 Kelly L. Walton
Kelly L. Walton Katharine E. Johnson
Katharine E. Johnson
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Pharmacol. , 14 July 2017
Sec. Cardiovascular and Smooth Muscle Pharmacology
Volume 8 - 2017 | https://doi.org/10.3389/fphar.2017.00461
This article is part of the Research Topic Novel Therapeutic Targets and Emerging Treatments for Fibrosis, Volume I View all 16 articles
Fibrosis occurs when there is an imbalance in extracellular matrix (ECM) deposition and degradation. Excessive ECM deposition results in scarring and thickening of the affected tissue, and interferes with tissue and organ homeostasis – mimicking an exaggerated “wound healing” response. Many transforming growth factor-β (TGF-β) ligands are potent drivers of ECM deposition, and additionally, have a natural affinity for the ECM, creating a concentrated pool of pro-fibrotic factors at the site of injury. Consequently, TGF-β ligands are upregulated in many human fibrotic conditions and, as such, are attractive targets for fibrosis therapy. Here, we will discuss the contribution of TGF-β proteins in the pathogenesis of fibrosis, and promising anti-fibrotic approaches that target TGF-β ligands.
In fibrotic disease, increased deposition of extracellular matrix (ECM) proteins compromises tissue architecture, and interferes with normal organ function. Fibrosis is frequently observed in the heart, liver, lungs, and kidneys, but can arise in any tissue that has suffered chronic insult. Triggers for fibrosis can be either biological (e.g., persistent bacterial/viral infections, genetic disorders or tissue injury) and/or environmental (e.g., pollutant/chemical exposure or allergens) (Macneal and Schwartz, 2012). Fibrosis is primarily driven by inflammatory cytokines including the interleukins (Nikolic-Paterson et al., 1996; O’Reilly et al., 2012), and members of the transforming growth factor-β (TGF-β) superfamily (Roberts et al., 1986; Meng et al., 2016), such as TGF-β1, activin A and activin B. Many of these ligands are expressed by infiltrating inflammatory cells, which migrate toward damage tissues. Of interest, these TGF-β ligands not only promote ECM deposition, but they also become concentrated within the accumulating matrix, thereby accelerating the pro-fibrotic response.
The TGF-β superfamily includes the TGF-β isoforms (TGF-β1, -β2 and -β3), activins and inhibins, growth differentiation factors (GDFs), bone morphogenetic proteins (BMPs), and anti-mullerian hormone (AMH). Although structurally similar, these ligands elicit distinct biological responses, based on their cell/tissue-specific expression, their interactions with inhibitory molecules and the unique combinations of receptors with which they complex. TGF-β ligands form receptor complexes with one of seven type I receptors (also termed activin-like kinase or ALK receptors) in combination with one of five type II receptors (ActRIIA, ActRIIB, TGFBRII, BMPRII, and AMHRII). For TGF-β1, ligand contact with the type II receptor TGFBRII leads to recruitment and phosphorylation of the type I receptor ALK-5. The BMPs, however, firstly contact their type I receptor (namely ALK3/ALK6), and then induce kinase activity toward the type II receptor BMPRII (reviewed in Miyazawa et al., 2002). In all instances, ligand-receptor complex formation leads to the activation of kinase domains within the receptors, which potentiate phosphorylation cascades involving SMAD transcription factors. The TGF-β isoforms and activins converge to induce intracellular signaling via SMAD-2/3 transcription factors. Activation of SMAD-2/3 regulates to the expression of several profibrotic genes, including collagens [COL1A1, COL3A1, COL5A2, COL6A1, COL6A3, COL7A1, (Verrecchia et al., 2001a,b)], plasminogen activator inhibitor-1 (PAI-1; Dennler et al., 1998; Hua et al., 1998), various proteoglycans (Schonherr et al., 1991; Romaris et al., 1995; Dadlani et al., 2008), integrins (Margadant and Sonnenberg, 2010), connective tissue growth factor (Chen et al., 2002), and matrix metalloproteases (MMPs; Yuan and Varga, 2001). BMPs via activation of SMAD-1/5/8, are capable of suppressing TGF-β mediated fibrotic gene expression (Wang and Hirschberg, 2003). As such, hyperactivation of activin/TGF-β-mediated SMAD-2/3 signaling promotes fibrosis, whereas increased BMP/SMAD-1/5/8 activity is likely anti-fibrotic. Here, we describe the contribution of TGF-β ligand induced SMAD signaling to the pathogenesis of human fibrosis, and emerging therapeutic strategies that target these ligands.
TGF-β isoforms – TGF-β ligands that activate the SMAD-2/3 intracellular pathway have been heavily implicated in fibrosis. In particular, TGF-β1 is considered a major driver of human fibrotic pathologies. Circulating or tissue levels of TGF-β1 are elevated in human hepatic (Nagy et al., 1991), renal (Ketteler et al., 1994), and pulmonary fibrosis (Molina-Molina et al., 2006), as well as during cardiac failure (Khan et al., 2014). Exogenous TGF-β1 in rodents is sufficient to induce fibrosis in the lungs (Sime et al., 1997), and kidneys (Clouthier et al., 1997). Tissue specific pro-fibrotic activities of TGF-β1 are outlined in Table 1.
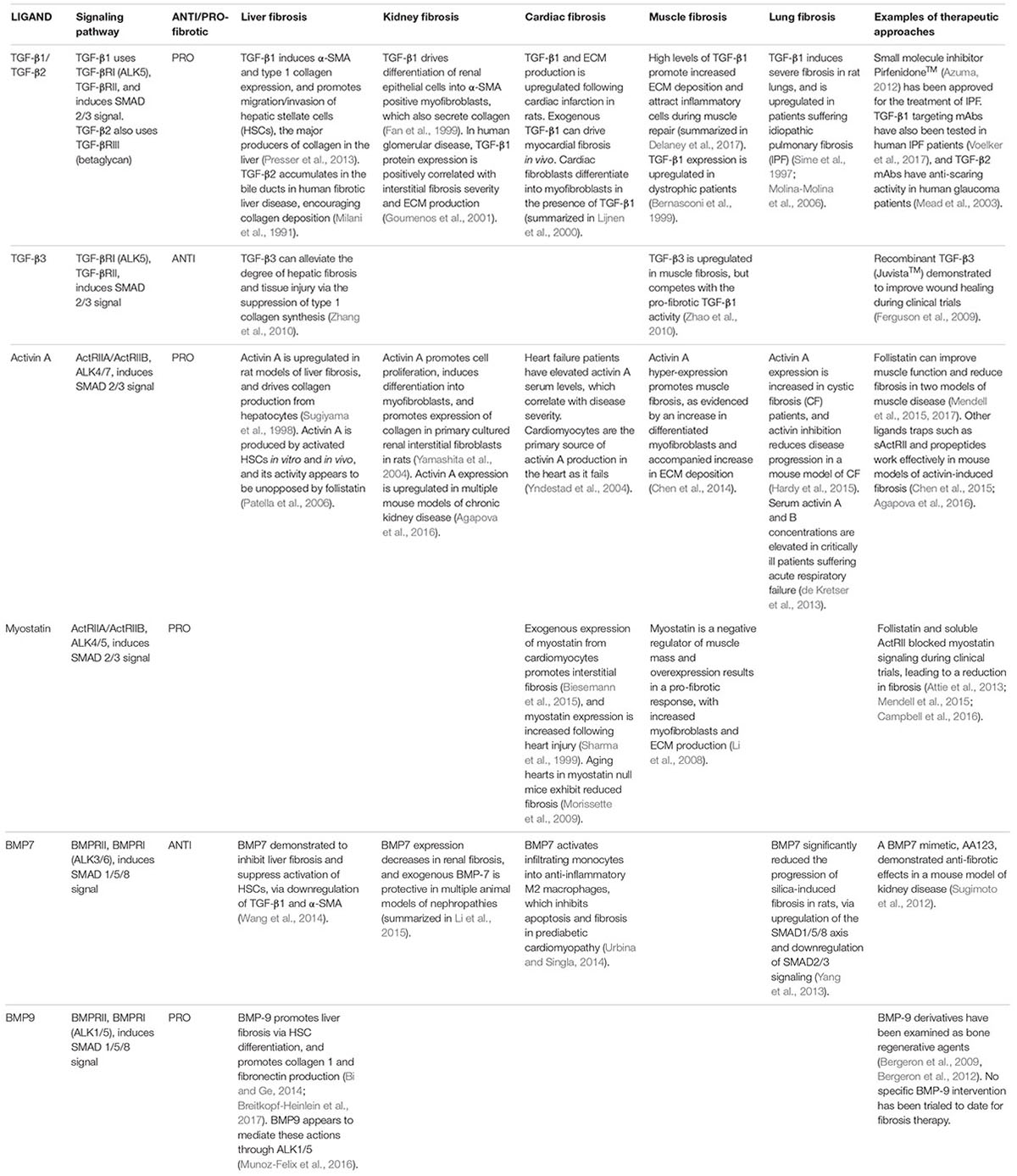
TABLE 1. Summary of evidence for TGF-β ligands in fibrosis.
Mechanistically, TGF-β1 promotes fibrosis by driving the differentiation of quiescent fibroblasts into matrix secreting myofibroblasts (Vaughan et al., 2000; Lijnen et al., 2003). Under duress fibroblasts differentiate into a proto-myofibroblast lineage, which in the presence of TGF-β1 (and ED-A fibronectin), become fully differentiated myofibroblasts (Gabbiani, 2003; Figure 1). Notably, TGF-β1 drives the production of α-smooth muscle (α-SM) actin, which gives the myofibroblasts their contractility. These cells represent activated fibroblasts, with a high synthetic capacity for ECM proteins (Ignotz and Massague, 1986). In response to heightened TGF-β1 signals, dominating myofibroblasts deposit excessive ECM, which compromises the local tissue architecture. TGF-β1 can further exacerbate this response by acting as a chemoattractant for some inflammatory cells (Wahl, 1992; Ludviksson and Gunnlaugsdottir, 2003), and is abundantly expressed by infiltrating macrophages in fibrotic tissue (Denholm and Rollins, 1993).
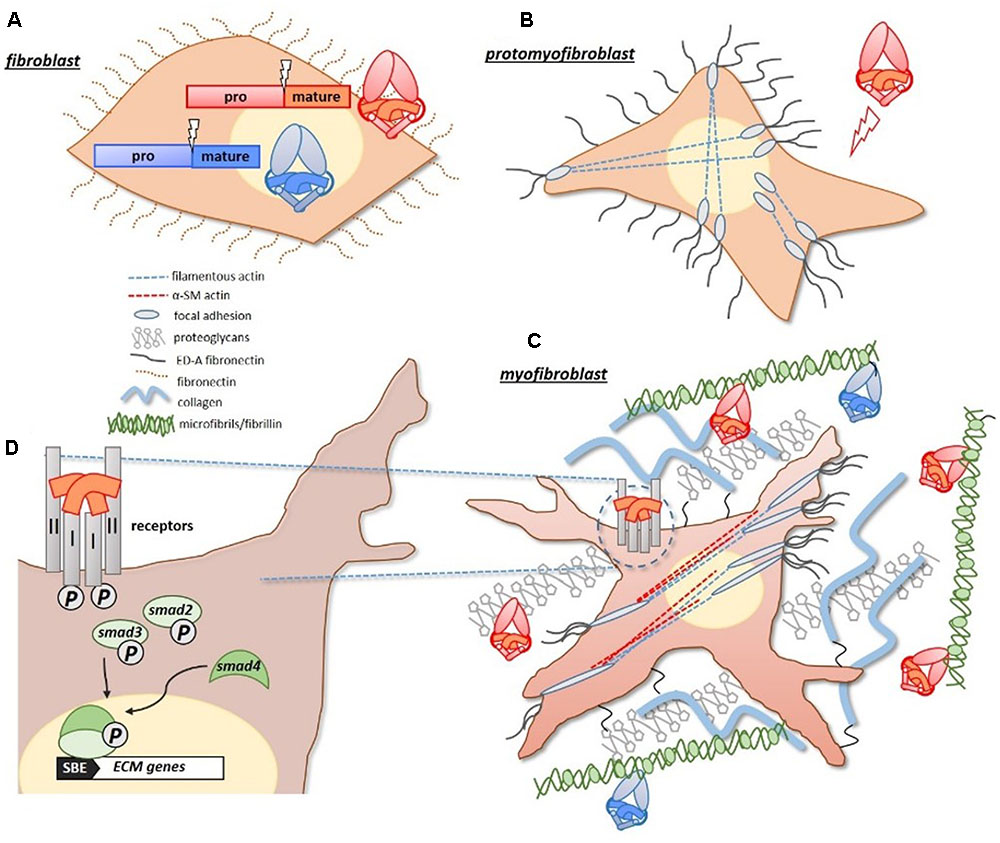
FIGURE 1. Pro-fibrotic activities of TGF-β proteins. (A) Pro-fibrotic TGF-β ligands, including TGF-β1 and activins (red/orange), and anti-fibrotic BMP ligands (blue) are similarly synthesized as large precursor proteins with pro-/mature-domains. (B) Under duress, quiescent fibroblasts differentiate into proto-myofibroblasts which express actin filaments that extend from a focal adhesion complex, and express extracellular ED-A fibronectin. (C) With increased exposure to TGF-β1, and ED-A fibronectin, proto-myofibroblasts become fully differentiated matrix-secreting myofibroblasts. The identifiable presence of α-smooth muscle actin (α-SMA), gives the myofibroblasts their measurable contractility. Both TGF-β1 and activins have a natural affinity for matrix proteins, such as fibrillin (microfibrils) and proteoglycans, and become concentrated in the expanding matrix of fibrotic tissue. The BMPs can also bind to fibrillin-containing microfibrils. (D) The activities of TGF-β1 and activins are initiated at the cell surface, where they form receptor complexes with type I/II receptors. Receptor activation leads to an intracellular phosphorylation cascade involving SMAD-2/3 transcription factors. SMAD-2/3 complex with SMAD-4, and translocate to the nucleus where they drive the expression of target matrix genes (via a Smad Binding Element, or SBE, in the promoter). Figure generated using Gabbiani (2003) as a guide.
TGF-β1 is ubiquitously expressed within the body, and its target receptors (ALK5 and TGFβRII) have been identified on most cell types. As such, elevated levels of TGF-β1, either locally or systemically, can result in widespread manifestations of fibrosis. Acting through the SMAD-2/3 axis, TGF-β1 drives the expression of key ECM genes, including collagens (Verrecchia et al., 2001a), fibronectin (Hocevar et al., 1999), and PAI-1 (Dennler et al., 1998). These proteins are key components of developing fibrotic tissue. Additionally, TGF-β1 has a natural affinity for the ECM, binding directly to fibrillin microfibrils (Taipale et al., 1996). As such, TGF-β1 becomes concentrated in the accumulating ECM, thereby exacerbating the fibrotic response. TGF-β1 activity is kept in-check in healthy tissues by its naturally affiliated propeptide (termed ‘latency associated peptide’), which prevents TGF-β1 from complexing with its target receptors (Bottinger et al., 1996). For signaling to be initiated, an activation mechanism is required to break the propetide’s hold. For TGF-β1, activators include integrins (namely αVβ6), thrombospondin-1 (TSP-1) and plasmin proteases (Lyons et al., 1990; Crawford et al., 1998; Munger et al., 1999; Morris et al., 2003). Significantly, in inflammatory conditions de novo expression of some/all of these activators increases (Zhang et al., 1999; Lopez-Dee et al., 2011), leading to activation and potentiation of TGF-β1 signaling (Popov et al., 2008). TGF-β1 activation is also increased in fibrotic tissue via biomechanical tissue stiffness, which causes force-dependant removal of the TGF-β1 propeptide (Wipff et al., 2007).
Although TGF-β1 is the best characterized pro-fibrotic factor within the family, TGF-β2 also displays potent fibrotic activity. TGF-β2 accumulates in the bile ducts in human fibrotic liver disease (Milani et al., 1991), and has been implicated in the fibrotic response associated with glaucoma (Wordinger et al., 2014). Remarkably, TGF-β3 appears to have anti-fibrotic activity in some tissues. TGF-β3 plays a key role in regulating epidermal and dermal cell motility during wound repair, a TGFβ-isoform-specific effect (Occleston et al., 2008). TGF-β3 is expressed at high concentrations during wound repair, and unlike TGF-β1 and -β2 isoforms, can promote wound healing without fibrotic scarring (Ferguson et al., 2009).
Activins – Similar to TGF-β1, activins can trigger a pro-fibrotic response in several tissues via activation of the SMAD-2/3 cascade. Activins promote the proliferation of fibroblasts, their differentiation into myofibroblasts (Ohga et al., 1996; Ota et al., 2003; Yamashita et al., 2004), and the accumulation of ECM (Yamashita et al., 2004; Murakami et al., 2006). Serum concentrations of activin are elevated in patients suffering cystic fibrosis (Hardy et al., 2015), acute respiratory failure (de Kretser et al., 2013), chronic kidney disease (Agapova et al., 2016), and heart failure (Yndestad et al., 2004). Indeed, increased serum activin is a hallmark of many human chronic conditions and can have catastrophic consequences for affected patients. Our studies have shown that activins can drive the multi-organ wasting syndrome, cachexia (Chen et al., 2014), and that high levels of activin can induce a marked fibrotic response in skeletal muscle and liver, characterized by an influx of differentiated myofibroblasts and accompanied ECM deposition. Significantly, we have shown that the fibrotic pathology induced by activins can be fully reversed (Chen et al., 2014, 2015), highlighting the potential of anti-activin therapy to treat muscular dystrophies, in which patients suffer severe muscular fibrosis.
Activin is one of four major TGF-β ligands that signal through the activin type II receptors, ActRIIA/ActRIIB. Activin A, activin B, myostatin (GDF-8), and GDF-11 can all complex with ActRIIA/ActRIIB and initiate SMAD-2/3 intracellular activity. Despite this, all four ligands have non-overlapping bioactivities in vivo owing to their cell/tissue specific expression and distinct preferences for type I receptors (ALK4, ALK5 or ALK7). Myostatin is expressed almost exclusively in skeletal muscle, and can mimic the pro-fibrotic response observed under high activin conditions (Li et al., 2008). Myostatin promotes the proliferation of fibroblasts in muscle, and induces the expression of ECM proteins including collagen and fibronectin both in vitro and in vivo (Li et al., 2008). Given the structural homology of GDF-11 with myostatin (Walker et al., 2017) and shared receptor contacts, it is predicted that at high local concentrations GDF-11 will also exhibit pro-fibrotic activity.
Unlike the TGF-β isoforms, activins are secreted in an ‘active’ form. To constrain their activity, activins (and myostatin) are regulated extracellularly by follistatin. Follistatin binds directly to activin-related ligands, shielding their receptor contact sites so as to limit their signaling potential. Follistatin has opposing activity to activins/myostatin in fibrosis; attenuating early liver fibrosis (Patella et al., 2006), and lung fibrosis (Aoki et al., 2005) in murine models.
Like the TGF-β isoforms, activins have a natural affinity for the ECM – binding to heparin-sulphated proteoglycans (HSPGs) such as perlecan (Li S. et al., 2010). HSPGs are upregulated in many human fibrotic conditions, including human idiopathic lung fibrosis (Jiang et al., 2010; Westergren-Thorsson et al., 2017), Duchenne’s Muscular Dystrophy (Alvarez et al., 2002), liver disease (Roskams et al., 1996) and kidney fibrosis (Ebefors et al., 2011). Additionally, heparanase, the enzyme that metabolizes the carbohydrate chains on these proteoglycans, is also upregulated in fibrotic pathologies (Lv et al., 2016). Increased heparanase activity drives the release and activation of growth factors like activin and TGF-β (Masola et al., 2014). Consequently, activins affinity for HSPGs ensures an enriched pool of bioactive growth factors in the accumulating matrix.
Whilst hyper-activation of the SMAD-2/3 pathway is reported to be pro-fibrotic, signaling through the alternate SMAD-1/5 pathway is anti-fibrotic. Of all the SMAD-1/5 activators, BMP-7 has the most well documented anti-fibrotic activity. BMP-7 is expressed only in select adult tissues, including the kidney, and its expression declines in rodent models of renal fibrosis. BMP-7 counteracts TGF-β1 induced induction of myofibroblasts and ECM in multiple models of organ fibrosis (Zeisberg et al., 2003; Weiskirchen and Meurer, 2013). Addition of exogenous BMP-7 can both prevent and even reverse fibrosis in models of kidney disease (reviewed in Weiskirchen and Meurer, 2013; Li et al., 2015).
BMP-7 mediates its anti-fibrotic activity through the type I receptor, ALK-3 [also termed BMPRIA, (Sugimoto et al., 2012)]. Expression of ALK-3 increases in the early stages of kidney disease, and loss of ALK-3 exacerbates TGF-β1 mediated fibrosis, suggesting that ALK-3 is protective for fibrosis. ALK-3 is also a docking receptor for related BMP ligands, BMP-2, BMP-4 and BMP-6. Both BMP-2 and BMP-6 can similarly attenuate kidney fibrosis in rodent models of kidney disease (Yang et al., 2009; Dendooven et al., 2011), and BMP-6 expression is increased in humans and mice suffering non-alcoholic fatty liver disease (NAFLD) and is protective for hepatic fibrosis (Arndt et al., 2015). Interestingly, loss of BMP-4 is speculated to drive opposing pro-fibrotic response in cardiac tissue (Sun et al., 2013), and increased BMP-9 in the liver induces a fibrotic-like response (Breitkopf-Heinlein et al., 2017).
Like activins, BMPs are secreted from cells in an activated state. Once secreted, many of the BMPs are sequestered to the ECM by propeptide-mediated binding to fibrillins. Fibrillin binding creates a local concentration of BMPs and is thought to facilitate release of the active BMP proteins (reviewed in Wohl et al., 2016). Notably, perturbed fibrillin/microfibril assembly is a feature of human fibrosis (Wohl et al., 2016), and likely alters the bioavailability of associated BMPs. BMP activity is also restricted by extracellular antagonists Gremlin, DAN, Chordin and Noggin (reviewed in Weiskirchen et al., 2009).
TGF-β targeted therapies in fibrosis are designed to reduce activin/TGF-β signaling via SMAD-2/3, or alternatively, promote a BMP-mediated SMAD-1/5 signal. Many approaches are currently being explored, and some have reached the clinic (e.g., TGF-β3, follistatin, BMP-7). Leading approaches and emerging new strategies targeted to TGF-β mediated SMAD signals are described here.
TGF-β targeted therapies – A plethora of TGF-β targeted therapies have been explored (reviewed in Akhurst and Hata, 2012), some of which have been designated specifically for fibrosis. These include, TGF-β1 antibodies for kidney fibrosis (Akhurst and Hata, 2012; Voelker et al., 2017), peptides (Llopiz et al., 2009), and receptor decoys for lung fibrosis (Yamada et al., 2007). Interventions for TGF-β2 mediated fibrosis include peptides for cardiac and skin fibrosis (Santiago et al., 2005; Hermida et al., 2009), and antibodies for glaucoma-related scarring (Mead et al., 2003). TGF-β3 in the form of JuvistaTM (Renova) was found to improve wound healing in phase I/II human clinical trials, but failed in phase III (Ferguson et al., 2009). However, few of these approaches have resulted in positive patient outcomes. Indeed, in a recent study testing a TGF-β1 specific antibody in a diabetic model of kidney fibrosis, disease progression was not improved (Voelker et al., 2017). Several factors likely impede the clinical effectiveness of these approaches, including the presence of multiple pro-fibrotic factors in advanced disease.
Despite their non-selectively, the small molecule inhibitors have advanced furthest owing to their economical production and ease of administration. Many of these approaches target the kinase activity of the TGF-β type I (TβRI, or ALK-5) and II (TGFβRII) receptors, including compounds GW788388 (Petersen et al., 2008) and SB-525334 (Grygielko et al., 2005), and have proven anti-fibrotic activities in vivo. Significantly, the small molecule inhibitor PirfenidoneTM (Azuma, 2012) has been approved for the treatment of lung fibrosis in humans. Though encouraging, the promiscuous nature of these small molecule inhibitors renders them more prone to side effects (Noble et al., 2011).
Interventions that target the intracellular phosphorylation of SMAD-2/3 proteins, can also reduce TGF-β triggered fibrosis (reviewed in Munoz-Felix et al., 2015). SMAD-7, which sequesters SMAD-2/3, protects against renal fibrosis upon viral gene-delivery in a mouse model of kidney fibrosis (Terada et al., 2002). SIS3, a selective compound that targets only SMAD-3 proved to reduce the expression of ECM proteins and delayed the progression of diabetic nephropathy in a mouse model (Li J. et al., 2010).
Activin and myostatin targeted therapies – Broad-spectrum TGF-β signaling inhibitors including SMAD-7, SIS3, and many of the small molecule kinase inhibitors can also block activin and myostatin induced signaling (Inman et al., 2002; Rodgers et al., 2014). The pleiotropic nature of these inhibitors is unfavorable for systemic use, and there is a pressing need for more tailored ligand therapies. ‘Ligand traps’ such as follistatin, soluble forms of the activin type II receptors (sActRII), and propeptides offer improved ligand selectivity, and their increased size favors serum retention (Wakefield et al., 1990).
Follistatin therapy is the leading approach for SMAD-2/3 blockade in activin/myostatin triggered fibrosis. Gene therapy approaches deploying follistatin have reached phase I/II clinical trials in humans (Mendell et al., 2015, 2017). Local expression of follistatin is sufficient to improve muscle function and reduce fibrosis in inflammatory myopathy (Mendell et al., 2017), and muscular dystrophy (Mendell et al., 2015). Similarly, the clinically relevant forms of sActRII (RAP-011 or ACE-011/sotatercept forms) can effectively reverse fibrosis in murine models of kidney disease (Agapova et al., 2016). However, clinical advancement of sActRII has been impeded by its apparent off-target vascular effects when used for human therapy (Attie et al., 2013; Campbell et al., 2016).
Propeptides, which are natural by-products of TGF-β assembly, and have an affinity for the ECM (Harrison et al., 2011), are attractive anti-fibrotic agents. We have shown that modified forms of the activin propeptides (propeptide-Fc fusion proteins) can both potently and specifically attenuate activin-mediated pathologies in mouse skeletal muscle (Chen et al., 2015, 2017). Similarly, we and others have found that the myostatin propeptide can revert muscle damage and attenuate fibrosis in mouse models (Qiao et al., 2008; Hamrick et al., 2010; Chen et al., 2017). This approach has also been demonstrated for the TGF-β propeptide, termed ‘latency associated propeptide’ or LAP, which has been shown to attenuate TGF-induced pathologies in vivo (Bottinger et al., 1996). However, as the TGF-β1 LAP has comparable affinity for all three TGF-β isoforms, and also GDF-8/-11, further modifications are required to improve its selectivity.
BMP-targeted therapies for fibrosis – The anti-fibrotic activity of BMP-7 encouraged its application as a human therapy. However, like many TGF-β proteins, BMP-7 is poorly made and processed in mammalian cells (Swencki-Underwood et al., 2008), and upon delivery, is likely rapidly cleared from the blood (Coffey et al., 1987). To address these limitations, Sugimoto et al. (2012), developed a small molecule BMP-7 mimetic (AA123), which has the same anti-fibrotic activity as recombinant BMP-7 in a mouse model of kidney disease. A similar BMP-7 mimetic is currently in phase II clinical trials for acute kidney injury (Thrasos Therapeutics, Canada).
Extracellular matrices provide a structural framework for cells and additionally serve as a scaffold for growth factors. Excessive ECM deposition, as observed in fibrosis, compromises tissue and organ structure and function and can lead to organ malfunction and failure. TGF-β proteins are major regulators of fibrosis, and the balanced activities of the pro-fibrotic and anti-fibrotic ligands ensure tissue homeostasis. TGF-β targeted therapies in fibrosis are designed to suppress the pro-fibrotic activity of TGF-β isoforms/activins/myostatin, or heighten the activity of anti-fibrotic BMPs. Promising anti-fibrotic TGF-β targeted therapies involve the use of ligand traps (follistatin, soluble receptors, propeptides) which sequester and deter activation of pro-fibrotic signals. Though encouraging, a major hurdle for clinical transition of these ligand traps is their in vivo stability, tissue-specificity, and minimisation of side effects. Indeed, one of the only drugs approved for TGF-β inhibition in lung fibrosis triggered gastrointestinal upset and the appearance of rashes (Noble et al., 2011) in treated patients, and activin blockade using the sActRII ligand trap caused vasculature complications (Attie et al., 2013; Campbell et al., 2016). Additionally, the importance of TGF-β ligands in non-fibrotic processes requires titrated treatments, so as not to ablate homeostatic functions. The ultimate goal for future TGF-β targeted fibrotic therapies is to identify ligand specific inhibitors with an extended signaling range that can act precisely within fibrotic tissue.
KW collected supporting evidence and wrote manuscript. KJ assisted with reference collection and collating. CH edited and provided guidance for the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer MC and handling Editor declared their shared affiliation, and the handling Editor states that the process met the standards of a fair and objective review.
Grant funding (1078907) from the National Health and Medical Research Council (NHMRC) Australia supported CH. An Early Career Seed Grant from the Victorian Cancer Agency supported KW.
Agapova, O. A., Fang, Y., Sugatani, T., Seifert, M. E., and Hruska, K. A. (2016). Ligand trap for the activin type IIA receptor protects against vascular disease and renal fibrosis in mice with chronic kidney disease. Kidney Int. 89, 1231–1243. doi: 10.1016/j.kint.2016.02.002
Akhurst, R. J., and Hata, A. (2012). Targeting the TGFbeta signalling pathway in disease. Nat. Rev. Drug Discov. 11, 790–811. doi: 10.1038/nrd3810
Alvarez, K., Fadic, R., and Brandan, E. (2002). Augmented synthesis and differential localization of heparan sulfate proteoglycans in Duchenne muscular dystrophy. J. Cell. Biochem. 85, 703–713. doi: 10.1002/jcb.10184
Aoki, F., Kurabayashi, M., Hasegawa, Y., and Kojima, I. (2005). Attenuation of bleomycin-induced pulmonary fibrosis by follistatin. Am. J. Respir. Crit. Care Med. 172, 713–720. doi: 10.1164/rccm.200412-1620OC
Arndt, S., Wacker, E., Dorn, C., Koch, A., Saugspier, M., Thasler, W. E., et al. (2015). Enhanced expression of BMP6 inhibits hepatic fibrosis in non-alcoholic fatty liver disease. Gut 64, 973–981. doi: 10.1136/gutjnl-2014-306968
Attie, K. M., Borgstein, N. G., Yang, Y., Condon, C. H., Wilson, D. M., Pearsall, A. E., et al. (2013). A single ascending-dose study of muscle regulator ace-031 in healthy volunteers. Muscle Nerve 47, 416–423. doi: 10.1002/mus.23539
Azuma, A. (2012). Pirfenidone treatment of idiopathic pulmonary fibrosis. Ther. Adv. Respir. Dis. 6, 107–114. doi: 10.1177/1753465812436663
Bergeron, E., Leblanc, E., Drevelle, O., Giguére, R., Beauvais, S., Grenier, G., et al. (2012). The evaluation of ectopic bone formation induced by delivery systems for bone morphogenetic protein-9 or its derived peptide. Tissue Eng. Part A. 18, 342–352. doi: 10.1089/ten.TEA.2011.0008
Bergeron, E., Senta, H., Mailloux, A., Park, H., Lord, E., and Faucheux, N. (2009). Murine preosteoblast differentiation induced by a peptide derived from bone morphogenetic proteins-9. Tissue Eng. Part A 15, 3341–3349. doi: 10.1089/ten.TEA.2009.0189
Bernasconi, P., Di Blasi, C., Mora, M., Morandi, L., Galbiati, S., Confalonieri, P., et al. (1999). Transforming growth factor-beta1 and fibrosis in congenital muscular dystrophies. Neuromuscul. Disord. 9, 28–33. doi: 10.1016/S0960-8966(98)00093-5
Bi, J., and Ge, S. (2014). Potential roles of BMP9 in liver fibrosis. Int. J. Mol. Sci. 15, 20656–20667. doi: 10.3390/ijms151120656
Biesemann, N., Mendler, L., Kostin, S., Wietelmann, A., Borchardt, T., and Braun, T. (2015). Myostatin induces interstitial fibrosis in the heart via TAK1 and p38. Cell Tissue Res. 361, 779–787. doi: 10.1007/s00441-015-2139-2
Bottinger, E. P., Factor, V. M., Tsang, M. L., Weatherbee, J. A., Kopp, J. B., Qian, S. W., et al. (1996). The recombinant proregion of transforming growth factor beta1 (latency-associated peptide) inhibits active transforming growth factor beta1 in transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 93, 5877–5882. doi: 10.1073/pnas.93.12.5877
Breitkopf-Heinlein, K., Meyer, C., Konig, C., Gaitantzi, H., Addante, A., Thomas, M., et al. (2017). BMP-9 interferes with liver regeneration and promotes liver fibrosis. Gut 66, 939–954. doi: 10.1136/gutjnl-2016-313314
Campbell, C., Mcmillan, H. J., Mah, J. K., Tarnopolsky, M., Selby, K., Mcclure, T., et al. (2016). Myostatin inhibitor ACE-031 treatment of ambulatory boys with Duchenne muscular dystrophy: results of a randomized, placebo-controlled clinical trial. Muscle Nerve 55, 458–464. doi: 10.1002/mus.25268
Chen, J. L., Walton, K. L., Al-Musawi, S. L., Kelly, E. K., Qian, H., La, M., et al. (2015). Development of novel activin-targeted therapeutics. Mol. Ther. 23, 434–444. doi: 10.1038/mt.2014.221
Chen, J. L., Walton, K. L., Hagg, A., Colgan, T. D., Johnson, K., Qian, H., et al. (2017). Specific targeting of TGF-beta family ligands demonstrates distinct roles in the regulation of muscle mass in health and disease. Proc. Natl. Acad. Sci. U.S.A. 26, E5266–E5275. doi: 10.1073/pnas.1620013114
Chen, J. L., Walton, K. L., Winbanks, C. E., Murphy, K. T., Thomson, R. E., Makanji, Y., et al. (2014). Elevated expression of activins promotes muscle wasting and cachexia. FASEB J. 28, 1711–1723. doi: 10.1096/fj.13-245894
Chen, Y., Blom, I. E., Sa, S., Goldschmeding, R., Abraham, D. J., and Leask, A. (2002). CTGF expression in mesangial cells: involvement of SMADs, MAP kinase, and PKC. Kidney Int. 62, 1149–1159. doi: 10.1111/j.1523-1755.2002.kid567.x
Clouthier, D. E., Comerford, S. A., and Hammer, R. E. (1997). Hepatic fibrosis, glomerulosclerosis, and a lipodystrophy-like syndrome in PEPCK-TGF-beta1 transgenic mice. J. Clin. Invest. 100, 2697–2713. doi: 10.1172/JCI119815
Coffey, R. J. Jr., Kost, L. J., Lyons, R. M., Moses, H. L., and Larusso, N. F. (1987). Hepatic processing of transforming growth factor beta in the rat. Uptake, metabolism, and biliary excretion. J. Clin. Invest. 80, 750–757. doi: 10.1172/JCI113130
Crawford, S. E., Stellmach, V., Murphy-Ullrich, J. E., Ribeiro, S. M. F., Lawler, J., Hynes, R. O., et al. (1998). Thrombospondin-1 is a major activator of TGF-β1 in vivo. Cell 93, 1159–1170. doi: 10.1016/S0092-8674(00)81460-9
Dadlani, H., Ballinger, M. L., Osman, N., Getachew, R., and Little, P. J. (2008). Smad and p38 MAP kinase-mediated signaling of proteoglycan synthesis in vascular smooth muscle. J. Biol. Chem. 283, 7844–7852. doi: 10.1074/jbc.M703125200
de Kretser, D. M., Bensley, J. G., Pettila, V., Linko, R., Hedger, M. P., Hayward, S., et al. (2013). Serum activin A and B levels predict outcome in patients with acute respiratory failure: a prospective cohort study. Crit. Care 17:R263. doi: 10.1186/cc13093
Delaney, K., Kasprzycka, P., Ciemerych, M. A., and Zimowska, M. (2017). The role of TGF-beta1 during skeletal muscle regeneration. Cell Biol. Int. 41, 706–715. doi: 10.1002/cbin.10725
Dendooven, A., Van Oostrom, O., Van Der Giezen, D. M., Leeuwis, J. W., Snijckers, C., Joles, J. A., et al. (2011). Loss of endogenous bone morphogenetic protein-6 aggravates renal fibrosis. Am. J. Pathol. 178, 1069–1079. doi: 10.1016/j.ajpath.2010.12.005
Denholm, E. M., and Rollins, S. M. (1993). Expression and secretion of transforming growth factor-beta by bleomycin-stimulated rat alveolar macrophages. Am. J. Physiol. 264, L36–L42.
Dennler, S., Itoh, S., Vivien, D., Ten Dijke, P., Huet, S., and Gauthier, J. M. (1998). Direct binding of Smad3 and Smad4 to critical TGF beta-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. 17, 3091–3100. doi: 10.1093/emboj/17.11.3091
Ebefors, K., Granqvist, A., Ingelsten, M., Molne, J., Haraldsson, B., and Nystrom, J. (2011). Role of glomerular proteoglycans in IgA nephropathy. PLoS ONE 6:e18575. doi: 10.1371/journal.pone.0018575
Fan, J. M., Ng, Y. Y., Hill, P. A., Nikolic-Paterson, D. J., Mu, W., Atkins, R. C., et al. (1999). Transforming growth factor-beta regulates tubular epithelial-myofibroblast transdifferentiation in vitro. Kidney Int. 56, 1455–1467. doi: 10.1046/j.1523-1755.1999.00656.x
Ferguson, M. W., Duncan, J., Bond, J., Bush, J., Durani, P., So, K., et al. (2009). Prophylactic administration of avotermin for improvement of skin scarring: three double-blind, placebo-controlled, phase I/II studies. Lancet 373, 1264–1274. doi: 10.1016/S0140-6736(09)60322-6
Gabbiani, G. (2003). The myofibroblast in wound healing and fibrocontractive diseases. J. Pathol. 200, 500–503. doi: 10.1002/path.1427
Goumenos, D. S., Tsamandas, A. C., Oldroyd, S., Sotsiou, F., Tsakas, S., Petropoulou, C., et al. (2001). Transforming growth factor-beta(1) and myofibroblasts: a potential pathway towards renal scarring in human glomerular disease. Nephron 87, 240–248. doi: 10.1159/000045921
Grygielko, E. T., Martin, W. M., Tweed, C., Thornton, P., Harling, J., Brooks, D. P., et al. (2005). Inhibition of gene markers of fibrosis with a novel inhibitor of transforming growth factor-beta type I receptor kinase in puromycin-induced nephritis. J. Pharmacol. Exp. Ther. 313, 943–951. doi: 10.1124/jpet.104.082099
Hamrick, M. W., Arounleut, P., Kellum, E., Cain, M., Immel, D., and Liang, L. F. (2010). Recombinant myostatin (GDF-8) propeptide enhances the repair and regeneration of both muscle and bone in a model of deep penetrant musculoskeletal injury. J. Trauma 69, 579–583. doi: 10.1097/TA.0b013e3181c451f4
Hardy, C. L., King, S. J., Mifsud, N. A., Hedger, M. P., Phillips, D. J., Mackay, F., et al. (2015). The activin A antagonist follistatin inhibits cystic fibrosis-like lung inflammation and pathology. Immunol. Cell Biol. 93, 567–574. doi: 10.1038/icb.2015.7
Harrison, C. A., Al-Musawi, S. L., and Walton, K. L. (2011). Prodomains regulate the synthesis, extracellular localisation and activity of TGF-beta superfamily ligands. Growth Fact. 29, 174–186. doi: 10.3109/08977194.2011.608666
Hermida, N., Lopez, B., Gonzalez, A., Dotor, J., Lasarte, J. J., Sarobe, P., et al. (2009). A synthetic peptide from transforming growth factor-beta1 type III receptor prevents myocardial fibrosis in spontaneously hypertensive rats. Cardiovasc. Res. 81, 601–609. doi: 10.1093/cvr/cvn315
Hocevar, B. A., Brown, T. L., and Howe, P. H. (1999). TGF-beta induces fibronectin synthesis through a c-Jun N-terminal kinase-dependent, Smad4-independent pathway. EMBO J. 18, 1345–1356. doi: 10.1093/emboj/18.5.1345
Hua, X., Liu, X., Ansari, D. O., and Lodish, H. F. (1998). Synergistic cooperation of TFE3 and smad proteins in TGF-beta-induced transcription of the plasminogen activator inhibitor-1 gene. Genes Dev. 12, 3084–3095. doi: 10.1101/gad.12.19.3084
Ignotz, R. A., and Massague, J. (1986). Transforming growth factor-beta stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J. Biol. Chem. 261, 4337–4345.
Inman, G. J., Nicolas, F. J., Callahan, J. F., Harling, J. D., Gaster, L. M., Reith, A. D., et al. (2002). SB-431542 is a potent and specific inhibitor of transforming growth factor-beta superfamily type I activin receptor-like kinase (ALK) receptors ALK 4, ALK5, and ALK7. Mol. Pharmacol. 62, 65–74. doi: 10.1124/mol.62.1.65
Jiang, D., Liang, J., Campanella, G. S., Guo, R., Yu, S., Xie, T., et al. (2010). Inhibition of pulmonary fibrosis in mice by CXCL10 requires glycosaminoglycan binding and syndecan-4. J. Clin. Invest. 120, 2049–2057. doi: 10.1172/JCI38644
Ketteler, M., Noble, N. A., and Border, W. A. (1994). Increased expression of transforming growth factor-beta in renal disease. Curr. Opin. Nephrol. Hypertens. 3, 446–452. doi: 10.1097/00041552-199407000-00011
Khan, S., Joyce, J., Margulies, K. B., and Tsuda, T. (2014). Enhanced bioactive myocardial transforming growth factor-beta in advanced human heart failure. Circ. J. 78, 2711–2718. doi: 10.1253/circj.CJ-14-0511
Li, J., Qu, X., Yao, J., Caruana, G., Ricardo, S. D., Yamamoto, Y., et al. (2010). Blockade of endothelial-mesenchymal transition by a Smad3 inhibitor delays the early development of streptozotocin-induced diabetic nephropathy. Diabetes Metab. Res. Rev. 59, 2612–2624. doi: 10.2337/db09-1631
Li, S., Shimono, C., Norioka, N., Nakano, I., Okubo, T., Yagi, Y., et al. (2010). Activin A binds to perlecan through its pro-region that has heparin/heparan sulfate binding activity. J. Biol. Chem. 285, 36645–36655. doi: 10.1074/jbc.M110.177865
Li, R. X., Yiu, W. H., and Tang, S. C. (2015). Role of bone morphogenetic protein-7 in renal fibrosis. Front. Physiol. 6:114. doi: 10.3389/fphys.2015.00114
Li, Z. B., Kollias, H. D., and Wagner, K. R. (2008). Myostatin directly regulates skeletal muscle fibrosis. J. Biol. Chem. 283, 19371–19378. doi: 10.1074/jbc.M802585200
Lijnen, P., Petrov, V., Rumilla, K., and Fagard, R. (2003). Transforming growth factor-beta 1 promotes contraction of collagen gel by cardiac fibroblasts through their differentiation into myofibroblasts. Methods Find. Exp. Clin. Pharmacol. 25, 79–86. doi: 10.1358/mf.2003.25.2.723680
Lijnen, P. J., Petrov, V. V., and Fagard, R. H. (2000). Induction of cardiac fibrosis by transforming growth factor-beta1. Mol. Genet. Metab. 71, 418–435. doi: 10.1006/mgme.2000.3032
Llopiz, D., Dotor, J., Casares, N., Bezunartea, J., Diaz-Valdes, N., Ruiz, M., et al. (2009). Peptide inhibitors of transforming growth factor-beta enhance the efficacy of antitumor immunotherapy. Int. J. Cancer 125, 2614–2623. doi: 10.1002/ijc.24656
Lopez-Dee, Z., Pidcock, K., and Gutierrez, L. S. (2011). Thrombospondin-1: multiple paths to inflammation. Mediators Inflamm. 2011:296069. doi: 10.1155/2011/296069
Ludviksson, B. R., and Gunnlaugsdottir, B. (2003). Transforming growth factor-beta as a regulator of site-specific T-cell inflammatory response. Scand. J. Immunol. 58, 129–138. doi: 10.1046/j.1365-3083.2003.01297.x
Lv, Q., Zeng, J., and He, L. (2016). The advancements of heparanase in fibrosis. Int. J. Mol. Epidemiol. Genet. 7, 137–140.
Lyons, R. M., Gentry, L. E., Purchio, A. F., and Moses, H. L. (1990). Mechanism of activation of latent recombinant transforming growth factor beta 1 by plasmin. J. Cell Biol. 110, 1361–1367. doi: 10.1083/jcb.110.4.1361
Macneal, K., and Schwartz, D. A. (2012). The genetic and environmental causes of pulmonary fibrosis. Proc. Am. Thorac. Soc. 9, 120–125. doi: 10.1513/pats.201112-055AW
Margadant, C., and Sonnenberg, A. (2010). Integrin-TGF-beta crosstalk in fibrosis, cancer and wound healing. EMBO Rep. 11, 97–105. doi: 10.1038/embor.2009.276
Masola, V., Zaza, G., Secchi, M. F., Gambaro, G., Lupo, A., and Onisto, M. (2014). Heparanase is a key player in renal fibrosis by regulating TGF-β expression and activity. Biochim. Biophys. Acta 1843, 2122–2128. doi: 10.1016/j.bbamcr.2014.06.005
Mead, A. L., Wong, T. T., Cordeiro, M. F., Anderson, I. K., and Khaw, P. T. (2003). Evaluation of anti-TGF-beta2 antibody as a new postoperative anti-scarring agent in glaucoma surgery. Invest. Ophthalmol. Vis. Sci. 44, 3394–3401. doi: 10.1167/iovs.02-0978
Mendell, J. R., Sahenk, Z., Al-Zaidy, S., Rodino-Klapac, L. R., Lowes, L. P., Alfano, L. N., et al. (2017). Follistatin gene therapy for sporadic inclusion body myositis improves functional outcomes. Mol. Ther. 25, 870–879. doi: 10.1016/j.ymthe.2017.02.015
Mendell, J. R., Sahenk, Z., Malik, V., Gomez, A. M., Flanigan, K. M., Lowes, L. P., et al. (2015). A phase 1/2a follistatin gene therapy trial for becker muscular dystrophy. Mol. Ther. 23, 192–201. doi: 10.1038/mt.2014.200
Meng, X. M., Nikolic-Paterson, D. J., and Lan, H. Y. (2016). TGF-beta: the master regulator of fibrosis. Nat. Rev. Nephrol. 12, 325–338. doi: 10.1038/nrneph.2016.48
Milani, S., Herbst, H., Schuppan, D., Stein, H., and Surrenti, C. (1991). Transforming growth factors beta 1 and beta 2 are differentially expressed in fibrotic liver disease. Am. J. Pathol. 139, 1221–1229.
Miyazawa, K., Shinozaki, M., Hara, T., Furuya, T., and Miyazono, K. (2002). Two major Smad pathways in TGF-beta superfamily signalling. Genes Cells 7, 1191–1204. doi: 10.1046/j.1365-2443.2002.00599.x
Molina-Molina, M., Lario, S., Luburich, P., Ramirez, J., Carrion, M. T., and Xaubet, A. (2006). Quantifying plasma levels of transforming growth factor beta1 in idiopathic pulmonary fibrosis. Arch. Bronconeumol. 42, 380–383. doi: 10.1157/13091645
Morissette, M. R., Stricker, J. C., Rosenberg, M. A., Buranasombati, C., Levitan, E. B., Mittleman, M. A., et al. (2009). Effects of myostatin deletion in aging mice. Aging Cell 8, 573–583. doi: 10.1111/j.1474-9726.2009.00508.x
Morris, D. G., Huang, X., Kaminski, N., Wang, Y., Shapiro, S. D., Dolganov, G., et al. (2003). Loss of integrin alpha(v)beta6-mediated TGF-beta activation causes Mmp12-dependent emphysema. Nature 422, 169–173. doi: 10.1038/nature01413
Munger, J. S., Huang, X., Kawakatsu, H., Griffiths, M. J., Dalton, S. L., Wu, J., et al. (1999). The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell 96, 319–328. doi: 10.1016/S0092-8674(00)80545-0
Munoz-Felix, J. M., Cuesta, C., Perretta-Tejedor, N., Subileau, M., Lopez-Hernandez, F. J., Lopez-Novoa, J. M., et al. (2016). Identification of bone morphogenetic protein 9 (BMP9) as a novel profibrotic factor in vitro. Cell. Signal. 28, 1252–1261. doi: 10.1016/j.cellsig.2016.05.015
Munoz-Felix, J. M., Gonzalez-Nunez, M., Martinez-Salgado, C., and Lopez-Novoa, J. M. (2015). TGF-beta/BMP proteins as therapeutic targets in renal fibrosis. Where have we arrived after 25 years of trials and tribulations? Pharmacol. Ther. 156, 44–58. doi: 10.1016/j.pharmthera.2015.10.003
Murakami, M., Ikeda, T., Saito, T., Ogawa, K., Nishino, Y., Nakaya, K., et al. (2006). Transcriptional regulation of plasminogen activator inhibitor-1 by transforming growth factor-beta, activin A and microphthalmia-associated transcription factor. Cell. Signal. 18, 256–265. doi: 10.1016/j.cellsig.2005.04.010
Nagy, P., Schaff, Z., and Lapis, K. (1991). Immunohistochemical detection of transforming growth factor-beta 1 in fibrotic liver diseases. Hepatology 14, 269–273. doi: 10.1002/hep.1840140211
Nikolic-Paterson, D. J., Main, I. W., Tesch, G. H., Lan, H. Y., and Atkins, R. C. (1996). Interleukin-1 in renal fibrosis. Kidney Int. Suppl. 54, S88–S90.
Noble, P. W., Albera, C., Bradford, W. Z., Costabel, U., Glassberg, M. K., Kardatzke, D., et al. (2011). Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet 377, 1760–1769. doi: 10.1016/S0140-6736(11)60405-4
Occleston, N. L., Laverty, H. G., O’kane, S., and Ferguson, M. W. (2008). Prevention and reduction of scarring in the skin by Transforming Growth Factor beta 3 (TGFbeta3): from laboratory discovery to clinical pharmaceutical. J. Biomater. Sci. Polym. Ed. 19, 1047–1063. doi: 10.1163/156856208784909345
Ohga, E., Matsuse, T., Teramoto, S., Katayama, H., Nagase, T., Fukuchi, Y., et al. (1996). Effects of activin A on proliferation and differentiation of human lung fibroblasts. Biochem. Biophys. Res. Commun. 228, 391–396. doi: 10.1006/bbrc.1996.1672
O’Reilly, S., Ciechomska, M., Cant, R., Hugle, T., and Van Laar, J. M. (2012). Interleukin-6, its role in fibrosing conditions. Cytokine Growth Factor. Rev. 23, 99–107. doi: 10.1016/j.cytogfr.2012.04.003
Ota, F., Maeshima, A., Yamashita, S., Ikeuchi, H., Kaneko, Y., Kuroiwa, T., et al. (2003). Activin A induces cell proliferation of fibroblast-like synoviocytes in rheumatoid arthritis. Arthritis Rheum. 48, 2442–2449. doi: 10.1002/art.11249
Patella, S., Phillips, D. J., Tchongue, J., De Kretser, D. M., and Sievert, W. (2006). Follistatin attenuates early liver fibrosis: effects on hepatic stellate cell activation and hepatocyte apoptosis. Am. J. Physiol. Gastrointest. Liver Physiol. 290, G137–G144.
Petersen, M., Thorikay, M., Deckers, M., Van Dinther, M., Grygielko, E. T., Gellibert, F., et al. (2008). Oral administration of GW788388, an inhibitor of TGF-beta type I and II receptor kinases, decreases renal fibrosis. Kidney Int. 73, 705–715. doi: 10.1038/sj.ki.5002717
Popov, Y., Patsenker, E., Stickel, F., Zaks, J., Bhaskar, K. R., Niedobitek, G., et al. (2008). Integrin alphavbeta6 is a marker of the progression of biliary and portal liver fibrosis and a novel target for antifibrotic therapies. J. Hepatol. 48, 453–464. doi: 10.1016/j.jhep.2007.11.021
Presser, L. D., Mcrae, S., and Waris, G. (2013). Activation of TGF-beta1 promoter by hepatitis C virus-induced AP-1 and Sp1: role of TGF-beta1 in hepatic stellate cell activation and invasion. PLoS ONE 8:e56367. doi: 10.1371/journal.pone.0056367
Qiao, C., Li, J., Jiang, J., Zhu, X., Wang, B., Li, J., et al. (2008). Myostatin propeptide gene delivery by adeno-associated virus serotype 8 vectors enhances muscle growth and ameliorates dystrophic phenotypes in mdx mice. Hum. Gene Ther. 19, 241–254. doi: 10.1089/hum.2007.159
Roberts, A. B., Sporn, M. B., Assoian, R. K., Smith, J. M., Roche, N. S., Wakefield, L. M., et al. (1986). Transforming growth factor type beta: rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proc. Natl. Acad. Sci. U.S.A. 83, 4167–4171. doi: 10.1073/pnas.83.12.4167
Rodgers, B. D., Wiedeback, B. D., Hoversten, K. E., Jackson, M. F., Walker, R. G., and Thompson, T. B. (2014). Myostatin stimulates, not inihibits, C2C12 myoblast proliferation. Endocrinology 155, 670–675. doi: 10.1210/en.2013-2107
Romaris, M., Bassols, A., and David, G. (1995). Effect of transforming growth factor-beta 1 and basic fibroblast growth factor on the expression of cell surface proteoglycans in human lung fibroblasts. Enhanced glycanation and fibronectin-binding of CD44 proteoglycan, and down-regulation of glypican. Biochem. J. 310(Pt 1), 73–81.
Roskams, T., Rosenbaum, J., De Vos, R., David, G., and Desmet, V. (1996). Heparan sulfate proteoglycan expression in chronic cholestatic human liver diseases. Hepatology 24, 524–532. doi: 10.1002/hep.510240310
Santiago, B., Gutierrez-Canas, I., Dotor, J., Palao, G., Lasarte, J. J., Ruiz, J., et al. (2005). Topical application of a peptide inhibitor of transforming growth factor-beta1 ameliorates bleomycin-induced skin fibrosis. J. Invest. Dermatol. 125, 450–455. doi: 10.1111/j.0022-202X.2005.23859.x
Schonherr, E., Jarvelainen, H. T., Sandell, L. J., and Wight, T. N. (1991). Effects of platelet-derived growth factor and transforming growth factor-beta 1 on the synthesis of a large versican-like chondroitin sulfate proteoglycan by arterial smooth muscle cells. J. Biol. Chem. 266, 17640–17647.
Sime, P. J., Xing, Z., Graham, F. L., Csaky, K. G., and Gauldie, J. (1997). Adenovector-mediated gene transfer of active transforming growth factor-beta1 induces prolonged severe fibrosis in rat lung. J. Clin. Invest. 100, 768–776. doi: 10.1172/JCI119590
Sugimoto, H., Lebleu, V. S., Bosukonda, D., Keck, P., Taduri, G., Bechtel, W., et al. (2012). Activin-like kinase 3 is important for kidney regeneration and reversal of fibrosis. Nat. Med. 18, 396–404. doi: 10.1038/nm.2629
Sugiyama, M., Ichida, T., Sato, T., Ishikawa, T., Matsuda, Y., and Asakura, H. (1998). Expression of activin A is increased in cirrhotic and fibrotic rat livers. Gastroenterology 114, 550–558. doi: 10.1016/S0016-5085(98)70539-6
Sharma, M., Kambadur, R., Matthews, K. G., Somers, W. G., Devlinm G. P., Conaglen, J. V., et al. (1999). Myostatin, a transforming growth factor-β superfamily member, is expressed in heart muscle and is upregulated in cardiomyocytes after infarct. J. Cell Physiol. 180, 1–9 doi: 10.1002/(SICI)1097-4652(199907)180:1<1::AID-JCP1>3.0.CO;2-V
Sun, B., Huo, R., Sheng, Y., Li, Y., Xie, X., Chen, C., et al. (2013). Bone morphogenetic protein-4 mediates cardiac hypertrophy, apoptosis, and fibrosis in experimentally pathological cardiac hypertrophy. Hypertension 61, 352–360. doi: 10.1161/HYPERTENSIONAHA.111.00562
Swencki-Underwood, B., Mills, J. K., Vennarini, J., Boakye, K., Luo, J., Pomerantz, S., et al. (2008). Expression and characterization of a human BMP-7 variant with improved biochemical properties. Protein Expr. Purif. 57, 312–319. doi: 10.1016/j.pep.2007.09.016
Taipale, J., Saharinen, J., Hedman, K., and Keski-Oja, J. (1996). Latent transforming growth factor-beta 1 and its binding protein are components of extracellular matrix microfibrils. J. Histochem. Cytochem. 44, 875–889. doi: 10.1177/44.8.8756760
Terada, Y., Hanada, S., Nakao, A., Kuwahara, M., Sasaki, S., and Marumo, F. (2002). Gene transfer of Smad7 using electroporation of adenovirus prevents renal fibrosis in post-obstructed kidney. Kidney Int. 61, S94–S98. doi: 10.1046/j.1523-1755.2002.0610s1094.x
Urbina, P., and Singla, D. K. (2014). BMP-7 attenuates adverse cardiac remodeling mediated through M2 macrophages in prediabetic cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 307, 762–772. doi: 10.1152/ajpheart.00367.2014
Vaughan, M. B., Howard, E. W., and Tomasek, J. J. (2000). Transforming growth factor-beta1 promotes the morphological and functional differentiation of the myofibroblast. Exp. Cell Res. 257, 180–189. doi: 10.1006/excr.2000.4869
Verrecchia, F., Chu, M. L., and Mauviel, A. (2001a). Identification of novel TGF-beta/Smad gene targets in dermal fibroblasts using a combined cDNA microarray/promoter transactivation approach. J. Biol. Chem. 276, 17058–17062.
Verrecchia, F., Vindevoghel, L., Lechleider, R. J., Uitto, J., Roberts, A. B., and Mauviel, A. (2001b). Smad3/AP-1 interactions control transcriptional responses to TGF-beta in a promoter-specific manner. Oncogene 20, 3332–3340.
Voelker, J., Berg, P. H., Sheetz, M., Duffin, K., Shen, T., Moser, B., et al. (2017). Anti-TGF-beta1 antibody therapy in patients with diabetic nephropathy. J. Am. Soc. Nephrol. 28, 953–962. doi: 10.1681/ASN.2015111230
Wahl, S. M. (1992). Transforming growth factor beta (TGF-β) in inflammation: a cause and a cure. J. Clin. Immunol. 12, 61–74.
Wakefield, L. M., Winokur, T. S., Hollands, R. S., Christopherson, K., Levinson, A. D., and Sporn, M. B. (1990). Recombinant latent transforming growth factor beta 1 has a longer plasma half-life in rats than active transforming growth factor beta 1, and a different tissue distribution. J. Clin. Invest. 86, 1976–1984. doi: 10.1172/JCI114932
Walker, R. G., Czepnik, M., Goebel, E. J., Mccoy, J. C., Vujic, A., Cho, M., et al. (2017). Structural basis for potency differences between GDF8 and GDF11. BMC Biol. 15:19. doi: 10.1186/s12915-017-0350-1
Wang, L. P., Dong, J. Z., Xiong, L. J., Shi, K. Q., Zou, Z. L., Zhang, S. N., et al. (2014). BMP-7 attenuates liver fibrosis via regulation of epidermal growth factor receptor. Int. J. Clin. Exp. Pathol. 7, 3537–3547.
Wang, S., and Hirschberg, R. (2003). BMP7 antagonizes TGF-beta -dependent fibrogenesis in mesangial cells. Am. J. Physiol. Renal Physiol. 284, F1006–F1013.
Weiskirchen, R., and Meurer, S. K. (2013). BMP-7 counteracting TGF-beta1 activities in organ fibrosis. Front. Biosci. 18:1407–1434. doi: 10.2741/4189
Weiskirchen, R., Meurer, S. K., Gressner, O. A., Herrmann, J., Borkham-Kamphorst, E., and Gressner, A. M. (2009). BMP-7 as antagonist of organ fibrosis. Front. Biosci. 14, 4992–5012. doi: 10.2741/3583
Westergren-Thorsson, G., Hedstrom, U., Nybom, A., Tykesson, E., Ahrman, E., Hornfelt, M., et al. (2017). Increased deposition of glycosaminoglycans and altered structure of heparan sulfate in idiopathic pulmonary fibrosis. Int. J. Biochem. Cell Biol. 83, 27–38. doi: 10.1016/j.biocel.2016.12.005
Wipff, P. J., Rifkin, D. B., Meister, J. J., and Hinz, B. (2007). Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J. Cell Biol. 179, 1311–1323. doi: 10.1083/jcb.200704042
Wohl, A. P., Troilo, H., Collins, R. F., Baldock, C., and Sengle, G. (2016). Extracellular regulation of bone morphogenetic protein activity by the microfibril component fibrillin-1. J. Biol. Chem. 291, 12732–12746. doi: 10.1074/jbc.M115.704734
Wordinger, R. J., Sharma, T., and Clark, A. F. (2014). The role of TGF-beta2 and bone morphogenetic proteins in the trabecular meshwork and glaucoma. J. Ocul. Pharmacol. Ther. 30, 154–162. doi: 10.1089/jop.2013.0220
Yamada, M., Kuwano, K., Maeyama, T., Yoshimi, M., Hamada, N., Fukumoto, J., et al. (2007). Gene transfer of soluble transforming growth factor type II receptor by in vivo electroporation attenuates lung injury and fibrosis. J. Clin. Pathol. 60, 916–920. doi: 10.1136/jcp.2006.039396
Yamashita, S., Maeshima, A., Kojima, I., and Nojima, Y. (2004). Activin A is a potent activator of renal interstitial fibroblasts. J. Am. Soc. Nephrol. 15, 91–101. doi: 10.1097/01.ASN.0000103225.68136.E6
Yang, G., Zhu, Z., Wang, Y., Gao, A., Niu, P., and Tian, L. (2013). Bone morphogenetic protein-7 inhibits silica-induced pulmonary fibrosis in rats. Toxicol. Lett. 220, 103–108. doi: 10.1016/j.toxlet.2013.04.017
Yang, Y. L., Liu, Y. S., Chuang, L. Y., Guh, J. Y., Lee, T. C., Liao, T. N., et al. (2009). Bone morphogenetic protein-2 antagonizes renal interstitial fibrosis by promoting catabolism of type I transforming growth factor-beta receptors. Endocrinology 150, 727–740. doi: 10.1210/en.2008-0090
Yndestad, A., Ueland, T., Oie, E., Florholmen, G., Halvorsen, B., Attramadal, H., et al. (2004). Elevated levels of activin A in heart failure: potential role in myocardial remodeling. Circulation 109, 1379–1385. doi: 10.1161/01.CIR.0000120704.97934.41
Yuan, W., and Varga, J. (2001). Transforming growth factor-beta repression of matrix metalloproteinase-1 in dermal fibroblasts involves Smad3. J. Biol. Chem. 276, 38502–38510. doi: 10.1074/jbc.M107081200
Zeisberg, M., Hanai, J., Sugimoto, H., Mammoto, T., Charytan, D., Strutz, F., et al. (2003). BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat. Med. 9, 964–968. doi: 10.1038/nm888
Zhang, L. P., Takahara, T., Yata, Y., Furui, K., Jin, B., Kawada, N., et al. (1999). Increased expression of plasminogen activator and plasminogen activator inhibitor during liver fibrogenesis of rats: role of stellate cells. J. Hepatol. 31, 703–711. doi: 10.1016/S0168-8278(99)80351-1
Zhang, Y., Liu, P., Gao, X., Qian, W., and Xu, K. (2010). rAAV2-TGF-beta(3) decreases collagen synthesis and deposition in the liver of experimental hepatic fibrosis rat. Dig. Dis. Sci. 55, 2821–2830. doi: 10.1007/s10620-009-1119-3
Keywords: fibrosis, TGF-β, activin, muscle, skeletal, myostatin, propeptide
Citation: Walton KL, Johnson KE and Harrison CA (2017) Targeting TGF-β Mediated SMAD Signaling for the Prevention of Fibrosis. Front. Pharmacol. 8:461. doi: 10.3389/fphar.2017.00461
Received: 28 April 2017; Accepted: 27 June 2017;
Published: 14 July 2017.
Edited by:
Tim David Hewitson, Royal Melbourne Hospital, AustraliaReviewed by:
Michael Cai, Royal Melbourne Hospital, AustraliaCopyright © 2017 Walton, Johnson and Harrison. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Craig A. Harrison, Y3JhaWcuaGFycmlzb25AbW9uYXNoLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.