 Rafael Franco
Rafael Franco Eva Martínez-Pinilla
Eva Martínez-Pinilla José L. Lanciego
José L. Lanciego Gemma Navarro
Gemma Navarro
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 31 March 2016
Sec. Experimental Pharmacology and Drug Discovery
Volume 7 - 2016 | https://doi.org/10.3389/fphar.2016.00076
Cell membrane receptors rarely work on isolation, often they form oligomeric complexes with other receptor molecules and they may directly interact with different proteins of the signal transduction machinery. For a variety of reasons, rhodopsin-like class A G-protein-coupled receptors (GPCRs) seem an exception to the general rule of receptor–receptor direct interaction. In fact, controversy surrounds their potential to form homo- hetero-dimers/oligomers with other class A GPCRs; in a sense, the field is going backward instead of forward. This review focuses on the convergent, complementary and telling evidence showing that homo- and heteromers of class A GPCRs exist in transfected cells and, more importantly, in natural sources. It is time to decide between questioning the occurrence of heteromers or, alternatively, facing the vast scientific and technical challenges that class A receptor-dimer/oligomer existence pose to Pharmacology and to Drug Discovery.
Historical views on any particular topic are both subjective and necessary. In the pre-G-protein-coupled receptor (GPCR) dimer era, pharmacological approaches of a variety of receptors attempted to know GPCR function while providing the tools for developing new drugs targeting GPCRs. Upon cloning, the classical pharmacological approach was challenged by functional studies in heterologous cells transfected with cDNAs coding for receptors. When the possibility of dimer occurrence was first suggested (Fuxe et al., 1983; Fuxe and Agnati, 1985) and, afterward, proved (vide infra), the previous working hypothesis and models were unsuitable to provide answers to the new questions. Intriguingly, the field is entering now in an unfruitful controversy instead of facing dimers to change the GPCR molecular physiology and pharmacological paradigms. Around 10 years after the (Prinster et al., 2005) comprehensive review on “specificity and functional significance” of GPCR hetero(di)merization and of the first IUPHAR recommendation on recognition and nomenclature of GPCR Hets, there is a need to reinforce the relevance of GPCR heteromerization (class A receptors included) for academic and for pharmaceutical purposes.
Although, GPCR homomerization provides advantages versus single/monomeric receptors (Banères and Parello, 2003; Corriden et al., 2014; Gherbi et al., 2015; Marsango et al., 2015), GPCR heteromerization gives added values for mammalians1, namely signaling versatility and diversity. For instance, whereas a dimer of a GPCR coupled to Gi would still be coupled to Gi, a heteromer constituted by two different receptors may couple to different signaling pathways than the individual receptors. Heteromerization in any context, i.e., T cell receptors, taste receptors, or adrenalin, dopamine, adenosine and opioid GPCRs, among others, entail selective advantages. As an example, receptor heteromers (Hets) are needed to taste many different flavors. Should not class A GPCRs heterodimers exist to provide a similar extra-added value (Franco, 2009)? One wonders why evolution could skip this straightforward signal decoding mechanism, but there is enough evidence to show that it is not the case. An exhaustive account of the selective advantages of class A GPCR heteromerization is out of the scope of the present article. From our laboratory we would select the adenosine A1–A2A receptor Het, which is a device able to sense the adenosine concentration and respond via Gi at low concentrations and via Gs at high concentrations (Ciruela et al., 2006; Ferré et al., 2007; Cristóvão-Ferreira et al., 2013). From other laboratories it is very difficult to choose but the coupling of the dopamine D1–D2 receptor Het to Gq (Rashid et al., 2007; So et al., 2007; Hasbi et al., 2009; George et al., 2014) when individual D1 or D2 receptors are coupled to, respectively, Gs or Gi, is worth mentioning. Also relevant is the finding of opioid receptor Hets that explain the strange pharmacology of opioid receptors and the atypical results obtained by drugs selectively targeting opioid receptor Hets, and that has helped to optimize the opioid receptor nomenclature (Gomes et al., 2000; Portoghese and Lunzer, 2003; Bhushan et al., 2004; Daniels et al., 2005; Waldhoer et al., 2005; van Rijn et al., 2010; Yekkirala et al., 2010; Gupta et al., 2014). Those few examples and the hundreds of already identified Hets contrast with the existing controversy on class A GPCR heteromerization.
Here, we will first compare the little but convincig evidence for taste receptor Hets with the similar but unconvincing evidence for heteromerization of two class A receptors (dopamine D1 and D2). Later the review selects a few Hets and a few techniques to build up examples of the varied, complementary and overwhelming evidence of class A GPCR heteromerization.
The three basic tastes, umami, sweet and bitter, are sensed by two types of specialized GPCRs, taste T1 and T22. T1 and T2 receptors are similar to, respectively, class C and class A GPCRs. IUPHAR indicates that taste T1 receptors are obligate Hets: “T1R3 acts as an obligate partner in T1R1/T1R3 and T1R2/T1R3 heterodimers, which sense umami or sweet, respectively. T1R1/T1R3 heterodimers respond to L-glutamic acid and may be positively allosterically modulated by 5′-nucleoside monophosphates, such as 5′-GMP [2]3. T1R2/T1R3 heterodimers respond to sugars, such as sucrose, and artificial sweeteners, such as saccharin (Nelson et al., 2001).” In this seminal paper referenced by IUPHAR, Nelson et al. (2001) using an heterologous expression system report “T1R2 and T1R3 combine to function as a sweet receptor.” Few years later, Hets for two class A dopamine receptors (D1 and D2) were identified using a quite similar experimental approach: “When dopamine D1 and D2 receptors were coactivated in D1-D2 receptor hetero-oligomeric complexes, a novel phospholipase C-mediated calcium signal was generated” (So et al., 2007). For both T1R1/R2/R3 and D1–D2 Hets, calcium mobilization was used as read-out, transfecting an engineered G protein in the case of taste receptors, and taking profit of endogenous Gq expressed in HEK-293T cells in the case of dopamine receptors. In the report of taste receptor heteromerization, transiently transfected cells were used, and in the report of dopamine receptor heteromerization, cells stably expressing D1 and D2 receptors were employed. Whereas taste T1 and class C GPCRs are considered Hets, class A receptor dimerization in general, or dopamine D1–D2 receptor Hets in particular are questioned in articles with titles such as: “GPCR dimers fall apart” (Lambert, 2010) or “Evidence against dopamine D1/D2 receptor heteromers” (Frederick et al., 2015). Reinforcing the idea of class A GPCR heteromerization is now due.
Class A taste T2 receptors are another example of the criteria used to accept dimerization. Although T2 receptors are often depicted as two molecules interacting together, IUPHAR does not support the dimeric view of these receptors. The main reason for such differential criteria is the big extracellular portion in T1. In fact, the most important difference between class A and C GPCRs is that the last ones have large extracellular agonist binding domains that may dimerize even in the absence of the rest of the receptor molecule (Kunishima et al., 2000; Romano et al., 2001; Kawahara et al., 2012). In contrast, the extracellular N-terminal domain of rhodopsin-like GPCRs is too short to be relevant for GPCR dimer formation. Historically, the discovery of class A GPCR dimers started by taking advantage of techniques used in the Immunology field for detecting interactions between membrane proteins, being the T-cell receptor one of the best studied (Figure 1). In this sense, antibody generation and co-immunoprecipitation constituted a revolution in Immunology and served to build up a molecular framework to understand antigen recognition and cell responses. Antibodies raised against GPCRs served to identify GPCRs by immunoblotting, and to label them in cells and tissues. They were also instrumental to identify homo- and heterodimers by immunoblotting and co-immunoprecipitation (Ciruela et al., 1995; Franco et al., 1996; Ginés et al., 2000; Gomes et al., 2000) and, more recently, by proximity ligation assays (Borroto-Escuela et al., 2011, 2012). Other techniques that are not based on antigen-antibody recognition have confirmed these early findings on GPCR homo- and heteromerization.
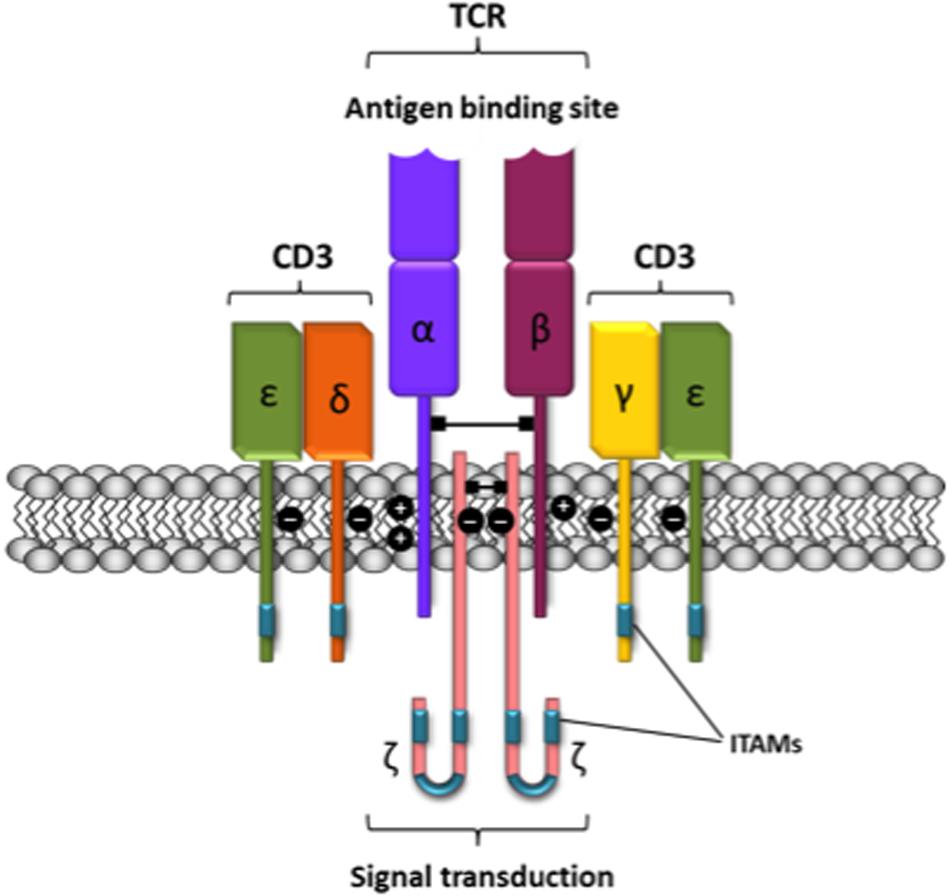
FIGURE 1. Schematic representation of the T-cell receptor-CD3 complex. The heterocomplex is formed by variable TCR-α and TCR-β chains coupled to three dimeric signaling transduction modules CD3δ/ε, CD3γ/ε and CD3ζ/ζ or CD247. CD3, Cluster of differentiation 3; CD247, cluster of differentiation 247 or CD3ζ/ζ; ITAM, immunoreceptor tyrosine-based activation motif; TCR, T-cell receptor.
The hypothesis of dimerization/oligomerization of GPCRs for neurotransmitters was put forward by Fuxe et al. (1983, 1987). However, technical difficulties prevented the discovery of GPCR dimers until the end of the 20th Century. Apart from diverse evidence on receptor homodimerization, and for demonstration that heteromerization was needed for cell surface expression and for GABAB receptor function (White et al., 1998), identification of first class A heterodimers came when Gomes et al. (2000) reported the first Het for two subtypes of opioid receptors μ and δ and when Ginés et al. (2000) identified the first Het of two receptors for two different neurotransmitter/neuromodulators, namely the adenosine A1 and dopamine D1 receptor Het.
As of 2014 the number of interactions between GPCRs was 537 (Borroto-Escuela et al., 2014). Proved contradictory results constitute approximately 1% of the total Hets, i.e., 5–10, and these exceptions have been reported in a given model but not found in a different model or using another assay type (Fuxe et al., 2014). This very low percentage of inconsistencies substantiates that GPCR and in particular class A GPCR receptor–receptor interactions appear as a robust discovery.
According to the rules of http://www.merriam-webster.com, the belief that GPCRs -or else- are monomers may be referred to as: monomerism. The Whorton et al. (2007) report was instrumental for a certain consolidation of this phenomenon. The authors showed that, in an artificial system, monomeric β2-adrenergic receptors may activate the coupled G protein. Such article has been frequently used in reviews and in thematic meetings to reinforce the idea that class A GPCRs should be monomers. On the one hand, it is likely that the lipoprotein particles used in the assays could not accommodate two GPCR molecules together to study differential properties of monomers versus homo- or hetero-dimers. On the other hand, an in vitro demonstration cannot be used to be certain about receptor operation in vivo. Neither the Whorton et al. (2007) data may scientifically substantiate authors’ assertion “cooperativity of agonist binding is due to G protein association to the receptor monomer and not receptor oligomerization.” Cooperativity in the binding of ligands to receptor monomers is an oxymoron since this phenomenon requires a dimer or a trimer or a higher-order macromolecule with more than one orthosteric site. In what concerns cooperativity by class A GPCR homomers, two reports using fluorescent ligands to adenosine A3 receptors in transfected cells, a challenging technological approach and a solid theoretical formulation and data analysis (May et al., 2011; Corriden et al., 2014), constitute solid grounds to interpret biphasic curves as cooperativity on ligand binding to orthosteric centers in a receptor dimer. Data from radioligand binding assays may provide not only evidence for homomer but for Het formation (Birdsall, 2010). The reluctance to accept class A GPCR dimers may be also challenged taking advantage of both structural information and interpretation of pharmacological data from similarly basic but powerful approaches.
The word “monomer,” often used in the GPCR research field is defined by [Encyclopædia Britannica, Inc.] as “a molecule of any of a class of compounds, mostly organic, that can react with other molecules to form very large molecules, or polymers. The essential feature of a monomer is polyfunctionality, the capacity to form chemical bonds to at least two other monomer molecules. Bifunctional monomers can form only linear, chainlike polymers, but monomers of higher functionality yield cross-linked, network polymeric products.” On the one hand, this definition of a monomer built on solid chemical knowledge should be considered in the field. On the other hand, even assuming that a given GPCR may act in “isolation” and far from other GPCRs, they are not monomers as they interact, at least, with heterotrimeric G proteins. Therefore, the real issue is whether or not class A GPCRs may interact with other class A GPCRs while they already interact with a whole bunch of other proteins. Furthermore, the nomenclature for GPCRs is confusing; probably an appropriate definition of “GPCR molecule” is needed.
The big N-terminal extracellular domain of class C receptors allows formation of crystals constituted by dimers of the extracellular domain; in contrast, the N-terminal domain of class A is very short and unable to form such dimers. This difference in the size of the N-terminal extracellular domain seems to be the only reason that dopamine receptors may not form dimers and that class A T2 taste receptors may not form dimers whereas class C T1 taste receptors do. Due to the overwhelming evidence for class A GPCR homo/heteromerization, it may well happen that dimerization is guided by transmembrane domains and not by extracellular domains. To our knowledge no study on the relative positions of extracellular versus transmebrane domains has been performed. In the absence of such studies, fixing the dimeric structure of the extracellular domains does not provide any clue on the position of the transmembrane domains of the interacting receptors, i.e., useful information on the quaternary structure of the overall class C GPCR dimer complexes is missing.
Difficulties arising from crystallization of membrane proteins have been sorted out for GPCRs using an approach that, in brief, consists of molecules containing transmembrane helices fused to proteins such as T4 lysozyme or thermostabilized apocytochrome b562, which favor crystal formation. Structures from X-ray diffraction data usually assume a monomer as the basic unit. Such interpretation was sanctioned by the finding that arrays of rhodopsin itself in crystals was antiparallel to the plane of the membrane (Edwards et al., 2004; Li et al., 2004), something that is not seemingly physiological but that was not found in other solved rhodopsin structures showing parallel arrangements (vide infra).
Subsequent studies, however, have developed crystals in which GPCR dimers exist in the correct -parallel- orientation. The obligate interfaces occurring in 3D GPCR structures (see Cordomí et al., 2015) may not reflect real inter-protomer interfaces of a dimer in its native conformation in the cell surface, but the finding of two neighbor molecules in GPCR crystals compatible with the arrangements of dimers in biological membranes should be considered as relevant. Different transmembrane helices, often in a head-to-head interacting mode (Cordomí et al., 2015) do participate in the parallel receptor dimer arrangement in a variety of class A GPCR crystals, from activated rhodopsin (Salom et al., 2006), to CXCR4 chemokine (Wu et al., 2010) and κ- or μ-opioid receptors (Manglik et al., 2012; Wu et al., 2012). Parallel association of receptors have also been described in crystals of β2-adrenoceptors (Cherezov et al., 2007). As Wu et al. (2012) pointed out: “While the existence of GPCR dimers in vivo and their physiological relevance remain highly debatable, several distinct potential dimer interfaces are starting to emerge from crystallographic and biochemical studies.”
Noteworthy, μ-opioid receptor bound to a morphinan antagonist crystalizes as a dimer with an interface containing a four-helix bundle motif formed by transmembrane domains 5 and 6 (Manglik et al., 2012). Shortly afterward, the 3D structure of ligand-free turkey β1-adrenergic receptors in a lipid membrane-like environment exhibited oligomers constituted by two alternating dimer interfaces, one involving transmembrane helices 4 and 5 and the other engaging transmembrane helices 1 and 2 and the first extracellular loop (Whorton et al., 2013). Last but not least, the seven transmembrane domain of the smoothened SMO receptor crystalizes as a parallel dimer. Despite its relatively small similarity with class A GPCRs, the SMO receptor shows a high degree of spatial conservation of the transmembrane bundle and, also, structural correspondences in the intracellular domains with the solved class A GPCR structures (Wang et al., 2013). These structural features in SMO and class A receptors have led to the suggestion to translate the class A numbering nomenclature (Ballesteros and Weinstein, 1995) to class F GPCRs (Wang et al., 2013).
Lacking yet is any structural data concerning Hets. Precisely, one of the challenges in the GPCR field is Het crystallization, irrespective of whether Hets are constituted by class A or class C receptors. In fact, class C heteromerization has a long way to go; as earlier-mentioned it is not known whether dimerization of the N-extracellular domain conditions the transmembrane intra-dimer interactions. It is possible that the hinge between the N-terminal and the first transmembrane domain loses its conformational flexibility upon dimerization. In other words, it is not known whether transmembrane domains may bring two class C receptors together independently of N-terminal-domain dimerization, i.e., may a given class C Hets have in the membrane more than one quaternary structure? May different dimer structures be established by simultaneously involving either transmembrane regions or the N-terminal domains? Actually, class C Hets raise more structure-related questions than putative class A Hets.
Adrenalin receptors were, and still are, a reference in the GPCR research field. Pioneering studies were centered on adrenergic receptors in natural sources, e.g., isolated heart membranes, but this methodology was almost completely displaced by work in heterologous systems. In fact, COS- or CHO-transfected cells expressing the GPCR have been widespread used to know the whereabouts of these receptors and the scientific advance has been paramount and deserving the 2012 Nobel Prize in Chemistry, awarded to Drs Lefkowitz and Kobilka.
Identification of GPCR dimers occurred in parallel in transfected cells and in samples from natural sources. Biophysical-based methodologies such as fluorescence (FRET) or bioluminescence (BRET) energy transfer have been instrumental to show homo- or hetero-dimers in living transfected cells. A combination of FRET and BRET [sequential resonance energy transfer (SRET); Carriba et al., 2008] has allowed detection of GPCR trimers, and a combination of BRET with molecular complementation, using GPCRs fused to complementary hemiproteins of donor/acceptor BRET pairs, permits detection of tetramers (Cristóvão-Ferreira et al., 2013; Bonaventura et al., 2015). One of the issues raised by monomerism is the high-amount of receptors present in transfected cells that may “force” dimer, trimer, etc. formation. By the same token, it could be argued that many of the relevant findings in the last 30 years should be questioned as they have been obtained in heterologous systems overexpressing the receptors. What is more relevant, membranes used in drug screening usually come from GPCR-overexpressing transfected cells that likely express dimer and higher-order structures, therefore, the screening is to, at least, homodimers. Consequently, one may wonder why tools available for receptor dimers, as the two-state dimer model (Franco et al., 2005, 2006; Casadó et al., 2007, 2009a,b), are not used for data analysis; it provides more robust parameters than alternative models such as the two-independent site, which assumes one high affinity and one low-affinity population of monomeric receptors. More importantly, should not the previous knowledge be revisited if, as monomerism predicts, GPCRs in the heterologous systems are forced to express dimers?
Remarkably, many of the studies describing dimers have attempted and succeeded in proving their occurrence in natural sources. The literature identifying and proving Hets in natural sources is extensive and this review is not intended to exhaustively address it. The occurrence of Hets in natural sources, however, should be taken into account when analyzing data from transgenic animals defective in a given GPCR. GPCRs are somewhat promiscuous in their interaction with other GPCRs4 (Borroto-Escuela et al., 2014). Therefore, any particular phenotype in one of these knockout animals is not simply due to the lack of the GPCR but to the lack of the Hets in which the GPCR participates. In this context, pharmacologists may stick to naïvely assign a phenotype to the lack of a monomer or take the lead in searching the tools needed to correlate a given phenotype with a particular receptor Het.
Activation of class A GPCRs results in signal transduction, meaning that agonist binding to the GPCR induces conformational changes that are transmitted toward the interior of the cell. Data from radioligand binding assays have confirmed one of the predictions of Het formation, namely the agonist-induced changes of affinity upon binding of a second agonist to the partner receptor in a receptor heterocomplex (Birdsall, 2010). In some cases, such modulation may be of physiological relevance as occurs in the serotonin 5HT2A and metabotropic glutamatergic 2 receptors (González-Maeso et al., 2008). In other cases the phenomenon may reflect dimer formation but the change in affinity is too small to be operating in vivo.
Agonist affinity cross-modulation suggests Het formation but it may not reflect a direct receptor–receptor interaction. As G proteins remain attached to GPCRs in the membrane preparations used for radioligand binding, agonist-induced conformational changes in a G protein coupled to one GPCR may indirectly affect a second receptor that is not directly interacting with the first one. By contrast, antagonists and inverse agonists are molecules that bind to GPCRs and inhibit agonist-induced signaling. Very consistently, the selective antagonist of a receptor blocks the signal transduction induced by the agonist binding to the partner heteroreceptor. This counter intuitive fact may be explained by heteromerization. The phenomenon has been described in a variety of GPCR Hets such as orexin/corticotropin-releasing factor receptor (Navarro et al., 2015), dopamine D1/histamine H3 (Moreno et al., 2011) or angiotensin II AT1/dopamine D2 (Martínez-Pinilla et al., 2015) receptor Hets. The Ockham’s razor or law of parsimony states “pluralitas non est ponenda sine necessitate.” Accordingly, if a convoluted indirect mechanism is not necessary, the simplest explanation is provided by a GPCR Het framework in which the antagonist fixes a non-productive conformational state in one receptor that also impedes the activation of the partner receptor in the Het. Indeed, cross-antagonism should be considered as a tool of heteromer identification. Usually, cross-antagonism is first demonstrated in heterologous cells co-expressing the two receptors and constitutes a so-called Het print. Subsequently, dimer occurrence may be investigated on looking for the Het print in samples from mammalian tissues/organs. GPCR-induced activation of the MAP kinase pathway(s) is a successful readout for detecting cross-antagonism, for instance in the central nervous system, both in fresh slices from brain regions and primary cultures of neurons or glia (Balenga et al., 2014; Martínez-Pinilla et al., 2014, 2015).
The affinity of a given agonist for a given GPCR may change if the receptor is forming Hets (Ferré et al., 2009). Het-selective ligands were then searched for both, detecting Hets in vivo and as conceptually novel therapeutic tools. One example is 6′-guanidinonaltrindole, a opioid-receptor-Het selective drug, that in preclinical studies showed analgesia only if administered in the parts on the central nervous system where δ–κ-opioid receptor Hets are expressed. The result was also a proof-of-concept for tissue-selective drug targeting (Waldhoer et al., 2005).
Another strategy of selectively targeting homo- or heterodimers is to synthesize bivalent compounds able to simultaneously bind the two receptors in a GPCR dimer. These molecules consist of two agonist/antagonist moieties separated by spacers of variable length. Therefore, two orthosteric sites may be bridged by a bivalent ligand if the two pharmacophores are appropriately designed. The capacity of simultaneous binding to two different orthosteric sites is an added value that may be used to detect receptor dimers (Figure 2). A very detailed review on the various possibilities of bivalent ligands for the characterization of GPCR dimers is provided by Hiller et al. (2013). Certainly, emerging structural information will help revisit data on bivalent compounds to be sure of whether the linkers have the appropriate length to allow simultaneous binding to the two receptors in a given homo- or heterodimer (see Glass et al., 2016).
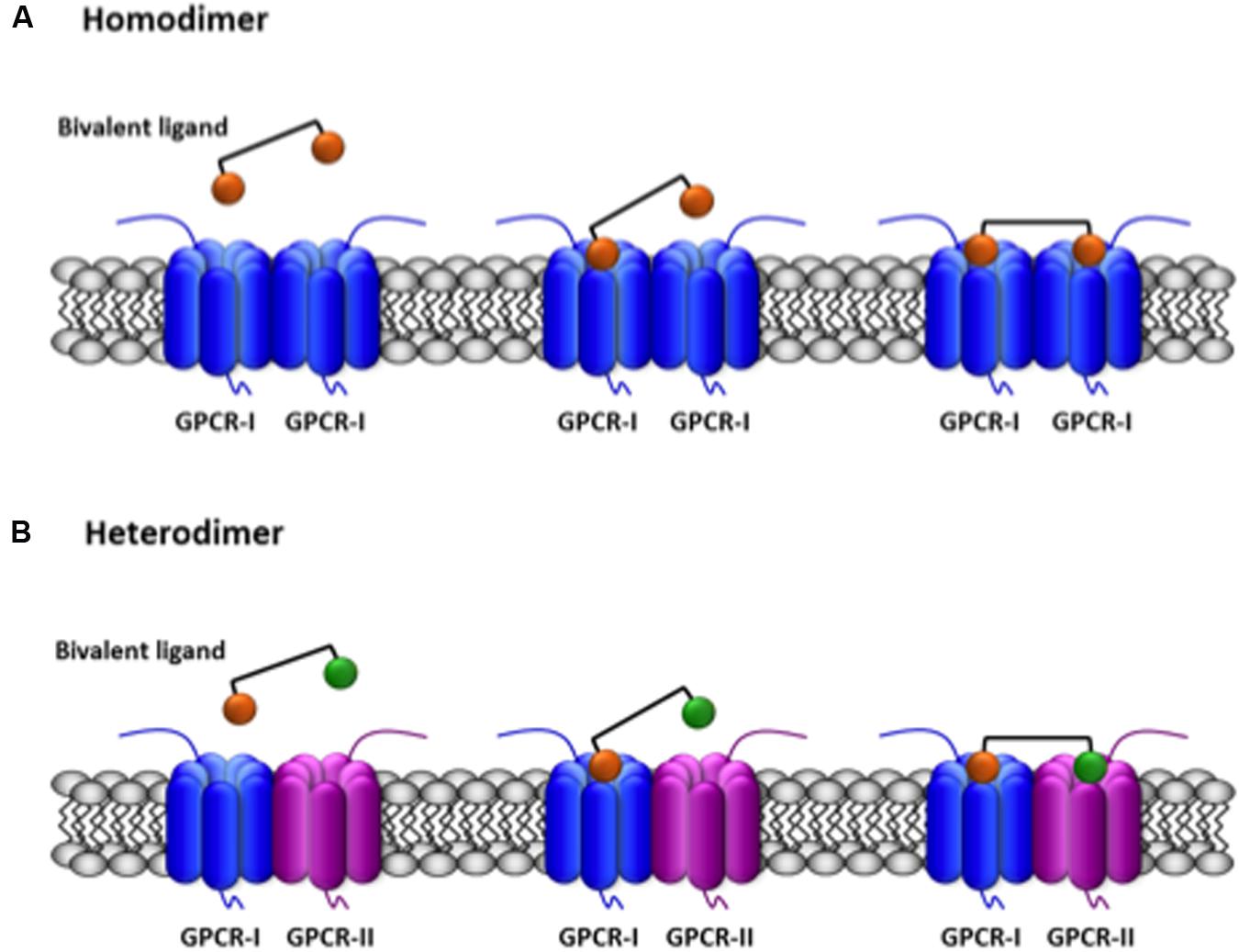
FIGURE 2. Scheme of the simultaneous binding of bivalent ligands with linkers of appropriate length to two receptors in a GPCR dimer. The agonist/antagonist moieties of the bivalent ligand are selective for their respective receptors (orange for GPCR-I, green for GPCR-II) and are linked by an spacer. (A) Binding to two equal GPCRs forming an homodimer. (B) Binding to two different GPCRs forming an heterodimer. A full account of the possibilities of binding of bivalent ligands to GPCRs is provided by Brogi et al. (2014).
Synthesis of bivalent ligands for GPCRs was performed in the pioneering report of Halazy et al. (1996), years before GPCR heterodimerization was reported. The authors concluded that their results support: “…the hypothesis that the important increase in potency of the serotonin dimers can be attributed to the presence of two serotonin pharmacophores in the same molecule.” Binding of those molecules to one orthosteric site cannot provide any increase in affinity or potency but dimers were not yet seriously considered at that time. It should be also pointed out that as early as in 1982 dimeric peptide enkephalins were synthesized and Shimohigashi et al. (1982) proved their interaction with two δ- but not with two μ-opioid receptors. To explain their results authors took advantage of another phenomenon that, by the way, may occur upon agonist activation, namely receptor clustering (Franco et al., 1996). As dimers for opioid receptors were not yet described, authors assumed that bivalent ligands were simultaneously acting in two close receptors within receptor clusters. Qualitatively similar results were obtained by Carrithers and Lerner (1996) that found bivalent ligands for alpha-melanocyte stimulating hormone receptors more potent than monovalents. Rationale for the study was that “multivalency can increase the apparent affinity of a ligand for its binding site” but the authors aimed at designing bivalent ligands “to bind to two adjacent receptor sites.” Multivalence of ligands together with dimer occurrence do fit with these pioneering results.
Russo et al. (2007) designed bivalents ligands that were able to bind, in transfected cells, to serotonin 5-HT4 constitutive receptor dimers identified by Berthouze et al. (2005). To our knowledge these compounds were not used to address the occurrence of direct 5-HT4 receptor–receptor interactions in natural sources. The differential binding to human dopamine D2 receptor of bivalent and monovalent ligands also suggested the occurrence of homodimers in transfected cells (Kühhorn et al., 2011a,c). As the theory of ligand binding to receptors would predict, the Hill coefficient of competition curves using a radiolabeled antagonist, [3H]spiperone, was 2 for (homo)bivalent and 1 for monovalent molecules (Kuhhorn et al., 2011b).
In Portoghese’s laboratory, the bivalent drug approach was successfully used in vitro -in living cells- and in natural sources, but also in behavioral models. Few years after the reporting of the first class A GPCR Hets, bivalent ligands containing δ- and κ-opioid antagonist moieties, provided evidence in the spinal cord of Hets formed by δ-κ-opioid receptors (Bhushan et al., 2004). Remarkably, these Hets explain the atypical pharmacology found for opioid GPCR subtypes in some areas of the central nervous system (Daniels et al., 2005), thus suggesting the occurrence of these receptor Hets in natural sources.
Also consistent with in vivo occurrence of μ-opioid/chemokine 5 receptor Hets, selective Het targeting may significantly reduce neuropathic and inflammatory pain (Akgün et al., 2013; Smeester et al., 2014). Although, the therapeutic use of bivalents to combat pain is limited due to the low chances of efficient crossing the blood brain barrier, these results are impotant to confirm Hets occurrence in vivo. Performing radioligand binding experiments in samples from tissues, and comparing competition curves of bi- and monovalent drugs could indeed prove or disprove the existence of dimers in mammals. In summary, heterobivalent ligands, i.e., molecules having two different moieties with a linker of appropriate length, are ad hoc probes for detecting GPCR Hets in natural sources.
In a detailed and careful study, Soriano et al. (2009) designed, synthesized and tested bivalent ligands containing both adenosine receptor agonist and dopamine receptor antagonist pharmacophores. Molecules with the appropriate spacer length, which was compatible with simultaneous binding to the two receptors, bound (in co-transfected cells) dopamine D2 and adenosine A2A receptors with higher affinity than their monovalent counterparts. This finding in cells coexpressing the two receptors was similarly detected in membranes from brain striatum. Those adenosine-dopamine bivalents did not bind with higher affinity to membranes expressing only one of the receptors. Modeling studies indicated that appropriate linker lengths were consistent with simultaneous binding to D2 and adenosine A2A receptor dimers. More importantly, the results from competition assays using specific radioligands (for adenosine or for dopamine receptors) can only be explained if there is simultaneous binding to dimers. Basic pharmacology indicates that simultaneous binding of bivalents to two binding sites, should be characterized by higher affinity than the monovalent control compounds. In fact, binding of one moiety to one binding site increases the effective local concentration available to the binding site in a partner receptor.
In summary, despite the field seems not yet mature to fully accept class A GPCR homo/heteromerization, data have been stubborn on providing evidence of the phenomenon. The models used have been very diverse and the techniques have been from co-immunoprecipitation all the way to powerful energy transfer in vitro and ex vivo assays and in vivo transactivation using transgenic mice (Rivero-Müller et al., 2010; Jonas et al., 2013; Grzesik et al., 2014). Class A receptor complexes have also been visualized in tissues by means of atomic force microcopy (Fotiadis et al., 2006; Cordomí and Perez, 2009), confocal fluorescence resonance energy transfer (So et al., 2007; Albizu et al., 2010; Perreault et al., 2010; Verma et al., 2010). Moreover, their occurrence, deduced from the avidity of peptides derived from class A GPCRs primary structure to bind together, has been detected by definitive methods5 such as mass spectrometry (Ciruela et al., 2004; Navarro et al., 2010), not to mention the positive results from surface plasmon resonance or recently-developed imaging techniques (e.g., fluorescence cross-correlation spectroscopy) able to detect dimer/oligomer-containing single particles.
RF: Contacting the Editor and contributing to the writing. JL: Discussing the sections, correcting, and contributing to the writing. GN: Contributing to the writing and searching for some specific references. EM-P: Contributing to the writing and preparing figures.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by grants SAF2012-39875-C02-01, BFU2012-37907, and BFU2015-64405-R –including FEDER- from the Spanish Ministry of Economy and Competitiveness (MINECO) and by grants 201413-30 (to RF) and 201413-31 (to JL) from the Fundació la Marató de TV3.
Akgün, E., Javed, M. I., Lunzer, M. M., Smeester, B. A., Beitz, A. J., and Portoghese, P. S. (2013). Ligands that interact with putative MOR-mGluR5 heteromer in mice with inflammatory pain produce potent antinociception. Proc. Natl. Acad. Sci. U.S.A. 110, 11595–11599. doi: 10.1073/pnas.1305461110
Albizu, L., Cottet, M., Kralikova, M., Stoev, S., Seyer, R., Brabet, I., et al. (2010). Time-resolved FRET between GPCR ligands reveals oligomers in native tissues. Nat. Chem. Biol. 6, 587–594. doi: 10.1038/nchembio.396
Balenga, N. A., Martínez-Pinilla, E., Kargl, J., Schröder, R., Peinhaupt, M., Platzer, W., et al. (2014). Heteromerization of GPR55 and cannabinoid CB2 receptors modulates signaling. Br. J. Pharmacol. 171, 1–64. doi: 10.1111/bph.12850
Ballesteros, J. A., and Weinstein, H. (1995). Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 25, 366–428. doi: 10.1016/S1043-9471(05)80049-7
Banères, J.-L., and Parello, J. (2003). Structure-based analysis of GPCR function: evidence for a novel pentameric assembly between the dimeric leukotriene B4 receptor BLT1 and the G-protein. J. Mol. Biol. 329, 815–829. doi: 10.1016/S0022-2836(03)00439-X
Berthouze, M., Ayoub, M., Russo, O., Rivail, L., Sicsic, S., Fischmeister, R., et al. (2005). Constitutive dimerization of human serotonin 5-HT4 receptors in living cells. FEBS Lett. 579, 2973–2980. doi: 10.1016/j.febslet.2005.04.040
Bhushan, R. G., Sharma, S. K., Xie, Z., Daniels, D. J., and Portoghese, P. S. (2004). A bivalent ligand (KDN-21) reveals spinal delta and kappa opioid receptors are organized as heterodimers that give rise to delta(1) and kappa(2) phenotypes. Selective targeting of delta-kappa heterodimers. J. Med. Chem. 47, 2969–2972. doi: 10.1021/jm0342358
Birdsall, N. J. M. (2010). Class a GPCR heterodimers: evidence from binding studies. Trends Pharmacol. Sci. 31, 499–508. doi: 10.1016/j.tips.2010.08.003
Bonaventura, J., Navarro, G., Casadó-Anguera, V., Azdad, K., Rea, W., Moreno, E., et al. (2015). Allosteric interactions between agonists and antagonists within the adenosine A2A receptor-dopamine D2 receptor heterotetramer. Proc. Natl. Acad. Sci. U.S.A. 112, E3609–E3618. doi: 10.1073/pnas.1507704112
Borroto-Escuela, D. O., Brito, I., Romero-Fernandez, W., Di Palma, M., Oflijan, J., Skieterska, K., et al. (2014). The G protein-coupled receptor heterodimer network (GPCR-HetNet) and its hub components. Int. J. Mol. Sci. 15, 8570–8590. doi: 10.3390/ijms15058570
Borroto-Escuela, D. O., Romero-Fernandez, W., Mudó, G., Pérez-Alea, M., Ciruela, F., Tarakanov, A. O., et al. (2012). Fibroblast growth factor receptor 1- 5-hydroxytryptamine 1A heteroreceptor complexes and their enhancement of hippocampal plasticity. Biol. Psychiatry 71, 84–91. doi: 10.1016/j.biopsych.2011.09.012
Borroto-Escuela, D. O., Van Craenenbroeck, K., Romero-Fernandez, W., Guidolin, D., Woods, A. S., Rivera, A., et al. (2011). Dopamine D2 and D4 receptor heteromerization and its allosteric receptor–receptor interactions. Biochem. Biophys. Res. Commun. 404, 928–934. doi: 10.1016/j.bbrc.2010.12.083
Brogi, S., Tafi, A., Désaubry, L., and Nebigil, C. G. (2014). Discovery of GPCR ligands for probing signal transduction pathways. Front. Pharmacol. 5:255. doi: 10.3389/fphar.2014.00255
Carriba, P., Navarro, G., Ciruela, F., Ferré, S., Casadó, V., Agnati, L., et al. (2008). Detection of heteromerization of more than two proteins by sequential BRET-FRET. Nat. Methods 5, 727–733. doi: 10.1038/nmeth.1229
Carrithers, M. D., and Lerner, M. R. (1996). Synthesis and characterization of bivalent peptide ligands targeted to G-protein-coupled receptors. Chem. Biol. 3, 537–542. doi: 10.1016/S1074-5521(96)90144-1
Casadó, V., Cortés, A., Ciruela, F., Mallol, J., Ferré, S., Lluis, C., et al. (2007). Old and new ways to calculate the affinity of agonists and antagonists interacting with G-protein-coupled monomeric and dimeric receptors: the receptor-dimer cooperativity index. Pharmacol. Ther. 116, 343–354. doi: 10.1016/j.pharmthera.2007.05.010
Casadó, V., Cortés, A., Mallol, J., Pérez-Capote, K., Ferré, S., Lluis, C., et al. (2009a). GPCR homomers and heteromers: a better choice as targets for drug development than GPCR monomers? Pharmacol. Ther. 124, 248–257. doi: 10.1016/j.pharmthera.2009.07.005
Casadó, V., Ferrada, C., Bonaventura, J., Gracia, E., Mallol, J., Canela, E. I., et al. (2009b). Useful pharmacological parameters for G-protein-coupled receptor homodimers obtained from competition experiments. Agonist-antagonist binding modulation. Biochem. Pharmacol. 78, 1456–1463. doi: 10.1016/j.bcp.2009.07.012
Cherezov, V., Rosenbaum, D. M., Hanson, M. A., Rasmussen, S. G. F., Thian, F. S., Kobilka, T. S., et al. (2007). High-resolution crystal structure of an engineered human b2 -adrenergic G protein-coupled receptor. Science 318, 1258–1265. doi: 10.1126/science.1150577
Ciruela, F., Burgueño, J., Casadó, V., Canals, M., Marcellino, D., Goldberg, S. R., et al. (2004). Combining mass spectrometry and pull-down techniques for the study of receptor heteromerization. Direct epitope-epitope electrostatic interactions between adenosine A2A and dopamine D2 receptors. Anal. Chem. 76, 5354–5363. doi: 10.1021/ac049295f
Ciruela, F., Casado, V., Mallol, J., Canela, E. I., Lluis, C., and Franco, R. (1995). Immunological identification of A1 adenosine receptors in brain cortex. J. Neurosci. Res. 42, 818–828. doi: 10.1002/jnr.490420610
Ciruela, F., Casadó, V., Rodrigues, R. J., Luján, R., Burgueño, J., Canals, M., et al. (2006). Presynaptic control of striatal glutamatergic neurotransmission by adenosine A1-A2A receptor heteromers. J. Neurosci. 26, 2080–2087. doi: 10.1523/JNEUROSCI.3574-05.2006
Cordomí, A., Navarro, G., Aymerich, M. S., and Franco, R. (2015). Structures for G-protein-coupled receptor tetramers in complex with G proteins. Trends Biochem. Sci. 40, 548–551. doi: 10.1016/j.tibs.2015.07.007
Cordomí, A., and Perez, J. J. (2009). Structural rearrangements of rhodopsin subunits in a dimer complex: a molecular dynamics simulation study. J. Biomol. Struct. Dyn. 27, 127–147. doi: 10.1080/07391102.2009.10507303
Corriden, R., Kilpatrick, L. E., Kellam, B., Briddon, S. J., and Hill, S. J. (2014). Kinetic analysis of antagonist-occupied adenosine-A3 receptors within membrane microdomains of individual cells provides evidence of receptor dimerization and allosterism. FASEB J. 28, 4211–4222. doi: 10.1096/fj.13-247270
Cristóvão-Ferreira, S., Navarro, G., Brugarolas, M., Pérez-Capote, K., Vaz, S. H., Fattorini, G., et al. (2013). A1R-A2AR heteromers coupled to Gs and G i/0 proteins modulate GABA transport into astrocytes. Purinergic Signal 9, 433–449. doi: 10.1007/s11302-013-9364-5
Daniels, D. J., Kulkarni, A., Xie, Z., Bhushan, R. G., and Portoghese, P. S. (2005). A bivalent ligand (KDAN-18) containing delta-antagonist and kappa-agonist pharmacophores bridges delta2 and kappa1 opioid receptor phenotypes. J. Med. Chem. 48, 1713–1716. doi: 10.1021/jm034234f
Edwards, P. C., Li, J., Burghammer, M., Hugh McDowell, J., Villa, C., Hargrave, P. A., et al. (2004). Crystals of native and modified bovine rhodopsins and their heavy atom derivatives. J. Mol. Biol. 343, 1439–1450. doi: 10.1016/j.jmb.2004.08.089
Ferré, S., Baler, R., Bouvier, M., Caron, M. G., Devi, L. A., Durroux, T., et al. (2009). Building a new conceptual framework for receptor heteromers. Nat. Chem. Biol. 5, 131–134. doi: 10.1038/nchembio0309-131
Ferré, S., Ciruela, F., Quiroz, C., Luján, R., Popoli, P., Cunha, R. A., et al. (2007). Adenosine receptor heteromers and their integrative role in striatal function. Sci. World J. 7, 74–85. doi: 10.1100/tsw.2007.211
Fotiadis, D., Jastrzebska, B., Philippsen, A., Müller, D. J., Palczewski, K., and Engel, A. (2006). Structure of the rhodopsin dimer: a working model for G-protein-coupled receptors. Curr. Opin. Struct. Biol. 16, 252–259. doi: 10.1016/j.sbi.2006.03.013
Franco, R. (2009). G-protein-coupled receptor heteromers or how neurons can display differently flavoured patterns in response to the same neurotransmitter. Br. J. Pharmacol. 158, 23–31. doi: 10.1111/j.1476-5381.2009.00181.x
Franco, R., Casadó, V., Ciruela, F., Mallol, J., Lluis, C., and Canela, E. I. (1996). The cluster-arranged cooperative model: a model that accounts for the kinetics of binding to A1 adenosine receptors. Biochemistry 35, 3007–3015. doi: 10.1021/bi952415g
Franco, R., Casadó, V., Mallol, J., Ferrada, C., Ferré, S., Fuxe, K., et al. (2006). The two-state dimer receptor model: a general model for receptor dimers. Mol. Pharmacol. 69, 1905–1912. doi: 10.1124/mol.105.020685
Franco, R., Casadó, V., Mallol, J., Ferré, S., Fuxe, K., Cortés, A., et al. (2005). Dimer-based model for heptaspanning membrane receptors. Trends Biochem. Sci. 30, 360–366. doi: 10.1016/j.tibs.2005.05.010
Frederick, A. L., Yano, H., Trifilieff, P., Vishwasrao, H. D., Biezonski, D., Mészáros, J., et al. (2015). Evidence against dopamine D1/D2 receptor heteromers. Mol. Psychiatry 20, 1373–1385. doi: 10.1038/mp.2014.166
Fuxe, K., and Agnati, L. F. (1985). Receptor-receptor interactions in the central nervous system. A new integrative mechanism in synapses. Med. Res. Rev. 5, 441–482. doi: 10.1002/med.2610050404
Fuxe, K., Agnati, L. F., Benfenati, F., Celani, M., Zini, I., Zoli, M., et al. (1983). Evidence for the existence of receptor–receptor interactions in the central nervous system. Studies on the regulation of monoamine receptors by neuropeptides. J. Neural Transm. Suppl. 18, 165–179.
Fuxe, K., Härfstrand, A., Agnati, L. F., Kalia, M., Fredholm, B., Svensson, T., et al. (1987). Central catecholamine-neuropeptide Y interactions at the pre- and postsynaptic level in cardiovascular centers. J. Cardiovasc. Pharmacol. 1(10 Suppl.), S1–S13. doi: 10.1097/00005344-198709002-00001
Fuxe, K., Tarakanov, A., Romero Fernandez, W., Ferraro, L., Tanganelli, S., Filip, M., et al. (2014). Diversity and bias through receptor-receptor interactions in GPCR heteroreceptor complexes. Focus on examples from dopamine D2 receptor heteromerization. Front. Endocrinol. (Lausanne) 5:71. doi: 10.3389/fendo.2014.00071
George, S. R., Kern, A., Smith, R. G., and Franco, R. (2014). Dopamine receptor heteromeric complexes and their emerging functions. Prog. Brain Res. 211, 183–200. doi: 10.1016/B978-0-444-63425-2.00008-8
Gherbi, K., May, L. T., Baker, J. G., Briddon, S. J., and Hill, S. J. (2015). Negative cooperativity across β1-adrenoceptor homodimers provides insights into the nature of the secondary low-affinity CGP 12177 β1-adrenoceptor binding conformation. FASEB J. 29, 2859–2871. doi: 10.1096/fj.14-265199
Ginés, S., Hillion, J., Torvinen, M., Le Crom, S., Casadó, V., Canela, E. I., et al. (2000). Dopamine D1 and adenosine A1 receptors form functionally interacting heteromeric complexes. Proc. Natl. Acad. Sci. U.S.A. 97, 8606–8611. doi: 10.1073/pnas.150241097
Glass, M., Govindpani, K., Furkert, D. P., Hurst, D. P., Reggio, P. H., and Flanagan, J. U. (2016). One for the price of two…are bivalent ligands targeting cannabinoid receptor dimers capable of simultaneously binding to both receptors? Trends Pharmacol. Sci. doi: 10.1016/j.tips.2016.01.010 [Epub ahead of print].
Gomes, I., Jordan, B. A., Gupta, A., Trapaidze, N., Nagy, V., and Devi, L. A. (2000). Heterodimerization of mu and delta opioid receptors: a role in opiate synergy. J. Neurosci. 20:RC110.
González-Maeso, J., Ang, R. L., Yuen, T., Chan, P., Weisstaub, N. V., López-Giménez, J. F., et al. (2008). Identification of a serotonin/glutamate receptor complex implicated in psychosis. Nature 452, 93–97. doi: 10.1038/nature06612
Grzesik, P., Teichmann, A., Furkert, J., Rutz, C., Wiesner, B., Kleinau, G., et al. (2014). Differences between lutropin-mediated and choriogonadotropin-mediated receptor activation. FEBS J. 281, 1479–1492. doi: 10.1111/febs.12718
Gupta, A., Mulder, J., Gomes, I., Rozenfeld, R., Bushlin, I., Ong, E., et al. (2014). Increased abundance of opioid receptor heteromers after chronic morphine administration. Sci. Signal 3:ra54. doi: 10.1126/scisignal.2000807
Halazy, S., Perez, M., Fourrier, C., Pallard, I., Pauwels, P. J., Palmier, C., et al. (1996). Serotonin dimers: application of the bivalent ligand approach to the design of new potent and selective 5-HT(1B/1D) agonists. J. Med. Chem. 39, 4920–4927. doi: 10.1021/jm960552l
Hasbi, A., Fan, T., Alijaniaram, M., Nguyen, T., Perreault, M. L., O’Dowd, B. F., et al. (2009). Calcium signaling cascade links dopamine D1-D2 receptor heteromer to striatal BDNF production and neuronal growth. Proc. Natl. Acad. Sci. U.S.A. 106, 21377–21382. doi: 10.1073/pnas.0903676106
Hiller, C., Kühhorn, J., and Gmeiner, P. (2013). Class A G-protein-coupled receptor (GPCR) dimers and bivalent ligands. J. Med. Chem. 56, 6542–6559. doi: 10.1021/jm4004335
Jonas, K. C., Rivero-Müller, A., Huhtaniemi, I. T., and Hanyaloglu, A. C. (2013). G protein-coupled receptor transactivation: from molecules to mice. Methods Cell Biol. 117, 433–450. doi: 10.1016/B978-0-12-408143-7.00023-2
Kawahara, K., Suenobu, M., Yoshida, A., Koga, K., Hyodo, A., Ohtsuka, H., et al. (2012). Intracerebral microinjection of interleukin-4/interleukin-13 reduces β-amyloid accumulation in the ipsilateral side and improves cognitive deficits in young amyloid precursor protein 23 mice. Neuroscience 207, 243–260. doi: 10.1016/j.neuroscience.2012.01.049
Kühhorn, J., Götz, A., Hübner, H., Thompson, D., Whistler, J., and Gmeiner, P. (2011a). Development of a bivalent dopamine D2 receptor agonist. J. Med. Chem. 54, 7911–7919. doi: 10.1021/jm2009919
Kühhorn, J., Hubner, H., and Gmeiner, P. (2011b). Bivalent dopamine D2 receptor ligands: synthesis and binding properties. J. Med. Chem. 54, 4896–4903. doi: 10.1021/jm2004859
Kühhorn, J., Hübner, H., Gmeiner, P., Kuhhorn, J., Hubner, H., and Gmeiner, P. (2011c). Bivalent dopamine D2 receptor ligands: synthesis and binding properties. J. Med. Chem. 54, 4896–4903. doi: 10.1021/jm2004859
Kunishima, N., Shimada, Y., Tsuji, Y., Sato, T., Yamamoto, M., Kumasaka, T., et al. (2000). Structural basis of glutamate recognition by a dimeric metabotropic glutamate receptor. Nature 407, 971–977. doi: 10.1038/35039564
Li, J., Edwards, P. C., Burghammer, M., Villa, C., and Schertler, G. F. X. (2004). Structure of bovine rhodopsin in a trigonal crystal form. J. Mol. Biol. 343, 1409–1438. doi: 10.1016/j.jmb.2004.08.090
Li, X., Tomita, M., Pilbeam, C. C., Breyer, R. M., and Raisz, L. G. (2002). Prostaglandin receptor EP2 mediates PGE2 stimulated hypercalcemia in mice in vivo. Prostaglandins Other Lipid Mediat. 67, 173–180. doi: 10.1016/S0090-6980(01)00186-1
Manglik, A., Kruse, A. C., Kobilka, T. S., Thian, F. S., Mathiesen, J. M., Sunahara, R. K., et al. (2012). Crystal structure of the μ-opioid receptor bound to a morphinan antagonist. Nature 485, 321–326. doi: 10.1038/nature10954
Marsango, S., Caltabiano, G., Pou, C., Varela Liste, M. J., and Milligan, G. (2015). Analysis of human dopamine D3 receptor quaternary structure. J. Biol. Chem. 290, 15146–15162. doi: 10.1074/jbc.M114.630681
Martínez-Pinilla, E., Reyes-Resina, I., Oñatibia-Astibia, A., Zamarbide, M., Ricobaraza, A., Navarro, G., et al. (2014). CB1 and GPR55 receptors are co-expressed and form heteromers in rat and monkey striatum. Exp. Neurol. 261, 44–52. doi: 10.1016/j.expneurol.2014.06.017
Martínez-Pinilla, E., Rodríguez-Pérez, A. I., Navarro, G., Aguinaga, D., Moreno, E., Lanciego, J. L., et al. (2015). Dopamine D2 and angiotensin II type 1 receptors form functional heteromers in rat striatum. Biochem. Pharmacol. 96, 131–142. doi: 10.1016/j.bcp.2015.05.006
May, L. T., Bridge, L. J., Stoddart, L. A., Briddon, S. J., and Hill, S. J. (2011). Allosteric interactions across native adenosine-A3 receptor homodimers: quantification using single-cell ligand-binding kinetics. FASEB J. 25, 3465–3476. doi: 10.1096/fj.11-186296
Moreno, E., Hoffmann, H., Gonzalez-Sepúlveda, M., Navarro, G., Casadó, V., Cortés, A., et al. (2011). Dopamine D1-histamine H3 receptor heteromers provide a selective link to MAPK signaling in GABAergic neurons of the direct striatal pathway. J. Biol. Chem. 286, 5846–5854. doi: 10.1074/jbc.M110.161489
Navarro, G., Ferre, S., Cordomi, A., Moreno, E., Mallol, J., Casado, V., et al. (2010). Interactions between intracellular domains as key determinants of the quaternary structure and function of receptor heteromers. J. Biol. Chem. 285, 27346–27359. doi: 10.1074/jbc.M110.115634
Navarro, G., Quiroz, C., Moreno-Delgado, D., Sierakowiak, A., McDowell, K., Moreno, E., et al. (2015). Orexin-corticotropin-releasing factor receptor heteromers in the ventral tegmental area as targets for cocaine. J. Neurosci. 35, 6639–6653. doi: 10.1523/JNEUROSCI.4364-14.2015
Nelson, G., Hoon, M. A., Chandrashekar, J., Zhang, Y., Ryba, N. J. P., and Zuker, C. S. (2001). Mammalian sweet taste receptors. Cell 106, 381–390. doi: 10.1016/S0092-8674(01)00451-2
Perreault, M. L., Hasbi, A., Alijaniaram, M., Fan, T., Varghese, G., Fletcher, P. J., et al. (2010). The dopamine D1-D2 receptor heteromer localizes in dynorphin/enkephalin neurons: increased high affinity state following amphetamine and in schizophrenia. J. Biol. Chem. 285, 36625–36634. doi: 10.1074/jbc.M110.159954
Portoghese, P. S., and Lunzer, M. M. (2003). Identity of the putative delta1-opioid receptor as a delta-kappa heteromer in the mouse spinal cord. Eur. J. Pharmacol. 467, 233–234. doi: 10.1016/S0014-2999(03)01599-1
Prinster, S. C., Hague, C., and Hall, R. A. (2005). Heterodimerization of g protein-coupled receptors: specificity and functional significance. Pharmacol. Rev. 57, 289–298. doi: 10.1124/pr.57.3.1
Rashid, A. J., So, C. H., Kong, M. M. C., Furtak, T., El-Ghundi, M., Cheng, R., et al. (2007). D1-D2 dopamine receptor heterooligomers with unique pharmacology are coupled to rapid activation of Gq/11 in the striatum. Proc. Natl. Acad. Sci. U.S.A. 104, 654–659. doi: 10.1073/pnas.0604049104
Rivero-Müller, A., Chou, Y.-Y., Ji, I., Lajic, S., Hanyaloglu, A. C., Jonas, K., et al. (2010). Rescue of defective G protein-coupled receptor function in vivo by intermolecular cooperation. Proc. Natl. Acad. Sci. U.S.A. 107, 2319–2324. doi: 10.1073/pnas.0906695106
Romano, C., Miller, J. K., Hyrc, K., Dikranian, S., Mennerick, S., Takeuchi, Y., et al. (2001). Covalent and non covalent interactions mediate metabotropic glutamate receptor mGlu5 dimerization. Mol. Pharmacol. 59, 46–53.
Russo, O., Berthouze, M., Giner, M., Soulier, J.-L., Rivail, L., Sicsic, S., et al. (2007). Synthesis of specific bivalent probes that functionally interact with 5-HT (4) receptor dimers. J. Med. Chem. 50, 4482–4492. doi: 10.1021/jm070552t
Salom, D., Lodowski, D. T., Stenkamp, R. E., Le Trong, I., Golczak, M., Jastrzebska, B., et al. (2006). Crystal structure of a photoactivated deprotonated intermediate of rhodopsin. Proc. Natl. Acad. Sci. U.S.A. 103, 16123–16128. doi: 10.1073/pnas.0608022103
Shimohigashi, Y., Costa, T., Chen, H. C., and Rodbard, D. (1982). Dimeric tetrapeptide enkephalins display extraordinary selectivity for the delta opiate receptor. Nature 297, 333–335. doi: 10.1038/297333a0
Smeester, B. A., Lunzer, M. M., Akgün, E., Beitz, A. J., and Portoghese, P. S. (2014). Targeting putative mu opioid/metabotropic glutamate receptor-5 heteromers produces potent antinociception in a chronic murine bone cancer model. Eur. J. Pharmacol. 743, 48–52. doi: 10.1016/j.ejphar.2014.09.008
So, C. H., Verma, V., O’Dowd, B. F., and George, S. R. (2007). Desensitization of the dopamine D1 and D2 receptor hetero-oligomer mediated calcium signal by agonist occupancy of either receptor. Mol. Pharmacol. 72, 450–462. doi: 10.1124/mol.107.034884
Soriano, A., Ventura, R., Molero, A., Hoen, R., Casadó, V., Cortés, A., et al. (2009). Adenosine A2A receptor-antagonist/dopamine D2 receptor-agonist bivalent ligands as pharmacological tools to detect A2A-D2 receptor heteromers. J. Med. Chem. 52, 5590–5602. doi: 10.1021/jm900298c
Uriano, G., and Gravatt, C. (1977). The role of reference materials and reference methods in chemical analysis. Crit. Rev. Anal. Chem. 6, 361–376. doi: 10.1080/10408347708085697
van Rijn, R. M., Whistler, J. L., and Waldhoer, M. (2010). Opioid-receptor-heteromer-specific trafficking and pharmacology. Curr. Opin. Pharmacol. 10, 73–79. doi: 10.1016/j.coph.2009.09.007
Verma, V., Hasbi, A., O’Dowd, B. F., and George, S. R. (2010). Dopamine D1-D2 receptor Heteromer-mediated calcium release is desensitized by D1 receptor occupancy with or without signal activation: dual functional regulation by G protein-coupled receptor kinase 2. J. Biol. Chem. 285, 35092–35103. doi: 10.1074/jbc.M109.088625
Waldhoer, M., Fong, J., Jones, R. M., Lunzer, M. M., Sharma, S. K., Kostenis, E., et al. (2005). A heterodimer-selective agonist shows in vivo relevance of G protein-coupled receptor dimers. Proc. Natl. Acad. Sci. U.S.A. 102, 9050–9055. doi: 10.1073/pnas.0501112102
Wang, C., Wu, H., Katritch, V., Han, G. W., Huang, X.-P., Liu, W., et al. (2013). Structure of the human smoothened receptor bound to an antitumour agent. Nature 497, 338–343. doi: 10.1038/nature12167
White, J. H., Wise, A., Main, M. J., Green, A., Fraser, N. J., Disney, G. H., et al. (1998). Heterodimerization is required for the formation of a functional GABA(B) receptor. Nature 396, 679–682. doi: 10.1038/25354
Whorton, M. R., Bokoch, M. P., Rasmussen, S. G. F., Huang, B., Zare, R. N., Kobilka, B., et al. (2007). A monomeric G protein-coupled receptor isolated in a high-density lipoprotein particle efficiently activates its G protein. Proc. Natl. Acad. Sci. U.S.A. 104, 7682–7687. doi: 10.1073/pnas.0611448104
Whorton, M. R., Bokoch, M. P., Rasmussen, S. G. F., Huang, B., Zare, R. N., Kobilka, B., et al. (2013). Crystal structure of oligomeric β1-adrenergic G protein-coupled receptors in ligand-free basal state. Nat. Struct. Mol. Biol. 20, 419–425. doi: 10.1038/nsmb.2504
Wu, B., Chien, E. Y. T., Mol, C. D., Fenalti, G., Liu, W., Katritch, V., et al. (2010). Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 330, 1066–1071. doi: 10.1126/science.1194396
Wu, H., Wacker, D., Katritch, V., Mileni, M., Han, G. W., Liu, W., et al. (2012). Structure of the human kappa opioid receptor in complex with JDTic. Nature 485, 327–332. doi: 10.1038/nature10939
Keywords: dimerization, dopamine receptor, GPCR, homodimer, heteromer, ligands, mammalian receptor, signal transduction taste receptor
Citation: Franco R, Martínez-Pinilla E, Lanciego JL and Navarro G (2016) Basic Pharmacological and Structural Evidence for Class A G-Protein-Coupled Receptor Heteromerization. Front. Pharmacol. 7:76. doi: 10.3389/fphar.2016.00076
Received: 17 December 2015; Accepted: 11 March 2016;
Published: 31 March 2016.
Edited by:
Xavier Deupi, Paul Scherrer Institute, SwitzerlandReviewed by:
Dasiel Oscar Borroto-Escuela, Karolinska Institutet, SwedenCopyright © 2016 Franco, Martínez-Pinilla, Lanciego and Navarro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eva Martínez-Pinilla, bWFydGluZXpwaW5pbGxhZXZhQGdtYWlsLmNvbQ==; Rafael Franco, cmZyYW5jbzEyM0BnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.