 Ping Li
Ping Li Xin Chen
Xin Chen- State Key Laboratory of Quality Research in Chinese Medicine, Institute of Chinese Medical Sciences, University of Macau, Macau, China
Although initially described as an anti-tumor mediator, tumor necrosis factor-alpha (TNF) is generally considered as the master pro-inflammatory cytokine. It plays a crucial role in the pathogenesis of inflammatory diseases, such as rheumatoid arthritis (RA), inflammatory bowel disease, ankylosing spondylitis (AS), and psoriasis. Consequently, anti-TNF therapy has become mainstay treatment for autoimmune diseases. Historically, anti-inflammatory agents were developed before the identification of TNF. Salicylates, the active components of Willow spp., were identified in the mid-19th century for the alleviation of pain, fever, and inflammatory responses. Study of this naturally occurring compound led to the discovery of aspirin, which was followed by the development of non-steroidal anti-inflammatory drugs (NSAIDs) due to the chemical advances in the 19th–20th centuries. Initially, the most of NSAIDs were organic acid, but the non-acidic compounds were also identified as NSAIDs. Although effective in the treatment of inflammatory diseases, NSAIDs have some undesirable and adverse effect, such as ulcers, kidney injury, and bleeding in the gastrointestinal tract. In the past two decades, anti-TNF biologics were developed. Drugs belong to this class include soluble TNF receptor 2 fusion protein and anti-TNF antibodies. The introduction of anti-TNF therapeutics has revolutionized the management of autoimmune diseases, such as RA, psoriatic arthritis (PsA), plaque psoriasis (PP), AS, CD and ulcerative colitis (UC). Nevertheless, up to 40% of patients have no response to anti-TNF treatment. Furthermore, this treatment is associated with some adverse effects such as increased risk of infection, and even triggered the de novo development of autoimmune diseases. Such harmful effect of anti-TNF treatment is likely caused by the global inhibition of TNF biological functions. Therefore, specific inhibition of TNF receptor (TNFR1 or TNFR2) may represent a safer and more effective treatment, as proposed by some recent studies. In this review article, the historical development of anti-inflammatory drugs after World War II as briefly described above will be reviewed and analyzed. The future trend in the development of novel TNF receptor-targeting therapeutics will be discussed in the context of latest progress in the research of TNF biology.
Introduction
Autoimmune inflammatory diseases affect approximately 7.6–9.4% of the world population, especially among the young and middle-aged women (Cooper et al., 2009; Bragazzi et al., 2016). Frequently accompanied by severe and chronic morbidity, autoimmune diseases are also leading causes of death all around world. The patients with autoimmune inflammatory diseases need intensive medical intervention, which imposes the huge burden on public health service and economy (Bragazzi et al., 2016). Excessive and prolonged activation of immune cells, such as T and B lymphocytes, and overexpression of the master pro-inflammatory cytokine tumor necrosis factor alpha (TNF), together with other mediators such as interlukin-6 (IL-6), interlukin-1 (IL-1), and interferon gamma (IFN-γ), play a central role in the pathogenesis of autoimmune inflammatory responses in rheumatoid arthritis (RA), inflammatory bowel disease (IBD), Crohn’s disease (CD), and ankylosing spondylitis (AS) (Moudgil and Choubey, 2011; Sticherling, 2016; Ellis and Braley-Mullen, 2017).
Non-steroidal anti-inflammatory drugs (NSAIDs), glucocorticoids, disease-modifying antirheumatic drugs (DMARDs) are traditionally used in the treatment of autoimmune inflammatory diseases. NSAIDs and glucocorticoids are effective in the alleviation of pain and inhibition of inflammation, while DMARDs have the capacity of reducing tissue and organ damage caused by inflammatory responses (Tabas and Glass, 2013). More recently, treatment for RA and other autoimmune diseases has been revolutionized with the discovery that TNF is critically important in the development of the diseases (Monaco et al., 2015). Anti-TNF biologics (such as infliximab, adalimumab, etanercept, golimumab, and certolizumab pepol) have markedly improved the outcome of the management of autoimmune inflammatory diseases (Meier et al., 2013). However, a considerable proportion of patients do not respond to anti-TNF treatment (Roda et al., 2016). Moreover, anti-TNF biologics are expensive, and are associated with some adverse effects. Recent studies indicate that specifically targeting of one of TNF receptors may represent a more effective and safer treatment for autoimmune disorders. In this review, the history of development of anti-inflammatory drugs, from small molecules to anti-TNF antibodies, will be discussed.
Non-Steroidal Anti-Inflammatory Drugs (NSAIDs)
The Historical Development of NSAIDs
Plant parts such as leaves of myrtle (Myrtus), bark of willow tree (Salix), bark of poplar (Populus), and meadow sweet (Spirea) were used in fork medicine for centuries in the treatment of fever, pain, and inflammatory responses (Hedner and Everts, 1998). These medicinal plants represented a primitive form of anti-inflammatory drugs. In the mid-19th century, salicylates were identified as active components of Willow spp. responsible for the anti-inflammatory activity, which laid the foundation for the mass synthesis of acetylsalicylic acid in 1899 (Vainio and Morgan, 1997; Vane, 2000). The progress in chemistry in the 19th–20th centuries promoted the fast development of NSAIDs. Initially, most of NSAIDs were the organic acid, but the non-acidic compounds were also discovered later. With outstanding safety profiles at dose ranges, ibuprofen was the first NSAIDs approved in the United Kingdom (Busson, 1986). After that, pharmaceutical companies began to develop NSAIDs with a series of chemical and biological properties (Rainsford, 2007). Overall, Post-World War II, the development of NSAIDs had experienced two periods: one was the pre-prostaglandin period (∼1970s) and another one was from 1970s to the end of the last century in which drugs were screened and evaluated partially based on their effect on the production of prostaglandin (Rainsford, 2007).
The Discovery of NSAIDs
Salicylic acid was synthesized by the Gerland in 1853 for the first time, and acetylsalicylic acid was synthesized by Charles Gerhardt in 1853 (Gerhardt, 1853; Gerland, 1853). Until 1876, salicylic acid was firstly used in clinic for the treatment of rheumatic disorders by two German physicians, Drs Streicher and Reiss (Hedner and Everts, 1998). Acetylsalicylic acid was re-discovered by Hoffman in 1897 (Hoffmann and Förster, 1987), and it became available worldwide in the treatment of rheumatic disorders and pain since then (Hedner and Everts, 1998). Acetyl-salicylate was first used as Aspirin in 1899 (Vainio and Morgan, 1997). The development of aspirin, a prototype of NSAIDs, was a landmark (Vainio and Morgan, 1997), which was followed by the development of phenylbutazone (1946) and indomethacin (1963) (Shen, 1982). The term of ‘non-steroidal anti-inflammatory drug’ was used for the first time when phenylbutazone was introduced 3 years later as an anti-inflammatory agent. Thus, aspirin, phenylbutazone, and indomethacin were founding members of NSAID family.
The Categories of NSAIDs
Non-steroidal anti-inflammatory drugs have the analgesic, antipyretic, and anti-inflammatory effect, frequently used for the treatment of conditions like arthritis and headaches (Rainsford, 2007). NSAIDs relieve pain through blocking cyclooxygenase (COX) enzymes (Simmons et al., 2004). COX promotes the production of prostaglandins, a mediator which causes inflammation and pain (Simmons et al., 2004). Although NSAIDs have different chemical structures, all of them have the similar therapeutic effect, e.g., inhibition of autoimmune inflammatory responses (Rainsford, 2007). In general, NSAIDs can be divided into two broad categories: traditional non-selective NSAIDs and selective cyclooxygenase-2 (COX-2) inhibitors (Antman et al., 2007).
Traditional Non-selective NSAIDs
Based on the chemical structure, the traditional non-selective NSAIDs can be classified into different sub-types (Antman et al., 2007): (1) salicylic acid derivatives: acetylsalicylic acid (aspirin), diflunisal and sulfasalazine; (2) para-aminophenol derivatives: acetaminophen; (3) fenamates: mefenamic acid, meclofenamate, flufenamic acid; (4) propionic acid derivatives: ibuprofen, naproxen, fenoprofen, ketoprofen, flurbiprofen, oxaprozin; and (5) enolic acid (oxicam) derivatives: piroxicam, tenoxicam. Most of these drugs were discovered in the pre-prostaglandins period and were developed in the 1960s. Their antipyretic, analgesic, and anti-inflammatory properties were discovered by animal studies, based on some biochemical experimental systems (Rainsford, 2007).
Selective COX-2 Inhibitors
The anti-inflammatory effect of NSAIDs is mainly based on the inhibition of activity of cyclooxygenase (COX) enzymes (Simmons et al., 2004). COX enzymes have two forms, e.g., COX-1 and COX-2. The COX-1 is constitutively expressed by the most of tissues and is responsible for the formation of prostaglandins and thromboxane A2, while the expression of COX-2 needs to be induced by inflammatory mediators (Antman et al., 2007). COX-1 plays an important role in some important physiological processes (Crofford, 1997). Therefore, complete inhibition of both COX-1 and COX-2 inevitably results in severe side effects (Cheng and Visco, 2012).
Inhibition of COX-1 by traditional non-selective NSAIDs causes various gastrointestinal toxicities. In order to reduce this harmful side effect, the selective COX-2 inhibitors were developed (Grosser et al., 2006). The selective COX-2 inhibitors can be further divided into two categories: selective COX-2 inhibitors and highly selective COX-2 inhibitors. The selective COX-2 inhibitors include meloxicam, salicylate, and nimesulide. These drugs more selectively inhibit COX-2, as compared to their action on COX-1. The highly selective COX-2 inhibitors characterized by replacing cis-stilbene with one of the pendant phenyl rings by different substitutes, termed as diarylheterocycles (Khanapure et al., 2003), including celecoxib, rofecoxib, valdecoxib, lumiracoxib, parecoxib, and etoricoxib (Antman et al., 2007). These coxibs have the same structure of diarylheterocycles which is decisively important for their highly potent inhibitory effect on COX-2 (Rainsford, 2007).
The Side Effects of NSAIDs
The traditional non-selective NSAIDs are able to inhibit both COX-1 and COX-2. These drugs also inhibit platelet aggregation and cause significant gastrointestinal disorders such as bleeding, ulcers, and perforation (Fujita et al., 2013). They also induce renal toxicity (Murray and Brater, 1993; Fujita et al., 2013). In contrast, the gastrointestinal adverse effect of selective COX-2 inhibitors is markedly reduced (Dajani and Islam, 2008). However, just like traditional non-selective NSAIDs, selective COX-2 inhibitor still have adverse effects on the cardiovascular system, including congestive heart failure, acute myocardial fraction, and even sudden death (Ray et al., 2002; Hermann and Ruschitzka, 2006).
Glucocorticoids
The glucocorticoids were discovered by Hench and Kendal in 1940s (Hench et al., 1949), through an observation that cortisone had a significant beneficial effect for patients with severe RA (Glyn, 1998; Ferreira et al., 2016). The synthetic development and commercial efforts on glucocorticoids (or corticosteroids) were made in 1950–1980s, and consequently, a number of drugs were yielded (Neeck, 2002). Since then, the synthetic glucocorticoids are extensively used in the treatment of RA, asthma and other inflammatory diseases (Rhen and Cidlowski, 2005). These drugs include prednisone/prednisolone, methylprednisolone, and the fluorinated glucocorticoids such as dexamethasone and betamethasone, which are more frequently used (Buttgereit et al., 2011). Prednisone is an inactive pro-drug and its active metabolite is prednisolone (Frey, 1987). Usually, the prednisolone is administered once a day, while the duration of action of other glucocorticoids is longer when administered into patients with rheumatic diseases (Laev and Salakhutdinov, 2015). The binding of glucocorticoids to its specific receptor results in the inhibition of cellular signaling pathway such as AP-1 and NF-κB, and consequently regulate the expression of cytokines and chemokines (Barnes, 1998). We for the first time found that immunosuppressive CD4+Foxp3+ regulatory T cells (Tregs) were relatively more resistant to glucocorticoid-induced cell death (Chen et al., 2004). Furthermore, glucocorticoid selectively inhibited the proliferation of effector T cells (Teff) while promoting Treg proliferation induced by IL-2 (Chen et al., 2006). This property of glucocorticoid was harnessed to induce immune tolerance which may represent a novel approach in the treatment of autoimmune diseases (Kang et al., 2008).
Although the glucocorticoids are highly effective in the treatment of chronic diseases, they also induce severe side effects, including gastrointestinal ulcers and bleeding, infection, immunosuppression, and bone damage (Ethgen et al., 2013). Currently, low-doses of glucocorticoids are usually used in the treatment of autoimmune inflammatory diseases, while the adverse effects remain a major concern for prolonged usage (Curtis et al., 2006; Rainsford, 2007).
Conventional Disease-Modifying Anti-Rheumatic Drugs (cDMARDs)
To relieve pain is a major goal in the management of RA and other arthritis. NSAIDs are able to alleviate pain and improve joint function through inhibition of inflammatory responses (Rainsford, 2007). However, NSAIDs alone do not reverse the pathological course of the autoimmune diseases (Tabas and Glass, 2013). Glucocorticoids can partially reduce pain symptom and suppress inflammation in the short-term (Laev and Salakhutdinov, 2015). Therefore, drugs that are capable of sustainably reducing inflammation with manageable side effects are highly desirable for the treatment of RA and other autoimmune diseases (Laev and Salakhutdinov, 2015). For this purpose, several drugs with different chemical structures, known as “disease-modifying anti-rheumatic drugs” (DMARDs), were developed.
Methotrexate
Methotrexate is an analog and antagonist of folic acid, which was synthesized firstly in 1947 (Swierkot and Szechinski, 2006). Methotrexate was firstly reported to be effective in the treatment of RA in 1962 (Kersley, 1968), and after that it was also widely used as an anti-inflammatory drug for the management of other autoimmune diseases, including psoriasis, CD, and UC (Nathan et al., 2008; Yelamos and Puig, 2015). Methotrexate has been used as the first-line DMARDs since it was developed, due to its long-term effectiveness, satisfying responsive rate, acceptable toxicity profile, and low cost (Visser and van der Heijde, 2009; Favalli et al., 2014). Fundamental mechanisms underlying the therapeutic effect of methotrexate have been elucidated. For example, methotrexate was found to competitively inhibit the activity of folate-dependent enzymes, and synthesis of purine and pyrimidine which are required for DNA and RNA synthesis, and consequently suppress lymphocyte proliferation and production of pro-inflammatory cytokines (Meier et al., 2013). Nevertheless, the methotrexate monotherapy is no longer considered as the gold standard for RA treatment, for the reason that DMARDs combination therapy is more effective (Kremer et al., 2004).
Leflunomide
Leflunomide was approved for the treatment of RA in the 1998 (Gabriel et al., 2001). Leflunomide is an isoxazole derivative that is capable of blocking the rate-limiting enzyme dihydroorotate dehydrogenase, thereby selectively inhibits the synthesis of de novo pyrimidine ribonucleotides such as rUMP (Fox et al., 1999). Leflunomide is a pro-drug that is metabolized into teriflunomide in vivo (Laev and Salakhutdinov, 2015). Teriflunomide is a potent inhibitor of NF-κB activation, and consequently suppresses the production of pro-inflammatory cytokines. Leflunomide also reduces the production of metalloproteinases in synovial tissue and prevents joint destruction (Laev and Salakhutdinov, 2015). The results of meta-analyses strongly suggest that leflunomide is a DMARDs and effective in the treatment of RA (Osiri et al., 2003). As shown by several studies, the therapeutic effect of leflunomide is comparable to that of methotrexate (Silverman et al., 2005). Leflunomide can improve all clinical outcomes and delay radiographic progression at 6 and 12 months of RA patients (Sharp et al., 2000). Therefore, leflunomide should be considered as first-line therapy if contraindications to methotrexate are present. Furthermore, leflunomide is very useful in the combination therapy (Kalden et al., 2005).
Other DMARDs
Except for methotrexate and leflunomide, other DMARDs, include gold compounds, sulfasalazine, azathioprine, cyclophosphamide, antimalarials, D-penicillamine, cyclosporine, are all effective in the treatment of autoimmune inflammatory diseases such as RA (Gabriel et al., 2001). The DMARDs commonly used for the treatment of RA include hydroxychloroquine, sulfasalazine, methotrexate and leflunomide, while D-penicillamine, azathioprine, gold compounds, and cyclosporine are used less frequently (Kim et al., 2013).
Parenteral organic gold compounds have been used for RA treatment since 1920s (Simon, 2004), and their effect is comparable with other DMARDs (Meier et al., 2013). Nonetheless, the toxicity of gold compounds, especially the adverse effect on skin and mucosa, is a major concern which limits their clinical application (Bendix and Bjelle, 1996).
Sulfasalazine was initially investigated for the treatment of IBD, including UC and CD (Box and Pullar, 1997). This drug was firstly reanimated in Europe where it has been used in early RA patients. The exact mechanism of sulfasalazine is still unclear, although it inhibits the production of pro-inflammatory cytokines, including TNF and IL-6 (Wahl et al., 1998). The main side effect of sulfasalazine is gastrointestinal upset or rash (Skosey, 1988).
Antimalarial drugs hydroxychloroquine and chloroquine have the capacity to inhibit the clinical progress of several autoimmune diseases, including RA and systemic lupus erythematosus (SLE) (Taherian et al., 2013). Although the exact molecular mechanism of its immunosuppressive effect remains to be fully understood, both drugs can inhibit the activation of T cells, B cells (Taherian et al., 2013) and the production of pro-inflammatory cytokines such as TNF, IL-6, and IL-1β (Jang et al., 2006). Both drugs almost have the same side effects, such as rare irreversible retinopathy and a more rarely form of myopathy (Costedoat-Chalumeau et al., 2015).
Combination Therapy with cDMARDs
Among cDMARDs drug class, the methotrexate is still an ‘anchor drug’ for RA therapy, and the first choice for the most of DMARD-naïve patients (Favalli et al., 2014). However, a considerable proportion of patients do not respond to methotrexate. Either anti-TNF biologics (which will be discussed later) or a combination therapy of cDMARDs should be considered for those patients (van Vollenhoven et al., 2012). Methotrexate has been used in combination with other drugs, including sulfasalazine, sulfasalazine and hydroxychloroquine, cyclosporine and biologic agents such as etanercept and infliximab (Kremer et al., 2004; Swierkot and Szechinski, 2006). For example, the concomitant treatment of methotrexate and leflunomide results in a synergistic effect in the treatment of patients with RA (Boers et al., 1997; Kremer et al., 2004). The triple combination of methotrexate, sulfasalazine and hydroxychloroquine represent an effective therapy regimen since 1996 (Meier et al., 2013), which is more effective and safer than the monotherapy with methotrexate, or methotrexate combination of sulfasalazine, or hydroxychloroquine (ODell et al., 1996, 2002). It has been reported that the combination of methotrexate and cyclosporine, or leflunomide and cyclosporine, are more effective than each drug alone (Karanikolas et al., 2006). However, the toxicity of cyclosporine, including reversible and irreversible renal disease, hypertension and heurism, remains an important issue in the combination therapy (Gabriel et al., 2001). Therefore, the ideal cDMARDs combination therapy should be one that is synergistic in their efficacy while lacking additive effects of toxicity.
Anti-TNF Biologics
In 1975, Lloyd and colleagues discovered an endotoxin-inducible serum factor named TNF had the capacity to cause necrosis of tumors (Carswell et al., 1975). It is one of subsequent studies of “Coley’s toxin,” a therapeutic regimen using killed bacteria to induce tumor regression in cancer patients, invented by Dr. William Coley in 1891 (Coley, 1891). Although initially identified as anti-tumor molecule, TNF is now considered as a pleiotropic cytokine which plays a major role in immune or inflammatory responses (Palladino et al., 2003; Efimov et al., 2009).
Feldman’s group showed that a number of pro-inflammatory cytokines such as IL-1, TNF, IL-6, GM-CSF, and IFN-γ were expressed at a high levels in synovium samples from active RA patients (Feldmann et al., 1996). Intriguingly, anti-TNF antibody inhibited the expression of other pro-inflammatory cytokines (IL-1, IL-6, and GM-CSF) in the synovial culture (Feldmann et al., 1996). Furthermore, anti-TNF antibodies such as infliximab and adalimumab suppressed antigen-induced IFN-γ production in blood (Wallis, 2007). These results provide the evidence that TNF plays a predominant role in the pathogenesis of autoimmune inflammatory diseases. IFN-γ is also an important mediator in the inflammatory responses, such as Schwartzman reactions (Billiau, 1988) and collagen-induced arthritis (Mauritz et al., 1988). It was also reported that IFN-γ promoted the production of TNF induced by LPS stimulation (Billiau, 1988). However, unlike the effect of anti-TNF biologics, treatment with antibody against IFN-γ did not markedly ameliorate the arthritic symptoms in RA patients (Sigidin et al., 2001; Schurgers et al., 2011).
TNF is now generally considered as a master pro-inflammatory cytokine that plays a critical role in the pathogenesis of autoimmune inflammatory diseases (Moudgil and Choubey, 2011). Consequently, anti-TNF biologics, which are designed to block the biological function of TNF, have been developed for the therapy of autoimmune inflammatory diseases (Meier et al., 2013). In the past two decades, five TNF-targeting drugs have been approved for clinical use, including infliximab, etanercept, adalimumab, golimumab, and certolizumab pegol, for the treatment of the autoimmune inflammatory diseases including RA, CD and AS (Monaco et al., 2015).
Infliximab
Infliximab is a recombinant chimeric antibody that is generated by mouse myeloma cells. The structure of infliximab contains a constant sequence of human IgG1 and variable regions of murine, which is specific for all forms of TNF in human and chimpanzees, and effectively blocks the binding of TNF to its soluble and membrane receptors (Monaco et al., 2015). The maximum serum concentration of infliximab can be reached within 1 h by intravenous administration (Atzeni et al., 2005), and the half-life is approximately 8–10 days (Chatzantoni and Mouzaki, 2006). After that, infliximab can be maintained by dosing every 8 weeks. Infliximab is capable of mediating both complement-dependent and antibody-dependent cytotoxicity on a cell line overexpressing TNF (Tracey et al., 2008). Infliximab is initially approved for the treatment of Crohn’s disease by the US Food and Drug Administration (FDA) and later also for RA. It was further approved for the treatment of AS, PsA, UC, CD, and chronic plaque psoriasis (Atzeni et al., 2005).
Etanercept
Etanercept comprises soluble TNF receptor 2 and Fc portion of IgG1, which is a human recombinant protein (Kerensky et al., 2012). After subcutaneous administration, etanercept has a half-life of 3–3.5 days (Chatzantoni and Mouzaki, 2006). Etanercept binds to and inactivates soluble form and membrane form of TNF and lymphotoxin, by blocking their interaction with the receptors (Tracey et al., 2008). Similar as infliximab, etanercept alleviate the signs and symptoms of arthritis and inhibits progress of RA in patients (Moreland et al., 1999). Furthermore, it is more effective when combined with methotrexate (Emery et al., 2008). Etanercept is also approved by FDA for the treatment of juvenile rheumatoid arthritis and PsA, RA, PP, and AS (Kerensky et al., 2012).
Adalimumab
Adalimumab is a fully humanized IgG1 monoclonal antibody, which can specific block human TNF binding to its receptors (Atzeni et al., 2005). Adalimumab needs a less frequent subcutaneous injection because of its relative longer half-life which is about 2 weeks (Simon, 2004). Furthermore, adalimumab shows the lower immunogenicity than infliximab (Rau, 2002).
Adalimumab is effective in alleviating the signs and symptoms of moderate to severe RA patients (van de Putte et al., 2004). Adalimumab is also effective in the treatment of patients with CD, with good tolerance and minimal immunogenicity, thus can be used in patients with allergic reactions to infliximab (Sandborn et al., 2004). Adalimumab has been approved by FDA for the treatment of autoimmune diseases including RA, PsA, AS, and CD (Lapadula et al., 2014).
Golimumab
Golimumab is also a fully humanized IgG1 monoclonal antibody, which has a high affinity and specificity for human TNF (Shealy et al., 2007). As shown by the preclinical data, golimumab has higher affinity and is more effective in the neutralization of soluble and trans-membrane forms of TNF, as compared with infliximab and adalimumab, thus it can potently neutralize TNF biological activity (Shealy et al., 2010). The half-life of golimumab is about 7–20 days (Zhou et al., 2007). Golimumab was approved by the FDA in 2009 for the treatment of moderate-to-severe RA when administrated in combination with methotrexate (Mazumdar and Greenwald, 2009). It was also approved for UC treatment by FDA and European Medicines Agency (EMA) in 2013 (Lowenberg and D’Haens, 2013).
Certolizumab Pegol
Certolizumab pegol (CDP870) is a humanized monoclonal antibody with the structure of polyethylene glycolated Fab fragment, which is a novel TNF inhibitor with a distinct mechanism of action compared with other TNF inhibitors (Pasut, 2014). The unique structure might be an explanation for certolizumab pegol with higher efficiency in comparison with other TNF inhibitors (Desai et al., 2012). PEGylation of certolizumab pegol improves its half-life to 2 weeks which may contribute to its high concentration in the inflamed tissues (Keystone et al., 2008).
Certolizumab pegol monotherapy could effectively control the symptoms of patients with RA (Choy et al., 2002). It is also effective the treatment of patients with moderate to severe CD (Schreiber et al., 2005). Certolizumab pegol has been approved for the treatment of CD and PsA by FDA (Deeks, 2016).
Biosimilars
Biosimilar has been defined as a biological medicinal product that has the similar quality, safety, and efficacy with the already approved biological medicine (Fiorino and Danese, 2014). Anti-TNF biosimilars that are extensively developed in recent years will help reduce the cost of anti-TNF treatment. To date, a number of anti-TNF biosimilar products (summarized in Table 1) have been marketed (Dorner and Kay, 2015). CT-13 is the first biosimilar monoclonal antibody against TNF. Comparing with the original TNF inhibitor infliximab, CT-13 is equally effective and safe (Yoo et al., 2013). It is approved in European for the treatment of RA, AS, PsA, PS, CD, and UC (Dorner et al., 2016).
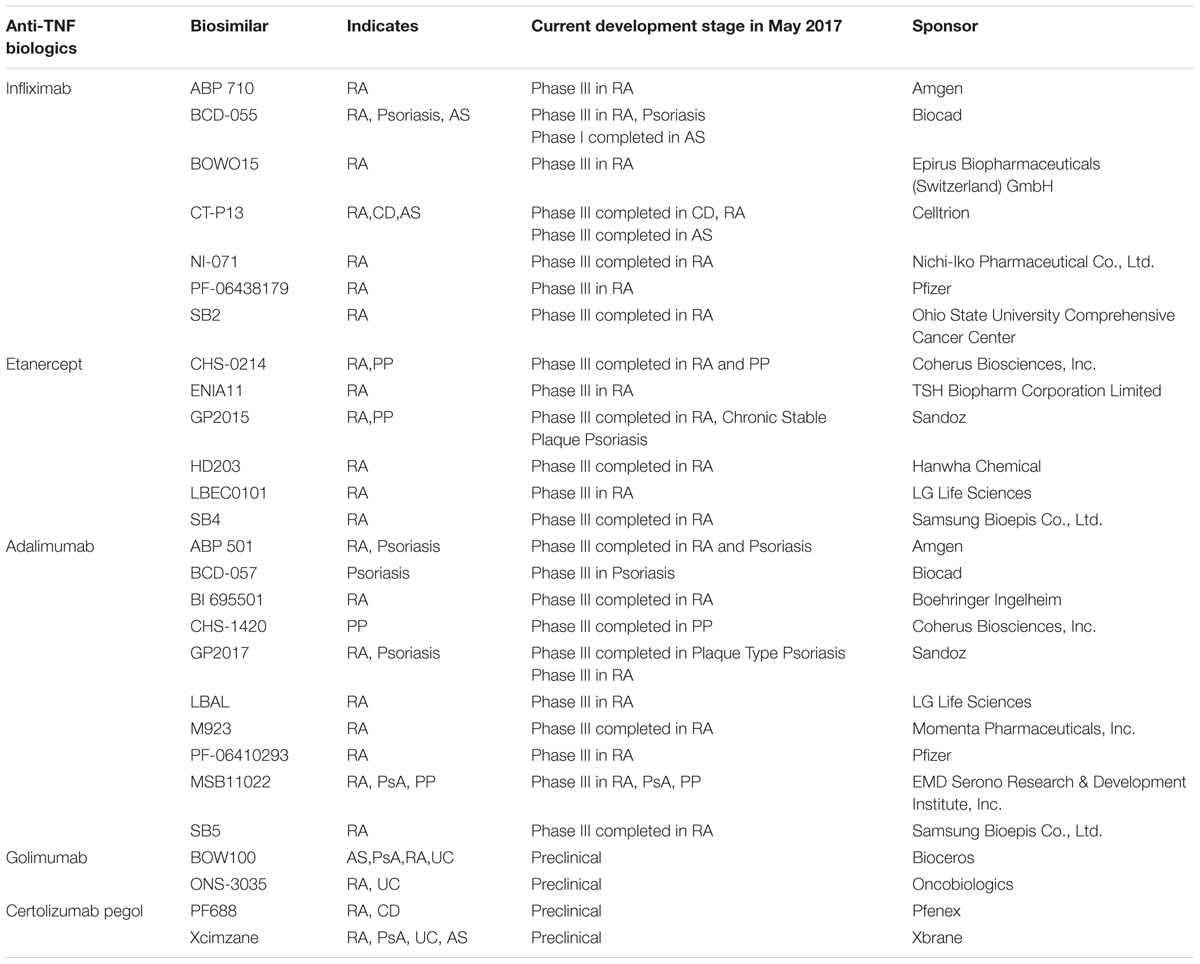
TABLE 1. Anti-TNF biosimilars in the development for the treatment of autoimmune inflammatory diseases.
The Side Effects of Anti-TNF Biologics
Although anti-TNF biologics are generally effective in the treatment of patients with different autoimmune inflammatory diseases (Willrich et al., 2015), not all patients respond equally well to the treatment. Up to 40% of patients have no response to anti-TNF treatment (Roda et al., 2016). Furthermore, several types of adverse effects have been associated with anti-TNF biologics.
It was reported recently that PBMCs isolated from patients responding to the treatment of adalimumab and etanercept can produce higher levels of TNF and soluble TNFR2 (sTNFR2) than those from patients responding to infliximab, and PBMCs isolated from patients who do not respond to infliximab produce higher levels of TNF and sTNFR2 than those from patients responding to infliximab (Gibellini et al., 2016). Therefore, anti-TNF biologics may have major and different effects on TNF expression and soluble TNFR2 levels in patients. It was reported that the TNF gene polymorphism (TNF-alpha 308G > A) is association with patients who do not respond to anti-TNF therapy (Wijnen et al., 2014). Furthermore, the polymorphism of tumor necrosis factor receptor superfamily member 1B, a gene encoding TNFR2 protein, is able to predict responses of patients with autoimmune diseases to anti-TNF therapy (Chen W. et al., 2015). Thus, aberrant expression of TNF and TNFR2, and their responses to TNF blockade, is responsible at least partially for the outcome of anti-TNF therapy.
TNF plays a crucial role in host defense to invading pathogens (Pfeffer, 2003). Anti-TNF biologics can inhibit IFN-γ expression in the blood (Wallis, 2007). In addition, TNF is a good inducer of nitric oxide (NO) in macrophage, and TNF also synergizes with IFN-γ in inducing inducible nitric oxide synthase (iNOS) (Vila-Del Sol et al., 2007), while iNOS is able to synthesize antimicrobial nitric oxide (Fang, 1997). This can explain why infections, such as tuberculosis and pneumonia, are common adverse event of patients treated with anti-TNF biologics (Antoni and Braun, 2002).
There is also some evidence that anti-TNF treatment is associated with increased risk of malignancy (Bongartz et al., 2006). However, this is not supported by the more recent studies (Minozzi et al., 2016; Shelton et al., 2016). In fact, the underlying inflammatory condition is likely to promote the development of cancer, while anti-inflammatory treatment has the beneficial effect in inhibition of malignancy (Lasry et al., 2016). For example, patient with RA and CD has a higher risk of lymphoma compared with the general population (Antoni and Braun, 2002). Therefore, patients with malignancy after treatment with anti-TNF biologics should be more carefully studied.
Anti-TNF treatment has been tried in patients with congestive cardiac failure, since the high levels of circulating TNF was found in such patients. However, infliximab, adalimumab, and etanercept have deleterious, rather than beneficial, effects on congestive cardiac failure patients (Balakumar and Singh, 2006). Anti-TNF biologics have been reported to have side effects on the neurological system. For example, anti-TNF treatment exacerbated diseases in almost all multiple sclerosis (MS) (Robinson et al., 2001). Paradoxically, some new autoimmune diseases can be de novo induced by anti-TNF therapy. For example, psoriasis can be induced in IBD patients treated with anti-TNF biologics (Guerra et al., 2012). Therefore, more effective and safer TNF-targeting treatment needs to be developed. Thorough understanding of the fundamental biology of TNF and it receptors is a prerequisite to reach this goal.
Anti-TNFR Therapeutics: Next Generation of TNF-Targeting Drug?
Tumor necrosis factor-alpha is a pleiotropic cytokine that is involved in the initiation and orchestration of inflammation and immunity (Aggarwal, 2003). Two receptors of TNF, namely TNFR1 (P55) and TNFR2 (P75), mediate different signaling pathways and induce diverse biological effects of TNF (Vandenabeele et al., 1995). TNFR1 is expressed by almost all cell types except erythrocyte, whereas TNFR2 is more strictly expressed by immune cells. The capacity of TNF to induce cell death is mediated by TNFR1. Conversely, TNFR2 signaling triggers cell survival pathways and promotes cell proliferation (Faustman and Davis, 2010). Initially, TNF is expressed on the cell surface as a transmembrane protein. It is then cleaved by a metalloprotease, called TNF-alpha converting enzyme (TACE) (Black et al., 1997). This process liberates a trimeric soluble protein, namely soluble TNF (sTNF). Both membrane-bound TNF and soluble TNF can bind to TNFR1 and TNFR2, but membrane-bound TNF preferentially binds with TNFR2 (Grell et al., 1995).
There is now compelling evidence that TNFR2 is constitutively expressed by immunosuppressive CD4+Foxp3+ regulatory T cells (Tregs) and TNF-TNFR2 interaction preferentially activates and expands naturally occurring Tregs (Chen et al., 2007, 2008, 2010, 2013; Nguyen and Ehrenstein, 2016). The immunosuppressive property of TNFR2 has been shown by many studies. For example, TNFR2 agonist is able to promote the expansion of Tregs more potently than TNF itself, and consequently inhibits autoimmune responses (Okubo et al., 2013). Selective blockade of TNFR1 also results in the proliferative expansion and activation Tregs (McCann et al., 2014), and inhibits clinical symptoms in CIA model (Shibata et al., 2009). It was shown recently that therapeutic anti-TNF antibody Infliximab binds to and promotes the expression of membrane bound TNF on monocytes from RA patients, and consequently enhances Treg activity through TNF-TNFR2 interaction (Chen and Oppenheim, 2016; Nguyen and Ehrenstein, 2016). These evidences favor the idea that biologic drugs which can selectively inhibit TNFR1, or selectively activate TNFR2, may be more effective and safer than those globally inhibit TNF. The expression of TNFR2 on cells and tissues is more limited than TNFR1. Thus, therapeutically targeting TNFR2 may have fewer side effects than targeting TNFR1. Expression of TNFR2 on CD4+Foxp3- effector T cells (Teffs) can be induced and upregulated by TCR stimulation, and TNFR2 expression on Teff cells has a functional consequence (Chen and Oppenheim, 2011; Govindaraj et al., 2013; Chen X. et al., 2015; Chen and Oppenheim, 2016; Chen et al., 2016; Zaragoza et al., 2016). Thus, the effect of TNFR2 on Teff function should be considered in the development of TNFR2-targeting therapeutics.
Other Biological Drugs for Autoimmune Inflammatory Diseases
In addition to anti-TNF agents, the biologics targeting other proinflammatory cytokines or immune competent molecules have also been extensively studied and actively developed. For example, abatacept, a fully humanized fusion protein of extracellular domain of CTLA-4 and Fc fraction of IgG1, has been approved for the RA patients with inadequate response to anti-TNF therapy (Genovese et al., 2005). The major immunological mechanism of abatacept is selective inhibition of co-stimulation pathway (CD80 and CD86) and activation of T cells. Tocilizumab, a humanized anti-IL-6 receptor monoclonal antibody was approved for RA patients intolerant to DMARDs and/or anti-TNF biologics (Okuda, 2008). This therapeutic mAb blocks the transmembrane signaling of IL-6 through binding with soluble and membrane forms of IL-6 receptor. Biological drugs targeting IL-1 (anakinra), Th1 immune responses (IL-12/IL-23, ustekinumab), Th17 immune responses (IL-17, secukinumab) and CD20 (rituximab) have also been approved for the treatment of autoimmune diseases, as summarized in the Table 2, as a complementary and alternative biological treatment of anti-TNF therapy.

TABLE 2. Alternative biologic in autoimmune inflammatory diseases.
Conclusion
Although TNF plays a central role in acute and chronic inflammation, the anti-inflammatory agents were developed before the identification of TNF. After the World War II, agents from small molecules to anti-TNF antibodies have been developed, including NSAIDs, glucocorticoids, DMSADs, and anti-TNF biologics. The traditional non-selective NSAIDs are associated with severe gastrointestinal disorders, attributable to simultaneous inhibition of COX-1 and COX-2, which prompted the development of selective NSAIDs. Glucocorticoids have potent anti-inflammatory activity, accompanied by some severe adverse effects. Methotrexate remains the first-line drug, while the efficacy can be enhanced and adverse effects can be reduced by DMARDs combination therapy. Currently, five anti-TNF biologics have been approved for patients with autoimmune disorders. With great success in treating autoimmune diseases, anti-TNF biologics also represent the most profitable drug class in the history, exceeding $US 25 billion total sale in 2012 (Monaco et al., 2015). Anti-TNF biosimilars bring the hope to reduce the medical costs and consequently improve the accessibility to this revolutionized treatment. Nevertheless, the effectiveness and safety of TNF-targeting treatment should be further improved, hopefully through the selective blockade of TNFR1 or activation of TNFR2. For those patients failed to respond appropriately to anti-TNF treatment, biologics targeting other pro-inflammatory cytokines and pathways provide an alternative therapy.
As compared to small molecule compounds, biologics have some clear advantages, such as higher safety profile or minimal toxicity, well-understood mechanisms, and more importantly, the target specificity (Tracey et al., 2008; Meier et al., 2013; Monaco et al., 2015). Due to these advantages, the biologics, including therapeutic monoclonal antibodies and antibody-drug conjugate (ADC), have become main stream therapeutics (Meier et al., 2013; Monaco et al., 2015). This class of drug represents the fastest growing segment of global pharmaceutical marked (based on the data of global sale and FDA approval) (Ioannidis et al., 2013; Monaco et al., 2015). Therefore, it is predictable that the development of new biologics for the treatment of autoimmune diseases will be the focus in the future, which will be facilitated by the more thorough understanding of molecular basis of autoimmune diseases.
Author Contributions
PL drafted the work; YZ and XC revised the manuscript. PL, YZ, and XC contributed substantially to the conception or design of the work, approved the final version to be published, and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Funding
This study is funded by the University of Macau under Grants MYRG2016-00023-ICMS-QRCM and MYRG2017-00120-ICMS and the Science and Technology Development Fund of Macau S.A.R. (FDCT) under grant 014/2015/A1 and grant SKL-QRCM-2014-2016.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aggarwal, B. B. (2003). Signalling pathways of the TNF superfamily: a double-edged sword. Nat. Rev. Immunol. 3, 745–756. doi: 10.1038/nri1184
Antman, E. M., Bennett, J. S., Daugherty, A., Furberg, C., Roberts, H., and Taubert, K. A. (2007). Use of nonsteroidal Antiinflammatory drugs an update for clinicians - A scientific statement from the American Heart Association. Circulation 115, 1634–1642. doi: 10.1161/Circulationaha.106.181424
Antoni, C., and Braun, J. (2002). Side effects of anti-TNF therapy: current knowledge. Clin. Exp. Rheumatol. 20(6 Suppl. 28), S152–S157.
Atzeni, F., Turiel, M., Capsoni, F., Doria, A., Meroni, P., and Sarzi-Puttini, P. (2005). Autoimmunity and anti-TNF-alpha agents. Ann. N. Y. Acad. Sci. 1051, 559–569. doi: 10.1196/annals.1361.100
Balakumar, P., and Singh, M. (2006). Anti-tumour necrosis factor-alpha therapy in heart failure: future directions. Basic Clin. Pharmacol. Toxicol. 99, 391–397. doi: 10.1111/j.1742-7843.2006.pto_508.x
Barnes, P. J. (1998). Anti-inflammatory actions of glucocorticoids: molecular mechanisms. Clin. Sci. 94, 557–572. doi: 10.1042/cs0940557
Bendix, G., and Bjelle, A. (1996). A 10 year follow up of parenteral gold therapy in patients with rheumatoid arthritis. Ann. Rheum. Dis. 55, 169–176. doi: 10.1136/Ard.55.3.169
Billiau, A. (1988). Gamma-interferon: the match that lights the fire? Immunol. Today 9, 37–40. doi: 10.1016/0167-5699(88)91256-X
Black, R. A., Rauch, C. T., Kozlosky, C. J., Peschon, J. J., Slack, J. L., Wolfson, M. F., et al. (1997). A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature 385, 729–733. doi: 10.1038/385729a0
Boers, M., Verhoeven, A. C., Markusse, H. M., vandeLaar, M. A. F. J., Westhovens, R., vanDenderen, J. C., et al. (1997). Randomised comparison of combined step-down prednisolone, methotrexate and sulphasalazine with sulphasalazine alone in early rheumatoid arthritis. Lancet 350, 309–318. doi: 10.1016/S0140-6736(97)01300-7
Bongartz, T., Sutton, A. J., Sweeting, M. J., Buchan, I., Matteson, E. L., and Montori, V. (2006). Anti-TNF antibody therapy in rheumatoid arthritis and the risk of serious infections and malignancies: systematic review and meta-analysis of rare harmful effects in randomized controlled trials. JAMA 295, 2275–2285. doi: 10.1001/jama.295.19.2275
Box, S. A., and Pullar, T. (1997). Sulphasalazine in the treatment of rheumatoid arthritis. Br. J. Rheumatol. 36, 382–386. doi: 10.1093/rheumatology/36.3.382
Bragazzi, N. L., Watad, A., Brigo, F., Adawi, M., Amital, H., and Shoenfeld, Y. (2016). Public health awareness of autoimmune diseases after the death of a celebrity. Clin. Rheumatol. doi: 10.1007/s10067-016-3513-5 [Epub ahead of print].
Busson, M. (1986). Update on ibuprofen: review article. J. Int. Med. Res. 14, 53–62. doi: 10.1177/030006058601400201
Buttgereit, F., Burmester, G. R., Straub, R. H., Seibel, M. J., and Zhou, H. (2011). Exogenous and endogenous glucocorticoids in rheumatic diseases. Arthritis Rheum. 63, 1–9. doi: 10.1002/art.30070
Campa, M., Mansouri, B., Warren, R., and Menter, A. (2016). A review of biologic therapies targeting IL-23 and IL-17 for use in moderate-to-severe plaque psoriasis. Dermatol. Ther. 6, 1–12. doi: 10.1007/s13555-015-0092-3
Carswell, E. A., Old, L. J., Kassel, R. L., Green, S., Fiore, N., and Williamson, B. (1975). An endotoxin-induced serum factor that causes necrosis of tumors. Proc. Natl. Acad. Sci. U.S.A. 72, 3666–3670. doi: 10.1073/pnas.72.9.3666
Chatzantoni, K., and Mouzaki, A. (2006). Anti-TNF-alpha antibody therapies in autoimmune diseases. Curr. Top. Med. Chem. 6, 1707–1714. doi: 10.2174/156802606778194217
Chen, W., Xu, H., Wang, X., Gu, J., Xiong, H., and Shi, Y. (2015). The tumor necrosis factor receptor superfamily member 1B polymorphisms predict response to anti-TNF therapy in patients with autoimmune disease: a meta-analysis. Int. Immunopharmacol. 28, 146–153. doi: 10.1016/j.intimp.2015.05.049
Chen, X., Baumel, M., Mannel, D. N., Howard, O. M., and Oppenheim, J. J. (2007). Interaction of TNF with TNF receptor type 2 promotes expansion and function of mouse CD4+CD25+ T regulatory cells. J. Immunol. 179, 154–161. doi: 10.4049/jimmunol.179.1.154
Chen, X., Murakami, T., Oppenheim, J. J., and Howard, O. M. (2004). Differential response of murine CD4+CD25+ and CD4+CD25- T cells to dexamethasone-induced cell death. Eur. J. Immunol. 34, 859–869. doi: 10.1002/eji.200324506
Chen, X., Nie, Y., Xiao, H., Bian, Z., Scarzello, A. J., Song, N. Y., et al. (2016). TNFR2 expression by CD4 effector T cells is required to induce full-fledged experimental colitis. Sci. Rep. 6:32834. doi: 10.1038/srep32834
Chen, X., and Oppenheim, J. J. (2011). Contrasting effects of TNF and anti-TNF on the activation of effector T cells and regulatory T cells in autoimmunity. FEBS Lett. 585, 3611–3618. doi: 10.1016/j.febslet.2011.04.025
Chen, X., and Oppenheim, J. J. (2016). Therapy: Paradoxical effects of targeting TNF signalling in the treatment of autoimmunity. Nat. Rev. Rheumatol. 12, 625–626. doi: 10.1038/nrrheum.2016.145
Chen, X., Oppenheim, J. J., Winkler-Pickett, R. T., Ortaldo, J. R., and Howard, O. M. (2006). Glucocorticoid amplifies IL-2-dependent expansion of functional FoxP3(+)CD4(+)CD25(+) T regulatory cells in vivo and enhances their capacity to suppress EAE. Eur. J. Immunol. 36, 2139–2149. doi: 10.1002/eji.200635873
Chen, X., Subleski, J. J., Hamano, R., Howard, O. M., Wiltrout, R. H., and Oppenheim, J. J. (2010). Co-expression of TNFR2 and CD25 identifies more of the functional CD4+FOXP3+ regulatory T cells in human peripheral blood. Eur. J. Immunol. 40, 1099–1106. doi: 10.1002/eji.200940022
Chen, X., Subleski, J. J., Kopf, H., Howard, O. M., Mannel, D. N., and Oppenheim, J. J. (2008). Cutting edge: expression of TNFR2 defines a maximally suppressive subset of mouse CD4+CD25+FoxP3+ T regulatory cells: applicability to tumor-infiltrating T regulatory cells. J. Immunol. 180, 6467–6471. doi: 10.4049/jimmunol.180.10.6467
Chen, X., Willette-Brown, J., Wu, X., Hu, Y., Howard, O. M., Hu, Y., et al. (2015). IKKalpha is required for the homeostasis of regulatory T cells and for the expansion of both regulatory and effector CD4 T cells. FASEB J. 29, 443–454. doi: 10.1096/fj.14-259564
Chen, X., Wu, X. Q., Zhou, Q., Howard, O. M. Z., Netea, M. G., and Oppenheim, J. J. (2013). TNFR2 Is critical for the stabilization of the CD4(+)Foxp3(+) regulatory T cell phenotype in the inflammatory environment. J. Immunol. 190, 1076–1084. doi: 10.4049/jimmunol.1202659
Cheng, D. S., and Visco, C. J. (2012). Pharmaceutical therapy for osteoarthritis. PM R 4(5 Suppl.), S82–S88. doi: 10.1016/j.pmrj.2012.02.009
Choy, E. H., Hazleman, B., Smith, M., Moss, K., Lisi, L., Scott, D. G., et al. (2002). Efficacy of a novel PEGylated humanized anti-TNF fragment (CDP870) in patients with rheumatoid arthritis: a phase II double-blinded, randomized, dose-escalating trial. Rheumatology 41, 1133–1137. doi: 10.1093/rheumatology/41.10.1133
Cooper, G. S., Bynum, M. L., and Somers, E. C. (2009). Recent insights in the epidemiology of autoimmune diseases: improved prevalence estimates and understanding of clustering of diseases. J. Autoimmun. 33, 197–207. doi: 10.1016/j.jaut.2009.09.008
Costedoat-Chalumeau, N., Dunogue, B., Leroux, G., Morel, N., Jallouli, M., Le Guern, V., et al. (2015). A critical review of the effects of hydroxychloroquine and chloroquine on the eye. Clin. Rev. Allergy Immunol. 49, 317–326. doi: 10.1007/s12016-015-8469-8
Crofford, L. J. (1997). COX-1 and COX-2 tissue expression: implications and predictions. J. Rheumatol. Suppl. 49, 15–19.
Curtis, J. R., Westfall, A. O., Allison, J., Bijlsma, J. W., Freeman, A., George, V., et al. (2006). Population-based assessment of adverse events associated with long-term glucocorticoid use. Arthritis Rheum. 55, 420–426. doi: 10.1002/art.21984
Dajani, E. Z., and Islam, K. (2008). Cardiovascular and gastrointestinal toxicity of selective cyclo-oxygenase-2 inhibitors in man. J. Physiol. Pharmacol. 59, 117–133.
Deeks, E. D. (2016). Certolizumab pegol: a review in inflammatory autoimmune diseases. BioDrugs 30, 607–617. doi: 10.1007/s40259-016-0197-y
Desai, R. J., Hansen, R. A., Rao, J. K., Wilkins, T. M., Harden, E. A., Yuen, A., et al. (2012). Mixed treatment comparison of the treatment discontinuations of biologic disease-modifying antirheumatic drugs in adults with rheumatoid arthritis. Ann. Pharmacother. 46, 1491–1505. doi: 10.1345/aph.1R203
Dorner, T., and Kay, J. (2015). Biosimilars in rheumatology: current perspectives and lessons learnt. Nat. Rev. Rheumatol. 11, 713–724. doi: 10.1038/nrrheum.2015.110
Dorner, T., Strand, V., Cornes, P., Goncalves, J., Gulacsi, L., Kay, J., et al. (2016). The changing landscape of biosimilars in rheumatology. Ann. Rheum. Dis. 75, 974–982. doi: 10.1136/annrheumdis-2016-209166
Efimov, G. A., Kruglov, A. A., Tillib, S. V., Kuprash, D. V., and Nedospasov, S. A. (2009). Tumor necrosis factor and the consequences of its ablation in vivo. Mol. Immunol. 47, 19–27. doi: 10.1016/j.molimm.2009.01.007
Ellis, J. S., and Braley-Mullen, H. (2017). Mechanisms by which B cells and regulatory t cells influence development of murine organ-specific autoimmune diseases. J. Clin. Med. 6:13. doi: 10.3390/jcm6020013
Emery, P., Breedveld, F. C., Hall, S., Durez, P., Chang, D. J., Robertson, D., et al. (2008). Comparison of methotrexate monotherapy with a combination of methotrexate and etanercept in active, early, moderate to severe rheumatoid arthritis (COMET): a randomised, double-blind, parallel treatment trial. Lancet 372, 375–382. doi: 10.1016/S0140-6736(08)61000-4
Ethgen, O., Esteves, F. D., Bruyere, O., and Reginster, J. Y. (2013). What do we know about the safety of corticosteroids in rheumatoid arthritis? Curr. Med. Res. Opin. 29, 1147–1160. doi: 10.1185/03007995.2013.818531
Fang, F. C. (1997). Perspectives series: host/pathogen interactions. Mechanisms of nitric oxide-related antimicrobial activity. J. Clin. Invest. 99, 2818–2825. doi: 10.1172/JCI119473
Faustman, D., and Davis, M. (2010). TNF receptor 2 pathway: drug target for autoimmune diseases. Nat. Rev. Drug Discov. 9, 482–493. doi: 10.1038/nrd3030
Favalli, E. G., Biggioggero, M., and Meroni, P. L. (2014). Methotrexate for the treatment of rheumatoid arthritis in the biologic era: still an ”anchor” drug? Autoimmun. Rev. 13, 1102–1108. doi: 10.1016/j.autrev.2014.08.026
Feldmann, M., Brennan, F. M., and Maini, R. N. (1996). Role of cytokines in rheumatoid arthritis. Annu. Rev. Immunol. 14, 397–440. doi: 10.1146/annurev.immunol.14.1.397
Ferreira, J. F., Ahmed Mohamed, A. A., and Emery, P. (2016). Glucocorticoids and rheumatoid arthritis. Rheum. Dis. Clin. North Am. 42, 33–46, vii. doi: 10.1016/j.rdc.2015.08.006
Fiorino, G., and Danese, S. (2014). The biosimilar road in inflammatory bowel disease: the right way? Best Pract. Res. Clin. Gastroenterol. 28, 465–471. doi: 10.1016/j.bpg.2014.04.006
Fleischmann, R. A. (2009). Safety of biologic therapy in rheumatoid arthritis and other autoimmune diseases: focus on rituximab. Semin. Arthritis Rheum. 38, 265–280. doi: 10.1016/j.semarthrit.2008.01.001
Fleischmann, R. M., Schechtman, J., Bennett, R., Handel, M. L., Burmester, G. R., Tesser, J., et al. (2003). Anakinra, a recombinant human interleukin-1 receptor antagonist (r-metHuIL-1ra), in patients with rheumatoid arthritis - A large, international, multicenter, placebo-controlled trial. Arthritis Rheum. 48, 927–934. doi: 10.1002/art.10870
Fox, R. I., Herrmann, M. L., Frangou, C. G., Wahl, G. M., Morris, R. E., Strand, V., et al. (1999). Mechanism of action for leflunomide in rheumatoid arthritis. Clin. Immunol. 93, 198–208. doi: 10.1006/clim.1999.4777
Frey, F. J. (1987). Kinetics and dynamics of prednisolone. Endocr. Rev. 8, 453–473. doi: 10.1210/edrv-8-4-453
Fujita, T., Kutsumi, H., Sanuki, T., Hayakumo, T., and Azuma, T. (2013). Adherence to the preventive strategies for nonsteroidal anti-inflammatory drug- or low-dose aspirin-induced gastrointestinal injuries. J. Gastroenterol. 48, 559–573. doi: 10.1007/s00535-013-0771-8
Gabriel, S. E., Coyle, D., and Moreland, L. W. (2001). A clinical and economic review of disease-modifying antirheumatic drugs. Pharmacoeconomics 19, 715–728. doi: 10.2165/00019053-200119070-00002
Genovese, M. C., Becker, J. C., Schiff, M., Luggen, M., Sherrer, Y., Kremer, J., et al. (2005). Abatacept for rheumatoid arthritis refractory to tumor necrosis factor alpha inhibition. N. Engl. J. Med. 353, 1114–1123. doi: 10.1056/NEJMoa050524
Gerhardt, M. C. (1853). X.—Researches on the constitution of organic acids. Q. J. Chem. Soc. 5, 127–131. doi: 10.1039/QJ8530500127
Gerland, H. (1853). XII.—New formation of salicylic acid. Q. J. Chem. Soc. 5, 133–136. doi: 10.1039/QJ8530500133
Gibellini, L., De Biasi, S., Bianchini, E., Bartolomeo, R., Fabiano, A., Manfredini, M., et al. (2016). Anti-TNF-alpha drugs differently affect the TNFalpha-sTNFR system and monocyte subsets in patients with psoriasis. PLoS ONE 11:e0167757. doi: 10.1371/journal.pone.0167757
Glyn, J. (1998). The discovery and early use of cortisone. J. R. Soc. Med. 91, 513–517. doi: 10.1177/014107689809101004
Govindaraj, C., Scalzo-Inguanti, K., Scholzen, A., Li, S., and Plebanski, M. (2013). TNFR2 expression on CD25(hi)FOXP3(+) T cells induced upon TCR stimulation of CD4 T cells identifies maximal cytokine-producing effectors. Front. Immunol. 4:233. doi: 10.3389/fimmu.2013.00233
Grell, M., Douni, E., Wajant, H., Lohden, M., Clauss, M., Maxeiner, B., et al. (1995). The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell 83, 793–802. doi: 10.1016/0092-8674(95)90192-2
Grosser, T., Fries, S., and FitzGerald, G. A. (2006). Biological basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunities. J. Clin. Invest. 116, 4–15. doi: 10.1172/JCI27291
Guerra, I., Algaba, A., Perez-Calle, J. L., Chaparro, M., Marin-Jimenez, I., Garcia-Castellanos, R., et al. (2012). Induction of psoriasis with anti-TNF agents in patients with inflammatory bowel disease: a report of 21 cases. J. Crohns Colitis 6, 518–523. doi: 10.1016/j.crohns.2011.10.007
Hedner, T., and Everts, B. (1998). The early clinical history of salicylates in rheumatology and pain. Clin. Rheumatol. 17, 17–25. doi: 10.1007/BF01450953
Hench, P. S., and Kendall, E. C., Slocumb, C. H., and Polley, H. F. (1949). The effect of a hormone of the adrenal cortex (17-hydroxy-11-dehydrocorticosterone; compound E) and of pituitary adrenocorticotropic hormone on rheumatoid arthritis. Proc. Staff Meet. Mayo Clin. 24, 181–197.
Hermann, M., and Ruschitzka, F. (2006). Coxibs, non-steroidal anti-inflammatory drugs and cardiovascular risk. Intern. Med. J. 36, 308–319. doi: 10.1111/j.1445-5994.2006.01056.x
Hoffmann, W., and Förster, W. (1987). Two years follow-up Cottbus reinfarction study with 30 and 60 mg acetylsalicylic acid. Prog. Clin. Biol. Res. 242, 393-397.
Ioannidis, J. P. A., Karassa, F. B., Druyts, E., Thorlund, K., and Mills, E. J. (2013). Biologic agents in rheumatology: unmet issues after 200 trials and $200 billion sales. Nat. Rev. Rheumatol. 9, 665–673. doi: 10.1038/nrrheum.2013.134
Jang, C. H., Choi, J. H., Byun, M. S., and Jue, D. M. (2006). Chloroquine inhibits production of TNF-alpha, IL-1beta and IL-6 from lipopolysaccharide-stimulated human monocytes/macrophages by different modes. Rheumatology 45, 703–710. doi: 10.1093/rheumatology/kei282
Kalden, J. R., Antoni, C., Alvaro-Gracia, J. M., Combe, B., Emery, P., Kremer, J. M., et al. (2005). Use of combination of leflunomide with biological agents in treatment of rheumatoid arthritis. J. Rheumatol. 32, 1620–1631.
Kang, Y., Xu, L., Wang, B., Chen, A., and Zheng, G. (2008). Cutting edge: immunosuppressant as adjuvant for tolerogenic immunization. J. Immunol. 180, 5172–5176. doi: 10.4049/jimmunol.180.8.5172
Karanikolas, G., Charalambopoulos, D., Andrianakos, A., Antoniades, C., and Katsilambros, N. (2006). Combination of cyclosporine and leflunomide versus single therapy in severe rheumatoid arthritis. J. Rheumatol. 33, 486–489.
Kerensky, T. A., Gottlieb, A. B., Yaniv, S., and Au, S. C. (2012). Etanercept: efficacy and safety for approved indications. Expert Opin. Drug Saf. 11, 121–139. doi: 10.1517/14740338.2012.633509
Kersley, G. D. (1968). Amethopterin (methotrexate) in connective tissue disease-psoriasis and polyarthritis. Ann. Rheum. Dis. 27, 64–66. doi: 10.1136/ard.27.1.64
Keystone, E., van der Heijde, D., Mason, D., Landewe, R., van Vollenhoven, R., Combe, B., et al. (2008). Certolizumab pegol plus methotrexate is significantly more effective than placebo plus methotrexate in active rheumatoid arthritis findings of a fifty-two-week, phase III, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Arthritis Rheum. 58, 3319–3329. doi: 10.1002/art.23964
Khanapure, S. P., Garvey, D. S., Young, D. V., Ezawa, M., Earl, R. A., Gaston, R. D., et al. (2003). Synthesis and structure-activity relationship of novel, highly potent metharyl and methcycloalkyl cyclooxygenase-2 (COX-2) selective inhibitors. J. Med. Chem. 46, 5484–5504. doi: 10.1021/jm030268b
Kim, S. C., Yelin, E., Tonner, C., and Solomon, D. H. (2013). Changes in use of disease-modifying antirheumatic drugs for rheumatoid arthritis in the united states during 1983-2009. Arthritis Care Res. 65, 1529–1533. doi: 10.1002/acr.21997
Koutruba, N., Emer, J., and Lebwohl, M. (2010). Review of ustekinumab, an interleukin-12 and interleukin-23 inhibitor used for the treatment of plaque psoriasis. Ther. Clin. Risk Manag. 6, 123–141.
Kremer, J., Genovese, M., Cannon, G. W., Caldwell, J., Cush, J., Furst, D. E., et al. (2004). Combination leflunomide and methotrexate (MTX) therapy for patients with active rheumatoid arthritis failing MTX monotherapy: open-label extension of a randomized, double-blind, placebo controlled trial. J. Rheumatol. 31, 1521–1531.
Laev, S. S., and Salakhutdinov, N. F. (2015). Anti-arthritic agents: progress and potential. Bioorg. Med. Chem. 23, 3059–3080. doi: 10.1016/j.bmc.2015.05.010
Lapadula, G., Marchesoni, A., Armuzzi, A., Blandizzi, C., Caporali, R., Chimenti, S., et al. (2014). Adalimumab in the treatment of immune-mediated diseases. Int. J. Immunopathol. Pharmacol. 27(1 Suppl.), 33–48. doi: 10.1177/03946320140270S103
Lasry, A., Zinger, A., and Ben-Neriah, Y. (2016). Inflammatory networks underlying colorectal cancer. Nat. Immunol. 17, 230–240. doi: 10.1038/ni.3384
Lowenberg, M., and D’Haens, G. (2013). Novel targets for inflammatory bowel disease therapeutics. Curr. Gastroenterol. Rep. 15:311. doi: 10.1007/s11894-012-0311-3
Mauritz, N. J., Holmdahl, R., Jonsson, R., Van der Meide, P. H., Scheynius, A., and Klareskog, L. (1988). Treatment with gamma-interferon triggers the onset of collagen arthritis in mice. Arthritis Rheum. 31, 1297–1304. doi: 10.1002/art.1780311012
McCann, F. E., Perocheau, D. P., Ruspi, G., Blazek, K., Davies, M. L., Feldmann, M., et al. (2014). Selective tumor necrosis factor receptor I blockade is antiinflammatory and reveals immunoregulatory role of tumor necrosis factor receptor II in collagen-induced arthritis. Arthritis Rheumatol. 66, 2728–2738. doi: 10.1002/art.38755
Meier, F. M. P., Frerix, M., Hermann, W., and Muller-Ladner, U. (2013). Current immunotherapy in rheumatoid arthritis. Immunotherapy 5, 955–974. doi: 10.2217/imt.13.94
Minozzi, S., Bonovas, S., Lytras, T., Pecoraro, V., Gonzalez-Lorenzo, M., Bastiampillai, A. J., et al. (2016). Risk of infections using anti-TNF agents in rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis: a systematic review and meta-analysis. Expert Opin. Drug Saf. 15, 11–34. doi: 10.1080/14740338.2016.1240783
Monaco, C., Nanchahal, J., Taylor, P., and Feldmann, M. (2015). Anti-TNF therapy: past, present and future. Int. Immunol. 27, 55–62. doi: 10.1093/intimm/dxu102
Moreland, L. W., Schiff, M. H., Baumgartner, S. W., Tindall, E. A., Fleischmann, R. M., Bulpitt, K. J., et al. (1999). Etanercept therapy in rheumatoid arthritis. A randomized, controlled trial. Ann. Intern. Med. 130, 478–486. doi: 10.7326/0003-4819-130-6-199903160-00004
Moudgil, K. D., and Choubey, D. (2011). Cytokines in autoimmunity: role in induction, regulation, and treatment. J. Interferon Cytokine Res. 31, 695–703. doi: 10.1089/jir.2011.0065
Murray, M. D., and Brater, D. C. (1993). Renal toxicity of the nonsteroidal anti-inflammatory drugs. Annu. Rev. Pharmacol. Toxicol. 33, 435–465. doi: 10.1146/annurev.pa.33.040193.002251
Nathan, D. M., Iser, J. H., and Gibson, P. R. (2008). A single center experience of methotrexate in the treatment of Crohn’s disease and ulcerative colitis: a case for subcutaneous administration. J. Gastroenterol. Hepatol. 23, 954–958. doi: 10.1111/j.1440-1746.2007.05006.x
Neeck, G. (2002). Fifty years of experience with cortisone therapy in the study and treatment of rheumatoid arthritis. Ann. N. Y. Acad. Sci. 966, 28–38. doi: 10.1111/j.1749-6632.2002.tb04199.x
Nguyen, D. X., and Ehrenstein, M. R. (2016). Anti-TNF drives regulatory T cell expansion by paradoxically promoting membrane TNF-TNF-RII binding in rheumatoid arthritis. J. Exp. Med. 213, 1241–1253. doi: 10.1084/jem.20151255
ODell, J. R., Haire, C. E., Erikson, N., Drymalski, W., Palmer, W., Eckhoff, P. J., et al. (1996). Treatment of rheumatoid arthritis with methotrexate alone, sulfasalazine and hydroxychloroquine, or a combination of all three medications. N. Engl. J. Med. 334, 1287–1291. doi: 10.1056/Nejm199605163342002
ODell, J. R., Leff, R., Paulsen, G., Haire, C., Mallek, J., Eckhoff, P. J., et al. (2002). Treatment of rheumatoid arthritis with methotrexate and hydroxychloroquine, methotrexate and sulfasalazine, or a combination of the three medications - Results of a two-year, randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 46, 1164–1170. doi: 10.1002/art.10228
Okubo, Y., Mera, T., Wang, L., and Faustman, D. L. (2013). Homogeneous expansion of human T-regulatory cells via tumor necrosis factor receptor 2. Sci. Rep. 3:3153. doi: 10.1038/srep03153
Okuda, Y. (2008). Review of tocilizumab in the treatment of rheumatoid arthritis. Biologics 2, 75–82. doi: 10.2147/btt.s1828
Osiri, M., Shea, B., Robinson, V., Suarez-Almazor, M., Strand, V., Tugwell, P., et al. (2003). Leflunomide for the treatment of rheumatoid arthritis: a systematic review and meta-analysis. J. Rheumatol. 30, 1182–1190.
Palladino, M. A., Bahjat, F. R., Theodorakis, E. A., and Moldawer, L. L. (2003). Anti-TNF-alpha therapies: the next generation. Nat. Rev. Drug Discov. 2, 736–746. doi: 10.1038/nrd1175
Pasut, G. (2014). Pegylation of biological molecules and potential benefits: pharmacological properties of certolizumab pegol. BioDrugs 28(Suppl. 1), S15–S23. doi: 10.1007/s40259-013-0064-z
Pfeffer, K. (2003). Biological functions of tumor necrosis factor cytokines and their receptors. Cytokine Growth Factor Rev. 14, 185–191. doi: 10.1016/S1359-6101(03)00022-4
Rainsford, K. D. (2007). Anti-inflammatory drugs in the 21st century. Subcell Biochem. 42, 3–27. doi: 10.1007/1-4020-5688-5_1
Rau, R. (2002). Adalimumab (a fully human anti-tumour necrosis factor alpha monoclonal antibody) in the treatment of active rheumatoid arthritis: the initial results of five trials. Ann. Rheum. Dis. 61, 70–73. doi: 10.1136/ard.61.suppl_2.ii70
Ray, W. A., Stein, C. M., Daugherty, J. R., Hall, K., Arbogast, P. G., and Griffin, M. R. (2002). COX-2 selective non-steroidal anti-inflammatory drugs and risk of serious coronary heart disease. Lancet 360, 1071–1073. doi: 10.1016/S0140-6736(02)11131-7
Rhen, T., and Cidlowski, J. A. (2005). Antiinflammatory action of glucocorticoids - New mechanisms for old drugs. N. Engl. J. Med. 353, 1711–1723. doi: 10.1056/Nejmra050541
Robinson, W. H., Genovese, M. C., and Moreland, L. W. (2001). Demyelinating and neurologic events reported in association with tumor necrosis factor alpha antagonism: by what mechanisms could tumor necrosis factor alpha antagonists improve rheumatoid arthritis but exacerbate multiple sclerosis? Arthritis Rheum. 44, 1977–1983. doi: 10.1002/1529-0131(200109)44:9<1977::AID-ART345>3.0.CO;2-6
Roda, G., Jharap, B., Neeraj, N., and Colombel, J. F. (2016). Loss of response to anti-TNFs: definition, epidemiology, and management. Clin. Transl. Gastroenterol. 7:e135. doi: 10.1038/ctg.2015.63
Sandborn, W. J., Hanauer, S., Loftus, E. V., Tremaine, W. J., Kane, S., Cohen, R., et al. (2004). An open-label study of the human anti-TNF monoclonal antibody adalimumab in subjects with prior loss of response or intolerance to infliximab for Crohn’s disease. Am. J. Gastroenterol. 99, 1984–1989. doi: 10.1111/j.1572-0241.2004.40462.x
Schreiber, S., Rutgeerts, P., Fedorak, R. N., Khaliq-Kareemi, M., Kamm, M. A., Boivin, M., et al. (2005). A randomized, placebo-controlled trial of certolizumab pegol (CDP870) for treatment of Crohn’s disease. Gastroenterology 129, 807–818. doi: 10.1053/j.gastro.2005.06.064
Schurgers, E., Billiau, A., and Matthys, P. (2011). Collagen-induced arthritis as an animal model for rheumatoid arthritis: focus on interferon-gamma. J. Interferon Cytokine Res. 31, 917–926. doi: 10.1089/jir.2011.0056
Sharp, J. T., Strand, V., Leung, H., Hurley, F., and Loew-Friedrich, I. (2000). Treatment with leflunomide slows radiographic progression of rheumatoid arthritis: results from three randomized controlled trials of leflunomide in patients with active rheumatoid arthritis. Leflunomide Rheumatoid Arthritis Investigators Group. Arthritis Rheum. 43, 495–505. doi: 10.1002/1529-0131(200003)43:3<495::AID-ANR4>3.0.CO;2-U
Shealy, D., Cai, A., Lacy, E., Nesspor, T., Staquet, K., Johns, L., et al. (2007). Characterization of golimumab (CNTO 148), a novel fully human monoclonal antibody specific for human TNFalpha. Ann. Rheum. Dis. 66, 151–151.
Shealy, D. J., Cai, A., Staquet, K., Baker, A., Lacy, E. R., Johns, L., et al. (2010). Characterization of golimumab, a human monoclonal antibody specific for human tumor necrosis factor alpha. MAbs 2, 428–439. doi: 10.4161/mabs.12304
Shelton, E., Laharie, D., Scott, F. I., Mamtani, R., Lewis, J. D., Colombel, J. F., et al. (2016). Cancer recurrence following immune-suppressive therapies in patients with immune-mediated diseases: a systematic review and meta-analysis. Gastroenterology 151, 97.e4–109.e4. doi: 10.1053/j.gastro.2016.03.037
Shen, T. Y. (1982). The discovery of indomethacin and the proliferation of nsaids. Semin. Arthritis Rheum. 12, 89–93. doi: 10.1016/0049-0172(82)90004-X
Shibata, H., Yoshioka, Y., Abe, Y., Ohkawa, A., Nomura, T., Minowa, K., et al. (2009). The treatment of established murine collagen-induced arthritis with a TNFR1-selective antagonistic mutant TNF. Biomaterials 30, 6638–6647. doi: 10.1016/j.biomaterials.2009.08.041
Sigidin, Y. A., Loukina, G. V., Skurkovich, B., and Skurkovich, S. (2001). Randomized, double-blind trial of anti-interferon-gamma antibodies in rheumatoid arthritis. Scand. J. Rheumatol. 30, 203–207. doi: 10.1080/030097401316909530
Silverman, E., Mouy, R., Spiegel, L., Jung, L. K., Saurenmann, R. K., Lahdenne, P., et al. (2005). Leflunomide or methotrexate for juvenile rheumatoid arthritis. N. Engl. J. Med. 352, 1655–1666. doi: 10.1056/NEJMoa041810
Simmons, D. L., Botting, R. M., and Hla, T. (2004). Cyclooxygenase isozymes: the biology of prostaglandin synthesis and inhibition. Pharmacol. Rev. 56, 387–437. doi: 10.1124/pr.56.3.3
Simon, L. S. (2004). The treatment of rheumatoid arthritis. Best Pract. Res. Clin. Rheumatol. 18, 507–538. doi: 10.1016/j.berh.2004.04.005
Skosey, J. L. (1988). Comparison of responses to and adverse effects of graded doses of sulfasalazine in the treatment of rheumatoid arthritis. J. Rheumatol. Suppl. 16, 5–8.
Sticherling, M. (2016). Psoriasis and autoimmunity. Autoimmun. Rev. 15, 1167–1170. doi: 10.1016/j.autrev.2016.09.004
Swierkot, J., and Szechinski, J. (2006). Methotrexate in rheumatoid arthritis. Pharmacol. Rep. 58, 473–492.
Tabas, I., and Glass, C. K. (2013). Anti-inflammatory therapy in chronic disease: challenges and opportunities. Science 339, 166–172. doi: 10.1126/science.1230720
Taherian, E., Rao, A., Malemud, C. J., and Askari, A. D. (2013). The biological and clinical activity of anti-malarial drugs in autoimmune disorders. Curr. Rheumatol. Rev. 9, 45–62. doi: 10.2174/1573397111309010010
Tracey, D., Klareskog, L., Sasso, E. H., Salfeld, J. G., and Tak, P. P. (2008). Tumor necrosis factor antagonist mechanisms of action: a comprehensive review. Pharmacol. Ther. 117, 244–279. doi: 10.1016/j.pharmthera.2007.10.001
Vainio, H., and Morgan, G. (1997). Aspirin for the second hundred years: new uses for an old drug. Pharmacol. Toxicol. 81, 151–152. doi: 10.1111/j.1600-0773.1997.tb02060.x
van de Putte, L. B., Atkins, C., Malaise, M., Sany, J., Russell, A. S., van Riel, P. L., et al. (2004). Efficacy and safety of adalimumab as monotherapy in patients with rheumatoid arthritis for whom previous disease modifying antirheumatic drug treatment has failed. Ann. Rheum. Dis. 63, 508–516. doi: 10.1136/ard.2003.013052
van Vollenhoven, R. F., Geborek, P., Forslind, K., Albertsson, K., Ernestam, S., Petersson, I. F., et al. (2012). Conventional combination treatment versus biological treatment in methotrexate-refractory early rheumatoid arthritis: 2 year follow-up of the randomised, non-blinded, parallel-group Swefot trial. Lancet 379, 1712–1720. doi: 10.1016/S0140-6736(12)60027-0
Vandenabeele, P., Declercq, W., Beyaert, R., and Fiers, W. (1995). Two tumour necrosis factor receptors: structure and function. Trends Cell Biol. 5, 392–399. doi: 10.1016/S0962-8924(00)89088-1
Vane, J. R. (2000). The fight against rheumatism: from willow bark to COX-1 sparing drugs. J. Physiol. Pharmacol. 51(4 Pt 1), 573–586.
Vila-Del Sol, V., Diaz-Munoz, M. D., and Fresno, M. (2007). Requirement of tumor necrosis factor at and nuclear factor-kappa B in the induction by IFN-gamma of inducible nitric oxide synthase in macrophages. J. Leukoc. Biol. 81, 272–283. doi: 10.1189/jlb.0905529
Visser, K., and van der Heijde, D. (2009). Optimal dosage and route of administration of methotrexate in rheumatoid arthritis: a systematic review of the literature. Ann. Rheum. Dis. 68, 1094–1099. doi: 10.1136/ard.2008.092668
Wahl, C., Liptay, S., Adler, G., and Schmid, R. M. (1998). Sulfasalazine: a potent and specific inhibitor of nuclear factor kappa B. J. Clin. Invest. 101, 1163–1174. doi: 10.1172/JCI992
Wallis, R. S. (2007). Reactivation of latent tuberculosis by TNF blockade: the role of interferon gamma. J. Invest. Dermatol. Symp. Proc. 12, 16–21. doi: 10.1038/sj.jidsymp.5650031
Wijnen, P. A., Cremers, J. P., Nelemans, P. J., Erckens, R. J., Hoitsma, E., Jansen, T. L., et al. (2014). Association of the TNF-alpha G-308A polymorphism with TNF-inhibitor response in sarcoidosis. Eur. Respir. J. 43, 1730–1739. doi: 10.1183/09031936.00169413
Willrich, M. A., Murray, D. L., and Snyder, M. R. (2015). Tumor necrosis factor inhibitors: clinical utility in autoimmune diseases. Transl. Res. 165, 270–282. doi: 10.1016/j.trsl.2014.09.006
Yelamos, O., and Puig, L. (2015). Systemic methotrexate for the treatment of psoriasis. Expert Rev. Clin. Immunol. 11, 553–563. doi: 10.1586/1744666X.2015.1026894
Yoo, D. H., Hrycaj, P., Miranda, P., Ramiterre, E., Piotrowski, M., Shevchuk, S., et al. (2013). A randomised, double-blind, parallel-group study to demonstrate equivalence in efficacy and safety of CT-P13 compared with innovator infliximab when coadministered with methotrexate in patients with active rheumatoid arthritis: the PLANETRA study. Ann. Rheum. Dis. 72, 1613–1620. doi: 10.1136/annrheumdis-2012-203090
Zaragoza, B., Chen, X., Oppenheim, J. J., Baeyens, A., Gregoire, S., Chader, D., et al. (2016). Suppressive activity of human regulatory T cells is maintained in the presence of TNF. Nat. Med. 22, 16–17. doi: 10.1038/nm.4019
Keywords: tumor necrosis factor alpha (TNF), autoimmune inflammatory diseases, non-steroidal anti-inflammatory drugs (NSAIDs), anti-TNF biologics, TNF receptor
Citation: Li P, Zheng Y and Chen X (2017) Drugs for Autoimmune Inflammatory Diseases: From Small Molecule Compounds to Anti-TNF Biologics. Front. Pharmacol. 8:460. doi: 10.3389/fphar.2017.00460
Received: 28 February 2017; Accepted: 27 June 2017;
Published: 12 July 2017.
Edited by:
Apostolos Zarros, Queen Mary University of London, United KingdomReviewed by:
Marinos Kallikourdis, Humanitas Università, ItalyDietmar Fuchs, Innsbruck Medical University, Austria
Copyright © 2017 Li, Zheng and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xin Chen, eGNoZW5AdW1hYy5tbw==