–3950. doi: 10.1007/s00018-014-1662-8
Gaddam, R. R., Fraser, R., Badiei, A., Chambers, S., Cogger, V. C., Le Couteur, D. G., et al. (2016). Cystathionine-gamma-lyase gene deletion protects mice against inflammation and liver sieve injury following polymicrobial sepsis. PLOS ONE 11:e0160521. doi: 10.1371/journal.pone.0160521
Gade, A. R., Kang, M., and Akbarali, H. I. (2013). Hydrogen sulfide as an allosteric modulator of ATP-sensitive potassium channels in colonic inflammation. Mol. Pharmacol. 83, 294–306. doi: 10.1124/mol.112.081596
Gambari, L., Lisignoli, G., Cattini, L., Manferdini, C., Facchini, A., and Grassi, F. (2014). Sodium hydrosulfide inhibits the differentiation of osteoclast progenitor cells via NRF2-dependent mechanism. Pharmacol. Res. 87, 99–112. doi: 10.1016/j.phrs.2014.06.014
Gao, X. H., Krokowski, D., Guan, B. J., Bederman, I., Majumder, M., Parisien, M., et al. (2015). Quantitative H2S-mediated protein sulfhydration reveals metabolic reprogramming during the integrated stress response. Elife 4:e10067. doi: 10.7554/eLife.10067
Guo, C., Liang, F., Shah Masood, W., and Yan, X. (2014). Hydrogen sulfide protected gastric epithelial cell from ischemia/reperfusion injury by Keap1 s-sulfhydration, MAPK dependent anti-apoptosis and NF-κB dependent anti-inflammation pathway. Eur. J. Pharmacol. 725, 70–78. doi: 10.1016/j.ejphar.2014.01.009
Han, W., Dong, Z., Dimitropoulou, C., and Su, Y. (2011). Hydrogen sulfide ameliorates tobacco smoke-induced oxidative stress and emphysema in mice. Antioxid. Redox Signal. 15, 2121–2134. doi: 10.1089/ars.2010.3821
Hara, M. R., Agrawal, N., Kim, S. F., Cascio, M. B., Fujimuro, M., Ozeki, Y., et al. (2005). S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat. Cell Biol. 7, 665–674. doi: 10.1038/ncb1268
Hosoki, R., Matsuki, N., and Kimura, H. (1997). The possible role of hydrogen sulfide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochem. Biophys. Res. Commun. 237, 527–531. doi: 10.1006/bbrc.1997.6878
Hourihan, J. M., Kenna, J. G., and Hayes, J. D. (2013). The gasotransmitter hydrogen sulfide induces nrf2-target genes by inactivating the keap1 ubiquitin ligase substrate adaptor through formation of a disulfide bond between cys-226 and cys-613. Antioxid. Redox Signal. 19, 465–481. doi: 10.1089/ars.2012.4944
Huang, P., Shen, Z., Liu, J., Huang, Y., Chen, S., Yu, W., et al. (2016). Hydrogen sulfide inhibits High-salt diet-induced renal oxidative stress and kidney injury in Dahl rats. Oxid. Med. Cell. Longev. 2016:2807490. doi: 10.1155/2016/2807490
Jaffrey, S. R., Erdjument-Bromage, H., Ferris, C. D., Tempst, P., and Snyder, S. H. (2001). Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat. Cell Biol. 3, 193–197. doi: 10.1038/35055104
Jarosz, A. P., Wei, W., Gauld, J. W., Auld, J., Özcan, F., Aslan, M., et al. (2015). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) is inactivated by S-sulfuration in vitro. Free Radic. Biol. Med. 89, 512–521. doi: 10.1016/j.freeradbiomed.2015.09.007
Jiang, B., Tang, G., Cao, K., Wu, L., and Wang, R. (2010). Molecular mechanism for H2S-induced activation of KATP channels. Antioxid. Redox Signal. 12, 1167–1178. doi: 10.1089/ars.2009.2894
Jornayvaz, F. R., and Shulman, G. I. (2010). Regulation of mitochondrial biogenesis. Essays Biochem. 47, 69–84. doi: 10.1042/bse0470069
Ju, Y., Untereiner, A., Wu, L., and Yang, G. (2015). H2S-induced S-sulfhydration of pyruvate carboxylase contributes to gluconeogenesis in liver cells. Biochim. Biophys. Acta 1850, 2293–2303. doi: 10.1016/j.bbagen.2015.08.003
Kabil, O., and Banerjee, R. (2014). Enzymology of H2S biogenesis, decay and signaling. Antioxid. Redox Signal. 20, 770–782. doi: 10.1089/ars.2013.5339
Kang, M., Hashimoto, A., Gade, A., and Akbarali, H. I. (2015). Interaction between hydrogen sulfide-induced sulfhydration and tyrosine nitration in the KATP channel complex. Am. J. Physiol. Gastrointest. Liver Physiol. 308, 532–539. doi: 10.1152/ajpgi.00281.2014
Kimura, H. (2015). Signaling molecules: hydrogen sulfide and polysulfide. Antioxid. Redox Signal. 22, 362–376. doi: 10.1089/ars.2014.6082
Krishnan, N., Fu, C., Pappin, D. J., and Tonks, N. K. (2011). H2S-Induced sulfhydration of the phosphatase PTP1B and its role in the endoplasmic reticulum stress response. Sci. Signal. 203, ra86. doi: 10.1126/scisignal.2002329
Li, S., and Yang, G. (2015). Hydrogen sulfide maintains mitochondrial DNA replication via demethylation of TFAM. Antioxid. Redox Signal. 23, 630–642. doi: 10.1089/ars.2014.6186
Li, X., Zhang, K. Y., Zhang, P., Chen, L. X., Wang, L., Xie, M., et al. (2014). Hydrogen sulfide inhibits formaldehyde-induced endoplasmic reticulum stress in PC12 cells by upregulation of SIRT-1. PLOS ONE 9:e89856. doi: 10.1371/journal.pone.0089856
Li, Y. L., Zhou, J., Zhang, H., Luo, Y., Long, L. H., Hu, Z. L., et al. (2016). Hydrogen sulfide promotes surface insertion of hippocampal AMPA receptor GluR1 subunit via phosphorylating at Serine-831/Serine-845 sites through a sulfhydration-dependent mechanism. CNS Neurosci. Ther. 22, 789–798. doi: 10.1111/cns.12585
Liu, Y., Yang, R., Liu, X., Zhou, Y., Qu, C., Kikuiri, T., et al. (2014). Hydrogen sulfide maintains mesenchymal stem cell function and bone homeostasis via regulation of Ca2+ channel sulfhydration. Cell Stem Cell 15, 66–78. doi: 10.1016/j.stem.2014.03.005
Longen, S., Richter, F., Köhler, Y., Wittig, I., Beck, K. F., and Pfeilschifter, J. (2016). Quantitative persulfide site identification (qPerS-SID) reveals protein targets of H2S releasing donors in Mammalian cells. Sci. Rep. 6:29808. doi: 10.1038/srep29808
Lu, C., Kavalier, A., Lukyanov, E., and Gross, S. S. (2013). S-sulfhydration/desulfhydration and S-nitrosylation/denitrosylation: a common paradigm for gasotransmitter signaling by H2S and NO. Methods 62, 177–181. doi: 10.1016/j.ymeth.2013.05.020
Marutani, E., Yamada, M., Ida, T., Tokuda, K., Ikeda, K., Kai, S., et al. (2015). Thiosulfate mediates cytoprotective effects of hydrogen sulfide against neuronal ischemia. J. Am. Heart Assoc. 4:e002125. doi: 10.1161/JAHA.115.002125
Meng, G., Xiao, Y., Ma, Y., Tang, X., Xie, L., Liu, J., et al. (2016). Hydrogen sulfide regulates krüppel-like factor 5 transcription activity via specificity protein 1 S-sulfhydration at Cys664 to prevent myocardial hypertrophy. J. Am. Heart Assoc. 9:e004160. doi: 10.1161/JAHA.116.004160
Módis, K., Ju, Y., Ahmad, A., Untereiner, A. A., Altaany, Z., Wu, L., et al. (2016). S-Sulfhydration of ATP synthase by hydrogen sulfide stimulates mitochondrial bioenergetics. Pharmacol. Res. 113, 116–124. doi: 10.1016/j.phrs.2016.08.023
Mustafa, A. K., Gadalla, M. M., Sen, N., Kim, S., Mu, W., Gazi, S. K., et al. (2009). H2S signals through protein S-sulfhydration. Sci. Signal. 96, ra72. doi: 10.1126/scisignal.2000464
Mustafa, A. K., Sikka, G., Gazi, S. K., Steppan, J., Jung, S. M., Bhunia, A. K., et al. (2011). Hydrogen sulfide as endothelium-derived hyperpolarizing factor sulfhydrates potassium channels. Circ. Res. 109, 1259–1268. doi: 10.1161/CIRCRESAHA.111.240242
Nagy, P. (2013). Kinetics and mechanisms of thiol-disulfide exchange covering direct substitution and thiol oxidation-mediated pathways. Antioxid. Redox Signal. 18, 1623–1641. doi: 10.1089/ars.2012.4973
Naik, J. S., Osmond, J. M., Walker, B. R., and Kanagy, N. L. (2016). Hydrogen sulfide-induced vasodilation mediated by endothelial TRPV4 channels. Am. J. Physiol. Heart Circ. Physiol. 311, H1437–H1444. doi: 10.1152/ajpheart.00465.2016
Nicholls, C., Li, H., and Liu, J. P. (2012). GAPDH: a common enzyme with uncommon functions. Clin. Exp. Pharmacol. Physiol. 8, 674–679. doi: 10.1111/j.1440-1681.2011.05599.x
Ohno, K., Okuda, K., and Uehara, T. (2015). Endogenous S-sulfhydration of PTEN helps protect against modification by nitric oxide. Biochem. Biophys. Res. Commun. 456, 245–249. doi: 10.1016/j.bbrc.2014.11.066
Pan, J., and Carroll, K. S. (2013). Persulfide reactivity in the detection of protein s-sulfhydration. ACS Chem. Biol. 8, 1110–1116. doi: 10.1021/cb4001052
Park, C. M., Macinkovic, I., Filipovic, M. R., and Xian, M. (2015). Use of the “tag-switch” method for the detection of protein S-sulfhydration. Methods Enzymol. 555, 39–56. doi: 10.1016/bs.mie.2014.11.033
Paul, B. D., and Snyder, S. H. (2012). H2S signalling through protein sulfhydration and beyond. Nat. Rev. Mol. Cell Biol. 13, 499–507. doi: 10.1038/nrm3391
Saha, S., Chakraborty, P. K., Xiong, X., Dwivedi, S. K., Mustafi, S. B., Leigh, N. R., et al. (2016). Cystathionine β-synthase regulates endothelial function via protein S-sulfhydration. FASEB J. 30, 441–456. doi: 10.1096/fj.15-278648
Sato, K., Iwasaki, T., Tamaki, I., Aoto, M., Tokmakov, A. A., and Fukami, Y. (1998). Involvement of protein-tyrosine phosphorylation and dephosphorylation in sperm-induced Xenopus egg activation. FEBS Lett. 424, 113–118. doi: 10.1016/S0014-5793(98)00123-9
Sen, N., Paul, B. D., Gadalla, M. M., Mustafa, A. K., Sen, T., Xu, R., et al. (2012). Hydrogen sulfide-linked sulfhydration of NF-κB mediates its antiapoptotic actions. Mol. Cell 45, 13–24. doi: 10.1016/j.molcel.2011.10.021
Shimada, S., Fukai, M., Wakayama, K., Ishikawa, T., Kobayashi, N., Kimura, T., et al. (2015). Hydrogen sulfide augments survival signals in warm ischemia and reperfusion of the mouse liver. Surg. Today 45, 892–903. doi: 10.1007/s00595-014-1064-4
Shindo, T., Manabe, I., Fukushima, Y., Tobe, K., Aizawa, K., Miyamoto, S., et al. (2002). Krüppel-like zinc-finger transcription factor KLF5/BTEB2 is a target for angiotensin II signaling and an essential regulator of cardiovascular remodeling. Nat. Med. 8, 856–863. doi: 10.1038/nm738
Su, Y., Liu, D., Liu, Y., Zhang, C., Wang, J., and Wang, S. (2015). Physiologic levels of endogenous hydrogen sulfide maintain the proliferation and differentiation capacity of periodontal ligament stem cells. J. Periodontol. 86, 1276–1286. doi: 10.1902/jop.2015.150240
Sun, J., Aponte, A. M., Menazza, S., Gucek, M., Steenbergen, C., and Murphy, E. (2016). Additive cardioprotection by pharmacological postconditioning with hydrogen sulfide and nitric oxide donors in mouse heart: S-sulfhydration vs. S-nitrosylation. Cardiovasc. Res. 110, 96–106. doi: 10.1093/cvr/cvw037
Sun, Y., Huang, Y., Yu, W., Chen, S., Yao, Q., Zhang, C., et al. (2017). Sulfhydration-associated phosphodiesterase 5A dimerization mediates vasorelaxant effect of hydrogen sulfide. Oncotarget 8, 31888–31900. doi: 10.18632/oncotarget.16649
Uesugi, S., Muroi, M., Kondoh, Y., Shiono, Y., Osada, H., and Kimura, K. I. (2017). Allantopyrone A activates Keap1-Nrf2 pathway and protects PC12 cells from oxidative stress-induced cell death. J. Antibiot. 70, 429–434. doi: 10.1038/ja.2016.99
Untereiner, A. A., Fu, M., Módis, K., Wang, R., Ju, Y., and Wu, L. (2016a). Stimulatory effect of CSE-generated H2S on hepatic mitochondrial biogenesis and the underlying mechanisms. Nitric Oxide 58, 67–76. doi: 10.1016/j.niox.2016.06.005
Untereiner, A. A., Oláh, G., Módis, K., Hellmich, M. R., and Szabo, C. (2017). H2S-induced S-sulfhydration of lactate dehydrogenase a (LDHA) stimulates cellular bioenergetics in HCT116 colon cancer cells. Biochem. Pharmacol. 136, 86–98. doi: 10.1016/j.bcp.2017.03.025
Untereiner, A. A., Wang, R., Ju, Y., and Wu, L. (2016b). Decreased gluconeogenesis in the absence of cystathionine gamma-lyase and the underlying mechanisms. Antioxid. Redox Signal. 24, 129–140. doi: 10.1089/ars.2015.6369
Vandiver, M. S., Paul, B. D., Xu, R., Karuppagounder, S., Rao, F., Snowman, A. M., et al. (2013). Sulfhydration mediates neuroprotective actions of parkin. Nat. Commun. 4, 1626. doi: 10.1038/ncomms2623
Wallace, J. L., Ferraz, J. G., and Muscara, M. N. (2012). Hydrogen sulfide: an endogenous mediator of resolution of inflammation and injury. Antioxid. Redox Signal. 17, 58–67. doi: 10.1089/ars.2011.4351
Wallace, J. L., Vong, L., McKnight, W., Dicay, M., and Martin, G. R. (2009). Endogenous and exogenous hydrogen sulfide promotes resolution of colitis in rats. Gastroenterology 137, 569–578. doi: 10.1053/j.gastro.2009.04.012
Wang, P., Wu, L., Ju, Y., Fu, M., Shuang, T., Qian, Z., et al. (2017). Age-dependent allergic asthma development and cystathionine gamma-lyase deficiency. Antioxid. Redox Signal. doi: 10.1089/ars.2016.6875 [Epub ahead of print].
Wang, Y., Zhao, X., Jin, H., Wei, H., Li, W., Bu, D., et al. (2009). Role of hydrogen sulfide in the development of atherosclerotic lesions in apolipoprotein E knockout mice. Arterioscler. Thromb. Vasc. Biol. 29, 173–179. doi: 10.1161/ATVBAHA.108.179333
Wang, Z., Liu, D. X., Wang, F. W., Zhang, Q., Du, Z. X., Zhan, J. M., et al. (2013). L-Cysteine promotes the proliferation and differentiation of neural stem cells via the CBS/H2S pathway. Neuroscience 237, 106–117. doi: 10.1016/j.neuroscience.2012.12.057
Wasik, U., Milkiewicz, M., Kempinska-Podhorodecka, A., and Milkiewicz, P. (2017). Protection against oxidative stress mediated by the Nrf2/Keap1 axis is impaired in primary biliary cholangitis. Sci. Rep. 7:44769. doi: 10.1038/srep44769
Wedmann, R., Onderka, C., Wei, S., Szijártó, I. A., Miljkovic, J. L., Mitrovic, A., et al. (2016). Improved tag-switch method reveals that thioredoxin acts as depersulfidase and controls the intracellular levels of protein persulfidation. Chem. Sci. 7, 3414–3426. doi: 10.1039/c5sc04818d
Wei, H. J., Xu, J. H., Li, M. H., Tang, J. P., Zou, W., Zhang, P., et al. (2014). Hydrogen sulfide inhibits homocysteine-induced endoplasmic reticulum stress and neuronal apoptosis in rat hippocampus via upregulation of the BDNF-TrkB pathway. Acta Pharmacol. Sin. 35, 707–715. doi: 10.1038/aps.2013.197
Whiteman, M., and Winyard, P. G. (2011). Hydrogen sulfide and inflammation: the good, the bad, the ugly and the promising. Expert Rev. Clin. Pharmacol. 4, 13–32. doi: 10.1586/ecp.10.134
Xie, L., Gu, Y., Wen, M., Zhao, S., Wang, W., Ma, Y., et al. (2016). Hydrogen sulfide induces Keap1 S-sulfhydration and suppresses diabetes-accelerated atherosclerosis via Nrf2 activation. Diabetes Metab. Res. Rev. 65, 3171–3184. doi: 10.2337/db16-0020
Xie, L., Tiong, C. X., and Bian, J. S. (2012). Hydrogen sulfide protects SH-SY5Y cells against 6-hydroxydopamine-induced endoplasmic reticulum stress. Am. J. Physiol. Cell Physiol. 303, 81–91. doi: 10.1152/ajpcell.00281.2011
Xie, Z. Z., Shi, M. M., Xie, L., Wu, Z. Y., Li, G., Hua, F., et al. (2014). Sulfhydration of p66Shc at cysteine59 mediates the antioxidant effect of hydrogen sulfide. Antioxid. Redox Signal. 21, 2531–2542. doi: 10.1089/ars.2013.5604
Yadav, V., Gao, X. H., Willard, B., Hatzoglou, M., Banerjee, R., and Kabil, O. (2017). Hydrogen sulfide modulates eukaryotic translation initiation factor 2α (eIF2α) phosphorylation status in the integrated stress response pathway. J. Biol. Chem. 292, 13143–13153. doi: 10.1074/jbc.M117.778654
Yang, G., Zhao, K., Ju, Y., Mani, S., Cao, Q., Puukila, S., et al. (2013). Hydrogen sulfide protects against cellular senescence via S-sulfhydration of Keap1 and activation of Nrf2. Antioxid. Redox Signal. 18, 1906–1919. doi: 10.1089/ars.2012.4645
Ying, R., Wang, X. Q., Yang, Y., Gu, Z. J., Mai, J. T., Qiu, Q., et al. (2016). Hydrogen sulfide suppresses endoplasmic reticulum stress-induced endothelial-to-mesenchymal transition through Src pathway. Life Sci. 144, 208–217. doi: 10.1016/j.lfs.2015.11.025
Yu, W., Liao, Y., Huang, Y., Chen, S. Y., Sun, Y., Sun, C., et al. (2017). Endogenous hydrogen sulfide enhances carotid sinus baroreceptor sensitivity by activating the transient receptor potential cation channel subfamily v member 1 (TRPV1) channel. J. Am. Heart Assoc. 6:e004971. doi: 10.1161/JAHA.116.004971
Zanardo, R. C., Brancaleone, V., Distrutti, E., Fiorucci, S., Cirino, G., and Wallace, J. L. (2006). Hydrogen sulfide is an endogenous modulator of leukocyte-mediated inflammation. FASEB J. 12, 2118–2120. doi: 10.1096/fj.06-6270fje
Zhang, D., Macinkovic, I., Devarie-Baez, N. O., Pan, J., Park, C. M., Carroll, K. S., et al. (2014). Detection of protein S-sulfhydration by a tag-switch technique. Angew. Chem. Int. Ed. Engl. 53, 575–581. doi: 10.1002/anie.201305876
Zhao, H., Lu, S., Chai, J., Zhang, Y., Ma, X., Chen, J., et al. (2017). Hydrogen sulfide improves diabetic wound healing in ob/ob mice via attenuating inflammation. J. Diabetes Complications 31, 1363–1369. doi: 10.1016/j.jdiacomp.2017.06.011
Zhao, K. X., Ju, Y. J., Li, S. S., Altaany, Z., Wang, R., and Yang, G. D. (2014a). S-sulfhydration of MEK1 leads to PARP-1 activation and DNA damage repair. EMBO Rep. 15, 792–800. doi: 10.1002/embr.201338213
Zhao, K. X., Li, S. S., Wu, L. Y., Lai, C., and Yang, G. D. (2014b). Hydrogen sulfide represses androgen receptor transactivation by targeting at the second zinc finger module. J. Biol. Chem. 289, 20824–20835. doi: 10.1074/jbc.M114.559518
Zhao, W., Zhang, J., Lu, Y., and Wang, R. (2001). The vasorelaxant effect of H2S as a novel endogenous gaseous KATP channel opener. EMBO J. 20, 6008–6016. doi: 10.1093/emboj/20.21.6008
Zheng, Y., Liao, F., Lin, X., Zheng, F., Fan, J., Cui, Q., et al. (2017). Cystathionine γ-lyase-hydrogen sulfide induces runt-related transcription factor 2 sulfhydration, thereby increasing osteoblast activity to promote bone fracture healing. Antioxid. Redox Signal. doi: 10.1089/ars.2016.6826 [Epub ahead of print].
Zhou, H., Ding, L., Wu, Z., Cao, X., Zhang, Q., Lin, L., et al. (2017). Hydrogen sulfide reduces RAGE toxicity through inhibition of its dimer formation. Free Radic. Biol. Med. 104, 262–271. doi: 10.1016/j.freeradbiomed.2017.01.026
Zuidema, M. Y., Yang, Y., Wang, M., Kalogeris, T., Liu, Y., Meininger, C. J., et al. (2010). Antecedent hydrogen sulfide elicits an anti-inflammatory phenotype in postischemic murine small intestine: role of BK channels. Am. J. Physiol. Heart Circ. Physiol. 299, 1554–1567. doi: 10.1152/ajpheart.01229.2009
Keywords: H2S, sulfhydration, protein, biological function, detecting method
Citation: Zhang D, Du J, Tang C, Huang Y and Jin H (2017) H2S-Induced Sulfhydration: Biological Function and Detection Methodology. Front. Pharmacol. 8:608. doi: 10.3389/fphar.2017.00608
Received: 21 May 2017; Accepted: 22 August 2017;
Published: 06 September 2017.
Edited by:
Alfonso Pompella, University of Pisa, ItalyReviewed by:
Ming Xian, Washington State University, United StatesCaroline Gaucher, Université de Lorraine, France
Copyright © 2017 Zhang, Du, Tang, Huang and Jin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yaqian Huang, eWFxaWFuaHVhbmdAMTI2LmNvbQ== Hongfang Jin, amluaG9uZ2Zhbmc1MUAxMjYuY29t