 Lieven D. Declercq
Lieven D. Declercq Rik Vandenberghe2
Rik Vandenberghe2 Guy Bormans
Guy Bormans
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 31 March 2016
Sec. Experimental Pharmacology and Drug Discovery
Volume 7 - 2016 | https://doi.org/10.3389/fphar.2016.00088
This article is part of the Research Topic In Vivo Imaging in Pharmacological Research View all 15 articles
Clinical trials aiming to develop disease-altering drugs for Alzheimer’s disease (AD), a neurodegenerative disorder with devastating consequences, are failing at an alarming rate. Poorly defined inclusion-and outcome criteria, due to a limited amount of objective biomarkers, is one of the major concerns. Non-invasive molecular imaging techniques, positron emission tomography and single photon emission (computed) tomography (PET and SPE(C)T), allow visualization and quantification of a wide variety of (patho)physiological processes and allow early (differential) diagnosis in many disorders. PET and SPECT have the ability to provide biomarkers that permit spatial assessment of pathophysiological molecular changes and therefore objectively evaluate and follow up therapeutic response, especially in the brain. A number of specific PET/SPECT biomarkers used in support of emerging clinical therapies in AD are discussed in this review.
The worldwide prevalence of AD is estimated at 35 million, a number expected to quadruple by 2050, due to the increasing lifespan of the world population (Brookmeyer et al., 2007). With an unfavorable prognosis and a life expectancy of approximately 8–10 years, AD is becoming one of the most costly diseases for society (Thies and Bleiler, 2013). In spite of increasing knowledge about the genetics, epidemiology, and histopathological features of AD there is, at this moment, only symptomatic treatment available. However, already at early clinical signs, intrinsic disease progression has developed for a long time; patients rapidly decline and develop irrevocable brain damage (Masters et al., 2006; Romano and Buratti, 2013). Therefore, there is a great need for efficient treatment that should be initiated in a very early phase of the disease. Strict definite diagnosis can still only be made post-mortem, on the basis of two pathological hallmarks: SP and NFTs (Hyman et al., 2012), although use of biomarkers is strongly advocated in research guidelines (Dubois et al., 2014). More than 90% of clinical trials aiming to intervene at the causative pathological elements have failed to produce disease altering effects. A major concern hereby is the lack of objective biomarkers assisting in the evaluation of inclusion- and outcome criteria of participating patients, as many participants turned out to be misdiagnosed, particularly in the early AD disease stages (Jack et al., 2010; Barthel et al., 2015; James et al., 2015). Currently five biomarkers for AD have been used for evaluation of disease and monitoring of disease progression (Jack et al., 2010): CSF levels of Aβ42, CSF levels of total tau and p-tau181 and p-tau231, structural imaging (CT and MRI) and functional imaging (PET with [18F]FDG). Elevated levels of CSF tau and reduced levels of CSF Aβ allow the prediction of, respectively, the NFT load and the SP deposits (Ahmed et al., 2014; de Souza et al., 2014; Wurtman, 2015). Therefore, these CSF levels appear to be useful biomarkers in the diagnosis of AD (Wallin et al., 2006; Weiner et al., 2015). Nonetheless, CSF sampling requires an invasive lumbar puncture, quantification of CSF levels is hampered by interlaboratory variability and CSF values do not provide regional information on tau- and Aβ deposits. The regional concentration of tau- and Aβ deposits is however essential for a differential diagnosis of AD, especially among the different tauopathies (Hampel and Teipel, 2004; Gozes et al., 2009; Hampel et al., 2010). Structural volume measurement can be used to measure regional cerebral atrophy. Although not highly specific for AD due to overlap with ‘normal’ aging, the degree of atrophy follows neuropathological progression in AD and severity of volume loss correlates well with disease progression (Masters et al., 2006; Jack et al., 2010). Regarding the biomarkers that can be visualized and quantified by the molecular imaging techniques PET and SPECT, there are three important applications to be considered, that could contribute to successful drug development in clinical trials. The first one is the ability of PET and SPECT to provide quantitative and spatial in vivo assessment of, for example, the amyloid- and tau burden in AD patients. By doing so, inclusion and exclusion criteria in clinical trials can be verified more objectively than what was possible now with the previous biomarkers. Indeed, the use of specific radiotracers, for various targets, may provide accurate differential diagnosis (even at early AD stages) and true confirmation of the availability of the drug target, which allows physicians to reliably select patients for clinical trials to evaluate novel AD therapeutics (Barthel et al., 2015). Another important role for these molecular imaging techniques is the assessment and quantitative follow-up of drugs aiming to intervene at the specific molecular pathophysiological processes. Using highly selective PET- or SPECT radioligands, the true biological effect of novel clinical candidates can be established and true quantitative assessment is possible (Barthel et al., 2015). Thirdly, molecular imaging can be applied to measure the dose-related occupancy of specific targets caused by drugs under test, which allows the characterization of the optimal therapeutic window and thus a more effective design of subsequent clinical drug trials (Broich et al., 1998; Passchier et al., 2002). Although PET is able to provide a much higher spatial resolution and dynamic scanning with higher temporal resolution and better quantification than SPECT, SPECT cameras are more widely available and cheaper than PET cameras. Both the availability and the economical aspect are important to consider when performing large multi-center clinical trials (Rahmim and Zaidi, 2008).
The most frequently used PET radiopharmaceutical is 2-[18F]fluoro-2-deoxy-D-glucose ([18F]FDG, Figure 1), a glucose derivative which allows measurement of brain glucose metabolism directly related with viability of brain tissue in AD (Barthel et al., 2015). This commercially available compound, with various clinical applications, has been well established in routine clinical practice, but also in the recruitment and follow-up of the majority of AD clinical trials which use PET as biomarker technique (Barthel et al., 2015). Indeed, several AD clinical trials are currently recruiting and following up patients with [18F]FDG (CTI: NCT02593318, NCT01561053, and NCT02560753). Although FDG-PET is able to provide information about regional glucose metabolism, which can aid in the detection and prognosis of MCI for further progression to AD, there is a great need for PET- and SPECT radiopharmaceuticals which deliver more target-specific information of a variety of (patho)physiological processes that are happening in AD. In this review we will therefore focus on some of the major molecular pathophysiological changes known to occur in AD, along with emerging pharmacological treatment approaches. Furthermore, specific attention will be attributed to the role that PET- and SPECT biomarkers (can) play during these clinical trials.
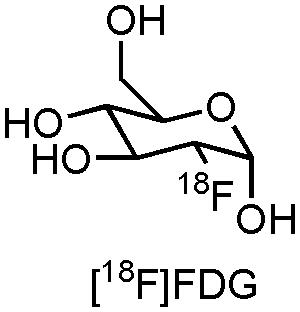
FIGURE 1. Structure of [18F]FDG.
The cholinergic hypothesis states that a decreased cholinergic neurotransmission, caused by a degeneration of cholinergic neurons in the basal forebrain, leads to several cognitive and functional conditions, associated with the symptoms of AD (Davies and Maloney, 1976; Bartus et al., 1982). Furthermore, the disruption of AChE seems to be associated with NFT- and Aβ deposits (Tavitian et al., 1993).
Acetylcholine, a neurotransmitter synthesized presynaptically by ChAT, is released in the synaptic cavity, where it is able to interact with nicotinic and muscarinic cholinergic receptors on pre- and postsynaptic membranes. Synaptic transmission is eventually stopped by the hydrolysis of ACh by AChE (Lleo et al., 2006). Both ChAT and AChE expression is reduced in cortical regions of AD patients (Davies and Maloney, 1976; Coyle et al., 1983; Vogels et al., 1990).
Inhibition of AChE was the first approach to treat AD, and this led to the FDA approval of eventually four AChE inhibitors: galantamine, rivastigmine, donepezil, and tacrine, though the latter one was largely discontinued due to hepatotoxicity issues (Wu et al., 2010). All inhibitors showed however only mild symptomatic improvement in patients with mild to moderate AD (Burns et al., 1999; Rosler et al., 1999; Farlow et al., 2000; Greenberg et al., 2000; Raskind et al., 2000; Tariot et al., 2000; Wilcock et al., 2000; Winblad et al., 2001). Several new agents are currently under development (Aprahamian et al., 2013), two of which have been evaluated in clinical trials: phenserine (Winblad et al., 2010; Darreh-Shori et al., 2014) and huperzine A (Rafii et al., 2011). Phenserine, structurally related to rivastigmine, showed a prolonged, but mild inhibition of AChE in AD patients. Researchers suggested an add-on therapy with donepezil to improve the clinical efficacy of this class of agents. Low dosage of huperzine A, a reversible AChE inhibitor, showed no significant improvement on the ADAS-cog [the primary cognitive outcome measure in mild to moderate AD patients (259)], in a phase II trial in mild to moderate AD patients. Higher dosage and a long term evaluation were suggested by the authors. A phase III trial (CTI:NCT01282619) using sustained-release huperzine A is currently ongoing (Ghezzi et al., 2013).
Visualization of the cholinergic system could be done by using radiolabeled ACh analogs or inhibitors of AChE (Tavitian et al., 1993; Irie et al., 1994; Iyo et al., 1997; Kilbourn et al., 1996). Both pathways have been pursued by researchers, but at present only two carbon-11 labeled compounds have been clinically evaluated on AD patients: [11C]MP4A and [11C]MP4P, two N-[11C]methylpiperidine esters (acetate and propionate, Figure 2). Moreover, both compounds were used to evaluate the effect of donepezil or rivastigmine in AD patients. Scans with [11C]MP4A or [11C]MP4P, taken before and after treatment with donepezil or rivastigmine, showed significant (up to 40%) cerebral cortical or frontal cortical inhibition of the AChE activity. Modest symptomatic improvement was recorded for all AD patients during these trials (Kuhl et al., 2000; Shinotoh et al., 2001; Kaasinen et al., 2002). These studies show the usefulness of both tracers for therapeutic monitoring of AChE inhibitors, as well as the possibility to evaluate newly developed drugs that target AChE (Shinotoh et al., 2004). Nonetheless, carbon-11 labeled compounds have the limitation that an on-site cyclotron is needed, which limits their widespread use. The lack of significant cognitive improvement and the fact that the cholinergic deficit is not an early event in the development of AD (Gilmor et al., 1999), has challenged the cholinergic hypothesis (Francis et al., 1999; Bartus, 2000; Terry and Buccafusco, 2003; Contestabile, 2011). Nevertheless, two decades after FDA approval of tacrine, AChE inhibitors remain (out of necessity) the mainstay for the current symptomatic treatment of AD.
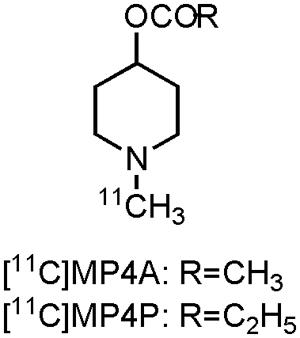
FIGURE 2. Structure of acetylcholinesterase PET tracers.
The presynaptic cholinergic signal is transmitted through the release of the neurotransmitter ACh, which can interact with both muscarinic and nicotinic ACh receptors (the latter one discussed in the next section). The pre-and postsynaptic muscarinic ACh receptor (mAChR) is a plasmamembrane, GTP binding protein coupled receptor, of which five subtypes exist (M1-M5) (Bonner et al., 1987; Peralta et al., 1987; Bonner, 1989). Due to the involvement in several neurological and psychiatric disorders, this target has been the topic of many research papers during the past few decades (Palacios et al., 1990; Tandon et al., 1991; Maziere, 1995). A comprehensive autoradiography study using tritium labeled compounds on the distribution of M1–M4 muscarinic receptors of histopathological diagnosed AD patients by Rodríguez-Puertas et al. (1997) showed that the decrease of M1 muscarinic receptors followed the general pattern of neurodegeneration as recorded in the Braak stages (Braak and Braak, 1991), while the M2 muscarinic receptor displayed significant reduction in the hippocampal area (up to 64%) and a significant increase in the striatum (up to 468% in the putamen), although this increase was not confirmed by other research groups (Eckelman, 2002). The density of M3-4 receptors on the other hand, was not altered compared to their density in brains of HCs. Interestingly, several research groups demonstrated that the stimulation of the M1- and M3 muscarinic receptors lead to an increase of the neuroprotective non-amyloidogenic pathway (formation of α-APP) (Buxbaum et al., 1992; Nitsch et al., 1992). Stimulation of these receptors could thus provide means for a decrease in Aβ production, a hypothesis which was confirmed in several studies (Wolf et al., 1995; Savonenko et al., 2005; Tsang et al., 2006). Little is known about the M5 muscarinic receptor, the latest receptor to be cloned (Bonner et al., 1988; Liao et al., 1989), although some research groups have demonstrated possible involvement in regulation of the cerebral blood flow and DA release (Yamada et al., 2001).
A number of clinical trials have been carried out to examine the potential role of mAChR agonists/antagonists on the clinical symptoms of AD patients. In the group of the selective M1 agonists, talsaclidine showed a significant decrease (up to 27% compared to placebo) in the Aβ-CSF levels in a randomized, double-blind, placebo controlled trial on AD patients with a MMSE score between 12 and 26. Results should, however, be interpreted with some caution, as suggested by the researchers, since assessment of the amyloid burden in AD patients by CSF has some flaws as biomarker, furthermore there was no mention on any cognitive improvement alongside the drop in Aβ CSF levels (Hock et al., 2000). Yet, another M1selective agonist, cevimeline (AF102B), an FDA approved drug for the treatment of dry mouth in Sjögren’s syndrome, did show cognitive improvement in the ADAS-cog and word recognition scales in a single-blind-placebo-controlled parallel group study with patients with probable AD (Fisher et al., 1996). Dual selectivity for both the M1- and M2-receptor on the other hand, as it is the case for RS-86, showed no consistent cognitive improvement in a double-blind, placebo-controlled trial on mild to moderate AD patients (Bruno et al., 1986). Palacios et al. (1986) hypothesized that RS-86’ failure could be due to the concomitant stimulation of the M1- and M2 receptor, where stimulation of M2 might inhibit the effects of the M1 receptor. On the other hand, milameline, a partial agonist for all five muscarinic receptor subtypes, demonstrated an effect on the rCBF in the frontal and subcortical regions of AD patients as part of an ‘add-on study’ during a Phase III clinical trial of this drug. AD participants were evaluated with SPECT, using the cerebral blood flow tracer 99mTc-exametazime (99mTc-HMPAO), during the performance of two cognitive tasks. Although a modest increase (of 26%) of rCBF was demonstrated, the authors suggested that there maybe neuropsychopharmacological effects associated with the intake of milameline during the performance of cognitive demanding tasks (Trollor et al., 2006). And finally, in a large-scale clinical trial, xanomeline, a M1- and M4-receptor agonist and M5 receptor antagonist (Grant and El-Fakahany, 2005), was evaluated in a randomized, double-blind, placebo-controlled trial on mild to moderate AD patients. Significant cognitive improvement was hereby shown in the ADAS-cog (drug vs. placebo; p ≤ 0.05), and the CIBIC+ scale (drug vs. placebo; p ≤ 0.02), demonstrating that a muscarinic receptor agonist can ameliorate cognitive symptoms in AD patients (Bodick et al., 1997).
Only one imaging agent, with affinity for the M1- and M4 muscarinic receptor (Piggott et al., 2002), has been clinically evaluated on AD patients thus far, namely [123I]I-quinuclidinyl benzilate ((R,R) [123I]I-QNB) (Figure 3). Eighteen mild to moderate AD patients and their age-matched HCs were evaluated with [123I]I-QNB. Significantly reduced uptake (p ≤ 0.001) was noted in the frontal rectal gyrus, right parahippocampal gyrus, left hippocampus, and regions of the left temporal lobe, compared to the HCs (Pakrasi et al., 2007). Nevertheless, conflicting results have been reported by other research groups with this tracer (Holman et al., 1985; Weinberger et al., 1991; Wyper et al., 1993; Kemp et al., 2003), although the sample sizes in these other studies were smaller. In another study, [123I]I-QNB was used as a biomarker to evaluate the density of the mAChR on 20 patients receiving the AChE inhibitor donepezil (Brown et al., 2003). No distinction could be made between donepezil responders and non-responders, furthermore no positive correlation was found between [123I]I-QNB scans and the extent of cognitive improvement on the ADAS-cog scale, apart from the insular cortex, were an inverse correlation was found. Researchers suggest that response to donepezil may thus be greater in patients with clear cholinergic deficits. Several efforts have been made to develop carbon-11 and fluorine-18 labeled tracers for muscarinergic receptors (Farde et al., 1996; Eckelman, 2001; Xie et al., 2004). A clinical trial in the US with3-(3-(3-([18F]fluoropropyl)thio)-1,2,5-thiadiazol-4-yl)-1,2,5,6-tetrahydro-1-methylpyridine ([18F]FP-TZTP) (Ravasi et al., 2012; Figure 3), which binds to the M2 receptor, on AD patients has been completed (CTI: NCT00001917), but results are yet to be published. Lastly, recruitment for a study with HCs and AD patients using an M4 positive allosteric modulator [11C]MK-6884 (structure not yet available), will soon start (CTI: NCT02621606). Future PET- and SPECT compounds hold promise to evaluate inclusion- and outcome criteria with novel drugs targeting the muscarinic receptor, as well as to establish therapeutic windows via dose-occupancy studies.
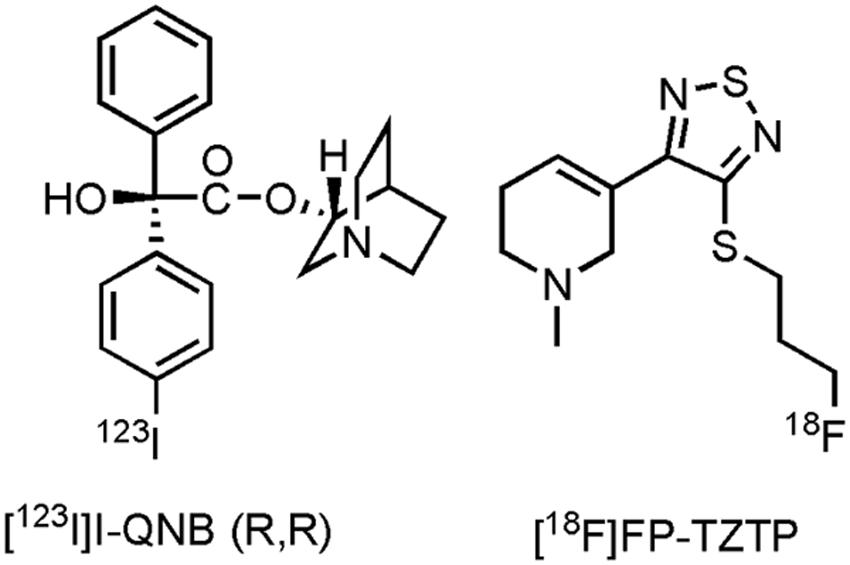
FIGURE 3. Structure of muscarinic PET/SPECT tracers.
nAChRs are ionotropic receptors, part of the ligand-gated ion channel superfamily (Cooper and Millar, 1997; Castelan et al., 2008; Criado et al., 2011; Valles and Barrantes, 2012). They consist of a hetero-or homopentameric structure, assembled from 17 possible subunits: α1-10, β1-4, γ, δ, and ε (Karlin, 2002; Gotti and Clementi, 2004). The main subtypes of the nAChRs in the human CNS are, however, α7, α4β2, and α3β2, although the latter one is not involved in the pathophysiology of AD (Wevers and Schroder, 1999; Pym et al., 2005). Reduction in nAChRs expression levels of several subtypes has indeed been revealed in regions with dense deposits of Aβ and NFTs (Pimlott et al., 2004; Oddo and Laferla, 2006; Buckingham et al., 2009). The α7 nAChR is mainly expressed in the hippocampus, whereas the α4β2 nAChR is homogenously expressed throughout brain (O’Brien et al., 2007; Valles and Barrantes, 2012). While loss of α4β2 nAChR can cause memory deficits in AD patients (Paterson and Nordberg, 2000; O’Brien et al., 2007; Kendziorra et al., 2011), a more complex relationship has been noted when evaluating the interaction between α7 nAChR and Aβ in AD (Oddo and Laferla, 2006). Aβ can either interact as agonist or as antagonist of α7 nAChR, depending on its concentration, where low concentrations may activate and high concentrations may inactivate α7 nAChR (Dineley et al., 2002; Puzzo and Arancio, 2013; Sadigh-Eteghad et al., 2014). Observations in an AD transgenic mouse model overexpressing APP, presenilin-1, and tau (3xTg-ADmice) (Oddo et al., 2003; Billings et al., 2005; Kitazawa et al., 2005) were consistent with the aforementioned in vitro conclusions for human AD, demonstrating an age dependent reduction of α7 nAChR, as higher Aβ may eventually block remaining α7 nAChRs (Hernandez et al., 2010). For a more detailed discussion about the many different roles of nAChRs in AD, readers are referred to several reviews on the subject (Oddo and Laferla, 2006; Jurgensen and Ferreira, 2010; Vandenberghe et al., 2010).
Since nicotine can induce the release of presynaptic ACh and is involved in the modulation of many other neurotransmitters, such as GABA, DA, norepinephrine, and serotonin (Levin, 1992), several clinical studies have investigated the effect of nicotine on AD patients (Jones et al., 1992; Wilson et al., 1995; Snaedal et al., 1996; White and Levin, 1999). However, as mentioned in a review by Oddo and Laferla (2006), these trails failed to demonstrate any cognitive improvement in AD patients; only an increase in attention could be determined. Yet, clinical trials with two nAChR agonists did show cognitive improvement in mild to moderate AD patients (Potter et al., 1999; Deardorff et al., 2015). The first of them was encenicline (EVP-6124), a partial agonist of α7 nAChR. This drug was well tolerated in Phase I and II trials, showing significant improvement in cognitive and functional domains (Deardorff et al., 2015). A currently ongoing Phase III trial involving mild to moderate AD patients, receiving or having already received AChE inhibitors, to assess the efficacy and tolerability of EVP-6124 (ECT: 2012-003209-92) in a large group of patients was halted due to severe gastrointestinal side-effects (Shugart, 2016). Another nAChR agonist, ABT-418, which has binding affinity for α4β2, α2β2, and α3β4 (Potter et al., 1999), showed some cognitive improvement in the acquisition and retention of verbal information of patients with early AD (Mean MMSE score of 21.4). It is, however, unclear whether this compound will be further pursued in large clinical trials. Two other trials with nAChR agonists were less convincing; efficacy of ispronicline (TC-1734 or AZD-3480) a selective agonist of α4β2 nAChR and α2β2 nAChR (Gatto et al., 2004) was investigated in a large Phase IIb dose-finding study on mild to moderate AD patients (MMSE score: 12–26). Despite the fact that ispronicline caused significant improvement on patients with age-associated memory impairment (Dunbar et al., 2007, 2011), no significant improvement could be shown on the ADAS-cog scale in the latest Phase IIb study, although secondary outcome measurements did show some improvement (Frolich et al., 2011). Additional Phase II trials on mild to moderate AD patients were halted, since no superiority over donepezil could be demonstrated. Finally, in a Phase II trial on mild to moderate AD patients (MMSE score of 26–26 and 14–20, respectively), no cognitive improvement could be observed with the α4β2 nAChR selective agonist varenicline (Kim et al., 2014). Researchers concluded that the dosing regimen was not optimal and overall longer (than 6 weeks) trials may be needed to show cognitive improvement. In the light of these two failed clinical trials with α4β2 nAChR agonists, another clarification could, however, be that the α4β2 nAChR subtype may only play a minor role on cognitive processes in AD, and α7 nAChR is therefore a more suitable target (Kim et al., 2014).
While nicotine was not really used as a therapeutic drug, as a carbon-11 labeled PET tracer it successfully showed reduction of nAChR in AD patients reflecting the loss of the nicotinic receptors during disease progression, in comparison with control subjects (Nordberg et al., 1990; Nordberg et al., 1995; Kadir et al., 2006). Additionally, a number of other clinical studies used [11C]nicotine PET imaging (Figure 4), to assess the efficacy of the AChEIs tacrine and rivastigmine (Nordberg et al., 1992; Nordberg et al., 1997; Nordberg et al., 1998; Kadir et al., 2007). Significant increase in nAChR expression compared to baseline in several cortical areas could be demonstrated, after treatment with both tacrine and rivastigmine. [11C]nicotine does, however, show high non-specific binding and rapid brain wash-out, making quantitative PET assessments of nAChR difficult. A few other PET radiotracers are currently under development for imaging of α7 and α4β2 nicotinic receptors (Toyohara et al., 2010; Meyer et al., 2014; Chalon et al., 2015); two structurally related compounds, one SPECT and one PET tracer, with affinity for the α4β2 nicotinic receptor have already been evaluated on AD patients (O’Brien et al., 2007; Ellis et al., 2008; Sabri et al., 2008). Reduced tracer uptake was noted in AD patients in the frontal lobe, striatum, right medial temporal lobe and the pons after scans with-5-[123I] iodo-3-[2(S)-2-azetidinylmethoxy]pyridine ([123I]5IA-85350) (Figure 4), consistent with known reductions of the α4β2 nicotinic receptor in AD (O’Brien et al., 2007). Its PET counterpart, 2-[18F]fluoro-3-(2(S)-azetidinylmethoxy)pyridine (2-[18F]FA-85380) (Figure 4), was able to demonstrate significant reduction (up to 75%) of the α4β2 nicotinic receptor in MCI patients, which later on converted to AD (Sabri et al., 2008). In another study with 2-[18F]FA-85380, the possible relationship between Aβ depositions and the reduction of the α4β2 nicotinic receptor was studied by evaluating early to moderate AD patients (Okada et al., 2013). A negative correlation between the presence of Aβ in the medial frontal cortex and the nucleus basalis magnocellularis, as assessed by [11C]Pittsburgh Compound B ([11C]PiB, an Aβ tracer), and the binding of 2-[18F]FA-85380 to the α4β2 nicotinic receptor could be established. Both α4β2 nicotinic receptor tracers suffer, however, from slow kinetics, leading to long scanning times, making routine application difficult (Meyer et al., 2014). Conversely, a novel α4β2 nAChR tracer, 2-{5-[2-[18F]fluoropyridin-4-yl]pyridin-3-yl}-7-methyl-7-azabicyclo[2.2.1]heptane ([18F]XTRA) (Meyer et al., 2014) (Figure 4), showed much faster pharmacokinetics, which allow scanning within a reasonable time frame. Recruitment for a clinical trial with [18F]XTRA on HCs, an AD or a MCI patient is currently ongoing (CTI:NCT01894646).
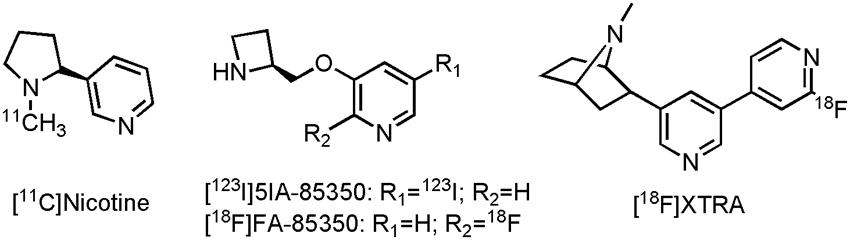
FIGURE 4. Structure of nicotinic PET/SPECT tracers.
As one of the pathological hallmarks in well over 20 neurodegenerative diseases (Lee et al., 2001), tau (tubulin associated unit), gained an increasing interest in the past few years, partly as a result of the large failure rate of clinical trials targeted toward the amyloid hypothesis (Barthel et al., 2015), but also due to recent availability of several tau specific PET ligands (Villemagne et al., 2015). As a member of the microtubule-associated protein (MAP) family, tau is mainly localized in the distal part of the neuronal axons (Binder et al., 1986). Consequently, the primary function of tau is stabilization and support of the microtubules (Weingarten et al., 1975). There are six different isoforms of tau, depending on alternative splicing of exon two, three, and ten of the MAPT. Localized on chromosome 17q32, this gene contains 16 exons (Neve et al., 1986). Exon two and three both encode a 29-aminoacid fragment at the N-terminal part of tau, yielding isoforms with none (0N), one (1N), or two inserts (2N). Exon ten on the other hand, encodes a 31-aminoacid fragment, which results in either three (3R) or four (4R) repeated binding domains at the C-terminus of the protein. The mature human brain contains thus six isoforms of tau: 0N3R, 1N3R, 2N3R, 0N4R, 1N4R, and 2N4R. Under normal conditions (and in AD) there is a 1:1 ratio of the 3R- and 4R isoforms, but this ratio somehow shifts in certain pathological conditions (Hong et al., 1998). The exact physiological role of these various isoforms remains to be elucidated, although 4R isoforms are better at promoting microtubuli assembly, and have greater binding affinity for microtubuli than the 3R isoforms (Goedert and Jakes, 1990; Butner and Kirschner, 1991). Under non-pathologic conditions, tau is a highly soluble protein with a limited secondary structure (Dunker et al., 2008), prone to several post-translational modifications. The most important one is phosphorylation on its serine and threonine residues, which modulates microtubule binding (Martin et al., 2011). Then again, in pathological conditions, such as AD, tau will become hyperphosphorylated, detaches from the microtubules and will self-aggregate into insoluble PHFs and NFTs, compromising neuronal cell function (Iqbal and Grundke-Iqbal, 2008). Different shapes and sizes of the aggregates can be found under diverse cognitive conditions, related to the presence of various isoforms, and post-translational modifications (Ballatore et al., 2007). In AD, the spreading of the tau pathology, which is thought to proceed in a prion-like manner (de Calignon et al., 2012), has been well-documented in the different Braak stages (Braak and Braak, 1991). Furthermore, several studies confirmed that this characteristic pattern of aggregated tau spread closely correlates to the clinical symptoms of AD, as measured by the MMSE (Bancher et al., 1993, 1996; Duyckaerts et al., 1997; Grober et al., 1999). This makes tau an interesting target for drug development. Due to the complexity of aggregated tau as a drug target, several tau-associated approaches have been investigated.
Glycogen synthase kinase 3β, being the dominant isoform of three GSK-3 variants (Jaworski et al., 2011), is the main kinase responsible for the hyperphosphorylation of tau, and hence an important potential target for disease-modification (Maqbool et al., 2016). Diverse GSK-3 inhibitors were reported the last few years by many research groups (Noble et al., 2011; Berg et al., 2012; Maqbool et al., 2016). Lithium and valproate were the first compounds to be clinically evaluated, but due to inconsistent and overall disappointing results, they were largely discontinued (Noble et al., 2005; Hampel et al., 2009; Tariot and Aisen, 2009; Tariot et al., 2011). Several other GSK-3 inhibitors have, however, been pursued (Maqbool et al., 2016), two of which, tideglusib (NP0311212) and AZD1080, entered clinical trials (King et al., 2014; Lovestone et al., 2015). Development of AZD1080 was, however, halted in Phase I due to nephrotoxicity problems (Eldar-Finkelman and Martinez, 2011) and no clinical benefit was seen with tidelusib on patients with mild to moderate AD in Phase II clinical trials. Dose finding studies and longer trials are now required with the latter drug to examine its possible long term benefit (Lovestone et al., 2015). Another tau-associated approach is the inhibition of tau-aggregation (Bulic et al., 2009, 2010). The first of this class to be pushed in Phase II clinical trials, methylthioninium chloride (methylene blue, MTC), a phenothiazine derivative, was able to stabilize disease progression over a period of 50 weeks in mild and moderate AD patients (Wischik et al., 2015). The brain bioavailability of this charged drug remains, however, to be elucidated. The pro-drug of MTC, leuco-methylthioninium (TRx0237 or LMTX), with a superior pharmacological profile (Wischik et al., 2014) will now be evaluated in three parallel Phase III trials on mild to moderate AD patients and patients with FTD (CTI: NCT01689246, NCT01689233, and NCT01626378). And finally, an increasing interest toward tau immunotherapy has been noted, as means of removing tau aggregation by the patients’ own immune system (Asuni et al., 2007). Two drugs of this kind, ACI-35 (ECT: 2015-000630-30) and AADvac1 (CTI: NCT02031198) are currently being evaluated in Phase I and II trials.
Despite the historical importance of tau as a pathological hallmark in AD (Graeber and Mehraein, 1999), only recently tau specific PET ligands have been developed. One of major issues during tau PET development is the lack of a representative tau-animal model, which may be explained by (ultra)structural differences between murine and humane tau (Duyckaerts et al., 2008). More than a few tracers are, however, currently being clinically evaluated. The first tau PET ligand to be reported was 2-(1-{6-[(2-[18F]fluoroethyl)(methyl)amino]-2-naphthyl}ethylidene)malononitrile ([18F]FDDNP) (Figure 5), although not specific for tau as such, high binding affinity was reported in several neurodegenerative diseases (Bresjanac et al., 2003; Small et al., 2006; Kepe et al., 2010; Nelson et al., 2011; Kepe et al., 2013; Small et al., 2013). Limited dynamic range of signal and relatively low affinity for tau, led to the development of novel tau directed ligands with similar structural moieties. Yet, the first real approach toward tau specific ligands was achieved by researchers of the Tohoku University in Japan with the development of 4-{6-[2-[18F]fluoroethoxy]quinolin-2-yl}aniline ([18F]THK523) (Fodero-Tavoletti et al., 2011) (Figure 5). While [18F]THK523 was able to visualize the known pattern of tau distribution in AD patients, high white matter binding and unfavorable pharmacokinetics (Villemagne et al., 2014) led to the development of three novel 2-arylquinoline derivatives: 1-({2-[4-(dimethylamino)phenyl]quinolin-6-yl}oxy)-3-[18F]fluoropropan-2-ol ([18F]THK5105) (Okamura et al., 2014a), 1-[18F]fluoro-3-({2-[4-(methylamino)phenyl]quinolin-6-yl}oxy)propan-2-ol ([18F]THK5117) (Ishiki et al., 2015) and the optically pure (2S)-1-[18F]fluoro-3-({2-[4-(methylamino)phenyl]quinolin-6-yl}oxy)propan-2-ol ([18F]THK5351) (Harada et al., 2016) (Figure 5). All three compounds showed high binding in AD patients, with radiotracer retention in sites known for their tau deposition. Of these three compounds, [18F]THK5351 showed superior pharmacokinetics, highest signal-to-noise ratio and the lowest white matter binding (Okamura et al., 2014b; Harada et al., 2016). Further clinical trials in Japan with [18F]THK5351 are underway (UMIN-CTR: UMIN000013929 and UMIN000018496). Often considered by many research groups as the current benchmark in tau PET development, 11-{4-[2-[18F]fluoroethyl]piperidin-1-yl}-1,8,10-triazatricyclo[7.4.0.02,7]trideca-2(7),3,5,8,10,12-hexaene ([18F]T808 or [18F]AV680), and 2-[18F]fluoro-5-{5H-pyrido[4,3-b]indol-7-yl}pyridine ([18F]T807 or [18F]AV1451) (Figure 5) have high affinity and selectivity for tau over Aβ (Zhang et al., 2012; Xia et al., 2013; Shah and Catafau, 2014). A small first-in-man study with [18F]T808 in eight AD patients (mean MMSE of 18) and their three age matched HCs, showed a rapid brain uptake and washout in HCs, and a tau pattern consistent with the Braak stages in the AD group (Chien et al., 2013). Interestingly, one of the AD patients who died a few weeks after his PET scan with [18F]T808, showed close correlation with his histopathological staining (Dani et al., 2015). Nonetheless, substantial bone uptake was observed with this compound (Villemagne et al., 2015), which led to the development of [18F]T807. In comparison to [18F]T808, [18F]T807 has slower kinetics and a relatively lower affinity for tau, but [18F]T807 does not show defluorination (Xia et al., 2013). Similar clinical findings as with [18F]T808 were demonstrated with [18F]T807 in a small first-in-man study on three HCs, in one patient with MCI (MMSE score of 26) and one severe AD patient (MMSE score of 7) in comparison with three HCs. Remarkably, the tracer retention was significantly lower in the patient with MCI, as compared to the patient with severe AD (Chien et al., 2012). A series of large clinical trials (ClinicalTrial.gov and EU Clinical Trials Register: search term: ‘T807’ OR ‘AV1451’ AND ‘PET’) is underway with [18F]T807 to evaluate its applicability not only in AD, but also in several other tauopathies. Being thus far the only compound to be able to visualize tau (and possibly different isoforms) in AD, but also in PSP and CBD (Maruyama et al., 2013), 2-((1E, 3E)-4-(6-([11C]methylamino)pyridin-3-yl)buta-1,3-dienyl)benzo[d]thiazol-6-ol ([11C]PBB3) received a lot of interest (Figure 5). Clinical studies on AD patients and a CBD patient, as compared to HCs, showed increased tracer uptake, consistent with the Braak stages (for the AD case), and higher retention in the basal ganglia (for the CBD case). Stability issues and a challenging radiosynthesis might, however, limit its commercial use. Several other tau directed PET ligands from Roche, such as [11C]RO6931643, [11C]RO6924963, and [18F]RO6958948 (structures not available) have been evaluated in a Phase I clinical trial, but data are yet to be published (CTI: NCT02187627) (Dani et al., 2015). Other clinical studies with [18F]MK-6240, [18F]MNI-798, and [18F]MNI-815 (structures not available) on AD cases are currently recruiting patients (CTI: NCT02562989, NCT02640092, and NCT02531360). For a more detailed discussion about the current state of tau PET development, readers are referred to some excellent reviews on the subject (Okamura et al., 2014b; Zimmer et al., 2014; Dani et al., 2015). An important question that remains to be elucidated is for which isoforms these tau PET tracers have affinity; a question which may have major implications on the differential diagnosis of closely related neurodegenerative tauopathies. Nevertheless, the substantial progress that has been made in this field will make it possible to allow in vivo detection of tau in AD and thus the reassessment of inclusion- and outcome criteria of clinical trials aiming to intervene at tau-aggregates.
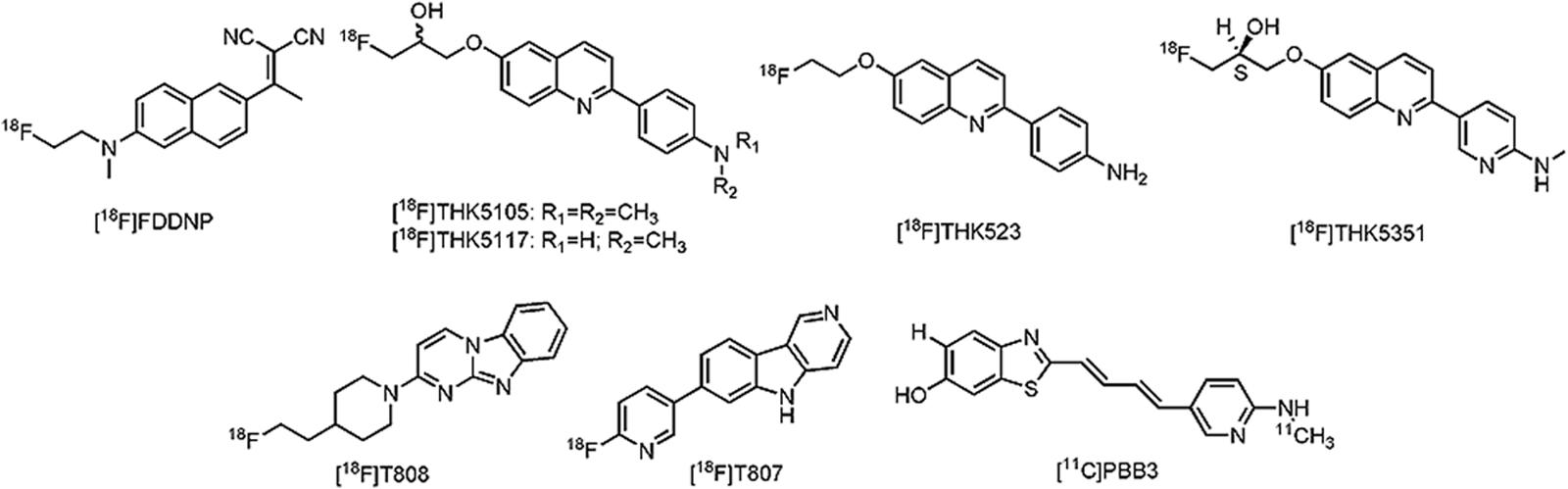
FIGURE 5. Structure of tau PET tracers.
For many decades, the amyloid hypothesis has been the main pathological model of AD (Hardy and Higgins, 1992; Korczyn, 2008), accepted by most researchers, and only recently contested (Hardy, 2006). It postulates that extensive deposits of amyloid in the human brain are the central lesions in the development of AD, responsible for a neurotoxic cascade of events, which ultimately leads to dementia (Barage and Sonawane, 2015). Aβ peptides, the main component of SP (Gomez-Isla et al., 1997), are 39–43 amino acid residues, formed during the sequential cleavage of the transmembrane APP by β-secretase 1 (also called BACE1) (Haass, 2004), followed by the action of the γ-secretase (Selkoe, 2001). Under ‘normal’ conditions APP is cleaved in a non-amyloidogenic pathway by the action of initially α-secretase, forming α-sAPP, which may have a neuroprotective function (Pagani and Eckert, 2011). α-sAPP is then further cleaved by γ-secretase to eventually produce P3. The function of APP itself is unknown, although a possible role in the Cu-homeostasis has been proposed (Barnham et al., 2004). There are two main isoforms of Aβ: Aβ40 and Aβ42, the latter one being more prone to aggregation and regarded as the main neurotoxic species (Ballard et al., 2011). Once formed, Aβ species will undergo several characteristic changes, from small oligomers into larger fibrils, which eventually form diffuse and later neuritic plaques. These plaques frequently trigger astrocytosis, activation of microglial cells, cytokine release, and a multi-protein neuroinflammatory response (Barage and Sonawane, 2015). Just like the NFTs, amyloid depositions follow a specific pattern, as recorded by Braak and Braak (1991). In contrast to the NFTs, however, there is a poor correlation between the extent of these plaques and the degree of cognitive impairment (Nelson et al., 2012), furthermore non-demented individuals can heave substantial loads of Aβ deposition without revealing any clinical symptoms (Villemagne et al., 2008). Recently, several studies pointed toward Aβ oligomers, and not amyloid plaques, as the main toxic species in AD (Haass and Selkoe, 2007; Minati et al., 2009). For a more extensive discussion about the neuropathological role Aβ plays in AD, readers are referred to other reviews (Haass and Selkoe, 2007; Hardy, 2009; Barage and Sonawane, 2015).
Huge efforts have been undertaken to develop disease altering drugs that target the amyloid deposition, but unfortunately many failed during clinical trials (Barthel et al., 2015). Some of the most recent ongoing trials targeting the amyloid deposits are summarized in Table 1. Various therapeutic approaches are to be considered when targeting Aβ (Barage and Sonawane, 2015). We will discuss here the most important tactics, together with some of their constraints, as it is imperative to know why so many trials fail in this area. One of many methods applied, is the reduction of Aβ production through inhibition of β-and/or γ-secretase or activation of α-secretase (Cummings, 2008). Indeed, the therapeutic potential of BACE1 inhibitors has been demonstrated in BACE1 knockout mice, which produced significantly (15-fold) less Aβ (Luo et al., 2001; Roberds et al., 2001). Nonetheless, inhibition of BACE1 causes several problems, since BACE1 has been shown to have many physiological roles, which might lead to toxicity problems when using BACE1 inhibitors. Furthermore, BACE1 inhibitors need to be quite bulky, due to the relatively large active site and this can cause BBB passage issues (Ghezzi et al., 2013). Inhibition of the multimeric γ-secretase complex encounters similar problems as the use of BACE1 inhibitors, as γ-secretase has many other physiological roles as well, especially cleavage of the Notch receptor, necessary for growth and development (Yiannopoulou and Papageorgiou, 2013). Brain penetration seems to be an issue as well in this area (Imbimbo and Giardina, 2011). Increasing the α-secretase activity, and thus promoting the non-amyloidogenic pathway, is another way to reduce the Aβ load. Less is however known about the possible physiological consequences of such an upregulation (Barage and Sonawane, 2015). Another way of interfering with the Aβ load is by modulation of Aβ aggregation, as increasing evidence suggests that soluble oligomers, which act as intermediates for the formation of aggregates, are the most toxic species in AD disease (Dahlgren et al., 2002; Hoshi et al., 2003; Kayed et al., 2003). Yet, the most promising small molecules ultimately failed due to their (toxic) pharmacological profile (Santa-Maria et al., 2007; Rishton, 2008; Yiannopoulou and Papageorgiou, 2013). Several studies also showed the relationship of APP and Aβ with mitochondrial dysfunction in AD (Anandatheerthavarada et al., 2003; Lustbader et al., 2004; Caspersen et al., 2005); interaction of both proteins with mitochondrial matrix proteins, such as Aβ-binding alcohol dehydrogenase and adenosine triphosphate synthase subunit alpha, could directly lead to mitochondrial toxicity, and thus oxidative stress (Devi et al., 2006; Reddy and Beal, 2008). Numerous antioxidant agents have been described and evaluated in clinical trials with MCI- and AD patients, and several studies are still ongoing (Mecocci and Polidori, 2012; Polidori and Nelles, 2014). Conflicting results were, however, reported and overall small cognitive benefit was seen during these trials. Long-term trails are now warranted in order to establish clinical benefits in AD. Then again, several promising antioxidative compounds are currently being investigated (Qosa et al., 2015; Rigacci, 2015). Still, the primary action in targeting amyloid came from monoclonal antibodies. First discovered by Schenk et al. (1999) to be very effective in reducing the Aβ load in mice, the mechanism of action of amyloid immunotherapy remains, however, to be fully elucidated (Yiannopoulou and Papageorgiou, 2013). Nevertheless, only a small fraction (0.1% of the injected dose) of antibodies seems to be able to pass the BBB in humans (Banks et al., 2002). A higher fraction of antibodies in the brain may thus be needed to be therapeutically effective. This hurdle was the topic of two recently reported reviews (Lemere, 2013; Spencer and Masliah, 2014). Despite the low BBB’s passage, one of the major concerns with immunotherapy is the development of serious side effects, for instance encephalitis (active immunization), microhemorrhages, or vasogenic edemas (passive immunization) (Orgogozo et al., 2003; Panza et al., 2012). Another important factor to consider is the time of intervention in the AD state; immunotherapy is probably most efficacious in early disease states, when there is more function to preserve (Barthel et al., 2015). Efficient biomarkers, that can predict the conversion from MCI to AD, are therefore of utter importance. Using longitudinal PET biomarkers to assess and follow up the amyloid burden in clinical trials would indeed allow a more confident formulation of inclusion, but also outcome criteria (Barthel et al., 2015). Several amyloid PET tracers are currently being used for these purposes (see Table 1).
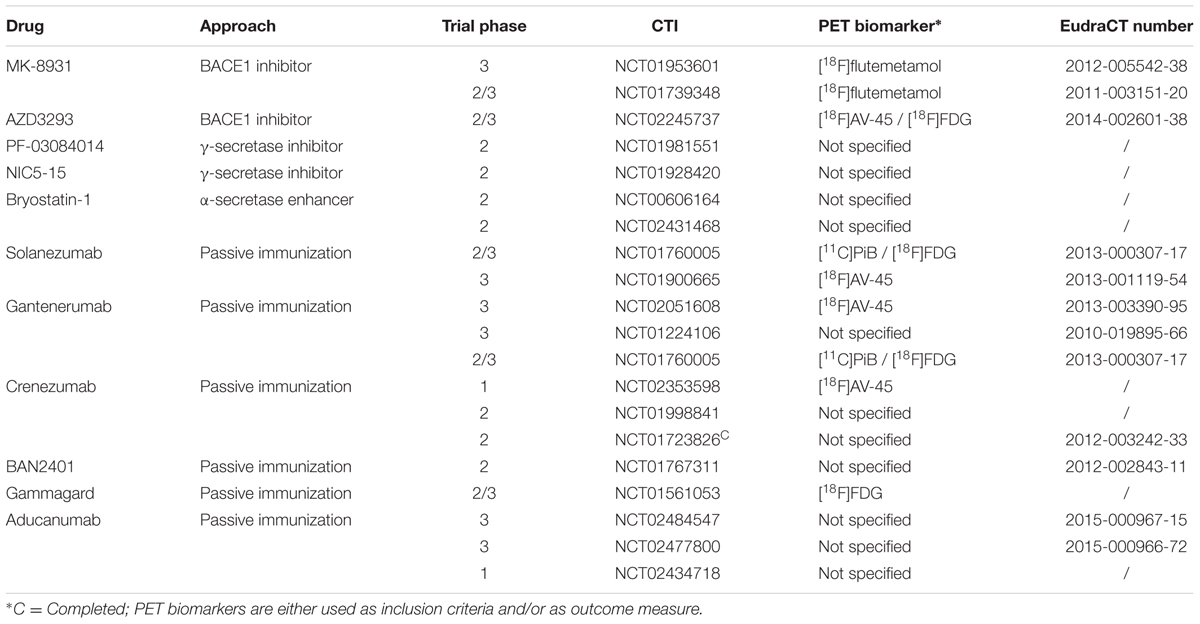
TABLE 1. Ongoing clinical trials with drugs targeting the amyloid hypothesis (Han and Mook-Jung, 2014; Wischik et al., 2014; Apter et al., 2015).
Although not FDA-approved, 2-{4-[[11C]methylamino]phenyl}-1,3-benzothiazol-6-ol ([11C]PiB) (Figure 6) has been used for many years as benchmark compound for in vivo imaging of the amyloid load in AD patients (Benadiba et al., 2012). Results of those trials have shown that clinically diagnosed AD cases have positive amyloid scans (Kemppainen et al., 2006; Jack et al., 2008, 2009; Lowe et al., 2009), and the ones that did not have positive scans, were most likely to be misdiagnosed (Rabinovici et al., 2007; Rabinovici et al., 2008). Furthermore, increased [11C]PiB binding is able to predict the conversion of MCI to AD (Okello et al., 2009). [11C]PiB has also been proven useful in the differential diagnosis of FTD and AD, as FTD patients typically have a normal [11C]PiB uptake (Rowe et al., 2007; Engler et al., 2008). Interestingly, a close correlation has been noted with CSF Aβ levels (Tolboom et al., 2009; Weigand et al., 2011), firmly establishing [11C]PiB as an Aβ biomarker. There are, however, a few limitations with [11C]PiB as an amyloid biomarker; [11C]PiB presumably binds to diffuse plaques and not to the more cognitive correlated neuritic plaques (Jack et al., 2013). Moreover, commercial use is excluded, due to the short half-life of carbon-11. Several attempts were thus undertaken to develop 18F-labeled analogs (Koo and Byun, 2013). Three of them: 4-[(E)-2-[6-(2-{2-[2-[18F]fluoroethoxy]ethoxy}ethoxy)pyridin-3-yl]ethenyl]-N-methylaniline ([18F]florbetapir or [18F]AV-45), 2-[3-[18F]fluoro-4-(methylamino)phenyl]-1,3-benzothiazol-6-ol ([18F]flutemetamol, [18F]GE-067 or [18F]AV-1) and 4-[(E)-2-[4-(2-{2-[2-[18F]fluoroethoxy]ethoxy}ethoxy)phenyl]ethenyl]-N-methylaniline([18F]florbetaben or [18F]BAY 97-9172) (Figure 6) have already been approved by the FDA and the EMA for their binding to neuritic plaques. Another one, 2-[2-[18F]fluoro-6-(methylamino)pyridin-3-yl]-1-benzofuran-6-ol ([18F]AZD-4694 or [18F]NAV4694) (Figure 6), is currently awaiting FDA-approval (Jack et al., 2013). Although all of the current 18F-labeled compounds show significant increased uptake in AD patients as compared to HCs in clinical trials (Rowe et al., 2008, 2013; Barthel et al., 2011; Villemagne et al., 2011), they suffer from high non-specific white matter binding, as compared to [11C]PiB (Benadiba et al., 2012; Rowe and Villemagne, 2013; Vandenberghe et al., 2013). Only [18F]AZD-4694 has a white matter uptake similar to [11C]PiB (Rowe et al., 2013). While a negative amyloid PET scan will exclude AD, a positive scan, on its own, is insuffient for the diagnosis of AD, as has been shown with [18F]florbetapir in clinic (Yang et al., 2012). Furthermore, limited reimbursement of these recently approved compounds limits their use in clinical practice (Barthel et al., 2015). The use of amyloid PET may, however, reveal true AD cases. Moreover, the amyloid tracers are able to predict the conversion from MCI to AD, and this can considerably influence decision making in AD related clinical trials (Rowe and Villemagne, 2013).
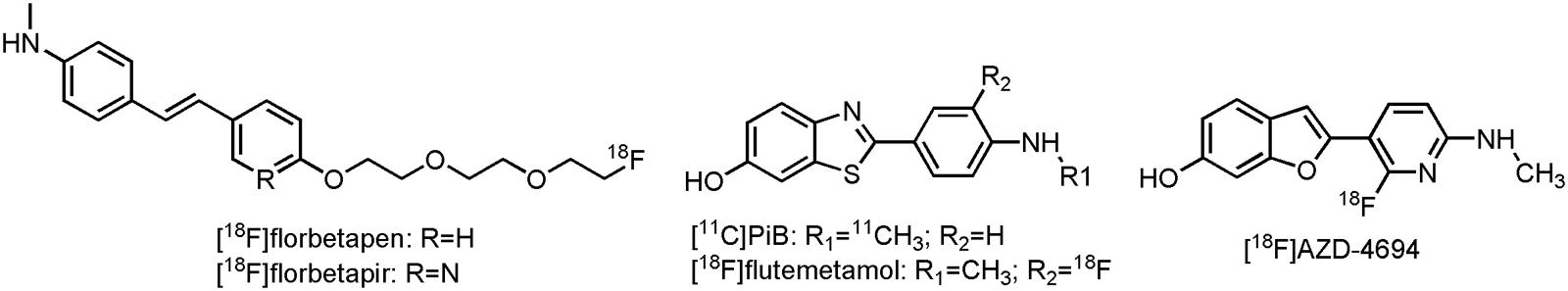
FIGURE 6. Structure of Aβ PET tracers.
The inhibitory GABA system in the CNS consists of three GABA receptor systems: GABAA, GABAB and GABAC (Chebib and Johnston, 1999). Since GABAB- and GABAC receptors have not been clinically evaluated in AD yet, focus will be toward the GABAA receptor. The GABAA receptor is a pentameric ligand gated ion channel, composed of a wide array of (possible) subunits: α1-6, β1-3, γ1-3, δ, ε, τ, π, and ρ1-3 (Mehta and Ticku, 1999). In order to be functional, the receptor seems to require the presence of at least one α- and one β-subunit. The most common composition is a pentamer composed of two α-, two β-, and one γ-subunit (Connolly et al., 1996). The GABA system plays an important role in AD, as it is one of the main culprits for the BPSD. Contributing to these BPSD, the GABA system is also known to modulate other neurotransmitters, such as serotonin, DA and ACh (Decker and McGaugh, 1991; Zorumski and Isenberg, 1991; Keverne, 1999). It has been a long standing, although contested (Lanctot et al., 2004), view that the GABA system undergoes little change during AD progression, due to dynamic plasticity of the system (Rissman et al., 2007). Recent findings suggest, however, otherwise and point to a severely altered GABAergic signaling in AD, with possible modulation of tau hyperphosphorylation (Lanctot et al., 2004; Limon et al., 2012; Nykanen et al., 2012). A more detailed discussion of the GABAsystem and its putative role in AD can be found in some extensive reviews (Marczynski, 1998; Lanctot et al., 2004; Rissman et al., 2007).
Benzodiazepines, which are allosteric modulators of the GABAA receptor (Hevers and Luddens, 1998), have long been used for the symptomatic treatment of anxiety and agitation in AD (Kirven and Montero, 1973; Covington, 1975; Sunderland et al., 1989; Zec and Burkett, 2008), nevertheless there is a need for randomized controlled trials to evaluate the true efficacy of these drugs in AD (Defrancesco et al., 2015). Caution is also to be advised with BZDs, as there are reports of rapid cognitive and functional decline in AD patients when taking these drugs for an extensive period of time (Zec and Burkett, 2008).
There have been numerous endeavors to developed radiotracers for in vivo imaging of the GABAA receptors (Katsifis and Kassiou, 2004; Andersson and Halldin, 2013). Ethyl 12-fluoro-8-[11C]methyl-9-oxo-2,4,8-triazatricyclo[8.4.0.022,6]tetradeca-1,3,5,10,12-pentaene-5-carboxylate ([11C]flumazenil) (Figure 7), a GABAAantagonist with affinity for the α1-3 and α5-subunit, is the most promising tracer thus far. Several clinical studies have been performed with [11C]flumazenil (Savic et al., 1988; Heiss et al., 2004; Frankle et al., 2009, 2012; Andersson and Halldin, 2013), one of which was carried out on early AD patients (Mean MMSE: 21.2) to evaluate the GABAA receptor density. Researches demonstrated a marked decrease in [11C]flumazenil binding, which correlated well with neuronal loss as evaluated by histopathological findings (Brun and Englund, 1981; Andersson and Halldin, 2013). The SPECT analog ethyl 11-[123I]iodo-8-methyl-9-oxo-2,4,8-triazatricyclo[8.4.0.02,6]tetradeca-1,3,5,10,12-pentaene-5-carboxylate ([123I]iomazenil) (Figure 7), showed significantly reduced uptake in the temporal, parietal end occipital cortex of moderate to severe AD patients (Soricelli et al., 1996; Fukuchi et al., 1997). In contrast to [11C]flumazenil though, [123I]iomazenil was not able to show significant changes in early AD patients (Pappata et al., 2010). Interestingly, in a direct PET-SPECT comparison study on healthy volunteers between [11C]flumazenil and [123I]iomazenil, the 123I-labeled variant came out as the better candidate, due to a better fit in compartmental modeling with SPECT (Bremner et al., 1999). These radiopharmaceuticals not only hold promise to be used as inclusion- and outcome criteria for drugs combatting BPSD symptoms in AD, but they could also be used in dose-occupancy studies to assess the (sometimes small) therapeutic window of BZDs.
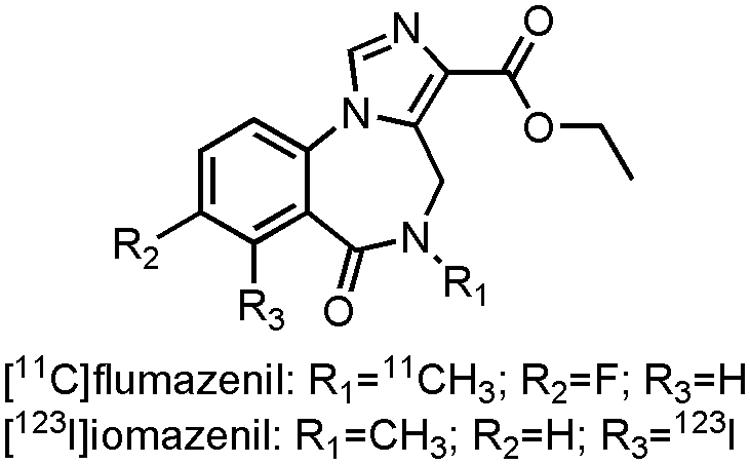
FIGURE 7. Structure of GABAergic PET/SPECT tracers.
Serotonin (5-HT), is a neurotransmitter that plays a complex role in the modulation of several psychological, emotional, and cognitive processes. Moreover, 5-HT affects long-term and short-term memory and cognitive function through the regulating of many other neurotransmitters, such as ACh, DA, GABA, and glutamate (Rodríguez et al., 2012). The principal 5-HT-source in the human brain comes from neurons in the raphe nuclei, with various projections throughout the CNS (Vertes, 1991; Vertes et al., 1999). There are seven main 5-HTRs, which can be divided into two major classes: the G-protein coupled receptors (5-HTR1,2,4-7) and the ligand-gated cation channels (5-HTR3), many of which have also several subcategories (Hoyer et al., 2002). For the ‘normal’ physiological and pharmacological role of these receptors, readers are referred to several reviews on the topic (Barnes and Sharp, 1999; Hoyer et al., 2002; Niesler et al., 2008). An overall reduction of the serotonergic system in AD pathology, likely reflecting the loss of serotonergic projections from the raphe nuclei, has been demonstrated (Bowen et al., 1983; Chen et al., 1996, 2000). Interestingly, loss of function seems more extensive in early onset AD than in later-onset AD, which may be due to compensating systems (Arai et al., 1992; Halliday et al., 1992). More specifically, marked reduction of the 5-HTR1A, which is expressed in brain areas known for their role in memory and learning, has been noted in the hippocampus and the frontal cortex during AD progression (Lai et al., 2003). This may, however, reflect a compensatory mechanism for reduction of cholinergic receptors in the AD brain, since inhibition of the 5-HTR1A has been implicated in the release of ACh (Millan et al., 2004; Kehr et al., 2010; Rodríguez et al., 2012). Another receptor that is affected during AD progression is 5-HTR2A, with reductions being noted in the frontal, temporal, parietal and enthorinal cortex and the hippocampus (Crow et al., 1984; Procter et al., 1988; Dewar et al., 1990). In a review by Rodríguez et al. (2012) it was suggested that a decrease in 5-HTR2A may effect cognitive functions in AD patients. A positive correlation between cognitive decline and 5-HTR2A related decrease in the frontal cortex has indeed been noted (Lai et al., 2005). Moreover, it was implied that decrease in 5-HTR2A density may be due to pathological accumulation of Aβ (Christensen et al., 2008; Holm et al., 2010). Stimulation of the 5-HTR4 may lead to an increase of the non-amyloidogenic pathway in vitro (Consolo et al., 1994; Robert and Benoit, 2008), indication for an important role in APP metabolism. Other 5-HTRs with marked reduction in AD are 5-HTR1B, 5-HTR1D, and 5-HTR6 (Garcia-Alloza et al., 2004; Lorke et al., 2006). Additionally, reduction of the former two correlates well with the cognitive decline in AD (Garcia-Alloza et al., 2004). Significant decrease (up to 25%) in binding sites of the 5-HTT during AD progression is also to be noted (Bowen et al., 1983; Ouchi et al., 2009). As part of the monoamine transporter family, the SERT is responsible for removal of serotonin from the synaptic cleft. There seems, however, no correlation between the reduced density of this transporter and BPSD symptoms, as seen in AD (Tsang et al., 2010).
Most drugs targeting the serotonergic system are used as adjuvant therapy, combatting BPSD symptoms by inhibition of SERT and/or the norepinephrine transporter. Recent meta-analyses have proven their efficacy in treating these behavioral symptoms in AD (Ballard and Corbett, 2010; Henry et al., 2011). Some serotonin reuptake inhibitors have, however, also been evaluated for their possible cognitive enhancement in AD patients (See Table 2). Likewise, increasing interest has been noted for 5-HTR drugs that are able to improve cognition and/or memory in AD. 5-HT1-, 5-HT4-, and 5-HT6 receptors are hereby of particular interest, due to their important role in learning and memory processes (Geldenhuys and Van der Schyf, 2011), effects which are most likely due to their modulation on glutamatergic and cholinergic transmission, or, in the case of 5-HT-4, due to an enhanced release of ACh upon stimulation of this receptor (Rodríguez et al., 2012). An overview of trials that have looked into the clinical benefit of serotonergic drugs on cognitive impairment in AD patients is given in Table 2.
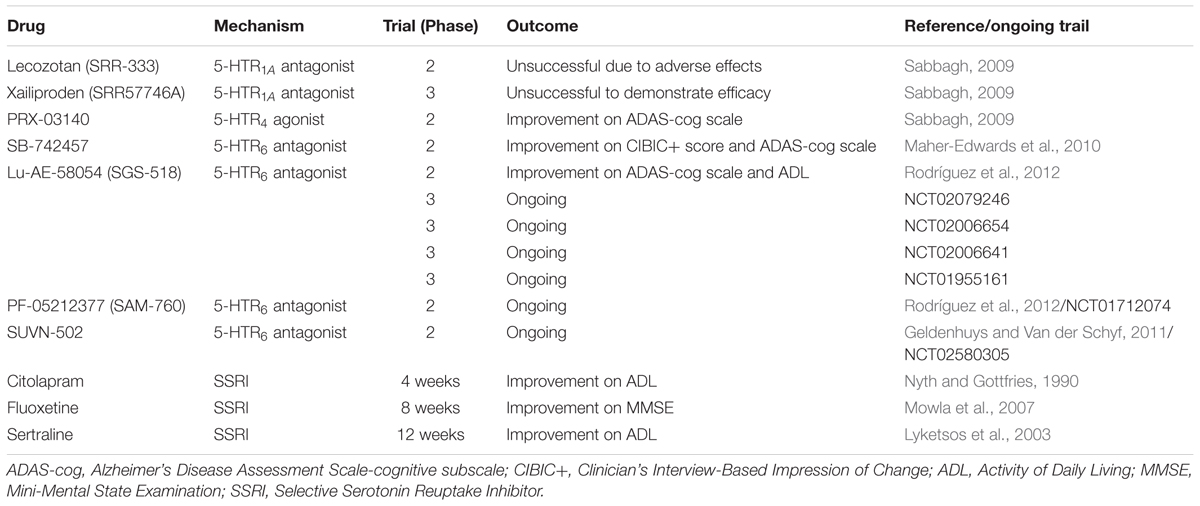
TABLE 2. Enhancement of cognitive functions in AD by drugs that modulate serotonergic neurotransmission (Geldenhuys and Van der Schyf, 2011; Ramirez et al., 2014).
Although much progress has been made in the development of PET- and SPECT radioligands for visualization of the serotonergic system (Paterson et al., 2013), only a few radiolabeled compounds have been evaluated on AD patients thus far. In the group of the 5-HT1AR, only one PET radioligand, 4-[18F]fluoro-N-{2-[4-(2-methoxyphenyl)-1-piperazinyl]ethyl}-N-(2-pyridinyl)benzamide ([18F]MPPF, Figure 8), a reversible, competitive 5-HT1AR antagonist, was investigated in patients with MCI and AD (Kepe et al., 2006; Truchot et al., 2008). Decrease of [18F]MPPF binding was noticed in the hippocampus and raphe nuclei of AD patients (as compared to HCs). Furthermore, loss of receptor density in the hippocampus was strongly correlated to a decline in the MMSE score. In patients with MCI, only a small loss of 5-HT1AR density was noticed, correlated to only small cognitive decline (Kepe et al., 2006). [18F]MPPF is one of many fluoro-analogs of [11C]WAY100635, the latter one being excessively studied in humans. Yet, no studies on AD patients were performed with [11C]WAY100635, mainly due its rapid metabolism, making kinetic modeling difficult (Paterson et al., 2013). [18F]MPPF does not suffer from these limitations, but is on the other hand a substrate of the P-gp, which could limit its further use in clinic (Kumar and Mann, 2014). Imaging of the 5-HT2R in AD patients was done by one SPECT- and three PET radiolabeled 5-HT2R antagonists, namely 4-amino-N-{1-[3-(4-fluorophenyl)propyl]-4-methylpiperidin-4-yl}-5-[123I]iodo-2-methoxybenzamide ([123I]-R91150), 6-(2-{4-[4-[18F]fluorobenzoyl]piperidin-1-yl}ethyl)-7-methyl-2H,3H,5H-[1,3]thiazolo[3,2-a]pyrimidin-5-one ([18F]setoperone), 3-(2-{4-[4-[18F]fluorobenzoyl]piperidin-1-yl}ethyl)-2-sulfanylidene-1,2,3,4-tetrahydroquinazolin-4-one ([18F]altanserin) and 3-(2-{4-[4-[18F]fluorobenzoyl]piperidin-1-yl}(2,2-2H2)ethyl)-2-sulfanylidene-1,2,3,4-tetrahydroquinazolin-4-one ([18F]deuteroaltanserin) (Figure 8). In agreement with previous postmortem studies, an overall significant reduction in the cerebral cortex was noted in mild to severe AD patients, compared to their age-matched controls (Blin et al., 1993; Versijpt et al., 2003; Santhosh et al., 2009; Marner et al., 2012). In the 5-HT4R class though, one PET ligand was evaluated on AD patients: [1-[11C]methylpiperidin-4-yl]methyl 8-amino-7-chloro-2,3-dihydro-1,4-benzodioxine-5-carboxylate ([11C]SB207145), a 5-HT4R antagonist (Figure 8). This radioligand did not display significant differences between mild AD cases and their HCs, although a positive correlation was found with the Aβ density (as measured by [11C]PiB). Moreover, a negative correlation was noticed between [11C]SB207145’s binding potential and the MMSE score. Authors suggested that upregulation of 5-HT4R may take place at a preclinical stage of AD (this in contrast to the other 5-HTRs) and that this may continue through the later AD stages (Madsen et al., 2011). Finally, (3-amino-4-(2-dimethylamino-methyl-phenylsulfanyl)-benzonitrile) ([11C]DASB, Figure 8), a SERT tracer, displayed a more outspoken decrease (25%) of binding in the subcortical serotonergic projection region in depressed, as compared to non-depressed AD patients (mean MMSE score of 18) (Ouchi et al., 2009). Yet, in another clinical study on patients with mild AD (not corrected for depression) no such reduction was found (Marner et al., 2012). Authors of the latter study suggest that this discrepancy may, however, lay in both differences in dementia severity as well as methodological differences between these studies (Marner et al., 2012). For a more detailed discussion about the current state of other 5-HT PET- and SPECT radioligands, readers are referred to some excellent reviews (Saulin et al., 2012; Paterson et al., 2013; Billard et al., 2014; Kumar and Mann, 2014). These compounds can be used for evaluation of inclusion- and outcome criteria, but also in dose-occupancy studies.
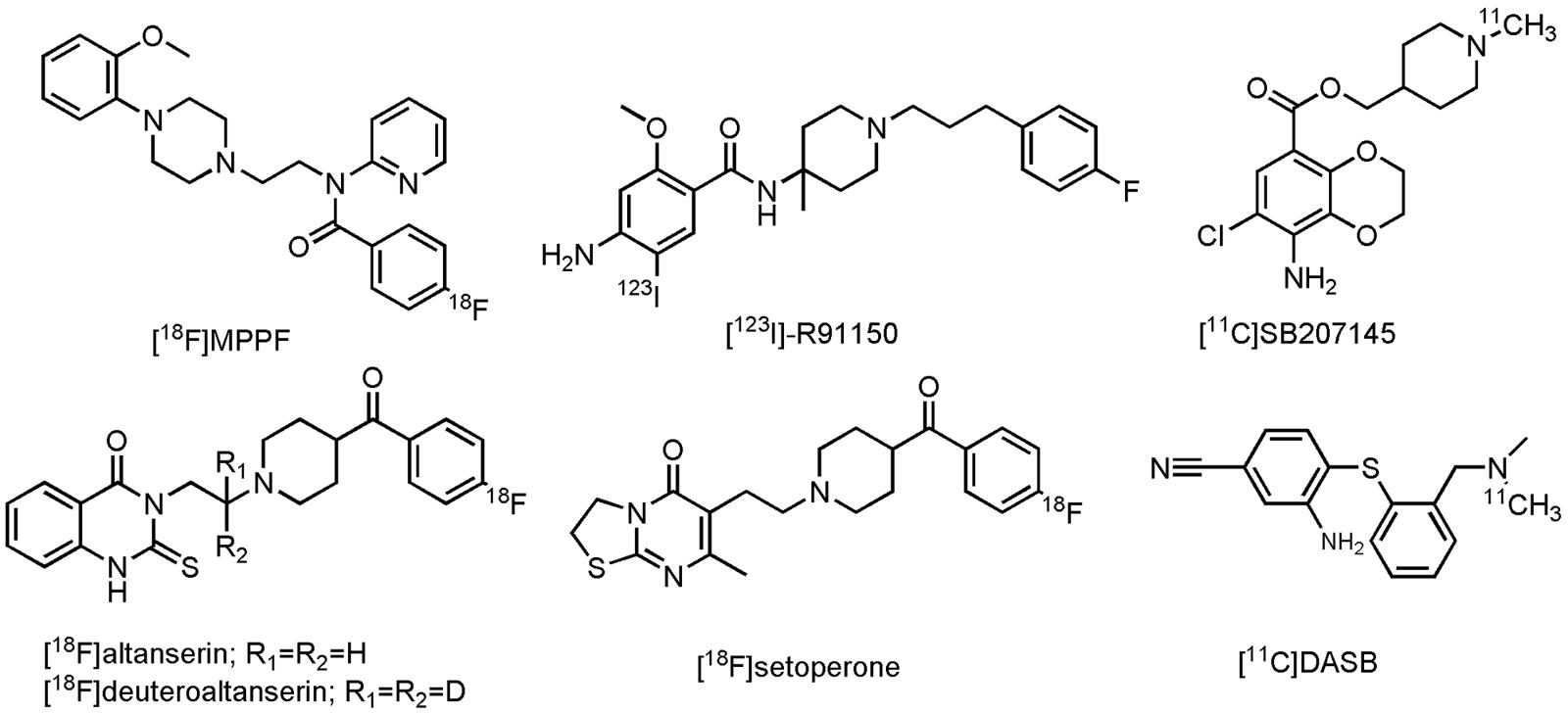
FIGURE 8. Structure of serotonergic PET/SPECT tracers.
The activity of DA, a catecholamine, is mediated through five dopaminergic, metabotropic, G-protein coupled receptors. They are divided into two classes: D1-like receptors (D1R and D5R) and D2-like receptors (D2-4R), depending on the downstream signaling cascade. Levels of DA are regulated through the activity of the presynaptic DAT, which removes DA from the synaptic cleft to terminate its activity (Mitchell et al., 2011). Dopaminergic neurons are largely located in the midbrain, with many projections throughout the brain (Martorana and Koch, 2014), where they are involved in various neurological processes. Of particular importance here is their role in motivation, cognition, and learning (Xu et al., 2012). Indeed, around 35–40% of AD patients exhibit extrapyramidal symptoms and more than 70% display extensive apathy (Lopez et al., 1997; Mitchell et al., 2011). These symptoms might be explained by the significantly reduced levels of DA and its precursor L-3,4-dihydroxyphenylalanine (L-DOPA) (Storga et al., 1996). Although large involvement of DA in AD is still under debate (Portet et al., 2009; Trillo et al., 2013), several noticeable changes have been documented in the DA receptor density. More specifically, a significant reduced expression of D1- and D2-like receptors has been documented in the prefrontal cortex and the hippocampus of AD patients (Kemppainen et al., 2003; Kumar and Patel, 2007). Furthermore, alterations of the D2R in AD seems positively correlated to BPSD and verbal memory performance (Kemppainen et al., 2003; Tanaka et al., 2003). Conflicting results are, however, reported for the D2R density in AD patients (see further) (Piggott et al., 1999; Piggott et al., 2007). Contradictory results are also reported for changes in the DAT levels in AD patients (Murray et al., 1995; Ceravolo et al., 2004). Despite some discrepancy, it is clear that there are important DA changes in the AD brains. Finally, it is to be noted that several in vivo experiments on mice, expressing AD like pathology, show that significant behavioral and cognitive deficits can be restored by administering DA reuptake inhibitors and L-DOPA (Ambree et al., 2009; Guzman-Ramos et al., 2012). Aβ oligomers may indeed have an early impact on catecholaminergic transmission (Mura et al., 2010).
There are several modes of interventions toward the failing dopaminergic system in AD patients, mostly used to address apathy (the most common BPSD symptom) and extrapyramidal symptoms. One of many approaches is the use of MAO-B inhibitors, which are discussed in Section “Monoamine Oxidase B” of this review. Another therapeutic method is modulation of the DAT transporter, and thus increasing synaptic DA levels. This was done by methylphenidate and dextroamphetamine in several clinical AD studies (Galynker et al., 1997; Herrmann et al., 2008; Lanctot et al., 2008). Although not selective for the dopaminergic system, an overall improvement was noted in symptoms of apathy on the Apathy Evaluation Scale (AES). There are, however, some concerns about the tolerability of methylphenidate (Padala et al., 2010). Other drugs that are frequently used to treat BPSD symptoms in clinical trials (and routine practice) involving AD patients are the antipsychotic drugs quetiapine, aripiprazole, olanzapine, and risperidone. As FDA- and EMA-approved drugs, these drugs act as partial DA receptor agonist or partial DA receptor antagonist (among often interaction with many other targets). Overall improvement on BPSD symptoms was recorded in a large meta-analysis of the use of antipsychotics in AD patients (Ballard and Waite, 2006). Nevertheless, caution was advised by the FDA with these drugs, as they were associated with an increase in risk of death, and other severe side effects, among elder people with dementia (Ballard and Waite, 2006; De Deyn et al., 2013). Yet another drug, rotigotine, a D2R- and D3R agonist, was able to show cognitive enhancement on probable AD patients, compared to their age-matched HCs by measuring the cortical excitability and central cholinergic transmission (Martorana et al., 2013).
Imaging of the dopaminergic system can be done by a number of PET- and SPECT radioligands. Conflicting results are, however, reported between several clinical studies on AD patients, using different PET- and/or SPECT tracers. In a combined PET study, reduced striatal expression of D1R, but not D2R was seen with D2R antagonist 3,5-dichloro-N-{[(2S)-1-ethylpyrrolidin-2-yl]methyl}-2-hydroxy-6-[11C]methoxybenzamide([11C]raclopride) and D1R antagonist (5R)-8-chloro-5-(2,3-dihydro-1-benzofuran-7-yl)-3-[11C]methyl-2,3,4,5-tetrahydro-1H-3-benzazepin-7-ol ([11C]NNC 756) in AD patients (Figure 9), compared to age-matched HCs (Kemppainen et al., 2000). Striatal uptake of 2-amino-3-[2-[18F]fluoro-4,5-dihydroxyphenyl]propanoic acid ([18F]FDOPA), a fluorinated form of L-DOPA (Figure 9), was also unchanged in AD patients, compared to HCs (Tyrrell et al., 1990). Conversely, decreased striatal expression of D2R with [11C]raclopride was demonstrated in AD patients (with BPSD symptoms) as compared to their HCs (Tanaka et al., 2003). Similar studies, using N-{[(2S)-1-ethylpyrrolidin-2-yl]methyl}-2-hydroxy-3-[123I]iodo-6-methoxybenzamide ([123I]IBZM) (Figure 9), a D2R antagonist, or methyl (2S,3S)-3-(4-fluorophenyl)-8-[11C]methyl-8-azabicyclo[3.2.1]octane-2-carboxylate ([11C]β-CFT) (Figure 9), a cocaine derivative which binds to DAT, showed, respectively, a reduced expression of D2R and a reduced DA reuptake (Pizzolato et al., 1996; Rinne et al., 1998). Reduction of DA reuptake sites, as measured by [11C]β-CFT, was hereby positively correlated to the severity of the extrapyramidal symptoms of AD patients, whereas in the study with [123I]IBZM, patients did not exhibit any extrapyramidal symptoms. Likewise, a decrease in [18F]FDOPA striatal uptake was noticed in another study on AD patients, a decrease which was correlated to the cognitive scores of the AD patients (Itoh et al., 1994). On the other hand, even more confusing, is the fact that in yet another clinical study involving AD patients an increase in D2R expression in the striatum was now measured with [11C]raclopride (Reeves et al., 2009). Discrepancies between these different studies might, however, be explained by different study populations, and the degree of dementia, since time-dependent dopaminergic receptor changes were also seen in patients with PD (Brooks, 1993). Another role for dopaminergic neuroimaging was displayed by methyl (2S,3S)-8-(3-fluoropropyl)-3-[4-[123I]iodophenyl]-8-azabicyclo[3.2.1]octane-2-carboxylate ([123I]FP-CIT) (Figure 9), an analog of [11C]β-CFT. [123I]FP-CIT was able to differentiate, with high accuracy, patients with AD, and patients with DLB (Colloby et al., 2008; Spehl et al., 2015). Overall reduced striatal uptake was noticed in both diseases, but lower binding potentials of [123I]FP-CIT were reported in DLB than in the case of the AD patients. These scans can greatly improve differential diagnosis between the different neurodegenerative diseases, which often display similar clinical presentations. [123I]FP-CIT SPECT scans are already used in clinical routine to distinguish DLB- from AD patients (Spehl et al., 2015). Finally, 5-[3-[18F]fluoropropyl]-2,3-dimethoxy-N-{[1-(prop-2-en-1-yl)pyrrolidin-2-yl]methyl}benzamide ([18F]fallypride) (Figure 9), a D2R/D3R antagonist, could be used to assess the ideal therapeutic window for the use of antipsychotic drugs (Clark-Papasavas et al., 2014), since, as mentioned before, elder people are very sensitive to these drugs. [18F]fallypride PET scans could consequently assist in antipsychotic dose-occupancy studies, and thus help to provide an ideal antipsychotic strategy in AD patients with extensive BPSD symptoms.
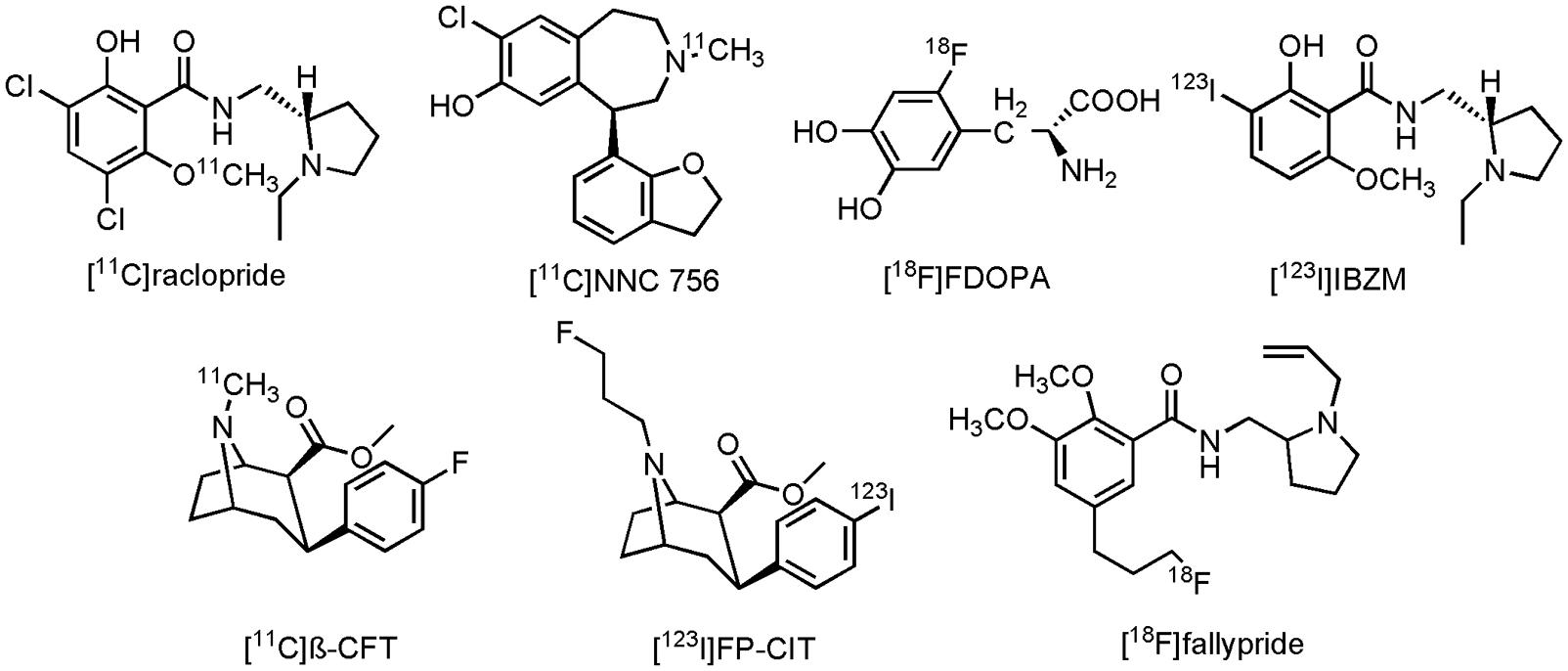
FIGURE 9. Structure of dopaminergic PET/SPECT tracers.
Formerly known as PBR, the 18 kDa TSPO, is located on the outer membrane of the mitochondria, predominantly in glial cells. As part of a multimeric complex, which is comprised of a VDAC and an adenine nucleotide carrier (McEnery et al., 1992; Casellas et al., 2002; Cosenza-Nashat et al., 2009), several functions are associated with TSPO (Midzak et al., 2015). They play an essential role in neurosteriod synthesis, by facilitating the transport of cholesterol from the outer to the inner membrane of the mitochondria (Papadopoulos et al., 2006a,b), and hence potentiate the GABAergic neurotransmission through allosteric modulation of the GABAA receptor by neurosteroids (Belelli and Lambert, 2005; Hosie et al., 2006; Rudolph and Mohler, 2006). Furthermore, TSPO may have a crucial function in a variety of cellular processes, such as cell proliferation (Miettinen et al., 1995; Hardwick et al., 1999), mitochondrial respiration (Hirsch et al., 1989) and cell apoptosis (Kugler et al., 2008). In light of TSPO’s association with the pathophysiology of neurodegenerative diseases, it has been well established that part of the neurotoxicity caused by tau and Aβ deposits in AD is induction of a neuroinflammatory response (McGeer and McGeer, 1995; Hoozemans et al., 2011), which triggers the upregulation of TSPO in activated microglia and astrocytes. Moreover, this upregulation clearly correlates with the degree of neuroinflammation, making TSPO a valuable target for drug monitoring (Venneti et al., 2006). Interestingly, in a review by Chua et al. (2014), it was suggested that TSPO ligands may provide effective tools for treatment of AD through activation of neuroprotective pathways of increased expression of astrocytes and microglial cells, since these mechanisms may have a protective phagocytic role in early AD (Morgan et al., 2005). Once a more progressed AD state has been reached, neuroinflammation turns chronic and becomes harmful (Hickman et al., 2008). This view is in contrast with numerous clinical trials, using anti-inflammatory drugs that failed to produce significant improvement in AD patients (Streit, 2010; Venigalla et al., 2015), although this failure may be attributed to a ‘wrong’ stage of the disease when therapy was initiated (Moreira et al., 2006). There are currently no drugs in clinical trials that interact with the TSPO receptor in AD. Drugs that are already described are mainly used for their use against BPSD symptoms (Rupprecht et al., 2009; Owen et al., 2011). TSPO is, however, an important marker for neuroinflammation, which makes it an interesting target for neuroimaging. PET tracers in this class will thus mainly be used to assess inclusion- and outcome criteria in clinical trials with anti-neuroinflammatory drugs in AD.
The most studied TSPO tracer in patients with CNS disorder is without a doubt N-[(2R)-butan-2-yl]-1-(2-chlorophenyl)-N-[11C]methylisoquinoline-3-carboxamide ([11C]PK11195) (Figure 10), despite its low specific binding and minimal brain uptake (Damont et al., 2013). Nonetheless, conflicting results are reported with [11C]PK11195, but also with several other clinical TPSO tracers, such as N-{[2-[11C]methoxyphenyl]methyl}-N-(4-phenoxypyridin-3-yl)acetamide ([11C]PBR28), N-(5-fluoro-2-phenoxyphenyl)-N-{[2-[11C]methoxy-5-methoxyphenyl]methyl}acetamide ([11C]DAA1106), (2-[11C])ethyl (15S,19S)-15-ethyl-1,11-diazapentacyclo[9.6.2.02,708,18.015,19]nonadeca-2,4,6,8(18),16-pentaene-17-carboxylate ([11C]vinpocetine), N-({2-[2-[18F]fluor-ethoxy]-5-methoxyphenyl}methyl)-N-[2-(4-methoxyphenoxy)pyridin-3-yl]acetamide ([18F]FEMPA), N-({2-[2-[18F]fluoroethoxy]-5-methoxyphenyl}methyl)-N-(2-phenoxyphenyl)acetamide ([18F]FEDAA1106) and N,N-diethyl-2-(2-{4-[2-[18F]fluoroethoxy]phenyl}-5,7-dimethylpyrazolo[1,5-a]pyrimidin-3-yl)acetamide ([18F]DPA-714) (Figure 10). While a majority of clinical trials was able to show significant tracer uptake in at least one brain area in AD patients, several other studies failed to differentiate MCI or even HCs from AD (Varley et al., 2015; Stefaniak and O’Brien, 2016). Apart from low signal-to-noise ratios and low brain uptake of some of these compounds, there are many possible explanations for the discrepancies in TSPO expression, as measured by PET in these clinical trials involving AD patients (Janssen et al., 2016). It is, however, important to realize, as suggested before (Janssen et al., 2016), that many patients exhibit different TSPO expression levels, depending on the specific polymorphism in the TSPO gene, resulting in intersubject variability in the binding affinities of TSPO PET tracers (Owen et al., 2012). Increasing efforts have therefore been focused toward compounds that are insensitive toward TSPO polymorphism, but also toward compounds for other neuroinflammatory targets (such as MAO-B, see Section Monoamine Oxidase B). Furthermore, overexpression of TSPO in both astrocytes and microglial cells make it difficult to differentiate MCI from AD patients (Ekonomou et al., 2015; Janssen et al., 2016). Nevertheless, [18F]DPA-714 is currently being used to assess the degree of neuroinflammation in AD patients in two clinical trials (CTI: NCT02377206 and NCT02062099). For a more detailed discussion of the current status of PET development for TSPO, or neuroinflammation in general, readers are referred to several other reviews (Ory et al., 2014; Varley et al., 2015; Janssen et al., 2016).
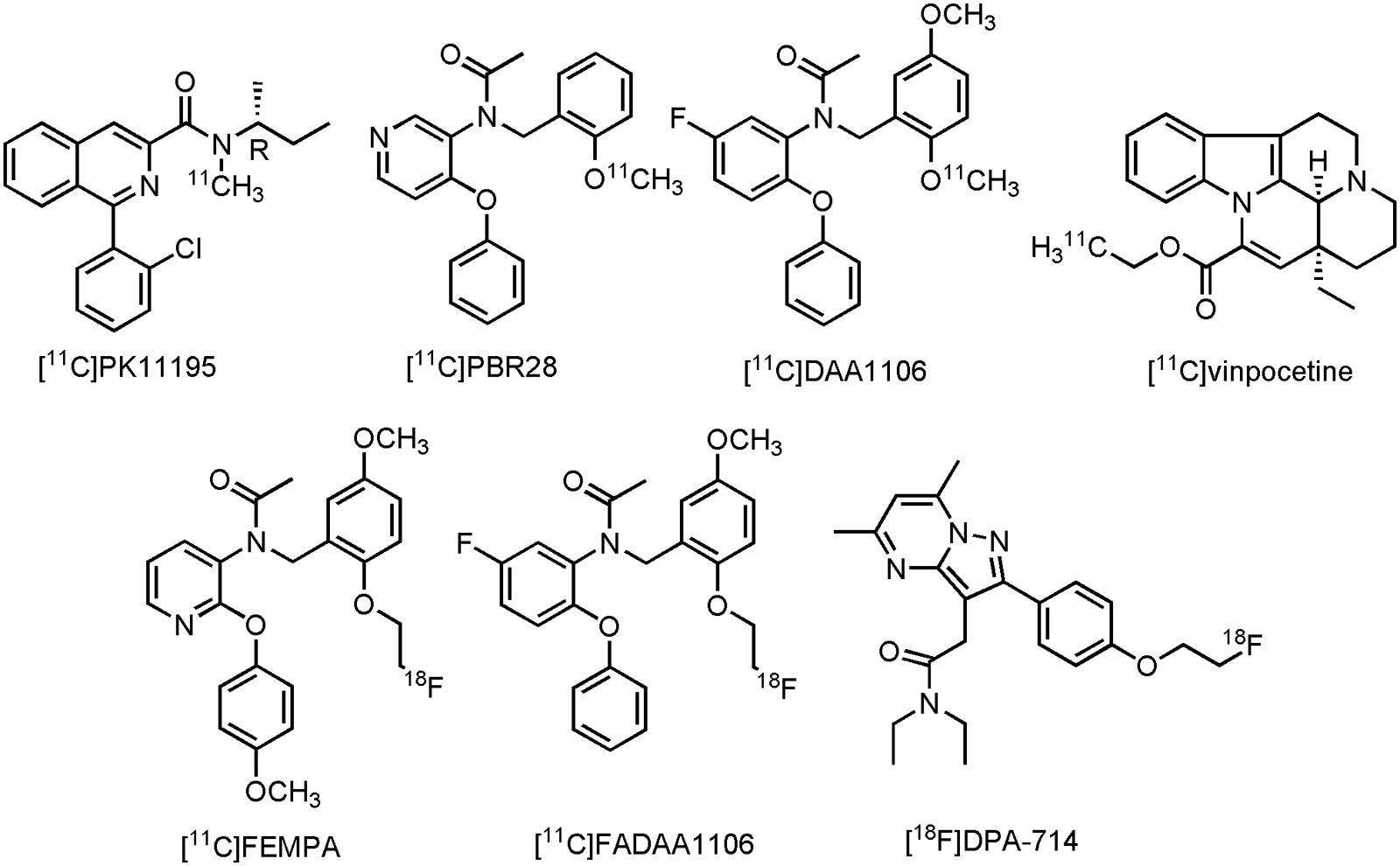
FIGURE 10. Structure of TSPO PET tracers.
Monoamine oxidases are mitochondrial bound enzymes, as a member of the flavin-containing amine oxidoreductases protein family, in the CNS primarily found in astrocytes. They are responsible for oxidative deamination of monamines of both endogenous and exogenous sources, regulating the physiological activity of neurotransmitters as serotonin, DA, and noradrenaline (Strolin and Dostert, 1989). There are two types of isoforms, MAO-A and MAO-B, which differ in inhibitor sensitivity and substrate selectivity, although there is an overlap to some degree (Bortolato et al., 2008). Increased MAO-B activity has been noted in AD in both brain and blood platelets (Adolfsson et al., 1980; Alexopoulos et al., 1987; Sparks et al., 1991), the severe upregulation in the brain mainly being a consequence of a plaque associated neuroinflammatory response by reactive astrocytes (Jossan et al., 1991; Saura et al., 1994). Furthermore, during their catalytic deamination, MAOs produce neurotoxic byproducts such as hydrogen peroxide, which are one of the main culprits in oxidative stress, contributing to the formation of amyloid plaques (Huang et al., 2012; Zheng et al., 2012). Since MAOs play a key role in the regulation of several important neurotransmitters, cognitive impairment, due to pathological upregulation of MAOs has also been demonstrated (Delumeau et al., 1994). MAO inhibitors may therefore have a significant neuroprotective role in AD. Since MAO-B is the main isoform present in brain (Riederer et al., 1978; Sonsalla and Golbe, 1988) and inhibition of MAO-B proved to be useful as therapeutic approach in PD, focus has mainly been targeted toward MAO-B inhibition in AD (Thomas, 2000). So far, five different drugs have inhibited MAOs in clinical trials involving AD patients (Cai, 2014). Two of them, selegiline (L-deprenyl) and rasagiline (Azilect), irreversible MAO-B selective inhibitors, are established drugs in the treatment of PD, delaying the need for DA replacement therapy (Birkmayer et al., 1977; Lees et al., 1977; Weinreb et al., 2010). While selegiline initially showed promise in clinical trials involving AD patients, demonstrating modest improvements on cognitive and behavioral functions (Filip and Kolibas, 1999), a comprehensive meta-analysis showed no justification for the use of selegiline in the treatment of AD, since there was a lack of overall significant benefit (Birks and Flicker, 2003). The beneficial effect of rasagiline is yet to be evaluated in AD patients. A Phase II proof of concept trial in patients with mild to moderate AD is, however, underway (CTI: NCT02359552). Interestingly, rasagiline formed the basis of two other multi-target drugs, ladogistil (TV3326), and M-30. The former drug is a MAO-B inhibitor and AChE inhibitor, the latter a MAO-A and MAO-B inhibitor (Youdim, 2013). Both compounds are thought to modulate APP expression levels (by stimulating the non-amyloidogenic pathway) and both may have neuroprotective and neurorestorative functions (Riederer et al., 2004; Youdim, 2013). Phase II trials with ladogistil on mild to moderate AD patients have been completed, but results are yet to be published (CTI: NCT01354691). These drugs may hold promise as multi-target approach for treatment of AD, being able to tackle various pathophysiological changes at once (Youdim, 2013). Finally, EVT 301 (RO4477478), a reversible MAO-B inhibitor was evaluated on four AD patients (MMSE score: not specified) in a dose-finding study, using [11C]deprenyl-D2 ([11C]DED) PET (See further, Figure 11) to assess MAO-B occupancy levels. Seven days of treatment resulted hereby in an almost complete dose-occupancy of MAO-B (Hirvonen et al., 2009). No further clinical trials, to our knowledge, have since been performed with this drug.

FIGURE 11. Structure of MAO-B PET tracer.
Only one MAO binding PET tracer has been elevated on AD patients thus far: [11C]methyl[(2R)-1-phenylpropan-2-yl][(1,1-2H2)prop-2-yn-1-yl]amine ([11C]DED), an irreversible MAO-B inhibitor (Hirvonen et al., 2009; Carter et al., 2012; Choo et al., 2014). Increased uptake of [11C]DED was observed in patients with MCI (who responded positively to a [11C]PIB scan), suggesting that astrocytocis may be an early event in the development of AD. Clinical AD studies with other emerging (fluorine-18 labeled) MAO radiotracers are yet to be published (Ory et al., 2014; Fowler et al., 2015).
This review demonstrates that there are multiple approaches to be considered when developing disease altering drugs for AD. Important to notice is that each of these pathways is linked to the pathophysiological processes of (often many) other targets, hence a multi-target approach, addressing various pathophysiological changes at once, will be the way forward. Concordant neuroimaging techniques, such as PET and SPECT, could hereby greatly improve therapeutic monitoring, but also significantly aid with the proposal of current inclusion- and outcome criteria in large clinical studies. Moreover, once a disease altering drug has been found, PET/SPECT could eventually be used as standard test to assess and follow up disease progression (Barthel et al., 2015). Still, there are a few important factors to take in consideration. One of them is the need for quantitative PET assessment, especially during evaluation of novel therapies, to allow quantitative and accurate evaluations. Visual inspection or simplified models (such as SUV) are indeed less robust, as they are influenced by several physiological and technical factors (Boellaard, 2009; Tomasi et al., 2012). The other side of the coin is, however, that quantitative PET assessment is very time-consuming, which limits capacity and throughput which are essential in large multi-center trials. Another crucial issue is the loss of BBB function during AD progression, which could greatly affect drug dosage and bioavailability of novel AD therapeutics. One way of monitoring the viability of the BBB is by looking at the P-gp function. Several promising PET candidates are now under development for this purpose (Syvanen and Eriksson, 2013), and a pilot study to assess the P-gp function in AD patients is currently ongoing (ECT: 2013-001724-19). Finally, there is a great need for a thorough preclinical evaluation of the mechanism of action. This could not be better demonstrated than by the rise and fall of dimebon. Initially developed as an antihistaminic drug in the former USSR, dimebon demonstrated significant improvement on the ADAS-cog-, MMSE-, and CIBIC+ scales in a 6-month Phase II trial on AD patients in Russia (Bezprozvanny, 2010). Consequently, a multi-national Phase III trial was launched, but this trial failed to show any significant improvement on mild to moderate AD patients, as compared to placebo (Bezprozvanny, 2010). In a synopsis by Bezprozvanny (Bezprozvanny, 2010), failure was largely attributed to poor understanding of the proper mechanism of action in preclinical studies and the lack of objective biomarkers to assess the true therapeutic response in clinical trials. In the last decade, most clinical trials aiming to find AD combatting drugs ultimately failed to produce (convincing) positive results. Although there may be many explanations for this overall failure, thorough preclinical assessment remains an important factor.
LD wrote the manuscript. RV, KVL, AV, and GB reviewed it, corrected it and made suggestions.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
5-HT, 5-hydroxytryptamine; 5-HTRs, 5-HT receptors; 5-HTT, 5-HT transporter; α-APP, α-amyloid precursor protein; Aβ, amyloid beta; ACh, acetylcholine; AChE, acetylcholinesterase; AD, Alzheimer’s disease; ADAS-cog, Alzheimer’s disease assessment scale-cognitive subscale; ALA, α-lipoic acid; APP, amyloid precursor protein; BACE1, β-site APP-cleaving enzyme 1; BBB, blood–brain barrier; BPSD, behavioral and psychological symptoms of dementia; BZDs, benzodiazepines; CBD, corticobasal degeneration; ChAT, choline acetyltransferase; CIBIC+, clinician’s interview-based impression of change; CNS, central nervous system; CSF, cerebrospinal fluid; CT, computerized tomography; CTI, clinical trial identifier; D1R, dopamine1-like receptors; DA, dopamine; DAT, dopamine transporter; DLB, dementia with Lewy bodies; ECT, EudraCT-number; EMA, European medicines agency; FDA, food and drug administration; FTD, frontal temporal dementia; GABA, gamma-amino butyric acid; GSK, glycogen synthase kinase; HCs, healthy controls; mAChR, muscarinic acetylcholine receptor; MAPT, microtubule-associated protein tau gene; MCI, mild cognitive impairment; MMSE, mini-mental state examination; MOAs, monoamine oxidases; MRI, magnetic resonance imaging; nAChRs, nicotinic acetylcholine receptors; NFTs, neurofibrillary tangles; PBR, peripheral benzodiazepine receptor; PET, positron emission tomography; P-gp, permeability glycoprotein; PHFs, paired helical filaments; PSP, progressive supranuclear palsy; p-tau, phosphorylated tau; rCBF, regional cerebral blood flow; SERT, 5-HT reuptake transporter; SP, senile plaques; SPE(C)T, single photon emission (computed) tomography; TSPO, translocator protein; VDAC, voltage dependent anion channel.
Adolfsson, R., Gottfries, C. G., Oreland, L., Wiberg, A., and Winblad, B. (1980). Increased activity of brain and platelet monoamine oxidase in dementia of Alzheimer type. Life Sci. 27, 1029–1034. doi: 10.1016/0024-3205(80)90025-9
Ahmed, R. M., Paterson, R. W., Warren, J. D., Zetterberg, H., O’Brien, J. T., Fox, N. C., et al. (2014). Biomarkers in dementia: clinical utility and new directions. J. Neurol. Neurosurg. Psychiatry 85, 1426–1434. doi: 10.1136/jnnp-2014-307662
Alexopoulos, G. S., Young, R. C., Lieberman, K. W., and Shamoian, C. A. (1987). Platelet MAO activity in geriatric patients with depression and dementia. Am. J. Psychiatry 144, 1480–1483. doi: 10.1176/ajp.144.11.1480
Ambree, O., Richter, H., Sachser, N., Lewejohann, L., Dere, E., de Souza Silva, M. A., et al. (2009). Levodopa ameliorates learning and memory deficits in a murine model of Alzheimer’s disease. Neurobiol. Aging 30, 1192–1204. doi: 10.1016/j.neurobiolaging.2007.11.010
Anandatheerthavarada, H. K., Biswas, G., Robin, M. A., and Avadhani, N. G. (2003). Mitochondrial targeting and a novel transmembrane arrest of Alzheimer’s amyloid precursor protein impairs mitochondrial function in neuronal cells. J. Cell Biol. 161, 41–54. doi: 10.1083/jcb.200207030
Andersson, J. D., and Halldin, C. (2013). PET radioligands targeting the brain GABAA /benzodiazepine receptor complex. J. Labelled Comp. Radiopharm. 56, 196–206. doi: 10.1002/jlcr.3008
Aprahamian, I., Stella, F., and Forlenza, O. V. (2013). New treatment strategies for Alzheimer’s disease: is there a hope? Indian J. Med. Res. 138, 449–460.
Apter, J. T., Shastri, K., and Pizano, K. (2015). Update on disease-modifying/preventive therapies in Alzheimer’s disease. Curr. Geriatr. Rep. 4, 312–317. doi: 10.1007/s13670-015-0141-x
Arai, H., Ichimiya, Y., Kosaka, K., Moroji, T., and Iizuka, R. (1992). Neurotransmitter changes in early- and late-onset Alzheimer-type dementia. Prog. Neuropsychopharmacol. Biol. Psychiatry 16, 883–890. doi: 10.1016/0278-5846(92)90106-O
Asuni, A. A., Boutajangout, A., Quartermain, D., and Sigurdsson, E. M. (2007). Immunotherapy targeting pathological tau conformers in a tangle mouse model reduces brain pathology with associated functional improvements. J. Neurosci. 27, 9115–9129. doi: 10.1523/JNEUROSCI.2361-07.2007
Ballard, C., and Corbett, A. (2010). Management of neuropsychiatric symptoms in people with dementia. CNS Drugs 24, 729–739. doi: 10.2165/11319240-000000000-00000
Ballard, C., Gauthier, S., Corbett, A., Brayne, C., Aarsland, D., and Jones, E. (2011). Alzheimer’s disease. Lancet 377, 1019–1031. doi: 10.1016/S0140-6736(10)61349-9
Ballard, C., and Waite, J. (2006). The effectiveness of atypical antipsychotics for the treatment of aggression and psychosis in Alzheimer’s disease. Cochrane Database Syst. Rev. 25:CD003476. doi: 10.1002/14651858.CD003476.pub2
Ballatore, C., Lee, V. M., and Trojanowski, J. Q. (2007). Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat. Rev. Neurosci. 8, 663–672. doi: 10.1038/nrn2194
Bancher, C., Braak, H., Fischer, P., and Jellinger, K. A. (1993). Neuropathological staging of Alzheimer lesions and intellectual status in Alzheimer’s and Parkinson’s disease patients. Neurosci. Lett. 162, 179–182. doi: 10.1016/0304-3940(93)90590-H
Bancher, C., Jellinger, K., Lassmann, H., Fischer, P., and Leblhuber, F. (1996). Correlations between mental state and quantitative neuropathology in the Vienna Longitudinal Study on Dementia. Eur. Arch. Psychiatry Clin. Neurosci. 246, 137–146. doi: 10.1007/BF02189115
Banks, W. A., Terrell, B., Farr, S. A., Robinson, S. M., Nonaka, N., and Morley, J. E. (2002). Passage of amyloid beta protein antibody across the blood-brain barrier in a mouse model of Alzheimer’s disease. Peptides 23, 2223–2226. doi: 10.1016/S0196-9781(02)00261-9
Barage, S. H., and Sonawane, K. D. (2015). Amyloid cascade hypothesis: pathogenesis and therapeutic strategies in Alzheimer’s disease. Neuropeptides 52, 1–18. doi: 10.1016/j.npep.2015.06.008
Barnes, N. M., and Sharp, T. (1999). A review of central 5-HT receptors and their function. Neuropharmacology 38, 1083–1152. doi: 10.1016/S0028-3908(99)00010-6
Barnham, K. J., Haeffner, F., Ciccotosto, G. D., Curtain, C. C., Tew, D., Mavros, C., et al. (2004). Tyrosine gated electron transfer is key to the toxic mechanism of Alzheimer’s disease beta-amyloid. FASEB J. 18, 1427–1429. doi: 10.1096/fj.04-1890fje
Barthel, H., Gertz, H. J., Dresel, S., Peters, O., Bartenstein, P., Buerger, K., et al. (2011). Cerebral amyloid-beta PET with florbetaben (18F) in patients with Alzheimer’s disease and healthy controls: a multicentre phase 2 diagnostic study. Lancet Neurol. 10, 424–435. doi: 10.1016/S1474-4422(11)700771
Barthel, H., Seibyl, J., and Sabri, O. (2015). The role of positron emission tomography imaging in understanding Alzheimer’s disease. Expert Rev. Neurother. 15, 395–406. doi: 10.1586/14737175.2015.1023296
Bartus, R. T. (2000). On neurodegenerative diseases, models, and treatment strategies: lessons learned and lessons forgotten a generation following the cholinergic hypothesis. Exp. Neurol. 163, 495–529. doi: 10.1006/exnr.2000.7397
Bartus, R. T., Dean, R. L. III, Beer, B., and Lippa, A. S. (1982). The cholinergic hypothesis of geriatric memory dysfunction. Science 217, 408–414. doi: 10.1126/science.7046051
Belelli, D., and Lambert, J. J. (2005). Neurosteroids: endogenous regulators of the GABA(A) receptor. Nat. Rev. Neurosci. 6, 565–575. doi: 10.1038/nrn1703
Benadiba, M., Luurtsema, G., Wichert-Ana, L., Buchpigel, C. A., and Busatto, F. G. (2012). New molecular targets for PET and SPECT imaging in neurodegenerative diseases. Rev. Bras. Psiquiatr. 34(Suppl. 2), S125–S136. doi: 10.1016/j.rbp.2012.07.002
Berg, S., Bergh, M., Hellberg, S., Hogdin, K., Lo-Alfredsson, Y., Soderman, P., et al. (2012). Discovery of novel potent and highly selective glycogen synthase kinase-3beta (GSK3beta) inhibitors for Alzheimer’s disease: design, synthesis, and characterization of pyrazines. J. Med. Chem. 55, 9107–9119. doi: 10.1021/jm201724m
Bezprozvanny, I. (2010). The rise and fall of Dimebon. Drug News Perspect 23, 518–523. doi: 10.1358/dnp.2010.23.8.1500435
Billard, T., Le, B. D., and Zimmer, L. (2014). PET radiotracers for molecular imaging of serotonin 5-HT1A receptors. Curr. Med. Chem. 21, 70–81. doi: 10.2174/09298673113209990215
Billings, L. M., Oddo, S., Green, K. N., McGaugh, J. L., and Laferla, F. M. (2005). Intraneuronal Abeta causes the onset of early Alzheimer’s disease-related cognitive deficits in transgenic mice. Neuron 45, 675–688. doi: 10.1016/j.neuron.2005.01.040
Binder, L. I., Frankfurter, A., and Rebhun, L. I. (1986). Differential localization of MAP-2 and tau in mammalian neurons in situ. Ann. N. Y. Acad. Sci. 466, 145–166. doi: 10.1111/j.1749-6632.1986.tb38392.x
Birkmayer, W., Riederer, P., Ambrozi, L., and Youdim, M. B. (1977). Implications of combined treatment with ’Madopar ’ and L-deprenil in Parkinson’s disease, A long-term study. Lancet 1, 439–443. doi: 10.1016/S0140-6736(77)91940-7
Birks, J., and Flicker, L. (2003). Selegiline for Alzheimer’s disease. Cochrane Database Syst. Rev. CD000442. doi: 10.1002/14651858.CD000442
Blin, J., Baron, J. C., Dubois, B., Crouzel, C., Fiorelli, M., Attar-Levy, D., et al. (1993). Loss of brain 5-HT2 receptors in Alzheimer’s disease. In vivo assessment with positron emission tomography and [18F]setoperone. Brain 116(Pt 3), 497–510. doi: 10.1093/brain/116.3.497
Bodick, N. C., Offen, W. W., Levey, A. I., Cutler, N. R., Gauthier, S. G., Satlin, A., et al. (1997). Effects of xanomeline, a selective muscarinic receptor agonist, on cognitive function and behavioral symptoms in Alzheimer disease. Arch. Neurol. 54, 465–473. doi: 10.1001/archneur.1997.00550160091022
Boellaard, R. (2009). Standards for PET image acquisition and quantitative data analysis. J. Nucl. Med. 50(Suppl. 1), 11S–20S. doi: 10.2967/jnumed.108.057182
Bonner, T. I. (1989). The molecular basis of muscarinic receptor diversity. Trends Neurosci. 12, 148–151. doi: 10.1016/0166-2236(89)90054-4
Bonner, T. I., Buckley, N. J., Young, A. C., and Brann, M. R. (1987). Identification of a family of muscarinic acetylcholine receptor genes. Science 237, 527–532. doi: 10.1126/science.3037705
Bonner, T. I., Young, A. C., Brann, M. R., and Buckley, N. J. (1988). Cloning and expression of the human and rat m5 muscarinic acetylcholine receptor genes. Neuron 1, 403–410. doi: 10.1016/0896-6273(88)90190-0
Bortolato, M., Chen, K., and Shih, J. C. (2008). Monoamine oxidase inactivation: from pathophysiology to therapeutics. Adv. Drug Deliv. Rev. 60, 1527–1533. doi: 10.1016/j.addr.2008.06.002
Bowen, D. M., Allen, S. J., Benton, J. S., Goodhardt, M. J., Haan, E. A., Palmer, A. M., et al. (1983). Biochemical assessment of serotonergic and cholinergic dysfunction and cerebral atrophy in Alzheimer’s disease. J. Neurochem. 41, 266–272. doi: 10.1111/j.1471-4159.1983.tb11838.x
Braak, H., and Braak, E. (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259. doi: 10.1007/BF00308809
Bremner, J. D., Baldwin, R., Horti, A., Staib, L. H., Ng, C. K., Tan, P. Z., et al. (1999). Quantitation of benzodiazepine receptor binding with PET [11C]iomazenil and SPECT [123I]iomazenil: preliminary results of a direct comparison in healthy human subjects. Psychiatry Res. 91, 79–91. doi: 10.1016/S0925-4927(99)00015-3
Bresjanac, M., Smid, L. M., Vovko, T. D., Petric, A., Barrio, J. R., and Popovic, M. (2003). Molecular-imaging probe 2-(1-[6-[(2-fluoroethyl)(methyl) amino]-2-naphthyl]ethylidene) malononitrile labels prion plaques in vitro. J. Neurosci. 23, 8029–8033.
Broich, K., Grunwald, F., Kasper, S., Klemm, E., Biersack, H. J., and Moller, H. J. (1998). D2-dopamine receptor occupancy measured by IBZM-SPECT in relation to extrapyramidal side effects. Pharmacopsychiatry 31, 159–162. doi: 10.1055/s-2007-979321
Brookmeyer, R., Johnson, E., Ziegler-Graham, K., and Arrighi, H. M. (2007). Forecasting the global burden of Alzheimer’s disease. Alzheimers Dement. 3, 186–191. doi: 10.1016/j.jalz.2007.04.381
Brooks, D. J. (1993). Functional imaging in relation to parkinsonian syndromes. J. Neurol. Sci. 115, 1–17. doi: 10.1016/0022-510X(93)90061-3
Brown, D., Chisholm, J. A., Owens, J., Pimlott, S., Patterson, J., and Wyper, D. (2003). Acetylcholine muscarinic receptors and response to anti-cholinesterase therapy in patients with Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 30, 296–300. doi: 10.1007/s00259-002-1028-6
Brun, A., and Englund, E. (1981). Regional pattern of degeneration in Alzheimer’s disease: neuronal loss and histopathological grading. Histopathology 5, 549–564. doi: 10.1111/j.1365-2559.1981.tb01818.x
Bruno, G., Mohr, E., Gillespie, M., Fedio, P., and Chase, T. N. (1986). Muscarinic agonist therapy of Alzheimer’s disease. A clinical trial of RS-86. Arch. Neurol. 43, 659–661. doi: 10.1001/archneur.1986.00520070017009
Buckingham, S. D., Jones, A. K., Brown, L. A., and Sattelle, D. B. (2009). Nicotinic acetylcholine receptor signalling: roles in Alzheimer’s disease and amyloid neuroprotection. Pharmacol. Rev. 61, 39–61. doi: 10.1124/pr.108.000562
Bulic, B., Pickhardt, M., Mandelkow, E. M., and Mandelkow, E. (2010). Tau protein and tau aggregation inhibitors. Neuropharmacology 59, 276–289. doi: 10.1016/j.neuropharm.2010.01.016
Bulic, B., Pickhardt, M., and Schmidt, B. (2009). Development of tau aggregation inhibitors for Alzheimer’s disease. Angew. Chem. Int. Ed. Engl. 48, 1740–1752. doi: 10.1002/anie.200802621
Burns, A., Rossor, M., Hecker, J., Gauthier, S., Petit, H., Moller, H. J., et al. (1999). The effects of donepezil in Alzheimer’s disease - results from a multinational trial. Dement. Geriatr. Cogn. Disord. 10, 237–244. doi: 10.1159/000017126
Butner, K. A., and Kirschner, M. W. (1991). Tau protein binds to microtubules through a flexible array of distributed weak sites. J. Cell Biol. 115, 717–730. doi: 10.1083/jcb.115.3.717
Buxbaum, J. D., Oishi, M., Chen, H. I., Pinkas-Kramarski, R., Jaffe, E. A., Gandy, S. E., et al. (1992). Cholinergic agonists and interleukin 1 regulate processing and secretion of the Alzheimer beta/A4 amyloid protein precursor. Proc. Natl. Acad. Sci. U.S.A. 89, 10075–10078. doi: 10.1073/pnas.89.21.10075
Cai, Z. (2014). Monoamine oxidase inhibitors: promising therapeutic agents for Alzheimer’s disease (Review). Mol. Med. Rep. 9, 1533–1541. doi: 10.3892/mmr.2014.2040
Carter, S. F., Scholl, M., Almkvist, O., Wall, A., Engler, H., Langstrom, B., et al. (2012). Evidence for astrocytosis in prodromal Alzheimer disease provided by 11C-deuterium-L-deprenyl: a multitracer PET paradigm combining 11C-Pittsburgh compound B and 18F-FDG. J. Nucl. Med. 53, 37–46. doi: 10.2967/jnumed.110.087031
Casellas, P., Galiegue, S., and Basile, A. S. (2002). Peripheral benzodiazepine receptors and mitochondrial function. Neurochem. Int. 40, 475–486. doi: 10.1016/S0197-0186(01)00118-8
Caspersen, C., Wang, N., Yao, J., Sosunov, A., Chen, X., Lustbader, J. W., et al. (2005). Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J. 19, 2040–2041. doi: 10.1096/fj.05-3735fje
Castelan, F., Castillo, M., Mulet, J., Sala, S., Sala, F., Dominguez Del, T. E., et al. (2008). Molecular characterization and localization of the RIC-3 protein, an effector of nicotinic acetylcholine receptor expression. J. Neurochem. 105, 617–627. doi: 10.1111/j.1471-4159.2007.05169.x
Ceravolo, R., Volterrani, D., Gambaccini, G., Bernardini, S., Rossi, C., Logi, C., et al. (2004). Presynaptic nigro-striatal function in a group of Alzheimer’s disease patients with parkinsonism: evidence from a dopamine transporter imaging study. J. Neural Transm. (Vienna) 111, 1065–1073.
Chalon, S., Vercouillie, J., Guilloteau, D., Suzenet, F., and Routier, S. (2015). PET tracers for imaging brain alpha7 nicotinic receptors: an update. Chem. Commun. (Camb.) 51, 14826–14831. doi: 10.1039/c5cc04536c
Chebib, M., and Johnston, G. A. (1999). The ‘ABC’ of GABA receptors: a brief review. Clin. Exp. Pharmacol. Physiol. 26, 937–940. doi: 10.1046/j.1440-1681.1999.03151.x
Chen, C. P., Alder, J. T., Bowen, D. M., Esiri, M. M., McDonald, B., Hope, T., et al. (1996). Presynaptic serotonergic markers in community-acquired cases of Alzheimer’s disease: correlations with depression and neuroleptic medication. J. Neurochem. 66, 1592–1598. doi: 10.1046/j.1471-4159.1996.66041592.x
Chen, C. P., Eastwood, S. L., Hope, T., McDonald, B., Francis, P. T., and Esiri, M. M. (2000). Immunocytochemical study of the dorsal and median raphe nuclei in patients with Alzheimer’s disease prospectively assessed for behavioural changes. Neuropathol. Appl. Neurobiol. 26, 347–355. doi: 10.1046/j.1365-2990.2000.00254.x
Chien, D. T., Bahri, S., Szardenings, A. K., Walsh, J. C., Mu, F., Su, M. Y., et al. (2012). Early clinical PET imaging results with the novel PHF-tau tadioligand [F-18]-T807. J. Alzheimers Dis. 34, 457–468. doi: 10.3233/JAD-122059
Chien, D. T., Szardenings, A. K., Bahri, S., Walsh, J. C., Mu, F., Xia, C., et al. (2013). Early clinical PET imaging results with the novel PHF-tau radioligand [F18]-T808. J. Alzheimers Dis. 38, 171–184. doi: 10.3233/JAD-130098
Choo, I. L., Carter, S. F., Scholl, M. L., and Nordberg, A. (2014). Astrocytosis measured by (1)(1)C-deprenyl PET correlates with decrease in gray matter density in the parahippocampus of prodromal Alzheimer’s patients. Eur. J. Nucl. Med. Mol. Imaging 41, 2120–2126. doi: 10.1007/s00259-014-2859-7
Christensen, R., Marcussen, A. B., Wortwein, G., Knudsen, G. M., and Aznar, S. (2008). Abeta(1-42) injection causes memory impairment, lowered cortical and serum BDNF levels, and decreased hippocampal 5-HT(2A) levels. Exp. Neurol. 210, 164–171. doi: 10.1016/j.expneurol.2007.10.009
Chua, S. W., Kassiou, M., and Ittner, L. M. (2014). The translocator protein as a drug target in Alzheimer’s disease. Expert Rev. Neurother. 14, 439–448. doi: 10.1586/14737175.2014.896201
Clark-Papasavas, C., Dunn, J. T., Greaves, S., Mogg, A., Gomes, R., Brownings, S., et al. (2014). Towards a therapeutic window of D2/3 occupancy for treatment of psychosis in Alzheimer’s disease, with [18F]fallypride positron emission tomography. Int. J. Geriatr. Psychiatry 29, 1001–1009. doi: 10.1002/gps.4090
Colloby, S. J., Firbank, M. J., Pakrasi, S., Lloyd, J. J., Driver, I., McKeith, I. G., et al. (2008). A comparison of 99mTc-exametazime and 123I-FP-CIT SPECT imaging in the differential diagnosis of Alzheimer’s disease and dementia with Lewy bodies. Int. Psychogeriatr. 20, 1124–1140. doi: 10.1017/S1041610208007709
Connolly, C. N., Krishek, B. J., McDonald, B. J., Smart, T. G., and Moss, S. J. (1996). Assembly and cell surface expression of heteromeric and homomeric gamma-aminobutyric acid type A receptors. J. Biol. Chem. 271, 89–96. doi: 10.1074/jbc.271.1.89
Consolo, S., Bertorelli, R., Russi, G., Zambelli, M., and Ladinsky, H. (1994). Serotonergic facilitation of acetylcholine release in vivo from rat dorsal hippocampus via serotonin 5-HT3 receptors. J. Neurochem. 62, 2254–2261. doi: 10.1046/j.1471-4159.1994.62062254.x
Contestabile, A. (2011). The history of the cholinergic hypothesis. Behav. Brain Res. 221, 334–340. doi: 10.1016/j.bbr.2009.12.044
Cooper, S. T., and Millar, N. S. (1997). Host cell-specific folding and assembly of the neuronal nicotinic acetylcholine receptor alpha7 subunit. J. Neurochem. 68, 2140–2151. doi: 10.1046/j.1471-4159.1997.68052140.x
Cosenza-Nashat, M., Zhao, M. L., Suh, H. S., Morgan, J., Natividad, R., Morgello, S., et al. (2009). Expression of the translocator protein of 18 kDa by microglia, macrophages and astrocytes based on immunohistochemical localization in abnormal human brain. Neuropathol. Appl. Neurobiol. 35, 306–328. doi: 10.1111/j.1365-2990.2008.01006.x
Covington, J. S. (1975). Alleviating agitation, apprehension, and related symptoms in geriatric patients: a double-blind comparison of a phenothiazine and a benzodiazepien. South. Med. J. 68, 719–724. doi: 10.1097/00007611-197506000-00015
Coyle, J. T., Price, D. L., and DeLong, M. R. (1983). Alzheimer’s disease: a disorder of cortical cholinergic innervation. Science 219, 1184–1190. doi: 10.1126/science.6338589
Criado, M., Mulet, J., Gerber, S., Sala, S., and Sala, F. (2011). A small cytoplasmic region adjacent to the fourth transmembrane segment of the alpha7 nicotinic receptor is essential for its biogenesis. FEBS Lett. 585, 2477–2480. doi: 10.1016/j.febslet.2011.06.028
Crow, T. J., Cross, A. J., Cooper, S. J., Deakin, J. F., Ferrier, I. N., Johnson, J. A., et al. (1984). Neurotransmitter receptors and monoamine metabolites in the brains of patients with Alzheimer-type dementia and depression, and suicides. Neuropharmacology 23, 1561–1569. doi: 10.1016/0028-3908(84)90100-X
Cummings, J. L. (2008). Optimizing phase II of drug development for disease-modifying compounds. Alzheimers Dement. 4, S15–S20. doi: 10.1016/j.jalz.2007.10.002
Dahlgren, K. N., Manelli, A. M., Stine, W. B. Jr., Baker, L. K., Krafft, G. A., and LaDu, M. J. (2002). Oligomeric and fibrillar species of amyloid-beta peptides differentially affect neuronal viability. J. Biol. Chem. 277, 32046–32053. doi: 10.1074/jbc.M201750200
Damont, A., Roeda, D., and Dolle, F. (2013). The potential of carbon-11 and fluorine-18 chemistry: illustration through the development of positron emission tomography radioligands targeting the translocator protein 18 kDa. J. Labelled Comp. Radiopharm. 56, 96–104. doi: 10.1002/jlcr.2992
Dani, M., Brooks, D. J., and Edison, P. (2015). Tau imaging in neurodegenerative diseases. Eur. J. Nucl. Med. Mol. Imaging doi: 10.1007/s00259-015-3231-2 [Epub ahead of print].
Darreh-Shori, T., Hosseini, S. M., and Nordberg, A. (2014). Pharmacodynamics of cholinesterase inhibitors suggests add-on therapy with a low-dose carbamylating inhibitor in patients on long-term treatment with rapidly reversible inhibitors. J. Alzheimers Dis. 39, 423–440. doi: 10.3233/JAD-130845
Davies, P., and Maloney, A. J. (1976). Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet 2:1403. doi: 10.1016/S0140-6736(76)91936-X
de Calignon, A., Polydoro, M., Suarez-Calvet, M., William, C., Adamowicz, D. H., Kopeikina, K. J., et al. (2012). Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron 73, 685–697. doi: 10.1016/j.neuron.2011.11.033
De Deyn, P. P., Drenth, A. F., Kremer, B. P., Oude Voshaar, R. C., and Van, D. D. (2013). Aripiprazole in the treatment of Alzheimer’s disease. Expert Opin. Pharmacother. 14, 459–474. doi: 10.1517/14656566.2013.764989
de Souza, L. C., Sarazin, M., Teixeira-Junior, A. L., Caramelli, P., Santos, A. E., and Dubois, B. (2014). Biological markers of Alzheimer’s disease. Arq. Neuropsiquiatr. 72, 227–231. doi: 10.1590/0004-282X20130233
Deardorff, W. J., Shobassy, A., and Grossberg, G. T. (2015). Safety and clinical effects of EVP-6124 in subjects with Alzheimer’s disease currently or previously receiving an acetylcholinesterase inhibitor medication. Expert Rev. Neurother. 15, 7–17. doi: 10.1586/14737175.2015.995639
Decker, M. W., and McGaugh, J. L. (1991). The role of interactions between the cholinergic system and other neuromodulatory systems in learning and memory. Synapse 7, 151–168. doi: 10.1002/syn.890070209
Defrancesco, M., Marksteiner, J., Fleischhacker, W. W., and Blasko, I. (2015). Use of benzodiazepines in Alzheimer’s disease: a systematic review of literature. Int. J. Neuropsychopharmacol. 18:pyv055. doi: 10.1093/ijnp/pyv055
Delumeau, J. C., Bentue-Ferrer, D., Gandon, J. M., Amrein, R., Belliard, S., and Allain, H. (1994). Monoamine oxidase inhibitors, cognitive functions and neurodegenerative diseases. J. Neural Transm. Suppl. 41, 259–266.
Devi, L., Prabhu, B. M., Galati, D. F., Avadhani, N. G., and Anandatheerthavarada, H. K. (2006). Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J. Neurosci. 26, 9057–9068. doi: 10.1523/JNEUROSCI.1469-06.2006
Dewar, D., Graham, D. I., and McCulloch, J. (1990). 5 HT2 receptors in dementia of Alzheimer type: a quantitative autoradiographic study of frontal cortex and hippocampus. J. Neural Transm. Park Dis. Dement. Sect. 2, 129–137. doi: 10.1007/BF02260900
Dineley, K. T., Bell, K. A., Bui, D., and Sweatt, J. D. (2002). beta -Amyloid peptide activates alpha 7 nicotinic acetylcholine receptors expressed in Xenopus oocytes. J. Biol. Chem. 277, 25056–25061. doi: 10.1074/jbc.M200066200
Dubois, B., Feldman, H. H., Jacova, C., Hampel, H., Molinuevo, J. L., Blennow, K., et al. (2014). Advancing research diagnostic criteria for Alzheimer’s disease: the IWG-2 criteria. Lancet Neurol. 13, 614–629. doi: 10.1016/S1474-4422(14)700900
Dunbar, G. C., Inglis, F., Kuchibhatla, R., Sharma, T., Tomlinson, M., and Wamsley, J. (2007). Effect of ispronicline, a neuronal nicotinic acetylcholine receptor partial agonist, in subjects with age associated memory impairment (AAMI). J. Psychopharmacol. 21, 171–178. doi: 10.1177/0269881107066855
Dunbar, G. C., Kuchibhatla, R. V., and Lee, G. (2011). A randomized double-blind study comparing 25 and 50 mg TC-1734 (AZD3480) with placebo, in older subjects with age-associated memory impairment. J. Psychopharmacol. 25, 1020–1029. doi: 10.1177/0269881110367727
Dunker, A. K., Silman, I., Uversky, V. N., and Sussman, J. L. (2008). Function and structure of inherently disordered proteins. Curr. Opin. Struct. Biol. 18, 756–764. doi: 10.1016/j.sbi.2008.10.002
Duyckaerts, C., Bennecib, M., Grignon, Y., Uchihara, T., He, Y., Piette, F., et al. (1997). Modeling the relation between neurofibrillary tangles and intellectual status. Neurobiol. Aging 18, 267–273. doi: 10.1016/S0197-4580(97)80306-5
Duyckaerts, C., Potier, M. C., and Delatour, B. (2008). Alzheimer disease models and human neuropathology: similarities and differences. Acta Neuropathol. 115, 5–38. doi: 10.1007/s00401-007-03128
Eckelman, W. C. (2001). Radiolabeled muscarinic radioligands for in vivo studies. Nucl. Med. Biol. 28, 485–491. doi: 10.1016/S0969-8051(01)00217-7
Eckelman, W. C. (2002). Accelerating drug discovery and development through in vivo imaging. Nucl. Med. Biol. 29, 777–782. doi: 10.1016/S0969-8051(02)00345-1
Ekonomou, A., Savva, G. M., Brayne, C., Forster, G., Francis, P. T., Johnson, M., et al. (2015). Stage-specific changes in neurogenic and glial markers in Alzheimer’s disease. Biol. Psychiatry 77, 711–719. doi: 10.1016/j.biopsych.2014.05.021
Eldar-Finkelman, H., and Martinez, A. (2011). GSK-3 inhibitors: preclinical and clinical focus on CNS. Front. Mol. Neurosci. 4:32. doi: 10.3389/fnmol.2011.00032
Ellis, J. R., Villemagne, V. L., Nathan, P. J., Mulligan, R. S., Gong, S. J., Chan, J. G., et al. (2008). Relationship between nicotinic receptors and cognitive function in early Alzheimer’s disease: a 2-[18F]fluoro-A-85380 PET study. Neurobiol. Learn. Mem. 90, 404–412. doi: 10.1016/j.nlm.2008.05.006
Engler, H., Santillo, A. F., Wang, S. X., Lindau, M., Savitcheva, I., Nordberg, A., et al. (2008). In vivo amyloid imaging with PET in frontotemporal dementia. Eur. J. Nucl. Med. Mol. Imaging 35, 100–106. doi: 10.1007/s00259-007-0523-1
Farde, L., Suhara, T., Halldin, C., Nyback, H., Nakashima, Y., Swahn, C. G., et al. (1996). PET study of the M1-agonists [11C]xanomeline and [11C]butylthio-TZTP in monkey and man. Dementia 7, 187–195.
Farlow, M., Anand, R., Messina, J. Jr., Hartman, R., and Veach, J. (2000). A 52-week study of the efficacy of rivastigmine in patients with mild to moderately severe Alzheimer’s disease. Eur. Neurol. 44, 236–241. doi: 10.1159/000008243
Filip, V., and Kolibas, E. (1999). Selegiline in the treatment of Alzheimer’s disease: a long-term randomized placebo-controlled trial. Czech and slovak senile dementia of alzheimer type study group. J. Psychiatry Neurosci. 24, 234–243.
Fisher, A., Heldman, E., Gurwitz, D., Haring, R., Karton, Y., Meshulam, H., et al. (1996). M1 agonists for the treatment of Alzheimer’s disease. Novel properties and clinical update. Ann. N. Y. Acad. Sci. 777, 189–196. doi: 10.1111/j.1749-6632.1996.tb34418.x
Fodero-Tavoletti, M. T., Okamura, N., Furumoto, S., Mulligan, R. S., Connor, A. R., McLean, C. A., et al. (2011). 18F-THK523: a novel in vivo tau imaging ligand for Alzheimer’s disease. Brain 134, 1089–1100. doi: 10.1093/brain/awr038
Fowler, J. S., Logan, J., Shumay, E., Alia-Klein, N., Wang, G. J., and Volkow, N. D. (2015). Monoamine oxidase: radiotracer chemistry and human studies. J. Labelled Comp. Radiopharm. 58, 51–64. doi: 10.1002/jlcr.3247
Francis, P. T., Palmer, A. M., Snape, M., and Wilcock, G. K. (1999). The cholinergic hypothesis of Alzheimer’s disease: a review of progress. J. Neurol. Neurosurg. Psychiatry 66, 137–147. doi: 10.1136/jnnp.66.2.137
Frankle, W. G., Cho, R. Y., Mason, N. S., Chen, C. M., Himes, M., Walker, C., et al. (2012). [11C]flumazenil binding is increased in a dose-dependent manner with tiagabine-induced elevations in GABA levels. PLoS ONE 7:e32443. doi: 10.1371/journal.pone.0032443
Frankle, W. G., Cho, R. Y., Narendran, R., Mason, N. S., Vora, S., Litschge, M., et al. (2009). Tiagabine increases [11C]flumazenil binding in cortical brain regions in healthy control subjects. Neuropsychopharmacology 34, 624–633. doi: 10.1038/npp.2008.104
Frolich, L., Ashwood, T., Nilsson, J., and Eckerwall, G. (2011). Effects of AZD3480 on cognition in patients with mild-to-moderate Alzheimer’s disease: a phase IIb dose-finding study. J. Alzheimers Dis. 24, 363–374. doi: 10.3233/JAD-2011-101554
Fukuchi, K., Hashikawa, K., Seike, Y., Moriwaki, H., Oku, N., Ishida, M., et al. (1997). Comparison of iodine-123-iomazenil SPECT and technetium-99m-HMPAO-SPECT in Alzheimer’s disease. J. Nucl. Med. 38, 467–470.
Galynker, I., Ieronimo, C., Miner, C., Rosenblum, J., Vilkas, N., and Rosenthal, R. (1997). Methylphenidate treatment of negative symptoms in patients with dementia. J. Neuropsychiatry Clin. Neurosci. 9, 231–239. doi: 10.1176/jnp.9.2.231
Garcia-Alloza, M., Hirst, W. D., Chen, C. P., Lasheras, B., Francis, P. T., and Ramirez, M. J. (2004). Differential involvement of 5-HT(1B/1D) and 5-HT6 receptors in cognitive and non-cognitive symptoms in Alzheimer’s disease. Neuropsychopharmacology 29, 410–416. doi: 10.1038/sj.npp.1300330
Gatto, G. J., Bohme, G. A., Caldwell, W. S., Letchworth, S. R., Traina, V. M., Obinu, M. C., et al. (2004). TC-1734: an orally active neuronal nicotinic acetylcholine receptor modulator with antidepressant, neuroprotective and long-lasting cognitive effects. CNS Drug Rev. 10, 147–166. doi: 10.1111/j.1527-3458.2004.tb00010.x
Geldenhuys, W. J., and Van der Schyf, C. J. (2011). Role of serotonin in Alzheimer’s disease: a new therapeutic target? CNS Drugs 25, 765–781. doi: 10.2165/11590190-000000000-00000
Ghezzi, L., Scarpini, E., and Galimberti, D. (2013). disease-modifying drugs in Alzheimer’s disease. Drug Des. Devel. Ther. 7, 1471–1478. doi: 10.2147/DDDT.S41431
Gilmor, M. L., Erickson, J. D., Varoqui, H., Hersh, L. B., Bennett, D. A., Cochran, E. J., et al. (1999). Preservation of nucleus basalis neurons containing choline acetyltransferase and the vesicular acetylcholine transporter in the elderly with mild cognitive impairment and early Alzheimer’s disease. J. Comp. Neurol. 411, 693–704. doi: 10.1002/(SICI)1096-9861(19990906)411:4<693::AID-CNE13>3.0.CO;2-D
Goedert, M., and Jakes, R. (1990). Expression of separate isoforms of human tau protein: correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J. 9, 4225–4230.
Gomez-Isla, T., Hollister, R., West, H., Mui, S., Growdon, J. H., Petersen, R. C., et al. (1997). Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann. Neurol. 41, 17–24. doi: 10.1002/ana.410410106
Gotti, C., and Clementi, F. (2004). Neuronal nicotinic receptors: from structure to pathology. Prog. Neurobiol. 74, 363–396. doi: 10.1016/j.pneurobio.2004.09.006
Gozes, I., Stewart, A., Morimoto, B., Fox, A., Sutherland, K., and Schmeche, D. (2009). Addressing Alzheimer’s disease tangles: from NAP to AL-108. Curr. Alzheimer Res. 6, 455–460. doi: 10.2174/156720509789207895
Graeber, M. B., and Mehraein, P. (1999). Reanalysis of the first case of Alzheimer’s disease. Eur. Arch. Psychiatry Clin. Neurosci. 249(Suppl. 3), 10–13. doi: 10.1007/PL00014167
Grant, M. K., and El-Fakahany, E. E. (2005). Persistent binding and functional antagonism by xanomeline at the muscarinic M5 receptor. J. Pharmacol. Exp. Ther. 315, 313–319. doi: 10.1124/jpet.105.090134
Greenberg, S. M., Tennis, M. K., Brown, L. B., Gomez-Isla, T., Hayden, D. L., Schoenfeld, D. A., et al. (2000). Donepezil therapy in clinical practice: a randomized crossover study. Arch. Neurol. 57, 94–99. doi: 10.1001/archneur.57.1.94
Grober, E., Dickson, D., Sliwinski, M. J., Buschke, H., Katz, M., Crystal, H., et al. (1999). Memory and mental status correlates of modified Braak staging. Neurobiol. Aging 20, 573–579. doi: 10.1016/S0197-4580(99)00063-9
Guzman-Ramos, K., Moreno-Castilla, P., Castro-Cruz, M., McGaugh, J. L., Martinez-Coria, H., Laferla, F. M., et al. (2012). Restoration of dopamine release deficits during object recognition memory acquisition attenuates cognitive impairment in a triple transgenic mice model of Alzheimer’s disease. Learn. Mem. 19, 453–460. doi: 10.1101/lm.026070.112
Haass, C. (2004). Take five–BACE and the gamma-secretase quartet conduct Alzheimer’s amyloid beta-peptide generation. EMBO J. 23, 483–488. doi: 10.1038/sj.emboj.7600061
Haass, C., and Selkoe, D. J. (2007). Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat. Rev. Mol. Cell Biol. 8, 101–112. doi: 10.1038/nrm2101
Halliday, G. M., McCann, H. L., Pamphlett, R., Brooks, W. S., Creasey, H., McCusker, E., et al. (1992). Brain stem serotonin-synthesizing neurons in Alzheimer’s disease: a clinicopathological correlation. Acta Neuropathol. 84, 638–650. doi: 10.1007/BF00227741
Hampel, H., Blennow, K., Shaw, L. M., Hoessler, Y. C., Zetterberg, H., and Trojanowski, J. Q. (2010). Total and phosphorylated tau protein as biological markers of Alzheimer’s disease. Exp. Gerontol. 45, 30–40. doi: 10.1016/j.exger.2009.10.010
Hampel, H., Ewers, M., Burger, K., Annas, P., Mortberg, A., Bogstedt, A., et al. (2009). Lithium trial in Alzheimer’s disease: a randomized, single-blind, placebo-controlled, multicenter 10-week study. J. Clin. Psychiatry 70, 922–931. doi: 10.4088/JCP.08m04606
Hampel, H., and Teipel, S. J. (2004). Total and phosphorylated tau proteins: evaluation as core biomarker candidates in frontotemporal dementia. Dement. Geriatr. Cogn. Disord. 17, 350–354. doi: 10.1159/000077170
Han, S. H., and Mook-Jung, I. (2014). Diverse molecular targets for therapeutic strategies in Alzheimer’s disease. J. Korean Med. Sci. 29, 893–902. doi: 10.3346/jkms.2014.29.7.893
Harada, R., Okamura, N., Furumoto, S., Furukawa, K., Ishiki, A., Tomita, N., et al. (2016). 18F-THK5351: a novel PET radiotracer for imaging neurofibrillary pathology in Alzheimer’s disease. J. Nucl. Med. 57, 208–214. doi: 10.2967/jnumed.115.164848
Hardwick, M., Fertikh, D., Culty, M., Li, H., Vidic, B., and Papadopoulos, V. (1999). Peripheral-type benzodiazepine receptor (PBR) in human breast cancer: correlation of breast cancer cell aggressive phenotype with PBR expression, nuclear localization, and PBR-mediated cell proliferation and nuclear transport of cholesterol. Cancer Res. 59, 831–842.
Hardy, J. (2006). Has the amyloid cascade hypothesis for Alzheimer’s disease been proved? Curr. Alzheimer Res. 3, 71–73. doi: 10.2174/156720506775697098
Hardy, J. (2009). The amyloid hypothesis for Alzheimer’s disease: a critical reappraisal. J. Neurochem. 110, 1129–1134. doi: 10.1111/j.1471-4159.2009.06181.x
Hardy, J. A., and Higgins, G. A. (1992). Alzheimer’s disease: the amyloid cascade hypothesis. Science 256, 184–185. doi: 10.1126/science.1566067
Heiss, W. D., Sobesky, J., Smekal, U., Kracht, L. W., Lehnhardt, F. G., Thiel, A., et al. (2004). Probability of cortical infarction predicted by flumazenil binding and diffusion-weighted imaging signal intensity: a comparative positron emission tomography/magnetic resonance imaging study in early ischemic stroke. Stroke 35, 1892–1898. doi: 10.1161/01.STR.0000134746.93535.9b
Henry, G., Williamson, D., and Tampi, R. R. (2011). Efficacy and tolerability of antidepressants in the treatment of behavioral and psychological symptoms of dementia, a literature review of evidence. Am. J. Alzheimers Dis. Other Demen. 26, 169–183. doi: 10.1177/1533317511402051
Hernandez, C. M., Kayed, R., Zheng, H., Sweatt, J. D., and Dineley, K. T. (2010). Loss of alpha7 nicotinic receptors enhances beta-amyloid oligomer accumulation, exacerbating early-stage cognitive decline and septohippocampal pathology in a mouse model of Alzheimer’s disease. J. Neurosci. 30, 2442–2453. doi: 10.1523/JNEUROSCI.5038-09.2010
Herrmann, N., Rothenburg, L. S., Black, S. E., Ryan, M., Liu, B. A., Busto, U. E., et al. (2008). Methylphenidate for the treatment of apathy in Alzheimer disease: prediction of response using dextroamphetamine challenge. J. Clin. Psychopharmacol. 28, 296–301. doi: 10.1097/JCP.0b013e318172b479
Hevers, W., and Luddens, H. (1998). The diversity of GABAA receptors. Pharmacological and electrophysiological properties of GABAA channel subtypes. Mol. Neurobiol. 18, 35–86. doi: 10.1007/BF02741459
Hickman, S. E., Allison, E. K., and El, K. J. (2008). Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. 28, 8354–8360. doi: 10.1523/JNEUROSCI.0616-08.2008
Hirsch, J. D., Beyer, C. F., Malkowitz, L., Beer, B., and Blume, A. J. (1989). Mitochondrial benzodiazepine receptors mediate inhibition of mitochondrial respiratory control. Mol. Pharmacol. 35, 157–163.
Hirvonen, J., Kailajarvi, M., Haltia, T., Koskimies, S., Nagren, K., Virsu, P., et al. (2009). Assessment of MAO-B occupancy in the brain with PET and [11C]-L-deprenyl-D2: a dose-finding study with a novel MAO-B inhibitor, EVT 301. Clin. Pharmacol. Ther. 85, 506–512. doi: 10.1038/clpt.2008.241
Hock, C., Maddalena, A., Heuser, I., Naber, D., Oertel, W., von der, K. H., et al. (2000). Treatment with the selective muscarinic agonist talsaclidine decreases cerebrospinal fluid levels of total amyloid beta-peptide in patients with Alzheimer’s disease. Ann. N. Y. Acad. Sci. 920, 285–291. doi: 10.1111/j.1749-6632.2000.tb06937.x
Holm, P., Ettrup, A., Klein, A. B., Santini, M. A., El-Sayed, M., Elvang, A. B., et al. (2010). Plaque deposition dependent decrease in 5-HT2A serotonin receptor in AbetaPPswe/PS1dE9 amyloid overexpressing mice. J. Alzheimers Dis. 20, 1201–1213. doi: 10.3233/JAD-2010-100117
Holman, B. L., Gibson, R. E., Hill, T. C., Eckelman, W. C., Albert, M., and Reba, R. C. (1985). Muscarinic acetylcholine receptors in Alzheimer’s disease. In vivo imaging with iodine 123-labeled 3-quinuclidinyl-4-iodobenzilate and emission tomography. JAMA 254, 3063–3066. doi: 10.1001/jama.254.21.3063
Hong, M., Zhukareva, V., Vogelsberg-Ragaglia, V., Wszolek, Z., Reed, L., Miller, B. I., et al. (1998). Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science 282, 1914–1917. doi: 10.1126/science.282.5395.1914
Hoozemans, J. J., Rozemuller, A. J., van Haastert, E. S., Eikelenboom, P., and van Gool, W. A. (2011). Neuroinflammation in Alzheimer’s disease wanes with age. J. Neuroinflammation 8, 171.
Hoshi, M., Sato, M., Matsumoto, S., Noguchi, A., Yasutake, K., Yoshida, N., et al. (2003). Spherical aggregates of beta-amyloid (amylospheroid) show high neurotoxicity and activate tau protein kinase I/glycogen synthase kinase-3beta. Proc. Natl. Acad. Sci. U.S.A. 100, 6370–6375. doi: 10.1073/pnas.1237107100
Hosie, A. M., Wilkins, M. E., da Silva, H. M., and Smart, T. G. (2006). Endogenous neurosteroids regulate GABAA receptors through two discrete transmembrane sites. Nature 444, 486–489. doi: 10.1038/nature05324
Hoyer, D., Hannon, J. P., and Martin, G. R. (2002). Molecular, pharmacological and functional diversity of 5-HT receptors. Pharmacol. Biochem. Behav. 71, 533–554. doi: 10.1016/S0091-3057(01)00746-8
Huang, L., Lu, C., Sun, Y., Mao, F., Luo, Z., Su, T., et al. (2012). Multitarget-directed benzylideneindanone derivatives: anti-beta-amyloid (Abeta) aggregation, antioxidant, metal chelation, and monoamine oxidase B (MAO-B) inhibition properties against Alzheimer’s disease. J. Med. Chem. 55, 8483–8492. doi: 10.1021/jm300978h
Hyman, B. T., Phelps, C. H., Beach, T. G., Bigio, E. H., Cairns, N. J., Carrillo, M. C., et al. (2012). National institute on aging-Alzheimer’s association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. 8, 1–13. doi: 10.1016/j.jalz.2011.10.007
Imbimbo, B. P., and Giardina, G. A. (2011). gamma-secretase inhibitors and modulators for the treatment of Alzheimer’s disease: disappointments and hopes. Curr. Top. Med. Chem. 11, 1555–1570. doi: 10.2174/156802611795860942
Iqbal, K., and Grundke-Iqbal, I. (2008). Alzheimer neurofibrillary degeneration: significance, etiopathogenesis, therapeutics and prevention. J. Cell Mol. Med. 12, 38–55. doi: 10.1111/j.1582-4934.2008.00225.x
Irie, T., Fukushi, K., Akimoto, Y., Tamagami, H., and Nozaki, T. (1994). Design and evaluation of radioactive acetylcholine analogs for mapping brain acetylcholinesterase (AchE) in vivo. Nucl. Med. Biol. 21, 801–808. doi: 10.1016/0969-8051(94)90159-7
Ishiki, A., Okamura, N., Furukawa, K., Furumoto, S., Harada, R., Tomita, N., et al. (2015). Longitudinal assessment of tau pathology in patients with Alzheimer’s disease using [18F]THK-5117 positron emission tomography. PLoS ONE 10:e0140311. doi: 10.1371/journal.pone.0140311
Itoh, M., Meguro, K., Fujiwara, T., Hatazawa, J., Iwata, R., Ishiwata, K., et al. (1994). Assessment of dopamine metabolism in brain of patients with dementia by means of 18F-fluorodopa and PET. Ann. Nucl. Med. 8, 245–251. doi: 10.1007/BF03165027
Iyo, M., Namba, H., Fukushi, K., Shinotoh, H., Nagatsuka, S., Suhara, T., et al. (1997). Measurement of acetylcholinesterase by positron emission tomography in the brains of healthy controls and patients with Alzheimer’s disease. Lancet 349, 1805–1809. doi: 10.1016/S0140-6736(96)091246
Jack, C. R. Jr., Barrio, J. R., and Kepe, V. (2013). Cerebral amyloid PET imaging in Alzheimer’s disease. Acta Neuropathol. 126, 643–657. doi: 10.1007/s00401-013-11857
Jack, C. R. Jr., Knopman, D. S., Jagust, W. J., Shaw, L. M., Aisen, P. S., Weiner, M. W., et al. (2010). Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 9, 119–128. doi: 10.1016/S1474-4422(09)702996
Jack, C. R. Jr., Lowe, V. J., Senjem, M. L., Weigand, S. D., Kemp, B. J., Shiung, M. M., et al. (2008). 11C PiB and structural MRI provide complementary information in imaging of Alzheimer’s disease and amnestic mild cognitive impairment. Brain 131, 665–680. doi: 10.1093/brain/awm336
Jack, C. R. Jr., Lowe, V. J., Weigand, S. D., Wiste, H. J., Senjem, M. L., Knopman, D. S., et al. (2009). Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer’s disease: implications for sequence of pathological events in Alzheimer’s disease. Brain 132, 1355–1365. doi: 10.1093/brain/awp062
James, O. G., Doraiswamy, P. M., and Borges-Neto, S. (2015). PET imaging of tau pathology in Alzheimer’s disease and tauopathies. Front. Neurol. 6:38. doi: 10.3389/fneur.2015.00038
Janssen, B., Vugts, D. J., Funke, U., Molenaar, G. T., Kruijer, P. S., van Berckel, B. N., et al. (2016). Imaging of neuroinflammation in Alzheimer’s disease, multiple sclerosis and stroke: recent developments in positron emission tomography. Biochim. Biophys. Acta 1862, 425–441. doi: 10.1016/j.bbadis.2015.11.011
Jaworski, T., Dewachter, I., Lechat, B., Gees, M., Kremer, A., Demedts, D., et al. (2011). GSK-3alpha/beta kinases and amyloid production in vivo. Nature 480, E4–E5. doi: 10.1038/nature10615
Jones, G. M., Sahakian, B. J., Levy, R., Warburton, D. M., and Gray, J. A. (1992). Effects of acute subcutaneous nicotine on attention, information processing and short-term memory in Alzheimer’s disease. Psychopharmacology (Berl.) 108, 485–494. doi: 10.1007/BF02247426
Jossan, S. S., Gillberg, P. G., Gottfries, C. G., Karlsson, I., and Oreland, L. (1991). Monoamine oxidase B in brains from patients with Alzheimer’s disease: a biochemical and autoradiographical study. Neuroscience 45, 1–12. doi: 10.1016/0306-4522(91)90098-9
Jurgensen, S., and Ferreira, S. T. (2010). Nicotinic receptors, amyloid-beta, and synaptic failure in Alzheimer’s disease. J. Mol. Neurosci. 40, 221–229. doi: 10.1007/s12031-009-92370
Kaasinen, V., Nagren, K., Jarvenpaa, T., Roivainen, A., Yu, M., Oikonen, V., et al. (2002). Regional effects of donepezil and rivastigmine on cortical acetylcholinesterase activity in Alzheimer’s disease. J. Clin. Psychopharmacol. 22, 615–620. doi: 10.1097/00004714-200212000-00012
Kadir, A., Almkvist, O., Wall, A., Langstrom, B., and Nordberg, A. (2006). PET imaging of cortical 11C-nicotine binding correlates with the cognitive function of attention in Alzheimer’s disease. Psychopharmacology (Berl.) 188, 509–520. doi: 10.1007/s00213-006-04477
Kadir, A., Darreh-Shori, T., Almkvist, O., Wall, A., Langstrom, B., and Nordberg, A. (2007). Changes in brain 11C-nicotine binding sites in patients with mild Alzheimer’s disease following rivastigmine treatment as assessed by PET. Psychopharmacology (Berl.) 191, 1005–1014. doi: 10.1007/s00213-007-0725-z
Karlin, A. (2002). Emerging structure of the nicotinic acetylcholine receptors. Nat. Rev. Neurosci. 3, 102–114. doi: 10.1038/nrn731
Katsifis, A., and Kassiou, M. (2004). Development of radioligands for in vivo imaging of GABA(A)-benzodiazepine receptors. Mini. Rev. Med. Chem. 4, 909–921. doi: 10.2174/1389557043403332
Kayed, R., Head, E., Thompson, J. L., McIntire, T. M., Milton, S. C., Cotman, C. W., et al. (2003). Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300, 486–489. doi: 10.1126/science.1079469
Kehr, J., Hu, X. J., Yoshitake, T., Wang, F. H., Osborne, P., Stenfors, C., et al. (2010). The selective 5-HT(1A) receptor antagonist NAD-299 increases acetylcholine release but not extracellular glutamate levels in the frontal cortex and hippocampus of awake rat. Eur. Neuropsychopharmacol. 20, 487–500. doi: 10.1016/j.euroneuro.2010.03.003
Kemp, P. M., Holmes, C., Hoffmann, S., Wilkinson, S., Zivanovic, M., Thom, J., et al. (2003). A randomised placebo controlled study to assess the effects of cholinergic treatment on muscarinic receptors in Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 74, 1567–1570. doi: 10.1136/jnnp.74.11.1567
Kemppainen, N., Laine, M., Laakso, M. P., Kaasinen, V., Nagren, K., Vahlberg, T., et al. (2003). Hippocampal dopamine D2 receptors correlate with memory functions in Alzheimer’s disease. Eur. J. Neurosci. 18, 149–154. doi: 10.1046/j.1460-9568.2003.02716.x
Kemppainen, N., Ruottinen, H., Nagren, K., and Rinne, J. O. (2000). PET shows that striatal dopamine D1 and D2 receptors are differentially affected in AD. Neurology 55, 205–209. doi: 10.1212/WNL.55.2.205
Kemppainen, N. M., Aalto, S., Wilson, I. A., Nagren, K., Helin, S., Bruck, A., et al. (2006). Voxel-based analysis of PET amyloid ligand [11C]PIB uptake in Alzheimer disease. Neurology 67, 1575–1580. doi: 10.1212/01.wnl.0000240117.55680.0a
Kendziorra, K., Wolf, H., Meyer, P. M., Barthel, H., Hesse, S., Becker, G. A., et al. (2011). Decreased cerebral alpha4beta2* nicotinic acetylcholine receptor availability in patients with mild cognitive impairment and Alzheimer’s disease assessed with positron emission tomography. Eur. J. Nucl. Med. Mol. Imaging 38, 515–525. doi: 10.1007/s00259-010-1644-5
Kepe, V., Barrio, J. R., Huang, S. C., Ercoli, L., Siddarth, P., Shoghi-Jadid, K., et al. (2006). Serotonin 1A receptors in the living brain of Alzheimer’s disease patients. Proc. Natl. Acad. Sci. U.S.A. 103, 702–707. doi: 10.1073/pnas.0510237103
Kepe, V., Bordelon, Y., Boxer, A., Huang, S. C., Liu, J., Thiede, F. C., et al. (2013). PET imaging of neuropathology in tauopathies: progressive supranuclear palsy. J. Alzheimers Dis. 36, 145–153. doi: 10.3233/JAD-130032
Kepe, V., Ghetti, B., Farlow, M. R., Bresjanac, M., Miller, K., Huang, S. C., et al. (2010). PET of brain prion protein amyloid in Gerstmann-Straussler-Scheinker disease. Brain Pathol. 20, 419–430. doi: 10.1111/j.1750-3639.2009.00306.x
Keverne, E. B. (1999). GABA-ergic neurons and the neurobiology of schizophrenia and other psychoses. Brain Res. Bull. 48, 467–473. doi: 10.1016/S0361-9230(99)00025-8
Kilbourn, M. R., Snyder, S. E., Sherman, P. S., and Kuhl, D. E. (1996). In vivo studies of acetylcholinesterase activity using a labeled substrate, N-[11C]methylpiperdin-4-yl propionate ([11C]PMP). Synapse 22, 123–131. doi: 10.1002/(SICI)1098-2396(199602)22:2<123::AID-SYN5>3.0.CO;2-F
Kim, S. Y., Choi, S. H., Rollema, H., Schwam, E. M., McRae, T., Dubrava, S., et al. (2014). Phase II crossover trial of varenicline in mild-to-moderate Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 37, 232–245. doi: 10.1159/000355373
King, M. K., Pardo, M., Cheng, Y., Downey, K., Jope, R. S., and Beurel, E. (2014). Glycogen synthase kinase-3 inhibitors: rescuers of cognitive impairments. Pharmacol. Ther. 141, 1–12. doi: 10.1016/j.pharmthera.2013.07.010
Kirven, L. E., and Montero, E. F. (1973). Comparison of thioridazine and diazepam in the control of nonpsychotic symptoms associated with senility: double-blind study. J. Am. Geriatr. Soc. 21, 546–551. doi: 10.1111/j.1532-5415.1973.tb01661.x
Kitazawa, M., Oddo, S., Yamasaki, T. R., Green, K. N., and Laferla, F. M. (2005). Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer’s disease. J. Neurosci. 25, 8843–8853. doi: 10.1523/JNEUROSCI.2868-05.2005
Koo, J., and Byun, Y. (2013). Current status of PET-imaging probes of beta-amyloid plaques. Arch. Pharm. Res. 36, 1178–1184. doi: 10.1007/s12272-013-01934
Korczyn, A. D. (2008). The amyloid cascade hypothesis. Alzheimers. Dement. 4, 176–178. doi: 10.1016/j.jalz.2007.11.008
Kugler, W., Veenman, L., Shandalov, Y., Leschiner, S., Spanier, I., Lakomek, M., et al. (2008). Ligands of the mitochondrial 18 kDa translocator protein attenuate apoptosis of human glioblastoma cells exposed to erucylphosphohomocholine. Cell Oncol. 30, 435–450.
Kuhl, D. E., Minoshima, S., Frey, K. A., Foster, N. L., Kilbourn, M. R., and Koeppe, R. A. (2000). Limited donepezil inhibition of acetylcholinesterase measured with positron emission tomography in living Alzheimer cerebral cortex. Ann. Neurol. 48, 391–395. doi: 10.1002/1531-8249(200009)48:3<391::AID-ANA17>3.3.CO;2-8
Kumar, J. S., and Mann, J. J. (2014). PET tracers for serotonin receptors and their applications. Cent. Nerv. Syst. Agents Med. Chem. 14, 96–112. doi: 10.2174/1871524914666141030124316
Kumar, U., and Patel, S. C. (2007). Immunohistochemical localization of dopamine receptor subtypes (D1R-D5R) in Alzheimer’s disease brain. Brain Res. 1131, 187–196. doi: 10.1016/j.brainres.2006.10.049
Lai, M. K., Tsang, S. W., Alder, J. T., Keene, J., Hope, T., Esiri, M. M., et al. (2005). Loss of serotonin 5-HT2A receptors in the postmortem temporal cortex correlates with rate of cognitive decline in Alzheimer’s disease. Psychopharmacology (Berl.) 179, 673–677. doi: 10.1007/s00213-004-2077-2
Lai, M. K., Tsang, S. W., Francis, P. T., Esiri, M. M., Keene, J., Hope, T., et al. (2003). Reduced serotonin 5-HT1A receptor binding in the temporal cortex correlates with aggressive behavior in Alzheimer disease. Brain Res. 974, 82–87. doi: 10.1016/S0006-8993(03)02554-X
Lanctot, K. L., Herrmann, N., Black, S. E., Ryan, M., Rothenburg, L. S., Liu, B. A., et al. (2008). Apathy associated with Alzheimer disease: use of dextroamphetamine challenge. Am. J. Geriatr. Psychiatry 16, 551–557. doi: 10.1097/JGP.0b013e318170a6d1
Lanctot, K. L., Herrmann, N., Mazzotta, P., Khan, L. R., and Ingber, N. (2004). GABAergic function in Alzheimer’s disease: evidence for dysfunction and potential as a therapeutic target for the treatment of behavioural and psychological symptoms of dementia. Can. J. Psychiatry 49, 439–453.
Lee, V. M., Goedert, M., and Trojanowski, J. Q. (2001). Neurodegenerative tauopathies. Annu. Rev. Neurosci. 24, 1121–1159. doi: 10.1146/annurev.neuro.24.1.1121
Lees, A. J., Shaw, K. M., Kohout, L. J., Stern, G. M., Elsworth, J. D., Sandler, M., et al. (1977). Deprenyl in Parkinson’s disease. Lancet 2, 791–795. doi: 10.1016/S0140-6736(77)90725-5
Lemere, C. A. (2013). Immunotherapy for Alzheimer’s disease: hoops and hurdles. Mol. Neurodegener. 8:36. doi: 10.1186/1750-1326-8-36
Levin, E. D. (1992). Nicotinic systems and cognitive function. Psychopharmacology (Berl.) 108, 417–431. doi: 10.1007/BF02247415
Liao, C. F., Themmen, A. P., Joho, R., Barberis, C., Birnbaumer, M., and Birnbaumer, L. (1989). Molecular cloning and expression of a fifth muscarinic acetylcholine receptor. J. Biol. Chem. 264, 7328–7337.
Limon, A., Reyes-Ruiz, J. M., and Miledi, R. (2012). Loss of functional GABA(A) receptors in the Alzheimer diseased brain. Proc. Natl. Acad. Sci. U.S.A. 109, 10071–10076. doi: 10.1073/pnas.1204606109
Lleo, A., Greenberg, S. M., and Growdon, J. H. (2006). Current pharmacotherapy for Alzheimer’s disease. Annu. Rev. Med. 57, 513–533. doi: 10.1146/annurev.med.57.121304.131442
Lopez, O. L., Wisnieski, S. R., Becker, J. T., Boller, F., and DeKosky, S. T. (1997). Extrapyramidal signs in patients with probable Alzheimer disease. Arch. Neurol. 54, 969–975. doi: 10.1001/archneur.1997.00550200033007
Lorke, D. E., Lu, G., Cho, E., and Yew, D. T. (2006). Serotonin 5-HT2A and 5-HT6 receptors in the prefrontal cortex of Alzheimer and normal aging patients. BMC Neurosci. 7:36. doi: 10.1186/1471-2202-7-36
Lovestone, S., Boada, M., Dubois, B., Hull, M., Rinne, J. O., Huppertz, H. J., et al. (2015). A phase II trial of tideglusib in Alzheimer’s disease. J. Alzheimers Dis. 45, 75–88. doi: 10.3233/JAD-141959
Lowe, V. J., Kemp, B. J., Jack, C. R. Jr., Senjem, M., Weigand, S., Shiung, M., et al. (2009). Comparison of 18F-FDG and PiB PET in cognitive impairment. J. Nucl. Med. 50, 878–886. doi: 10.2967/jnumed.108.058529
Luo, Y., Bolon, B., Kahn, S., Bennett, B. D., Babu-Khan, S., Denis, P., et al. (2001). Mice deficient in BACE1, the Alzheimer’s beta-secretase, have normal phenotype and abolished beta-amyloid generation. Nat. Neurosci. 4, 231–232. doi: 10.1038/85059
Lustbader, J. W., Cirilli, M., Lin, C., Xu, H. W., Takuma, K., Wang, N., et al. (2004). ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease. Science 304, 448–452. doi: 10.1126/science.1091230
Lyketsos, C. G., DelCampo, L., Steinberg, M., Miles, Q., Steele, C. D., Munro, C., et al. (2003). Treating depression in Alzheimer disease: efficacy and safety of sertraline therapy, and the benefits of depression reduction: the DIADS. Arch. Gen. Psychiatry 60, 737–746. doi: 10.1001/archpsyc.60.7.737
Madsen, K., Neumann, W. J., Holst, K., Marner, L., Haahr, M. T., Lehel, S., et al. (2011). Cerebral serotonin 4 receptors and amyloid-beta in early Alzheimer’s disease. J. Alzheimers Dis. 26, 457–466. doi: 10.3233/JAD-2011-110056
Maher-Edwards, G., Zvartau-Hind, M., Hunter, A. J., Gold, M., Hopton, G., Jacobs, G., et al. (2010). Double-blind, controlled phase II study of a 5-HT6 receptor antagonist, SB-742457, in Alzheimer’s disease. Curr. Alzheimer Res. 7, 374–385. doi: 10.2174/156720510791383831
Maqbool, M., Mobashir, M., and Hoda, N. (2016). Pivotal role of glycogen synthase kinase-3: a therapeutic target for Alzheimer’s disease. Eur. J. Med. Chem. 107, 63–81. doi: 10.1016/j.ejmech.2015.10.018
Marczynski, T. J. (1998). GABAergic deafferentation hypothesis of brain aging and Alzheimer’s disease revisited. Brain Res. Bull. 45, 341–379. doi: 10.1016/S0361-9230(97)00347-X
Marner, L., Frokjaer, V. G., Kalbitzer, J., Lehel, S., Madsen, K., Baare, W. F., et al. (2012). Loss of serotonin 2A receptors exceeds loss of serotonergic projections in early Alzheimer’s disease: a combined [11C]DASB and [18F]altanserin-PET study. Neurobiol. Aging 33, 479–487. doi: 10.1016/j.neurobiolaging.2010.03.023
Martin, L., Latypova, X., and Terro, F. (2011). Post-translational modifications of tau protein: implications for Alzheimer’s disease. Neurochem. Int. 58, 458–471. doi: 10.1016/j.neuint.2010.12.023
Martorana, A., Di, L. F., Esposito, Z., Lo, G. T., Bernardi, G., Caltagirone, C., et al. (2013). Dopamine D(2)-agonist rotigotine effects on cortical excitability and central cholinergic transmission in Alzheimer’s disease patients. Neuropharmacology 64, 108–113. doi: 10.1016/j.neuropharm.2012.07.015
Martorana, A., and Koch, G. (2014). Is dopamine involved in Alzheimer’s disease? Front. Aging Neurosci. 6:252. doi: 10.3389/fnagi.2014.00252
Maruyama, M., Shimada, H., Suhara, T., Shinotoh, H., Ji, B., Maeda, J., et al. (2013). Imaging of tau pathology in a tauopathy mouse model and in Alzheimer patients compared to normal controls. Neuron 79, 1094–1108. doi: 10.1016/j.neuron.2013.07.037
Masters, C. L., Cappai, R., Barnham, K. J., and Villemagne, V. L. (2006). Molecular mechanisms for Alzheimer’s disease: implications for neuroimaging and therapeutics. J. Neurochem. 97, 1700–1725. doi: 10.1111/j.1471-4159.2006.03989.x
Maziere, M. (1995). Cholinergic neurotransmission studied in vivo using positron emission tomography or single photon emission computerized tomography. Pharmacol. Ther. 66, 83–101. doi: 10.1016/0163-7258(95)00003-Y
McEnery, M. W., Snowman, A. M., Trifiletti, R. R., and Snyder, S. H. (1992). Isolation of the mitochondrial benzodiazepine receptor: association with the voltage-dependent anion channel and the adenine nucleotide carrier. Proc. Natl. Acad. Sci. U.S.A. 89, 3170–3174. doi: 10.1073/pnas.89.8.3170
McGeer, P. L., and McGeer, E. G. (1995). The inflammatory response system of brain: implications for therapy of Alzheimer and other neurodegenerative diseases. Brain Res. Brain Res. Rev. 21, 195–218. doi: 10.1016/0165-0173(95)00011-9
Mecocci, P., and Polidori, M. C. (2012). Antioxidant clinical trials in mild cognitive impairment and Alzheimer’s disease. Biochim. Biophys. Acta 1822, 631–638. doi: 10.1016/j.bbadis.2011.10.006
Mehta, A. K., and Ticku, M. K. (1999). An update on GABAA receptors. Brain Res. Brain Res. Rev. 29, 196–217. doi: 10.1016/S0165-0173(98)00052-6
Meyer, P. M., Tiepolt, S., Barthel, H., Hesse, S., and Sabri, O. (2014). Radioligand imaging of alpha4beta2* nicotinic acetylcholine receptors in Alzheimer’s disease and Parkinson’s disease. Q. J. Nucl. Med. Mol. Imaging 58, 376–386.
Midzak, A., Zirkin, B., and Papadopoulos, V. (2015). Translocator protein: pharmacology and steroidogenesis. Biochem. Soc. Trans. 43, 572–578. doi: 10.1042/BST20150061
Miettinen, H., Kononen, J., Haapasalo, H., Helen, P., Sallinen, P., Harjuntausta, T., et al. (1995). Expression of peripheral-type benzodiazepine receptor and diazepam binding inhibitor in human astrocytomas: relationship to cell proliferation. Cancer Res. 55, 2691–2695.
Millan, M. J., Gobert, A., Roux, S., Porsolt, R., Meneses, A., Carli, M., et al. (2004). The serotonin1A receptor partial agonist S15535 [4-(benzodioxan-5-yl)1-(indan-2-yl)piperazine] enhances cholinergic transmission and cognitive function in rodents: a combined neurochemical and behavioral analysis. J. Pharmacol. Exp. Ther. 311, 190–203. doi: 10.1124/jpet.104.069625
Minati, L., Edginton, T., Bruzzone, M. G., and Giaccone, G. (2009). Current concepts in Alzheimer’s disease: a multidisciplinary review. Am. J. Alzheimers. Dis. Other Demen. 24, 95–121. doi: 10.1177/1533317508328602
Mitchell, R. A., Herrmann, N., and Lanctot, K. L. (2011). The role of dopamine in symptoms and treatment of apathy in Alzheimer’s disease. CNS Neurosci. Ther. 17, 411–427. doi: 10.1111/j.1755-5949.2010.00161.x
Moreira, P. I., Zhu, X., Nunomura, A., Smith, M. A., and Perry, G. (2006). Therapeutic options in Alzheimer’s disease. Expert Rev. Neurother. 6, 897–910. doi: 10.1586/14737175.6.6.897
Morgan, D., Gordon, M. N., Tan, J., Wilcock, D., and Rojiani, A. M. (2005). Dynamic complexity of the microglial activation response in transgenic models of amyloid deposition: implications for Alzheimer therapeutics. J. Neuropathol. Exp. Neurol. 64, 743–753. doi: 10.1097/01.jnen.0000178444.33972.e0
Mowla, A., Mosavinasab, M., and Pani, A. (2007). Does fluoxetine have any effect on the cognition of patients with mild cognitive impairment? A double-blind, placebo-controlled, clinical trial. J. Clin. Psychopharmacol. 27, 67–70. doi: 10.1097/JCP.0b013e31802e0002
Mura, E., Lanni, C., Preda, S., Pistoia, F., Sara, M., Racchi, M., et al. (2010). Beta-amyloid: a disease target or a synaptic regulator affecting age-related neurotransmitter changes? Curr. Pharm. Des. 16, 672–683. doi: 10.2174/138161210790883723
Murray, A. M., Weihmueller, F. B., Marshall, J. F., Hurtig, H. I., Gottleib, G. L., and Joyce, J. N. (1995). Damage to dopamine systems differs between Parkinson’s disease and Alzheimer’s disease with parkinsonism. Ann. Neurol. 37, 300–312. doi: 10.1002/ana.410370306
Nelson, L. D., Siddarth, P., Kepe, V., Scheibel, K. E., Huang, S. C., Barrio, J. R., et al. (2011). Positron emission tomography of brain beta-amyloid and tau levels in adults with Down syndrome. Arch. Neurol. 68, 768–774. doi: 10.1001/archneurol.2011.104
Nelson, P. T., Alafuzoff, I., Bigio, E. H., Bouras, C., Braak, H., Cairns, N. J., et al. (2012). Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J. Neuropathol. Exp. Neurol. 71, 362–381. doi: 10.1097/NEN.0b013e31825018f7
Neve, R. L., Harris, P., Kosik, K. S., Kurnit, D. M., and Donlon, T. A. (1986). Identification of cDNA clones for the human microtubule-associated protein tau and chromosomal localization of the genes for tau and microtubule-associated protein 2. Brain Res. 387, 271–280. doi: 10.1016/0169-328X(86)90033-1
Niesler, B., Kapeller, J., Hammer, C., and Rappold, G. (2008). Serotonin type 3 receptor genes: HTR3A, B, C, D, E. Pharmacogenomics 9, 501–504. doi: 10.2217/14622416.9.5.501
Nitsch, R. M., Slack, B. E., Wurtman, R. J., and Growdon, J. H. (1992). Release of Alzheimer amyloid precursor derivatives stimulated by activation of muscarinic acetylcholine receptors. Science 258, 304–307. doi: 10.1126/science.1411529
Noble, W., Planel, E., Zehr, C., Olm, V., Meyerson, J., Suleman, F., et al. (2005). Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc. Natl. Acad. Sci. U.S.A. 102, 6990–6995. doi: 10.1073/pnas.0500466102
Noble, W., Pooler, A. M., and Hanger, D. P. (2011). Advances in tau-based drug discovery. Expert Opin. Drug Discov. 6, 797–810. doi: 10.1517/17460441.2011.586690
Nordberg, A., Amberla, K., Shigeta, M., Lundqvist, H., Viitanen, M., Hellstrom-Lindahl, E., et al. (1998). Long-term tacrine treatment in three mild Alzheimer patients: effects on nicotinic receptors, cerebral blood flow, glucose metabolism, EEG, and cognitive abilities. Alzheimer Dis. Assoc. Disord. 12, 228–237. doi: 10.1097/00002093-199809000-00017
Nordberg, A., Hartvig, P., Lilja, A., Viitanen, M., Amberla, K., Lundqvist, H., et al. (1990). Decreased uptake and binding of 11C-nicotine in brain of Alzheimer patients as visualized by positron emission tomography. J. Neural Transm. Park. Dis. Dement. Sect. 2, 215–224. doi: 10.1007/BF02257652
Nordberg, A., Lilja, A., Lundqvist, H., Hartvig, P., Amberla, K., Viitanen, M., et al. (1992). Tacrine restores cholinergic nicotinic receptors and glucose metabolism in Alzheimer patients as visualized by positron emission tomography. Neurobiol. Aging 13, 747–758. doi: 10.1016/0197-4580(92)90099-J
Nordberg, A., Lundqvist, H., Hartvig, P., Andersson, J., Johansson, M., Hellstrom-Lindahi, E., et al. (1997). Imaging of nicotinic and muscarinic receptors in Alzheimer’s disease: effect of tacrine treatment. Dement. Geriatr. Cogn. Disord. 8, 78–84. doi: 10.1159/000106611
Nordberg, A., Lundqvist, H., Hartvig, P., Lilja, A., and Langstrom, B. (1995). Kinetic analysis of regional (S)(-)11C-nicotine binding in normal and Alzheimer brains–in vivo assessment using positron emission tomography. Alzheimer Dis. Assoc. Disord. 9, 21–27. doi: 10.1097/00002093-199505000-00006
Nykanen, N. P., Kysenius, K., Sakha, P., Tammela, P., and Huttunen, H. J. (2012). gamma-Aminobutyric acid type A (GABAA) receptor activation modulates tau phosphorylation. J. Biol. Chem. 287, 6743–6752. doi: 10.1074/jbc.M111.309385
Nyth, A. L., and Gottfries, C. G. (1990). The clinical efficacy of citalopram in treatment of emotional disturbances in dementia disorders. A Nordic multicentre study. Br. J. Psychiatry 157, 894–901. doi: 10.1192/bjp.157.6.894
O’Brien, J. T., Colloby, S. J., Pakrasi, S., Perry, E. K., Pimlott, S. L., Wyper, D. J., et al. (2007). Alpha4beta2 nicotinic receptor status in Alzheimer’s disease using 123I-5IA-85380 single-photon-emission computed tomography. J. Neurol. Neurosurg. Psychiatry 78, 356–362. doi: 10.1136/jnnp.2006.108209
Oddo, S., Caccamo, A., Shepherd, J. D., Murphy, M. P., Golde, T. E., Kayed, R., et al. (2003). Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39, 409–421. doi: 10.1016/S0896-6273(03)00434-3
Oddo, S., and Laferla, F. M. (2006). The role of nicotinic acetylcholine receptors in Alzheimer’s disease. J. Physiol. Paris 99, 172–179. doi: 10.1016/j.jphysparis.2005.12.080
Okada, H., Ouchi, Y., Ogawa, M., Futatsubashi, M., Saito, Y., Yoshikawa, E., et al. (2013). Alterations in alpha4beta2 nicotinic receptors in cognitive decline in Alzheimer’s aetiopathology. Brain 136, 3004–3017. doi: 10.1093/brain/awt195
Okamura, N., Furumoto, S., Fodero-Tavoletti, M. T., Mulligan, R. S., Harada, R., Yates, P., et al. (2014a). Non-invasive assessment of Alzheimer’s disease neurofibrillary pathology using 18F-THK5105 PET. Brain 137, 1762–1771. doi: 10.1093/brain/awu064
Okamura, N., Harada, R., Furumoto, S., Arai, H., Yanai, K., and Kudo, Y. (2014b). Tau PET imaging in Alzheimer’s disease. Curr. Neurol. Neurosci. Rep. 14:500. doi: 10.1007/s11910-014-0500_6
Okello, A., Koivunen, J., Edison, P., Archer, H. A., Turkheimer, F. E., Nagren, K., et al. (2009). Conversion of amyloid positive and negative MCI to AD over 3 years: an 11C-PIB PET study. Neurology 73, 754–760. doi: 10.1212/WNL.0b013e3181b23564
Orgogozo, J. M., Gilman, S., Dartigues, J. F., Laurent, B., Puel, M., Kirby, L. C., et al. (2003). Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology 61, 46–54. doi: 10.1212/01.WNL.0000073623.84147.A8
Ory, D., Celen, S., Verbruggen, A., and Bormans, G. (2014). PET radioligands for in vivo visualization of neuroinflammation. Curr. Pharm. Des. 20, 5897–5913. doi: 10.2174/1381612820666140613120212
Ouchi, Y., Yoshikawa, E., Futatsubashi, M., Yagi, S., Ueki, T., and Nakamura, K. (2009). Altered brain serotonin transporter and associated glucose metabolism in Alzheimer disease. J. Nucl. Med. 50, 1260–1266. doi: 10.2967/jnumed.109.063008
Owen, D. R., Lewis, A. J., Reynolds, R., Rupprecht, R., Eser, D., Wilkins, M. R., et al. (2011). Variation in binding affinity of the novel anxiolytic XBD173 for the 18 kDa translocator protein in human brain. Synapse 65, 257–259. doi: 10.1002/syn.20884
Owen, D. R., Yeo, A. J., Gunn, R. N., Song, K., Wadsworth, G., Lewis, A., et al. (2012). An 18-kDa translocator protein (TSPO) polymorphism explains differences in binding affinity of the PET radioligand PBR28. J. Cereb. Blood Flow Metab. 32, 1–5. doi: 10.1038/jcbfm.2011.147
Padala, P. R., Burke, W. J., Shostrom, V. K., Bhatia, S. C., Wengel, S. P., Potter, J. F., et al. (2010). Methylphenidate for apathy and functional status in dementia of the Alzheimer type. Am. J. Geriatr. Psychiatry 18, 371–374. doi: 10.1097/JGP.0b013e3181cabcf6
Pagani, L., and Eckert, A. (2011). Amyloid-Beta interaction with mitochondria. Int. J. Alzheimers Dis. 2011, 925050. doi: 10.4061/2011/925050
Pakrasi, S., Colloby, S. J., Firbank, M. J., Perry, E. K., Wyper, D. J., Owens, J., et al. (2007). Muscarinic acetylcholine receptor status in Alzheimer’s disease assessed using (R, R) 123I-QNB SPECT. J. Neurol. 254, 907–913. doi: 10.1007/s00415-006-04738
Palacios, J. M., Bolliger, G., Closse, A., Enz, A., Gmelin, G., and Malanowski, J. (1986). The pharmacological assessment of RS 86 (2-ethyl-8-methyl-2,8-diazaspiro-[4,5]-decan-1,3-dion hydrobromide). A potent, specific muscarinic acetylcholine receptor agonist. Eur. J. Pharmacol. 125, 45–62. doi: 10.1016/0014-2999(86)90082-8
Palacios, J. M., Mengod, G., Vilaro, M. T., Wiederhold, K. H., Boddeke, H., Alvarez, F. J., et al. (1990). Cholinergic receptors in the rat and human brain: microscopic visualization. Prog. Brain Res. 84, 243–253. doi: 10.1016/S0079-6123(08)60909-7
Panza, F., Frisardi, V., Solfrizzi, V., Imbimbo, B. P., Logroscino, G., Santamato, A., et al. (2012). Immunotherapy for Alzheimer’s disease: from anti-beta-amyloid to tau-based immunization strategies. Immunotherapy 4, 213–238. doi: 10.2217/imt.11.170
Papadopoulos, V., Baraldi, M., Guilarte, T. R., Knudsen, T. B., Lacapere, J. J., Lindemann, P., et al. (2006a). Translocator protein (18kDa): new nomenclature for the peripheral-type benzodiazepine receptor based on its structure and molecular function. Trends Pharmacol. Sci. 27, 402–409. doi: 10.1016/j.tips.2006.06.005
Papadopoulos, V., Lecanu, L., Brown, R. C., Han, Z., and Yao, Z. X. (2006b). Peripheral-type benzodiazepine receptor in neurosteroid biosynthesis, neuropathology and neurological disorders. Neuroscience 138, 749–756. doi: 10.1016/j.neuroscience.2005.05.063
Pappata, S., Varrone, A., Vicidomini, C., Milan, G., De, F. C., Sansone, V., et al. (2010). SPECT imaging of GABA(A)/benzodiazepine receptors and cerebral perfusion in mild cognitive impairment. Eur. J. Nucl. Med. Mol. Imaging 37, 1156–1163. doi: 10.1007/s00259-010-1409-1
Passchier, J., Gee, A., Willemsen, A., Vaalburg, W., and van, W. A. (2002). Measuring drug-related receptor occupancy with positron emission tomography. Methods 27, 278–286. doi: 10.1016/S1046-2023(02)00084-1
Paterson, D., and Nordberg, A. (2000). Neuronal nicotinic receptors in the human brain. Prog. Neurobiol. 61, 75–111. doi: 10.1016/S0301-0082(99)00045-3
Paterson, L. M., Kornum, B. R., Nutt, D. J., Pike, V. W., and Knudsen, G. M. (2013). 5-HT radioligands for human brain imaging with PET and SPECT. Med. Res. Rev. 33, 54–111. doi: 10.1002/med.20245
Peralta, E. G., Ashkenazi, A., Winslow, J. W., Smith, D. H., Ramachandran, J., and Capon, D. J. (1987). Distinct primary structures, ligand-binding properties and tissue-specific expression of four human muscarinic acetylcholine receptors. EMBO J. 6, 3923–3929.
Piggott, M., Owens, J., O’Brien, J., Paling, S., Wyper, D., Fenwick, J., et al. (2002). Comparative distribution of binding of the muscarinic receptor ligands pirenzepine, AF-DX 384, (R,R)-I-QNB and (R,S)-I-QNB to human brain. J. Chem. Neuroanat. 24, 211–223. doi: 10.1016/S0891-0618(02)00066-2
Piggott, M. A., Ballard, C. G., Rowan, E., Holmes, C., McKeith, I. G., Jaros, E., et al. (2007). Selective loss of dopamine D2 receptors in temporal cortex in dementia with Lewy bodies, association with cognitive decline. Synapse 61, 903–911. doi: 10.1002/syn.20441
Piggott, M. A., Marshall, E. F., Thomas, N., Lloyd, S., Court, J. A., Jaros, E., et al. (1999). Striatal dopaminergic markers in dementia with Lewy bodies, Alzheimer’s and Parkinson’s diseases: rostrocaudal distribution. Brain 122(Pt 8), 1449–1468. doi: 10.1093/brain/122.8.1449
Pimlott, S. L., Piggott, M., Owens, J., Greally, E., Court, J. A., Jaros, E., et al. (2004). Nicotinic acetylcholine receptor distribution in Alzheimer’s disease, dementia with Lewy bodies, Parkinson’s disease, and vascular dementia: in vitro binding study using 5-[(125)i]-a-85380. Neuropsychopharmacology 29, 108–116. doi: 10.1038/sj.npp.1300302
Pizzolato, G., Chierichetti, F., Fabbri, M., Cagnin, A., Dam, M., Ferlin, G., et al. (1996). Reduced striatal dopamine receptors in Alzheimer’s disease: single photon emission tomography study with the D2 tracer [123I]-IBZM. Neurology 47, 1065–1068. doi: 10.1212/WNL.47.4.1065
Polidori, M. C., and Nelles, G. (2014). Antioxidant clinical trials in mild cognitive impairment and Alzheimer’s disease - challenges and perspectives. Curr. Pharm. Des. 20, 3083–3092. doi: 10.2174/13816128113196660706
Portet, F., Scarmeas, N., Cosentino, S., Helzner, E. P., and Stern, Y. (2009). Extrapyramidal signs before and after diagnosis of incident Alzheimer disease in a prospective population study. Arch. Neurol. 66, 1120–1126. doi: 10.1001/archneurol.2009.196
Potter, A., Corwin, J., Lang, J., Piasecki, M., Lenox, R., and Newhouse, P. A. (1999). Acute effects of the selective cholinergic channel activator (nicotinic agonist) ABT-418 in Alzheimer’s disease. Psychopharmacology (Berl.) 142, 334–342. doi: 10.1007/s002130050897
Procter, A. W., Lowe, S. L., Palmer, A. M., Francis, P. T., Esiri, M. M., Stratmann, G. C., et al. (1988). Topographical distribution of neurochemical changes in Alzheimer’s disease. J. Neurol. Sci. 84, 125–140. doi: 10.1016/0022-510X(88)90118-9
Puzzo, D., and Arancio, O. (2013). Amyloid-beta peptide: Dr. Jekyll or Mr. Hyde? J. Alzheimers Dis. 33(Suppl. 1), S111–S120.
Pym, L., Kemp, M., Raymond-Delpech, V., Buckingham, S., Boyd, C. A., and Sattelle, D. (2005). Subtype-specific actions of beta-amyloid peptides on recombinant human neuronal nicotinic acetylcholine receptors (alpha7, alpha4beta2, alpha3beta4) expressed in Xenopus laevis oocytes. Br. J. Pharmacol. 146, 964–971. doi: 10.1038/sj.bjp.0706403
Qosa, H., Batarseh, Y. S., Mohyeldin, M. M., El Sayed, K. A., Keller, J. N., and Kaddoumi, A. (2015). Oleocanthal enhances amyloid-beta clearance from the brains of TgSwDI mice and in vitro across a human blood-brain barrier model. ACS Chem. Neurosci. 6, 1849–1859. doi: 10.1021/acschemneuro.5b00190
Rabinovici, G. D., Furst, A. J., O’Neil, J. P., Racine, C. A., Mormino, E. C., Baker, S. L., et al. (2007). 11C-PIB PET imaging in Alzheimer disease and frontotemporal lobar degeneration. Neurology 68, 1205–1212. doi: 10.1212/01.wnl.0000259035.98480.ed
Rabinovici, G. D., Jagust, W. J., Furst, A. J., Ogar, J. M., Racine, C. A., Mormino, E. C., et al. (2008). Abeta amyloid and glucose metabolism in three variants of primary progressive aphasia. Ann. Neurol. 64, 388–401. doi: 10.1002/ana.21451
Rafii, M. S., Walsh, S., Little, J. T., Behan, K., Reynolds, B., Ward, C., et al. (2011). A phase II trial of huperzine A in mild to moderate Alzheimer disease. Neurology 76, 1389–1394. doi: 10.1212/WNL.0b013e318216eb7b
Rahmim, A., and Zaidi, H. (2008). PET versus SPECT: strengths, limitations and challenges. Nucl. Med. Commun. 29, 193–207. doi: 10.1097/MNM.0b013e3282f3a515
Ramirez, M. J., Lai, M. K., Tordera, R. M., and Francis, P. T. (2014). Serotonergic therapies for cognitive symptoms in Alzheimer’s disease: rationale and current status. Drugs 74, 729–736. doi: 10.1007/s40265-014-02175
Raskind, M. A., Peskind, E. R., Wessel, T., and Yuan, W. (2000). Galantamine in AD: a 6-month randomized, placebo-controlled trial with a 6-month extension. The Galantamine USA-1 Study Group. Neurology 54, 2261–2268. doi: 10.1212/WNL.54.12.2261
Ravasi, L., Tokugawa, J., Nakayama, T., Seidel, J., Sokoloff, L., Eckelman, W. C., et al. (2012). Imaging of the muscarinic acetylcholine neuroreceptor in rats with the M2 selective agonist [18F]FP-TZTP. Nucl. Med. Biol. 39, 45–55. doi: 10.1016/j.nucmedbio.2011.06.003
Reddy, P. H., and Beal, M. F. (2008). Amyloid beta, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol. Med. 14, 45–53. doi: 10.1016/j.molmed.2007.12.002
Reeves, S., Brown, R., Howard, R., and Grasby, P. (2009). Increased striatal dopamine (D2/D3) receptor availability and delusions in Alzheimer disease. Neurology 72, 528–534. doi: 10.1212/01.wnl.0000341932.21961.f3
Riederer, P., Danielczyk, W., and Grunblatt, E. (2004). Monoamine oxidase-B inhibition in Alzheimer’s disease. Neurotoxicology 25, 271–277. doi: 10.1016/S0161-813X(03)001062
Riederer, P., Youdim, M. B., Rausch, W. D., Birkmayer, W., Jellinger, K., and Seemann, D. (1978). On the mode of action of L-deprenyl in the human central nervous system. J. Neural Transm. 43, 217–226. doi: 10.1007/BF01246958
Rigacci, S. (2015). Olive oil phenols as promising multi-targeting agents against Alzheimer’s disease. Adv. Exp. Med. Biol. 863, 1–20. doi: 10.1007/978-3-319-18365-7_1
Rinne, J. O., Sahlberg, N., Ruottinen, H., Nagren, K., and Lehikoinen, P. (1998). Striatal uptake of the dopamine reuptake ligand [11C]beta-CFT is reduced in Alzheimer’s disease assessed by positron emission tomography. Neurology 50, 152–156. doi: 10.1212/WNL.50.1.152
Rishton, G. M. (2008). Aggregator compounds confound amyloid fibrillization assay. Nat. Chem. Biol. 4, 159–160. doi: 10.1038/nchembio0308-159
Rissman, R. A., De Blas, A. L., and Armstrong, D. M. (2007). GABA(A) receptors in aging and Alzheimer’s disease. J. Neurochem. 103, 1285–1292. doi: 10.1111/j.1471-4159.2007.04832.x
Roberds, S. L., Anderson, J., Basi, G., Bienkowski, M. J., Branstetter, D. G., Chen, K. S., et al. (2001). BACE knockout mice are healthy despite lacking the primary beta-secretase activity in brain: implications for Alzheimer’s disease therapeutics. Hum. Mol. Genet. 10, 1317–1324. doi: 10.1093/hmg/10.12.1317
Robert, P. H., and Benoit, M. (2008). Neurochemistry of cognition: serotonergic and adrenergic mechanisms. Handb. Clin. Neurol. 88, 31–40. doi: 10.1016/S0072-9752(07)88002-X
Rodriguez, J. J., Noristani, H. N., and Verkhratsky, A. (2012). The serotonergic system in ageing and Alzheimer’s disease. Prog. Neurobiol. 99, 15–41. doi: 10.1016/j.pneurobio.2012.06.010
Rodríguez-Puertas, R., Pascual, J., Vilaro, T., and Pazos, A. (1997). Autoradiographic distribution of M1, M2, M3, and M4 muscarinic receptor subtypes in Alzheimer’s disease. Synapse 26, 341–350. doi: 10.1002/(SICI)1098-2396(199708)26:4<341::AID-SYN2>3.0.CO;2-6
Romano, M., and Buratti, E. (2013). Florbetapir F 18 for brain imaging of beta-amyloid plaques. Drugs Today (Barc.) 49, 181–193. doi: 10.1358/dot.2013.49.3.1937428
Rosler, M., Anand, R., Cicin-Sain, A., Gauthier, S., Agid, Y., Dal-Bianco, P., et al. (1999). Efficacy and safety of rivastigmine in patients with Alzheimer’s disease: international randomised controlled trial. BMJ 318, 633–638. doi: 10.1136/bmj.318.7184.633
Rowe, C. C., Ackerman, U., Browne, W., Mulligan, R., Pike, K. L., O’Keefe, G., et al. (2008). Imaging of amyloid beta in Alzheimer’s disease with 18F-BAY94-9172, a novel PET tracer: proof of mechanism. Lancet Neurol. 7, 129–135. doi: 10.1016/S1474-4422(08)70001-2
Rowe, C. C., Ng, S., Ackermann, U., Gong, S. J., Pike, K., Savage, G., et al. (2007). Imaging beta-amyloid burden in aging and dementia. Neurology 68, 1718–1725. doi: 10.1212/01.wnl.0000261919.22630.ea
Rowe, C. C., Pejoska, S., Mulligan, R. S., Jones, G., Chan, J. G., Svensson, S., et al. (2013). Head-to-head comparison of 11C-PiB and 18F-AZD4694 (NAV4694) for beta-amyloid imaging in aging and dementia. J. Nucl. Med. 54, 880–886. doi: 10.2967/jnumed.112.114785
Rowe, C. C., and Villemagne, V. L. (2013). Brain amyloid imaging. J. Nucl. Med. Technol. 41, 11–18. doi: 10.2967/jnumed.110.076315
Rudolph, U., and Mohler, H. (2006). GABA-based therapeutic approaches: GABAA receptor subtype functions. Curr. Opin. Pharmacol. 6, 18–23. doi: 10.1016/j.coph.2005.10.003
Rupprecht, R., Rammes, G., Eser, D., Baghai, T. C., Schule, C., Nothdurfter, C., et al. (2009). Translocator protein (18 kD) as target for anxiolytics without benzodiazepine-like side effects. Science 325, 490–493. doi: 10.1126/science.1175055
Sabbagh, M. N. (2009). Drug development for Alzheimer’s disease: where are we now and where are we headed? Am. J. Geriatr. Pharmacother. 7, 167–185. doi: 10.1016/j.amjopharm.2009.06.003
Sabri, O., Kendziorra, K., Wolf, H., Gertz, H. J., and Brust, P. (2008). Acetylcholine receptors in dementia and mild cognitive impairment. Eur. J. Nucl. Med. Mol. Imaging 35(Suppl. 1), S30–S45. doi: 10.1007/s00259-007-0701-1
Sadigh-Eteghad, S., Majdi, A., Farhoudi, M., Talebi, M., and Mahmoudi, J. (2014). Different patterns of brain activation in normal aging and Alzheimer’s disease from cognitional sight: meta analysis using activation likelihood estimation. J. Neurol. Sci. 343, 159–166. doi: 10.1016/j.jns.2014.05.066
Santa-Maria, I., Hernandez, F., Del, R. J., Moreno, F. J., and Avila, J. (2007). Tramiprosate, a drug of potential interest for the treatment of Alzheimer’s disease, promotes an abnormal aggregation of tau. Mol. Neurodegener. 2:17. doi: 10.1186/1750-1326-2-17
Santhosh, L., Estok, K. M., Vogel, R. S., Tamagnan, G. D., Baldwin, R. M., Mitsis, E. M., et al. (2009). Regional distribution and behavioral correlates of 5-HT(2A) receptors in Alzheimer’s disease with [(18)F]deuteroaltanserin and PET. Psychiatry Res. 173, 212–217. doi: 10.1016/j.pscychresns.2009.03.007
Saulin, A., Savli, M., and Lanzenberger, R. (2012). Serotonin and molecular neuroimaging in humans using PET. Amino Acids 42, 2039–2057. doi: 10.1007/s00726-011-10789
Saura, J., Luque, J. M., Cesura, A. M., Da, P. M., Chan-Palay, V., Huber, G., et al. (1994). Increased monoamine oxidase B activity in plaque-associated astrocytes of Alzheimer brains revealed by quantitative enzyme radioautography. Neuroscience 62, 15–30. doi: 10.1016/0306-4522(94)90311-5
Savic, I., Persson, A., Roland, P., Pauli, S., Sedvall, G., and Widen, L. (1988). In-vivo demonstration of reduced benzodiazepine receptor binding in human epileptic foci. Lancet 2, 863–866. doi: 10.1016/S0140-6736(88)92468-3
Savonenko, A., Xu, G. M., Melnikova, T., Morton, J. L., Gonzales, V., Wong, M. P., et al. (2005). Episodic-like memory deficits in the APPswe/PS1dE9 mouse model of Alzheimer’s disease: relationships to beta-amyloid deposition and neurotransmitter abnormalities. Neurobiol. Dis. 18, 602–617. doi: 10.1016/j.nbd.2004.10.022
Schenk, D., Barbour, R., Dunn, W., Gordon, G., Grajeda, H., Guido, T., et al. (1999). Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 400, 173–177. doi: 10.1038/22124
Shah, M., and Catafau, A. M. (2014). Molecular imaging insights into neurodegeneration: focus on tau PET radiotracers. J. Nucl. Med. 55, 871–874. doi: 10.2967/jnumed.113.136069
Shinotoh, H., Aotsuka, A., Fukushi, K., Nagatsuka, S., Tanaka, N., Ota, T., et al. (2001). Effect of donepezil on brain acetylcholinesterase activity in patients with AD measured by PET. Neurology 56, 408–410. doi: 10.1212/WNL.56.3.408
Shinotoh, H., Fukushi, K., Nagatsuka, S., and Irie, T. (2004). Acetylcholinesterase imaging: its use in therapy evaluation and drug design. Curr. Pharm. Des. 10, 1505–1517. doi: 10.2174/1381612043384763
Shugart, J. (2016). Rare but Severe Side Effects Sideline Some Phase 3 Encenicline Trials. Alzforum. Available at: http://www.alzforum.org/news/research-news/rare-severe-side-effects-sideline-some-phase-3-encenicline-trials5-2-2016
Small, G. W., Kepe, V., Ercoli, L. M., Siddarth, P., Bookheimer, S. Y., Miller, K. J., et al. (2006). PET of brain amyloid and tau in mild cognitive impairment. N. Engl. J. Med. 355, 2652–2663. doi: 10.1056/NEJMoa054625
Small, G. W., Kepe, V., Siddarth, P., Ercoli, L. M., Merrill, D. A., Donoghue, N., et al. (2013). PET scanning of brain tau in retired national football league players: preliminary findings. Am. J. Geriatr. Psychiatry 21, 138–144. doi: 10.1016/j.jagp.2012.11.019
Snaedal, J., Johannesson, T., Jonsson, J. E., and Gylfadottir, G. (1996). The effects of nicotine in dermal plaster on cognitive functions in patients with Alzheimer’s disease. Dementia 7, 47–52.
Sonsalla, P. K., and Golbe, L. I. (1988). Deprenyl as prophylaxis against Parkinson’s disease? Clin. Neuropharmacol. 11, 500Ű511.
Soricelli, A., Postiglione, A., Grivet-Fojaja, M. R., Mainenti, P. P., Discepolo, A., Varrone, A., et al. (1996). Reduced cortical distribution volume of iodine-123 iomazenil in Alzheimer’s disease as a measure of loss of synapses. Eur. J. Nucl. Med. 23, 1323–1328. doi: 10.1007/BF01367587
Sparks, D. L., Woeltz, V. M., and Markesbery, W. R. (1991). Alterations in brain monoamine oxidase activity in aging, Alzheimer’s disease, and Pick’s disease. Arch. Neurol. 48, 718–721. doi: 10.1001/archneur.1991.00530190064017
Spehl, T. S., Frings, L., Hellwig, S., Weiller, C., Hull, M., Meyer, P. T., et al. (2015). Role of semiquantitative assessment of regional binding potential in 123I-FP-CIT SPECT for the differentiation of frontotemporal dementia, dementia with Lewy bodies, and Alzheimer’s dementia. Clin. Nucl. Med. 40, e27–e33. doi: 10.1097/RLU.0000000000000554
Spencer, B., and Masliah, E. (2014). Immunotherapy for Alzheimer’s disease: past, present and future. Front. Aging Neurosci. 6:114. doi: 10.3389/fnagi.2014.00114
Stefaniak, J., and O’Brien, J. (2016). Imaging of neuroinflammation in dementia: a review. J. Neurol. Neurosurg. Psychiatry 87, 21–28. doi: 10.1136/jnnp-2015-311336
Storga, D., Vrecko, K., Birkmayer, J. G., and Reibnegger, G. (1996). Monoaminergic neurotransmitters, their precursors and metabolites in brains of Alzheimer patients. Neurosci. Lett. 203, 29–32. doi: 10.1016/0304-3940(95)12256-7
Streit, W. J. (2010). Microglial activation and neuroinflammation in Alzheimer’s disease: a critical examination of recent history. Front. Aging Neurosci. 2:22. doi: 10.3389/fnagi.2010.00022
Strolin, B. M., and Dostert, P. (1989). Monoamine oxidase, brain ageing and degenerative diseases. Biochem. Pharmacol. 38, 555–561. doi: 10.1016/0006-2952(89)90198-6
Sunderland, T., Weingartner, H., Cohen, R. M., Tariot, P. N., Newhouse, P. A., Thompson, K. E., et al. (1989). Low-dose oral lorazepam administration in Alzheimer subjects and age-matched controls. Psychopharmacology (Berl.) 99, 129–133. doi: 10.1007/BF00634466
Syvanen, S., and Eriksson, J. (2013). Advances in PET imaging of P-glycoprotein function at the blood-brain barrier. ACS Chem. Neurosci. 4, 225–237. doi: 10.1021/cn3001729
Tanaka, Y., Meguro, K., Yamaguchi, S., Ishii, H., Watanuki, S., Funaki, Y., et al. (2003). Decreased striatal D2 receptor density associated with severe behavioral abnormality in Alzheimer’s disease. Ann. Nucl. Med. 17, 567–573. doi: 10.1007/BF03006670
Tandon, R., Shipley, J. E., Greden, J. F., Mann, N. A., Eisner, W. H., and Goodson, J. A. (1991). Muscarinic cholinergic hyperactivity in schizophrenia. Relationship to positive and negative symptoms. Schizophr. Res. 4, 23–30. doi: 10.1016/0920-9964(91)90006-D
Tariot, P. N., and Aisen, P. S. (2009). Can lithium or valproate untie tangles in Alzheimer’s disease? J. Clin. Psychiatry 70, 919–921. doi: 10.4088/JCP.09com05331
Tariot, P. N., Schneider, L. S., Cummings, J., Thomas, R. G., Raman, R., Jakimovich, L. J., et al. (2011). Chronic divalproex sodium to attenuate agitation and clinical progression of Alzheimer disease. Arch. Gen. Psychiatry 68, 853–861. doi: 10.1001/archgenpsychiatry.2011.72
Tariot, P. N., Solomon, P. R., Morris, J. C., Kershaw, P., Lilienfeld, S., and Ding, C. (2000). A 5-month, randomized, placebo-controlled trial of galantamine in AD. The Galantamine USA-10 Study Group. Neurology 54, 2269–2276. doi: 10.1212/WNL.54.12.2269
Tavitian, B., Pappata, S., Bonnot-Lours, S., Prenant, C., Jobert, A., Crouzel, C., et al. (1993). Positron emission tomography study of [11C]methyl-tetrahydroaminoacridine (methyl-tacrine) in baboon brain. Eur. J. Pharmacol. 236, 229–238. doi: 10.1016/0014-2999(93)90593-7
Terry, A. V. Jr., and Buccafusco, J. J. (2003). The cholinergic hypothesis of age and Alzheimer’s disease-related cognitive deficits: recent challenges and their implications for novel drug development. J. Pharmacol. Exp. Ther. 306, 821–827. doi: 10.1124/jpet.102.041616
Thies, W., and Bleiler, L. (2013). 2013 Alzheimer’s disease facts and figures. Alzheimers Dement. 9, 208–245. doi: 10.1016/j.jalz.2013.02.003
Thomas, T. (2000). Monoamine oxidase-B inhibitors in the treatment of Alzheimer’s disease. Neurobiol. Aging 21, 343–348. doi: 10.1016/S0197-4580(00)00100-7
Tolboom, N., van der Flier, W. M., Yaqub, M., Boellaard, R., Verwey, N. A., Blankenstein, M. A., et al. (2009). Relationship of cerebrospinal fluid markers to 11C-PiB and 18F-FDDNP binding. J. Nucl. Med. 50, 1464–1470. doi: 10.2967/jnumed.109.064360
Tomasi, G., Turkheimer, F., and Aboagye, E. (2012). Importance of quantification for the analysis of PET data in oncology: review of current methods and trends for the future. Mol. Imaging Biol. 14, 131–146. doi: 10.1007/s11307-011-05142
Toyohara, J., Wu, J., and Hashimoto, K. (2010). Recent development of radioligands for imaging alpha7 nicotinic acetylcholine receptors in the brain. Curr. Top. Med. Chem. 10, 1544–1557. doi: 10.2174/156802610793176828
Trillo, L., Das, D., Hsieh, W., Medina, B., Moghadam, S., Lin, B., et al. (2013). Ascending monoaminergic systems alterations in Alzheimer’s disease. translating basic science into clinical care. Neurosci. Biobehav. Rev. 37, 1363–1379. doi: 10.1016/j.neubiorev.2013.05.008
Trollor, J. N., Sachdev, P. S., Haindl, W., Brodaty, H., Wen, W., and Walker, B. M. (2006). Combined cerebral blood flow effects of a cholinergic agonist (milameline) and a verbal recognition task in early Alzheimer’s disease. Psychiatry Clin. Neurosci. 60, 616–625. doi: 10.1111/j.1440-1819.2006.01567.x
Truchot, L., Costes, N., Zimmer, L., Laurent, B., Le, B. D., Thomas-Anterion, C., et al. (2008). A distinct [18F]MPPF PET profile in amnestic mild cognitive impairment compared to mild Alzheimer’s disease. Neuroimage 40, 1251–1256. doi: 10.1016/j.neuroimage.2008.01.030
Tsang, S. W., Keene, J., Hope, T., Spence, I., Francis, P. T., Wong, P. T., et al. (2010). A serotoninergic basis for hyperphagic eating changes in Alzheimer’s disease. J. Neurol. Sci. 288, 151–155. doi: 10.1016/j.jns.2009.08.066
Tsang, S. W., Lai, M. K., Kirvell, S., Francis, P. T., Esiri, M. M., Hope, T., et al. (2006). Impaired coupling of muscarinic M1 receptors to G-proteins in the neocortex is associated with severity of dementia in Alzheimer’s disease. Neurobiol. Aging 27, 1216–1223. doi: 10.1016/j.neurobiolaging.2005.07.010
Tyrrell, P. J., Sawle, G. V., Ibanez, V., Bloomfield, P. M., Leenders, K. L., Frackowiak, R. S., et al. (1990). Clinical and positron emission tomographic studies in the ‘extrapyramidal syndrome’ of dementia of the Alzheimer type. Arch. Neurol. 47, 1318–1323. doi: 10.1001/archneur.1990.00530120062011
Valles, A. S., and Barrantes, F. J. (2012). Chaperoning alpha7 neuronal nicotinic acetylcholine receptors. Biochim. Biophys. Acta 1818, 718–729. doi: 10.1016/j.bbamem.2011.10.012
Vandenberghe, R., Adamczuk, K., and Van, L. K. (2013). The interest of amyloid PET imaging in the diagnosis of Alzheimer’s disease. Curr. Opin. Neurol. 26, 646–655. doi: 10.1097/WCO.0000000000000036
Vandenberghe, R., Van, L. K., Ivanoiu, A., Salmon, E., Bastin, C., Triau, E., et al. (2010). 18F-flutemetamol amyloid imaging in Alzheimer disease and mild cognitive impairment: a phase 2 trial. Ann. Neurol. 68, 319–329. doi: 10.1002/ana.22068
Varley, J., Brooks, D. J., and Edison, P. (2015). Imaging neuroinflammation in Alzheimer’s disease and other dementias: recent advances and future directions. Alzheimers Dement. 11, 1110–1120. doi: 10.1016/j.jalz.2014.08.105
Venigalla, M., Gyengesi, E., Sharman, M. J., and Munch, G. (2015). Novel promising therapeutics against chronic neuroinflammation and neurodegeneration in Alzheimer’s disease. Neurochem. Int. doi: 10.1016/j.neuint.2015.10.011 [Epub ahead of print].
Venneti, S., Lopresti, B. J., and Wiley, C. A. (2006). The peripheral benzodiazepine receptor (Translocator protein 18kDa) in microglia: from pathology to imaging. Prog. Neurobiol. 80, 308–322. doi: 10.1016/j.pneurobio.2006.10.002
Versijpt, J., Van Laere, K. J., Dumont, F., Decoo, D., Vandecapelle, M., Santens, P., et al. (2003). Imaging of the 5-HT2A system: age-, gender-, and Alzheimer’s disease-related findings. Neurobiol. Aging 24, 553–561. doi: 10.1016/S0197-4580(02)00137-9
Vertes, R. P. (1991). A PHA-L analysis of ascending projections of the dorsal raphe nucleus in the rat. J. Comp Neurol. 313, 643–668. doi: 10.1002/cne.903130409
Vertes, R. P., Fortin, W. J., and Crane, A. M. (1999). Projections of the median raphe nucleus in the rat. J. Comp Neurol. 407, 555–582. doi: 10.1002/(SICI)1096-9861(19990517)407:4<555::AID-CNE7>3.0.CO;2-E
Villemagne, V. L., Fodero-Tavoletti, M. T., Masters, C. L., and Rowe, C. C. (2015). Tau imaging: early progress and future directions. Lancet Neurol. 14, 114–124. doi: 10.1016/S1474-4422(14)702522
Villemagne, V. L., Furumoto, S., Fodero-Tavoletti, M. T., Mulligan, R. S., Hodges, J., Harada, R., et al. (2014). In vivo evaluation of a novel tau imaging tracer for Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 41, 816–826. doi: 10.1007/s00259-013-2681-7
Villemagne, V. L., Ong, K., Mulligan, R. S., Holl, G., Pejoska, S., Jones, G., et al. (2011). Amyloid imaging with (18)F-florbetaben in Alzheimer disease and other dementias. J. Nucl. Med. 52, 1210–1217. doi: 10.2967/jnumed.111.089730
Villemagne, V. L., Pike, K. E., Darby, D., Maruff, P., Savage, G., Ng, S., et al. (2008). Abeta deposits in older non-demented individuals with cognitive decline are indicative of preclinical Alzheimer’s disease. Neuropsychologia 46, 1688–1697. doi: 10.1016/j.neuropsychologia.2008.02.008
Vogels, O. J., Broere, C. A., ter Laak, H. J., ten Donkelaar, H. J., Nieuwenhuys, R., and Schulte, B. P. (1990). Cell loss and shrinkage in the nucleus basalis Meynert complex in Alzheimer’s disease. Neurobiol. Aging 11, 3–13. doi: 10.1016/0197-4580(90)90056-6
Wallin, A. K., Blennow, K., Andreasen, N., and Minthon, L. (2006). CSF biomarkers for Alzheimer’s disease: levels of beta-amyloid, tau, phosphorylated tau relate to clinical symptoms and survival. Dement. Geriatr. Cogn Disord. 21, 131–138. doi: 10.1159/000090631
Weigand, S. D., Vemuri, P., Wiste, H. J., Senjem, M. L., Pankratz, V. S., Aisen, P. S., et al. (2011). Transforming cerebrospinal fluid Abeta42 measures into calculated Pittsburgh Compound B units of brain Abeta amyloid. Alzheimers. Dement. 7, 133–141. doi: 10.1016/j.jalz.2010.08.230
Weinberger, D. R., Gibson, R., Coppola, R., Jones, D. W., Molchan, S., Sunderland, T., et al. (1991). The distribution of cerebral muscarinic acetylcholine receptors in vivo in patients with dementia. A controlled study with 123IQNB and single photon emission computed tomography. Arch. Neurol. 48, 169–176. doi: 10.1001/archneur.1991.00530140061018
Weiner, M. W., Veitch, D. P., Aisen, P. S., Beckett, L. A., Cairns, N. J., Cedarbaum, J., et al. (2015). Impact of the Alzheimer’s disease Neuroimaging Initiative, 2004 to 2014. Alzheimers. Dement. 11, 865–884. doi: 10.1016/j.jalz.2015.04.005
Weingarten, M. D., Lockwood, A. H., Hwo, S. Y., and Kirschner, M. W. (1975). A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. U.S.A. 72, 1858–1862. doi: 10.1073/pnas.72.5.1858
Weinreb, O., Amit, T., Bar-Am, O., and Youdim, M. B. (2010). Rasagiline: a novel anti-Parkinsonian monoamine oxidase-B inhibitor with neuroprotective activity. Prog. Neurobiol. 92, 330–344. doi: 10.1016/j.pneurobio.2010.06.008
Wevers, A., and Schroder, H. (1999). Nicotinic acetylcholine receptors in Alzheimer’s disease. J. Alzheimers Dis. 1, 207–219.
White, H. K., and Levin, E. D. (1999). Four-week nicotine skin patch treatment effects on cognitive performance in Alzheimer’s disease. Psychopharmacology (Berl.) 143, 158–165. doi: 10.1007/s002130050931
Wilcock, G. K., Lilienfeld, S., and Gaens, E. (2000). Efficacy and safety of galantamine in patients with mild to moderate Alzheimer’s disease: multicentre randomised controlled trial. Galantamine International-1 Study Group. BMJ 321, 1445–1449.
Wilson, A. L., Langley, L. K., Monley, J., Bauer, T., Rottunda, S., McFalls, E., et al. (1995). Nicotine patches in Alzheimer’s disease: pilot study on learning, memory, and safety. Pharmacol. Biochem. Behav. 51, 509–514. doi: 10.1016/0091-3057(95)00043-V
Winblad, B., Engedal, K., Soininen, H., Verhey, F., Waldemar, G., Wimo, A., et al. (2001). A 1-year, randomized, placebo-controlled study of donepezil in patients with mild to moderate AD. Neurology 57, 489–495. doi: 10.1212/WNL.57.3.489
Winblad, B., Giacobini, E., Frolich, L., Friedhoff, L. T., Bruinsma, G., Becker, R. E., et al. (2010). Phenserine efficacy in Alzheimer’s disease. J. Alzheimers Dis. 22, 1201–1208. doi: 10.3233/JAD-2010-101311
Wischik, C. M., Harrington, C. R., and Storey, J. M. (2014). Tau-aggregation inhibitor therapy for Alzheimer’s disease. Biochem. Pharmacol. 88, 529–539. doi: 10.1016/j.bcp.2013.12.008
Wischik, C. M., Staff, R. T., Wischik, D. J., Bentham, P., Murray, A. D., Storey, J. M., et al. (2015). Tau aggregation inhibitor therapy: an exploratory phase 2 study in mild or moderate Alzheimer’s disease. J. Alzheimers. Dis. 44, 705–720. doi: 10.3233/JAD-142874
Wolf, B. A., Wertkin, A. M., Jolly, Y. C., Yasuda, R. P., Wolfe, B. B., Konrad, R. J., et al. (1995). Muscarinic regulation of Alzheimer’s disease amyloid precursor protein secretion and amyloid beta-protein production in human neuronal NT2N cells. J. Biol. Chem. 270, 4916–4922. doi: 10.1074/jbc.270.9.4916
Wu, T. Y., Chen, C. P., and Jinn, T. R. (2010). Alzheimer’s disease: aging, insomnia and epigenetics. Taiwan. J. Obstet. Gynecol. 49, 468–472. doi: 10.1016/S1028-4559(10)60099-X
Wurtman, R. (2015). Biomarkers in the diagnosis and management of Alzheimer’s disease. Metabolism 64, S47–S50. doi: 10.1016/j.metabol.2014.10.034
Wyper, D. J., Brown, D., Patterson, J., Owens, J., Hunter, R., Teasdale, E., et al. (1993). Deficits in iodine-labelled 3-quinuclidinyl benzilate binding in relation to cerebral blood flow in patients with Alzheimer’s disease. Eur. J. Nucl. Med. 20, 379–386. doi: 10.1007/BF00208995
Xia, C. F., Arteaga, J., Chen, G., Gangadharmath, U., Gomez, L. F., Kasi, D., et al. (2013). [(18)F]T807, a novel tau positron emission tomography imaging agent for Alzheimer’s disease. Alzheimers Dement. 9, 666–676. doi: 10.1016/j.jalz.2012.11.008
Xie, G., Gunn, R. N., Dagher, A., Daloze, T., Plourde, G., Backman, S. B., et al. (2004). PET quantification of muscarinic cholinergic receptors with [N-11C-methyl]-benztropine and application to studies of propofol-induced unconsciousness in healthy human volunteers. Synapse 51, 91–101. doi: 10.1002/syn.10292
Xu, Y., Yan, J., Zhou, P., Li, J., Gao, H., Xia, Y., et al. (2012). Neurotransmitter receptors and cognitive dysfunction in Alzheimer’s disease and Parkinson’s disease. Prog. Neurobiol. 97, 1–13. doi: 10.1016/j.pneurobio.2012.02.002
Yamada, M., Lamping, K. G., Duttaroy, A., Zhang, W., Cui, Y., Bymaster, F. P., et al. (2001). Cholinergic dilation of cerebral blood vessels is abolished in M(5) muscarinic acetylcholine receptor knockout mice. Proc. Natl. Acad. Sci. U.S.A. 98, 14096–14101. doi: 10.1073/pnas.251542998
Yang, L., Rieves, D., and Ganley, C. (2012). Brain amyloid imaging–FDA approval of florbetapir F18 injection. N. Engl. J. Med. 367, 885–887. doi: 10.1056/NEJMp1208061
Yiannopoulou, K. G., and Papageorgiou, S. G. (2013). Current and future treatments for Alzheimer’s disease. Ther. Adv. Neurol. Disord. 6, 19–33. doi: 10.1177/1756285612461679
Youdim, M. B. (2013). Multi target neuroprotective and neurorestorative anti-Parkinson and anti-Alzheimer drugs ladostigil and m30 derived from rasagiline. Exp. Neurobiol. 22, 1–10. doi: 10.5607/en.2013.22.1.1
Zec, R. F., and Burkett, N. R. (2008). Non-pharmacological and pharmacological treatment of the cognitive and behavioral symptoms of Alzheimer disease. NeuroRehabilitation 23, 425–438.
Zhang, W., Arteaga, J., Cashion, D. K., Chen, G., Gangadharmath, U., Gomez, L. F., et al. (2012). A highly selective and specific PET tracer for imaging of tau pathologies. J. Alzheimers Dis. 31, 601–612. doi: 10.3233/JAD-2012-120712
Zheng, H., Fridkin, M., and Youdim, M. B. (2012). From antioxidant chelators to site-activated multi-target chelators targeting hypoxia inducing factor, beta-amyloid, acetylcholinesterase and monoamine oxidase A/B. Mini Rev. Med. Chem. 12, 364–370. doi: 10.2174/138955712800493898
Zimmer, E. R., Leuzy, A., Gauthier, S., and Rosa-Neto, P. (2014). Developments in tau PET imaging. Can. J. Neurol. Sci. 41, 547–553. doi: 10.1017/cjn.2014.15
Keywords: Alzheimer’s disease, PET, SPECT, drug development, biomarkers
Citation: Declercq LD, Vandenberghe R, Van Laere K, Verbruggen A and Bormans G (2016) Drug Development in Alzheimer’s Disease: The Contribution of PET and SPECT. Front. Pharmacol. 7:88. doi: 10.3389/fphar.2016.00088
Received: 05 February 2016; Accepted: 16 March 2016;
Published: 31 March 2016.
Edited by:
Albert D. Windhorst, VU University Medical Center, NetherlandsReviewed by:
Bashir M. Rezk, Southern University at New Orleans, USACopyright © 2016 Declercq, Vandenberghe, Van Laere, Verbruggen and Bormans. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guy Bormans, Z3V5LmJvcm1hbnNAcGhhcm0ua3VsZXV2ZW4uYmU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.