 Xiaohong Tian
Xiaohong Tian Yang He
Yang He Jinming Zhou
Jinming Zhou
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol., 24 March 2015
Sec. Experimental Pharmacology and Drug Discovery
Volume 6 - 2015 | https://doi.org/10.3389/fphar.2015.00057
This article is part of the Research TopicMutations and Drug ResistanceView all 4 articles
Androgen receptor (AR) plays a critical role in the development and progression of prostate cancer (PCa). Current clinically used antiandrogens such as flutamide, bicalutamide, and newly approved enzalutamide mainly target the hormone binding pocket (HBP) of AR. However, over time, drug resistance invariably develops and switches these antiandrogens from antagonist to agonist of the AR. Accumulated evidence indicates that AR mutation is an important cause for the drug resistance. This review will give an overview of the mutation based resistance of the current clinically used antiandrogens and the rational drug design to overcome the resistance, provides a promising strategy for the development of the new generation of antiandrogens targeting HBP.
Prostate cancer (PCa) is one of the most common cancer and the second leading cause of cancer death in men in the western countries (Jemal et al., 2011). AR, a member of nuclear receptor family that is activated by binding of androgens (Roy et al., 1999), plays an important role in promoting the development of PCa (Dong et al., 2005). Moreover, it has been commonly agreed that AR expression and signaling remains intact as the disease evolves from androgen-sensitive cancer to castration-resistant prostate cancer (CRPC) which are still dependent on AR signaling axis (Jenster, 1999; Taplin, 2007). Thus, AR has become the most important therapeutic target for the treatment of PCa (Aragon-Ching, 2014; Carver, 2014; Culig, 2014).
Through blocking AR signaling, androgen-deprivation therapy (ADT) (via surgical or chemical castration) has been remaining the mainstay for the treatment of advanced PCa since 1940s. (Ruckle and Oesterling, 1993; Lubeck et al., 2001; Ryan and Small, 2006; Cannata et al., 2012). Normally, ADT reduces 95% of testosterone levels. However the androgen stimulus is still persisting as a result of circulating androgens produced by intracrine steroidogenesis (Montgomery et al., 2008). Thus, by using antiandrogens as an adjuvant treatment of ATD to block the intracrine steroidogenesis, may eventually delay or prevent the progression to CRPC. AR antiandrogens prevent androgens from carrying out their biological activity by directly binding and blocking the AR LBD, or by inducing repressive activity (Maeda and Usami, 2002; Gillatt, 2006). Current clinically used antiandrogens such as flutamide (Goldspiel and Kohler, 1990), bicalutamide (Blackledge et al., 1997), and newly approved enzalutamide (Semenas et al., 2013) mainly target the HBP of the AR LBD. These approved antiandrogens have greatly improved the survival and life quality of the PCa patients. However, after the initially effective response, most tumors progress to CRPC under the treatment of antiandrogens, and no curative therapy is available nowadays.
It has been wildly accepted that the acquired AR mutation is an important cause for the drug resistance of PCa toward antiandrogens. For example, T877A mutant would turn flutamide into AR agonist (Bohl et al., 2005a), and W741C would turn bicalutamide into AR agonist. (Bohl et al., 2005b). Therefore, the development of novel antiandrogens to circumvent the mutation based resistance is highly demanded. Fortunately, up to now, several effective strategies, especially the rational drug design, have been applied in the design of the antiandrogens targeting HBP of the AR, and several promising agents have been obtained (Trendel, 2013). This review will give an overview of the mutation based resistance of the current clinically used antiandrogens and the rational drug design to overcome the resistance, which provides a promising strategy for the development of the new generation of antiandrogens targeting the AR HBP.
As a member of nuclear receptor family, AR possesses a modular organization characteristic to all of the nuclear receptors. There is only one AR gene identified in human so far, consisting of 8 exons encoding the AR with a typical size of 919 amino acids (Werner et al., 2006; Gao, 2010). AR is comprised of an N-terminal domain (NTD), a central DNA binding domain (DBD), a short-hinge region, and a C-terminal LBD (Figure 1A). Among these domains, the NTD, as an intrinsically disordered region, is the least conserved domain in AR, which contains a transcription activation domain: activation function 1 (AF1) that regulates gene transcription in a ligand-independent fashion (Yuan et al., 2001). The adjacent DBD is the most conserved domain, composed of two cysteine-rich zinc-finger motifs, mediating AR binding to recognition elements of specific genes in DNA. The hinge region bridges between the DBD and the LBD and harbors a nuclear localization signal (NLS). The C-terminal LBD (Figure 1B) comprises a 12 helical structure that encloses a central hormone binding pocket (HBP), a second activation function domain (AF2) that is located at the carboxy-terminal end of the LBD and mediates ligand dependent transactivation, and a recently discovered binding site, Binding function 3 (BF3). Helix 12 (H12) is the most flexible part of AR, and conformational changes of H12 are unambiguously associated with the molecular mechanism of action of ligands bound to the HBP (Caboni and Lloyd, 2013). The HBP is primarily composed of hydrophobic residues that can form strong nonpolar interactions with ligands. The protein-ligand anchoring can be additionally stabilized by a network of hydrogen bonds involving polar residues such as R752, Q711, N705, and T877 (Figure 1C).
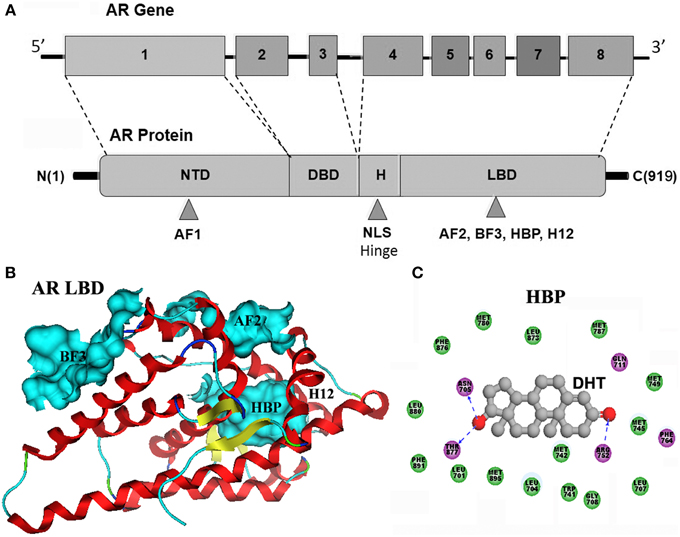
Figure 1. (A) AR gene consists of 8 exons encoding the androgen receptor with a gene product of typical size of 919 amino acids. AR is comprised of an N-terminal domain (NTD), a central DNA binding domain (DBD), a short-hinge region, and a C-terminal LBD. (B) LBD comprises a 12 helical structure that encloses a central hormone binding pocket (HBP), a second activation function domain (AF2) that is located at the carboxy-terminal end of the LBD, and a recently discovered binding site, Binding function 3 (BF3). The adopted conformation of H12 are unambiguously associated with the molecular mechanism of action of ligands bound to the HBP. (C) As shown in compex structure of dihydrotestosterone (DHT) and AR-LBD, the AR HBP is primarily composed of hydrophobic residues (green ball) that can form strong nonpolar interactions with DHT. The protein-ligand anchoring can be additionally stabilized by a network of hydrogen bonds (blue dashed line) involving R752, Q711, N705, and T877 polar residues.
Androgen exerts its biological effects through binding the HBP of AR. Upon binding, H12 is repositioned to cover the HBP, triggering agonist-induced conformational change in the LBD, results in the formation of AF2. The AR dissociates from heat shock proteins (HSPs), homodimerizes and translocats into the nucleus where it binds ARE sites of DNA, directly regulates targeting genes transcription in the presence of coactivator which binds to AF2, promoting the recruitment of RNA polymerase II, triggering the transcription process (Pratt and Toft, 1997; Heinlein and Chang, 2002; Shang et al., 2002). In addition to the classic genomic actions, the nongenomic actions of the androgens have also been observed in various tissues and characterized by the lack of immediate activation of transcription/translation processes, due to the rapidity of action that is mainly mediated by the activation of cytoplasmic and/or plasma membrane-associated receptors and downstream signaling pathways (Norman et al., 2004), which contributes to the overall effects of androgen stimulation, along with the classic genomic actions.
AR plays an important role in promoting the development of PCa (Dong et al., 2005). Accumulated evidence shows that the AR signaling still contributes to CRPC through several mechanisms, including AR protein overexpression, acquired mutations of AR which disables the antagonistic activity of antiandrogen, aberrant expression of AR co-regulators and alternative AR activation by cytokines and growth factors in the absence of androgens, and the expression of AR splice variants (ARvs) lacking LBD such as AR-V7 and ARv567. In addition, the TMPRSS-ERG fusion also plays an important role in PCa progression by disrupting the AR lineage-specific differentiation through gene rearrangements, which leads to an EZH2-mediated de-differentiation of cells. The most common one is the fusion of the 3′ region of ERG with the 5′ region of the highly AR-regulated TMPRSS2 gene. ERG rearrangements have been identified in 40–60% of PCa (Tomlins et al., 2005).
Current clinically used antiandrogens mainly target the HBP. There are two types of antiandrogens: the steroidal antiandrogens and the non-steroidal antiandrogens. Several steroidal anti-androgens (cyproterone acetate, megestrol acetate, and medroxyprogesterone acetate) are initially used for the androgen blockade in patients. However, severe drawbacks such as hepatotoxicity, interference with libido and potency, cardiovascular side effects and low efficacy have limited their clinical use. The later developed non-steroidal antiandrogens including the first-generation antiandrogens such as flutamide, bicalutamide, nilutamide, and the second-generation compounds: enzalutamide and ARN509. These non-steroidal antiandrogens which avoid the typical constraints of the steroidal antiandrogens, have been widely used in clinical nowadays. However, after initially effective response, approximately 50% of patients whose cancer started to grow again under the treatment of antiandrogens, the cancer has been observed to regress by simply stopping the antiandrogen, which referred to the Anti-Androgen Withdrawal Response (AAWR) (Paul and Breul, 2000; Sartor et al., 2008). It has been widely accepted that the acquired mutations of AR in PCa under the pressure of the antiandrogen is responsible for such phenomenon, which turns antiandrogens from AR antagonist to AR agonist and causing drug resistance (Figure 2A).
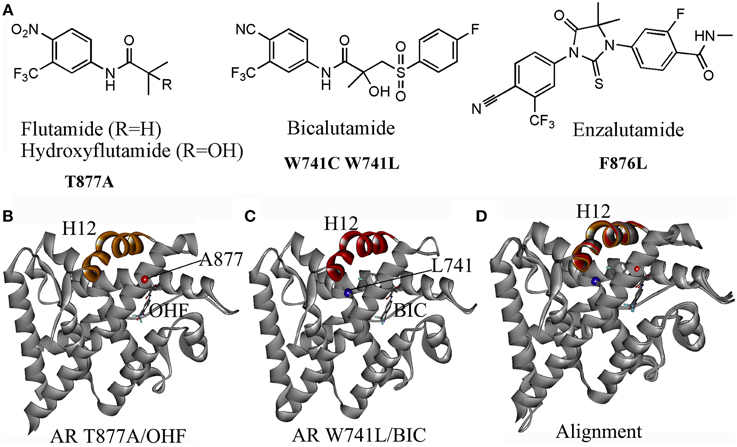
Figure 2. (A) Several acquired mutations drive drug resistance: T877A mutant turns hydroxyflutamide (OHF), an active metabolite of antiandrogen flutamide in AR agonist; W741C or W741L converts bicalutamide (BIC) into AR agonist; a novel mutant F876L was identified inducing the resistance to enzalutamide and ARN509. (B) The crystal structure (PDB-ID: 2ax6) of T877A LBD/ hydroxyflutamide (OHF) shows adopting a agonistic conformation; (C) the crystal structure (PDB-ID: 1z95) of W741L LBD/ bicalutamide (BIC) shows adopting a agonistic conformation; (D) A superimposition of T877A LBD/OHF and W741L LBD/ BIC structures with the LBD structure bound with DHT (PDB-ID: 1i37) by sequence conferred a perfect alignment of the H12 with the RMSD value of 0.28 and 0.48 Å, respectively, which confirmed the mutant-driven conversion from antiandrogen to AR agonist.
One particular AR mutation is the T877A in the LBD of AR, which actually results in paradoxical activation by hydroxyflutamide, an active metabolite of flutamide. Several variations of the 877 mutations have been discovered: T877S, T877C, and T877G. The T877A mutation along with H874Y mutation allows AR to be activated by cortisol. The W741C and W741L AR mutant are activated by bicalutamide. Another mutation, L701H, enhances cell proliferation upon stimulation of IL-6. Additionally, V715M, along with T877A, L701H, and H874Y, were identified in LNCaP cells treated with bisphenol A. Besides, a mutation at Q640S produces a truncated AR resulting in constitutive activation in the absence of ligand (Grasso et al., 2012; Trendel, 2013). Recently, a novel mutant F876L which turned enzalutamide and ARN509 from AR antagonist to AR agonist was identified in preclinical models and in the patients being treated with ARN509 (Joseph et al., 2013).
The mechanisms of mutant-driven conversion of antiandrogen from AR antagonist to AR agonist have been verified by the biological structural data. The crystal structure of T877A AR-LBD in complex with OHF (PDB-ID, 2ax6) demonstrates that the H12 adopts the agonistic conformation, which elucidates that the T877A mutation converts the OHF from an antagonist to an agonist (Figure 2B) (Bohl et al., 2005a). Another crystal structure (PDB-ID, 1z95) provides the structural evidence that W741L AR mutant switches bicalutamide to an agonist (Figure 2C) (Bohl et al., 2005b). A structural superimposition of these two structures with the AR-LBD structure bound with DHT conferred perfect alignments of the H12 with the RMSD (root mean square deviation) values of 0.28 and 0.48 Å, respectively, which confirmed that the mutant-driven conversion from the AR antagonist to AR agonist (Figure 2D). However, the mechanism under the antagonist-agonist conversion of ligands remains elusive.
As mentioned previously, PCa is continually undergoing AR mutations that switch the antiandrogen from AR antagonist to AR agonist and eventually relapses to lethal CRPC. Thus the rational development of novel antiandrogens based on the AR acquired mutations driven drug resistant seems like ongoing running race and remains a big challenge (Josan and Katzenellenbogen, 2013). Up to now, several effective strategies, especially the rational drug design, have been applied in the development of the antiandrogens targeting HBP of AR (Figure 3).
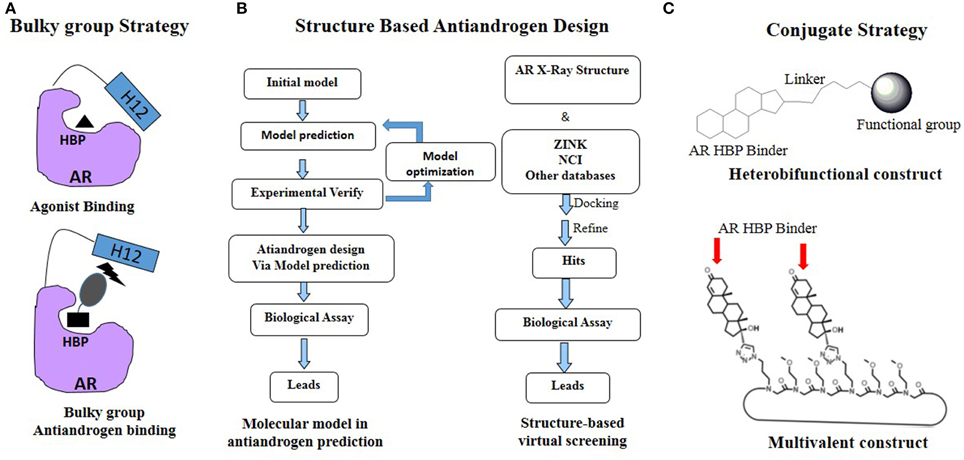
Figure 3. Rational antiandrogen design strategy to combat the mutation driven drug resistance: (A) Bulky group strategy; (B) Structure based antiandrogen design; (C) Conjugate strategy.
Considerating that the agonistic conformation of H12 is like a lid enclosing the HBP, designing compounds bearing an extended bulky arm to displace H12 (Figure 3A) is an effective strategy. For example, based on the structure of bicalutamide, several derivatives with an extended aryl sulfone core were designed and prepared. Among them, compounds 1–3 (Figure 4) showed potent antagonistic activity in all three mutations (T877A, W741L, and W741C) as well as wild-type AR (McGinley and Koh, 2007). Similarly, a series of flutamide analogs with bulky groups were also obtained and some of them circumvent the resistant mutation to their parent compound (Duke et al., 2011). Other bulky group antiandrogens with novel scaffolds were also reported. Zhou et al have developed a novel antiandrogen 4 (Figure 4) with two bulky side chains which shown low micromolar cytotoxicity in a panel of five PCa cell lines and potently suppressed DHT-induced transactivation of the WT and the T877A, W741C, and H874Y mutated ARs. Molecular modeling indicated that 4 adopts a “Y”-shape conformation and forms multiple hydrogen bonds with AR backbone in HBP (Zhou et al., 2009). Besides, via the SAR studies of the lead compound DIMN, Yang et al have synthesized a series of nicotinamides with extended linear scaffold bearing sterically bulky alkoxy groups on isoquinoline end and identified compounds 5 and 6 (Figure 4) as promising candidates of second generation antiandrogen for advanced PCa (Yang et al., 2013).
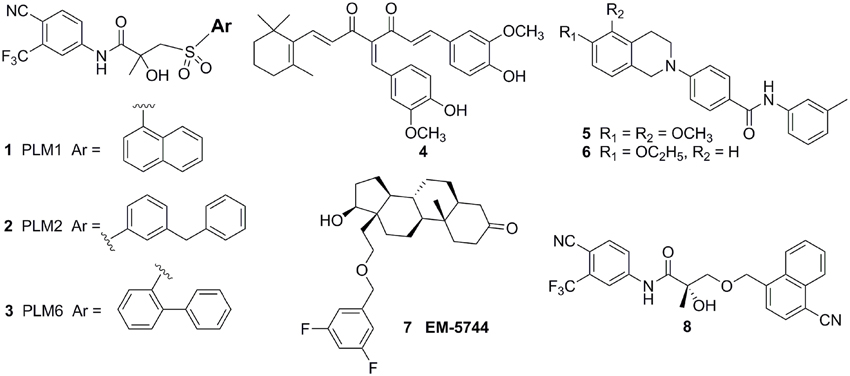
Figure 4. Chemical structures of AR Antagonists and Agonists designed by using bulky group strategy.
Interestingly, Endo et al have developed a series of carborane substituted antiandrogens (Fujii et al., 2005; Goto et al., 2005, 2010; Ohta et al., 2008). Carborane (dicarba-closo-dodecaborane, C2B10H12) is an icosahedral boron cluster, has a bulky spherical structure, exhibits remarkable thermal stability, and has high hydrophobicity. (Armstrong and Valliant, 2007). By replacing the hydrophobic ring such as the steroidal skeleton or the phenyl ring in hydroxyflutamide or bicalutamide by carborane cage, thus disposition of the helix-12 by using steric repulsion between the bulky carborane cage and several amino acid residues, mainly M895 and F876, in the hydrophobic pocket of the AR LBD. Several carborane containing antiandrogens showed the comparable potency to the known antiandrogens hydroxyflutamide or bicalutamide and exhibit pure AR full antagonistic property.
However, although there were a couple of successful examples, the bulky group strategy was somehow casted doubt (Duke et al., 2011). For example, several DHT-derived molecules bearing one bulky chain surprisingly turned out to be potent agonists of the AR. The complex structure of the AR LBD and one of the compounds termed EM5744 (Figure 4) (PDB-ID, 2pnu) was solved, which indicates the H12 of AR adopts the agonistic position (Cantin et al., 2007). Besides, a crystal structure (PDB-ID: 3rll) of compound 8 (Figure 4) complexed with AR (T877A) shown that receptor accommodated the added bulky groups such as phenyl to naphthyl substitution (Duke et al., 2011). The failure of the bulky ligand strategy might due to the flexibility of HBP, since it was reported that the volume of HBP would be hugely increased upon the binding of the ligands and the recruitment of coactivators (Xu et al., 2011).
Recently, structural based molecular modeling has been developed to predict target mutation-induced drug resistance. Meanwhile, various structural based design strategies, including targeting protein backbone, targeting highly conserved residues and dual/multiple targeting, have been used to design novel inhibitors for combating the drug resistance (Hao et al., 2012). To date, there are 89 AR LBD structures deposited in Protein Database Bank (PDB, www.pdb.org), which facilitate the discovery and development of novel antiandrogens to combat the drug resistance by using “structure-based” drug design (Figure 3B).
The molecular modeling techniques, especially molecular dynamics, have been widely used to predict the conformational displacement of H12 when the mutant occurs. Zhou et al investigated the impact of the T877A mutation on ligand-induced helix-12 positioning by replica-exchange molecular dynamics (REMD) simulations and proposed a REMD based methodology to predict agonist/antagonist potency of a ligand. According to the simulation results, a novel flutamide derivative called SC333 (Figure 5) was designed and predicted to be a pure antagonist of the T877A mutant, which was further experimentally confirmed as a pan-antiandrogen against the wild type AR and the T877A and W741C mutated ARs (Zhou et al., 2010). In additional, Osguthorpe et al predicated that bicalutamide antagonizes AR by accessing an additional binding pocket (B-site) adjacent to the HBP via molecular dynamics, induced by displacing H12(Osguthorpe and Hagler, 2011). These molecular modeling studies based on crystal structure shed light on the mechanism of the mutant-driven antagonist/agonist conversion and provide a structural framework for the design of novel antiandrogens (Osguthorpe and Hagler, 2011). An impressive work was reported by Dr Sawyers and the collaborators recently (Balbas et al., 2013). They performed molecular dynamics simulations of antiandrogen-AR complexes and suggested the mechanism that the F876L substitution alleviates antagonism through repositioning of the coactivator recruiting H12. Based on the mechanism, a focused chemical screening was performed and three novel compounds, including the most potent compound DR103 (Figure 5), were identified effectively antagonizing AR F876L (and AR WT) to suppress the growth of PCa cells lines resistant to enzalutamide (Balbas et al., 2013).
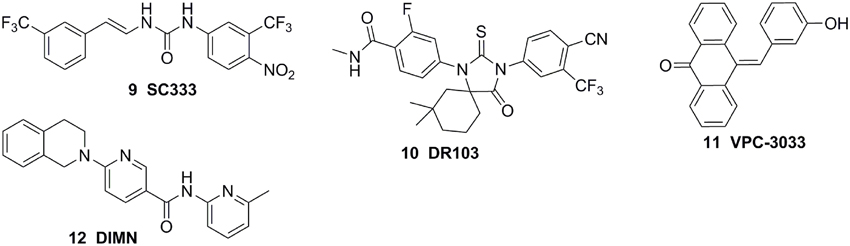
Figure 5. Chemical structures of AR Antagonists based on Structure of HBP.
Structure-based virtual screening is another effective way to obtain the lead compound of antiandrogens. For example, combining virtual screening and further biological assay, a lead compound, VPC-3033 (Figure 5), was identified to demonstrated strong androgen displacement potency which effectively inhibited AR transcriptional activity possess, profoundly degraded AR and significantly suppressed enzalutamide resistance PCa cells (Li et al., 2013a). Moreover, through structure-based virtual screening using the FlexX docking model, 54 candidates were selected and further screened for AR antagonism via cell-based tests. One compound, DIMN (Figure 5), showed antagonistic effect specific to AR with comparable potency to that of hydroxyflutamide and bicalutamide (Song et al., 2012). In another virtual screening work, the database was firstly filtered through a pharmacophore models built according to the structures of the reported antiandrogens, and the hits were subjected to structure-based docking evaluations using an AR homology model and cross-docked into the variant AR crystal structure bound to bicalutamide using the Surflex suite. A series of structural distinct competitive AR antagonists were obtained, which belonged to six chemotypes. Among these compounds, chemotype A compounds functioned as AR antagonists in vivo in normal male mice and suppressed AR activity and tumor cell proliferation in human CRPC xenografts (Shen et al., 2012). In addition, the molecular docking was also applied to establish the binding mode of the antiandrogens in HBP of the AR LBD, which would be beneficial to further optimization of the lead compounds (Zhou et al., 2009; Pepe et al., 2013; Guerrini et al., 2014).
Cross coupling and conjugation strategies are wildly applied to develop the modulator with multivalent and heterobifunctional constructs, which exhibit high affinity and specificity to the biomolecular target (Lambert, 2013; Washburn et al., 2013) (Figure 3C). To date, there are limited examples targeting the AR with steroidal conjugates (Levine et al., 2014). Utilizing conjugates dubbed PROteolysis TArgeting Chimeric moleculeS (PROTACS), the first steroid conjugate PROTAC-5 (Figure 6) to selectively induce AR degradation was developed, which consist of three components: a targeting moiety (DHT), a linker, and a recognition element for E3. Initial ex vivo studies showed that PROTAC-5 successfully degraded AR without compromising normal cell viability at a concentration of 25 μ M 9 (Schneekloth et al., 2004). In addition, Hashimoto lab has developed Specific and Nongenetic IAPs-dependent Protein ERasers (SNIPERs) that consist of a targeting moiety (DHT), linker, and a recognition element for IAPs. In human mammary tumor (MCF-7) cells that express AR, SNIPER 13 (Figure 6), an AR targeting compound, decreased AR protein levels at a concentration of 30 μ M. Recent studies from the Hannon group have discovered the first metallo-based chemotherapeutic conjugates targeting AR. Ethisterone was conjugated to pyridines, quinolines, and isoquinolines utilizing Sonogashira cross-coupling conditions. Subsequent coordination to platinum (II) complexes yielded metallo-based bifunctional agents. Initial evaluation of the cytotoxic effects of the two most promising metallo-based bifunctional agents 15 and 16 (Figure 6), in the cell lines that express AR revealed promising biological activity (IC50 = 15.9 μ M) (Huxley et al., 2010). Essigmann's group has developed heterobifunctional DNA-damaging agent 17 (Figure 6) in which a alkylating agent N,N-bis-2-chloroethylaniline was linked to a steroid hormone that targets AR, allowing the conjugate to simultaneously bind to AR and DNA, resulting in the blockade of DNA repair enzymes in PCa cell lines that overexpressing AR, subsequently leading to the disruption of AR-mediated transcription and signaling (Marquis et al., 2005). An emerging avenue in molecular pharmacology is the development of multivalent therapeutic agents. Therefore, the Kirshenbaum lab designed multivalent ethisterone conjugates to specifically target the AR LDB and modulate AR activity via different mechanisms of action. Ethisterone was conjugated at the 17-α position to the peptoid scaffold via highly stable triazole linakges. Two conjugates 18 and 19 (Figure 6) exhibited potent anti-proliferative properties in proliferation studies of LNCaP-abl cell lines and no cytotoxicity in PC-3 and HEK293 cell lines, establishing that conjugates selectively target AR (Levine et al., 2012).
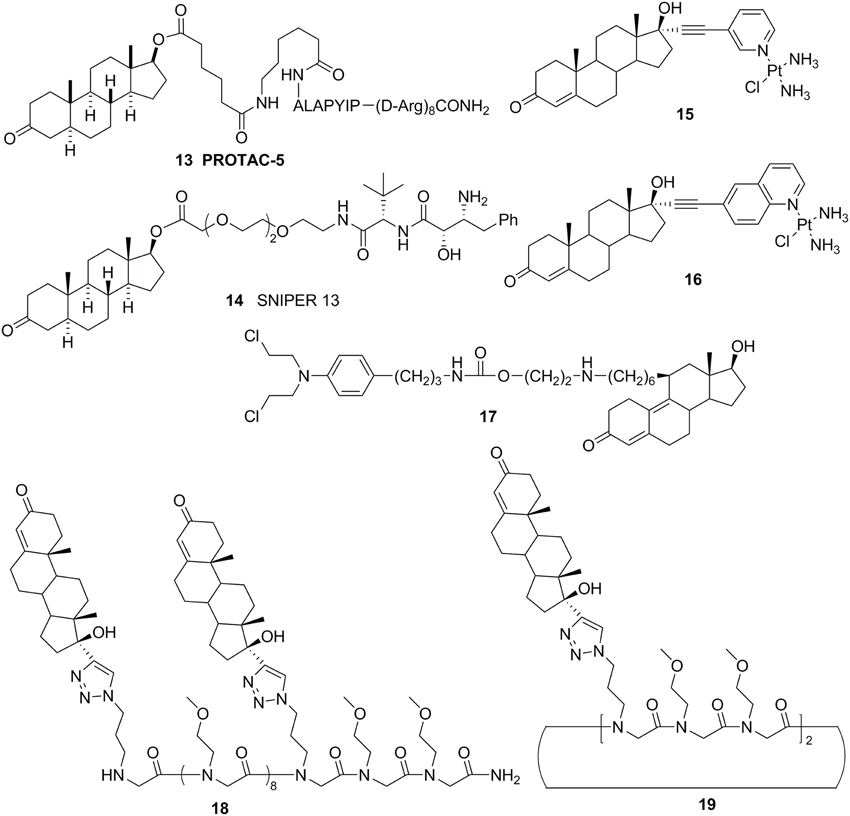
Figure 6. Chemical structures of steroid conjugate AR Antagonists.
Except for steroid conjugates, there are several representative examples of promising strategies that have been used to target AR with non-steroidal conjugates. Recently, the Oyelere's lab reported a non-steroidal heterobifunctional conjugate 20 (Figure 7) outfitted with histone deacetylase inhibitors that exhibit higher potency in modulation of AR activity than clinically used anti-androgens (Gryder et al., 2013). In similar studies, the Koch lab reported a non-steroidal heterobifunctional conjugate 21 (Figure 7) containing doxorubicin, a non-selective cytotoxic therapeutic DNA intercalator. The antiandrogen conjugate successfully delivered the doxorubicin-formaldehyde Schiff base to cells overexpressing AR (Cogan and Koch, 2003). The El-Sayed lab introduced the first non-steroidal multivalent conjugate 22 (Figure 7) that selectively target membrane-associated AR. Bicalutamide was conjugated to gold nanoparticles. The multivalent compounds enhanced potency by one order of magnitude, in comparison to the monovalent ligand, in PCa cells (Dreaden et al., 2012).
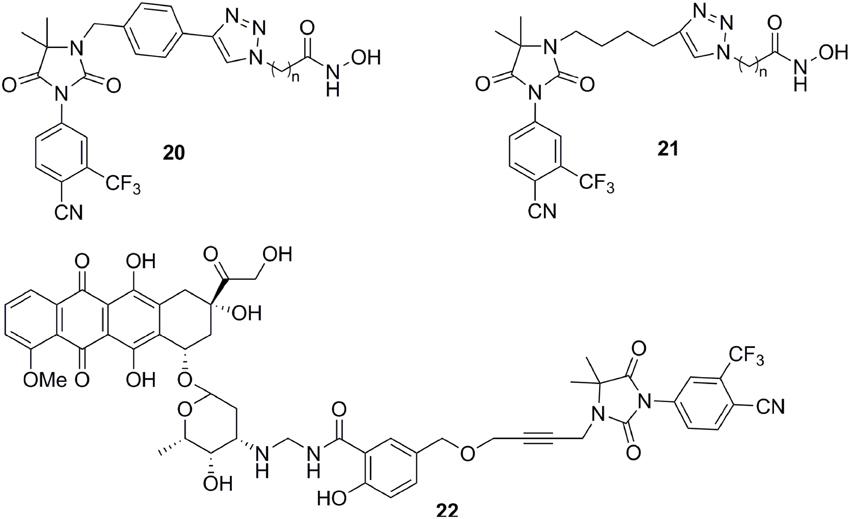
Figure 7. Chemical structures of non-steroid conjugate AR Antagonists.
Despite the great improvements h ave been made in the development of antiandrogen circumventing mutation-based resistance, it remains a big challenge. First of all, there is still no antagonistic or apo AR LBD structure available, current LBD actually are all agonistic, which add the hurdle of structure-based antiandrogen design. Up to now, the adopted antagonistic AR LBD model included the homology model based on the antagonistic LBD of other nuclear receptor such as ER, GR and PR, the model with the H12 simply deleted from the agonistic AR LBD, and the model equilibrated via molecular dynamics from the started structure of the agonistic AR LBD. Thus, the antagonistic or apo AR LBD structure is highly in demand to provide more accurate model in structural based drug design of antiandogens. However, as the antagonistic or apo AR LBD protein is formidable to be prepared and crystallized due to its flexibility and tight association with the bacterial chaperonin (Bohl et al., 2005b), to solve the 3D structure of the antagonistic or apo AR LBD is still a huge challenge. Secondly, besides of acquired the mutation of AR to induce drug resistance, there are other mechanisms including aberrant expression of AR co-regulators and alternative AR activation by cytokines and growth factors, and the expression of AR splice variants lacking LBD. As an example, the cells with AR gene rearrangements expressing both full-length and AR-Vs are identified androgen independent and enzalutamide resistant, and selective knock-down of AR-Vs expression inhibited androgen-independent growth and restored responsiveness to androgens and antiandrogens (Li et al., 2013b). Moreover, it was proposed that the resistance to enzalutamide via the activation of AR and its splice variants may be mediated by NF-kB2/p52 (Nadiminty et al., 2013). Recent work revealed the enzalutamide resistance could also be achieved through the activation of GR signaling (Arora et al., 2013). Therefore, the drug resistance is associated with multiple factors, and still a long trudge is left to circumvent the antiandrogen resistance in prostate cancer.
Besides of HBP, several other binding sites on AR like AF2, BF3, the DNA binding site, and AF1 have been attracted attentions in novel antiandrogen development (Haendler and Cleve, 2012). To date, several ligands have been identified to bind to these sites, and exhibiting potent activities in antagonizing the AR signaling, which inhibit the proliferation of AR dependent prostate cancer cells (Andersen et al., 2010; Axerio-Cilies et al., 2011; Ravindranathan et al., 2013; Li et al., 2014; Munuganti et al., 2014). Some of these active agents demonstrated significant antiandrogen potency against enzalutamide-resistant prostate cancer cell lines (Li et al., 2014; Munuganti et al., 2014). Especially, the agents that target the DNA binding site or AF1 could effectively inhibit the growth of enzalutamide-resistant cells as well as block the transcriptional activity of constitutively active AR splice variants like AR-V7, ARv567 (Li et al., 2014). Thus, targeting these sites other than HBP provides an another feasible avenue to develop the therapeutic agents for prostate cancer. As it has been beyond the scope of current review, several comprehensive reviews are recommended here (Nyronen and Soderholm, 2010; Haendler and Cleve, 2012; Lallous et al., 2013; Culig, 2014; Tan et al., 2015; Lorente et al., 2015).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was in part supported by the Nature Science Foundation of China (81311120299, 81271844) and the McGill Integrated Cancer Research Training Program (MICRTP) Postdoctoral Fellowship 2012.
Andersen, R. J., Mawji, N. R., Wang, J., Wang, G., Haile, S., Myung, J. K., et al. (2010). Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell 17, 535–546. doi: 10.1016/j.ccr.2010.04.027
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Aragon-Ching, J. B. (2014). The evolution of prostate cancer therapy: targeting the androgen receptor. Front. Oncol. 4:295. doi: 10.3389/fonc.2014.00295
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Armstrong, A. F. and Valliant, J. F. (2007). The bioinorganic and medicinal chemistry of carboranes: from new drug discovery to molecular imaging and therapy. Dalton Trans. 4240–4251. doi: 10.1039/b709843j
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Arora, V. K., Schenkein, E., Murali, R., Subudhi, S. K., Wongvipat, J., Balbas, M. D., et al. (2013). Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell 155, 1309–1322. doi: 10.1016/j.cell.2013.11.012
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Axerio-Cilies, P., Lack, N. A., Nayana, M. R., Chan, K. H., Yeung, A., Leblanc, E., et al. (2011). Inhibitors of androgen receptor activation function-2 (AF2) site identified through virtual screening. J. Med. Chem. 54, 6197–6205. doi: 10.1021/jm200532b
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Balbas, M. D., Evans, M. J., Hosfield, D. J., Wongvipat, J., Arora, V. K., Watson, P. A., et al. (2013). Overcoming mutation-based resistance to antiandrogens with rational drug design. Elife 2:e00499. doi: 10.7554/eLife.00499
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Blackledge, G. R., Cockshott, I. D., and Furr, B. J. (1997). Casodex (bicalutamide): overview of a new antiandrogen developed for the treatment of prostate cancer. Eur. Urol. 31(Suppl. 2), 30–39.
Bohl, C. E., Gao, W., Miller, D. D., Bell, C. E., and Dalton, J. T. (2005b). Structural basis for antagonism and resistance of bicalutamide in prostate cancer. Proc. Natl. Acad. Sci. U.S.A. 102, 6201–6206. doi: 10.1073/pnas.0500381102
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bohl, C. E., Miller, D. D., Chen, J., Bell, C. E., and Dalton, J. T. (2005a). Structural basis for accommodation of nonsteroidal ligands in the androgen receptor. J. Biol. Chem. 280, 37747–37754. doi: 10.1074/jbc.M507464200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Caboni, L., and Lloyd, D. G. (2013). Beyond the ligand-binding pocket: targeting alternate sites in nuclear receptors. Med. Res. Rev. 33, 1081–1118. doi: 10.1002/med.21275
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cannata, D. H., Kirschenbaum, A., and Levine, A. C. (2012). Androgen deprivation therapy as primary treatment for prostate cancer. J. Clin. Endocrinol. Metab. 97, 360–365. doi: 10.1210/jc.2011-2353
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cantin, L., Faucher, F., Couture, J. F., de Jesus-Tran, K. P., Legrand, P., Ciobanu, L. C., et al. (2007). Structural characterization of the human androgen receptor ligand-binding domain complexed with EM5744, a rationally designed steroidal ligand bearing a bulky chain directed toward helix 12. J. Biol. Chem. 282, 30910–30919. doi: 10.1074/jbc.M705524200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Carver, B. S. (2014). Strategies for targeting the androgen receptor axis in prostate cancer. Drug Discov. Today 19, 1493–1497. doi: 10.1016/j.drudis.2014.07.008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cogan, P. S., and Koch, T. H. (2003). Rational design and synthesis of androgen receptor-targeted nonsteroidal anti-androgen ligands for the tumor-specific delivery of a doxorubicin-formaldehyde conjugate. J. Med. Chem. 46, 5258–5270. doi: 10.1021/jm0303305
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Culig, Z. (2014). Targeting the androgen receptor in prostate cancer. Expert Opin. Pharmacother. 15, 1427–1437. doi: 10.1517/14656566.2014.915313
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dong, Y., Zhang, H., Gao, A. C., Marshall, J. R., and Ip, C. (2005). Androgen receptor signaling intensity is a key factor in determining the sensitivity of prostate cancer cells to selenium inhibition of growth and cancer-specific biomarkers. Mol. Cancer Ther. 4, 1047–1055. doi: 10.1158/1535-7163.MCT-05-0124
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dreaden, E. C., Gryder, B. E., Austin, L. A., Tene Defo, B. A., Hayden, S. C., Pi, M., et al. (2012). Antiandrogen gold nanoparticles dual-target and overcome treatment resistance in hormone-insensitive prostate cancer cells. Bioconjug. Chem. 23, 1507–1512. doi: 10.1021/bc300158k
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Duke, C. B., Jones, A., Bohl, C. E., Dalton, J. T., and Miller, D. D. (2011). Unexpected binding orientation of bulky-B-ring anti-androgens and implications for future drug targets. J. Med. Chem. 54, 3973–3976. doi: 10.1021/jm2000097
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fujii, S., Hashimoto, Y., Suzuki, T., Ohta, S., and Endo, Y. (2005). A new class of androgen receptor antagonists bearing carborane in place of a steroidal skeleton. Bioorg. Med. Chem. Lett. 15, 227–230. doi: 10.1016/j.bmcl.2004.09.091
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gao, W. (2010). Androgen receptor as a therapeutic target. Adv. Drug Deliv. Rev. 62, 1277–1284. doi: 10.1016/j.addr.2010.08.002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gillatt, D. (2006). Antiandrogen treatments in locally advanced prostate cancer: are they all the same? J. Cancer Res. Clin. Oncol. 132(Suppl. 1), S17–S26. doi: 10.1007/s00432-006-0133-5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Goldspiel, B. R., and Kohler, D. R. (1990). Flutamide: an antiandrogen for advanced prostate cancer. DICP 24, 616–623.
Goto, T., Ohta, K., Fujii, S., Ohta, S., and Endo, Y. (2010). Design and synthesis of androgen receptor full antagonists bearing a p-carborane cage: promising ligands for anti-androgen withdrawal syndrome. J. Med. Chem. 53, 4917–4926. doi: 10.1021/jm100316f
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Goto, T., Ohta, K., Suzuki, T., Ohta, S., and Endo, Y. (2005). Design and synthesis of novel androgen receptor antagonists with sterically bulky icosahedral carboranes. Bioorg. Med. Chem. 13, 6414–6424. doi: 10.1016/j.bmc.2005.06.061
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Grasso, C. S., Wu, Y.-M., Robinson, D. R., Cao, X., Dhanasekaran, S. M., et al. (2012). The Mutational landscape of lethal castrate resistant prostate Cancer. Nature 487, 239–243. doi: 10.1038/nature11125
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gryder, B. E., Akbashev, M. J., Rood, M. K., Raftery, E. D., Meyers, W. M., Dillard, P., et al. (2013). Selectively targeting prostate cancer with antiandrogen equipped histone deacetylase inhibitors. ACS Chem. Biol. 8, 2550–2560. doi: 10.1021/cb400542w
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Guerrini, A., Tesei, A., Ferroni, C., Paganelli, G., Zamagni, A., Carloni, S., et al. (2014). A new avenue toward androgen receptor pan-antagonists: C2 sterically hindered substitution of hydroxy-propanamides. J. Med. Chem. 57, 7263–7279. doi: 10.1021/jm5005122
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Haendler, B., and Cleve, A. (2012). Recent developments in antiandrogens and selective androgen receptor modulators. Mol. Cell Endocrinol. 352, 79–91. doi: 10.1016/j.mce.2011.06.002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hao, G. F., Yang, G. F., and Zhan, C. G. (2012). Structure-based methods for predicting target mutation-induced drug resistance and rational drug design to overcome the problem. Drug Discov. Today 17, 1121–1126. doi: 10.1016/j.drudis.2012.06.018
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Heinlein, C. A., and Chang, C. (2002). Androgen receptor (AR) coregulators: an overview. Endocr. Rev. 23, 175–200. doi: 10.1210/edrv.23.2.0460
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Huxley, M., Sanchez-Cano, C., Browning, M. J., Navarro-Ranninger, C., Quiroga, A. G., Rodger, A., et al. (2010). An androgenic steroid delivery vector that imparts activity to a non-conventional platinum(II) metallo-drug. Dalton Trans. 39, 11353–11364. doi: 10.1039/c0dt00838a
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jemal, A., Bray, F., Center, M. M., Ferlay, J., Ward, E., and Forman, D. (2011). Global cancer statistics. CA A Cancer J. Clin. 61, 69–90. doi: 10.3322/caac.20107
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jenster, G. (1999). The role of the androgen receptor in the development and progression of prostate cancer. Semin. Oncol. 26, 407–421.
Josan, J. S., and Katzenellenbogen, J. A. (2013). Designer antiandrogens join the race against drug resistance. Elife 2:e00692. doi: 10.7554/eLife.00692
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Joseph, J. D., Lu, N., Qian, J., Sensintaffar, J., Shao, G., Brigham, D., et al. (2013). A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discov. 3, 1020–1029. doi: 10.1158/2159-8290.CD-13-0226
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lallous, N., Dalal, K., Cherkasov, A., and Rennie, P. S. (2013). Targeting alternative sites on the androgen receptor to treat castration-resistant prostate cancer. Int. J. Mol. Sci. 14, 12496–12519. doi: 10.3390/ijms140612496
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lambert, J. M. (2013). Drug-conjugated antibodies for the treatment of cancer. Br. J. Clin. Pharmacol. 76, 248–262. doi: 10.1111/bcp.12044
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Levine, P. M., Garabedian, M. J., and Kirshenbaum, K. (2014). Targeting the androgen receptor with steroid conjugates. J. Med. Chem. 57, 8224–8237. doi: 10.1021/jm500101h
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Levine, P. M., Imberg, K., Garabedian, M. J., and Kirshenbaum, K. (2012). Multivalent peptidomimetic conjugates: a versatile platform for modulating androgen receptor activity. J. Am. Chem. Soc. 134, 6912–6915. doi: 10.1021/ja300170n
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Li, H., Ban, F., Dalal, K., Leblanc, E., Frewin, K., Ma, D., et al. (2014). Discovery of small-molecule inhibitors selectively targeting the DNA-binding domain of the human androgen receptor. J. Med. Chem. 57, 6458–6467. doi: 10.1021/jm500802j
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Li, H., Hassona, M. D., Lack, N. A., Axerio-Cilies, P., Leblanc, E., Tavassoli, P., et al. (2013a). Characterization of a new class of androgen receptor antagonists with potential therapeutic application in advanced prostate cancer. Mol. Cancer Ther. 12, 2425–2435. doi: 10.1158/1535-7163.MCT-13-0267
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Li, Y., Chan, S. C., Brand, L. J., Hwang, T. H., Silverstein, K. A., and Dehm, S. M. (2013b). Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res. 73, 483–489. doi: 10.1158/0008-5472.CAN-12-3630
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lorente, D., Mateo, J., Zafeiriou, Z., Smith, A., D., Sandhu, S., et al. (2015). Switching and withdrawing hormonal agents for castration-resistant prostate cancer. Nat. Rev. Urol. 12, 37–47. doi: 10.1038/nrurol.2014.345
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lubeck, D. P., Grossfeld, G. D., and Carroll, P. R. (2001). The effect of androgen deprivation therapy on health-related quality of life in men with prostate cancer. Urology 58, 94–100. doi: 10.1016/S0090-4295(01)01250-X
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Maeda, O., and Usami, M. (2002). [Antiandrogen in prostate cancer]. Nihon Rinsho 60(Suppl. 11), 188–192.
Marquis, J. C. Hillier, S. M., Dinaut, A. N., Rodrigues, D., Mitra, K., Essigmann, J. M., and Croy, R. G. (2005). Disruption of gene expression and induction of apoptosis in prostate cancer cells by a DNA-damaging agent tethered to an androgen receptor ligand. Chem. Biol. 12, 779–787. doi: 10.1016/j.chembiol.2005.05.009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
McGinley, P. L., and Koh, J. T. (2007). Circumventing anti-androgen resistance by molecular design. J. Am. Chem. Soc. 129, 3822–3823. doi: 10.1021/ja0701154
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Montgomery, R. B., Mostaghel, E. A., Vessella, R., Hess, D. L., Kalhorn, T. F., Higano, C. S., et al. (2008). Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res. 68, 4447–4454. doi: 10.1158/0008-5472.CAN-08-0249
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Munuganti, R. S., Hassona, M. D., Leblanc, E., Frewin, K., Singh, K., Ma, D., et al. (2014). Identification of a Potent Antiandrogen that Targets the BF3 Site of the Androgen Receptor and Inhibits Enzalutamide-Resistant Prostate Cancer. Chem. Biol. 21, 1476–1485. doi: 10.1016/j.chembiol.2014.09.012
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nadiminty, N., Tummala, R., Liu, C., Yang, J., Lou, W., Evans, C. P., et al. (2013). NF-kappaB2/p52 induces resistance to enzalutamide in prostate cancer: role of androgen receptor and its variants. Mol. Cancer Ther. 12, 1629–1637. doi: 10.1158/1535-7163.MCT-13-0027
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Norman, A. W., Mizwicki, M. T., and Norman, D. P. (2004). Steroid-hormone rapid actions, membrane receptors and a conformational ensemble model. Nat. Rev. Drug Discov. 3, 27–41. doi: 10.1038/nrd1283
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nyronen, T. H., and Soderholm, A. A. (2010). Structural basis for computational screening of non-steroidal androgen receptor ligands. Expert Opin. Drug Discov. 5, 5–20. doi: 10.1517/17460440903468680
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ohta, K., Goto, T., Fijii, S., Suzuki, T., Ohta, S., and Endo, Y. (2008). Design and synthesis of carborane-containing androgen receptor (AR) antagonist bearing a pyridine ring. Bioorg. Med. Chem. 16, 8022–8028. doi: 10.1016/j.bmc.2008.07.055
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Osguthorpe, D. J., and Hagler, A. T. (2011). Mechanism of androgen receptor antagonism by bicalutamide in the treatment of prostate cancer. Biochemistry 50, 4105–4113. doi: 10.1021/bi102059z
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Paul, R., and Breul, J. (2000). Antiandrogen withdrawal syndrome associated with prostate cancer therapies: incidence and clinical significance. Drug Saf. 23, 381–390. doi: 10.2165/00002018-200023050-00003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pepe, A., Pamment, M., Kim, Y. S., Lee, S., Lee, M. J., Beebe, K., et al. (2013). Synthesis and structure-activity relationship studies of novel dihydropyridones as androgen receptor modulators. J. Med. Chem. 56, 8280–8297. doi: 10.1021/jm301714s
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pratt, W. B., and Toft, D. O. (1997). Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr. Rev. 18, 306–360.
Ravindranathan, P., Lee, T. K., Yang, L., Centenera, M. M., Butler, L., Tilley, W. D., et al. (2013). Peptidomimetic targeting of critical androgen receptor-coregulator interactions in prostate cancer. Nat. Commun. 4:1923. doi: 10.1038/ncomms2912
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Roy, A. K., Lavrovsky, Y., Song, C. S., Chen, S., Jung, M. H., Velu, N. K., et al. (1999). Regulation of androgen action. Vitam. Horm. 55, 309–352. doi: 10.1016/S0083-6729(08)60938-3
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ruckle, H. C., and Oesterling, J. E. (1993). Prostate-specific antigen and androgen deprivation therapy. World J. Urol. 11, 227–232. doi: 10.1007/BF00185075
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ryan, C. J., and Small, E. J. (2006). Androgen deprivation therapy in locally advanced and metastatic prostate cancer. Minerva Urol. Nefrol. 58, 119–126.
Sartor, A. O., Tangen, C. M., Hussain, M. H., Eisenberger, M. A., Parab, M., Fontana, J. A., et al. (2008). Antiandrogen withdrawal in castrate-refractory prostate cancer: a Southwest Oncology Group trial (SWOG 9426). Cancer 112, 2393–2400. doi: 10.1002/cncr.23473
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schneekloth, J. S. Jr., Fonseca, F. N., Koldobskiy, M., Mandal, A., Deshaies, R., Sakamoto, K., et al. (2004). Chemical genetic control of protein levels: selective in vivo targeted degradation. J. Am. Chem. Soc. 126, 3748–3754. doi: 10.1021/ja039025z
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Semenas, J., Dizeyi, N., and Persson, J. L. (2013). Enzalutamide as a second generation antiandrogen for treatment of advanced prostate cancer. Drug Des. Devel. Ther. 7, 875–881. doi: 10.2147/DDDT.S45703
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shang, Y., Myers, M., and Brown, M. (2002). Formation of the androgen receptor transcription complex. Mol. Cell 9, 601–610. doi: 10.1016/S1097-2765(02)00471-9
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shen, H. C., Shanmugasundaram, K., Simon, N. I., Cai, C., Wang, H., Chen, S., et al. (2012). In silico discovery of androgen receptor antagonists with activity in castration resistant prostate cancer. Mol. Endocrinol. 26, 1836–1846. doi: 10.1210/me.2012-1222
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Song, C. H., Yang, S. H., Park, E., Cho, S. H., Gong, E. Y., Khadka, D. B., et al. (2012). Structure-based virtual screening and identification of a novel androgen receptor antagonist. J. Biol. Chem. 287, 30769–30780. doi: 10.1074/jbc.M112.379107
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tan, M. E., Li, J., Xu, H. E., Melcher, K., and Yong, E. L. (2015). Androgen receptor: structure, role in prostate cancer and drug discovery. Acta Pharmacol. Sin. 36, 3–23. doi: 10.1038/aps.2014.18
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Taplin, M. E. (2007). Drug insight: role of the androgen receptor in the development and progression of prostate cancer. Nat. Clin. Pract. Oncol. 4, 236–244. doi: 10.1038/ncponc0765
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tomlins, S. A., Rhodes, D. R., Perner, S., Dhanasekaran, S. M., Mehra, R., Sun, X. W., et al. (2005). Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 310, 644–648. doi: 10.1126/science.1117679
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Trendel, J. A. (2013). The hurdle of antiandrogen drug resistance: drug design strategies. Expert Opin. Drug Discov. 8, 1491–1501. doi: 10.1517/17460441.2013.855194
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Washburn, N. R., Prata, J. E., Friedrich, E. E., Ramadan, M. H., Elder, A. N., and Sun, L. T. (2013). Polymer-conjugated inhibitors of tumor necrosis factor-alpha for local control of inflammation. Biomatter 3:e25597. doi: 10.4161/biom.25597
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Werner, R., Holterhus, P. M., Binder, G., Schwarz, H. P., Morlot, M., Struve, D., et al. (2006). The A645D mutation in the hinge region of the human androgen receptor (AR) gene modulates AR activity, depending on the context of the polymorphic glutamine and glycine repeats. J. Clin. Endocrinol. Metab. 91, 3515–3520. doi: 10.1210/jc.2006-0372
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Xu, X., Yang, W., Wang, X., Li, Y., Wang, Y., and Ai, C. (2011). Dynamic communication between androgen and coactivator: mutually induced conformational perturbations in androgen receptor ligand-binding domain. Proteins 79, 1154–1171. doi: 10.1002/prot.22951
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yang, S. H., Song, C. H., Van, H. T., Park, E., Khadka, D. B., Gong, E. Y., et al. (2013). SAR based design of nicotinamides as a novel class of androgen receptor antagonists for prostate cancer. J. Med. Chem. 56, 3414–3418. doi: 10.1021/jm3014103
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yuan, X., Lu, M. L., Li, T., and Balk, S. P. (2001). SRY interacts with and negatively regulates androgen receptor transcriptional activity. J. Biol. Chem. 276, 46647–46654. doi: 10.1074/jbc.M108404200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhou, J., Geng, G., Shi, Q., Sauriol, F., and Wu, J. H. (2009). Design and synthesis of androgen receptor antagonists with bulky side chains for overcoming antiandrogen resistance. J. Med. Chem. 52, 5546–5550. doi: 10.1021/jm801218k
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhou, J., Liu, B., Geng, G., and Wu, J. H. (2010). Study of the impact of the T877A mutation on ligand-induced helix-12 positioning of the androgen receptor resulted in design and synthesis of novel antiandrogens. Proteins 78, 623–637. doi: 10.1002/prot.22592
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: androgen receptor, antiandrogen, drug resistance, mutation, rational drug design
Citation: Tian X, He Y and Zhou J (2015) Progress in antiandrogen design targeting hormone binding pocket to circumvent mutation based resistance. Front. Pharmacol. 6:57. doi: 10.3389/fphar.2015.00057
Received: 31 December 2014; Paper pending published: 10 February 2015;
Accepted: 05 March 2015; Published: 24 March 2015.
Edited by:
Rongtuan Lin, McGill University, CanadaReviewed by:
Vincenzo Arena, Università Cattolica del Sacro Cuore, ItalyCopyright © 2015 Tian, He and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jinming Zhou, Immunology, Institute of Medicinal Biotechnology Chinese Academy of Medical Science, Tian Tan Xi Li 1, Beijing 100050, ChinaemhvdWppbm1pbmdAaW1iLnB1bWMuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.