 Yan Liang
Yan Liang Farah Sheikh
Farah Sheikh
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Pharmacol. , 29 February 2016
Sec. Cardiovascular and Smooth Muscle Pharmacology
Volume 7 - 2016 | https://doi.org/10.3389/fphar.2016.00037
This article is part of the Research Topic Regulation of protein kinase signaling in cardiovascular disease View all 11 articles
The mitogen activated protein kinase (MAPK)-extracellular regulated kinase 1/2 (ERK1/2) pathway is a central downstream signaling pathway that is activated in cardiac muscle cells during mechanical and agonist-mediated hypertrophy. Studies in genetic mouse models deficient in ERK-associated MAPK components pathway have further reinforced a direct role for this pathway in stress-induced cardiac hypertrophy and disease. However, more recent studies have highlighted that these signaling pathways may exert their regulatory functions in a more compartmentalized manner in cardiac muscle. Emerging data has uncovered specific MAPK scaffolding proteins that tether MAPK/ERK signaling specifically at the sarcomere and plasma membrane in cardiac muscle and show that deficiencies in these scaffolding proteins alter ERK activity and phosphorylation, which are then critical in altering the cardiac myocyte response to stress-induced hypertrophy and disease progression. In this review, we provide insights on ERK-associated scaffolding proteins regulating cardiac myofilament function and their impact on cardiac hypertrophy and disease.
The mitogen activated protein kinase (MAPK)/extracellular regulated kinase (ERK) signaling pathway is one of the first blueprints describing how extracellular cues/factors (growth factors and hormones) generate intracellular signals and converge on the nucleus to drive gene expression and a wide range of cellular functions (Pearson et al., 2001). How this single pathway regulates diverse functions and achieves specificity in function in different cell types has been a longstanding question in the field. Studies have suggested that scaffolding proteins are emerging as playing an essential role in regulating the MAPK signaling response (Brown and Sacks, 2009). MAPK scaffold proteins are dynamic entities that (i) directly interact with multiple components of the MAPK signaling complex, (ii) consolidate or sequester protein interactions to physically insulate the MAPK pathway to specific cellular locations as well as (iii) regulate signal strength and stimulus-specific responses to efficiently transmit MAPK signals in a spatiotemporal manner and narrow its actions (Brown and Sacks, 2009). Notable MAPK scaffolds that regulate ERK signaling include, kinase suppressor of Ras (KSR) (Kornfeld et al., 1995; Sundaram and Han, 1995; Therrien et al., 1995), MAPK organizer 1, MORG1 (Vomastek et al., 2004), MEK partner 1, MP1 (Schaeffer et al., 1998), paxillin (Ishibe et al., 2003), β-arrestin (Raabo et al., 1992), and MEKK1 (Karandikar et al., 2000). KSR is one of the best-characterized mammalian MAPK scaffold proteins (Kornfeld et al., 1995). KSR is critical in recruiting the Raf/MEK/ERK complex to the plasma membrane as well as regulating MAPK signaling strength and duration at this location (Brown and Sacks, 2009). Its importance was most notable in Caenorhabditis elegans development as well as immune and cancer cells since in vivo models deficient in KSR-1 exhibited deficiencies in ERK activation in a stimulus-specific manner, resulting in resistance to inflammatory diseases and Ras-mediated cancers (Nguyen et al., 2002; Lozano et al., 2003; Fusello et al., 2006).
Recently, MAPK scaffold proteins have been identified in the heart and shown to play a pivotal role in pathophysiological cardiac hypertrophy (Figure 1). Cardiac hypertrophy is the main way in which cardiomyocytes initially adapt to increased mechanical workload and neurohormonal stimuli to increase cardiac output and compensate for adverse effects on cardiac pump function (Bernardo et al., 2010). Physiological cardiac hypertrophy (i.e., the ‘athlete’s heart’) is characterized by normal cardiac organization and normal or enhanced cardiac function. However, pathological cardiac hypertrophy is a key risk factor for heart failure and is associated with increased interstitial fibrosis, apoptosis, and cardiac dysfunction (McMullen and Jennings, 2007). More recently studies have linked Ras-Raf1-MEK1/2-ERK1/2 signaling pathway in pathological cardiac hypertrophy (Lorenz et al., 2009b). Both constitutive activation of Ras (H-Ras-V12) and overexpression of MEK1 in the mouse heart displayed characteristic features of exaggerated cardiac hypertrophy (Bueno et al., 2000; Zheng et al., 2004). Inhibition of the ERK pathway via dominant negative Raf or cardiac specific deletion of c-raf-1 attenuated pathological cardiac hypertrophy (Yamaguchi et al., 2004; Goldsmith and Dhanasekaran, 2007). The recent identification of a new autophosphorylation site of ERK2 at Thr188 (Thr208 in ERK1) demonstrated a specific role for ERK signaling in the induction of pathological cardiac hypertrophy in response to various hypertrophic stimuli (Lorenz et al., 2009a). Interference with ERKThr188 phosphorylation attenuated ERK-mediated pathological hypertrophy, but not physiological cardiac hypertrophy (Ruppert et al., 2013). More recent efforts have been devoted to understanding how MAPK scaffold proteins regulate ERK signaling strength and duration as well as their role in cardiac muscle in the context of hypertrophy. This review will highlight the subcellular functions and actions of ERK-associated MAPK scaffolds in cardiac muscle and pathological cardiac hypertrophy, with a particular focus on evidence gained from genetic mouse models (Table 1).
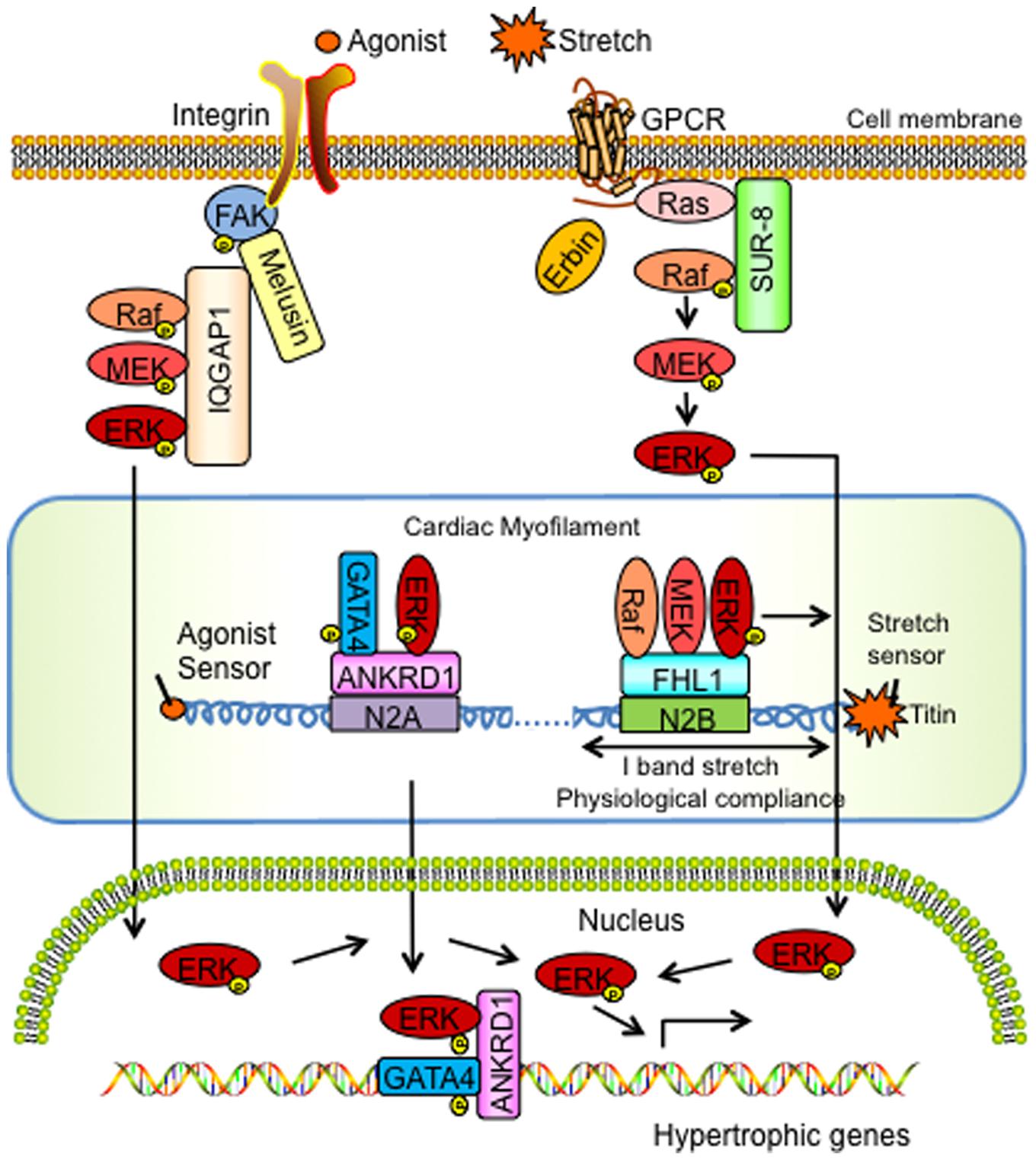
FIGURE 1. Schemata of the signaling actions of MAPK scaffolding proteins in cardiac hypertrophy and disease. ANKRD1, Ankyrin Repeat Domain 1; ERK, extracellular signal-regulated kinase; FAK, focal adhesion kinase; FHL1, Four and a half LIM domain protein-1; GATA4, GATA binding protein-4; GPCR, G Protein Coupled Receptor; IQGAP1, IQ motif-containing guanine-5′-triphosphate hydrolase activating protein; MEK, mitogen-activated protein kinase kinase; SUR-8, Suppressor of Ras-8.
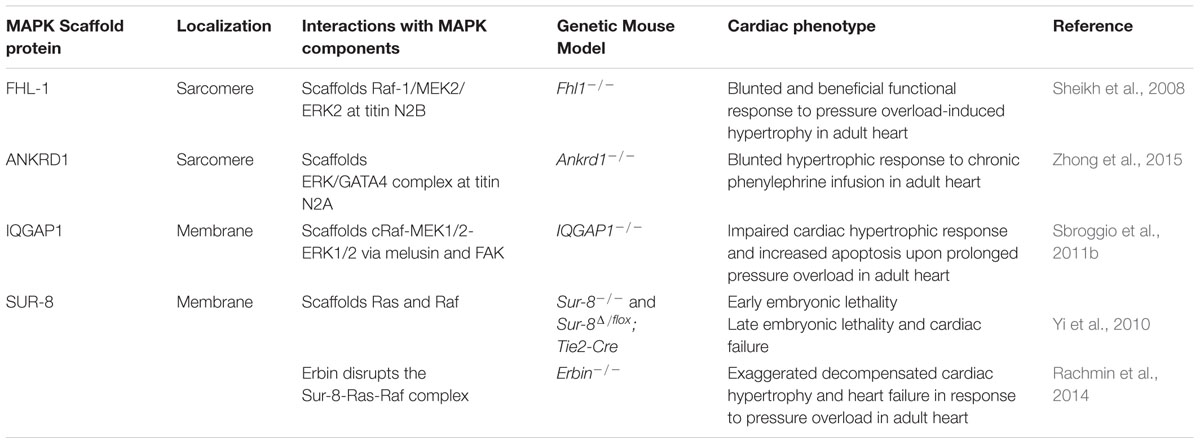
TABLE 1. Genetic mouse models targeting MAPK scaffold proteins and ERK-mediated signaling in cardiac muscle and their role in cardiac hypertrophy.
Studies from our laboratory have reported FHL1 as a novel ERK associated MAPK scaffold protein that mediates MAPK/ERK signaling responses at the sarcomere during stress-induced cardiac hypertrophy (Sheikh et al., 2008; Raskin et al., 2012). FHL1 is first detected at embryonic day (E) 8.5 in the mouse heart during early development (Chu et al., 2000). Although FHL1 is ubiquitously expressed, it is found to be highly abundant in mammalian striated muscle (Chu et al., 2000). Various studies have shown that FHL1 interacts with multiple binding partners, including structural proteins, signal transducers, transcription regulators, receptors, and channels, to regulate different biological processes (Shathasivam et al., 2010). However, most notably in the heart, FHL1 becomes highly upregulated during hypertrophic conditions in mice and humans (Hwang et al., 1997, 2000; Chu et al., 2000; Lim et al., 2001). Studies in FHL1 knockout (Fhl1-/-) mice demonstrated that FHL1 is dispensable for survival and basal heart function (Sheikh et al., 2008). However, under conditions of pressure overload, adult Fhl1-/- hearts displayed a blunted response to cardiac hypertrophy (Sheikh et al., 2008). Most notably, Fhl1-/- mice subjected to transverse aortic constriction (TAC) maintained cardiac function comparable to sham-operated control mice, and unlike control mice, which exhibited pathological cardiac hypertrophy and dysfunction (Sheikh et al., 2008), highlighting that loss of FHL1 is essential for preservation of cardiac function in pressure overload.
In vivo studies performed in transgenic mice overexpressing a constitutively active form of Gαq selectively in the heart, highlighted that FHL1 intersected with Gαq signaling pathways (Sheikh et al., 2008). The Gαq signaling pathway has also been shown to be critical for pressure-overload induced cardiac hypertrophy (D’Angelo et al., 1997; Wettschureck et al., 2001). Interestingly, deletion of Fhl1 was shown to be sufficient to prevent the cardiomyopathy in Gαq overexpressing transgenic mice (Sheikh et al., 2008). Yeast two-hybrid and co-immunoprecipitation based assays further identified that FHL1 could directly interact with MAPK components, Raf1, MEK2, and ERK2 in vitro and in in vivo (Sheikh et al., 2008). Immunofluorescence microscopy studies further showed that the FHL1/Raf-1/ MEK2/ERK2 complex all localized to the sarcomeric I band, and thus highlighted a MAPK scaffolding function for FHL1 at the titin N2B region of the sarcomere (Sheikh et al., 2008).
In terms of downstream effects on MAPK/ERK signaling, FHL1 overexpression was sufficient to increase ERK1/2 phosphorylation in cardiomyocytes in vitro (Sheikh et al., 2008). In vivo studies performed in FHL1 deficient settings such as TAC subjected Fhl1-/- adult hearts as well as double Fhl1-/-/Gαq constitutively overexpressing adult hearts demonstrated a blunted response to ERK1/2 phosphorylation, suggesting that FHL1 can positively regulate Raf-1/MEK/ERK-mediated signaling in a Gαq stimulus-specific manner (Sheikh et al., 2008). Convergence of these pathways to titin N2B function was shown in isolated adult Fhl1-/- cardiac muscles subjected to a maximum extension of 15–20% stretch (Sheikh et al., 2008; Raskin et al., 2012). Stretched Fhl1-/- cardiac muscles displayed a loss of upregulation of Elk-1 mRNA (transcriptional target of ERK1/2 signaling) and diastolic stress when compared with controls, further demonstrating an impact of loss of FHL1 and MAPK-ERK signaling on cardiac muscle compliance (Sheikh et al., 2008; Raskin et al., 2012).
Phosphorylation of titin N2B has been reported to be pivotal in mediating muscle compliance in cardiomyocytes (Fukuda et al., 2005). Interestingly, titin N2B isoform expression was increased in adult Fhl1-/- cardiac muscles and these muscles were also found to be insensitive to titin N2B phosphorylation-mediated diastolic reduction, as they intrinsically exhibited functions associated with maximal titin N2B phosphorylation (Raskin et al., 2012). In vitro studies demonstrated that ERK2 can directly phosphorylate titin N2B (Ser-3873, Ser-3915, Ser-3965) and that FHL1 functioned to interfere with ERK2- mediated titin N2B phosphorylation (Raskin et al., 2012). Thus, in FHL1 deficient settings titin N2B phosphorylation was thought to be hyper or maximally phosphorylated correlating with increased compliance. These studies lead to a model whereby FHL1 acts to scaffold MAPK components at titin N2B in order to regulate cardiac muscle compliance (via titin N2B phosphorylation) and hypertrophic signaling under conditions of stretch/hypertrophic stress/Gq (Raskin et al., 2012). Interestingly in FHL1 deficient settings, a unique role for the MAPK component, ERK2, was identified as it was no longer tethered to titin N2B but instead “open” to directly phosphorylate titin N2B to increase compliance of cardiac muscle as opposed to being sequestered to participate in hypertrophic signaling (Raskin et al., 2012). These results suggest that FHL1 functions as a MAPK scaffold by directly interacting with Raf-1/MEK2/ ERK2 to physically insulate the MAPK pathway to the titin N2B region of the sarcomere in a Gq stimulus-specific manner and efficiently transmit MAPK signals to regulate cardiac hypertrophy (Figure 1).
Recent studies also suggested that ANKRD1 [also known as cardiac adriamycin responsive protein (CARP)] is an ERK associated protein that mediates ERK signaling responses at the sarcomere (titin N2A) during phenylephrine-induced hypertrophy (Zhong et al., 2015). ANKRD1 is a transcriptional cofactor and highly expressed in embryonic cardiomyocytes (Mikhailov and Torrado, 2008). ANKRD1 was shown to negatively regulate gene expression of cardiac-specific genes in fetal cardiomyocytes in vitro (Jeyaseelan et al., 1997; Zou et al., 1997; Kuo et al., 1999). In adult cardiac muscle ANKRD1 expression is developmentally downregulated but was reported to increase in animal models of cardiac hypertrophy and human patients with various forms of cardiomyopathy leading to heart failure (Arber et al., 1997; Kuo et al., 1999; Aihara et al., 2000; Maeda et al., 2002; Zolk et al., 2003). Genetic mutations in ANKRD1 were also found to be causative for hypertrophic and dilated cardiomyopathy (Arimura et al., 2009; Duboscq-Bidot et al., 2009; Moulik et al., 2009). Based on the direct interaction between ANKRD1 and titin N2A region, it was thought that ANKRD1 may play a critical role in linking stretch-induced signaling pathways and muscle gene expression (Miller et al., 2003). However, in vivo studies performed in Ankrd1 knockout mice (Ankrd1-/-) highlighted an indispensable role for ANKRD1 in the heart at basal conditions (Bang et al., 2014). In addition, Ankrd1-/- mice subjected to pressure overload via TAC, were also able to maintain a normal cardiac hypertrophic response, similar to control mice (Bang et al., 2014). These results were supported by triple (Ankrd1/Ankrd2/Ankrd23) MARP knockout mice, which also mounted a normal cardiac hypertrophic response to TAC similar to controls (Bang et al., 2014).
In vitro and in vivo studies have suggested that ANKRD1 may be coupled to the α-adrenergic associated hypertrophic pathway (Maeda et al., 2002; Zhong et al., 2015). Ankrd1-/- mouse hearts and Ankrd1 knockdown by shRNA was shown to attenuate the hypertrophic response to the α-adrenergic agonist, phenylephrine (PE) (Zhong et al., 2015). Specifically, PE infused Ankrd1-/- mice displayed reduced heart size similar to vehicle controls (Zhong et al., 2015). Co-immunoprecipitation and immunostaining studies showed that ANKRD1, ERK, and GATA4 formed a complex at the sarcomeric I band and nucleus (Zhong et al., 2015). Cardiomyocytes showed that cardiomyocytes transfected with Ankrd1 siRNA resulted in diffuse cytoplasmic staining of both ERK1/2 and GATA4, suggesting that ANKRD1 may serve as a scaffolding protein to anchor ERK and GATA4 (Zhong et al., 2015). GATA4, which is a zinc finger transcription factor, plays a pivotal role in cardiac development and cardiac hypertrophy (Oka et al., 2006). Recent studies highlight that ERK phosphorylates GATA4 at S105, leading to enhanced GATA4-DNA binding and activation of ERK1/2-induced cardiac hypertrophy (van Berlo et al., 2011). These findings suggested that ANKRD1 acts as a scaffold to enhance GATA4 phosphorylation via ERK1/2 in the sarcomere and that the ANKRD1-ERK-GATA4 complex is translocated to the nucleus upon PE stimulation to regulate hypertrophic responses (Figure 1). In vitro findings support this mechanism as cardiomyocytes transfected with Ankrd1 siRNA were shown to have an attenuated increase in PE-mediated pERK1/2 and pGATA4 as well as signals related to the nuclear ERK1/2-GATA4 complex (Zhong et al., 2015).
IQGAP1 also acts as a membrane-bound MAPK scaffold to regulate responses associated with cardiac hypertrophy. IQGAP1 is a highly conserved cytoplasmic scaffold protein that is ubiquitously expressed (Weissbach et al., 1994). It interacts with many binding partners and is involved in cytoskeleton, cell-cell adhesion, protein trafficking, and cell signaling (Smith et al., 2015). In vivo studies performed in IQGAP1 null mice, demonstrated a dispensable role for IQGAP1 in heart function at basal conditions (Sbroggio et al., 2011b). Also, during chronic pressure overload induced by TAC, IQGAP1 null mice initially displayed a compensatory hypertrophic response identical to control mice. However, upon prolonged pressure overload, IQGAP1 null mice displayed unfavorable cardiac remodeling, contractile dysfunction, impaired cardiac hypertrophy, and increased apoptosis when compared with control mice (Sbroggio et al., 2011b). The deletion of IQGAP1 did not affect global MEK-ERK1/2 signaling in the heart, but selectively impaired the second wave of ERK1/2 activation which peaked at 4 days in response to pressure overload (Sbroggio et al., 2011b).
Mechanisms underlying the effects of IQGAP1 in cardiac hypertrophy may be due to a direct interaction with melusin. Melusin is a muscle specific protein that binds to the cytoplasmic domain of β1 integrin, thought to act as a mechanical stretch sensor (Brancaccio et al., 1999). Melusin expression increases with various hypertrophic stimuli (cyclic mechanical stretch, angiotensin-II, endothelin-1, and phenelyphrine) in neonatal ventricular cardiomyocytes in vitro (Aro et al., 2013), suggesting a direct role in cardiac hypertrophy. In vivo studies performed with Melusin-null mice subjected to pressure overload demonstrated reduced left ventricular hypertrophy, accelerated development of dilated cardiomyopathy and contractile dysfunction (Brancaccio et al., 2003). Biphasic effects of melusin were demonstrated via in vivo studies involving transgenic mice that selectively overexpressed melusin in the heart (De Acetis et al., 2005). Cardiac-specific overexpression of melusin induced cardiomyocyte hypertrophy during basal conditions and retained compensatory hypertrophy during acute pressure overload; however, the transition toward heart failure through activation of ERK phosphorylation was prevented in response to prolonged pressure overload (De Acetis et al., 2005). Studies performed using transgenic mice overexpressing melusin also demonstrated cardioprotective effects of melusin during myocardial infarction as well as ERK-dependent cardioprotective effects of melusin during ischemia-reperfusion injury (Penna et al., 2014; Unsold et al., 2014).
Cross-talk between IQGAP1 and melusin was shown in hearts of double IQGAP1 null/melusin overexpressing mice, which demonstrated that the absence of IQGAP1 abrogated melusin-induced ERK1/2 hyper-phophorylation in both basal conditions and in response to pressure overload (Sbroggio et al., 2011a). Co-immunoprecipitation studies in the heart demonstrated that melusin, IQGAP1 and cRaf-MEK1/2-ERK1/2 formed a supercomplex with focal adhesion kinase (FAK) at the membrane (Sbroggio et al., 2011a).
Focal adhesion kinase is a well-known downstream target of integrin, which is activated by biomechanical stress and agonists as well as mediates ERK1/2 pathway activation (Lal et al., 2007). Studies performed in FAK deficient settings showed that depletion of FAK does not affect the phenotype of non-stimulated cardiomyocytes, but blocked the up-regulation of hypertrophic gene markers after pressure overload (Marin et al., 2008). Furthermore, in vivo studies showed that depletion of global myocardial FAK by RNA interference or cardiac-specific FAK knockout mice prevented left ventricular hypertrophy and the deterioration of cardiac structure after TAC and angiotensin II infusion (DiMichele et al., 2006; Clemente et al., 2007). These studies further suggested an important requirement for FAK in melusin-dependent ERK activation and cardiomyocyte hypertrophy during pressure overload. Cardiomyocytes treated with the FAK inhibitor (PF573228) or knockdown (FAK shRNA) demonstrated an abolishment of melusin-dependent hypertrophy and reduced ERK1/2 phosphorylation (Sbroggio et al., 2011a). Thus, FAK, melusin and IQGAP1 were all shown to be required for ERK1/2 activation in response to pressure overload (Sbroggio et al., 2011a). These data altogether suggested a model whereby IQGAP1 scaffolds MAPK components (c-Raf, MEK1/2 and ERK1/2) at the plasma membrane via Melusin and FAK to form a supercomplex to regulate MAPK-ERK signaling and cardiac hypertrophy (Figure 1).
Suppressor of ras-8, a conserved leucine-rich repeat protein, was identified as a positive regulator of Ras-mediated signal transduction in C. elegans (Sieburth et al., 1998). Studies show that SUR-8 functions as a MAPK scaffold that forms a ternary complex with Ras and Raf, which then specifically enhances Ras-induced ERK activation by facilitating the interaction between Ras and Raf, without any effect on AKT or JNK activity (Li et al., 2000). Sur-8 conventional knockout mice were embryonic lethal at early stages (around E8.5) of mouse development (Yi et al., 2010). Endothelial-specific Sur-8 knockout mice displayed embryonic lethality at later stages of mouse development as well as multiple cardiac defects (Yi et al., 2010).
Although the mechanisms underlying how SUR-8 regulates MAPK signaling in the heart are not well understood, recent data suggests an intersection with Erbin and cardiac hypertrophy. Erbin is a LAP [leucine-rich repeat (LRR) and PDZ domain] family protein (Dillon et al., 2002). Recent studies demonstrated that LRR- and PDZ-domain-containing proteins play a key role in assembling protein complexes for signal transduction, suggesting a signaling role for Erbin (Birrane et al., 2003). Erbin has been reported to inhibit Ras-mediated activation of the MAPK pathway via an effect on SUR-8. Specifically, the LRR domain of Erbin was shown to bind to SUR-8 and inhibit the interaction of SUR-8 with Ras and Raf (Dai et al., 2006). Interestingly, Erbin inhibited Raf activation, but had no effect on Ras activation (Dai et al., 2006). Furthermore, Erbin overexpression was sufficient to inhibit the interaction between SUR-8 and Ras and Raf (Dai et al., 2006). Suppression of Erbin by shRNA increased the interaction of SUR-8 with Ras and Raf, resulting in enhanced Raf and ERK activation (Dai et al., 2006). Co-immunoprecipitation studies showed that Erbin can form an endogenous complex with SUR-8 in adult mouse hearts and cardiomyocytes and that this interaction was lost in Erbin deficient (Erbin-/-) hearts, which now resulted in p-Raf binding to SUR-8 and thus, ERK1/2 phosphorylation (Rachmin et al., 2014). Interestingly, Erbin expression is downregulated in adult mouse hearts subjected to hypertrophic agonists or TAC (Rachmin et al., 2014). Studies performed in Erbin-/- mice demonstrated that Erbin is dispensable for the heart during basal conditions; however, when these mice were subjected to pressure overload they displayed an exaggerated decompensated cardiac hypertrophy and failure when compared with controls (Rachmin et al., 2014). In terms of MAPK-ERK signaling, Erbin-/- mouse hearts exhibited a twofold increase in p-ERK when compared to controls at baseline and this effect was even more pronounced (threefold) under hypertrophic conditions (Rachmin et al., 2014). These data altogether suggested that Erbin is a negative regulator of the MAPK scaffold, SUR-8, and can disrupt the Sur-8-Raf-Ras complex to regulate cardiac hypertrophic signaling (Rachmin et al., 2014) (Figure 1).
Mitogen activated protein kinase scaffold proteins are emerging as important players in shaping and localizing the MAPK-ERK signaling response in cardiac muscle in order to simplify the response to distinct hypertrophic stimuli (e. g., pressure overload, Gαq, phenylephrine, etc.). Interestingly, loss of function studies in mouse models deficient in MAPK scaffold proteins bound to the sarcomere, such as FHL1 and ANKRD1, suggest a detrimental role in Gαq and phenylephrine-induced cardiac hypertrophy, respectively, while studies on MAPK scaffold proteins bound to the plasma membrane, such as IQGAP1 and Sur-8, suggest a protective role in mechanical induced-cardiac hypertrophy. These studies highlight that targeting distinct scaffolds may allow for more specific MAPK signaling modulation in pathological states associated with cardiac hypertrophy. Given the complexity and pleiotropic actions of the MAPK pathway in cardiac muscle, future studies should also be directed at determining (i) whether other hypertrophic associated MAPK scaffolds existed in cardiac muscle, (ii) how MAPK scaffolds insulated themselves from one another so that cardiac hypertrophic signaling is stringently regulated and (iii) how MAPK scaffolds regulated their hypertrophic function in cardiac muscle in a spatiotemporal manner. Studies focused on developing selective compounds that target these scaffolds may also provide a means to harness MAPK signaling to clinically intervene with the deleterious effects associated with cardiac hypertrophy and failure.
YL contributed to drafting the content, figure, and table in the review article. FS contributed to the design as well as finalizing the content, figure, and table in the review article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
FS is supported by grants from NIH/NHLBI (HL09780), American Heart Association (GRNT22940045), and Tobacco-Related Disease Research program (24RT-22).
Aihara, Y., Kurabayashi, M., Saito, Y., Ohyama, Y., Tanaka, T., Takeda, S., et al. (2000). Cardiac ankyrin repeat protein is a novel marker of cardiac hypertrophy: role of M-CAT element within the promoter. Hypertension 36, 48–53. doi: 10.1161/01.HYP.36.1.48
Arber, S., Hunter, J. J., Ross, J. Jr., Hongo, M., Sansig, G., Borg, J., et al. (1997). MLP-deficient mice exhibit a disruption of cardiac cytoarchitectural organization, dilated cardiomyopathy, and heart failure. Cell 88, 393–403. doi: 10.1016/S0092-8674(00)81878-4
Arimura, T., Bos, J. M., Sato, A., Kubo, T., Okamoto, H., Nishi, H., et al. (2009). Cardiac ankyrin repeat protein gene (ANKRD1) mutations in hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 54, 334–342. doi: 10.1016/j.jacc.2008.12.082
Aro, J., Tokola, H., Ronkainen, V. P., Koivisto, E., Tenhunen, O., Ilves, M., et al. (2013). Regulation of cardiac melusin gene expression by hypertrophic stimuli in the rat. Acta Physiol. (Oxf.) 207, 470–484. doi: 10.1111/apha.12044
Bang, M. L., Gu, Y., Dalton, N. D., Peterson, K. L., Chien, K. R., and Chen, J. (2014). The muscle ankyrin repeat proteins CARP, Ankrd2, and DARP are not essential for normal cardiac development and function at basal conditions and in response to pressure overload. PLoS ONE 9:e93638. doi: 10.1371/journal.pone.0093638
Bernardo, B. C., Weeks, K. L., Pretorius, L., and McMullen, J. R. (2010). Molecular distinction between physiological and pathological cardiac hypertrophy: experimental findings and therapeutic strategies. Pharmacol. Ther. 128, 191–227. doi: 10.1016/j.pharmthera.2010.04.005
Birrane, G., Chung, J., and Ladias, J. A. (2003). Novel mode of ligand recognition by the Erbin PDZ domain. J. Biol. Chem. 278, 1399–1402. doi: 10.1074/jbc.C200571200
Brancaccio, M., Fratta, L., Notte, A., Hirsch, E., Poulet, R., Guazzone, S., et al. (2003). Melusin, a muscle-specific integrin beta1-interacting protein, is required to prevent cardiac failure in response to chronic pressure overload. Nat. Med. 9, 68–75. doi: 10.1038/nm805
Brancaccio, M., Guazzone, S., Menini, N., Sibona, E., Hirsch, E., De Andrea, M., et al. (1999). Melusin is a new muscle-specific interactor for beta(1) integrin cytoplasmic domain. J. Biol. Chem. 274, 29282–29288. doi: 10.1074/jbc.274.41.29282
Brown, M. D., and Sacks, D. B. (2009). Protein scaffolds in MAP kinase signalling. Cell. Signal. 21, 462–469. doi: 10.1016/j.cellsig.2008.11.013
Bueno, O. F., De Windt, L. J., Tymitz, K. M., Witt, S. A., Kimball, T. R., Klevitsky, R., et al. (2000). The MEK1-ERK1/2 signaling pathway promotes compensated cardiac hypertrophy in transgenic mice. EMBO J. 19, 6341–6350. doi: 10.1093/emboj/19.23.6341
Chu, P. H., Ruiz-Lozano, P., Zhou, Q., Cai, C., and Chen, J. (2000). Expression patterns of FHL/SLIM family members suggest important functional roles in skeletal muscle and cardiovascular system. Mech. Dev. 95, 259–265. doi: 10.1016/S0925-4773(00)00341-5
Clemente, C. F., Tornatore, T. F., Theizen, T. H., Deckmann, A. C., Pereira, T. C., Lopes-Cendes, I., et al. (2007). Targeting focal adhesion kinase with small interfering RNA prevents and reverses load-induced cardiac hypertrophy in mice. Circ. Res. 101, 1339–1348. doi: 10.1161/CIRCRESAHA.107.160978
Dai, P., Xiong, W. C., and Mei, L. (2006). Erbin inhibits RAF activation by disrupting the sur-8-Ras-Raf complex. J. Biol. Chem. 281, 927–933. doi: 10.1074/jbc.M507360200
D’Angelo, D. D., Sakata, Y., Lorenz, J. N., Boivin, G. P., Walsh, R. A., Liggett, S. B., et al. (1997). Transgenic Galphaq overexpression induces cardiac contractile failure in mice. Proc. Natl. Acad. Sci. U.S.A. 94, 8121–8126. doi: 10.1073/pnas.94.15.8121
De Acetis, M., Notte, A., Accornero, F., Selvetella, G., Brancaccio, M., Vecchione, C., et al. (2005). Cardiac overexpression of melusin protects from dilated cardiomyopathy due to long-standing pressure overload. Circ. Res. 96, 1087–1094. doi: 10.1161/01.RES.0000168028.36081.e0
Dillon, C., Creer, A., Kerr, K., Kumin, A., and Dickson, C. (2002). Basolateral targeting of ERBB2 is dependent on a novel bipartite juxtamembrane sorting signal but independent of the C-terminal ERBIN-binding domain. Mol. Cell. Biol. 22, 6553–6563. doi: 10.1128/MCB.22.18.6553-6563.2002
DiMichele, L. A., Doherty, J. T., Rojas, M., Beggs, H. E., Reichardt, L. F., Mack, C. P., et al. (2006). Myocyte-restricted focal adhesion kinase deletion attenuates pressure overload-induced hypertrophy. Circ. Res. 99, 636–645. doi: 10.1161/01.RES.0000240498.44752.d6
Duboscq-Bidot, L., Charron, P., Ruppert, V., Fauchier, L., Richter, A., Tavazzi, L., et al. (2009). Mutations in the ANKRD1 gene encoding CARP are responsible for human dilated cardiomyopathy. Eur. Heart J. 30, 2128–2136. doi: 10.1093/eurheartj/ehp225
Fukuda, N., Wu, Y., Nair, P., and Granzier, H. L. (2005). Phosphorylation of titin modulates passive stiffness of cardiac muscle in a titin isoform-dependent manner. J. Gen. Physiol. 125, 257–271. doi: 10.1085/jgp.200409177
Fusello, A. M., Mandik-Nayak, L., Shih, F., Lewis, R. E., Allen, P. M., and Shaw, A. S. (2006). The MAPK scaffold kinase suppressor of Ras is involved in ERK activation by stress and proinflammatory cytokines and induction of arthritis. J. Immunol. 177, 6152–6158. doi: 10.4049/jimmunol.177.9.6152
Goldsmith, Z. G., and Dhanasekaran, D. N. (2007). G protein regulation of MAPK networks. Oncogene 26, 3122–3142. doi: 10.1038/sj.onc.1210407
Hwang, D. M., Dempsey, A. A., Lee, C. Y., and Liew, C. C. (2000). Identification of differentially expressed genes in cardiac hypertrophy by analysis of expressed sequence tags. Genomics 66, 1–14. doi: 10.1006/geno.2000.6171
Hwang, D. M., Dempsey, A. A., Wang, R. X., Rezvani, M., Barrans, J. D., Dai, K. S., et al. (1997). A genome-based resource for molecular cardiovascular medicine: toward a compendium of cardiovascular genes. Circulation 96, 4146–4203. doi: 10.1161/01.CIR.96.12.4146
Ishibe, S., Joly, D., Zhu, X., and Cantley, L. G. (2003). Phosphorylation-dependent paxillin-ERK association mediates hepatocyte growth factor-stimulated epithelial morphogenesis. Mol. Cell 12, 1275–1285. doi: 10.1016/S1097-2765(03)00406-4
Jeyaseelan, R., Poizat, C., Baker, R. K., Abdishoo, S., Isterabadi, L. B., Lyons, G. E., et al. (1997). A novel cardiac-restricted target for doxorubicin. CARP, a nuclear modulator of gene expression in cardiac progenitor cells and cardiomyocytes. J. Biol. Chem. 272, 22800–22808. doi: 10.1074/jbc.272.36.22800
Karandikar, M., Xu, S., and Cobb, M. H. (2000). MEKK1 binds raf-1 and the ERK2 cascade components. J. Biol. Chem. 275, 40120–40127. doi: 10.1074/jbc.M005926200
Kornfeld, K., Hom, D. B., and Horvitz, H. R. (1995). The ksr-1 gene encodes a novel protein kinase involved in Ras-mediated signaling in C. elegans. Cell 83, 903–913. doi: 10.1016/0092-8674(95)90206-6
Kuo, H., Chen, J., Ruiz-Lozano, P., Zou, Y., Nemer, M., and Chien, K. R. (1999). Control of segmental expression of the cardiac-restricted ankyrin repeat protein gene by distinct regulatory pathways in murine cardiogenesis. Development 126, 4223–4234.
Lal, H., Verma, S. K., Smith, M., Guleria, R. S., Lu, G., Foster, D. M., et al. (2007). Stretch-induced MAP kinase activation in cardiac myocytes: differential regulation through beta1-integrin and focal adhesion kinase. J. Mol. Cell Cardiol. 43, 137–147. doi: 10.1016/j.yjmcc.2007.05.012
Li, W., Han, M., and Guan, K. L. (2000). The leucine-rich repeat protein SUR-8 enhances MAP kinase activation and forms a complex with Ras and Raf. Genes Dev. 14, 895–900.
Lim, D. S., Roberts, R., and Marian, A. J. (2001). Expression profiling of cardiac genes in human hypertrophic cardiomyopathy: insight into the pathogenesis of phenotypes. J. Am. Coll. Cardiol. 38, 1175–1180. doi: 10.1016/S0735-1097(01)01509-1
Lorenz, K., Schmitt, J. P., Schmitteckert, E. M., and Lohse, M. J. (2009a). A new type of ERK1/2 autophosphorylation causes cardiac hypertrophy. Nat. Med. 15, 75–83. doi: 10.1038/nm.1893
Lorenz, K., Schmitt, J. P., Vidal, M., and Lohse, M. J. (2009b). Cardiac hypertrophy: targeting Raf/MEK/ERK1/2-signaling. Int. J. Biochem. Cell Biol. 41, 2351–2355. doi: 10.1016/j.biocel.2009.08.002
Lozano, J., Xing, R., Cai, Z., Jensen, H. L., Trempus, C., Mark, W., et al. (2003). Deficiency of kinase suppressor of Ras1 prevents oncogenic ras signaling in mice. Cancer Res. 63, 4232–4238.
Maeda, T., Sepulveda, J., Chen, H. H., and Stewart, A. F. (2002). Alpha(1)-adrenergic activation of the cardiac ankyrin repeat protein gene in cardiac myocytes. Gene 297, 1–9. doi: 10.1016/S0378-1119(02)00924-1
Marin, T. M., Clemente, C. F., Santos, A. M., Picardi, P. K., Pascoal, V. D., Lopes-Cendes, I., et al. (2008). Shp2 negatively regulates growth in cardiomyocytes by controlling focal adhesion kinase/Src and mTOR pathways. Circ. Res. 103, 813–824. doi: 10.1161/CIRCRESAHA.108.179754
McMullen, J. R., and Jennings, G. L. (2007). Differences between pathological and physiological cardiac hypertrophy: novel therapeutic strategies to treat heart failure. Clin. Exp. Pharmacol. Physiol. 34, 255–262. doi: 10.1111/j.1440-1681.2007.04585.x
Mikhailov, A. T., and Torrado, M. (2008). The enigmatic role of the ankyrin repeat domain 1 gene in heart development and disease. Int. J. Dev. Biol. 52, 811–821. doi: 10.1387/ijdb.082655am
Miller, M. K., Bang, M. L., Witt, C. C., Labeit, D., Trombitas, C., Watanabe, K., et al. (2003). The muscle ankyrin repeat proteins: CARP, ankrd2/Arpp and DARP as a family of titin filament-based stress response molecules. J. Mol. Biol. 333, 951–964. doi: 10.1016/j.jmb.2003.09.012
Moulik, M., Vatta, M., Witt, S. H., Arola, A. M., Murphy, R. T., McKenna, W. J., et al. (2009). ANKRD1, the gene encoding cardiac ankyrin repeat protein, is a novel dilated cardiomyopathy gene. J. Am. Coll. Cardiol. 54, 325–333. doi: 10.1016/j.jacc.2009.02.076
Nguyen, A., Burack, W. R., Stock, J. L., Kortum, R., Chaika, O. V., Afkarian, M., et al. (2002). Kinase suppressor of Ras (KSR) is a scaffold which facilitates mitogen-activated protein kinase activation in vivo. Mol. Cell. Biol. 22, 3035–3045. doi: 10.1128/MCB.22.9.3035-3045.2002
Oka, T., Maillet, M., Watt, A. J., Schwartz, R. J., Aronow, B. J., Duncan, S. A., et al. (2006). Cardiac-specific deletion of Gata4 reveals its requirement for hypertrophy, compensation, and myocyte viability. Circ. Res. 98, 837–845. doi: 10.1161/01.RES.0000215985.18538.c4
Pearson, G., Robinson, F., Beers Gibson, T., Xu, B. E., Karandikar, M., Berman, K., et al. (2001). Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr. Rev. 22, 153–183. doi: 10.1210/er.22.2.153
Penna, C., Brancaccio, M., Tullio, F., Rubinetto, C., Perrelli, M. G., Angotti, C., et al. (2014). Overexpression of the muscle-specific protein, melusin, protects from cardiac ischemia/reperfusion injury. Basic Res. Cardiol. 109:418. doi: 10.1007/s00395-014-0418-9
Raabo, E., Bartels, P. D., Blom, M., Christensen, N. J., Hansen, M. S., Jorgensen, P. J., et al. (1992). [Quality assurance of results of clinical chemical analyses in Danish hospital laboratories]. Ugeskr Laeger 154, 1627–1631.
Rachmin, I., Tshori, S., Smith, Y., Oppenheim, A., Marchetto, S., Kay, G., et al. (2014). Erbin is a negative modulator of cardiac hypertrophy. Proc. Natl. Acad. Sci. U.S.A. 111, 5902–5907. doi: 10.1073/pnas.1320350111
Raskin, A., Lange, S., Banares, K., Lyon, R. C., Zieseniss, A., Lee, L. K., et al. (2012). A novel mechanism involving four-and-a-half LIM domain protein-1 and extracellular signal-regulated kinase-2 regulates titin phosphorylation and mechanics. J. Biol. Chem. 287, 29273–29284. doi: 10.1074/jbc.M112.372839
Ruppert, C., Deiss, K., Herrmann, S., Vidal, M., Oezkur, M., Gorski, A., et al. (2013). Interference with ERK(Thr188) phosphorylation impairs pathological but not physiological cardiac hypertrophy. Proc. Natl. Acad. Sci. U.S.A. 110, 7440–7445. doi: 10.1073/pnas.1221999110
Sbroggio, M., Bertero, A., Velasco, S., Fusella, F., De Blasio, E., Bahou, W. F., et al. (2011a). ERK1/2 activation in heart is controlled by melusin, focal adhesion kinase and the scaffold protein IQGAP1. J. Cell Sci. 124, 3515–3524. doi: 10.1242/jcs.091140
Sbroggio, M., Carnevale, D., Bertero, A., Cifelli, G., De Blasio, E., Mascio, G., et al. (2011b). IQGAP1 regulates ERK1/2 and AKT signalling in the heart and sustains functional remodelling upon pressure overload. Cardiovasc. Res. 91, 456–464. doi: 10.1093/cvr/cvr103
Schaeffer, H. J., Catling, A. D., Eblen, S. T., Collier, L. S., Krauss, A., and Weber, M. J. (1998). MP1: a MEK binding partner that enhances enzymatic activation of the MAP kinase cascade. Science 281, 1668–1671. doi: 10.1126/science.281.5383.1668
Shathasivam, T., Kislinger, T., and Gramolini, A. O. (2010). Genes, proteins and complexes: the multifaceted nature of FHL family proteins in diverse tissues. J. Cell Mol. Med. 14, 2702–2720. doi: 10.1111/j.1582-4934.2010.01176.x
Sheikh, F., Raskin, A., Chu, P. H., Lange, S., Domenighetti, A. A., Zheng, M., et al. (2008). An FHL1-containing complex within the cardiomyocyte sarcomere mediates hypertrophic biomechanical stress responses in mice. J. Clin. Invest. 118, 3870–3880. doi: 10.1172/JCI34472
Sieburth, D. S., Sun, Q., and Han, M. (1998). SUR-8, a conserved Ras-binding protein with leucine-rich repeats, positively regulates Ras-mediated signaling in C. elegans. Cell 94, 119–130. doi: 10.1016/S0092-8674(00)81227-1
Smith, J. M., Hedman, A. C., and Sacks, D. B. (2015). IQGAPs choreograph cellular signaling from the membrane to the nucleus. Trends Cell Biol. 25, 171–184. doi: 10.1016/j.tcb.2014.12.005
Sundaram, M., and Han, M. (1995). The C. elegans ksr-1 gene encodes a novel Raf-related kinase involved in Ras-mediated signal transduction. Cell 83, 889–901. doi: 10.1016/0092-8674(95)90205-8
Therrien, M., Chang, H. C., Solomon, N. M., Karim, F. D., Wassarman, D. A., and Rubin, G. M. (1995). KSR, a novel protein kinase required for RAS signal transduction. Cell 83, 879–888. doi: 10.1016/0092-8674(95)90204-X
Unsold, B., Kaul, A., Sbroggio, M., Schubert, C., Regitz-Zagrosek, V., Brancaccio, M., et al. (2014). Melusin protects from cardiac rupture and improves functional remodelling after myocardial infarction. Cardiovasc. Res. 101, 97–107. doi: 10.1093/cvr/cvt235
van Berlo, J. H., Elrod, J. W., Aronow, B. J., Pu, W. T., and Molkentin, J. D. (2011). Serine 105 phosphorylation of transcription factor GATA4 is necessary for stress-induced cardiac hypertrophy in vivo. Proc. Natl. Acad. Sci. U.S.A. 108, 12331–12336. doi: 10.1073/pnas.1104499108
Vomastek, T., Schaeffer, H. J., Tarcsafalvi, A., Smolkin, M. E., Bissonette, E. A., and Weber, M. J. (2004). Modular construction of a signaling scaffold: MORG1 interacts with components of the ERK cascade and links ERK signaling to specific agonists. Proc. Natl. Acad. Sci. U.S.A. 101, 6981–6986. doi: 10.1073/pnas.0305894101
Weissbach, L., Settleman, J., Kalady, M. F., Snijders, A. J., Murthy, A. E., Yan, Y. X., et al. (1994). Identification of a human rasGAP-related protein containing calmodulin-binding motifs. J. Biol. Chem. 269, 20517–20521.
Wettschureck, N., Rutten, H., Zywietz, A., Gehring, D., Wilkie, T. M., Chen, J., et al. (2001). Absence of pressure overload induced myocardial hypertrophy after conditional inactivation of Galphaq/Galpha11 in cardiomyocytes. Nat. Med. 7, 1236–1240. doi: 10.1038/nm1101-1236
Yamaguchi, O., Watanabe, T., Nishida, K., Kashiwase, K., Higuchi, Y., Takeda, T., et al. (2004). Cardiac-specific disruption of the c-raf-1 gene induces cardiac dysfunction and apoptosis. J. Clin. Invest. 114, 937–943. doi: 10.1172/JCI200420317
Yi, J., Chen, M., Wu, X., Yang, X., Xu, T., Zhuang, Y., et al. (2010). Endothelial SUR-8 acts in an ERK-independent pathway during atrioventricular cushion development. Dev. Dyn. 239, 2005–2013. doi: 10.1002/dvdy.22343
Zheng, M., Dilly, K., Dos Santos Cruz, J., Li, M., Gu, Y., Ursitti, J. A., et al. (2004). Sarcoplasmic reticulum calcium defect in Ras-induced hypertrophic cardiomyopathy heart. Am. J. Physiol. Heart Circ. Physiol. 286, H424–H433. doi: 10.1152/ajpheart.00110.2003
Zhong, L., Chiusa, M., Cadar, A. G., Lin, A., Samaras, S., Davidson, J. M., et al. (2015). Targeted inhibition of ANKRD1 disrupts sarcomeric ERK-GATA4 signal transduction and abrogates phenylephrine-induced cardiomyocyte hypertrophy. Cardiovasc. Res. 106, 261–271. doi: 10.1093/cvr/cvv108
Zolk, O., Marx, M., Jackel, E., El-Armouche, A., and Eschenhagen, T. (2003). Beta-adrenergic stimulation induces cardiac ankyrin repeat protein expression: involvement of protein kinase A and calmodulin-dependent kinase. Cardiovasc. Res. 59, 563–572. doi: 10.1016/S0008-6363(03)00476-0
Keywords: MAPK and ERK signaling, cardiac myocyte, scaffolding proteins, mouse models, cardiac hypertrophy
Citation: Liang Y and Sheikh F (2016) Scaffold Proteins Regulating Extracellular Regulated Kinase Function in Cardiac Hypertrophy and Disease. Front. Pharmacol. 7:37. doi: 10.3389/fphar.2016.00037
Received: 19 December 2015; Accepted: 11 February 2016;
Published: 29 February 2016.
Edited by:
Kristina Lorenz, University of Wuerzburg, GermanyReviewed by:
Bin-Nan Wu, Kaohsiung Medical University, TaiwanCopyright © 2016 Liang and Sheikh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Farah Sheikh, ZmFzaGVpa2hAdWNzZC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.