 Anji Xiong
Anji Xiong Yi Liu
Yi Liu- Department of Rheumatology and Immunology, West China Hospital, Sichuan University, Chengdu, China
Fibrosis, characterized by increased extracellular matrix (ECM) deposition, and widespread vasculopathy, has the prominent trait of chronic hypoxia. Hypoxia inducible factors-1α (HIF-1α), a key transcriptional factor in response to this chronic hypoxia, is involved in fibrotic disease, such as Systemic sclerosis (SSc). The implicated function of HIF-1α in fibrosis include stimulation of excessive ECM, vascular remodeling, and futile angiogenesis with further exacerbation of chronic hypoxia and deteriorate pathofibrogenesis. This review will focus on the molecular biological behavior of HIF-1α in regulating progressive fibrosis. Better understanding of the role for HIF-1α-regulated pathways in fibrotic disease will accelerate development of novel therapeutic strategies that target HIF-1α. Such new therapeutic strategies may be particularly effective for treatment of the prototypic, multisystem fibrotic, autoimmune disease SSc.
Introduction
Fibrotic disease is a kind of chronic hypoxia related disease with pathogenesis that includes increased extracellular matrix (ECM) deposition, and widespread vasculopathy (Gabrielli et al., 2009). Fibrosis is increasingly seen as the result of deregulated tissue repair in response to chronic hypoxia that results in the excessive accumulation of ECM. Severe chronic hypoxia is overt in involved tissues of fibrotic disease patients (Distler et al., 2004). There are a variety of mechanisms leading to persistent chronic hypoxia. First, continuous and extensive microangiopathy caused by inflammation (Kahaleh, 2004; Cheung et al., 2017) or metabolic stress (Petersen et al., 2017; Wang et al., 2017) is regarded as an early and possibly the earliest pathogenic event in the fibrotic disease (Kahaleh et al., 1979) that leads to chronic hypoxia. Chronic hypoxia in turn induces vascular remodeling ultimately giving rise to progressive luminal narrowing and blockage (Flavahan et al., 2003) resulting in progressive exacerbation of the chronic hypoxic state. Moreover, excessive deposition of ECM, the hallmark of fibrosis (Bhattacharyya et al., 2012), further worsens hypoxia by increasing diffusion distances between blood vessels and tissue cells and increased tissue pressure. Extensive microangiopathy, vascular remodeling, and ECM deposition leads to vascular rarefaction and chronic hypoxia that directly contributes to progressive amplification of fibrosis. Increasing evidence has demonstrated that chronic hypoxia is actively involved in the pathogenesis of fibrosis (Ho et al., 2014) by stimulating the production of ECM including fibronectin-1, IGF-binding protein 3 (Distler et al., 2007), collagens, and collagen-modifying enzymes such as COL4A1, COL4A2, COL5A1, COL9A1, COL18A1, procollagen prolyl hydroxylases (P4HA1 and P4HA2), and lysyl hydroxylases (procollagen lysyl hydroxylase and procollagen lysyl hydroxylase 2)(Manalo et al., 2005). Hence, persistent and extensive chronic hypoxia is a distinctive feature of fibrotic disease that definitely aggravates tissue fibrosis.
Hypoxia inducible factors (HIFs) are regarded as the “master regulators” (Imtiyaz and Simon, 2010) in response to the hypoxic environment and are essential for mediating adaptive reactions to hypoxia (Appelhoff et al., 2004; Farahani et al., 2012). HIFs are in a family of basic–helix-loop-helix/Per-ARNT-Sim (bHLH/PAS) DNA binding transcription factors (Greer et al., 2012) and are heterodimers composed of two different subunits: HIF-α, that is oxygen regulated, and HIF-β, that is expressed constitutively in the nucleus (Wang et al., 1995; Semenza, 2003). There are at least three α subunits-HIF-1α, HIF-2α, and HIF-3α, that accumulate in the cytoplasm and translocate into the nucleus to form heterodimers with a β subunit. After translocating to the nucleus, the HIF heterodimers associate with co-activators and bind to hypoxia response elements (HREs) in gene promoters to initiate gene transcription (Kaelin and Ratcliffe, 2008; Semenza, 2009). Hypoxia induces stabilization and nuclear translocation of HIF-α subunits and their transcriptional activity (Kaelin and Ratcliffe, 2008) by inhibiting the activity of both prolyl hydroxylases and factor-inhibiting HIF1. Hypoxia increases the half-life of HIF-1α from 5 min to approximately 60 min (Huang et al., 1998).
Compelling evidence indicates that HIF-1α plays a key role in vascular remodeling under hypoxic conditions (Yu et al., 1999). The extensive and cumulative vascular remodeling in arterioles that accompanies chronic hypoxia results in multiple internal organ fibrosis and pulmonary hypertension (PH). Of note, PH associated with pulmonary fibrosis is the major cause of mortality in individuals suffering from fibrotic disease, such as SSc, and accumulating evidence has revealed that HIF-1α is implicated in producing excessive ECM which was the underlying cause of fibrosis (Distler et al., 2007; Higgins et al., 2007; Halberg et al., 2009; Ueno et al., 2009; Zhou et al., 2009; Gilkes et al., 2013; Nayak et al., 2016). Fibrosis is typically characterized by prolonged and/or exaggerated activation of fibroblasts (Ho et al., 2014). Strong and stable expression of HIF-1α was found in fibrotic dermal fibroblasts cultured under hypoxic conditions, 1% oxygen, equivalent to a PO2 value of 7 mmHg, which is close to the 10th percentile measured in involved dermal areas of fibrotic disease patients (Hong et al., 2006; Distler et al., 2007). Furthermore, increased expression of HIF-1α occurred in subcutaneous fibroblasts from healthy skin (Modarressi et al., 2010) and fibrotic skin (Hattori et al., 2015) exposed to hypoxic conditions in vitro. Fibroblasts isolated from human arteries also exhibited a remarkable up-regulation of HIF-1α under hypoxic conditions (Krick et al., 2005). In a more detailed study, HIF-1α completely translocated from the cytosol into the nucleus (Wenger, 2002) in dermal fibroblasts from fibrotic disease patients after hypoxic exposure (Distler et al., 2004). HIF-1α expression is elevated in a number of fibrotic diseases (Fine et al., 1998; Baan et al., 2003; Zhang et al., 2003, 2004) and overt up-regulation of HIF-1α in the skin of naïve SSc patients was observed compared with normal skin (Ioannou et al., 2013) further suggesting that HIF1α is involved in the pathogenesis of fibrotic disease, particularly in SSc (Wipff et al., 2009). In addition, HIF-1α is particularly related to subgroups of SSc patients with prominent vascular manifestations (Wipff et al., 2009). Inhibition of HIF-1α is therefore a rational strategy for novel therapeutic development since effective therapies are not yet available for fibrotic disease, such as SSc.
HIF-1α and ECM (Figure 1A)
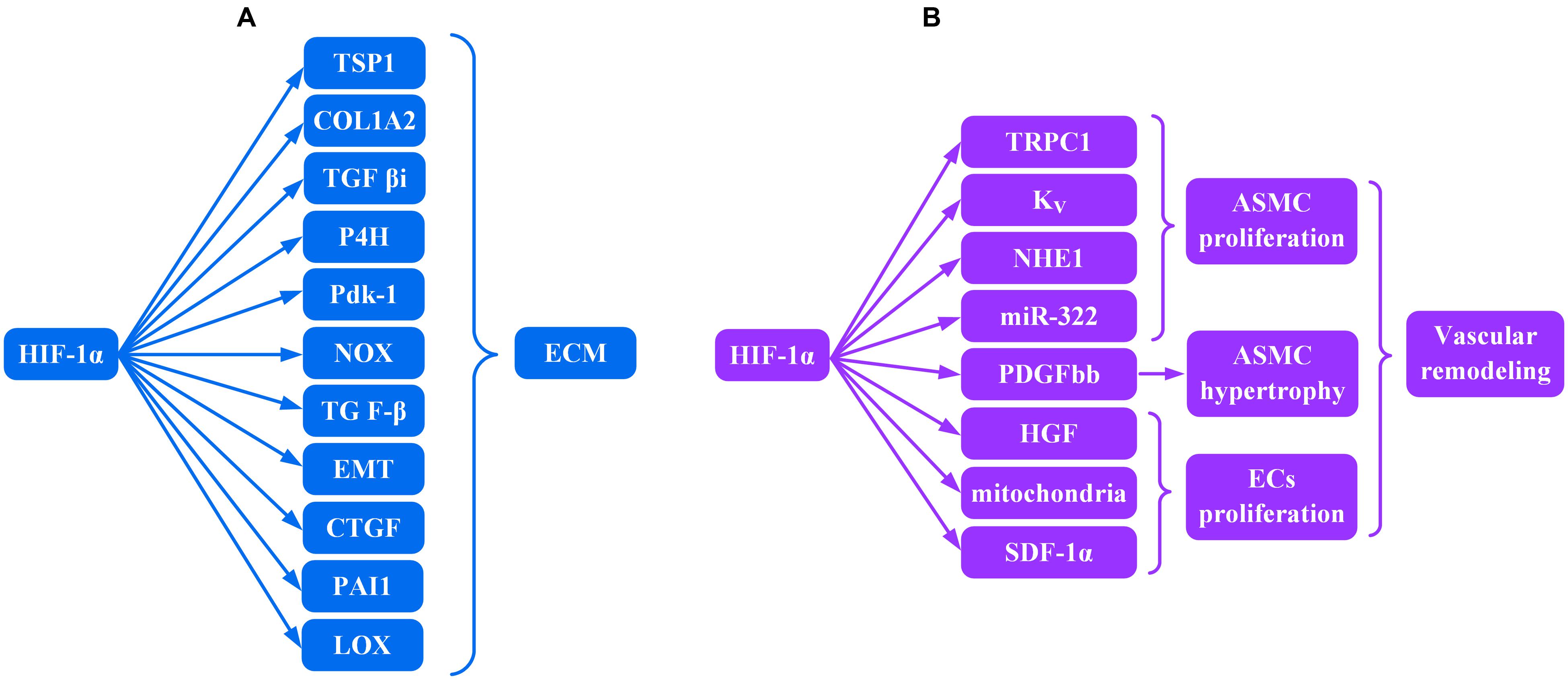
FIGURE 1. Role of HIF-lα transcription factor in the production of (A) ECM and (B) vascular remodeling.
Fibrosis is characterized by excessive deposition of ECM in organs or tissues including different kinds of collagens, hyaluronic acid, fibronectin, and proteoglycans (Ho et al., 2014). HIF-1α contributed to the up-regulated gene expression for several ECM and non-ECM in fibroblast cultures in vitro.
Increased expression of pro α2 (I) collagen (COL1A2), thrombospondin (TSP) 1, and transforming growth factor β–induced protein (TGF βi) were observed in both mouse embryonic and human dermal fibroblasts under hypoxic conditions (Distler et al., 2007). Bentovim et al. (2012) demonstrated that HIF-1α induced collagen hydroxylation and normal collagen secretion in the hypoxic milieu by directly activating transcription of the collagen prolyl 4-hydroxylase enzyme (P4H) and pyruvate dehydrogenase kinase 1 (Pdk1). HIF-1α deficiency resulted in impaired collagen secretion in the presence of hypoxia. Similarly, HIF-1α mediates ECM accumulation through NADPH oxidase (NOX) in vitro in cultured renal mesangial cells (Nayak et al., 2016). Microarray genome expression profiling from skin biopsies of fibrotic disease patients revealed that a prominent alteration in gene expression underlying fibrosis is within the transforming growth factor β (TGF-β) pathway (Whitfield et al., 2003), and TGF-β was closely involved in the induction of ECM (Falanga et al., 1987). However, HIF-1α is upstream of TGF-β production, and hypoxia-induced TGF-β production requires HIF-1α (Zhou et al., 2009). Qian et al. (2015) demonstrated that inhibition of HIF-1α reduced TGF-β expression in vivo as well.
Epithelial-to-mesenchymal transition (EMT) can be characterized by acquisition of mesenchymal markers such as α-smooth muscle actin (α-SMA). EMT results in the production of more ECM including α-SMA and vimentin (Strutz et al., 1995; Zeisberg and Kalluri, 2004) and requires HIF-1α expression (Zhou et al., 2009). Higgins et al. (2007) demonstrated that increased HIF-1α expression may promote fibrogenesis by facilitating EMT. Plasminogen activator inhibitor-1 (PAI-1), found in the ECM (Podor and Loskutoff, 1992) and a key inhibitor of fibrinolysis (Collen and Lijnen, 1991), inhibited proteolytic processes that were linked with fibrosis (Eddy et al., 1995). HIF-1α heterodimers with HIF-1β induced by hypoxia in vivo bind HRE in the PAI-1 promoter and induce PAI-1 expression (Kietzmann et al., 1999). Lysyl oxidase (LOX) is important for normal synthesis of collagen and elastin (Giampuzzi et al., 2000; Oleggini et al., 2007). LOX is a transcriptional target for HIF-1α-HIF-1β heterodimers (Halberg et al., 2009) that translocate into the nuclear compartment of fibrogenic cells (Li et al., 1997) and is up-regulated during fibrogenesis. Higgins et al. (2007), Halberg et al. (2009) showed that HIF-1α could up-regulate the expression of LOX in vivo and in vitro, leading to the accumulation of collagen and other components involved in establishing and remodeling the ECM. Halberg et al. (2009) pinpointed LOX as a key player in HIF-1α mediated deposition of ECM. Furthermore, connective tissue growth factor (CTGF) has been reported to enhance cell proliferation and ECM production in fibroblasts (Frazier et al., 1996). Mounting evidence has demonstrated that expression of CTGF is upregulated during fibrotic disorders (Igarashi et al., 1996; Ito et al., 1998; Leask et al., 2002; Higgins et al., 2003), and in hypoxia, the induction of CTGF is directly mediated by HIF-1α-HIF-1β heterodimer binding to the CTGF associated HRE (Higgins et al., 2004). In summary, HIF-1α is ubiquitous in many different tissues (Wang et al., 1995) and in fibrotic disease contributes to persistent pathofibrogenesis in multiple organs by stimulating production of excessive ECM.
HIF-1α and Vascular Remodeling (Figure 1B)
Vascular remodeling is primarily composed of dysregulated proliferation of endothelial cells (ECs) and an increase in the number (hyperplasia) and volume (hypertrophy) of arterial smooth muscle cells (ASMC) resulting in progressive vascular occlusion and chronic hypoxia. High expression of HIF-1α within endothelial plexiform lesions (Tuder et al., 2001) and ASMC (Bonnet et al., 2006) suggests a strong correlation between HIF-1α and proliferative vasculopathy.
Arterial smooth muscle cells hyperproliferation in the media of the artery was suggested to be the key event in vascular remodeling (Cheng et al., 2017). Transient receptor potential channel (TRPC) 1, a non-selective cation channel, is permeable to Ca2+ ions. Increase in levels of TRPC1 mediated by bone morphogenetic protein4 (BMP4) (Wang et al., 2015) was HIF-1α dependent in ASMC (Wang et al., 2006). Reduction in voltage-gated K+ currents, resulting in membrane depolarization and activation of voltage-dependent Ca2+ channels and subsequently increasing Ca2+ influx, was regulated by HIF-1α as well (Shimoda et al., 2001). Both voltage-gated K+ (Kv) channels and TRPC1, mediated by HIF-1α, contributed to an increase in cytosolic free Ca2+ which was a major trigger for ASMC proliferation (Veith et al., 2016). ASMC proliferation may be a consequence of up-regulated aquaporin 1 as a result of the increased cytosolic free Ca2+ (Yun et al., 2015). Furthermore, both TRPC1 silencing by small interfering RNA (siRNA) and TRPC1 knockout impaired hypoxia-induced ASMC proliferation in vitro, and TRPC1-/- mice had less vascular muscularization compared with wild type mice (Malczyk et al., 2013). In addition, hypoxic induction of the Na+/H+ exchanger isoform 1 (NHE1) expression and alkalinization of intracellular pH were regulated by HIF-1α (Shimoda et al., 2006). Both activation of the Na+/H+ exchanger and alkalinization of intracellular pH were necessary for ASMC proliferation (Quinn et al., 1996). Zeng et al. (2015) demonstrated that HIF-1α transcriptionally upregulated the expression of miR-322 in hypoxia, which led to proliferative responses of ASMC due to direct targeting of BMPR1a and smad5. Similarly, Platelet derived growth factor bb (PDGFbb) can induce proliferation of ASMC in vitro and in vivo (Schermuly et al., 2005). PDGFbb-induced signaling gave rise to the hypertrophy of ASMC both in vitro and in vivo (Ke et al., 2016) via excessive deposition of hyaluronic acid (HA) in smooth muscle cells (Pullen et al., 2001). The possible mechanism is through tyrosine 31 (Y31) and 118 (Y118) phosphorylation of paxillin, which was attenuated by HIF-1α knockdown (Veith et al., 2014).
Similarly, HIF-1α is also involved in the proliferation of ECs. Abnormally proliferating ECs are characterized by low numbers of mitochondria (Xu et al., 2007). Knockdown of HIF-1α increased the numbers of mitochondria in ECs in vitro (Fijalkowska et al., 2010) and suggests that the reduced mitochondria number in abnormally proliferating ECs may be a consequence, at least in part, of increased HIF-1α expression. HIF -1α inducible factors include hepatocyte growth factor (HGF) (Kitajima et al., 2008) and stromal-derived factor-1a (SDF-1a) (Ceradini et al., 2004). A special kind of hematopoietic endothelial stem cell, CD34+CD133+hemangioblast, may promote angioproliferative vascular remodeling (Asosingh et al., 2008). Local production of chemoattractants, such as SDF-1α and HGF, by diseased endothelium can recruit substantial numbers of CD34+CD133+hemangioblasts to sites of angioproliferative vascular remodeling (Farha et al., 2011). Both signal transducers and activators of transcription (STAT) 3 (Xu and Erzurum, 2011) and chloride intracellular channel 4 (CLIC4) (Wojciak-Stothard et al., 2014) contribute to the hyperproliferative pathology of ECs invoking another important role for HIF-1α in vascular fibrosis.
Targeting HIF-1α in Fibrosis
Studies to date indicate that HIF-1α is intimately involved in persistent pathofibrogenesis, vascular remodeling, and PH in fibrotic disease. Severe, multiple organ fibrosis associated with the continuous accumulation of HIF-1α, caused by chronic or prolonged hypoxia in fibrotic disease, suggests that HIF-1α maybe a promising target for novel fibrotic disease treatments, such as SSc.
Recently, hypoxic prodrugs, projecting to be specifically activated in the low O2 milieu, deliver the active agent to hypoxic tissues through reduction of the prodrug by cellular reductases (Phillips, 2016). These hypoxic prodrug agents may significantly alleviate off-target effects of the biological therapy by limiting active drug to hypoxic tissue and only inhibiting HIF-1α in hypoxic tissues. Gene therapy targeting HIF-1α may also be effective for therapy in hypoxia-related diseases as well (Tal et al., 2008; Wang et al., 2008; del Rey et al., 2009; Chen et al., 2016). In addition, the therapeutic benefits of HIF-1α inhibitors would be maximized in the presence of delivery carriers that eliminate pharmacokinetic and stability problems and minimize potential systemic toxicity. For example, liposomes and nanoscale-based drug delivery systems may be applied as a delivery assistant for HIF-1α gene therapy (Wang et al., 2008; Chen et al., 2016). The most successful example of a successful liposomal drug delivery system may be that for Amphotericin B, which has been widely applied in the clinic for treating invasive fungal infections. Amphotericin B is a highly effective drug but with potential severe toxic side effects (Barratt and Bretagne, 2007; Wasko et al., 2012). Amphotericin B encapsulated in liposome has significantly reduced toxicity as well as increased therapeutic benefit when administered systemically encapsulated within liposomes (Torchilin, 2005; Allen and Cullis, 2013). Antisense oligonucleotides targeted to HIF-1α mRNA combined with doxorubicin were successfully delivered to oncocytes by poly (ethylene glycol) polymer (PEGylated) liposomes as drug carriers (Wang et al., 2008). Furthermore, YC-1 [3-(5′-hydroxymethyl-2′-furyl)-1-benzyl indazole], a HIF-1α inhibitor, reduced ECM accumulation in vivo (Nayak et al., 2016). Trichostatin A, identified indirectly to down-regulate HIF-1α, has been applied in clinical trials in patients with cancers (Kim et al., 2001) and has been shown to reduce the release of collagen from fibrotic dermal fibroblasts in vitro (Huber et al., 2005). In conclusion, a viable therapy option for fibrotic disease may include agents that target and inhibit HIF-1α since delivery vehicles may help reduce off-target effects and enhance therapeutic efficiency (Sercombe et al., 2015).
On the other hand, HIF-1α has been repeatedly observed to assist wound healing through inflammation, angiogenesis, vasculargenesis, and fibroplasia in acute injury (Semenza, 1998, 1999). The most prominent contraindication for systemic administration of HIF-1α inhibitors, therefore, is trauma. An analogy for adverse effects that may accompany biological therapy to inhibit HIF-1α may be tumor necrosis factor-α (TNF-α) inhibitors in rheumatic diseases. TNF-α inhibitors inhibit inflammation that is necessary for tissue repair. HIF-1α is required for repair in acute injury as well (Darby and Hewitson, 2016). In particular, we need to be vigilant about physiological repair events such as menstruation within the context of HIF-1α inhibitor administration.
Persistent and remarkable up-regulation of vascular endothelial growth factor (VEGF) has been observed in all stages of fibrotic disease (Distler et al., 2004) and actively proliferating ECs of plexiform lesions (Tuder et al., 2001). VEGF is the predominant proangiogenic factor regulated by HIF-1α in other hypoxia related diseases, but VEGF up-regulation and consequent massive and extensive microangiopathy in fibrotic disease is HIF-1α independent, even with hypoxia (Distler et al., 2004). HIF-1α expression did not correlate with up-regulated VEGF in affected tissues from patients suffering from fibrotic disease (Distler et al., 2004). Since HIF-1α is critical for VEGF up-regulation in other hypoxia related diseases, consideration must be given to the question of whether deceased VEGF after systemic HIF-1α inhibition may severely decrease and impair neoangiogenesis. The worry may be unnecessary for fibrotic disease and its associated massive and extensive microangiopathy in affected tissues and organs. Up-regulated expression of VEGF is also driven by interleukin-1β, PDGF, and TGF-β, all of which are up-regulated in fibrotic disease and can stimulate the expression of VEGF (Pertovaara et al., 1994; Kissin and Korn, 2003). Moreover, the role played by HIF-2α and HIF-3α in the over-expression of VEGF has not yet been extensively investigated. Above all, sufficient tissue vascularization depends on strict regulation of VEGF expression rather than on persistent up-regulated expression of VEGF (Distler et al., 2004). The formation of chaotic vessels, feathered with glomeruloid and haemangioma-like morphology, was partly due to chronic and uncontrolled over-expression of VEGF (Drake and Little, 1995; Sundberg et al., 2001). Dor et al. (2002) designed an animal model system in which a source of VEGF could be specifically induced and steadily maintained for a desired duration and then subsequently switched off. Time-dependent regulation of VEGF expression was necessary for adequate and normal vascularization (Dor et al., 2002). Persistent, uninterrupted exposure to VEGF led to formation of irregularly shaped, sac-like vessels resulting in decreased blood flow compared to normal, mature, functional blood vessel formation after short-term over-expression of VEGF (Dor et al., 2002). Irregularly shaped, sac-like vessels observed in nailfold, a prominent character of the prototypic fibrotic disease-SSc (LeRoy, 1996), may also suggest that persistent up-regulated expression of VEGF is involved in fibrotic disease, and is harmful rather than beneficial, regardless of whether VEGF is HIF-1α independent in fibrosis (Distler et al., 2004). Other angiogenic factors contribute less to neovascularization and have no effect on irregular and sac-like vessels in the presence of persistent and remarkable up-regulation of VEGF. The US FDA approved FTY720 inhibits HIF-1α accumulation by inhibiting the S1P signaling pathway. FTY720 transformed a chaotic vascular network to vascular normalization while simultaneously and subsequently redressing hypoxia in vivo and in vitro (Gstalder et al., 2016). That result gives further credence to the suggestion that HIF-1α is implicated in chaotic angiogenesis. This result also suggests that targeting HIF-1α would be a viable strategy for fibrotic disease, such as SSc, without impairing normal angiogenesis.
Conclusion
HIF-1α per se is helpful in repairing injury and correcting hypoxia via multiple mechanisms, however, prolonged exposure to HIF-1α is harmful and contributes to persistent pathofibrogenesis in fibrotic disease (Figure 2). Furthermore, fibrosis in organs resulting in organ failure accounts for much of the morbidity and mortality associated with fibrotic disease. SSc is prototypic multisystem fibrotic disease and present immunosuppressive therapy exhibits intolerable side effects without selectively targeting the immunopathogenic mechanisms responsible for SSc. In addition, fibrosis in SSc is not restricted to a single organ, but rather involves multiple internal organs and skin. Biotherapy targeting HIF-1α, therefore, is a promising therapeutic alternative that is more likely to confer therapeutic benefits specific to fibrotic disease, particularly to SSc, by attenuating fibrosis and terminating or delaying vascular remodeling.
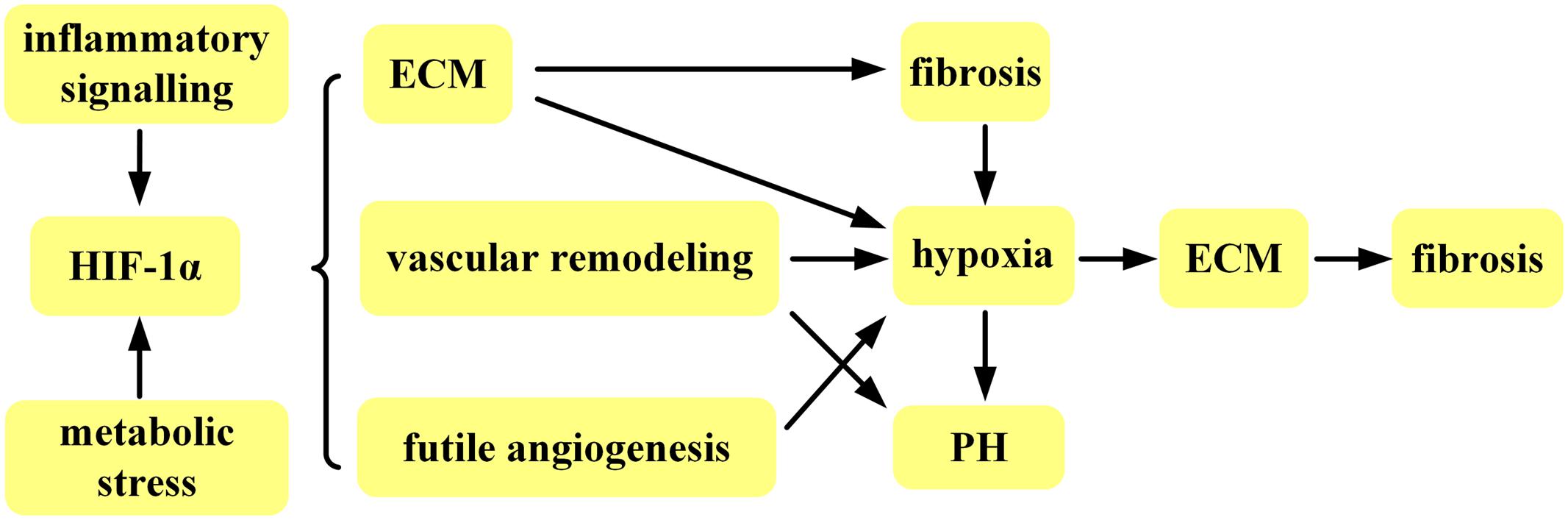
FIGURE 2. Simplified overview of HIF-1α implicated in fibrotic disease.
Author Contributions
AX wrote and assembled the manuscript. YL prepared the figures and revised the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
The authors acknowledge Dr. Tony Marion for critical reading and editing of the manuscript.
References
Allen, T. M., and Cullis, P. R. (2013). Liposomal drug delivery systems: From concept to clinical applications. Adv. Drug Deliv. Rev. 65, 36–48. doi: 10.1016/j.addr.2012.09.037
Appelhoff, R. J., Tian, Y. M., Raval, R. R., Turley, H., Harris, A. L., Pugh, C. W., et al. (2004). Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J. Biol. Chem. 279, 38458–38465. doi: 10.1074/jbc.M406026200
Asosingh, K., Aldred, M. A., Vasanji, A., Drazba, J., Sharp, J., Farver, C., et al. (2008). Circulating angiogenic precursors in idiopathic pulmonary arterial hypertension. Am. J. Pathol. 172, 615–627. doi: 10.2353/ajpath.2008.070705
Baan, C., van Gelder, T., Peeters, A., Mol, W., Niesters, H., Weimar, W., et al. (2003). Living kidney donors and hypoxia-inducible factor-1 alpha. Transplantation 75, 570–571. doi: 10.1097/01.tp.0000034241.55602.ad
Barratt, G., and Bretagne, S. (2007). Optimizing efficacy of Amphotericin B through nanomodification. Int. J. Nanomed. 2, 301–313.
Bentovim, L., Amarilio, R., and Zelzer, E. (2012). HIF1 alpha is a central regulator of collagen hydroxylation and secretion under hypoxia during bone development. Development 139, 4473–4483. doi: 10.1242/dev.083881
Bhattacharyya, S., Wei, J., and Varga, J. (2012). Understanding fibrosis in systemic sclerosis: shifting paradigms, emerging opportunities. Nat. Rev. Rheumatol. 8, 42–54. doi: 10.1038/nrrheum.2011.149
Bonnet, S., Michelakis, E. D., Porter, C. J., Andrade-Navarro, M. A., Thebaud, B., Bonnet, S., et al. (2006). An abnormal mitochondrial-hypoxia inducible factor-1 alpha-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats - Similarities to human pulmonary arterial hypertension. Circulation 113, 2630–2641. doi: 10.1161/circulationaha.105.609008
Ceradini, D. J., Kulkarni, A. R., Callaghan, M. J., Tepper, O. M., Bastidas, N., Kleinman, M. E., et al. (2004). Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat. Med. 10, 858–864. doi: 10.1038/nm1075
Chen, Z., Zhang, T., Wu, B., and Zhang, X. (2016). Insights into the therapeutic potential of hypoxia-inducible factor-1 alpha small interfering RNA in malignant melanoma delivered via folate-decorated cationic liposomes. Int. J. Nanomed. 11, 991–1002. doi: 10.2147/ijn.s101872
Cheng, G., Wang, X., Li, Y., and He, L. (2017). Let-7a-transfected mesenchymal stem cells ameliorate monocrotaline-induced pulmonary hypertension by suppressing pulmonary artery smooth muscle cell growth through STAT3-BMPR2 signaling. Stem cell Res. Ther. 8:34. doi: 10.1186/s13287-017-0480-y
Cheung, A. C., Lazaridis, K. N., LaRusso, N. F., and Gores, G. J. (2017). Emerging pharmacologic therapies for primary sclerosing cholangitis. Curr. Opin. Gastroenterol. 33, 149–157. doi: 10.1097/mog.0000000000000352
Collen, D., and Lijnen, H. R. (1991). Basic and clinical aspects of fibrinolysis and thrombolysis. Blood 78, 3114–3124.
Darby, I. A., and Hewitson, T. D. (2016). Hypoxia in tissue repair and fibrosis. Cell Tissue Res. 365, 553–562. doi: 10.1007/s00441-016-2461-3
del Rey, M. J., Izquierdo, E., Caja, S., Usategui, A., Santiago, B., Galindo, M., et al. (2009). Human inflammatory synovial fibroblasts induce enhanced myeloid cell recruitment and angiogenesis through a hypoxia-inducible transcription factor 1 alpha/vascular endothelial growth factor-mediated pathway in immunodeficient mice. Arthritis Rheumatol. 60, 2926–2934. doi: 10.1002/art.24844
Distler, J. H. W., Juengel, A., Pileckyte, M., Zwerina, J., Michel, B. A., Gay, R. E., et al. (2007). Hypoxia-induced increase in the production of extracellular matrix proteins in systemic sclerosis. Arthritis Rheum. 56, 4203–4215. doi: 10.1002/art.23074
Distler, O., Distler, J. H. W., Scheid, A., Acker, T., Hirth, A., Rethage, J., et al. (2004). Uncontrolled expression of vascular endothelial growth factor and its receptors leads to insufficient skin angiogenesis in patients with systemic sclerosis. Circ. Res. 95, 109–116. doi: 10.1161/01.res.0000134644.89917.96
Dor, Y., Djonov, V., Abramovitch, R., Itin, A., Fishman, G. I., Carmeliet, P., et al. (2002). Conditional switching of VEGF provides new insights into adult neovascularization and pro-angiogenic therapy. Embo J. 21, 1939–1947. doi: 10.1093/emboj/21.8.1939
Drake, C. J., and Little, C. D. (1995). Exogenous vascular endothelial growth-factor induces malformed and hyperfused vessels during embryonic neovascularization. Proc. Natl. Acad. Sci. U.S.A. 92, 7657–7661. doi: 10.1073/pnas.92.17.7657
Eddy, A. A., Giachelli, C. M., McCulloch, L., and Liu, E. (1995). Renal expression of genes that promote interstitial inflammation and fibrosis in rats with protein-overload proteinuria. Kidney Int. 47, 1546–1557. doi: 10.1038/ki.1995.218
Falanga, V., Tiegs, S. L., Alstadt, S. P., Roberts, A. B., and Sporn, M. B. (1987). Transforming growth-factor-beta - selective increase in glycosaminoglycan synthesis by cultures of fibroblasts from patients with progressive systemic-sclerosis. J. Invest. Dermatol. 89, 100–104. doi: 10.1111/1523-1747.ep12580445
Farahani, R. M., Sarrafpour, B., Simonian, M., Li, Q., and Hunter, N. (2012). Directed glia-assisted angiogenesis in a mature neurosensory structure: pericytes mediate an adaptive response in human dental pulp that maintains blood-barrier function. J. Compar. Neurol. 520, 3803–3826. doi: 10.1002/cne.23162
Farha, S., Asosingh, K., Xu, W., Sharp, J., George, D., Comhair, S., et al. (2011). Hypoxia-inducible factors in human pulmonary arterial hypertension: a link to the intrinsic myeloid abnormalities. Blood 117, 3485–3493. doi: 10.1182/blood-2010-09-306357
Fijalkowska, I., Xu, W., Comhair, S. A. A., Janocha, A. J., Mavrakis, L. A., Krishnamachary, B., et al. (2010). Hypoxia inducible-factor1 alpha regulates the metabolic shift of pulmonary hypertensive endothelial cells. Am. J. Pathol. 176, 1130–1138. doi: 10.2353/ajpath.2010.090832
Fine, L. G., Orphanides, C., and Norman, J. T. (1998). Progressive renal disease: the chronic hypoxia hypothesis. Kidney Int. 65, S74–S78.
Flavahan, N. A., Flavahan, S., Mitra, S., and Chotani, M. A. (2003). The vasculopathy of Raynaud’s phenomenon and scleroderma. Rheum. Dis. Clin. North Am. 29, 275–91, vi. doi: 10.1016/s0889-857x(03)00021-8
Frazier, K., Williams, S., Kothapalli, D., Klapper, H., and Grotendorst, G. R. (1996). Stimulation of fibroblast cell growth, matrix production, and granulation tissue formation by connective tissue growth factor. J. Invest. Dermatol. 107, 404–411. doi: 10.1111/1523-1747.ep12363389
Gabrielli, A., Avvedimento, E. V., and Krieg, T. (2009). Mechanisms of disease: scleroderma. N. Engl. J. Med. 360, 1989–2003. doi: 10.1056/NEJMra0806188
Giampuzzi, M., Botti, G., Di Duca, M., Arata, L., Ghiggeri, G., Gusmano, R., et al. (2000). Lysyl oxidase activates the transcription activity of human collagene III promoter - Possible involvement of Ku antigen. J. Biol. Chem. 275, 36341–36349. doi: 10.1074/jbc.M003362200
Gilkes, D. M., Bajpai, S., Chaturvedi, P., Wirtz, D., and Semenza, G. L. (2013). Hypoxia-inducible factor 1 (HIF-1) promotes extracellular matrix remodeling under hypoxic conditions by inducing P4HA1, P4HA2, and PLOD2 expression in fibroblasts. J. Biol. Chem. 288, 10819–10829. doi: 10.1074/jbc.M112.442939
Greer, S. N., Metcalf, J. L., Wang, Y., and Ohh, M. (2012). The updated biology of hypoxia-inducible factor. Embo J. 31, 2448–2460. doi: 10.1038/emboj.2012.125
Gstalder, C., Ader, I., and Cuvillier, O. (2016). FTY720 (Fingolimod) inhibits HIF1 and HIF2 signaling, promotes vascular remodeling, and chemosensitizes in renal cell carcinoma animal model. Mol. Cancer Ther. 15, 2465–2474. doi: 10.1158/1535-7163.mct-16-0167
Halberg, N., Khan, T., Trujillo, M. E., Wernstedt-Asterholm, I., Attie, A. D., Sherwani, S., et al. (2009). Hypoxia-inducible factor 1 alpha induces fibrosis and insulin resistance in white adipose tissue. Mol. Cell. Biol. 29, 4467–4483. doi: 10.1128/mcb.00192-09
Hattori, M., Yokoyama, Y., Hattori, T., Motegi, S.-I., Amano, H., Hatada, I., et al. (2015). Global DNA hypomethylation and hypoxia-induced expression of the ten eleven translocation (TET) family, TET1, in scleroderma fibroblasts. Exp. Dermatol. 24, 841–846. doi: 10.1111/exd.12767
Higgins, D. F., Biju, M. P., Akai, Y., Wutz, A., Johnson, R. S., and Haase, V. H. (2004). Hypoxic induction of Ctgf is directly mediated by Hif-1. Am. J. Physiol. Renal Physiol. 287, F1223–F1232. doi: 10.1152/ajprenal.00245.2004
Higgins, D. F., Kimura, K., Bernhardt, W. M., Shrimanker, N., Akai, Y., Hohenstein, B., et al. (2007). Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J. Clin. Invest. 117, 3810–3820. doi: 10.1172/jci30487
Higgins, D. F., Lappin, D. W. P., Kieran, N. E., Anders, H. J., Watson, R. W. G., Strutz, F., et al. (2003). DNA oligonucleotide microarray technology identifies fisp-12 among other potential fibrogenic genes following murine unilateral ureteral obstruction (UUO): modulation during epithelial-mesenchymal transition. Kidney Int. 64, 2079–2091. doi: 10.1046/j.1523-1755.2003.00306.x
Ho, Y. Y., Lagares, D., Tager, A. M., and Kapoor, M. (2014). Fibrosis-a lethal component of systemic sclerosis. Nat. Rev. Rheumatol. 10, 390–402. doi: 10.1038/nrrheum.2014.53
Hong, K. H., Yoo, S. A., Kang, S. S., Choi, J. J., Kim, W. U., and Cho, C. S. (2006). Hypoxia induces expression of connective tissue growth factor in scleroderma skin fibroblasts. Clin. Exp. Immunol. 146, 362–370. doi: 10.1111/j.1365-2249.2006.03199.x
Huang, L. E., Gu, J., Schau, M., and Bunn, H. F. (1998). Regulation of hypoxia-inducible factor 1 alpha is mediated by an O-2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc. Natl. Acad. Sci. U.S.A. 95, 7987–7992. doi: 10.1073/pnas.95.14.7987
Huber, L. C., Distler, J. H. W., Michel, B. A., Gay, R. E., Kalden, J. R., Matucci-Cerinic, M., et al. (2005). Inhibition of histone deacetylases reduces the TGF beta-stimulated production of extracellular matrix proteins of skin fibroblasts from patients with systemic sclerosis (SSC). Arthritis Rheum. 52, S463–S463.
Igarashi, A., Nashiro, K., Kikuchi, K., Sato, S., Ihn, H., Fujimoto, M., et al. (1996). Connective tissue growth factor gene expression in tissue sections from localized scleroderma, keloid, and other fibrotic skin disorders. J. Invest. Dermatol. 106, 729–733. doi: 10.1111/1523-1747.ep12345771
Imtiyaz, H. Z., and Simon, M. C. (2010). “Hypoxia-inducible factors as essential regulators of inflammation,” in Diverse Effects of Hypoxia on Tumor Progression, ed. M. C. Simon (Berlin: Springer).
Ioannou, M., Pyrpasopoulou, A., Simos, G., Paraskeva, E., Nikolaidou, C., Venizelos, I., et al. (2013). Upregulation of VEGF expression is associated with accumulation of HIF-1 alpha in the skin of naive scleroderma patients. Mod. Rheumatol. 23, 1245–1248. doi: 10.1007/s10165-012-0787-6
Ito, Y., Aten, J., Bende, R. J., Oemar, B. S., Rabelink, T. J., Weening, J. J., et al. (1998). Expression of connective tissue growth factor in human renal fibrosis. Kidney Int. 53, 853–861. doi: 10.1046/j.1523-1755.1998.00820.x
Kaelin, W. G. Jr., and Ratcliffe, P. J. (2008). Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol. Cell. 30, 393–402. doi: 10.1016/j.molcel.2008.04.009
Kahaleh, M. B. (2004). Raynaud phenomenon and the vascular disease in scleroderma. Curr. Opin. Rheumatol. 16, 718–722. doi: 10.1097/01.bor.0000138677.88694.a4
Kahaleh, M. B., Sherer, G. K., and Leroy, E. C. (1979). Endothelial injury in scleroderma. J. Exp. Med. 149, 1326–1335. doi: 10.1084/jem.149.6.1326
Ke, M.-W., Cheng, C.-C., Huang, W.-C., Wann, S.-R., Shu, C.-W., Tseng, C.-J., et al. (2016). Suppression of HIF-1 alpha-mediated PDGF(BB) signaling inhibits the hypertrophy of pulmonary arterial smooth muscle cell in vitro and in vivo. Circ. Res. 119, E170–E170.
Kietzmann, T., Roth, U., and Jungermann, K. (1999). Induction of the plasminogen activator inhibitor-1 gene expression by mild hypoxia via a hypoxia response element binding the hypoxia-inducible factor-1 in rat hepatocytes. Blood 94, 4177–4185.
Kim, M. S., Kwon, H. J., Lee, Y. M., Baek, J. H., Jang, J. E., Lee, S. W., et al. (2001). Histone deacetylases induce angiogenesis by negative regulation of tumor suppressor genes. Nat. Med. 7, 437–443. doi: 10.1038/86507
Kissin, E. Y., and Korn, J. H. (2003). Fibrosis in scleroderma. Rheum. Dis. Clin. North Am. 29, 351–369. doi: 10.1016/s0889-857x(03)00018-8
Kitajima, Y., Ide, T., Ohtsuka, T., and Miyazaki, K. (2008). Induction of hepatocyte growth factor activator gene expression under hypoxia activates the hepatocyte growth factor/c-Met system via hypoxia inducible factor-1 in pancreatic cancer. Cancer Sci. 99, 1341–1347. doi: 10.1111/j.1349-7006.2008.00828.x
Krick, S., Hanze, J., Eul, B., Savai, R., Seay, U., Grimminger, F., et al. (2005). Hypoxia-driven proliferation of human pulmonary artery fibroblasts: cross-talk between HIF-1 alpha and an autocrine angiotensin system. FASEB J. 19, 857–859. doi: 10.1096/fj.04-2890fje
Leask, A., Holmes, A., and Abraham, D. J. (2002). Connective tissue growth factor: a new and important player in the pathogenesis of fibrosis. Curr. Rheumatol. Rep. 4, 136–142. doi: 10.1007/s11926-002-0009-x
LeRoy, E. C. (1996). Systemic sclerosis - A vascular perspective. Rheum. Dis. Clin. North Am. 22, 675–694. doi: 10.1016/S0889-857X(05)70295-7
Li, W. D., Nellaiappan, K., Strassmaier, T., Graham, L., Thomas, K. M., and Kagan, H. M. (1997). Localization and activity of lysyl oxidase within nuclei of fibrogenic cells. Proc. Natl. Acad. Sci. U.S.A. 94, 12817–12822. doi: 10.1073/pnas.94.24.12817
Malczyk, M., Veith, C., Fuchs, B., Hofmann, K., Storch, U., Schermuly, R. T., et al. (2013). Classical transient receptor potential channel 1 in hypoxia-induced pulmonary hypertension. Am. J. Respir. Crit. Care Med. 188, 1451–1459. doi: 10.1164/rccm.201307-1252OC
Manalo, D. J., Rowan, A., Lavoie, T., Natarajan, L., Kelly, B. D., Ye, S. Q., et al. (2005). Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood 105, 659–669. doi: 10.1182/blood-2004-07-2958
Modarressi, A., Pietramaggiori, G., Godbout, C., Vigato, E., Pittet, B., and Hinz, B. (2010). Hypoxia impairs skin myofibroblast differentiation and function. J. Investig. Dermatol. 130, 2818–2827. doi: 10.1038/jid.2010.224
Nayak, B. K., Shanmugasundaram, K., Friedrichs, W. E., Cavaglierii, R. C., Patel, M., Barnes, J., et al. (2016). HIF-1 mediates renal fibrosis in OVE26 type 1 diabetic mice. Diabetes Metab. Res. Rev. 65, 1387–1397. doi: 10.2337/db15-0519
Oleggini, R., Gastaldo, N., and Di Donato, A. (2007). Regulation of elastin promoter by lysyl oxidase and growth factors: cross control of lysyl oxidase on TGF-beta 1 effects. Matrix Biol. 26, 494–505. doi: 10.1016/j.matbio.2007.02.003
Pertovaara, L., Kaipainen, A., Mustonen, T., Orpana, A., Ferrara, N., Saksela, O., et al. (1994). Vascular endothelial growth-factor is induced in response to transforming growth-factor-beta in fibroblastic and epithelial-cells. J. Biol. Chem. 269, 6271–6274.
Petersen, P. S., Lei, X., Wolf, R. M., Rodriguez, S., Tan, S. Y., Little, H. C., et al. (2017). CTRP7 deletion attenuates obesity-linked glucose intolerance, adipose tissue inflammation, and hepatic stress. Am. J. Physiol. Endocrinol. Metab. 312, E309–E325. doi: 10.1152/ajpendo.00344.2016
Phillips, R. M. (2016). Targeting the hypoxic fraction of tumours using hypoxia-activated prodrugs. Cancer Chemother. Pharmacol. 77, 441–457. doi: 10.1007/s00280-015-2920-7
Podor, T. J., and Loskutoff, D. J. (1992). Immunoelectron microscopic localization of type 1 plasminogen activator inhibitor in the extracellular matrix of transforming growth factor-β-activated endothelial cells. Ann. N.Y. Acad. Sci. 667, 46–49. doi: 10.1111/j.1749-6632.1992.tb51595.x
Pullen, M. A., Thomas, K., Wu, H. L., and Nambi, P. (2001). Stimulation of hyaluronan synthetase by platelet-derived growth factor bb in human prostate smooth muscle cells. Pharmacology 62, 103–106. doi: 10.1159/000056079
Qian, F., He, M., Duan, W., Mao, L., Li, Q., Yu, Z., et al. (2015). Cross regulation between hypoxia-inducible transcription factor-1 alpha (HIF-1 alpha) and transforming growth factor (TGF)-beta 1 mediates nickel oxide nanoparticles (NiONPs)-induced pulmonary fibrosis. Am. J. Trans. Res. 7, 2364–2378.
Quinn, D. A., Dahlberg, C. G. W., Bonventre, J. P., Scheid, C. R., Honeyman, T., Joseph, P. M., et al. (1996). The role of Na+/H+ exchange and growth factors in pulmonary artery smooth muscle cell proliferation. Am. J. Respir. Cell Mol. Biol. 14, 139–145. doi: 10.1165/ajrcmb.14.2.8630263
Schermuly, R. T., Dony, E., Ghofrani, H. A., Pullamsetti, S., Savai, R., Roth, M., et al. (2005). Reversal of experimental pulmonary hypertension by PDGF inhibition. J. Clin. Investig. 115, 2811–2821. doi: 10.1172/jci24838
Semenza, G. L. (1998). Hypoxia-inducible factor 1: master regulator of O-2 homeostasis. Curr. Opin. Genet. Dev. 8, 588–594. doi: 10.1016/S0959-437X(98)80016-6
Semenza, G. L. (1999). Regulation of mammalian O-2 homeostasis by hypoxia- inducible factor 1. Annu. Rev. Cell Dev. Biol. 15, 551–578. doi: 10.1146/annurev.cellbio.15.1.551
Semenza, G. L. (2003). Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 3, 721–732. doi: 10.1038/nrc1187
Semenza, G. L. (2009). Regulation of vascularization by hypoxia-inducible factor 1. Ann. N.Y. Acad. Sci. 1177, 2–8. doi: 10.1111/j.1749-6632.2009.05032.x
Sercombe, L., Veerati, T., Moheimani, F., Wu, S. Y., Sood, A. K., and Hua, S. (2015). Advances and challenges of liposome assisted drug delivery. Front. Pharmacol. 6:286. doi: 10.3389/fphar.2015.00286
Shimoda, L. A., Fallon, M., Pisarcik, S., Wang, J., and Semenza, G. L. (2006). HIF-1 regulates hypoxic induction of NHE1 expression and alkalinization of intracellular pH in pulmonary arterial myocytes. Am. J. Physiol. Lung Cell. Mol. Physiol. 291, L941–L949. doi: 10.1152/ajplung.00528.2005
Shimoda, L. A., Manalo, D. J., Sham, J. S. K., Semenza, G. L., and Sylvester, J. T. (2001). Partial HIF-1 alpha deficiency impairs pulmonary arterial myocyte electrophysiological responses to hypoxia. Am. J. Physiol. Lung Cell. Mol. Physiol. 281, L202–L208.
Strutz, F., Okada, H., Lo, C. W., Danoff, T., Carone, R. L., Tomaszewski, J. E., et al. (1995). Identification and characterization of a fibroblast marker - FSP1. J. Cell Biol. 130, 393–405. doi: 10.1083/jcb.130.2.393
Sundberg, C., Nagy, J. A., Brown, L. F., Feng, D., Eckelhoefer, I. A., Manseau, E. J., et al. (2001). Glomeruloid microvascular proliferation follows adenoviral vascular permeability factor/vascular endothelial growth factor-164 gene delivery. Am. J. Pathol. 158, 1145–1160. doi: 10.1016/s0002-9440(10)64062-x
Tal, R., Shaish, A., Rofe, K., Feige, E., Varda-Bloom, N., Afek, A., et al. (2008). Endothelial-targeted gene transfer of hypoxia-inducible factor-1 alpha augments ischemic neovascularization following systemic administration. Mol. Ther. 16, 1927–1936. doi: 10.1038/mt.2008.191
Torchilin, V. P. (2005). Recent advances with liposomes as pharmaceutical carriers. Nat. Rev. Drug Discov. 4, 145–160. doi: 10.1038/nrd1632
Tuder, R. M., Chacon, M., Alger, L., Wang, J., Taraseviciene-Stewart, L., Kasahara, Y., et al. (2001). Expression of angiogenesis-related molecules in plexiform lesions in severe pulmonary hypertension: evidence for a process of disordered angiogenesis. J. Pathol. 195, 367–374. doi: 10.1002/path.953
Ueno, M., Maeno, T., Nomura, M., Ikeda, K., Aizawa, T., Yamaguchi, S., et al. (2009). HIF-1a promotes pulmonary fibrosis through PAI-1 production. Am. J. Respir. Crit. Care Med 179:A3423. doi: 10.1164/ajrccm-conference.2009.179.1_meetingabstracts.a3423
Veith, C., Schermuly, R. T., Brandes, R. P., and Weissmann, N. (2016). Molecular mechanisms of hypoxia-inducible factor-induced pulmonary arterial smooth muscle cell alterations in pulmonary hypertension. J. Physiol. London 594, 1167–1177. doi: 10.1113/jp270689
Veith, C., Zakrzewicz, D., Dahal, B. K., Balint, Z., Murmann, K., Wygrecka, M., et al. (2014). Hypoxia- or PDGF-BB-dependent paxillin tyrosine phosphorylation in pulmonary hypertension is reversed by HIF-1 alpha depletion or imatinib treatment. Thromb. Haemost. 112, 1288–1303. doi: 10.1160/th13-12-1031
Wang, G. L., Jiang, B. H., Rue, E. A., and Semenza, G. L. (1995). Hypoxia-inducible factor-1 is a basic-helix-loop-helix-pas heterodimer regulated by cellular o-2 tension. Proc. Natl. Acad. Sci. U.S.A. 92, 5510–5514. doi: 10.1073/pnas.92.12.5510
Wang, J., Fu, X., Yang, K., Jiang, Q., Chen, Y., Jia, J., et al. (2015). Hypoxia inducible factor-1-dependent up-regulation of BMP4 mediates hypoxia-induced increase of TRPC expression in PASMCs. Cardiovasc. Res. 107, 108–118. doi: 10.1093/cvr/cvv122
Wang, J., Weigand, L., Lu, W. J., Sylvester, J. T., Semenza, G. L., and Shimoda, L. A. (2006). Hypoxia inducible factor 1 mediates hypoxia-induced TRPC expression and elevated intracellular Ca2+ in pulmonary arterial smooth muscle cells. Circ. Res. 98, 1528–1537. doi: 10.1161/01.res.0000227551.68124.98
Wang, L., Bell, P., Morizono, H., He, Z., Pumbo, E., Yu, H., et al. (2017). AAV gene therapy corrects OTC deficiency and prevents liver fibrosis in aged OTC-knock out heterozygous mice. Mol. Genet. Metab. 120, 299–305. doi: 10.1016/j.ymgme.2017.02.011
Wang, Y., Saad, M., Pakunlu, R. I., Khandare, J. J., Garbuzenko, O. B., Garbuzenko, O. B., et al. (2008). Nonviral nanoscale-based delivery of antisense oligonucleotides targeted to hypoxia-inducible factor 1 alpha enhances the efficacy of chemotherapy in drug-resistant tumor. Clin. Cancer Res. 14, 3607–3616. doi: 10.1158/1078-0432.ccr-07-2020
Wasko, P., Luchowski, R., Tutaj, K., Grudzinski, W., Adamkiewicz, P., and Gruszecki, W. I. (2012). Toward understanding of toxic side effects of a polyene antibiotic amphotericin B: fluorescence spectroscopy reveals widespread formation of the specific supramolecular structures of the drug. Mol. Pharm. 9, 1511–1520. doi: 10.1021/mp300143n
Wenger, R. H. (2002). Cellular adaptation to hypoxia: O-2-sensing protein hydroxylases, hypoxia-inducible transcription factors, and O-2-regulated gene expression. FASEB J. 16, 1151–1162. doi: 10.1096/fj.01-0944rev
Whitfield, M. L., Finlay, D. R., Murray, J. I., Troyanskaya, O. G., Chi, J. T., Pergamenschikov, A., et al. (2003). Systemic and cell type-specific gene expression patterns in scleroderma skin. Proc. Natl. Acad. Sci. U.S.A. 100, 12319–12324. doi: 10.1073/pnas.1635114100
Wipff, J., Dieude, P., Avouac, J., Tiev, K., Hachulla, E., Granel, B., et al. (2009). Association of hypoxia-inducible factor 1A (HIF1A) gene polymorphisms with systemic sclerosis in a French European Caucasian population. Scand. J. Rheumatol. 38, 291–294. doi: 10.1080/03009740802629432
Wojciak-Stothard, B., Abdul-Salam, V. B., Lao, K. H., Tsang, H., Irwin, D. C., Lisk, C., et al. (2014). Aberrant chloride intracellular channel 4 expression contributes to endothelial dysfunction in pulmonary arterial hypertension. Circulation 129, 1770–1780. doi: 10.1161/circulationaha.113.006797
Xu, W., and Erzurum, S. C. (2011). Endothelial cell energy metabolism, proliferation, and apoptosis in pulmonary hypertension. Compr. Physiol. 1, 357–372. doi: 10.1002/cphy.c090005
Xu, W., Koeck, T., Lara, A. R., Neumann, D., DiFilippo, F. P., Koo, M., et al. (2007). Alterations of cellular bioenergetics in pulmonary artery endothelial cells. Proc. Natl. Acad. Sci. U.S.A. 104, 1342–1347. doi: 10.1073/pnas.0605080104
Yu, A. Y., Shimoda, L. A., Iyer, N. V., Huso, D. L., Sun, X., McWilliams, R., et al. (1999). Impaired physiological responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1 alpha. J. Clin. Investig. 103, 691–696. doi: 10.1172/jci5912
Yun, X., Maman, S., Jiang, H., and Shimoda, L. (2015). HIF upregulates AQP1 expression in pulmonary arterial smooth muscle cells (PASMCs) in a calcium-dependent manner. FASEB J. 29(Suppl. 1031.8).
Zeisberg, M., and Kalluri, R. (2004). The role of epithelial-to-mesenchymal transition in renal fibrosis. J. Mol. Med. 82, 175–181. doi: 10.1007/s00109-003-0517-9
Zeng, Y., Liu, H., Kang, K., Wang, Z., Hui, G., Zhang, X., et al. (2015). Hypoxia inducible factor-1 mediates expression of miR-322: potential role in proliferation and migration of pulmonary arterial smooth muscle cells. Sci. Rep. 5:12098. doi: 10.1038/srep12098
Zhang, Q. Z., Wu, Y. D., Ann, D. K., Messadi, D. V., Tuan, T. L., Kelly, A. P., et al. (2003). Mechanisms of hypoxic regulation of plasminogen activator inhibitor-1 gene expression in keloid fibroblasts. J. Investig. Dermatol. 121, 1005–1012. doi: 10.1046/j.1523-1747.2003.12564.x
Zhang, Q. Z., Wu, Y. D., Chau, C. H., Ann, D. K., Bertolami, C. N., and Le, A. D. (2004). Crosstalk of hypoxia-mediated signaling pathways in upregulating plasminogen activator inhibitor-1 expression in keloid fibroblasts. J. Cell. Physiol. 199, 89–97. doi: 10.1002/jcp.10452
Keywords: fibrosis, chronic hypoxia, hypoxia inducible factor, targeted therapy, systemic sclerosis
Citation: Xiong A and Liu Y (2017) Targeting Hypoxia Inducible Factors-1α As a Novel Therapy in Fibrosis. Front. Pharmacol. 8:326. doi: 10.3389/fphar.2017.00326
Received: 15 March 2017; Accepted: 16 May 2017;
Published: 30 May 2017.
Edited by:
Chrishan S. Samuel, Monash University, AustraliaReviewed by:
Timothy E. Cooney, UPMC Hamot, United StatesGaetano Santulli, Columbia University, United States
Xiao-jun Du, Baker IDI Heart and Diabetes Institute, Australia
Copyright © 2017 Xiong and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yi Liu, bGl1eWlodWF4aXlpeXVhbkAxMjYuY29t