
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 19 December 2014
Sec. Integrative and Regenerative Pharmacology
Volume 5 - 2014 | https://doi.org/10.3389/fphar.2014.00270
This article is part of the Research Topic Fetal Therapies and Maternal-Fetal Tolerance View all 11 articles
 Durrgah L. Ramachandra1Steven S. W. Shaw2,3,4Panicos Shangaris4Stavros Loukogeorgakis1Pascale V. Guillot1,5Paolo De Coppi1Anna L. David4*
Durrgah L. Ramachandra1Steven S. W. Shaw2,3,4Panicos Shangaris4Stavros Loukogeorgakis1Pascale V. Guillot1,5Paolo De Coppi1Anna L. David4*Congenital diseases are responsible for over a third of all pediatric hospital admissions. Advances in prenatal screening and molecular diagnosis have allowed the detection of many life-threatening genetic diseases early in gestation. In utero transplantation (IUT) with stem cells could cure affected fetuses but so far in humans, successful IUT using allogeneic hematopoietic stem cells (HSCs), has been limited to fetuses with severe immunologic defects and more recently IUT with allogeneic mesenchymal stem cell transplantation, has improved phenotype in osteogenesis imperfecta. The options of preemptive treatment of congenital diseases in utero by stem cell or gene therapy changes the perspective of congenital diseases since it may avoid the need for postnatal treatment and reduce future costs. Amniotic fluid stem (AFS) cells have been isolated and characterized in human, mice, rodents, rabbit, and sheep and are a potential source of cells for therapeutic applications in disorders for treatment prenatally or postnatally. Gene transfer to the cells with long-term transgenic protein expression is feasible. Recently, pre-clinical autologous transplantation of transduced cells has been achieved in fetal sheep using minimally invasive ultrasound guided injection techniques. Clinically relevant levels of transgenic protein were expressed in the blood of transplanted lambs for at least 6 months. The cells have also demonstrated the potential of repair in a range of pre-clinical disease models such as neurological disorders, tracheal repair, bladder injury, and diaphragmatic hernia repair in neonates or adults. These results have been encouraging, and bring personalized tissue engineering for prenatal treatment of genetic disorders closer to the clinic.
Congenital diseases attributed to about 510,000 deaths globally in 2010 (Lozano et al., 2012), and are estimated to contribute to over a third of pediatric admissions to the hospital and up to 50% of the total costs of pediatric hospital treatment (McCandless et al., 2004). Prenatal diagnosis of many congenital diseases are performed using traditional invasive techniques such as amniocentesis or chorionic villus sampling (CVS), but increasingly non-invasive methods using circulating fetal DNA in the maternal blood are feasible and available for prenatal diagnosis early in gestation (Danzer et al., 2012; Danzer and Johnson, 2014). The current options for most parents facing congenital diseases following prenatal diagnosis are either to terminate or continue with a known affected pregnancy.
Progress over the last two decades have resulted in fetal therapy being available for a small number of congenital structural anomalies such as spina bifida, identical twin placental complications, and congenital diaphragmatic hernia, using open surgical or fetoscopic interventions (Pearson and Flake, 2013). These options are currently restricted to the treatment of fetal pathophysiology and are usually performed in the second half of gestation, when pathology is already evident. There are almost no therapeutic options however for life-threatening genetic disorders which have pathology beginning in utero. Success with in utero transplantation (IUT) using allogeneic hematopoietic stem cells (HSCs), has been limited to fetuses with severe immunologic defects where there is an effective lack of immune response to allogeneic cells, and transplanted genetically normal cells have a proliferative advantage (Tiblad and Westgren, 2008). Mesenchymal stem cells (MSCs) appear to be less immunogenic than their hematopoietic counterparts (O’Donoghue and Fisk, 2004) and have shown to reduce fracture rate in a mouse model (Guillot et al., 2008) and engraft in human fetuses with osteogenesis imperfecta in an allogeneic setting (Horwitz et al., 2002). Attempts to treat diseases such as sickle cell disease (Westgren et al., 1996) with in utero HSC transplantation, have been unsuccessful, even where a suitably matched donor has been available. Mouse studies suggest that the immune barrier to allogeneic in utero HSC transplantation may be stronger than previously thought (Peranteau et al., 2007). Transplantation of autologous progenitor cells, which have been corrected for the disease, could avoid the fetal immune barrier and may prove more successful than allogenic progenitors.
Autologous progenitors can be obtained from the fetus itself. Both proliferative and differentiation potentials of amniotic fluid stem (AFS) cells has been demonstrated in vitro and in vivo (De Coppi et al., 2007; Ditadi et al., 2009). Studies exploring the potential of this stem cell source for the use in autologous or allogenic prenatal therapy of congenital diseases have been conducted in large animal models (Shaw et al., 2014). In this review, we explore the latest developments in the field of in utero therapy for congenital disorders such as stem cell transplantation and gene transfer using AFS and their potential clinical applications.
Amniotic fluid (AF) consists of cells of fetal origin such as the amnion, skin, and respiratory system (Prusa and Hengstschläger, 2002; Tsai et al., 2004) and it can be obtained by routine clinical amniocentesis during pregnancy, a minimally invasive procedure used for prenatal diagnosis that usually takes place from 15 weeks of gestation (Gosden, 1983; Prusa and Hengstschläger, 2002; Delo et al., 2006). AF can also be collected during therapeutic amniodrainage procedures or even at cesarean section surgeries. Other fetal stem cell sources include the placenta, which can be accessed via ultrasound-guided CVS from 11 weeks of gestation or after birth yields epithelial, hematopoietic, and MSC types (Pipino et al., 2013; Jones et al., 2014). Fetal blood and the HSCs therein can also be collected from the umbilical cord in the first trimester of pregnancy by thin-gauge embryo fetoscopic-directed or ultrasound-guided blood sampling, although the long-term outcome following this procedure is not known (Chan et al., 2008).
In recent years, AFS cells have been explored in many clinical applications such as tissue engineering, cell transplantation, and gene therapy (Kaviani et al., 2001, 2003; Fauza, 2004; Tsai et al., 2004). C-Kit+ cells can be successfully isolated from AF, expanded with good population doublings and possess a very well characterized phenotype (Tsai et al., 2004; Delo et al., 2006; De Coppi et al., 2007; Ghionzoli et al., 2010). The surface antigen c-Kit (CD117) is known to be the receptor of stem cell factor and plays an essential role in gametogenesis, melanogenesis, and hematopoiesis (Fleischman, 1993; De Coppi et al., 2007). The successful expansion of AFS c-Kit+ cells has led to the finding of unique cell types such as mesenchymal (AFMSCs) and hematopoietic progenitors (Prusa and Hengstschläger, 2002; Delo et al., 2006; De Coppi et al., 2007; Ditadi et al., 2009).
Human AFS cells give rise to a variety of cell types such as osteogenic, myogenic, adipogenic, endothelial, hepatic, and neuronal origin; differentiation which has been be validated by the expression of mRNAs in lineage specific genes (Tsai et al., 2004; De Coppi et al., 2007). Rodent and murine AFS cells, like human AFS cells share similar growth properties and differentiation potential in vitro as well as the expression of embryonic and adult stem cell markers, respectively (De Coppi et al., 2007). AFS cells derived from human, mice, and sheep can be easily transduced without losing their characteristics (De Coppi et al., 2007; Ditadi et al., 2009; Mehta et al., 2011; Shaw and Bollini, 2011), and they possess privileged immunological characteristics that make them an ideal and reliable source for therapeutic transplantation (Ditadi et al., 2009).
The immune modulatory properties of AFS have shown resistance to natural killer (NK) cytotoxicity by inflammatory priming of AFS with interferon gamma (IFN-γ) and tumor necrosis factor alpha (TNF-α) and its ability to modulate lymphocyte proliferation according to its gestational age (Di Trapani et al., 2014). For instance, IFN-γ increases both MHC Class I and MHC Class II expression, indicating that like MSCs, they may not strongly contribute to rejection responses in allogeneic hosts (Moorefield et al., 2011). Moreover, AFS have shown to release high levels of cytokines including IL-6, MCP-2, MIP-3α, and MIP-1α when activated suggesting that they possess alternative molecular mechanisms to modulate immune response and regulation (Perin et al., 2010; Moorefield et al., 2011).
AFS cells have an estimated doubling time of 36 h and are grown without feeder layers (De Coppi et al., 2007). MSC subpopulations of AFS cells (AFMSCs), like other MSCs, maintain their spindle-shaped fibroblast-like morphology, their proliferation rate as well as their differentiation potential. Growth kinetics assays have shown that AFMSCs have a higher proliferation rate with an average doubling time of 25–38 h compared to bone marrow (BM) derived-MSCs that have an average doubling time of 30–90 h (Kaviani et al., 2001; Roubelakis et al., 2007). In addition, AFMSCs have a greater clonogenic potential compared to BM-MSCs (86 ± 4.3 versus 70 ± 5.1 colonies; Nadri and Soleimani, 2007). Despite the high proliferative rate of AFMSCs, they are still able to retain a normal karyotype with no evidence of tumorigenicity (Roubelakis et al., 2007). Human AFS cells express both markers of mesenchymal and pluripotent stem cells origin, such as stage-specific embryonic antigen (SSEA)-4 and Oct-4 (De Coppi et al., 2007). Once cultured in adherence however, they do not express markers of hematopoietic lineage such as CD45, CD34, and CD133 and express CD29, CD44, CD73, CD90, and CD105 (De Coppi, 2013). Interestingly, their plasticity, which is superior to adult stem cells, allow reprogramming into AFS derived induced pluripotent stem cells (iPS) with the change of the culture conditions they are exposed to (Lu et al., 2012; Moschidou et al., 2012, 2013; Pipino et al., 2014). This is particularly relevant as AFS cells can be utilized for cell banking of patient-specific pluripotent cells for potential applications in allogeneic cellular replacement therapies, pharmaceutical screening, and disease modeling (Moschidou et al., 2012, 2013).
In addition AFS cells, similarly to other fetal cells may represent the ideal source for therapy because, similarly to ES cells, they are easy to expand, and, in common with the adult counterparts, they are less controversial, not tumorigenic, readily cryopreserved for cell banking and their use can be accomplished on an autologous setting (De Coppi, 2013). The latter is particularly important in neonatal surgery, in the context of congenital malformations.
To be a successful autologous therapeutic resource for correcting genetic disease, AFS cells must be easily transduced to give high levels of therapeutic transgenic protein expression. The transduction of human AFS cells with vectors was explored with recombinant adenovirus vectors containing reporter genes such as AdHCMVsp1LacZ and AdCMV.eGFP (Grisafi et al., 2008). Human AFS cells presented a transduction efficiency of 100% when infected with 50 pfu/cell. Transduced human AFS cells maintained stemness features such as the expression of stem cell markers (SSEA4 and OCT4), adhesion and stromal molecules (CD29 and CD73) as well as adipogenic and osteogenic differentiation potential after infection (Grisafi et al., 2008). However, a decrease in SSEA4 expression, no expression of lipoprotein lipase (Lpl), an important adipogenic gene during differentiation and the expression of transcription factors Cbfa1 and PPaRγ detected only during early stages of differentiation suggests a slowdown in the differentiative progression and pluripotency after transduction.
Recently, we have shown that sheep AFS cells have the ability to be transduced using a lentivirus vector encoding the HIV-1 central polypurine tract element, the spleen focus forming virus LTR promoter, and the marker gene eGFP (63.2% efficiency). They have the ability to maintain the expression of MSC markers (CD44, CD58, and CD166) but were negative for hematopoietic, and endothelial markers (CD14, CD31, and CD45) as well as differentiate into adipogenic and osteogenic lineages (Shaw and Bollini, 2011; Shaw et al., 2014).
IUT involves the transplantation of cells to the fetus in utero (Muench, 2005) with the aim of treating congenital disorders by providing the correct stem cells (IUSCT) or gene corrected stem cells (IUSCGT). The benefits of an in utero approach to correcting genetic disease includes the prevention of pathology when it arises antenatally in those genetic diseases that cause irreversible damage to organs in utero, targeting of stem cell progenitors that are abundant and accessible in the fetus as well as dose scaling where the small size of the fetus allows relatively high doses of cells to be delivered. There is an opportunity for engraftment of donor cells without the need for myeloablation due to the immature status of the fetal immune system prior to thymic processing of self-antigen, a normal event in hematopoietic ontogeny (Waddington et al., 2007; David and Peebles, 2008; Shaw et al., 2011). Furthermore, immunological tolerance would allow postnatal reinfusion of cells to boost the effect after birth, as demonstrated recently in two children with osteogenesis imperfecta treated by IUT using MSCs (Götherström et al., 2014). Postnatal reinfusion with more MSCs from the original infusion source resulted in improved growth rate after birth.
As with any new therapeutic modality, the risks of IUT are not well characterized and the efficacy is still to be determined for some diseases. For in utero gene therapy, where vectors are given directly to the fetus for correction of genetic disease there has been direct guidance given by the NIH Recombinant DNA advisory committee report (RAC, 2000) on a pre-proposal for the initial application. The recommendations included that treatment should be limited only to: diseases that carry serious morbidity and mortality risks for the fetus either in utero or postnatally, do not have an effective postnatal therapy, or have a poor outcome using available postnatal therapies, can be definitively diagnosed in utero and have a well-defined genotype/phenotype relationship, have an animal model for in utero gene transfer that recapitulates the human disease and that the therapy would correct all serious abnormalities. It was recognized that a direct fetal vector injection approach would be difficult to justify given the above.
A combination IUSCGT approach however seems more likely to be acceptable. The UK Gene Therapy Advisory Committee (GTAC) considered this in their broader judgments about gene therapy in utero (Eckstein, 2003). The New and Emerging Technologies subgroup of GTAC found that the use of genetically modified stem cells in stem cell transplantation to the fetus was a possibility stating “such ex vivo modification would be unlikely to carry with it any higher risk to the germ line than the trials of postnatal somatic gene therapy which have already been approved.”
The fetal immune system is commonly regarded as immature and unresponsive despite reports showing its functional immune response (Mold and McCune, 2012). IUT relies crucially on the concept that the developing fetal immune system might accept a foreign cell or antigen and become tolerant to it. The presence of human NK cells have been detected as early as gestational week 6 in the fetal liver and in the fetal spleen at gestational week 15 (Phillips et al., 1992). Fetal NK cells have the ability to differentiate early in utero and are highly responsive to cytokines and antibody-mediated stimulation, and have shown to be functionally immature compared to adult NK cells (Phillips et al., 1992; Ivarsson et al., 2013).
The immune system during early gestation undergoes a process of self-education that occurs in the thymus. The positive and negative selection of pre-lymphocytes for the recognition of “self” major histocompatibility complex (MHC) antigen allows a repertoire of lymphocytes to be capable of direct and indirect antigen presentation which results in the deletion of alloreactive T-cells, regulatory T cells (Tregs) enrichment and creates donor-specific immune tolerance (Nijagal et al., 2013). Thus, to prevent limited engraftment, transplants should be introduced prior to the appearance of mature T-cells in the fetal thymus (Peranteau et al., 2006; Roybal et al., 2010; Nijagal et al., 2013).
In the human fetus, the immune system develops from 12 to 14 weeks of gestation, when profound increases in circulating T lymphocytes can be observed (Darrasse-Jèze et al., 2005; Takahama, 2006). Delivery of gene therapy may be required before this gestational age, which currently could limit the routes of application that can be safely used, although advances in engineering and imaging is leading to large improvements in fetal imaging and injection systems. It was demonstrated that the human fetus may have developed a functional immune system during the second trimester of gestation (Tse et al., 2005). Hematological compositions of human fetal blood and liver between 8 and 17 weeks gestation showed an increase in fetal red blood cell, white blood cell, and platelet counts with advancing gestation reflecting hematologic development (Pahal et al., 2000). An increase in circulating and hepatic T lymphocytes showed the presence of thymic maturation before the 13th week of gestation while the proportion of circulating primitive hematopoietic stem and progenitor cells decreased after each successive gestational week. These findings support the concept of introducing IUT before the 13th week of gestation to induce actively acquired specific tolerance to the foreign antigen (Pahal et al., 2000). Thus, IUT could be performed to the corresponding hematopoietic compartments or systemically depending on the gestational age the transplantation occurs (Tavian and Peault, 2005).
Studies in mice strongly support there being an immune barrier to allogeneic engraftment after IUHCT. Transplantation of allogenic HSCs at day 14 post conception gave initially similar results to IUT with congenic HSCs at 1 week of age (100%) but after 6 months, engraftment dropped rapidly (19% allogenic versus 100% congenic; Muench, 2005; Tse et al., 2005; Peranteau et al., 2007; Shaw and Bollini, 2011). Strategies to improve engraftment of allogenic HSC in utero have include the use of busulfan, cotransplantation of LLME-treated, MHC-sensitized donor lymphocytes, CD26 inhibition and using haploidentical HSC sources (Hayashi et al., 2004; Ashizuka et al., 2006; Vrecenak et al., 2014). For instance, low-levels of allogeneic chimerism could be enhanced to near-complete donor chimerism in murine models by postnatal minimally myeloablative total body irradiation (TBI) followed by same-donor BM transplantation (Peranteau et al., 2002). Due to the concerns with toxicity, minimally toxic postnatal regimens such as busulfan conditioning have been studied and shown to improve therapeutic levels of allogeneic engraftment (Ashizuka et al., 2006). Mice with <1 and >1% chimerism, had 60 and 100% enhanced engraftment, respectively (Ashizuka et al., 2006).
Maternal T cells play a key role in the success in utero therapy by being a barrier to engraftment (Nijagal et al., 2011). There were no differences observed in engraftment of syngeneic and allogeneic fetal recipients when cells were matched to the mother in a murine model. It is believed that the immune barrier may result from maternal pathogenic immune responses as a result of pro-inflammatory signals released during fetal intervention (Nijagal et al., 2011). Recent studies of canine IUT with HSCs in utero were encouraging. A time of 40 days gestation (term 63 days) was chosen for these experiments since it was at the initiation of thymic selection, and prior to BM hematopoiesis, therefore being optimal for engraftment. Intracardiac injection was the most efficient delivery method giving much higher levels of donor cell engraftment than intraperitoneal injection. The authors achieved stable long-term multilineage engraftment in 21 of 24 surviving recipients with an average level of initial chimerism of 11.7% (range 3–39%) without conditioning and with no evidence of graft versus host disease (GVHD). Donor cell chimerism remained stable for up to 2 years and was associated with donor specific tolerance for renal transplantation (Vrecenak et al., 2014). Intracardiac injection early in gestation currently carries an increased risk of miscarriage in clinical practice compared to intraperitoneal injection, but these findings suggest that clinically relevant levels of engraftment might be achievable using this approach and research is underway to evaluate safety and feasibility in relevant pre-clinical animal models prior to the first human studies. Using stem cells that are matched to the fetus, i.e., autologous cells, is an alternative approach which is discussed further on in this review.
Severe combined immunodeficiency (SCID) has been successfully corrected by ultrasound guided intraperitoneal or intravenous fetal injection of HSCs derived from the paternal BM or an allogeneic fetal liver (Flake et al., 1996; Westgren et al., 2002). X-linked SCID is an immunodeficiency caused by the mutation of IL2RG, which encodes the cytokine-receptor γ chain that results in a block in T-cell development and a severe deficiency of mature T cells (Flake et al., 1996). After IUSCT, stable split chimerism with the T-cell lineage of donor origin and all other lineages of host origin was seen postnatally in treated individuals as evidence of immune system reconstitution (Flake, 2004). For most patients, the diagnosis of SCID is only made in the neonatal period meaning that postnatal treatment is the only option. Rapid advances in fetal medicine, such as the availability of non-invasive prenatal diagnosis in the first trimester is likely to make prenatal screening a reality. IUSCT currently is an option for affected families that have a one in four risk of recurrence, where first trimester prenatal diagnosis can be made by CVS leaving time to perform stem cell transplantation using allogeneic stem cells. The most common treatment for SCID patients is a postnatal BM transplant where a matched donor is required. More recently, where a suitable donor is not available, a stem cell gene therapy approach has used gene corrected autologous BM transplantation with great success (Demaison et al., 2002; Gaspar and Thrasher, 2005; Thrasher et al., 2006; Gaspar et al., 2011; Montiel-Equihua et al., 2012). For instance, in 2012, around 30 patients had been treated most of whom had experienced clinical benefit with the absence of any vector-related complications (Gaspar and Thrasher, 2005). There is a chance of insertional mutagenesis occurring during retroviral and lentiviral vector integration into host-cell chromosomes as well as the development of lymphoproliferative disease in individuals with SCID (Gaspar and Thrasher, 2005; Yáñez-Muñoz et al., 2006; Howe et al., 2008). It is important for clinical therapies to achieve stable transgene expression while minimizing insertional mutagenesis (Baum et al., 2003). Integration-deficient lentiviral vectors and self-inactivating (SIN) gammaretroviral vectors have a low risk and in cellular and in vivo models of SCID can mediate stable transduction (Yáñez-Muñoz et al., 2006; Thornhill et al., 2008).
Inherited blood disorders such as the hemoglobinopathies or clotting disorders would be a relatively simple target for IUT as the fetal circulation can be reached through the umbilical vein (UV) at the placental cord insertion or the intrahepatic UV, or even via the peritoneal cavity, a route used successfully to transfuse anemic fetuses.
Prenatal screening and diagnostic services for congenital hemoglobinopathies are available in many countries making them an attractive option for an in utero therapeutic approach (David and Waddington, 2012). Prenatal diagnosis can be achieved currently from 11 weeks of gestation using CVS, or amniocentesis from 15 weeks, but increasingly there are advances in non-invasive prenatal screening and diagnosis using circulating fetal DNA detected in the maternal plasma allows the diagnosis of congenital disorders as early as 7 weeks (Lo et al., 1998). Since AF or chorionic villus samples are accessible relatively easily and early in pregnancy, they would provide the potential for therapeutic use after clinical prenatal diagnosis have been performed.
Inherited abnormalities of hemoglobin (Hb), a tetramer of two α-like and two β-like globin chains, are a common and global problem. Over 330,000 affected infants are born annually worldwide, 83% with sickle cell disorders and 17% with thalassemias (Modell and Darlison, 2008). Current treatment of β-thalassemia is by postnatal allogeneic hematopoietic stem cell transplantation (HSCT) which can cure the condition with recent results of 90% survival and 80% thalassemia-free survival (Angelucci et al., 2000). However, this option is only available in approximately 30% of cases due to the lack of a suitable matched donor (Lucarelli, 2002), and it is associated with complications such as GVHD. For children where HSCT is unavailable, they are dependent on blood transfusions that result in iron overload, and the need for iron chelation therapy. In alpha-thalassemia, some individuals who make very little or no α globin chains, have severe anemia, termed Hb Bart’s hydrops fetalis syndrome which is commonly diagnosed prenatally and if untreated causes death in the neonatal period (Harteveld and Higgs, 2010). Current treatment of sickle cell relies on a number of strategies such as the use of prophylactic antibiotics, pneumococcal vaccination and good hydration, and effective crisis management such as using oxygen and pain-relief (Meremikwu and Okomo, 2011).
Attempts to cure thalassemia and sickle cell disease using gene therapy have been hampered by the large globin gene and globin promoters that are difficult to accommodate within vector systems. Amelioration or even cure of mouse models of human sickle cell disease (Pawliuk et al., 2001) and β-thalassemia major (Pawliuk et al., 2001; Persons et al., 2003; Puthenveetil et al., 2004) has been achieved using lentivirus vectors that contain complex regulatory sequences from the LCR region. Recent advances in vector design have improved gene transfer for the hemoglobinopathies such as the ubiquitous chromatin opening element (UCOE) augmented spleen focus forming virus (SFFV) promoter/enhancer which provides lentivirus vectors with a natural tropism for the hematopoietic system (Antoniou et al., 2003; Williams et al., 2005) resulting in reproducible and stable function in BM and all differentiated peripheral hematopoietic cell lineages (Zhang et al., 2007).
Clotting disorders are caused by deficiencies in coagulation factors, for example, hemophilia B, which is due to mutations in the factor IX (F9) gene resulting in a deficiency in the blood clotting protein human factor IX (hFIX; Waddington et al., 2004b). The current treatment offered to patients with inherited coagulopathies includes lifelong recombinant protein infusions, which is required to avoid major pathology, and it is an expensive and limited resource. In some patients, protein infusions can also lead to the formation of antibodies to the infused product, which prevents its use. Gene therapy cure of inherited coagulopathies has come closer to reality with the use of adeno-associated virus vectors (AAV). Animal experiments have shown AAV to be a promising vector system and this has led to the first human trials for this disease by applying AAV-hFIX intramuscularly to eight adult patients with severe hemophilia B which showed a small increase in hFIX plasma levels and a reduction in exogenous protein requirement (Kay et al., 2000; Manno et al., 2003). More recently, one trial used a self-complimentary AAV-hFIX vector that gives higher levels of transgenic protein expression in vivo than earlier single-stranded vectors. A single peripheral vein infusion of a serotype-8-pseudotyped, self-complementary AAV vector expressing a codon-optimized hFIX transgene in six patients with severe hemophilia B (FIX activity, <1% of normal values) gave FIX expression at 2–11% of normal levels in all participants. A short course of glucocorticoid therapy normalized raised liver enzyme levels that were observed in two patients (Nathwani et al., 2011). AAV vectors with hFVIII and hFVII are becoming available and are being tested in pre-clinical studies (Binny et al., 2012; McIntosh et al., 2013).
Proof of principle studies have shown long-term expression of hFIX proteins at therapeutic levels and induction of immune tolerance (Waddington et al., 2007) after in utero gene therapy using lentiviral vectors in mice (Waddington et al., 2004a,b). More recently, using the same self-complementary AAV8 vector expressing the human factor IX (hFIX) gene used for the clinical trials, long-term hFIX expression was observed after ultrasound guided intraperitoneal injection of fetal sheep in early and late gestation (Nathwani et al., 2006; David et al., 2011). No functional antibodies could be detected against the vector or transgene product and no liver toxicity was observed. Antibodies to the therapeutic gene were detectable when the animals were challenged at 6 months of age postnatally with the hFIX recombinant protein, showing that induction of immune tolerance was not achieved. This was probably due to the fall in hFIX expression that was undetectable by 1 year after birth. UV delivery in fetal non-human primates of a 10-fold higher dose of the same self-complementary AAV system in late gestation produced clinically relevant levels of hFIX sustained for over a year, with liver-specific expression and a non-neutralizing immune response (Mattar et al., 2011).
In comparison to the relative success of postnatal transplantation for blood disorders, results of clinical cases of IUT to cure blood disorders have been disappointing (Nijagal et al., 2012; Pearson and Flake, 2013). For instance, the transplantation of CD34+ cells from either fetal liver or adult BM in cases of hemoglobinopathies, showed no evidence of engraftment in all 22 cases with a clinical outcome of lifelong blood transfusion or disease-related mortality (Tiblad and Westgren, 2008). Attempts to treat other diseases such as sickle cell or metabolic storage disorders have been unsuccessful after fetal liver-derived stem cell transplantation, even wherein a suitably matched donor has been available (D’Azzo, 2003; Westgren, 2006).
Transplantation of autologous derived fetal liver stem cells has been attempted in the fetal sheep. Fetal liver stem cells collected from first trimester preimmune sheep fetuses using ultrasound-guided hepatic sampling were labeled with PKH26 and then transplanted intraperitoneally into allogeneic and autologous fetal recipients. Engraftment of donor cells was equivalent after autologous or allogeneic transplantation (up to 4.7% in fetal liver, spleen, BM, blood, and thymus) but the fetal loss rate was high (29% allogeneic and 73% autologous transplantation) making this technique difficult to justify in clinical practice (Schoeberlein et al., 2004).
For coagulopathies, transplantation of MSCs may be a feasible therapeutic option. In a sheep model of hemophilia A that recapitulates the human condition (spontaneous bleeds and debilitating hemarthroses), encouraging results were found after postnatal intraperitoneal infusion of paternally derived MSCs transduced with a porcine FVIII-encoding lentiviral vector. Infusions of factor VIII were no longer required and damaged joints were fully recovered. However, a sharp increase in pre-existent antibodies occurred with time following transplantation which decreased the effectiveness and limited the duration of therapy (Porada et al., 2011). This emphasizes the need for an IUSCT approach for this condition.
Given the concerns around in utero application of gene therapy directly to the fetus, our group have been studying whether a combination of autologous transplantation with gene corrected AFS might be a potential therapeutic approach. We have studied the functional hematopoietic potential of transduced green fluorescent protein (GFP)+ sheep AF-derived stem cells, before and after autologous IUSCT. First trimester sheep AF was collected by ultrasound-guided amniocentesis or at post mortem examination. We used a novel sheep CD34+ antibody that allows flow cytometric detection of sheep HSC/progenitors present within BM, cord blood, and mobilized peripheral blood. This antibody also enriches for HSC/progenitors with enhanced in vitro colony-forming potential (Porada et al., 2008). Sheep CD34+ AF or adult BM cells were selected and transduced overnight with an HIV lentivirus vector containing eGFP. Transduced fresh or frozen CD34+ AF, or BM cells, were injected intravenously into NOD-SCID-gamma (NSG) mice. GFP+ cells were detected in the hematopoietic organs and peripheral blood of NSG mice primary and secondary recipients 3 months later (Figure 1). Autologous IUSCT was performed in fetal sheep using ultrasound-guided intraperitoneal injection of fresh transduced GFP+ CD34+AF cells. GFP+ cells were detected in the peripheral blood of injected lambs up to 6 months postnatally (Figure 2) and 3 months after secondary transplantation of BM from autologous IUSCT lambs into NSG mice, GFP+ cells were detected in hematopoietic organs. This demonstration of autologous IUSCT of CD34+AF cells in a large animal model supports the concept for clinical translation to treat congenital hematopoietic diseases in utero (Shaw et al., 2014).
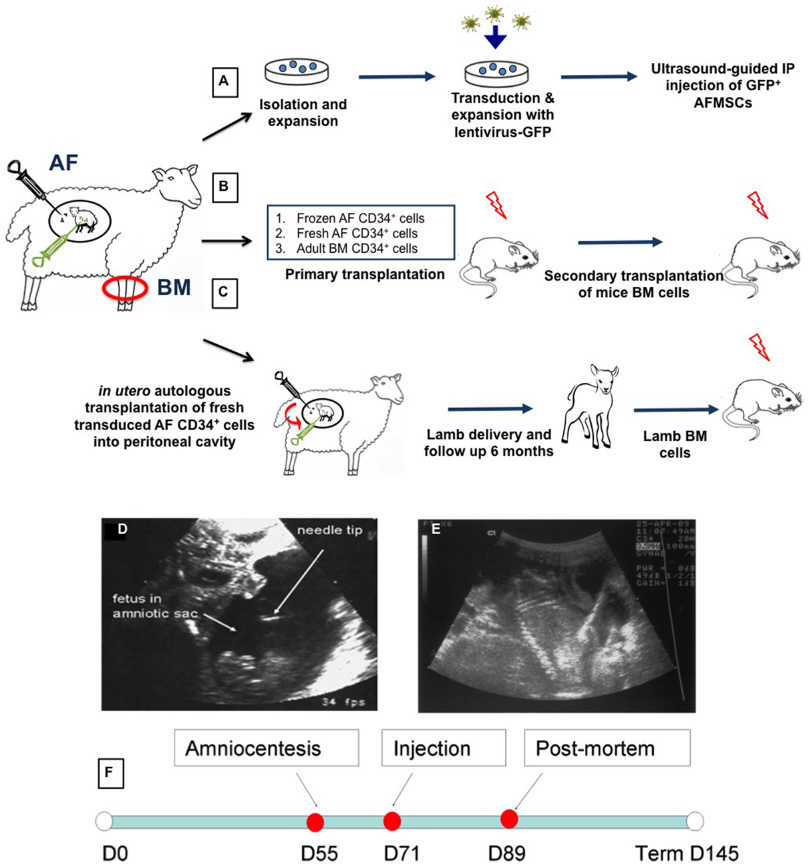
Figure 1. Experimental design and injection procedure. After amniocentesis collection, amniotic fluid mesenchymal stem cells (AFMSCs) were cultured in adherence in defined conditions. Cells were transfected with lentivirus GFP and re-injected into the peritoneal cavity of the fetal donor (A). Transduced sheep eGFP+ CD34+ selected from fresh or frozen AF and adult BM cells were transplanted into immunocompromised NSG mice (primary and secondary xenogeneic transplantation) (B). Transduced sheep eGFP+CD34+ fresh AF were also injected into donor sheep fetuses (in utero autologous transplantation) that were subsequently delivered and followed for up to 3 months of age. Bone marrow from these primary sheep recipients was then used to perform xenogeneic secondary transplantation into NSG mice (C). AF, amniotic fluid; BM, bone marrow. Sonograms showing ultrasound guided amniocentesis (D) and intraperitoneal injection (E). Amniocentesis was performed using a 22 gauge needle to collect 10 ml amniotic fluid from the amniotic cavity around a fetal sheep at 58 days of gestation. For intraperitoneal injection of transduced expanded amniotic fluid cells we used a 20 gauge needle inserted through the anterior abdominal wall of a sheep fetus at 76 days of gestation. Echogenicity can be seen throughout the peritoneal cavity after injection of cells. Scale bars: 5 cm. (F) Timeline of the experiment.
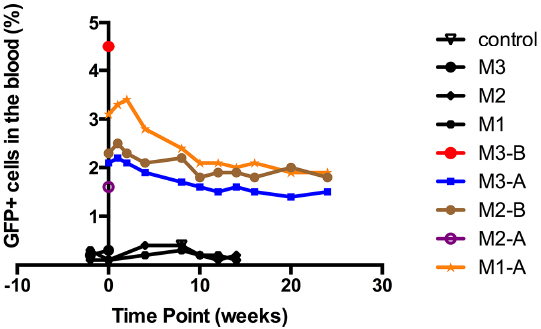
Figure 2. In utero autologous intraperitoneal transplantation of sheep amniotic fluid CD34+ cells in fetal sheep with long-term follow up. Engraftment in the peripheral blood after in utero transplantation of autologous sheep CD34+eGFP+ AF cells. All five born lambs showed eGFP+ cells in the peripheral blood at birth (M1-A, M2-A, M3-A, M2-B, and M3-B), and all three survivors revealed persistent levels of engraftment of around 2% that persisted up to the last sampling point at 6 months of age (M1-A, M2-B, and M3-A). Negative control: peripheral blood from uninjected sheep. M1, M2, and M3: the three ewes that showed negativity for eGFP signal.
Human β-thalassemia iPS have now been generated from AFS using a single excisable lentiviral stem cell cassette vector. AFS from the prenatal diagnosis of a β-thalassemia patient were reprogrammed by expression of the four human reprogramming factors Oct4, KLF4, SOX2, and c-MYC using a doxycycline lentiviral system and demonstrated teratoma formation (Fan et al., 2012). There are concerns that these iPS cells may be more likely to develop teratomas than AFS cells that have a low risk of this complication. This type of cell manipulation however, may provide clinicians with corrected autologous patient-specific iPS cells to use in a combination IUSCGT approach for the treatment of thalassemia (Fan et al., 2012).
IUT is a possible treatment strategy for congenital disorders that affect organ systems other than the blood. These include myelomeningocele (MMC; Danzer et al., 2012) which represents the most severe form of spina bifida, cystic fibrosis (CF), lysosomal storage diseases such as acute neuronopathic (type II) Gaucher disease, neuronal ceroid lipofuscinoses, and Niemann–Pick disease type C, ornithine transcarbamylase deficiency (OTC), as well as muscular dystrophy (David et al., 2003). Many of these diseases and organ systems would be amenable to IUT using MSCs, which compared to HSCs, are less immunologically competent and may result in less transplantation related rejection (O’Donoghue and Fisk, 2004).
Human BM-MSCs have shown to have long-term engraftment and have the ability to differentiate into various tissues when transplanted into fetal sheep (Mackenzie and Flake, 2001). The therapeutic potential for combining surgical repair and transplantation of MSCs in utero has been demonstrated recently for the treatment of spina bifida in a rat model (Li et al., 2012). IUT of first trimester human fetal blood MSC ameliorates the skeletal disorder in a mouse model of osteogenesis imperfecta (Guillot et al., 2008). IUT of fetal MSCs reduced fracture rates and skeletal abnormalities (Guillot et al., 2008). Two cases of IUT using allogenic fetal liver MSC in the third trimester had encouraging results with a successful engraftment which demonstrated 7.4% chimerism at 9 months of age in one case and good long-term outcomes (Le Blanc et al., 2005; Götherström et al., 2014).
High fetal survival was found after intraperitoneal injection of autologous AFMSCs in the sheep (Mehta et al., 2011; Shaw and Bollini, 2011). AF was collected under ultrasound-guided amniocentesis in early gestation pregnant sheep (n = 9, 58 days of gestation, term = 145 days) and AFMSCs were isolated, expanded, and transduced using an HIV vector encoding enhanced GFP with 63.2% (range 38.3–96.2%) transduction efficiency rate (Figure 1). Transduced AFMSCs were injected into the peritoneal cavity of each donor fetal sheep at 76 days under ultrasound guidance with a 78% overall survival rate for the full procedure. After 2 weeks, GFP+ cells and protein was detected in fetal tissues including liver, heart, placenta, membrane, umbilical cord, adrenal gland, and muscle and this was further confirmed by cytofluorimetric and immunofluorescence analysis (Figure 3).
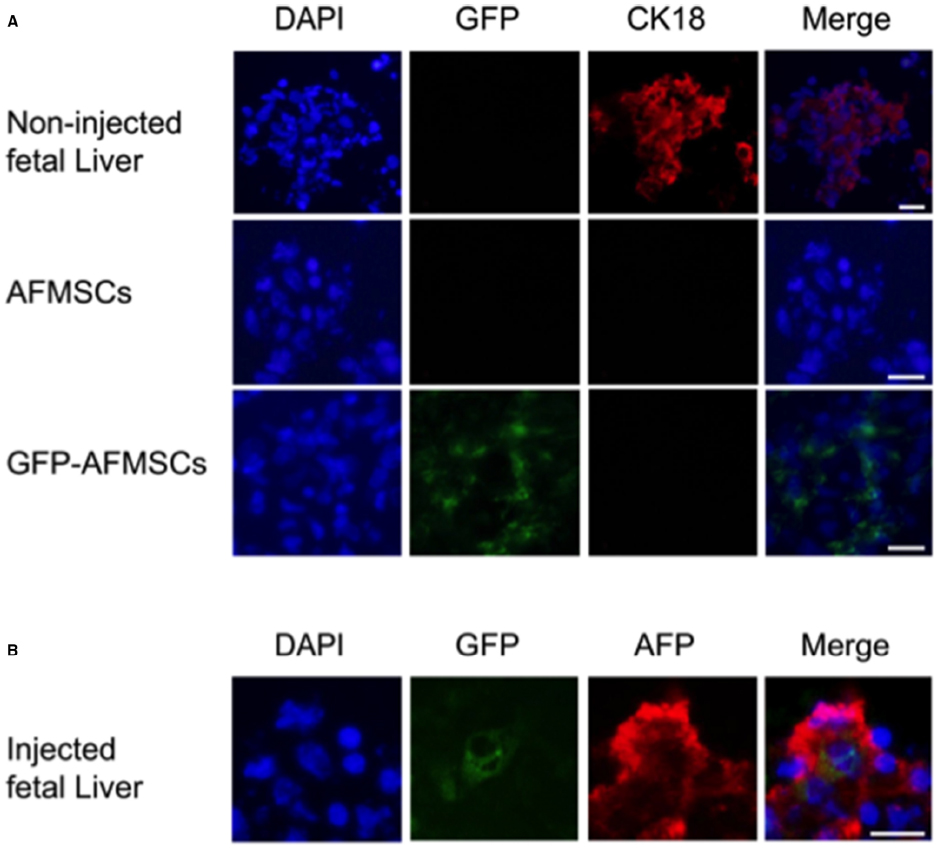
Figure 3. Immunofluorescence for CK18 and AFP expression in fetal liver and amniotic fluid stem cells. Panel (A) shows positive CK18 expression in cells cultured from a control fetal sheep liver but no expression in amniotic fluid mesenchymal stem cells (AFMSCs) or transduced cells (GFP-AFMSCs) before injection. An uninjected sheep fetus of comparable gestational age was used as the control. Panel (B) shows co-expression of GFP with expression of Alpha fetoprotein (AFP) another liver specific marker in the fetal liver after transplantation of transduced AFMSCs. Scale bars: 20 µm.
AFS cells intraperitoneally injected in a necrotizing enterocolitis rat model had shown improved survival, clinical status, gut structure, and function (Eaton et al., 2013; Zani et al., 2014). These findings suggest that the use of AFS cells as a source of cells for in utero therapy could be an alternative way of ameliorating prenatal congenital disease.
Preclinical testing in animal models of disease will be an important step before clinical translation is realized. There is no ideal animal model and a balance is needed, taking into consideration the gestational development of the organ to be targeted and how that relates to its development in the human, the type of placentation, fetal size, number and lifespan, parturition, and the fetal and maternal immune response (Mehta et al., 2012; Mehta and Abi-Nader, 2012). An assessment of the safety, accessibility, transduction efficiency, and behavior of various stem cells (i.e., cord blood, placenta, AF, fetal tissue) in vitro as well as in the fetal environment are required to evaluate proof-of-principle strategies based on gene transfer or cell transplantation into the fetus to ensure accurate organ-directed manipulation and delivery (Moreno et al., 2012). Thus, the efficacy of treatment can be evaluated from murine models to large animal models such as sheep and primates (Mehta et al., 2012; Mehta and Abi-Nader, 2012).
Toxicology studies will be needed using animals such as the pregnant rabbit, in which reproductive toxicology is commonly performed, with good historical datasets and a model that is understood by the regulators. A variety of guidelines and regulations such as those described by the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency will need to be taken into consideration when planning preclinical study protocols. These could include for example, the guidelines on the non-clinical testing for inadvertent germline transmission of gene transfer vectors (EU, 2006) or on the non-clinical studies required before first clinical use of gene therapy medicinal products (European Medicines Agency, 2007).
In addition to animal studies, the safety of gene therapy vectors has to be evaluated. Integration site analysis has become a critical tool to measure the “vector-on-host” and “host-on-vector” effects in gene therapy (Biasco et al., 2011). Also, models such as the human placenta can be utilized in vitro as it would provide a wealth of data on the physiology of normal and pathological human placentae and may be useful in measuring the spread of vector from the fetus to the mother or vice versa.
Advances in prenatal screening and molecular diagnosis have provided the ability of detecting the majority of genetic diseases early in gestation. Early diagnosis provides the option of possible treatment options that can be explored either at the prenatal or postnatal period depending on the condition. The option of preemptive treatment of congenital diseases in utero by stem cell or gene therapy are encouraging as it changes the perspective of congenital diseases. However, further work focusing on the safety and ethical issues need to be addressed before clinical applications can be considered.
Durrgah L. Ramachandra, Steven S. W. Shaw, Panicos Shangaris, Stavros Loukogeorgakis, Pascale V. Guillot, Paolo De Coppi, and Anna L. David made substantial contributions to the conception and design of the work. Durrgah L. Ramachandra and Panicos Shangaris drafted the work and Paolo De Coppi and Anna L. David revised it critically for important intellectual content. Durrgah L. Ramachandra, Steven S. W. Shaw, Panicos Shangaris, Stavros Loukogeorgakis, Pascale V. Guillot, Paolo De Coppi, and Anna L. David gave final approval of the version to be published and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Anna L. David is an unpaid consultant and director of Magnus Growth, part of Magnus Life Science, which is aiming to take to market a novel treatment for fetal growth restriction.
Anna L. David is supported by funding from the National Institute for Health Research University College London Hospitals Biomedical Research Centre. Paolo De Coppi is supported by the Great Ormond Street Hospital Children’s Charity. Durrgah L. Ramachandra is supported by M’Asia9. Panicos Shangaris is supported by Wellcome Trust Sparks Research Training Fellowship number 099083/Z/11/Z.
Angelucci, E., Brittenham, G. M., McLaren, C. E., Ripalti, M., Baronciani, D., Giardini, C., et al. (2000). Hepatic iron concentration and total body iron stores in thalassemia major. N. Engl. J. Med. 343, 327–331. doi: 10.1056/NEJM200008033430503
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Antoniou, M., Harland, L., Mustoe, T., Williams, S., Holdstock, J., Yague, E., et al. (2003). Transgenes encompassing dual-promoter CpG islands from the human TBP and HNRPA2B1 loci are resistant to heterochromatin-mediated silencing. Genomics 82, 269–279. doi: 10.1016/S0888-7543(03)00107-1
Ashizuka, S., Peranteau, W. H., Hayashi, S., and Flake, A. W. (2006). Busulfan-conditioned bone marrow transplantation results in high-level allogeneic chimerism in mice made tolerant by in utero hematopoietic cell transplantation. Exp. Hematol. 34, 359–368. doi: 10.1016/j.exphem.2005.11.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Baum, C., Düllmann, J., Li, Z., Fehse, B., Meyer, J., Williams, D. A., et al. (2003). Side effects of retroviral gene transfer into hematopoietic stem cells. Blood 101, 2099–2114. doi: 10.1182/blood-2002-07-2314
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Biasco, L. A., Ambrosi, A., Pellin, D., Bartholomae, C., Brigida, I., Roncarlo, M. G., et al. (2011). Integration profile of retroviral vector in gene therapy treated patients is cell-specific according to gene expression and chromatin conformation of target cell. EMBO Mol. Med. 3, 89–101. doi: 10.1002/emmm.201000108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Binny, C., McIntosh, J., Peruta, M., Kymalainen, H., Tuddenham, E., Waddington, S., et al. (2012). AAV-mediated gene transfer in the perinatal period results in expression of FVII at levels that protect against fatal spontaneous hemorrhage. Blood 119, 957–966. doi: 10.1182/blood-2011-09-377630
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chan, J., Kumar, S., and Fisk, N. M. (2008). First trimester embryo-fetoscopic and ultrasound-guided fetal blood sampling for ex vivo viral transduction of cultured human fetal mesenchymal stem cells. Hum. Reprod. (Oxford, England) 23, 2427–2437. doi: 10.1093/humrep/den302
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Danzer, E., and Johnson, M. P. (2014). Fetal surgery for neural tube defects. Semin. Fetal Neonatal Med. 19, 2–8. doi: 10.1016/j.siny.2013.09.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Danzer, E., Johnson, M. P., and Adzick, N. S. (2012). Fetal surgery for myelomeningocele: progress and perspectives. Dev. Med. Child Neurol. 54, 8–14. doi: 10.1111/j.1469-8749.2011.04049.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Darrasse-Jèze, G., Marodon, G., Salomon, B. L., Catala, M., and Klatzmann, D. (2005). Ontogeny of CD4+CD25+ regulatory/suppressor T cells in human fetuses. Blood 105, 4715–4721. doi: 10.1182/blood-2004-10-4051
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
David, A. L., McIntosh, J., Peebles, D. M., Cook, T., Waddington, S., Weisz, B., et al. (2011). Recombinant adeno-associated virus-mediated in utero gene transfer gives therapeutic transgene expression in the sheep. Hum. Gene Ther. 22, 419–426. doi: 10.1089/hum.2010.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
David, A. L., and Peebles, D. (2008). Gene therapy for the fetus: is there a future? Best Pract. Res. Clin. Obstet. Gynaecol. 22, 203–218. doi: 10.1016/j.bpobgyn.2007.08.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
David, A. L., Themis, M., Waddington, S. N., Gregory, L., Buckley, S. M. K., Nivsarkar, M., et al. (2003). The current status and future direction of fetal gene therapy. Gene Ther. Mol. Biol. 7, 181–209.
David, A. L., and Waddington, S. N. (2012). Candidate diseases for prenatal gene therapy. Methods Mol. Biol. (Clifton, NJ) 891, 9–39. doi: 10.1007/978-1-61779-873-3_2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
D’Azzo, A. (2003). Gene transfer strategies for correction of lysosomal storage disorders. Acta Haematol. 110, 71–85. doi: 10.1159/000072456
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
De Coppi, P. (2013). Regenerative medicine for congenital malformations. J. Pediatr. Surg. 48, 273–280. doi: 10.1016/j.jpedsurg.2012.11.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
De Coppi, P., Bartsch, G. Jr, Siddiqui, M. M., Xu, T., Santos, C. C., Perin, L., et al. (2007). Isolation of amniotic stem cell lines with potential for therapy. Nat. Biotechnol. 25, 100–106. doi: 10.1038/nbt1274
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Delo, D. M., De Coppi, P., and Bartsch, G. (2006). Amniotic fluid and placental stem cells. Methods Enzymol. 419, 426–438. doi: 10.1016/S0076-6879(06)19017-5
Demaison, C., Parsley, K., Brouns, G., Scherr, M., Battmer, K., Kinnon, C., et al. (2002). High-level transduction and gene expression in hematopoietic repopulating cells using a human immunodeficiency virus type 1-based lentiviral vector containing an internal spleen focus forming virus promoter. Hum. Gene Ther. 13, 803–813. doi: 10.1089/10430340252898984
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ditadi, A., de Coppi, P., Picone, O., Gautreau, L., Smati, R., Six, E., et al. (2009). Human and murine amniotic fluid c-kit+Lin-cells display hematopoietic activity. Blood 113, 3953–3960. doi: 10.1182/blood-2008-10-182105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Di Trapani, M., Bassi, G., Fontana, E., Giacomello, L., Pozzobon, M., Guillot, P. V., et al. (2014). Immune regulatory properties of cd117(pos) amniotic fluid stem cells vary according to gestational age. Stem Cells Dev. doi: 10.1089/scd.2014.0234 [Epub ahead of print].
Eaton, S., Zani, A., Pierro, A., and De Coppi, P. (2013). Stem cells as a potential therapy for necrotizing enterocolitis. Expert Opin. Biol. Ther. 13, 1683–1689. doi: 10.1517/14712598.2013.849690
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Eckstein, S. (2003). “Report on the potential use of gene therapy in utero,” in Manual for Research Ethics Committees: Centre of Medical Law and Ethics, King’s College London, ed. S. Eckstein (London: Cambridge University Press), 329–333. doi: 10.1017/CBO9780511550089.051
EU. (2006). Committee for the Medicinal Products for Human Use (CHMP). Guideline on Non-Clinical Testing for Inadvertent Germline Transmission of Gene Transfer Vectors. EMEA/273974/2005. London: European Medicines Agency.
European Medicines Agency. (2007). Guideline on Human Cell-Based Medicinal Products. Draft Committee for Human Medicinal Product (CHMP). London: European Medicines Agency.
Fan, Y., Luo, Y., Chen, X., Li, Q., and Sun, X. (2012). Generation of human β-thalassemia induced pluripotent stem cells from amniotic fluid cells using a single excisable lentiviral stem cell cassette. J. Reprod. Dev. 58, 404–409. doi: 10.1262/jrd.2011-046
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fauza, D. (2004). Amniotic fluid and placental stem cells. Best Pract. Res. Clin. Obstet. Gynaecol. 18, 877–891. doi: 10.1016/j.bpobgyn.2004.07.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Flake, A. W. (2004). In utero stem cell transplantation. Best Pract. Res. Clin. Obstet. Gynaecol. 18, 941–58. doi: 10.1016/j.bpobgyn.2004.06.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Flake, A. W., Roncarolo, M. G., Puck, J. M., Almeida-Porada, G., Evans, M. I., Johnson, M. P., et al. (1996). Treatment of X-linked severe combined immunodeficiency by in utero transplantation of paternal bone marrow. N. Engl. J. Med. 335, 1806–1810. doi: 10.1056/NEJM199612123352404
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fleischman, R. A. (1993). From white spots to stem cells: the role of the kit receptor in mammalian development. Trends Genet. 9, 285–290. doi: 10.1016/0168-9525(93)90015-A
Gaspar, H. B., Cooray, S., Gilmour, K. C., Parsley, K. L., Zhang, F., Adams, S., et al. (2011). Hematopoietic stem cell gene therapy for adenosine deaminase-deficient severe combined immunodeficiency leads to long-term immunological recovery and metabolic correction. Sci. Transl. Med. 3, 97ra80. doi: 10.1126/scitranslmed.3002716
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gaspar, H. B., and Thrasher, A. J. (2005). Gene therapy for severe combined immunodeficiencies. Expert Opin. Biol. Ther. 5, 1175–1182. doi: 10.1517/14712598.5.9.1175
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ghionzoli, M., Cananzi, M., Zani, A., Rossi, C. A., Leon, F. F., Pierro, A., et al. (2010). Amniotic fluid stem cell migration after intraperitoneal injection in pup rats: implication for therapy. Pediatr. Surg. Int. 26, 79–84. doi: 10.1007/s00383-009-2504-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Götherström, C., Westgren, M., Shaw, S. W., Aström, E., Biswas, A., Byers, P. H., et al. (2014). Pre- and postnatal transplantation of fetal mesenchymal stem cells in osteogenesis imperfecta: a two-center experience. Stem Cells Transl. Med. 3, 255–264. doi: 10.5966/sctm.2013-0090
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Grisafi, D., Piccoli, M., Pozzobon, M., Ditadi, A., Zaramella, P., Chiandetti, L., et al. (2008). High transduction efficiency of human amniotic fluid stem cells mediated by adenovirus vectors. Stem Cells Dev. 17, 953–962. doi: 10.1089/scd.2007.0188
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Guillot, P. V., Abass, O., Bassett, J. H., Shefelbine, S. J., Bou-Gharios, G., Chan, J., et al. (2008). Intrauterine transplantation of human fetal mesenchymal stem cells from first-trimester blood repairs bone and reduces fractures in osteogenesis imperfecta mice. Blood 111, 1717–1725. doi: 10.1182/blood-2007-08-105809
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Harteveld, C. L., and Higgs, D. R. (2010). Alpha-thalassaemia. Orphanet. J. Rare Dis. 5, 1–21. doi: 10.1186/1750-1172-5-13
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hayashi, S., Hsieh, M., Peranteau, W. H., Ashizuka, S., and Flake, A. W. (2004). Complete allogeneic hematopoietic chimerism achieved by in utero hematopoietic cell transplantation and cotransplantation of LLME-treated, MHC-sensitized donor lympho,cytes. Exp. Hematol. 32, 290–299. doi: 10.1016/j.exphem.2003.12.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Horwitz, E. M., Gordon, P. L., Koo, W. K., Marx, J. C., Neel, M. D., McNall, R. Y., et al. (2002). Isolated allogeneic bone marrow-derived mesenchymal cells engraft and stimulate growth in children with osteogenesis imperfecta: implications for cell therapy of bone. Proc. Natl Acad. Sci. U.S.A. 99, 8932–8937. doi: 10.1073/pnas.132252399
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Howe, S. J., Mansour, M. R., Schwarzwaelder, K., Bartholomae, C., Hubank, M., Kempski, H., et al. (2008). Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J. Clin. Invest. 118, 3143–3150. doi: 10.1172/JCI35798
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ivarsson, M. A., Loh, L., Marquardt, N., Kekäläinen, E., Berglin, L., Björkström, N. K., et al. (2013). Differentiation and functional regulation of human fetal NK cells. J. Clin. Invest. 123, 3889–3901. doi: 10.1172/JCI68989
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jones, G. N., Moschidou, D., Abdulrazzak, H., Kalirai, B. S., Vanleene, M., Osatis, S., et al. (2014). Potential of human fetal chorionic stem cells for the treatment of osteogenesis imperfecta. Stem Cells Dev. 23, 262–276. doi: 10.1089/scd.2013.0132
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kaviani, A., Guleserian, K., Perry, T. E., Jennings, R. W., Ziegler, M. M., Fauza, D. O., et al. (2003). Fetal tissue engineering from amniotic fluid. J. Am. Coll. Surg. 196, 592–597. doi: 10.1016/S1072-7515(02)01834-3
Kaviani, A., Perry, T. E., Dzakovic, A., Jennings, R. W., Ziegler, M. M., and Fauza, D. O. (2001). The amniotic fluid as a source of cells for fetal tissue engineering. J. Pediatr. Surg. 36, 1662–1665. doi: 10.1053/jpsu.2001.27945
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kay, M. A., Manno, C. S., Ragni, M. V., Larson, P. J., Couto, L. B., McClelland, A., et al. (2000). Evidence for gene transfer and expression of factor IX in haemophilia B patients treated with an AAV vector. Nat. Genet. 24, 257–261. doi: 10.1038/73464
Le Blanc, K., Götherström, C., Ringdén, O., Hassan, M., McMahon, R., McMahon, R., et al. (2005). Fetal mesenchymal stem-cell engraftment in bone after in utero transplantation in a patient with severe osteogenesis imperfecta. Transplantation 79, 1607–1614. doi: 10.1097/01.TP.0000159029.48678.93
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, H., Gao, F., Ma, L., Jiang, J., Miao, J., Jiang, M., et al. (2012). Therapeutic potential of in utero mesenchymal stem cell (MSCs) transplantation in rat foetuses with spina bifida aperta. J. Cell. Mol. Med. 16, 1606–1617. doi: 10.1111/j.1582-4934.2011.01470.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lo, Y. M., Tein, M. S., Lau, T. K., Haines, C. J., Leung, T. N., Poon, P. M., et al. (1998). Quantitative analysis of fetal DNA in maternal plasma and serum: implications for noninvasive prenatal diagnosis. Am. J. Hum. Genet. 62, 768–775. doi: 10.1086/301800
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lozano, R., Naghavi, M., Foreman, K., Lim, S., Shibuya, K., Aboyans, V., et al. (2012). Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 380, 2095–2128. doi: 10.1016/S0140-6736(12)61728-0
Lu, H. E., Yang, Y.-C., Chen, S.-M., Su, H.-L., Huang, P.-C., Tsai, M.-S., et al. (2012). Modeling neurogenesis impairment in Down syndrome with induced pluripotent stem cells from trisomy 21 amniotic fluid cells. Exp. Cell Res. 319, 498–505. doi: 10.1016/j.yexcr.2012.09.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lucarelli, G. (2002). The cure of thalassemia by bone marrow transplantation. Blood Rev. 16, 81–85. doi: 10.1054/blre.2002.0192
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mackenzie, T. C., and Flake, A. W. (2001). Human mesenchymal stem cells persist, demonstrate site-specific multipotential differentiation, and are present in sites of wound healing and tissue regeneration after transplantation into fetal sheep. Blood Cells Mol. Dis. 27, 601–604. doi: 10.1006/bcmd.2001.0424
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Manno, C. S., Chew, A. J., Hutchison, S., Larson, P. J., Herzog, R. W., Arruda, V. R., et al. (2003). AAV-mediated factor IX gene transfer to skeletal muscle in patients with severe hemophilia B. Blood 101, 2963–2972. doi: 10.1182/blood-2002-10-3296
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mattar, C. N., Nathwani, A. C., Waddington, S. N., Dighe, N., Kaeppel, C., Nowrouzi, A., et al. (2011). Stable human FIX expression after 0.9G intrauterine gene transfer of self-complementary adeno-associated viral vector 5 and 8 in macaques. Mol. Ther. 19, 1950–1960. doi: 10.1038/mt.2011.107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McCandless, S. E., Brunger, J. W., and Cassidy, S. B. (2004). The burden of genetic disease on inpatient care in a children’s hospital. Am. J. Hum. Genet. 74, 121–127. doi: 10.1086/381053
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McIntosh, J., Lenting, P. J., Rosales, C., Lee, D., Rabbanian, S., Raj, D., et al. (2013). Therapeutic levels of FVIII following a single peripheral vein administration of rAAV vector encoding a novel human factor VIII variant. Blood 121, 3335–3344. doi: 10.1182/blood-2012-10-462200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mehta, V., and Abi-Nader, K. N. (2012). Monitoring for potential adverse effects of prenatal gene therapy: use of large animal models with relevance to human application. Methods Mol. Biol. (Clifton, NJ) 891, 291–328. doi: 10.1007/978-1-61779-873-3_14
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mehta, V., Nader, K. A., Waddington, S., and David, A. L. (2011). Organ targeted prenatal gene therapy—how far are we? Prenat. Diagn. 31, 720–734. doi: 10.1002/pd.2787
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mehta, V., Peebles, D., and David, A. L. (2012). Animal models for prenatal gene therapy: choosing the right model. Methods Mol. Biol. 891, 183–200. doi: 10.1007/978-1-61779-873-3_9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Modell, B., and Darlison, M. (2008). Global epidemiology of haemoglobin disorders and derived service indicators. Bull. World Health Organ. 86, 480–487. doi: 10.2471/BLT.06.036673
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mold, J. E., and McCune, J. M. (2012). Immunological tolerance during fetal development: from mouse to man. Adv. Immunol. 115, 73–111. doi: 10.1016/B978-0-12-394299-9.00003-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Montiel-Equihua, C. A., Thrasher, A. J., and Gaspar, H. B. (2012). Gene therapy for severe combined immunodeficiency due to adenosine deaminase deficiency. Curr. Gene Ther. 12, 57–65. doi: 10.2174/156652312799789253
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Moorefield, E. C., McKee, E. E., Solchaga, L., Orlando, G., Yoo, J. J., Walker, S., et al. (2011). Cloned, CD117 selected human amniotic fluid stem cells are capable of modulating the immune response. PLoS ONE 6:e26535. doi: 10.1371/journal.pone.0026535
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Moreno, R., Martínez-González, I., Rosal, M., Nadal, M., Petriz, J., and Gratacós, E. (2012). Fetal liver-derived mesenchymal stem cell engraftment after allogeneic in utero transplantation into rabbits. Stem Cells Dev. 21, 284–295. doi: 10.1089/scd.2010.0483
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Moschidou, D., Mukherjee, S., Blundell, M. P., Drews, K., Jones, G. N., Abdulrazzak, H., et al. (2012). Valproic acid confers functional pluripotency to human amniotic fluid stem cells in a transgene-free approach. Mol. Ther. 20, 1953–1967. doi: 10.1038/mt.2012.117
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Moschidou, D., Mukherjee, S., Blundell, M. P., Jones, G. N., Atala, A. J., Thrasher, A. J., et al. (2013). Human mid-trimester amniotic fluid stem cells cultured under embryonic stem cell conditions with valproic acid acquire pluripotent characteristics. Stem Cells Dev. 22, 444–458. doi: 10.1089/scd.2012.0267
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Muench, M. O. (2005). In utero transplantation: baby steps towards an effective therapy. Bone Marrow Transpl. 35, 537–547. doi: 10.1038/sj.bmt.1704811
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nadri, S., and Soleimani, M. (2007). Comparative analysis of mesenchymal stromal cells from murine bone marrow and amniotic fluid. Cytotherapy 9, 729–737. doi: 10.1080/14653240701656061
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nathwani, A. C., Gray, J. T., Ng, C. Y., Zhou, J., Spence, Y., Waddington, S. N., et al. (2006). Self-complementary adeno-associated virus vectors containing a novel liver-specific human factor IX expression cassette enable highly efficient transduction of murine and nonhuman primate liver. Blood 107, 2653–2661. doi: 10.1182/blood-2005-10-4035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nathwani, A. C., Tuddenham, E. G. D., Rangarajan, S., Rosales, C., McIntosh, J., Linch, D. C., et al. (2011). Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N. Engl. J. Med. 365, 2357–2365. doi: 10.1056/NEJMoa1108046
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nijagal, A., Derderian, C., Le, T., Jarvis, E., Nguyen, L., Tang, T., et al. (2013). Direct and indirect antigen presentation lead to deletion of donor-specific T cells after in utero hematopoietic cell transplantation in mice. Blood 121, 4595–4602. doi: 10.1182/blood-2012-10-463174
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nijagal, A., Flake, A. W., and MacKenzie, T. C. (2012). In utero hematopoietic cell transplantation for the treatment of congenital anomalies. Clin. Perinatol. 39, 301–310. doi: 10.1016/j.clp.2012.04.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nijagal, A., Wegorzewska, M., Jarvis, E., Le, T., Tang, Q., MacKenzie, T. C., et al. (2011). Maternal T cells limit engraftment after in utero hematopoietic cell transplantation in mice. J. Clin. Invest. 121, 582–592. doi: 10.1172/JCI44907
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
O’Donoghue, K., and Fisk, N. M. (2004). Fetal stem cells. Best Pract. Res. Clin. Obstet. Gynaecol. 18, 853–875. doi: 10.1016/j.bpobgyn.2004.06.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pahal, G. S., Auniaux, E., Kinnon, C., Thrasher, A. J., and Rodeck, C. H. (2000). Normal development of human fetal hematopoiesis between eight and seventeen weeks’ gestation. Am. J. Obstet. Gynecol. 183, 1029–1034. doi: 10.1067/mob.2000.106976
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pawliuk, R., Westerman, K. A., Fabry, M. E., Payen, E., Tighe, R., Bouhassira, E. E., et al. (2001). Correction of sickle cell disease in transgenic mouse models by gene therapy. Science 294, 2368–2371. doi: 10.1126/science.1065806
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pearson, E. G., and Flake, A. W. (2013). Stem cell and genetic therapies for the fetus. Semin. Pediatr. Surg. 22, 56–61. doi: 10.1053/j.sempedsurg.2012.10.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Peranteau, W. H., Endo, M., Adibe, O. O., and Flake, A. W. (2007). Evidence for an immune barrier after in utero hematopoietic-cell transplantation. Blood 109, 1331–1333. doi: 10.1182/blood-2006-04-018606
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Peranteau, W. H., Endo, M., Adibe, O. O., Merchant, A., Zoltick, P. W., and Flake, A. W. (2006). CD26 inhibition enhances allogeneic donor-cell homing and engraftment after in utero hematopoietic-cell transplantation. Blood 108, 4268–4274. doi: 10.1182/blood-2006-04-018986
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Peranteau, W. H., Hayashi, S., Hsieh, M., Shaaban, A. F., and Flake, A. W. (2002). High-level allogeneic chimerism achieved by prenatal tolerance induction and postnatal nonmyeloablative bone marrow transplantation. Blood 100, 2225–2234. doi: 10.1182/blood-2002-01-0166
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Perin, L., Sedrakyan, S., Giuliani, S., Da Sacco, S., Carraro, G., Shiri, L., et al. (2010). Protective effect of human amniotic fluid stem cells in an immunodeficient mouse model of acute tubular necrosis. PLoS ONE 5:e9357. doi: 10.1371/journal.pone.0009357
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Persons, D. A., Allay, E. R., Sawai, N., Hargrove, P. W., Brent, T. P., Hanawa, H., et al. (2003). Successful treatment of murine beta-thalassemia using in vivo selection of genetically modified, drug-resistant hematopoietic stem cells. Blood 102, 506–513. doi: 10.1182/blood-2003-03-0677
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Phillips, J. H., Hori, T., Nagler, A., Bhat, N., Spits, H., and Lanier, L. L. (1992). Ontogeny of human natural killer (NK) cells: fetal NK cells mediate cytolytic function and express cytoplasmic CD3 epsilon, delta proteins. J. Exp. Med. 175, 1055–1066. doi: 10.1084/jem.175.4.1055
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pipino, C., Mukherjee, S., David, A. L., Blundell, M. P., Shaw, S. W., Sung, P., et al. (2014). Trisomy 21 mid-trimester amniotic fluid induced pluripotent stem cells maintain genetic signatures during reprogramming: implications for disease modeling and cryobanking. Cell. Reprogram. 16, 331–344. doi: 10.1089/cell.2013.0091
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pipino, C., Shangaris, P., Resca, E., Zia, S., Deprest, J., Sebire, N. J., et al. (2013). Placenta as a reservoir of stem cells: an underutilized resource? Br. Med. Bull. 105, 43–68. doi: 10.1093/bmb/lds033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Porada, C. D., Harrison-Findik, D. D., Sanada, C., Valiente, V., Thain, D., Simmons, P. J., et al. (2008). Development and characterization of a novel CD34 monoclonal antibody that identifies sheep hematopoietic stem/progenitor cells. Exp. Hematol. 36, 1739–1749. doi: 10.1016/j.exphem.2008.09.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Porada, C. D., Sanada, C., Kuo, C.-J., Colletti, E., Mandeville, W., Hasenau, J., et al. (2011). Phenotypic correction of hemophilia A in sheep by postnatal intraperitoneal transplantation of FVIII-expressing MSC. Exp. Hematol. 39, 1124–1135. doi: 10.1016/j.exphem.2011.09.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Prusa, A. R., and Hengstschläger, M. (2002). Amniotic fluid cells and human stem cell research: a new connection. Signature 8, 253–257. doi: 10.1093/humrep/deg279
Puthenveetil, G., Scholes, J., Carbonell, D., Qureshi, N., Xia, P., Zeng, L., et al. (2004). Successful correction of the human beta-thalassemia major phenotype using a lentiviral vector. Blood 104, 3445–3453. doi: 10.1182/blood-2004-04-1427
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
RAC. (2000). Regulatory issues: prenatal gene transfer: scientific, medical, and ethical issues. A report of the Recombinant DNA Advisory Committee. Hum. Gene Ther. 11, 1211–1229.
Roubelakis, M. G., Pappa, K. I., Bitsika, V., Zagoura, D., Vlahou, A., Papadaki, H. A., et al. (2007). Molecular and proteomic characterization of human mesenchymal stem cells derived from amniotic fluid: comparison to bone marrow mesenchymal stem cells. Stem Cells Dev. 16, 931–952. doi: 10.1089/scd.2007.0036
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Roybal, J. L., Santore, M. T., and Flake, A. W. (2010). Stem cell and genetic therapies for the fetus. Semin. Fetal Neonatal Med. 15, 46–51. doi: 10.1016/j.siny.2009.05.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schoeberlein, A., Holzgreve, W., Dudler, L., Hahn, S., and Surbek, D. V. (2004). In utero transplantation of autologous and allogeneic fetal liver stem cells in ovine fetuses. Am. J. Obstet. Gynecol. 191, 1030–1036. doi: 10.1016/j.ajog.2004.06.042
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shaw, S. W. S., Blundell, M. P., Pipino, C., Shangaris, P., Maghsoudlou, P., Ramachandra, D. L., et al. (2014). Sheep CD34+ amniotic fluid cells have haematopoietic potential and engraft after autologous in utero transplantation. Stem Cells. doi: 10.1002/stem.1839 [Epub ahead of print].
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shaw, S. W. S., and Bollini, S. (2011). Autologous transplantation of amniotic fluid-derived mesenchymal stem cells into sheep fetuses. Cell Transplant. 20, 1015–1031. doi: 10.3727/096368910X543402
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shaw, S. W. S., David, A. L., and De Coppi, P. (2011). Clinical applications of prenatal and postnatal therapy using stem cells retrieved from amniotic fluid. Curr. Opin. Obstet. Gynecol. 23, 109–116. doi: 10.1097/GCO.0b013e32834457b1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Takahama, Y. (2006). Journey through the thymus: stromal guides for T-cell development and selection. Nat. Rev. Immunol. 6, 127–135. doi: 10.1038/nri1781
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tavian, M., and Peault, B. (2005). Embryonic development of the human hematopoietic system. Int. J. Dev. Biol. 49, 243–250. doi: 10.1387/ijdb.041957mt
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Thornhill, S. I., Schambach, A., Howe, S. J., Ulaganathan, M., Grassman, E., Williams, D., et al. (2008). Self-inactivating gammaretroviral vectors for gene therapy of X-linked severe combined immunodeficiency. Mol. Ther. 16, 590–598. doi: 10.1038/sj.mt.6300393
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Thrasher, A. J., Gaspar, H. B., Baum, C., Modlich, U., Schambach, A., Candotti, F., et al. (2006). Gene therapy: X-SCID transgene leukaemogenicity. Nature 443, E5–E7. doi: 10.1038/nature05219
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tiblad, E., and Westgren, M. (2008). Fetal stem-cell transplantation. Best Pract. Res. Clin. Obstet. Gynaecol. 22, 189–201. doi: 10.1016/j.bpobgyn.2007.07.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tsai, M. S., Hwang, S.-M., Tsai, Y.-L., Cheng, F.-C., Lee, J.-L., and Chang, Y.-J. (2004). Clonal amniotic fluid-derived stem cells express characteristics of both. Am. J. Obstet. Gnecol. 191, 309–314. doi: 10.1095/biolreprod.105.046029
Tse, D. B., Ching, E., Yousefzadeh, N., Roque, H., and Young, B. K. (2005). Heterogeneity in fetal immunocompetence during the second trimester of gestation: implications for treatment of nonimmune genetic disorders by in utero transplantation. Fetal Diagn. Ther. 20, 175–181. doi: 10.1159/000083900
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vrecenak, J. D., Pearson, E. G., Santore, M. T., Todorow, C. A., Li, H., Radu, A., et al. (2014). Stable long-term mixed hematopoietic chimerism achieved in a canine model of allogeneic in utero hematopoietic cell transplantation. Blood 124, 1987–1995. doi: 10.1182/blood-2013-11-537571
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Waddington, S. N., Buckley, S. M., David, A. L., Peebles, D. M., Rodeck, C. H., and Coutelle, C. (2007). Fetal gene transfer. Curr. Opin. Mol. Ther. 9, 432–438.
Waddington, S. N., Kennea, N. L., Buckley, S. M., Gregory, L. G., Themis, M., Coutelle, C., et al. (2004a). Fetal and neonatal gene therapy: benefits and pitfalls. Gene Ther. 11(Suppl. 1), S92–S97. doi: 10.1038/sj.gt.3302375
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Waddington, S. N., Nivsarkar, M. S., Mistry, A. R., Buckley, S. M., Kemball-Cook, G., Mosley, K. L., et al. (2004b). Permanent phenotypic correction of hemophilia B in immunocompetent mice by prenatal gene therapy. Blood 104, 2714–2721. doi: 10.1182/blood-2004-02-0627
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Westgren, M. (2006). In utero stem cell transplantation. Semin. Reprod. Med. 24, 348–357. doi: 10.1055/s-2006-952156
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Westgren, M., Ringdén, O., Bartmann, P., Bui, T. H., Lindton, B., Mattsson, J., et al. (2002). Prenatal T-cell reconstitution after in utero transplantation with fetal liver cells in a patient with X-linked severe combined immunodeficiency. Am. J. Obstet. Gynecol. 187, 475–482. doi: 10.1067/mob.2002.123602
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Westgren, M., Ringden, O., Eik-Nes, S., Ek, S., Anvret, M., Brubakk, A.-M., et al. (1996). Lack of evidence of permanent engraftment after in utero fetal stem cell transplantation in congenital hemoglobinopathies. Transplantation 61, 1176–1179. doi: 10.1097/00007890-199604270-00010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Williams, S., Mustoe, T., Mulcahy, T., Griffiths, M., Simpson, D., Antoniou, M., et al. (2005). CpG-island fragments from the HNRPA2B1/CBX3 genomic locus reduce silencing and enhance transgene expression from the hCMV promoter/enhancer in mammalian cells. BMC Biotechnol. 5, 1–9. doi: 10.1186/1472-6750-5-17
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yáñez-Muñoz, R. J., Balaggan, K. S., MacNeil, A., Howe, S. J., Schmidt, M., Smith, A. J., et al. (2006). Effective gene therapy with nonintegrating lentiviral vectors. Nat. Med. 12, 348–353. doi: 10.1038/nm1365
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zani, A., Cananzi, M., Fascetti-Leon, F., Lauriti, G., Smith, V. V., Bollini, S., et al. (2014). Amniotic fluid stem cells improve survival and enhance repair of damaged intestine in necrotising enterocolitis via a COX-2 dependent mechanism. Gut 63, 300–309. doi: 10.1136/gutjnl-2012-303735
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, F., Thornhill, S. I., Howe, S. J., Ulaganathan, M., Schambach, A., Sinclair, J., et al. (2007). Lentiviral vectors containing an enhancer-less ubiquitously acting chromatin opening element (UCOE) provide highly reproducible and stable transgene expression in hematopoietic cells. Blood 110, 1448–1457. doi: 10.1182/blood-2006-12-060814
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: congenital disease, in utero therapy, stem cells, gene therapy, amniotic fluid
Citation: Ramachandra DL, Shaw SSW, Shangaris P, Loukogeorgakis S, Guillot PV, De Coppi P and David AL (2014) In utero therapy for congenital disorders using amniotic fluid stem cells. Front. Pharmacol. 5:270. doi: 10.3389/fphar.2014.00270
Received: 30 September 2014; Paper pending published: 14 October 2014;
Accepted: 18 November 2014; Published online: 19 December 2014.
Edited by:
Graca Almeida-Porada, Institute for Regenerative Medicine, USAReviewed by:
Christopher D. Porada, Wake Forest Institute for Regenerative Medicine, USACopyright © 2014 Ramachandra, Shaw, Shangaris, Loukogeorgakis, Guillot, De Coppi and David. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anna L. David, Perinatal Therapy, Institute for Women’s Health, University College London, 86-96 Chenies Mews, London WC1E 6HX, UK e-mail:YS5kYXZpZEB1Y2wuYWMudWs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.