 Elizabeth M. Gordon
Elizabeth M. Gordon Debbie M. Figueroa
Debbie M. Figueroa Amisha V. Barochia
Amisha V. Barochia Stewart J. Levine
Stewart J. Levine
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Pharmacol. , 21 September 2016
Sec. Experimental Pharmacology and Drug Discovery
Volume 7 - 2016 | https://doi.org/10.3389/fphar.2016.00323
This article is part of the Research Topic Apolipoprotein AI and high-density lipoproteins in the prevention and treatment of disease View all 16 articles
Apolipoprotein A-I (apoA-I) and high-density lipoproteins (HDL) mediate reverse cholesterol transport out of cells. Furthermore, HDL has additional protective functions, which include anti-oxidative, anti-inflammatory, anti-apoptotic, and vasoprotective effects. In contrast, HDL can become dysfunctional with a reduction in both cholesterol efflux and anti-inflammatory properties in the setting of disease or the acute phase response. These paradigms are increasingly being recognized to be active in the pulmonary system, where apoA-I and HDL have protective effects in normal lung health, as well as in a variety of disease states, including acute lung injury (ALI), asthma, chronic obstructive pulmonary disease, lung cancer, pulmonary arterial hypertension, pulmonary fibrosis, and viral pneumonia. Similar to observations in cardiovascular disease, however, HDL may become dysfunctional and contribute to disease pathogenesis in respiratory disorders. Furthermore, synthetic apoA-I mimetic peptides have been shown to have protective effects in animal models of ALI, asthma, pulmonary hypertension, and influenza pneumonia. These findings provide evidence to support the concept that apoA-I mimetic peptides might be developed into a new treatment that can either prevent or attenuate the manifestations of lung diseases, such as asthma. Thus, the lung is positioned to take a page from the cardiovascular disease playbook and utilize the protective properties of HDL and apoA-I as a novel therapeutic approach.
High-density lipoprotein particles (HDL) and its major protein, apolipoprotein A-I (apoA-I), play key roles in attenuating the risk of atherosclerotic cardiovascular disease (Navab et al., 2011; Kingwell et al., 2014; Rader and Hovingh, 2014; Rosenson et al., 2016). A key mechanism by which HDL decreases atherosclerosis is by mediating reverse cholesterol efflux out of cells. ApoA-I, which is primarily produced in the liver and small intestine, can form nascent, discoidal HDL particles by complexing with unesterified cholesterol and phospholipids that have been effluxed via the ATP-binding cassette subfamily A member 1 (ABCA1) transporter (Kingwell et al., 2014). Following esterification and transport of cholesterol to the particle core by lecithin-cholesterol acyltransferase, spheroidal HDL particles can efflux additional cholesterol and phospholipids from cells via ABCG1 and scavenger receptor class B member 1 (SRB1). In addition, HDL has anti-oxidative, anti-inflammatory, anti-apoptotic, and vasoprotective properties (Navab et al., 2011; Rosenson et al., 2016). An anti-inflammatory function of HDL is its ability to reduce the cholesterol content of immune cells by decreasing receptor localization and signaling within lipid rafts (Catapano et al., 2014). However, in the setting of disease or the acute-phase response, HDL can become dysfunctional, with diminished cholesterol efflux and anti-inflammatory properties (Kingwell et al., 2014; Rosenson et al., 2016). It is increasingly recognized that both HDL (Figure 1) and apoA-I (Figure 2) similarly, play important roles in modulating normal lung health and disease. Thus, paradigms that have long been applied to atherosclerotic cardiovascular disease may also be relevant for pulmonary disorders, including acute lung injury (ALI), asthma, chronic obstructive pulmonary disease, lung cancer, pulmonary fibrosis, pulmonary hypertension, and viral pneumonia.
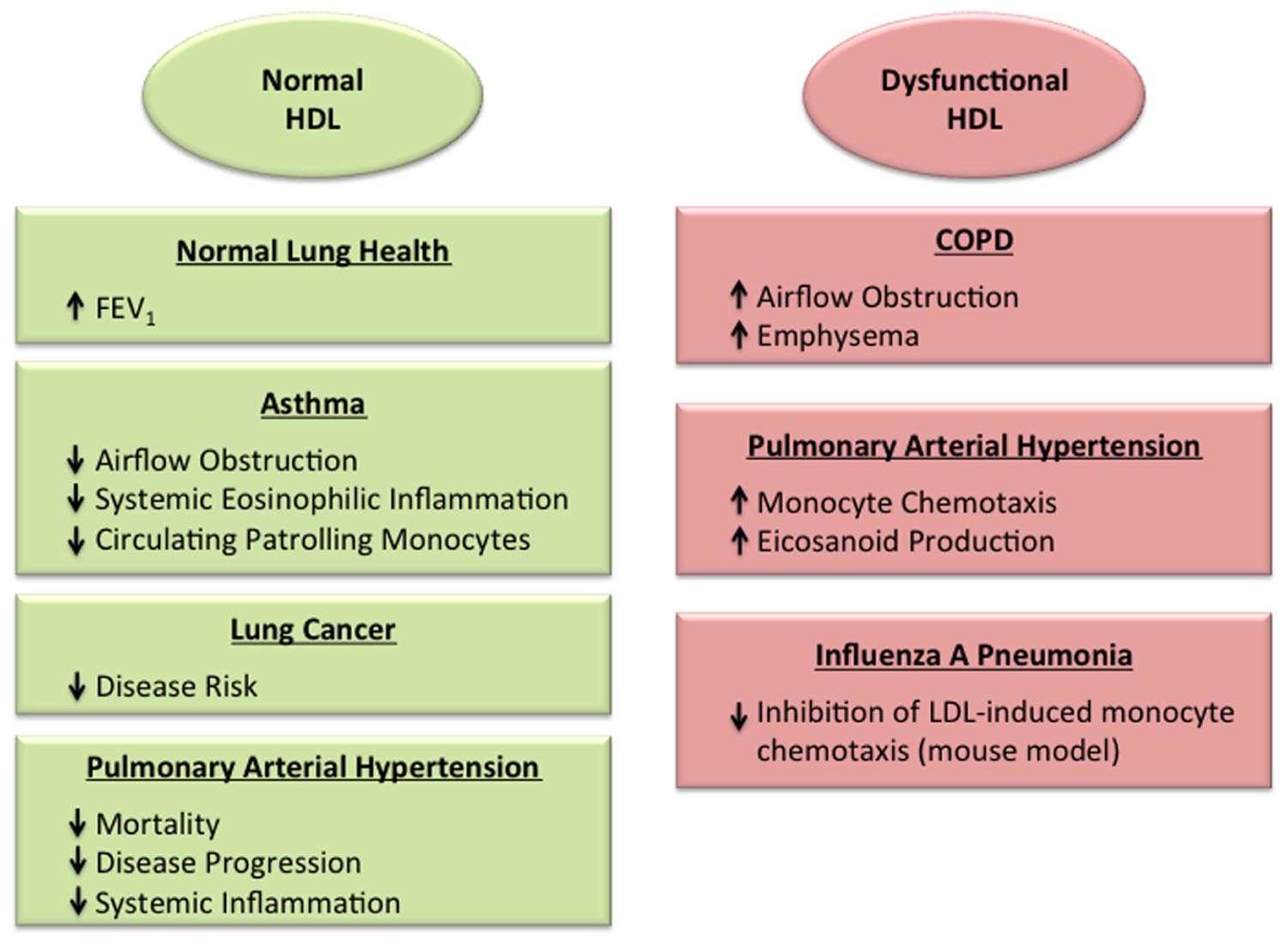
FIGURE 1. Roles of normal and dysfunctional high-density lipoproteins (HDL) in the lung. HDL has been associated with protective effects in the normal lung (Cirillo et al., 2002), as well as in the setting of asthma (Barochia et al., 2015, 2016; Rastogi et al., 2015) and lung cancer (Kucharska-Newton et al., 2008; Chi et al., 2014; Chandler et al., 2016), which may reflect its anti-inflammatory and anti-oxidant properties. In contrast, dysfunctional HDL particles may promote disease severity in the setting of chronic obstructive pulmonary disease (Burkart et al., 2014) and viral pneumonia (Van Lenten et al., 2001). HDL particles in pulmonary arterial hypertension have been found to have either protective effects (Heresi et al., 2010; Sharma et al., 2014), or dysfunctional, pro-inflammatory properties (Ross et al., 2015).
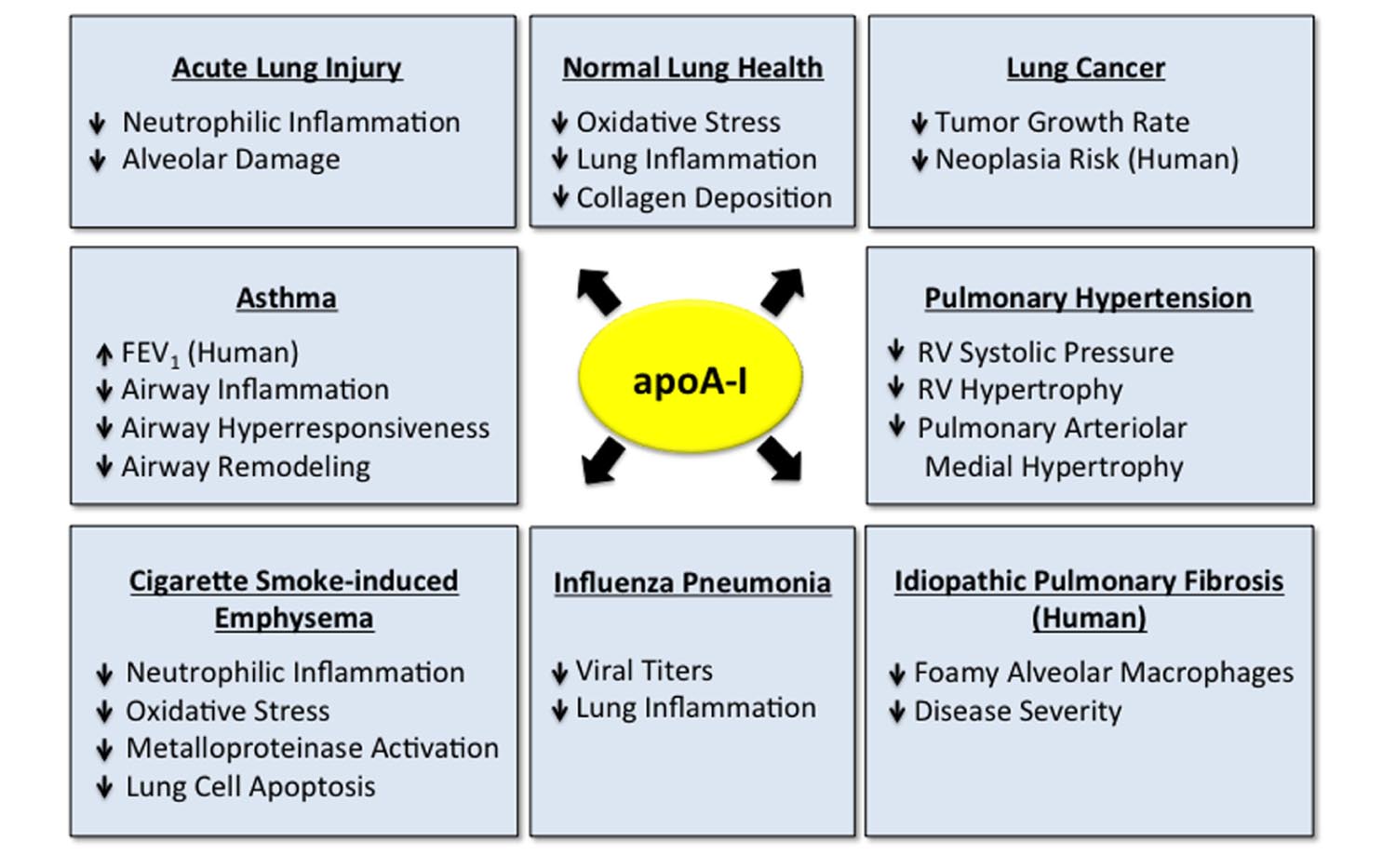
FIGURE 2. Apolipoprotein A-I (apoA-I) has multiple protective functions in the normal and diseased lung. Apolipoprotein A-I has multiple protective and beneficial functions in the normal lung (Wang et al., 2010) and in a variety of pulmonary disorders, including acute lung injury (ALI) (Yan et al., 2006; Jiao and Wu, 2008; Li et al., 2008; Van Linthout et al., 2011; Kwon et al., 2012; Madenspacher et al., 2012; Sharifov et al., 2013), asthma (Nandedkar et al., 2011; Dai et al., 2012; Yao et al., 2011; Park et al., 2013), cigarette smoke-induced emphysema (Kim et al., 2016), influenza pneumonia (Van Lenten et al., 2002, 2004), lung cancer (Zamanian-Daryoush et al., 2013), pulmonary hypertension (Sharma et al., 2014) and idiopathic pulmonary fibrosis (IPF) (Kim et al., 2010; Lee et al., 2013). Data addressing the role of apoA-I in lung disease have primarily been generated in experimental animal models, although several studies have suggested that apoA-I may be associated with decreased disease severity in subjects with asthma (Barochia et al., 2015, 2016) and IPF (Kim et al., 2010), which are indicated.
Levels of mRNA encoding apoA-I, apoA-II, and apoH, are expressed at higher levels in the murine fetal lung than the adult lung, which suggests that pulmonary apolipoprotein production modulates lung maturation via lipid metabolism and transport during embryonic development (Provost et al., 2009). Apolipoprotein A-I and apoA-II are primarily expressed by capillary-like structures during gestational days 15.5–18.5 (Cote et al., 2011). In the adult lung, apoA-I is expressed by alveolar epithelial cells and alveolar macrophages, while the ABCA1 transporter is expressed by type I and type II alveolar epithelial cells, alveolar macrophages, pulmonary vascular endothelial cells (PVECs), and airway smooth muscle cells (Bortnick et al., 2003; Bates et al., 2008; Delvecchio et al., 2008; Kim et al., 2010; Dai et al., 2012, 2014; Park et al., 2013).
The apoA-I/ABCA1 pathway maintains normal lipid homeostasis in the lung by mediating the release of lipids from type I alveolar epithelial cells to apoA-I (Bates et al., 2008). Similarly, ABCA1 expression by type II alveolar epithelial cells facilitates the removal of cholesterol mass and reduces basal rates of surfactant secretion (Bortnick et al., 2003). The important role of ABCA1 in maintaining normal lipid homeostasis in the lung is further highlighted by the finding that alveolar macrophages and type II alveolar epithelial cells from Abca1-deficient mice are enriched with cholesterol (McNeish et al., 2000; Bates et al., 2005). Furthermore, Abca1-deficient mice have abnormal lung morphology characterized by alveolar proteinosis, as well as hypertrophy and hyperplasia of type II alveolar epithelial cells.
Studies utilizing Apoa1-deficient mice have shown an important role for apoA-I in attenuating basal levels of inflammation and oxidative stress in the lung. For example, HDL from Apoa1-deficient mice oxidizes at a faster rate than HDL from wild-type mice (Wang et al., 2010). Furthermore, Apoa1-deficient mice display increased systemic oxidative stress, lung inflammation, and collagen deposition. Basal levels of airway hyperresponsiveness (AHR) are also increased in Apoa1-deficient mice, while pulmonary artery vasodilatation is impaired. Thus, apoA-I plays an important role in maintaining normal lung health.
Acute respiratory distress syndrome describes a clinical entity of severe lung dysfunction that reflects an acute inflammatory lung injury secondary to a variety of causative factors, such as bacterial infection (Bellani et al., 2016). A beneficial property of apoA-I is its ability to directly bind and neutralize lipopolysaccharide (LPS), which is derived from the cell walls of gram-negative bacteria, as well as lipoteichoic acid (LTA), which is a cell wall component of gram-positive bacteria (Emancipator et al., 1992; Ma et al., 2004; Jiao and Wu, 2008; Henning et al., 2011; Van Linthout et al., 2011; Sharifov et al., 2013). The domains that mediate LPS binding are located in the carboxy-terminus of apoA-I (Henning et al., 2011). Apolipoprotein A-I can also associate with the lipopolysaccharide-binding protein, which binds and neutralizes LPS (Wurfel et al., 1994).
Both murine and human studies have provided evidence for a protective function of apoA-I in ALI and ARDS. Apoa1-deficient mice have increased neutrophil recruitment to the lung following inhalation of LPS or direct instillation of CXCL1, which can be attenuated by administration of the L-4F apoA-I mimetic peptide by a mechanism involving inhibition of CXCR2-mediated neutrophil chemotaxis (Madenspacher et al., 2012). Similarly, administration of apoA-I mimetic peptides, human apoA-I, or over-expression of human apoA-I via a recombinant adenoviral vector, protected rodents from developing neutrophilic airway inflammation and ALI in models of LPS- or LTA-mediated systemic inflammation (Yan et al., 2006; Jiao and Wu, 2008; Li et al., 2008; Van Linthout et al., 2011; Kwon et al., 2012; Sharifov et al., 2013). Treatment of LPS-stimulated human blood with the L-4F apoA-I mimetic peptide has suppressed endotoxin activity and IL-6 secretion, as well as preserved paraoxonase function (Sharifov et al., 2013). The L-4F apoA-I mimetic peptide also inhibited cellular activation and superoxide formation when human neutrophils were exposed to serum obtained from subjects with ARDS. Administration of human HDL to mice has also attenuated LPS-induced ALI (Xiao et al., 2008).
Human translational studies have suggested a correlation between the APOA1 gene and the risk of sepsis-associated ALI. Carriage of the APOA1 -75 AA genotype in the APOA1 promoter, which has reduced promoter activity as compared to the major G allele, has been associated with a higher risk of ALI following cardiopulmonary bypass surgery (Smith et al., 1992; Tu et al., 2013). The rs11216153 SNP in the APOA1 gene has also been associated with ALI risk in subjects with sepsis, with the GG genotype and G allele being more common among ALI patients than controls (Hao and He, 2014). However, since this SNP is located in a non-coding region of the APOA1 gene, it is unclear if the association is with the APOA1 gene itself or variants from nearby genes.
Murine studies have demonstrated a protective role for an apoA-I/ABCA1 pathway in the pathogenesis of asthma. Apoa1-deficient mice that were sensitized and challenged with ovalbumin developed augmented neutrophilic airway inflammation that was primarily mediated by the increased production of granulocyte colony stimulating factor (G-CSF) by alveolar macrophages (AMϕs) and PVECs, as well as by the increased expression of IL-17A, TNF-α, CXCL5, and VCAM-1 (Dai et al., 2012). Furthermore, G-CSF can increase the number of tissue neutrophils by attenuating apoptosis, which prolongs neutrophil survival in the lung (Matute-Bello et al., 1997). Similarly, transgenic mice that expressed human ABCA1 under the control of the Tie2 promoter in both PVECs and AMϕs had decreased ovalbumin-induced neutrophilic airway inflammation that was in part mediated by the reduced expression of G-CSF (Dai et al., 2014).
Murine studies have further demonstrated that apoA-I mimetic peptides can suppress allergen-mediated airway inflammation. Administration of the 5A apoA-I mimetic peptide inhibited allergen-mediated increases in neutrophilic airway inflammation in Apoa1-deficient mice (Dai et al., 2012). Similarly, administration of the 5A apoA-I mimetic peptide to wild-type mice attenuated the development of allergen-induced airway inflammation and AHR, and also reduced airway remodeling responses, including mucous cell metaplasia and collagen gene expression (Yao et al., 2011). Likewise, administration of the D-4F apoA-I mimetic peptide suppressed oxidative stress, eosinophilic airway inflammation, AHR, and total immunoglobulin E levels in allergen-challenged wild-type mice (Nandedkar et al., 2011). Administration of human apoA-I to allergen-challenged wild-type mice similarly, suppressed airway inflammation, AHR, dendritic cell migration to the lung, and reduced the release of IL-25, IL-33, and TSLP (thymic stromal lymphopoietin) by airway epithelial cells (Park et al., 2013). In addition, apoA-I administration promoted the recovery of disrupted airway epithelial tight junction proteins and increased the pro-resolution lipid mediator, lipoxin A4, in both mice and humans. Human translational studies have also supported a role for apoA-I in asthma. For example, apoA-I levels in bronchoalveolar lavage fluid (BALF) from mild-to-moderate asthmatics are five-fold lower than in BALF from normal control subjects (Park et al., 2013). Similarly, apoA-I levels are reduced in BALF from ovalbumin-challenged wild-type mice as compared to control mice that were sensitized and challenged with saline (Dai et al., 2012).
Clinical studies have shown that higher systemic levels of HDL and apoA-I are associated with less severe airflow obstruction in both healthy individuals and asthmatics. For example, in an analysis of subjects without respiratory disease who participated in the Third National Health and Nutrition Survey, both HDL and apoA-I were positively associated with FEV1 (Cirillo et al., 2002). Similarly, both HDL and apoA-I were positively correlated with FEV1 in a cohort of atopic asthmatics at the National Heart, Lung, and Blood Institute (Barochia et al., 2015). Furthermore, the positive association with FEV1 in atopic asthmatics was mediated by the subset of large HDLNMR particles, as quantified by nuclear magnetic resonance (NMR) spectroscopy. Higher circulating levels of HDL have also been correlated with lower blood eosinophil counts in asthma, which is a biomarker of type 2 inflammation (Barochia et al., 2016). The negative association between serum HDL and absolute blood eosinophil counts was again mediated by the subset of large HDLNMR particles. Of note, large HDLNMR particles have also been associated with a lower risk of incident cardiovascular disease and type 2 diabetes in women (Mora et al., 2009, 2010). Another correlation between HDL and inflammation was identified in obese, adolescent asthmatics who had an inverse association with the number of patrolling monocytes, which is an innate immune cell population that can be rapidly recruited to the lung during inflammation (Rastogi et al., 2015). Collectively, these studies suggest that apoA-I and HDL may modulate both airflow obstruction and airway inflammation in asthma.
Chronic obstructive pulmonary disease is caused by inhalation of noxious particles or gases, such as cigarette smoke, with resultant chronic inflammation and airflow limitation that is not fully reversible (Celli et al., 2015). ApoA-I and HDL have context-dependent effects in COPD. For example, apoA-I has a protective role in preventing cigarette-smoke induced emphysema. In a murine model of cigarette smoke-induced lung disease, transgenic mice that conditionally over-expressed the human APOA1 gene in alveolar epithelial cells were protected from developing emphysema (Kim et al., 2016). In particular, apoA-I over-expression attenuated cigarette smoke-induced increases in lung inflammation, oxidative stress, metalloproteinase activation, and lung cell apoptosis by a mechanism that involved the reduced translocation of Fas to lipid rafts, which decreased the formation of death-inducing signaling complexes and caspase-8 activation. Cigarette smoke exposure also decreased the amount of apoA-I in the lungs of wild-type mice, which suggests that the loss of its protective function may contribute to emphysema pathogenesis (Kim et al., 2016). Similarly, lung tissue from patients with moderate emphysema contained reduced amounts of apoA-I as compared to lung tissue from non-smoking subjects. Furthermore, the amount of apoA-I was reduced in induced sputum samples of COPD patients as compared to healthy smokers (Nicholas et al., 2010). These studies suggest that the reduction in lung apoA-I levels may contribute to lung disease in COPD.
High-density lipoproteins may also have a therapeutic role in emphysema caused by alpha-1-antitrypsin deficiency. Alpha-1-antitrypsin is a component of HDL that confers anti-protease properties that inhibit leukocyte elastase (Ortiz-Munoz et al., 2009). Intravenous administration of HDL enriched with alpha-1-antitrypsin to cigarette smoke-exposed mice prevented the development of pulmonary emphysema and also reduced the numbers of BALF neutrophils and macrophages, as well as BALF levels of IL-6, TNF-α (tumor necrosis factor), and MCP-1 (monocyte chemoattractant protein-1) (Moreno et al., 2014). BALF fibronectin degradation and matrix metalloproteinase (MMP-2 and MMP-9) activity were also reduced. The protective effects of intravenous administration of HDL complexed with alpha-1-antitrypsin were superior to those of HDL or alpha-1-antitrypsin alone, which suggested that combining HDL with alpha-1-antitrypsin might be more effective than the current strategy of alpha-1-antitrypsin augmentation therapy.
In contrast to the ability of apoA-I to attenuate the induction of cigarette smoke-induced emphysema in murine models, HDL has been associated with increased disease severity in human subjects with emphysema. In the MESA (Multi-Ethnic Study of Atherosclerosis) COPD study, higher plasma HDL levels were associated with a lower FEV1/FVC ratio, which indicated more severe airflow obstruction (Burkart et al., 2014). Furthermore, higher HDL levels were associated with greater percent emphysema on chest computed tomography scans. This suggests that HDL may not be protective in the setting of established emphysema, but instead may have dysfunctional properties (Navab et al., 2011; Kingwell et al., 2014; Rader and Hovingh, 2014; Rosenson et al., 2016).
Potential anti-tumorigenic effects of apoA-I have been identified using murine neoplasia models that showed a dose-dependent inhibition of Lewis lung tumor growth rates with increasing apoA-I levels (Zamanian-Daryoush et al., 2013). Potential anti-neoplastic effects of apoA-I included inhibition of angiogenesis and MMP-9 activity, accumulation of myeloid-derived suppressor cells in tumor beds, and expression of the anti-apoptotic protein, survivin. In addition apoA-I, was found to promote the anti-neoplastic effects of tumor-associated CD11b+ macrophages.
The prospective Atherosclerosis Risk in Communities Study found an inverse association between serum HDL-C levels and incident lung cancer over a 13 years period in former smokers, but not in current smokers (Kucharska-Newton et al., 2008). Similarly, an inverse association between low serum HDL-C and neoplasia was seen with other cancers, which suggests a common mechanism by which low HDL-C levels may promote an environment that is permissive to neoplasia. Another study of non-small cell lung cancer (NSCLC) subjects found that serum HDL-C levels were significantly decreased as compared to normal controls (Chi et al., 2014). Furthermore, NSCLC subjects with low levels of HDL-C had significantly reduced cumulative 5-year survival rates as compared to individuals with normal HDL-C levels. The Women’s Health Study prospectively associated incident cancer rates and cancer mortality with circulating lipid biomarkers in health professionals (Chandler et al., 2016). This study found that higher circulating levels of apoA-I and HDL were inversely associated with the risk of both total cancer and lung cancer. Collectively, these data support an anti-neoplastic role for apoA-I and HDL in lung cancer.
Apolipoprotein A-I and HDL may also have context-dependent effects in pulmonary hypertension. For example, administration of the 4F apoA-I mimetic peptide rescued preexisting pulmonary hypertension in rodent models by increasing the expression of the microRNA, mmu-miR-193, via the retinoid X receptor α (Sharma et al., 2014). Induction of mmu-miR-193 inhibited the expression of insulin-like growth factor-1 receptor and lipoxygenases (e.g., ALOX5, ALOX12, and ALOX15), which suppressed pulmonary artery smooth muscle cell proliferation.
Human translational studies have shown that HDL may be associated with disease severity and outcome in pulmonary arterial hypertension (PAH). For example, a study of PAH subjects at the Cleveland Clinic found significantly lower serum levels of HDL-C as compared to control subjects (Heresi et al., 2010). Furthermore, when subjects were stratified by HDL-C levels, those with HDL-C greater than 35 mg/dl had no mortality and a longer time to clinical worsening over a period of 1.5 years, which was independent of underlying cardiovascular risk factors, insulin resistance, or PAH disease severity. Furthermore, PAH subjects with HDL-C levels that exceeded 35 mg/dl had less systemic inflammation, as indicated by lower levels of serum C-reactive protein, TNF-α, IL-17A, CCL2, and ICAM-1. In contrast, another study of subjects with “idiopathic” PAH (IPAH) or PAH “associated” with underlying etiologies (APAH) found that HDL had been converted to a dysfunctional, pro-inflammatory particle which increased the chemotaxis of peripheral blood monocytes as compared to normal HDL (Ross et al., 2015). Ex vivo treatment with the 4F apoA-I mimetic peptide significantly improved, but did not completely attenuate, the dysfunctional pro-inflammatory HDL phenotype. In addition, HDL fractions from IPAH and APAH subjects had increased levels of multiple eicosanoids. Therefore, additional studies will be needed to further define the roles of HDL in PAH.
Idiopathic pulmonary fibrosis (IPF) is a disease of chronic, progressive fibrosing interstitial pneumonia that occurs primarily in older adults (Raghu et al., 2015). Reduced amounts of apoA-I have been found in BALF from subjects with IPF as compared to normal controls (Kim et al., 2010). Furthermore, apoA-I levels in BALF from IPF patients were inversely correlated with both clinical severity scores and the percentage of foamy macrophages. The role of apoA-I in pulmonary fibrotic disease was subsequently assessed in murine models, where intranasal administration of human apoA-I protein suppressed the number of BALF inflammatory cells, histologic evidence of inflammatory cell infiltration, and lung collagen deposition. Furthermore, apoA-I levels were reduced in BALF from bleomycin-challenged mice. Similarly, transgenic mice that over-expressed human apoA-I in alveolar epithelial cells were protected from silica-induced lung inflammation and fibrotic nodule formulation by a mechanism that might involve increased expression of lipoxin A4 (Lee et al., 2013). Collectively, these data suggest that apoA-I has a protective effect in IPF.
Apolipoprotein A-I and HDL also modulate the pathogenesis of influenza A infection. HDL isolated from mice infected with influenza A is dysfunctional with reduced anti-inflammatory and anti-oxidant properties that, in part, reflected decreased activity of paraoxonase and platelet-activating factor acetylhydrolase, as well as increased ceruloplasmin, which is a potent oxidant (Van Lenten et al., 2001). In contrast, apolipoprotein J levels in HDL were increased, which might promote cholesterol efflux from macrophages. Administration of the D-4F apoA-I mimetic peptide to mice infected with influenza A attenuated lung inflammation, suppressed IL-6 production, and prevented macrophage trafficking into arteries (Van Lenten et al., 2002). The D-4F apoA-I mimetic peptide also had antiviral activity that reduced influenza titers by more than 50%. Similarly, the D-4F apoA-I mimetic peptide inhibited the production of oxidized phospholipids, IFN-α/β, and IL-6 by an alveolar epithelial cell line and also suppressed viral titers and caspase activity (Van Lenten et al., 2004). Collectively, these findings suggest that apoA-I mimetic peptides may have therapeutic potential for the treatment of influenza A pneumonia.
An increasing body of evidence supports the concept that HDL, and particularly apoA-I, modulate the pathogenesis and severity of lung disease. This may involve both the systemic effects of HDL in the circulation, as well as the local production of apoA-I in the lung. Although normal HDL has multiple protective effects, HDL may become dysfunctional in the setting of lung disease and no longer attenuate disease manifestations. Furthermore, data from murine models has suggested that small apoA-I mimetic peptides, which are based upon the α-helical structure of apoA-I, may be developed into a novel therapy for lung disease. In particular, administration of 5A and D-4F apoA-I mimetic peptides has protective effects in animal models of ALI, asthma, pulmonary hypertension, and influenza pneumonia (Yao et al., 2011; Van Lenten et al., 2002; Dai et al., 2012; Kwon et al., 2012; Madenspacher et al., 2012; Sharifov et al., 2013; Sharma et al., 2014). Furthermore, the D-4F and L-4F apoA-I mimetic peptides have already been evaluated in human subjects with cardiovascular disease and have been well-tolerated (Dunbar et al., 2007; Bloedon et al., 2008; Watson et al., 2011). This provides evidence to support the concept that inhalational administration of apoA-I mimetic peptides to provide site-directed delivery to the lung may attenuate the manifestations of pulmonary disease. Consistent with this concept, our laboratory is currently working toward advancing an inhaled formulation of the 5A apoA-I mimetic peptide to clinical trials that will assess its safety and role for the treatment of asthma. Therefore, the lung may benefit from taking a page from the cardiovascular disease playbook and apply the protective properties of HDL and apoA-I as a novel therapeutic approach for pulmonary disease.
All authors listed, have made substantial, direct and intellectual contribution to the work, and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was funded by the Intramural Research Program of the National Heart, Lung, and Blood Institute, NIH, Bethesda, Maryland.
Barochia, A. V., Kaler, M., Cuento, R., Gordon, E. M., Theard, P., Figueroa, D., et al. (2016). Serum high density lipoprotein cholesterol and large HDL particles are negatively correlated with blood eosinophils in atopic asthma. Am. J. Resp. Crit. Care Med. 193, A1451.
Barochia, A. V., Kaler, M., Cuento, R. A., Gordon, E. M., Weir, N. A., Sampson, M., et al. (2015). Serum apolipoprotein A-I and large high-density lipoprotein particles are positively correlated with FEV1 in atopic asthma. Am. J. Resp. Crit. Care Med. 191, 990–1000. doi: 10.1164/rccm.201411-1990OC
Bates, S. R., Tao, J. Q., Collins, H. L., Francone, O. L., and Rothblat, G. H. (2005). Pulmonary abnormalities due to abca1 deficiency in mice. Am. J. Physiol. Lung Cell Mol. Physiol. 289, L980–L989. doi: 10.1152/ajplung.00234.2005
Bates, S. R., Tao, J. Q., Yu, K. J., Borok, Z., Crandall, E. D., Collins, H. L., et al. (2008). Expression and biological activity of ABCA1 in alveolar epithelial cells. Am. J. Respir. Cell Mol. Biol. 38, 283–292. doi: 10.1165/rcmb.2007-0020OC
Bellani, G., Laffey, J. G., Pham, T., Fan, E., Brochard, L., Esteban, A., et al. (2016). Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA 315, 788–800. doi: 10.1001/jama.2016.0291
Bloedon, L. T., Dunbar, R., Duffy, D., Pinell-Salles, P., Norris, R., DeGroot, B. J., et al. (2008). Safety, pharmacokinetics, and pharmacodynamics of oral apoa-i mimetic peptide D-4F in high-risk cardiovascular patients. J. Lipid Res. 49, 1344–1352. doi: 10.1194/jlr.P800003-JLR200
Bortnick, A. E., Favari, E., Tao, J. Q., Francone, O. L., Reilly, M., Zhang, Y., et al. (2003). Identification and characterization of rodent ABCA1 in isolated type II pneumocytes. Am. J. Physiol. Lung Cell Mol. Physiol. 285, L869–L878. doi: 10.1152/ajplung.00077.2003
Burkart, K. M., Manichaikul, A., Wilk, J. B., Ahmed, F. S., Burke, G. L., Enright, P., et al. (2014). ApoM and high-density lipoprotein cholesterol are associated with lung function and per cent emphysema. Eur. Respir. J. 43, 1003–1017. doi: 10.1183/09031936.00147612
Catapano, A. L., Pirillo, A., Bonacina, F., and Norata, G. D. (2014). HDL in innate and adaptive immunity. Cardiovasc. Res. 103, 372–383. doi: 10.1093/cvr/cvu150
Celli, B. R., Decramer, M., Wedzicha, J. A., Wilson, K. C., Agusti, A., Criner, G. J., et al. (2015). An official american thoracic society/european respiratory society statement: research questions in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 191, e4–e27. doi: 10.1164/rccm.201501-0044ST
Chandler, P. D., Song, Y., Lin, J., Zhang, S., Sesso, H. D., Mora, S., et al. (2016). Lipid biomarkers and long-term risk of cancer in the women’s health study. Am. J. Clin. Nutr. 103, 1397–1407. doi: 10.3945/ajcn.115.124321
Chi, P. D., Liu, W., Chen, H., Zhang, J. P., Lin, Y., Zheng, X., et al. (2014). High-density lipoprotein cholesterol is a favorable prognostic factor and negatively correlated with c-reactive protein level in non-small cell lung carcinoma. PLoS ONE 9:e91080. doi: 10.1371/journal.pone.0091080
Cirillo, D. J., Agrawal, Y., and Cassano, P. A. (2002). Lipids and pulmonary function in the third national health and nutrition examination survey. Am. J. Epidemiol. 155, 842–848. doi: 10.1093/aje/155.9.842
Cote, M., Provost, P. R., and Tremblay, Y. (2011). Apolipoprotein A-I, A-II, and H mRNA and protein accumulation sites in the developing lung in late gestation. BMC Res. Notes 4:235. doi: 10.1186/1756-0500-4-235
Dai, C., Yao, X., Keeran, K. J., Zywicke, G. J., Qu, X., Yu, Z. X., et al. (2012). Apolipoprotein A-I attenuates ovalbumin-induced neutrophilic airway inflammation via a granulocyte colony-stimulating factor-dependent mechanism. Am. J. Respir. Cell Mol. Biol. 47, 186–195. doi: 10.1165/rcmb.2011-0322OC
Dai, C., Yao, X., Vaisman, B., Brenner, T., Meyer, K. S., Gao, M., et al. (2014). ATP-binding cassette transporter 1 attenuates ovalbumin-induced neutrophilic airway inflammation. Am. J. Respir. Cell Mo.l Biol. 51, 626–636. doi: 10.1165/rcmb.2013-0264OC
Delvecchio, C. J., Bilan, P., Nair, P., and Capone, J. P. (2008). LXR-induced reverse cholesterol transport in human airway smooth muscle is mediated exclusively by ABCA1. Am. J. Physiol. Lung. Cell Mol. Physiol. 295, L949–L957. doi: 10.1152/ajplung.90394.2008
Dunbar, R. L., Bloedon, L. T., Duffy, D., Norris, R. B., Movva, R., Navab, M., et al. (2007). Daily oral administration of the apolipoprotein A-I mimetic peptide D-4F in patients with heart disease or equivalent risk improves high-density lipoprotein anti-inflammatory function (abstract). J. Am. Coll. Cardiol. 49, 366A.
Emancipator, K., Csako, G., and Elin, R. J. (1992). In vitro inactivation of bacterial endotoxin by human lipoproteins and apolipoproteins. Infect. Immun. 60, 596–601.
Hao, J., and He, X. D. (2014). Haplotype analysis of apoAI gene and sepsis-associated acute lung injury. Lipids Health Dis. 13:79. doi: 10.1186/1476-511X-13-79
Henning, M. F., Herlax, V., and Bakas, L. (2011). Contribution of the c-terminal end of apolipoprotein AI to neutralization of lipopolysaccharide endotoxic effect. Innate Immun. 17, 327–337. doi: 10.1177/1753425910370709
Heresi, G. A., Aytekin, M., Newman, J., DiDonato, J., and Dweik, R. A. (2010). Plasma levels of high-density lipoprotein cholesterol and outcomes in pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 182, 661–668. doi: 10.1164/rccm.201001-0007OC
Jiao, Y. L., and Wu, M. P. (2008). Apolipoprotein A-I diminishes acute lung injury and sepsis in mice induced by lipoteichoic acid. Cytokine 43, 83–87. doi: 10.1016/j.cyto.2008.04.002
Kim, C., Lee, J. M., Park, S. W., Kim, K. S., Lee, M. W., Paik, S., et al. (2016). Attenuation of cigarette smoke-induced emphysema in mice by apolipoprotein A-1 overexpression. Am. J. Respir. Cell Mol. Biol. 54, 91–102. doi: 10.1165/rcmb.2014-0305OC
Kim, T. H., Lee, Y. H., Kim, K. H., Lee, S. H., Cha, J. Y., Shin, E. K., et al. (2010). Role of lung apolipoprotein A-I in idiopathic pulmonary fibrosis: antiinflammatory and antifibrotic effect on experimental lung injury and fibrosis. Am. J. Respir. Crit. Care Med. 182, 633–642. doi: 10.1164/rccm.200905-0659OC
Kingwell, B. A., Chapman, M. J., Kontush, A., and Miller, N. E. (2014). HDL-targeted therapies: progress, failures and future. Nat. Rev. Drug Discov. 13, 445–464. doi: 10.1038/nrd4279
Kucharska-Newton, A. M., Rosamond, W. D., Schroeder, J. C., McNeill, A. M., Coresh, J., Folsom, A. R., et al. (2008). HDL-cholesterol and the incidence of lung cancer in the atherosclerosis risk in communities (ARIC) study. Lung Cancer 61, 292–300. doi: 10.1016/j.lungcan.2008.01.015
Kwon, W. Y., Suh, G. J., Kim, K. S., Kwak, Y. H., and Kim, K. (2012). 4F, apolipoprotein AI mimetic peptide, attenuates acute lung injury and improves survival in endotoxemic rats. J. Trauma Acute Care Surg. 72, 1576–1583. doi: 10.1097/TA.0b013e3182493ab4
Lee, E., Lee, E. J., Kim, H., Jang, A., Koh, E., Uh, S. T., et al. (2013). Overexpression of apolipoprotein A1 in the lung abrogates fibrosis in experimental silicosis. PLoS ONE 8:e55827. doi: 10.1371/journal.pone.0055827
Li, Y., Dong, J. B., and Wu, M. P. (2008). Human apoA-I overexpression diminishes LPS-induced systemic inflammation and multiple organ damage in mice. Eur. J. Pharmacol. 590, 417–422. doi: 10.1016/j.ejphar.2008.06.047
Ma, J., Liao, X. L., Lou, B., and Wu, M. P. (2004). Role of apolipoprotein A-I in protecting against endotoxin toxicity. Acta Biochim. Biophys. Sin. 36, 419–424. doi: 10.1093/abbs/36.6.419
Madenspacher, J. H., Azzam, K. M., Gong, W., Gowdy, K. M., Vitek, M. P., Laskowitz, D. T., et al. (2012). Apolipoproteins and apolipoprotein mimetic peptides modulate phagocyte trafficking through chemotactic activity. J. Biol. Chem. 287, 43730–43740. doi: 10.1074/jbc.M112.377192
Matute-Bello, G., Liles, W. C., Radella, F. II, Steinberg, K. P., Ruzinski, J. T., Jonas, M., et al. (1997). Neutrophil apoptosis in the acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 156, 1969–1977. doi: 10.1164/ajrccm.156.6.96-12081
McNeish, J., Aiello, R. J., Guyot, D., Turi, T., Gabel, C., Aldinger, C., et al. (2000). High density lipoprotein deficiency and foam cell accumulation in mice with targeted disruption of ATP-binding cassette transporter-1. Proc. Natl. Acad. Sci. U.S.A. 97, 4245–4250. doi: 10.1073/pnas.97.8.4245
Mora, S., Otvos, J. D., Rifai, N., Rosenson, R. S., Buring, J. E., and Ridker, P. M. (2009). Lipoprotein particle profiles by nuclear magnetic resonance compared with standard lipids and apolipoproteins in predicting incident cardiovascular disease in women. Circulation 119, 931–939. doi: 10.1161/CIRCULATIONAHA.108.816181
Mora, S., Otvos, J. D., Rosenson, R. S., Pradhan, A., Buring, J. E., and Ridker, P. M. (2010). Lipoprotein particle size and concentration by nuclear magnetic resonance and incident type 2 diabetes in women. Diabetes 59, 1153–1160. doi: 10.2337/db09-1114
Moreno, J. A., Ortega-Gomez, A., Rubio-Navarro, A., Louedec, L., Ho-Tin-Noe, B., Caligiuri, G., et al. (2014). High-density lipoproteins potentiate alpha1-antitrypsin therapy in elastase-induced pulmonary emphysema. Am. J. Respir. Cell Mol. Biol. 51, 536–549. doi: 10.1165/rcmb.2013-0103OC
Nandedkar, S. D., Weihrauch, D., Xu, H., Shi, Y., Feroah, T., Hutchins, W., et al. (2011). D-4F, an apoA-1 mimetic, decreases airway hyperresponsiveness, inflammation, and oxidative stress in a murine model of asthma. J. Lipid Res. 52, 499–508. doi: 10.1194/jlr.M012724
Navab, M., Reddy, S. T., Van Lenten, B. J., and Fogelman, A. M. (2011). HDL and cardiovascular disease: atherogenic and atheroprotective mechanisms. Nat. Rev. Cardiol. 8, 222–232. doi: 10.1038/nrcardio.2010.222
Nicholas, B. L., Skipp, P., Barton, S., Singh, D., Bagmane, D., Mould, R., et al. (2010). Identification of lipocalin and apolipoprotein A1 as biomarkers of chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 181, 1049–1060. doi: 10.1164/rccm.200906-0857OC
Ortiz-Munoz, G., Houard, X., Martin-Ventura, J. L., Ishida, B. Y., Loyau, S., Rossignol, P., et al. (2009). HDL antielastase activity prevents smooth muscle cell anoikis, a potential new antiatherogenic property. FASEB J. 23, 3129–3139. doi: 10.1096/fj.08-127928
Park, S. W., Lee, E. H., Lee, E. J., Kim, H. J., Bae, D. J., Han, S., et al. (2013). Apolipoprotein a1 potentiates lipoxin a4 synthesis and recovery of allergen-induced disrupted tight junctions in the airway epithelium. Clin. Exp. Allergy 43, 914–927. doi: 10.1111/cea.12143
Provost, P. R., Boucher, E., and Tremblay, Y. (2009). Apolipoprotein A-I, A-II, C-II, and H expression in the developing lung and sex difference in surfactant lipids. J. Endocrinol. 200, 321–330. doi: 10.1677/JOE-08-0238
Rader, D. J., and Hovingh, G. K. (2014). HDL and cardiovascular disease. Lancet 384, 618–625. doi: 10.1016/S0140-6736(14)61217-4
Raghu, G., Rochwerg, B., Zhang, Y., Garcia, C. A., Azuma, A., Behr, J., et al. (2015). An official ATS/ERS/JRS/ALAT clinical practice guideline: treatment of idiopathic pulmonary fibrosis. An update of the 2011 clinical practice guideline. Am. J. Respir. Crit. Care Med. 192, e3–e19. doi: 10.1164/rccm.201506-1063ST
Rastogi, D., Fraser, S., Oh, J., Huber, A. M., Schulman, Y., Bhagtani, R. H., et al. (2015). Inflammation, metabolic dysregulation, and pulmonary function among obese urban adolescents with asthma. Am. J. Respir. Crit. Care Med. 191, 149–160. doi: 10.1164/rccm.201409-1587OC
Rosenson, R. S., Brewer, H. B. Jr., Ansell, B. J., Barter, P., Chapman, M. J., Heinecke, J. W., et al. (2016). Dysfunctional HDL and atherosclerotic cardiovascular disease. Nat. Rev. Cardiol. 13, 48–60. doi: 10.1038/nrcardio.2015.124
Ross, D. J., Hough, G., Hama, S., Aboulhosn, J., Belperio, J. A., Saggar, R., et al. (2015). Proinflammatory high-density lipoprotein results from oxidized lipid mediators in the pathogenesis of both idiopathic and associated types of pulmonary arterial hypertension. Pulm. Circ. 5, 640–648. doi: 10.1086/683695
Sharifov, O. F., Xu, X., Gaggar, A., Grizzle, W. E., Mishra, V. K., Honavar, J., et al. (2013). Anti-inflammatory mechanisms of apolipoprotein A-I mimetic peptide in acute respiratory distress syndrome secondary to sepsis. PLoS ONE 8:e64486. doi: 10.1371/journal.pone.0064486
Sharma, S., Umar, S., Potus, F., Iorga, A., Wong, G., Meriwether, D., et al. (2014). Apolipoprotein A-I mimetic peptide 4F rescues pulmonary hypertension by inducing microrna-193-3p. Circulation 130, 776–785. doi: 10.1161/CIRCULATIONAHA.114.007405
Smith, J. D., Brinton, E. A., and Breslow, J. L. (1992). Polymorphism in the human apolipoprotein A-I gene promoter region. Association of the minor allele with decreased production rate in vivo and promoter activity in vitro. J. Clin. Invest. 89, 1796–1800. doi: 10.1172/JCI115783
Tu, J., Zhang, B., Chen, Y., Liang, B., Liang, D., Liu, G., et al. (2013). Association of apolipoprotein a1 -75 G/A polymorphism with susceptibility to the development of acute lung injury after cardiopulmonary bypass surgery. Lipids Health Dis. 12:172. doi: 10.1186/1476-511X-12-172
Van Lenten, B. J., Wagner, A. C., Anantharamaiah, G. M., Garber, D. W., Fishbein, M. C., Adhikary, L., et al. (2002). Influenza infection promotes macrophage traffic into arteries of mice that is prevented by D-4F, an apolipoprotein A-I mimetic peptide. Circulation 106, 1127–1132. doi: 10.1161/01.CIR.0000030182.35880.3E
Van Lenten, B. J., Wagner, A. C., Navab, M., Anantharamaiah, G. M., Hui, E. K., Nayak, D. P., et al. (2004). D-4F, an apolipoprotein a-I mimetic peptide, inhibits the inflammatory response induced by influenza A infection of human type II pneumocytes. Circulation 110, 3252–3258. doi: 10.1161/01.CIR.0000147232.75456.B3
Van Lenten, B. J., Wagner, A. C., Nayak, D. P., Hama, S., Navab, M., and Fogelman, A. M. (2001). High-density lipoprotein loses its anti-inflammatory properties during acute influenza A infection. Circulation 103, 2283–2288. doi: 10.1161/01.CIR.103.18.2283
Van Linthout, S., Spillmann, F., Graiani, G., Miteva, K., Peng, J., Van Craeyveld, E., et al. (2011). Down-regulation of endothelial TLR4 signalling after apo A-I gene transfer contributes to improved survival in an experimental model of lipopolysaccharide-induced inflammation. J. Mol. Med. 89, 151–160. doi: 10.1007/s00109-010-0690-6
Wang, W., Xu, H., Shi, Y., Nandedkar, S., Zhang, H., Gao, H., et al. (2010). Genetic deletion of apolipoprotein a-i increases airway hyperresponsiveness, inflammation, and collagen deposition in the lung. J. Lipid Res. 51, 2560–2570. doi: 10.1194/jlr.M004549
Watson, C. E., Weissbach, N., Kjems, L., Ayalasomayajula, S., Zhang, Y., Chang, I., et al. (2011). Treatment of patients with cardiovascular disease with L-4F, an apo-A1 mimetic, did not improve select biomarkers of HDL function. J. Lipid Res. 52, 361–373. doi: 10.1194/jlr.M011098
Wurfel, M. M., Kunitake, S. T., Lichenstein, H., Kane, J. P., and Wright, S. D. (1994). Lipopolysaccharide (LPS)-binding protein is carried on lipoproteins and acts as a cofactor in the neutralization of LPS. J. Exp. Med. 180, 1025–1035. doi: 10.1084/jem.180.3.1025
Xiao, G. L., Luo, Z. Q., Xiao, G., Li, C., Xiong, X. D., Yang, Y., et al. (2008). High-density lipoprotein attenuates lipopolysaccharide-induced acute lung injury in mice. Sheng Li Xue Bao 60, 403–408.
Yan, Y. J., Li, Y., Lou, B., and Wu, M. P. (2006). Beneficial effects of apoA-I on LPS-induced acute lung injury and endotoxemia in mice. Life Sci. 79, 210–215. doi: 10.1016/j.lfs.2006.02.011
Yao, X., Dai, C., Fredriksson, K., Dagur, P. K., McCoy, J. P., Qu, X., et al. (2011). 5A, an apolipoprotein A-I mimetic peptide, attenuates the induction of house dust mite-induced asthma. J. Immunol. 186, 576–583. doi: 10.4049/jimmunol.1001534
Keywords: high-density lipoprotein, Apolipoprotein A-I, lung diseases, apolipoprotein mimetic peptides
Citation: Gordon EM, Figueroa DM, Barochia AV, Yao X and Levine SJ (2016) High-density Lipoproteins and Apolipoprotein A-I: Potential New Players in the Prevention and Treatment of Lung Disease. Front. Pharmacol. 7:323. doi: 10.3389/fphar.2016.00323
Received: 08 July 2016; Accepted: 05 September 2016;
Published: 21 September 2016.
Edited by:
Norman Eric Miller, University of Oxford, UKReviewed by:
Lina Badimon, Cardiovascular Research Center (CSIC-ICCC), SpainThis work is authored by Elizabeth M. Gordon, Debbie M. Figueroa, Amisha V. Barochia, Xianglan Yao and Stewart J. Levine on behalf of the US Government and, as regards Drs. Gordon, Figueroa, Barochia, Yao and Levine and the US Government, is not subject to copyright protection in the United States. Foreign and other copyrights may apply. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stewart J. Levine, bGV2aW5lc0BuaGxiaS5uaWguZ292
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.