 Laura Rinaldi
Laura Rinaldi
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 04 September 2015
Sec. Experimental Pharmacology and Drug Discovery
Volume 6 - 2015 | https://doi.org/10.3389/fphar.2015.00177
This article is part of the Research Topic Frontiers in the Pharmacological Manipulation of Intracellular cAMP Levels View all 18 articles
Phosphorylation waves drive the propagation of signals generated in response to hormones and growth factors in target cells. cAMP is an ancient second messenger implicated in key biological functions. In mammals, most of the effects elicited by cAMP are mediated by protein kinase A (PKA). Activation of the kinase by cAMP results in the phosphorylation of a variety of cellular substrates, leading to differentiation, proliferation, survival, metabolism. The identification of scaffold proteins, namely A-Kinase Anchor proteins (AKAPs), that localize PKA in specific cellular districts, provided critical cues for our understanding of the role played by cAMP in cell biology. Multivalent complexes are assembled by AKAPs and include signaling enzymes, mRNAs, adapter molecules, receptors and ion channels. A novel development derived from the molecular analysis of these complexes nucleated by AKAPs is represented by the presence of components of the ubiquitin-proteasome system (UPS). More to it, the AKAP complex can be regulated by the UPS, eliciting relevant effects on downstream cAMP signals. This represents a novel, yet previously unpredicted interface between compartmentalized signaling and the UPS. We anticipate that impairment of these regulatory mechanisms could promote cell dysfunction and disease. Here, we will focus on the reciprocal regulation between cAMP signaling and UPS, and its relevance to human degenerative and proliferative disorders.
Since the discovery of cyclic adenosine 3′,5′-monophosphate (cAMP) in the late 1950s, significant advances have been made to better understand the link between the cAMP and the regulation of downstream signaling and cellular homeostasis. The principal elements of the cAMP cascade have been intensively studied, both at functional and structural side, delineating a complex and finely regulated network of signaling scaffolds and regulatory proteins (Walsh and Van Patten, 1994). cAMP levels are tightly regulated through the balance between two classes of enzymes: the adenylyl cyclases (ACs) and the cyclic nucleotide phosphodiesterases (PDEs). The main effector of cAMP is protein kinase A (PKA), whose role is fundamental in the propagation of the signal downstream to target substrates/effectors (Taylor et al., 2005). The duration and the amplitude of the propagating signal are controlled by a combination of different classes of ACs, protein kinases, PDE, phosphatases (PPs) and scaffold proteins (Figure 1).
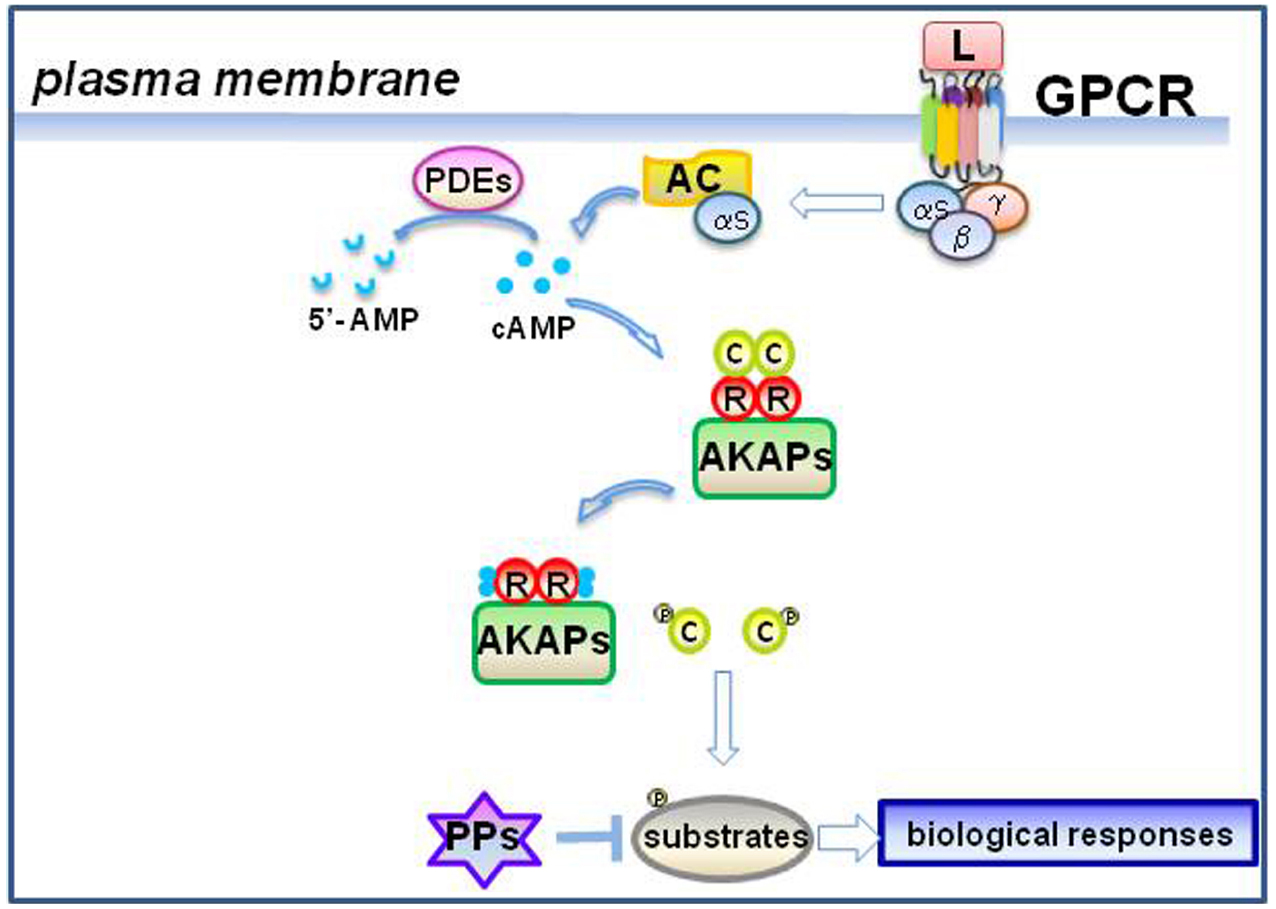
Figure 1. GPCR stimulation and cAMP signaling. Ligand-induced activation of a GPCR dissociates heterotrimeric G proteins and activates the adenylyl cyclase (AC) through Gαs subunit (Rosenbaum et al., 2009). AC converts ATP into cAMP. cAMP binding to regulatory (R) subunits of AKAP-assembled PKA dissociates the holoenzyme and activates the catalytic (C) subunits (Taylor et al., 2005). Phosphorylation of cellular substrates by C evokes plenty of biological responses. Phosphodiesterases (PDEs) converts the cAMP in 5′-AMP and decrease cAMP signaling (Maurice et al., 2014). Dephosphorylation of substrates by protein phosphatases (PPs) contributes to attenuate the signal (Zhang et al., 2013).
G protein–coupled receptors (GPCRs) constitute a large family of membrane proteins that transduce signals from the extracellular microenvironment to inside cell (Rosenbaum et al., 2009). The binding of extracellular ligand to its cognate GPCR at the cell membrane activates AC, which in turn generates cAMP at discrete points along the plasma membrane. The mammalian ACs are encoded by nine independent genes differentially expressed in several cell types and tissues (Iyengar, 1993). The cAMP-generating activity of ACs is stimulated by the interaction with the stimulatory α subunit of the G-proteins (Gαs). In the absence of ligand, Gαs forms a heterotrimeric complex with the β and the γ subunits. Once activated, the GPCR causes the dissociation of heterotrimeric G-proteins, with consequent activation of ACs by the Gαs subunit (Cooper and Tabbasum, 2014). Continuous or repeated pulses of hormone stimulation downregulate GPCR activation. This phenomenon, called receptor desensitization (Reiter and Lefkowitz, 2006), includes two phases: (1) acute desensitization, which involves the recruitment of β-arrestin to the activated GPCR, impairing the coupling between the receptor and G-proteins; (2) long-term desensitization which consists in the internalization and lysosomal degradation of the receptors (Sibley and Lefkowitz, 1985; Bouvier et al., 1989; Moore et al., 2007). Although stimulation by Gαs is the major mechanism of AC activation, different isoforms of ACs can receive signals from a variety of sources, as kinases (PKA, PKC and Calmodulin kinase) or Ca2+, supporting and integrating distinct signal transduction pathways (Timofeyev et al., 2013; Zhang et al., 2013).
The cAMP-PDEs are enzymes that hydrolize the 3′,5′ phosphodiester bond in the second messenger cAMP, producing 5′-AMP (Maurice et al., 2014). By reducing the levels of cAMP, PDEs regulate the duration and amplitude of the cyclic nucleotide signaling. PDEs are encoded by 21 genes that generate 11 different families (PDE 1–11) that share structural similarities, but different substrate specificity, regulatory mechanisms and kinetics (Maurice et al., 2014). PDEs can hydrolyze cAMP (PDE4, PDE7, and PDE8), cGMP (PDE5, PDE6, and PDE9) or both cyclic nucleotides (PDE1, PDE2, PDE3, PDE10, and PDE11; Francis et al., 2011). The N-terminal regulatory region of PDEs controls the subcellular localization of the enzymes (Kenan et al., 2000). The differential distribution of PDEs within the cell generates intracellular microdomains of the second messenger that locally enhance the sensitivity and specificity of the signals carried out by cAMP (Lomas and Zaccolo, 2014). In this context, the use of cAMP biosensors, such as those utilizing fluorescence resonance energy transfer (FRET), contributed to dissect and visualize compartmentalized pools of cAMP that are generated in response to GPCR stimulation (Stefan et al., 2007). The establishment of the so-called “signalosome” is based on the protein–protein interaction network among the unique combinations of cyclic nucleotides generators (AC), effectors (PKA, EPAC, and cAMP-gated ion channels), degrading PDEs and scaffolds proteins (A-Kinase Anchor Proteins, AKAPs).
In eukaryotes, most of the effects elicited by cAMP depend on the activation of PKA. This kinase consists of a tetramer composed of two regulatory (R) and two catalytic (C) subunits. The binding of cAMP to R subunit dissociates the PKA holoenzyme and releases the active C subunit, which in turn phosphorylates a wide array of cellular substrates, controlling different aspects of cell physiology (Taylor et al., 2008). The biochemical and functional features of PKA holoenzymes are largely determined by the structure, properties and relative abundance of the R subunits. The analysis of the kinetics of PKA activation/de-activation cycles contributed to understand the mechanisms of cAMP action on the effector kinase (Knighton et al., 1991). PKA stimulation by cAMP is followed by a refractory phase where a coordinated activation of Ser/Thr PPs, PKA inhibitors (PKIs) and changes in the ratio between R and C subunits eventually attenuate the signal (Armstrong et al., 1995; Canettieri et al., 2003).
The localization of PKA in the cell is mediated by scaffolding proteins, namely A-kinase anchoring proteins (AKAPs). AKAPs belong to a group of structurally different proteins that share the common feature to target the PKA holoenzyme in close proximity of its substrate (Michel and Scott, 2002). Each AKAP contains a PKA-binding motif that binds the R subunit of PKA and a targeting domain that directs the kinase to specific subcellular compartments. Biochemical and structural studies identified a conserved PKA-binding domain of AKAPs that forms an amphipathic helical wheel composed of 14–18-residues (Carr et al., 1992). The helical wheel binds with high affinity the N-terminal docking/dimerization (D/D) domain of the PKA-R dimer (Newlon et al., 1999, 2001). In particular, the hydrophobic residues of the helical wheel are located in the interior face, while charged residues align on the exterior surface. Although most of the AKAPs bind to RII subunit (Herberg et al., 2000; Carnegie and Scott, 2003), several RI-specific AKAPs have been characterized (Huang et al., 1997; Angelo and Rubin, 1998; Means et al., 2011; Burgers et al., 2012). The residues determining binding specificity of AKAPs to RI and RII have been partially defined (Alto et al., 2003). Disruption of the amphipathic helical wheel abrogates the binding to R subunits, both in vitro and in vivo (Welch et al., 2010). By modulating the dissemination of cAMP signals inside the cell, AKAPs control important biological responses, such as hormone secretion, metabolism, differentiation, cell growth and survival, synaptic transmission, learning and memory (Rubin, 1994; Alto et al., 2002; Tasken and Aandahl, 2004). AKAPs form a macromolecular complex, named transduceosome, that assembles components of cAMP generating systems (receptors and ACs), effectors (PKA and Epac) and attenuating enzymes (PDEs and PPs). This implies that complexes nucleated by AKAPs create intracellular domains where distinct signaling pathways converge and are locally attenuated or amplified, optimizing the biological response to extracellular stimuli (Feliciello et al., 2001, 2005; Dell’Acqua et al., 2006; Welch et al., 2010).
The ubiquitin-proteasome system (UPS) is emerging as an important control mechanism of cell growth, survival and metabolism. Degradation of a protein via UPS involves modification of the substrate protein by the covalent attachment of multiple ubiquitin molecules. The ubiquitin-tagged protein is eventually degraded through the proteasome (Ciechanover, 2005). Defects of the UPS may represent the trigger of several important human disorders (Wang and Hill, 2015; Dantuma and Bott, 2014; Ortega and Lucas, 2014; Schmidt and Finley, 2014). The ubiquitylation is mediated by the attachment of ubiquitin to the ε-amine of lysine residues of target proteins. This process requires a series of ATP-dependent enzymatic steps catalyzed by E1 (ubiquitin activating), E2 (ubiquitin conjugating), and E3 (ubiquitin ligating) enzymes (Ciechanover, 2003). The result of this sequential cascade of events is the covalent attachment of ubiquitin molecules to lysine residues on the target protein. These modifications can involve either a single ubiquitin (mono-ubiquitylation) or a chain of ubiquitin (poly-ubiquitylation; Ramanathan and Ye, 2012). Poly-ubiquitylation of a substrate is mostly related to protein degradation through the proteasome (Ciechanover, 2005). By modulating the protein levels, the UPS influences many cellular processes. Polyubiquitylated proteins can also follow a non-degradative pathway (De Bie and Ciechanover, 2011). This mechanism may control the intracellular trafficking of the target protein or its activity (Bonifacino and Weissman, 1998). In this case, de-ubiquitinating enzymes (DUBs), by removing the ubiquitin moieties, can restore the localization/activity of the modified protein (De Bie and Ciechanover, 2011).
The cAMP-PKA signaling is regulated by- and can regulate the UPS at different steps, giving rise to a complex interactive and regulatory network that controls different aspects of cell fate (Figure 2).
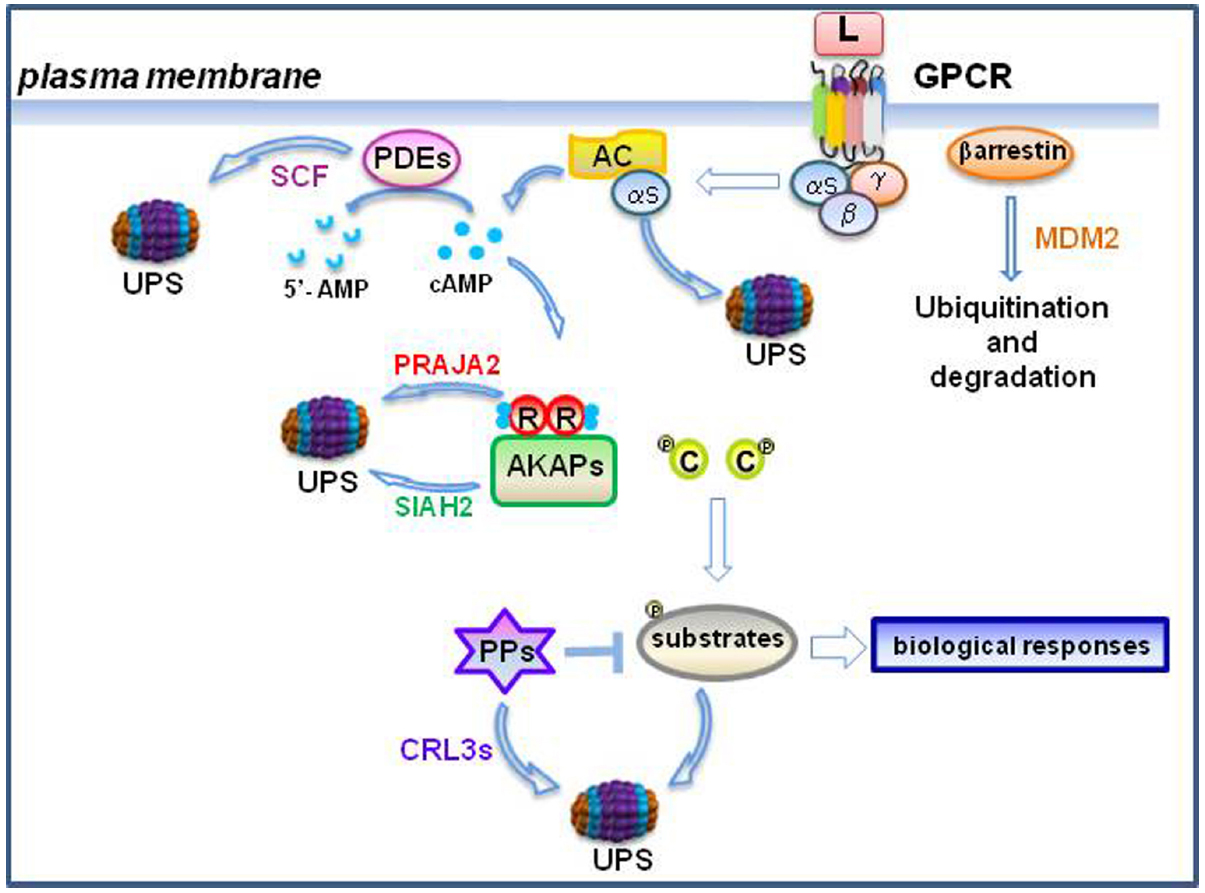
Figure 2. Feed-back and feed-forward control of cAMP by the ubiquitin-proteasome system (UPS). In the burst phase, ubiquitylation of cAMP-phosphodiesterases (PDEs) through the E3 ligase SCF complex contributes to modulate cAMP levels (Zhu et al., 2010). R subunits undergo to proteolysis by the praja2-UPS pathway. Loss of R subunits sustains PKA signaling (Lignitto et al., 2011). Moreover, ubiquitylation of protein phosphatases (PPs) by cullin E3 ligases (CRL3s) further modulates phosphorylation-dependent downstream signaling (Xu et al., 2014). During the desensitization phase, agonist-induced ubiquitylation of both receptor and b-arrestins promotes receptor endocytosis and degradation, attenuating downstream signaling (Reiter and Lefkowitz, 2006). Gαs subunits are ubiquitylated and degraded by the UPS (Zha et al., 2015). During hypoxia, Siah2-mediated ubiquitylation and proteolysis of AKAP121 modulates mitochondrial activity (Carlucci et al., 2008a).
At cell membrane, the ubiquitylation and consequent proteolysis of receptors by the UPS contributes to post-stimulus receptor desensitization (Bonifacino and Weissman, 1998). As example, following β adrenergic receptor 2 (β-2AR) stimulation, the adaptor protein ARRDC3 (arrestin domain containing 3) recruits the E3 ligase NEDD4 (neural precursor development downregulated protein 4) close to β-2AR. Concomitant inhibition of the deubiquitinase USP20 (Ubiquitin-specific-processing protease 20) by PKA favors ubiquitylation and degradation of the receptor by NEDD4 (Nabhan et al., 2010; Kommaddi et al., 2015). Agonist-induced ubiquitylation of both receptor and β-arrestins (β-receptor regulatory proteins) also contributes to regulate receptor endocytosis. Internalized GPCRs can undergo to degradation or be recycled back to the cell surface (Shenoy et al., 2001). β-adrenergic signal transduction is the major pathway involved in the maintenance of cardiac muscle contraction. Reduced response to β-adrenergic stimulation and pathological cardiac hypertrophy are hallmark of heart failure (Port and Bristow, 2001; Tilley, 2011) In this context, blunted response to agonist might be a consequence of decreased levels of Gαs subunit (Tang et al., 2008). Accordingly, recent evidence indicates that in hypertrophic hearts Gαs undergoes to extensive ubiquitylation with suppression of its downstream signaling. This eventually leads to cardiac contractility dysfunction (Jenie et al., 2013). Gβ subunits can also become a target of the UPS, contributing to feed-back regulation of GPCR signaling. Thus, Gβ2 binds to DDB1 (DNA damage-binding protein 1), a core component of CUL4B-based E3 ubiquitin ligase complex, and targets the GPCR kinase 2 (GRK2) to ubiquitylation by the DDB1-CUL4A-ROC1 ubiquitin ligase complex. Following GPCR activation, PKA phosphorylates DDB1 and induces its dissociation from Gβ2, increasing the levels of GRK2 and promoting receptor desensitization. Deletion of Cul4a gene resulted in cardiac hypertrophy and this phenotype can be partially rescued by concomitant deletion of GRK2 (Zha et al., 2015). These results unvealed a novel mechanism of feedback regulation of GPCR signaling based on a non-canonical function of Gβ2 protein, that acts as a component of the ubiquitin ligase complex that targets GRK2 for degradation.
It emerged that PDEs can be regulated by the UPS. Ubiquitin conjugation and proteasomal degradation of PDE4D by a cullin 1-containing E(3) ubiquitin ligase complex is induced through concomitant phosphorylation of PDE4D by casein kinase 1 (CK1) and glycogen synthase kinase 3β (GSK3β). A phospho-degron motif within the PDE4D was identified as responsible of ubiquitin-mediated proteolysis of the enzyme. Interestingly, protein PPs calcineurin (CaN) counteracts the effects of the SCF complex on PDE4D stability (Zhu et al., 2010), unveiling a complex regulatory mechanism of signal integration between PPs and kinases involved in the control of cAMP pathway.
The UPS can also regulate the PKA stability and signaling. praja2 is a widely expressed mammalian RING-H2 protein with intrinsic E3 ligase activity (Yu et al., 2002). praja2 acts as an AKAP that binds and targets PKA holoenzyme to the cell membrane, perinuclear region and cellular organelles. Co-localization of praja2-PKA complexes with PKA substrate/effector molecules ensures efficient integration and propagation of the locally generated cAMP to distinct target sites. In course of agonist stimulation, praja2 couples ubiquitylation to proteolysis of the R subunits of PKA. By decreasing the ratio between R/C levels, praja2 sustains downstream signals carried out by PKA, positively impacting on specialized cell functions (Lignitto et al., 2011).
As major regulators of cAMP signaling, AKAPs can be regulated at post-translational level by the UPS. Thus, under normoxic conditions, mitochondrial AKAP121 assembles a multienzyme scaffold complex on the outer mitochondrial membrane that ensures efficient propagation of cAMP and src signals from sites of signal generation to mitochondria, enhancing oxidative phosphorylation, mitochondria remodeling, calcium homeostasis and cell survival (Cardone et al., 2004; Livigni et al., 2006; Dickey and Strack, 2011; Scorziello et al., 2013). Under hypoxic conditions, the RING E3 ligase seven in-absentia homolog 2 (Siah2) binds to- and ubiquitylates AKAP121. Ubiquitylated AKAP121 undergoes to proteasomal degradation. Disappearance of AKAP121 is accompanied by a significant drop of mitochondrial metabolic activity, leading to mitochondrial fission and cell death (Carlucci et al., 2008a,b; Merrill et al., 2011). In the ischemic mouse heart, infarct size and degree of cell death were blunted by genetic knock-out of Siah2. In hatching Caenorhabditis elegans, inhibiting Siah2 reduces life span, highlighting a role of the UPS-AKAP-PKA axis in the control of essential aspects of nematode aging (Kim et al., 2011).
Besides auto-regulatory mechanisms, cAMP can also control the activity of E3 ligases. By modulating the ubiquitin pathway, PKA controls the biological activity of a wide number of cellular substrates, integrating signals generated by distinct hormones/growth factors. As example, p300 acts as scaffold and co-activator for transcription factors, facilitating chromatin remodeling and gene expression. p300 controls important biological functions, as cell proliferation, differentiation, apoptosis, and senescence (Rack et al., 2014). In lung cancer cells, the levels of p300 are tightly regulated post-translationally by the cAMP signaling. Thus, agonist-induced rise of cAMP levels promotes ubiquitin-dependent proteolysis of p300, downregulating nuclear gene transcription (Jeong et al., 2013). The Ca2+/Calmodulin-dependent protein kinase III (CAMKIII), inhibits the elongation phase of translation by phosphorylating eukaryotic elongation factor-2 (eEF-2; Heise et al., 2014). Interestingly, CAMKIII protein levels are negatively regulated by isoproterenol stimulation of cAMP cascade. Degradation of the kinase requires the proteasome activity, linking the UPS to cAMP-dependent facilitation of protein translation (Wiseman et al., 2013).
A relevant role of cAMP-PKA axis in the epigenetic control of gene expression has been proposed. In eukaryotic cells, histone proteins are involved in the control of chromatin structure and remodeling. These are important mechanism(s) that cells adopt to regulate gene transcription (Tessarz and Kouzarides, 2014). Histones undergo to reversible post-translational modifications, such as acetylation. Acetylation/deacetylation cycles of histones are essential processes underlying gene expression and are catalyzed by families of histone acetyltransferases (HATs) and deacetylases (HDACs and sirtuins), respectively (Dekker and Haisma, 2009; Stasevich et al., 2014). Sirtuins have been implicated in a wide range of biological processes, such as transcription, DNA damage repair, and metabolism (Etchegaray et al., 2013). Sirtuin-6 (SIRT6) is a stress-induced gene that belongs to NAD+-dependent Class III of histone deacetylases and controls the maintenance of telomere structure and length. By deacetylating histones, SIRT6 regulates genome stability and cell viability (Tennen et al., 2010). Loss of SIRT6 gene induces premature lethality and aging-related degeneration (Mostoslavsky et al., 2006). A link between sirtuins and cAMP signaling has been recently identified. Thus, cAMP stimulates ubiquitylation of SIRT6 protein and its consequent degradation through the proteasome. By reducing the levels of SIRT6, cAMP sustains radiation-induced apoptosis of lung cancer cells (Kim and Juhnn, 2015).
In neurons, cAMP-PKA signaling controls a variety of biological cues, as neurite outgrowth, morphogenesis and synaptic transmission and plasticity (Tasken and Aandahl, 2004). Most of the effects elicited by cAMP are mediated by a transcriptional control of gene expression. A post-translational mechanism of neurite extension which involves the UPS has been recently identified. Thus, neurotrophin-induced activation of PKA promotes praja2-dependent ubiquitylation and degradation of the neurite outgrowth inhibitor NOGO-A. By removing the inhibitory constrain of neurite extension imposed by NOGO-A, PKA-UPS drives a signaling circuit that promotes and sustains neuronal differentiation and synaptic activity (Sepe et al., 2014). In course of neuronal differentiation, phosphorylation of E3 ligases by PKA could also affect substrate recognition, switching the target selectivity between proteins with opposing functions. Thus, neurotrophin-stimulated phosphorylation of Smad ubiquitylation regulator factor 1 (Smurf1), a key component of TGF-β/BMP pathway, reduces degradation of polarity protein Par6 and enhances proteolysis of growth-inhibiting RhoA factor, eventually leading to axon outgrowth (Cheng et al., 2011).Similarly, in cisplatin-treated cancer cells, PKA phosphorylation of Smurf1 prevents degradation of the pro-apoptotic protein Nur77, triggering the mitochondrial apoptotic machinery (Lin et al., 2014).
Dephosphorylation of cellular substrates is mediated by distinct families of protein PPs, (Zhang et al., 2013). Among PPs, protein phosphatase 2 (PP2A) is a conserved Serine/Threonine phosphatase that regulates a wide number of signaling pathways. PP2A is composed of a dimeric core enzyme (structural A and catalytic C subunits), and a regulatory B subunit. In eukaryotes, C and A subunits of PP2A show high degree of sequence similarity, while the regulatory B subunits are highly heterogeneous. The assembly of the three subunits generates different PP2A holoenzymes, whose substrate specificity and intracellular localization are controlled by B subunits (Kiely and Kiely, 2015). PP2A dephosphorylates a variety of substrates, including cAMP-response element-binding protein (CREB). De-phosphorylation of CREB by PP2A attenuates cAMP-induced gene transcription (Wadzinski et al., 1993). PP2A is also involved in a variety of cell functions, as proliferation, differentiation and cell death (Tsuchiya et al., 2014). During apoptosis, PP2A/C subunit is post-translationally regulated by the UPS. Thus, stimulation with tumor-necrosis factor-related apoptosis-inducing ligand (TRAIL) promotes the recruitment of PP2A/C, caspase-8 and Cullin3, a subunit of the cullin family of E3 ligases, into the death-inducing signaling complex (DISC). Within the complex, Cul3 targets PP2A/C for ubiquitylation and degradation by the proteasome. Downregulation of PP2A/C signaling and downstream gene transcription may account for the activation of the apoptotic machinery induced by TRAIL (Xu et al., 2014).
cAMP signaling is involved in a variety of different biological responses (Formosa and Vassallo, 2014). The complexity of the pathway and the high number of components involved ensure an efficient and a fine regulation of the signal transmission from the site of generation to downstream effectors. Genetic mutations or altered expression of any component of this sophisticated signaling cascade may lead to dys-regulation of the signaling, contributing to the onset and progression of human diseases.
In neurons, cAMP balance is crucial for physiological events underlying learning, memory and loco-motor activity (Gomez et al., 2002; Dell’Acqua et al., 2006). Several studies confirmed the pathogenic role of deranged cAMP signaling in neurodegenerative phenotypes (Satoh et al., 2009; Poppinga et al., 2014). As example, Huntington’s disease (HD) is a genetic neurological disorder characterized by alteration of motor coordination that eventually leads to mental decline and behavioral symptoms. HD is caused by the expansion of a CAG repeat in the Huntington (HTT) gene, which induces accumulation of poly(Q)-expanded mutant HTT protein (mHTT; Labbadia and Morimoto, 2013). Accumulation of mHTT within neurons downregulates cAMP signaling, inhibits CREB-dependent gene transcription and profoundly affects neuronal activity and cell survival (Jeong et al., 2012). Recent evidence pointed to a role of mHTT in UPS-cAMP pathway. In particular, mHTT-mediated proteasome impairment inhibits the proteolytic turnover of R subunits within the striatum, increasing the R/C ratio and favoring reconstitution of inactive PKA holoenzyme. By limiting local activation of PKA, mHTT alters the stability of several proteins and impacts on neurons and loco-motor activity. Under these conditions, forced activation of PKA promotes phosphorylation of components of the proteasome (Rpt6) and rescues the impaired proteasome activity, favoring the removal of mHTT aggregates and improving loco-motor activity (Lin et al., 2013).
The UPS is an important control mechanism of cell growth, survival and metabolism. Removal of tumor suppressors or pro-apoptotic factors could, thus, play an important role in tumor growth. Changes in the levels, subcellular targeting or catalytic activity of the E3 ligases may exert major effect on cell growth and survival. Accordingly, dys-regulation of the UPS has been found in a wide array of human cancer (Landis et al., 1989; Weinstein et al., 1991; Palmer et al., 2000). Recent findings demonstrated that praja2, which regulates R subunit turnover, ubiquitylates and degrades MOB1, a core component of NDR/LATS kinase and positive regulator of the tumor-suppressor Hippo cascade (Hergovich, 2011; Lignitto et al., 2013). Removal of MOB1 by the praja2-UPS pathway attenuates the Hippo cascade and sustains glioblastoma growth in vivo (Lignitto et al., 2013). These findings uncover the existence of an intricate interplay between GPCR-cAMP signaling, UPS and tumor suppressor pathways in the control of cell proliferation and tumor growth.
In the last decades, cumulative evidence uncovered a major role of PKA pathway in the control of important biological activities, ranging from differentiation, growth, metabolism, survival to more sophisticated brain activities. Derangement of the cAMP-PKA pathway has been pathogenically linked to the onset and progression of several neurodegenerative and proliferative disorders. So far, most of the cAMP-PKA effects have been attributed to phosphorylation/dephosphorylation events occurring at distal sites of cAMP generation. Emerging data suggest the existence of a cAMP-driven UPS circuitry that controls the turnover/stability of key elements of metabolic and proliferative pathways. At the same time, mounting evidence indicates that UPS by regulating the stability of components of the cAMP cascade controls directly the strength and duration of cAMP-PKA signals. Dys-regulation of this intricate interface between the cAMP and the UPS may underpin the pathogenesis of human diseases. Therefore, efforts are needed to discover new targets and mechanism(s) connecting the UPS to cAMP-PKA signaling, but also to construct a network that is able to predict and quantify the biological outcome (for example, degenerative versus proliferative phenotypes) of human genetic mutations affecting key elements of these transduction pathways. Understanding the complexity of such regulatory mechanisms and exploring further the biological significance of this kinase-ligase network will help to design novel tools and drugs that selectively restore a perturbed cAMP cascade in various human phenotypes.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by a grant from “Associazione Italiana per la Ricerca sul Cancro” (IG15264). The authors apologize to those whose work was not cited owing to space limitations.
Alto, N., Carlisle Michel, J. J., Dodge, K. L., Langeberg, L. K., and Scott, J. D. (2002). Intracellular targeting of protein kinases and phosphatases. Diabetes Metab. Res. Rev 51(Suppl. 3), S385–S388. doi: 10.2337/diabetes.51.2007.s385
Alto, N. M., Soderling, S. H., Hoshi, N., Langeberg, L. K., Fayos, R., Jennings, P. A., et al. (2003). Bioinformatic design of A-kinase anchoring protein-in silico: a potent and selective peptide antagonist of type II protein kinase A anchoring. Proc. Natl. Acad. Sci. U.S.A. 100, 4445–4450. doi: 10.1073/pnas.0330734100
Angelo, R., and Rubin, C. S. (1998). Molecular characterization of an anchor protein (AKAPCE) that binds the RI subunit (RCE) of type I protein kinase A from Caenorhabditis elegans. J. Biol. Chem. 273, 14633–14643. doi: 10.1074/jbc.273.23.14633
Armstrong, R., Wen, W., Meinkoth, J., Taylor, S., and Montminy, M. (1995). A refractory phase in cyclic AMP-responsive transcription requires down regulation of protein kinase A. Mol. Cell. Biol. 15, 1826–1832.
Bonifacino, J. S., and Weissman, A. M. (1998). Ubiquitin and the control of protein fate in the secretory and endocytic pathways. Annu. Rev. Cell Dev. Biol. 14, 19–57. doi: 10.1146/annurev.cellbio.14.1.19
Bouvier, M., Collins, S., O’dowd, B. F., Campbell, P. T., De Blasi, A., Kobilka, B. K., et al. (1989). Two distinct pathways for cAMP-mediated down-regulation of the β2-adrenergic receptor. Phosphorylation of the receptor and regulation of its mRNA level. J. Biol. Chem. 264, 16786–16792.
Burgers, P. P., Ma, Y., Margarucci, L., Mackey, M., Van Der Heyden, M. A., Ellisman, M., et al. (2012). A small novel A-kinase anchoring protein (AKAP) that localizes specifically protein kinase A-regulatory subunit I (PKA-RI) to the plasma membrane. J. Biol. Chem. 287, 43789–43797. doi: 10.1074/jbc.M112.395970
Canettieri, G., Morantte, I., Guzman, E., Asahara, H., Herzig, S., Anderson, S. D., et al. (2003). Attenuation of a phosphorylation-dependent activator by an HDAC-PP1 complex. Nat. Struct. Biol. 10, 175–181. doi: 10.1038/nsb895
Cardone, L., Carlucci, A., Affaitati, A., Livigni, A., Decristofaro, T., Garbi, C., et al. (2004). Mitochondrial AKAP121 binds and targets protein tyrosine phosphatase D1, a novel positive regulator of src signaling. Mol. Cell. Biol. 24, 4613–4626. doi: 10.1128/MCB.24.11.4613-4626.2004
Carlucci, A., Adornetto, A., Scorziello, A., Viggiano, D., Foca, M., Cuomo, O., et al. (2008a). Proteolysis of AKAP121 regulates mitochondrial activity during cellular hypoxia and brain ischaemia. EMBO J. 27, 1073–1084. doi: 10.1038/emboj.2008.33
Carlucci, A., Lignitto, L., and Feliciello, A. (2008b). Control of mitochondria dynamics and oxidative metabolism by cAMP, AKAPs and the proteasome. Trends Cell Biol. 18, 604–613. doi: 10.1016/j.tcb.2008.09.006
Carnegie, G. K., and Scott, J. D. (2003). A-kinase anchoring proteins and neuronal signaling mechanisms. Genes Dev. 17, 1557–1568. doi: 10.1101/gad.1095803
Carr, D. W., Stofko-Hahn, R. E., Fraser, I. D., Cone, R. D., and Scott, J. D. (1992). Localization of the cAMP-dependent protein kinase to the postsynaptic densities by A-kinase anchoring proteins. Characterization of AKAP 79. J. Biol. Chem. 267, 16816–16823.
Cheng, P. L., Lu, H., Shelly, M., Gao, H., and Poo, M. M. (2011). Phosphorylation of E3 ligase Smurf1 switches its substrate preference in support of axon development. Neuron 69, 231–243. doi: 10.1016/j.neuron.2010.12.021
Ciechanover, A. (2003). The ubiquitin proteolytic system and pathogenesis of human diseases: a novel platform for mechanism-based drug targeting. Biochem. Soc. Trans. 31, 474–481. doi: 10.1042/BST0310474
Ciechanover, A. (2005). Proteolysis: from the lysosome to ubiquitin and the proteasome. Nat. Rev. Mol. Cell Biol. 6, 79–87. doi: 10.1038/nrm1552
Cooper, D. M., and Tabbasum, V. G. (2014). Adenylate cyclase-centred microdomains. Biochem J. 462, 199–213. doi: 10.1042/BJ20140560
Dantuma, N. P., and Bott, L. C. (2014). The ubiquitin-proteasome system in neurodegenerative diseases: precipitating factor, yet part of the solution. Front. Mol. Neurosci. 7:70. doi: 10.3389/fnmol.2014.00070
De Bie, P., and Ciechanover, A. (2011). Ubiquitination of E3 ligases: self-regulation of the ubiquitin system via proteolytic and non-proteolytic mechanisms. Cell Death. Differ. 18, 1393–1402. doi: 10.1038/cdd.2011.16
Dekker, F. J., and Haisma, H. J. (2009). Histone acetyl transferases as emerging drug targets. Drug Discov. Today 14, 942–948. doi: 10.1016/j.drudis.2009.06.008
Dell’Acqua, M. L., Smith, K. E., Gorski, J. A., Horne, E. A., Gibson, E. S., and Gomez, L. L. (2006). Regulation of neuronal PKA signaling through AKAP targeting dynamics. Eur. J. Cell Biol. 85, 627–633. doi: 10.1016/j.ejcb.2006.01.010
Dickey, A. S., and Strack, S. (2011). PKA/AKAP1 and PP2A/Bβ2 regulate neuronal morphogenesis via Drp1 phosphorylation and mitochondrial bioenergetics. J. Neurosci. 31, 15716–15726. doi: 10.1523/JNEUROSCI.3159-11.2011
Etchegaray, J. P., Zhong, L., and Mostoslavsky, R. (2013). The histone deacetylase SIRT6: at the crossroads between epigenetics, metabolism and disease. Curr. Top. Med. Chem. 13, 2991–3000. doi: 10.2174/15680266113136660213
Feliciello, A., Gottesman, M. E., and Avvedimento, E. V. (2001). The biological functions of A-kinase anchor proteins. J. Mol. Biol. 308, 99–114. doi: 10.1006/jmbi.2001.4585
Feliciello, A., Gottesman, M. E., and Avvedimento, E. V. (2005). cAMP-PKA signaling to the mitochondria: protein scaffolds, mRNA and phosphatases. Cell. Signal. 17, 279–287. doi: 10.1016/j.cellsig.2004.09.009
Formosa, R., and Vassallo, J. (2014). cAMP signalling in the normal and tumorigenic pituitary gland. Mol. Cell. Endocrinol. 392, 37–50. doi: 10.1016/j.mce.2014.05.004
Francis, S. H., Blount, M. A., and Corbin, J. D. (2011). Mammalian cyclic nucleotide phosphodiesterases: molecular mechanisms and physiological functions. Physiol. Rev. 91, 651–690. doi: 10.1152/physrev.00030.2010
Gomez, L. L., Alam, S., Smith, K. E., Horne, E., and Dell’Acqua, M. L. (2002). Regulation of A-kinase anchoring protein 79/150-cAMP-dependent protein kinase postsynaptic targeting by NMDA receptor activation of calcineurin and remodeling of dendritic actin. J. Neurosci. 22, 7027–7044.
Heise, C., Gardoni, F., Culotta, L., Di Luca, M., Verpelli, C., and Sala, C. (2014). Elongation factor-2 phosphorylation in dendrites and the regulation of dendritic mRNA translation in neurons. Front. Cell Neurosci. 8:35. doi: 10.3389/fncel.2014.00035
Herberg, F. W., Maleszka, A., Eide, T., Vossebein, L., and Tasken, K. (2000). Analysis of A-kinase anchoring protein (AKAP) interaction with protein kinase A (PKA) regulatory subunits: PKA isoform specificity in AKAP binding. J. Mol. Biol. 298, 329–339. doi: 10.1006/jmbi.2000.3662
Hergovich, A. (2011). Mob control: reviewing a conserved family of kinase regulators. Cell. Signal. 23, 1433–1440. doi: 10.1016/j.cellsig.2011.04.007
Huang, L. J., Durick, K., Weiner, J. A., Chun, J., and Taylor, S. S. (1997). Identification of a novel protein kinase A anchoring protein that binds both type I and type II regulatory subunits. J. Biol. Chem. 272, 8057–8064. doi: 10.1074/jbc.272.12.8057
Iyengar, R. (1993). Molecular and functional diversity of mammalian Gs-stimulated adenylyl cyclases. FASEB J. 7, 768–775.
Jenie, R. I., Nishimura, M., Fujino, M., Nakaya, M., Mizuno, N., Tago, K., et al. (2013). Increased ubiquitination and the crosstalk of G protein signaling in cardiac myocytes: involvement of Ric-8B in Gs suppression by Gq signal. Genes Cells 18, 1095–1106. doi: 10.1111/gtc.12099
Jeong, H., Cohen, D. E., Cui, L., Supinski, A., Savas, J. N., Mazzulli, J. R., et al. (2012). Sirt1 mediates neuroprotection from mutant huntingtin by activation of the TORC1 and CREB transcriptional pathway. Nat. Med. 18, 159–165. doi: 10.1038/nm.2559
Jeong, M. J., Kim, E. J., Cho, E. A., Ye, S. K., Kang, G. H., and Juhnn, Y. S. (2013). cAMP signalling decreases p300 protein levels by promoting its ubiquitin/proteasome dependent degradation via Epac and p38 MAPK in lung cancer cells. FEBS Lett. 587, 1373–1378. doi: 10.1016/j.febslet.2013.03.010
Kenan, Y., Murata, T., Shakur, Y., Degerman, E., and Manganiello, V. C. (2000). Functions of the N-terminal region of cyclic nucleotide phosphodiesterase 3 (PDE 3) isoforms. J. Biol. Chem. 275, 12331–12338. doi: 10.1074/jbc.275.16.12331
Kiely, M., and Kiely, P. A. (2015). PP2A: the wolf in sheep’s clothing? Cancers (Basel), 7, 648–669. doi: 10.3390/cancers7020648
Kim, E. J., and Juhnn, Y. S. (2015). Cyclic AMP signaling reduces sirtuin 6 expression in non-small cell lung cancer cells by promoting ubiquitin-proteasomal degradation via inhibition of the Raf-MEK-ERK (Raf/mitogen-activated extracellular signal-regulated kinase/extracellular signal-regulated kinase) pathway. J. Biol. Chem. 290, 9604–9613. doi: 10.1074/jbc.M114.633198
Kim, H., Scimia, M. C., Wilkinson, D., Trelles, R. D., Wood, M. R., Bowtell, D., et al. (2011). Fine-tuning of Drp1/Fis1 availability by AKAP121/Siah2 regulates mitochondrial adaptation to hypoxia. Mol. Cell. 44, 532–544. doi: 10.1016/j.molcel.2011.08.045
Knighton, D. R., Zheng, J. H., Ten Eyck, L. F., Ashford, V. A., Xuong, N. H., Taylor, S. S., et al. (1991). Crystal structure of the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science 253, 407–414. doi: 10.1126/science.1862342
Kommaddi, R. P., Jean-Charles, P. Y., and Shenoy, S. K. (2015). Phosphorylation of the deubiquitinase USP20 by protein kinase A regulates post-endocytic trafficking of β2 adrenergic receptors to autophagosomes during physiological stress. J. Biol. Chem. 290, 8888–8903. doi: 10.1074/jbc.M114.630541
Labbadia, J., and Morimoto, R. I. (2013). Huntington’s disease: underlying molecular mechanisms and emerging concepts. Trends Biochem. Sci. 38, 378–385. doi: 10.1016/j.tibs.2013.05.003
Landis, C. A., Masters, S. B., Spada, A., Pace, A. M., Bourne, H. R., and Vallar, L. (1989). GTPase inhibiting mutations activate the α chain of Gs and stimulate adenylyl cyclase in human pituitary tumours. Nature 340, 692–696. doi: 10.1038/340692a0
Lignitto, L., Arcella, A., Sepe, M., Rinaldi, L., Delle Donne, R., Gallo, A., et al. (2013). Proteolysis of MOB1 by the ubiquitin ligase praja2 attenuates Hippo signalling and supports glioblastoma growth. Nat. Commun. 4, 1822. doi: 10.1038/ncomms2791
Lignitto, L., Carlucci, A., Sepe, M., Stefan, E., Cuomo, O., Nistico, R., et al. (2011). Control of PKA stability and signalling by the RING ligase praja2. Nat. Cell Biol. 13, 412–422. doi: 10.1038/ncb2209
Lin, H., Lin, Q., Liu, M., Lin, Y., Wang, X., Chen, H., et al. (2014). PKA/Smurf1 signaling-mediated stabilization of Nur77 is required for anticancer drug cisplatin-induced apoptosis. Oncogene 33, 1629–1639. doi: 10.1038/onc.2013.116
Lin, J. T., Chang, W. C., Chen, H. M., Lai, H. L., Chen, C. Y., Tao, M. H., et al. (2013). Regulation of feedback between protein kinase A and the proteasome system worsens Huntington’s disease. Mol. Cell. Biol. 33, 1073–1084. doi: 10.1128/MCB.01434-12
Livigni, A., Scorziello, A., Agnese, S., Adornetto, A., Carlucci, A., Garbi, C., et al. (2006). Mitochondrial AKAP121 links cAMP and src signaling to oxidative metabolism. Mol. Biol. Cell 17, 263–271. doi: 10.1091/mbc.E05-09-0827
Lomas, O., and Zaccolo, M. (2014). Phosphodiesterases maintain signaling fidelity via compartmentalization of cyclic nucleotides. Physiology (Bethesda) 29, 141–149. doi: 10.1152/physiol.00040.2013
Maurice, D. H., Ke, H., Ahmad, F., Wang, Y., Chung, J., and Manganiello, V. C. (2014). Advances in targeting cyclic nucleotide phosphodiesterases. Nat. Rev. Drug Discov. 13, 290–314. doi: 10.1038/nrd4228
Means, C. K., Lygren, B., Langeberg, L. K., Jain, A., Dixon, R. E., Vega, A. L., et al. (2011). An entirely specific type I A-kinase anchoring protein that can sequester two molecules of protein kinase A at mitochondria. Proc. Natl. Acad. Sci. U.S.A. 108, E1227–35. doi: 10.1073/pnas.1107182108
Merrill, R. A., Dagda, R. K., Dickey, A. S., Cribbs, J. T., Green, S. H., Usachev, Y. M., et al. (2011). Mechanism of neuroprotective mitochondrial remodeling by PKA/AKAP1. PLoS Biol. 9:e1000612. doi: 10.1371/journal.pbio.1000612
Michel, J. J., and Scott, J. D. (2002). AKAP mediated signal transduction. Annu. Rev. Pharmacol. Toxicol. 42, 235–257. doi: 10.1146/annurev.pharmtox.42.083101.135801
Moore, R. H., Millman, E. E., Godines, V., Hanania, N. A., Tran, T. M., Peng, H., et al. (2007). Salmeterol stimulation dissociates β2-adrenergic receptor phosphorylation and internalization. Am. J. Respir. Cell Mol. Biol. 36, 254–261. doi: 10.1165/rcmb.2006-0158OC
Mostoslavsky, R., Chua, K. F., Lombard, D. B., Pang, W. W., Fischer, M. R., Gellon, L., et al. (2006). Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 124, 315–329. doi: 10.1016/j.cell.2005.11.044
Nabhan, J. F., Pan, H., and Lu, Q. (2010). Arrestin domain-containing protein 3 recruits the NEDD4 E3 ligase to mediate ubiquitination of the β2-adrenergic receptor. EMBO Rep. 11, 605–611. doi: 10.1038/embor.2010.80
Newlon, M. G., Roy, M., Morikis, D., Carr, D. W., Westphal, R., Scott, J. D., et al. (2001). A novel mechanism of PKA anchoring revealed by solution structures of anchoring complexes. EMBO J. 20, 1651–1662. doi: 10.1093/emboj/20.7.1651
Newlon, M. G., Roy, M., Morikis, D., Hausken, Z. E., Coghlan, V., Scott, J. D., et al. (1999). The molecular basis for protein kinase A anchoring revealed by solution NMR. Nat Struct Biol. 6, 222–227. doi: 10.1038/6663
Ortega, Z., and Lucas, J. J. (2014). Ubiquitin-proteasome system involvement in Huntington’s disease. Front. Mol. Neurosci. 7:77. doi: 10.3389/fnmol.2014.00077
Palmer, J. S., Duffy, D. L., Box, N. F., Aitken, J. F., O’gorman, L. E., Green, A. C., et al. (2000). Melanocortin-1 receptor polymorphisms and risk of melanoma: is the association explained solely by pigmentation phenotype? Am. J. Hum. Genet. 66, 176–186. doi: 10.1086/302711
Poppinga, W. J., Munoz-Llancao, P., Gonzalez-Billault, C., and Schmidt, M. (2014). A-kinase anchoring proteins: cAMP compartmentalization in neurodegenerative and obstructive pulmonary diseases. Br. J. Pharmacol. 171, 5603–5623. doi: 10.1111/bph.12882
Port, J. D., and Bristow, M. R. (2001). Altered β-adrenergic receptor gene regulation and signaling in chronic heart failure. J. Mol. Cell Cardiol. 33, 887–905. doi: 10.1006/jmcc.2001.1358
Rack, J. G., Lutter, T., Kjaereng Bjerga, G. E., Guder, C., Ehrhardt, C., Varv, S., et al. (2014). The PHD finger of p300 influences its ability to acetylate histone and non-histone targets. J. Mol. Biol. 426, 3960–3972. doi: 10.1016/j.jmb.2014.08.011
Ramanathan, H. N., and Ye, Y. (2012). Cellular strategies for making monoubiquitin signals. Crit. Rev. Biochem. Mol. Biol 47, 17–28. doi: 10.3109/10409238.2011.620943
Reiter, E., and Lefkowitz, R. J. (2006). GRKs and β-arrestins: roles in receptor silencing, trafficking and signaling. Trends Endocrinol. Metab. 17, 159–165. doi: 10.1016/j.tem.2006.03.008
Rosenbaum, D. M., Rasmussen, S. G., and Kobilka, B. K. (2009). The structure and function of G-protein-coupled receptors. Nature 459, 356–363. doi: 10.1038/nature08144
Rubin, C. S. (1994). A kinase anchor proteins and the intracellular targeting of signals carried by cyclic AMP. Biochim. Biophys. Acta 1224, 467–479.
Satoh, J., Tabunoki, H., and Arima, K. (2009). Molecular network analysis suggests aberrant CREB-mediated gene regulation in the Alzheimer disease hippocampus. Dis. Markers 27, 239–252. doi: 10.1155/2009/618619
Schmidt, M., and Finley, D. (2014). Regulation of proteasome activity in health and disease. Biochim. Biophys. Acta 1843, 13–25. doi: 10.1016/j.bbamcr.2013.08.012
Scorziello, A., Savoia, C., Sisalli, M. J., Adornetto, A., Secondo, A., Boscia, F., et al. (2013). NCX3 regulates mitochondrial Ca2+ handling through the AKAP121-anchored signaling complex and prevents hypoxia-induced neuronal death. J. Cell Sci. 126, 5566–5577. doi: 10.1242/jcs.129668
Sepe, M., Lignitto, L., Porpora, M., Delle Donne, R., Rinaldi, L., Belgianni, G., et al. (2014). Proteolytic control of neurite outgrowth inhibitor NOGO-A by the cAMP/PKA pathway. Proc. Natl. Acad. Sci. U.S.A. 111, 15729–15734. doi: 10.1073/pnas.1410274111
Shenoy, S. K., Mcdonald, P. H., Kohout, T. A., and Lefkowitz, R. J. (2001). Regulation of receptor fate by ubiquitination of activated β2-adrenergic receptor and β-arrestin. Science 294, 1307–1313. doi: 10.1126/science.1063866
Sibley, D. R., and Lefkowitz, R. J. (1985). Molecular mechanisms of receptor desensitization using the β-adrenergic receptor-coupled adenylate cyclase system as a model. Nature 317, 124–129. doi: 10.1038/317124a0
Stasevich, T. J., Hayashi-Takanaka, Y., Sato, Y., Maehara, K., Ohkawa, Y., Sakata-Sogawa, K., et al. (2014). Regulation of RNA polymerase II activation by histone acetylation in single living cells. Nature 516, 272–275. doi: 10.1038/nature13714
Stefan, E., Aquin, S., Berger, N., Landry, C. R., Nyfeler, B., Bouvier, M., et al. (2007). Quantification of dynamic protein complexes using Renilla luciferase fragment complementation applied to protein kinase A activities in vivo. Proc. Natl. Acad. Sci. U.S.A. 104, 16916–16921. doi: 10.1073/pnas.0704257104
Tang, T., Gao, M. H., Miyanohara, A., and Hammond, H. K. (2008). Gαq reduces cAMP production by decreasing Gαs protein abundance. Biochem. Biophys. Res. Commun. 377, 679–684. doi: 10.1016/j.bbrc.2008.10.054
Tasken, K., and Aandahl, E. M. (2004). Localized effects of cAMP mediated by distinct routes of protein kinase A. Physiol. Rev. 84, 137–167. doi: 10.1152/physrev.00021.2003
Taylor, S. S., Kim, C., Cheng, C. Y., Brown, S. H., Wu, J., and Kannan, N. (2008). Signaling through cAMP and cAMP-dependent protein kinase: diverse strategies for drug design. Biochim. Biophys. Acta 1784, 16–26. doi: 10.1016/j.bbapap.2007.10.002
Taylor, S. S., Kim, C., Vigil, D., Haste, N. M., Yang, J., Wu, J., et al. (2005). Dynamics of signaling by PKA. Biochim. Biophys. Acta 1754, 25–37. doi: 10.1016/j.bbapap.2005.08.024
Tennen, R. I., Berber, E., and Chua, K. F. (2010). Functional dissection of SIRT6: identification of domains that regulate histone deacetylase activity and chromatin localization. Mech. Ageing Dev. 131, 185–192. doi: 10.1016/j.mad.2010.01.006
Tessarz, P., and Kouzarides, T. (2014). Histone core modifications regulating nucleosome structure and dynamics. Nat. Rev. Mol. Cell Biol. 15, 703–708. doi: 10.1038/nrm3890
Tilley, D. G. (2011). G protein-dependent and G protein-independent signaling pathways and their impact on cardiac function. Circ. Res. 109, 217–230. doi: 10.1161/CIRCRESAHA.110.231225
Timofeyev, V., Myers, R. E., Kim, H. J., Woltz, R. L., Sirish, P., Heiserman, J. P., et al. (2013). Adenylyl cyclase subtype-specific compartmentalization: differential regulation of L-type Ca2+ current in ventricular myocytes. Circ. Res. 112, 1567–1576. doi: 10.1161/CIRCRESAHA.112.300370
Tsuchiya, A., Kanno, T., Shimizu, T., Nakao, S., Tanaka, A., Tabata, C., et al. (2014). A novel PP2A enhancer induces caspase-independent apoptosis of MKN28 gastric cancer cells with high MEK activity. Cancer Lett. 347, 123–128. doi: 10.1016/j.canlet.2014.01.034
Wadzinski, B. E., Wheat, W. H., Jaspers, S., Peruski, L. F., Jr., Lickteig, R. L., et al. (1993). Nuclear protein phosphatase 2A dephosphorylates protein kinase A-phosphorylated CREB and regulates CREB transcriptional stimulation. Mol. Cell. Biol. 13, 2822–2834.
Walsh, D. A., and Van Patten, S. M. (1994). Multiple pathway signal transduction by the cAMP-dependent protein kinase. FASEB J. 8, 1227–1236.
Wang, Z. V., and Hill, J. A. (2015). Protein quality control and metabolism: bidirectional control in the heart. Cell Metab. 21, 215–226. doi: 10.1016/j.cmet.2015.01.016
Weinstein, L. S., Shenker, A., Gejman, P. V., Merino, M. J., Friedman, E., and Spiegel, A. M. (1991). Activating mutations of the stimulatory G protein in the McCune-Albright syndrome. N. Engl. J. Med. 325, 1688–1695. doi: 10.1056/NEJM199112123252403
Welch, E. J., Jones, B. W., and Scott, J. D. (2010). Networking with AKAPs: context-dependent regulation of anchored enzymes. Mol. Interv. 10, 86–97. doi: 10.1124/mi.10.2.6
Wiseman, S. L., Shimizu, Y., Palfrey, C., and Nairn, A. C. (2013). Proteasomal degradation of eukaryotic elongation factor-2 kinase (EF2K) is regulated by cAMP-PKA signaling and the SCFβTRCP ubiquitin E3 ligase. J. Biol. Chem. 288, 17803–17811. doi: 10.1074/jbc.M113.477182
Xu, J., Zhou, J. Y., Xu, Z., Kho, D. H., Zhuang, Z., Raz, A., et al. (2014). The role of Cullin3-mediated ubiquitination of the catalytic subunit of PP2A in TRAIL signaling. Cell Cycle 13, 3750–3758. doi: 10.4161/15384101.2014.965068
Yu, P., Chen, Y., Tagle, D. A., and Cai, T. (2002). PJA1, encoding a RING-H2 finger ubiquitin ligase, is a novel human X chromosome gene abundantly expressed in brain. Genomics 79, 869–874. doi: 10.1006/geno.2002.6770
Zha, Z., Han, X., Smith, M. D., Liu, Y., Giguere, P. M., Kopanja, D., et al. (2015). A Non-Canonical Function of Gβ as a Subunit of E3 Ligase in Targeting GRK2 Ubiquitylation. Mol. Cell. 58, 794–803. doi: 10.1016/j.molcel.2015.04.017
Zhang, M., Patriarchi, T., Stein, I. S., Qian, H., Matt, L., Nguyen, M., et al. (2013). Adenylyl cyclase anchoring by a kinase anchor protein AKAP5 (AKAP79/150) is important for postsynaptic β-adrenergic signaling. J. Biol. Chem. 288, 17918–17931. doi: 10.1074/jbc.M112.449462
Zhang, M., Yogesha, S. D., Mayfield, J. E., Gill, G. N., Zhang, Y. (2013). Viewing serine/threonine protein phosphatases through the eyes of drug designers. FEBS J. 280, 4739–60. doi: 10.1111/febs.12481
Keywords: cyclic AMP, PKA signaling, proteasome, ubiquitination, AKAP
Citation: Rinaldi L, Sepe M, Delle Donne R, and Feliciello A (2015) A dynamic interface between ubiquitylation and cAMP signaling. Front. Pharmacol. 6:177. doi: 10.3389/fphar.2015.00177
Received: 23 June 2015; Accepted: 06 August 2015;
Published: 04 September 2015.
Edited by:
Apostolos Zarros, University of Glasgow, UKReviewed by:
Catarina Oliveira, University of Coimbra, PortugalCopyright © 2015 Rinaldi, Sepe, Delle Donne and Feliciello. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Antonio Feliciello, Dipartimento di Medicina Molecolare e Biotecnologie Mediche, University of Naples Federico II, via S. Pansini 5, 80131, Naples, Italy,ZmVsaWNpZWxAdW5pbmEuaXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.