 Eric E. Schadt
Eric E. Schadt Sean Buchanan
Sean Buchanan Kristen J. Brennand
Kristen J. Brennand Kalpana M. Merchant
Kalpana M. Merchant- 1Icahn Institute for Genomics and Multiscale Biology, Icahn School of Medicine at Mount Sinai, New York, NY, USA
- 2Department of Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai, New York, NY, USA
- 3Lilly Research Laboratories, Eli Lilly and Company, Indianapolis, IN, USA
- 4Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, NY, USA
A disruptive approach to therapeutic discovery and development is required in order to significantly improve the success rate of drug discovery for central nervous system (CNS) disorders. In this review, we first assess the key factors contributing to the frequent clinical failures for novel drugs. Second, we discuss cancer translational research paradigms that addressed key issues in drug discovery and development and have resulted in delivering drugs with significantly improved outcomes for patients. Finally, we discuss two emerging technologies that could improve the success rate of CNS therapies: human induced pluripotent stem cell (hiPSC)-based studies and multiscale biology models. Coincident with advances in cellular technologies that enable the generation of hiPSCs directly from patient blood or skin cells, together with methods to differentiate these hiPSC lines into specific neural cell types relevant to neurological disease, it is also now possible to combine data from large-scale forward genetics and post-mortem global epigenetic and expression studies in order to generate novel predictive models. The application of systems biology approaches to account for the multiscale nature of different data types, from genetic to molecular and cellular to clinical, can lead to new insights into human diseases that are emergent properties of biological networks, not the result of changes to single genes. Such studies have demonstrated the heterogeneity in etiological pathways and the need for studies on model systems that are patient-derived and thereby recapitulate neurological disease pathways with higher fidelity. In the context of two common and presumably representative neurological diseases, the neurodegenerative disease Alzheimer’s Disease, and the psychiatric disorder schizophrenia, we propose the need for, and exemplify the impact of, a multiscale biology approach that can integrate panomic, clinical, imaging, and literature data in order to construct predictive disease network models that can (i) elucidate subtypes of syndromic diseases, (ii) provide insights into disease networks and targets and (iii) facilitate a novel drug screening strategy using patient-derived hiPSCs to discover novel therapeutics for CNS disorders.
Introduction
The disease burden on society is increasing at a dramatic rate. Focusing specifically on central nervous system (CNS disorders), the prevalence is growing at an alarming rate, with one in sixty-eight in the U.S. having some form of autism (Baoi, 2014), 1.8 million estimated to suffer from schizophrenia (SZ), and Alzheimer’s Disease (AD) affecting more than five million in the U.S. today, with projections of a 40% increase in the number of AD cases in the next 10 years (Hebert et al., 2013). The costs of these diseases are staggering, both in financial and human terms. In 2002, the overall estimated cost of SZ was $62.7 billion with 36% attributed directly to health care expenses, though most costs are non-health care related, such as decreased productivity. By 2050, if healthcare costs for AD remain unchanged, the entire Medicare budget will be consumed by the treatment of those with AD (Alzheimer’s Association, 2014).
Considerable effort and resources are being expended on drug discovery research aimed at developing novel therapeutics that would address the unmet medical need across a broad spectrum of diseases. However, less than one out of every eleven drug discovery programs makes it to market (Cummings et al., 2014). The success rate for CNS disorders is even lower. Though many factors may contribute to the high rate of attrition, the major drivers for CNS disorders, are inadequate efficacy or margins of safety (Kola and Landis, 2004). In this review, we will exemplify the issues of CNS drug discovery and provide a perspective for changing the current paradigms within the context of the neurodegenerative disease, AD, and the psychiatric disorder, SZ.
For SZ, existing treatments target a very limited number of putative mechanisms, which treat some of the symptoms of SZ in some of the patients some of the time. Although several new pharmacologic interventions have been tested clinically in the last decade, none have shown medically relevant efficacy required for approval by regulatory authorities. Similarly, the field of therapeutic development to slow the progression of AD is littered with clinical failures of multiple pharmacological mechanisms and treatment modalities. These failures have had somewhat sobering effect on the field, particularly since many of the interventions were founded on human genetics- and pathology-informed amyloid hypothesis. A bright spot and beacon of hope may be the field of cancer therapeutics, where personalized treatments with far superior efficacy than traditional chemotherapies have been developed successfully. It behooves us to understand the primary reasons for the emerging successes in cancer treatments in order to adapt the paradigm to drug discovery for CNS disorders.
We will begin this review by discussing key factors that have likely contributed to the clinical failures for novel drugs, discuss the cancer research paradigms that have led to drugs with superior efficacy and diagnostic tests, and offer a perspective on the application of emerging technologies and tools that could improve the success rate of novel therapies. Specifically, we will focus on two potentially game-changing paradigms: (1) advances in human induced pluripotent stem cell (hiPSC)-based disease models and (2) multiscale predictive modeling. Together, these approaches can enable a more integrative biology approach to deriving insights into disease mechanisms, upon which drug screening and development may be founded in the near future. Although these approaches may be adapted broadly to many CNS indications, this review will focus primarily on SZ and AD.
Challenges for Drug Discovery in Neuroscience
First, the selection of a target for novel drug therapies requires an in-depth understanding of disease biology, from the etiological factors to pathophysiological mechanisms, and their relationship to disease progression and duration. It is insufficient to move forward with only the knowledge that a particular target is expressed in the brain or that a specific DNA variant is associated to a neurological disease. For common, complex trait diseases, a more systems oriented view is emerging in which human diseases are demonstrated to be emergent properties of biological networks, not the result of changes to single genes (Schadt, 2009; Schadt et al., 2009; Califano et al., 2012). Hence, rather than repeating the mistakes of the past, it is imperative to understand the biological context in which the susceptibility gene/gene networks and gene products operate to give rise to the disease, before beginning high throughput drug discovery screens. As the next step, we will need insights into the effect of the implicated gene network on cellular/physiological pathways in order to determine whether a novel therapy should augment or suppress, either fully or partially, the function of the disease-associated network.
Secondly, other than rare or orphan diseases that are caused by Mendelian mutations, common CNS disorders are syndromic diseases diagnosed primarily by non-specific and blunt clinical diagnostic tools, often based on patient reported symptoms. Thus, both the so-called clinically diagnosed SZ or AD patient population represents a diverse patient population with respect to etiologies and associated pathophysiological mechanisms, giving rise to similar sets of clinical symptoms but with distinct rates of disease progression. The clinical diagnostic tools, such as the Diagnostic and Statistical Manual (DSM) criteria for mental disorders or Mini-Mental State Examination (MMSE) for dementia, have served us well and led to approval of several drugs. However, the poor specificity and sensitivity of these tools require that they be supplemented with objective, diagnostic biomarkers with which to classify or enrich patient populations that are more homogenous in either their etiological or pathophysiological factors, or disease stage, being targeted by drugs.
Third, chronic diseases have an additional inherent issue related to adaptive biological mechanisms that emerge with chronicity of the disease or drug treatment. For neurodegenerative disorders, there is also the complicated factor of loss of resiliency in surviving neurons affected by the disease process. Thus, interventional strategies have to be designed to be specific to the stage of the disease, and when possible, to target primary prevention. Therefore, an understanding of adaptive molecular mechanisms as well as objective biomarkers that can monitor the biologic processes associated with the disease stage or progression and adaptive processes induced by drugs or disease will be required to improve drug discovery and development.
Finally, drug screening paradigms need to evolve so that disease biology mechanisms are monitored in cellular and animal models that more faithfully recapitulate human disease biology. Similarly, the systems biology/disease network approach will require that end-points of drug screens may have to be multi-parameter or phenotypic in nature, rather than those based on ease or throughput considerations alone. Thus, we must facilitate the development of cell-based systems derived from patients with disease as well as normal controls, in which the cell types are directly relevant to those implicated in human disease, so that we may garner insights into disease biology and design effective screening paradigms. Given that the large-scale generation and integration of panomic data has enabled the construction of complex gene networks that provide a new framework for understanding the molecular basis of disease (Ideker et al., 2002; van’t Veer et al., 2002; Schadt et al., 2003; Barabasi and Oltvai, 2004; Bystrykh et al., 2005; Ghazalpour et al., 2005; Schadt, 2005; Lum et al., 2006; Wang et al., 2006; Chen et al., 2008; Emilsson et al., 2008; Zhu et al., 2008), it is now possible to take a data driven, network-based view of diseases, which in turn enables the elaboration of a network-based view of drug discovery and development, one that is fundamentally different from current methods (Chen et al., 2008; Emilsson et al., 2008; Yang et al., 2009; Zhong et al., 2010a,b; Califano et al., 2012; Zhu et al., 2012; Zhang et al., 2013a; Kidd et al., 2014; Figure 1). Systems biology approaches seek to presume less knowledge, capture more information, and interpret the information in a more data driven way.
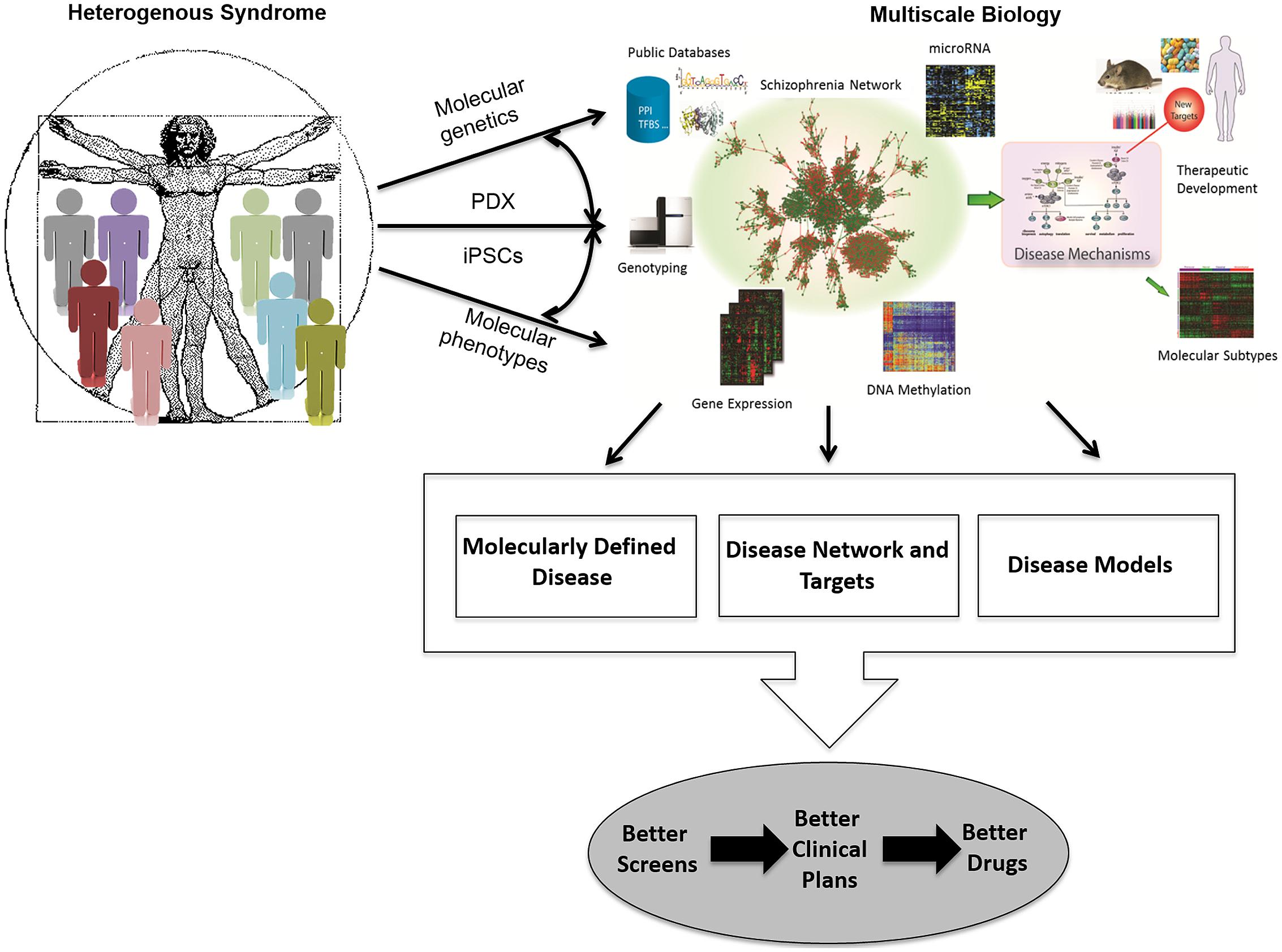
FIGURE 1. Schematic for how multiscale biology can lead to therapeutic development. A network-driven framework to integrate diverse types of data that lead to multiscale models of disease that can then be used to elucidate disease mechanisms, stratify patient populations, and develop novel therapeutics. PDX, patient-derived tumor grafts; iPSCs, induced pluripotent stem cells.
The four points discussed above require translational approaches that begin with patient-centered research to derive insights into disease biology. Of all the major therapeutic areas, cancer drug discovery research has led the way in translating the explosion of human genomic data into new therapeutics despite each clinically diagnosed malignancy being a heterogeneous mixture. Hence, we begin by reviewing the cancer drug discovery paradigm.
Cancer as an Emerging Model of Modern Day Drug Discovery
Drug hunters have been able to take advantage of two unique features of cancer biology. Firstly, the accessibility of tumor tissues facilitate their study through genomic and phenotypic approaches as well as provide patient-derived pre-clinical in vitro and in vivo model systems, which are being shown to more faithfully recapitulate important aspects (notably uncontrolled proliferation) of the disease, and in many cases can be formatted for high throughput screening (Cheung et al., 2011b; Barretina et al., 2012; Dar et al., 2012; Garnett et al., 2012; Hirabayashi et al., 2013). This has resulted in a second distinguishing feature of cancer biology: although cancer encompasses a large diversity of distinct malignancies, each can be defined by an even larger diversity of driving mutations. Importantly, the driver mutations are not random, but instead reflect different core biological processes and signaling pathways central to the onset and progression of the tumors and thereby provide a rich source of new drug targets. Thus the taxonomy of cancers is being redefined on the basis of molecular markers.
Multi-center genome sequencing endeavors (Stratton, 2011) have revealed 100s of cancer causing mutations, discoveries which shed welcome light on vital nodes in the otherwise largely cryptic underlying disrupted networks (Eifert and Powers, 2012; Alexandrov et al., 2013; Cancer Genome Atlas Research Network, 2013, 2014; Cancer Genome Atlas Research Network et al., 2013a,b; Kandoth et al., 2013; Zhang et al., 2014a). These growing lists of cancer genes become immediate opportunities for therapeutic intervention. When the encoded product of an oncogenic mutation belongs to a conventionally druggable protein class, this information can lead to very rapid development of novel and effective drugs. For example, translocations fusing EML4 to the tyrosine kinase gene, ALK, were discovered in lung cancer biopsies in Rikova et al. (2007), Soda et al. (2007). The EML4-ALK fusion protein results in a constitutively activated catalytic domain and is a key driver of the uncontrolled proliferation of cancer cells with this lesion. So it was postulated that ALK-directed tyrosine kinase inhibitors might be able to nullify the oncoprotein. Crizotinib, a tyrosine kinase inhibitor already in clinical testing for MET kinase-driven tumors, was known to inhibit also ALK kinase and trials were therefore extended to target EML4-ALK-positive non-small cell lung cancer (NSCLC) patients. Efficacy results in these patients were dramatic, leading to FDA approval of crizotinib in 2011, an unprecedented short duration (less than 4 years) after the first reports of the fusion in patients. Cancer sequencing projects have revealed driving mutations in genes encoding other druggable oncoproteins and, much like crizotinib, drugs targeting several of these have shown considerable promise in clinical trials for patients whose tumors bear the relevant mutation (Table 1) although, as described below, the development of resistance has plagued each of these drugs.
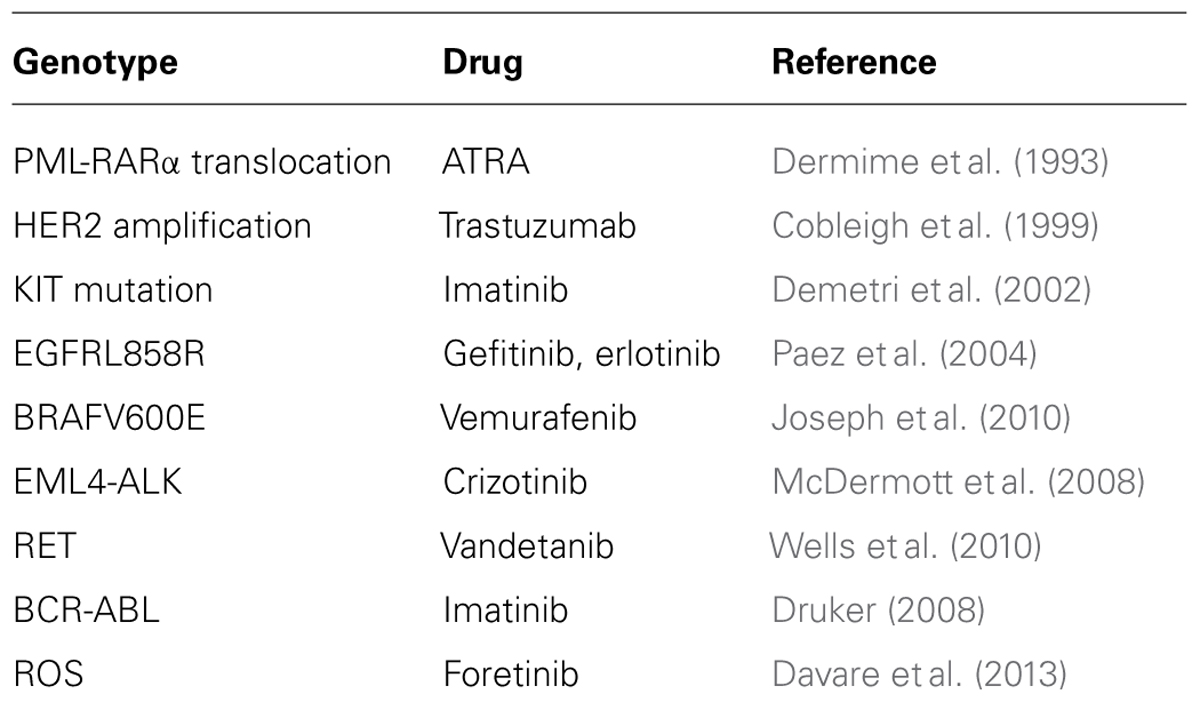
TABLE 1. Driver genes identified from cancer sequencing studies have led directly to drug targets and subsequent therapeutics that have shown significant promise in clinical trials.
Some of the most frequently mutated cancer genes, however, including oncogenes such as MYC and RAS, as well as tumor suppressor genes, do not encode readily druggable proteins. In principle, it may simply be a matter of following the known signaling pathways from the cancer gene product until we reach a tractable drug target, but the further we stray from the mutated node the more likely the intervention will not be effective due to redundancies and divergent branches in the relevant networks. Inhibitors of the druggable GPCR target, Smoothened, have shown encouraging promise in cancers with loss of function mutations in the tumor suppressor PTCH, which encodes an upstream regulator of Smoothened (Berman et al., 2002; Rudin et al., 2009; Von Hoff et al., 2009). Similarly, MEK inhibitors have proven effective in treating BRAF-mutant melanoma, where the defective oncogene is directly upstream of, and activates, MEK in the MAPK pathway. Conversely, however, MEK inhibitors have not thus far proven very effective in treating cancers with mutations that activate the oncoprotein, RAS, just one extra step further upstream of RAF in the canonical pathway. What has become evident now is that a comprehensive understanding of the entire defective network, rather than a single canonical pathway, is necessary to identify new drug targets for cancers harboring these undruggable oncogenes and tumor suppressors (Solit et al., 2006; Lito et al., 2014; Schmit et al., 2014; Sun et al., 2014).
Molecular networks can be interrogated with empirical approaches that take advantage of the fact that cells from cancer biopsies can be propagated as established cell cultures, as rodent xenografts, or as patient-derived tumor grafts (PDX). Genetically characterized cancer cell lines and tumor grafts have been established from human disease specimens and generally preserve the genomic features observed in the disease. In many cases, mutated oncogenes and tumor suppressors that drive disease in animal models are also known to be critical for the growth and survival, in culture, of human cancer cell lines with the same mutations. This offered the opportunity to use proliferation assays as a facile but disease biology-relevant phenotypic assay on human cancer cell lines to identify vulnerabilities particular to the network reconfigured by specific mutations. A large number of in vitro and in vivo screens based on this principle have reported potential new targets, which take advantage of synthetic lethality, non-oncogene addiction and co-lateral vulnerabilities in cancer (Whitehurst et al., 2007; Turner et al., 2008; Luo et al., 2009a,b; Astsaturov et al., 2010; Rehman et al., 2010; Cheung et al., 2011b; Muellner et al., 2011; Kumar et al., 2012; Muller et al., 2012; Toyoshima et al., 2012; Riabinska et al., 2013; Mair et al., 2014). Such approaches have revealed vulnerabilities in cancer cells whose primary driving mutations are not directly druggable. For example PARP inhibitors were demonstrated to be synthetic lethal in vitro with mutations in the tumor suppressors BRCA1 and BRCA2 (Farmer et al., 2005; McCabe et al., 2005) and clinical testing has established the same genetic dependence on tumor sensitivity to the PARP inhibitor olaparib in patients (Fong et al., 2009).
Using high throughput screening methodologies coupled with statistical methods, large panels of genetically characterized tumor cell lines can be assembled to identify gene–drug interactions in an unbiased manner and thereby identify either drugs or drug–drug combinations effective for the treatment of cancers with the relevant mutation (Barretina et al., 2012; Garnett et al., 2012). In an analogous manner, tumor cell panels are being tested to identify the context of dependencies on the entire human transcriptome using a hairpin dropout whole genome RNAi screen (Cheung et al., 2011b).
These developments over the last decade have demonstrated that the ‘one gene – one drug’ paradigm is leading to progress in the treatment of multiple cancers and is likely to continue to bear some fruit as new mutations and drugs emerge. However, it is also clear that the efficacy of new targeted therapeutics in cancer is too often short-lived due to the eventual, sometimes rapid, emergence of drug resistance due to adaptive biological processes induced by the disease or drug treatment (Yauch et al., 2009; Barber et al., 2013; Das Thakur and Stuart, 2013; Lord and Ashworth, 2013; Niederst and Engelman, 2013). The mechanisms of resistance to the modern cancer pharmacopeia are being deciphered. Hence, one approach to tackle the drug resistance problem is to continue along the same path, looking to exploit new vulnerabilities that emerge in the resistant clones (O’Hare et al., 2011; Cortes et al., 2012; Yadav et al., 2012; Friboulet et al., 2014; Hata et al., 2014; Traer et al., 2014). Resistance to imatinib, a very effective inhibitor of BCR-ABL for the treatment of chronic myelogenous leukemia, most often occurs due to point mutations in the target kinase that prevent drug binding. Three drugs, designed specifically to inhibit these imatinib-resistant enzymes, have since been approved and CML is now a well-managed disease for the majority of patients (O’Hare et al., 2011). However, in the case of resistance to epidermal growth factor receptor (EGFR) and BRAF inhibitors, distinct mechanisms of resistance can emerge in different clones from the same primary tumor – a situation that likely will not be feasibly solved with gene–drug pairs, even if drugs directly targeting the new mutations that arise in each metastasis could be discovered. Instead, the disrupted networks need to be more completely understood to identify downstream or parallel pathways common to all the resistant clones. Here again, cell lines and PDX models derived from drug-resistant tumors can be used to screen for drugs or by RNAi methods, to identify targets that will be effective in dealing with multiple mechanisms of resistance (Huang et al., 2012; Konieczkowski et al., 2014; Sun et al., 2014). High throughput, unbiased drug–drug combination screens are also feasible, at least in vitro, to identify drug cocktails that are predicted to more effectively treat and prevent the emergence of drug resistance (Bajrami et al., 2012; Roller et al., 2012).
Other ways to address the rapid ability of cancer cells to evolve drug resistance are emerging. Targeting cancer by avoiding the cancer cells altogether and instead developing drugs that target the other physiologic processes essential to cancer progression has shown considerable promise. Thus discovery of drugs targeting angiogenesis, inflammation, and immune checkpoints does not benefit from the wealth of smoking gun targets informed by somatic mutation data in tumor cells, and preclinical models of these aspects of disease are more complex than proliferation-based assays. But exciting progress has been made, nevertheless, with spectacular results in some cases (Robert et al., 2011; Topalian et al., 2014) and unbiased screening approaches are being explored in the search for new targets in each of these areas (e.g., Zhou et al., 2014). In addition, work done in fruit fly to demonstrate multifactorial targeting of tumors by simultaneously hitting multiple pathways that serve as key drivers of the cancer has not only demonstrated that targeting of individual signaling pathways in a given cancer will have short-lived efficacy, but that multifactorial targeting of the tumor can greatly diminish chances that tumor cells will evolve to defeat the cocktail of drugs that get used in these cases (Cagan, 2013; Das and Cagan, 2013). Here, the use of PDX models provides a more holistic way of studying how tumors may evolve in response to different types of therapy. In this way, a more systems oriented approach can be employed in which PDX models are used as patient avatars to establish the most effective combination of treatment specific to that individual’s tumor. These more progressive approaches that seek to model a patient’s tumor in systems that can be rapidly screened for therapies that will be most effective for that individual, represent the new generation of precision medicine strategies that hold promise in transforming how we diagnose and treat disease (National Research Council (US) Committee on A Framework for Developing a New Taxonomy of Disease, 2011).
These same model systems can be used to reveal the adaptive processes the can follow drug treatment, and often dampen drug efficacy, and to therefore suggest combination therapies that counteract them. For example, BRAF inhibitors can be very effective in melanoma patients with BRAF mutant tumors, but have been much less effective in BRAF mutant colorectal cancer and in Ras mutant tumors. Activation of c-Raf caused by BRAF inhibitors in tumors with activated Ras has been shown, paradoxically, to stimulate, rather than inhibit MAPK pathway signaling and is suspected of causing new skin cancers that have been observed as a frequent side effect of BRAF inhibitors (Hall-Jackson et al., 1999; Hatzivassiliou et al., 2010; Heidorn et al., 2010; Poulikakos et al., 2010). This model predicts that nullifying signaling downstream of c-Raf with MEK inhibitors should prevent the skin lesion side effect and also improve efficacy. The combination of the MEK inhibitor, trametinib, and BRAF inhibitor, dabrafenib, was approved for the treatment of melanoma in 2014. In BRAF mutant colorectal cancer it has been learned that negative feedback pathways are stimulated by the constitutive activation of the MAPK pathway conferred by BRAF activating mutations and that these pathways act to diminish signaling through the EGFR. A consequence of inhibition of BRAF, therefore, is reactivation of EGFR mediated signaling which diminishes drug efficacy (Corcoran et al., 2012; Prahallad et al., 2012). Based on these discoveries, clinical trials are underway testing combined inhibition of BRAF and EGFR in colorectal cancer (NCT01750918). Similarly MEK inhibitors are only poorly effective in KRAS mutant cancers. RNAi-based screens were used to identify new targets that, in concert with MEK inhibition, augment the antiproliferative activity of MEK inhibitors in KRAS mutant colorectal cancer cell lines, revealing that c-Raf knockdown, or c-Raf inhibitors, were able to potentiate the activity of MEK inhibitors in KRAS mutant tumor cells (Lito et al., 2012; Lamba et al., 2014). Approved Raf kinase inhibitors, such as dabrafenib and vemurafenib, are ineffective inhibitors of c-Raf and so clinical testing of this hypothesis will have to await the emergence of true c-Raf inhibitor drugs. These approaches are beginning to reveal the adaptive processes the can follow drug treatment, and often dampen drug efficacy, and suggest combination therapies to counteract them.
To summarize, the transformative success of the cancer drug discovery and development may be attributable to four key factors: (i) de-risking target selection through identification of driver mutations or disease networks through multi-center studies on patient-derived tumor specimens, (ii) reducing patient heterogeneity by implementing molecular definition of disease taxonomy rather than clinical diagnosis alone, (iii) addressing drug resistance by targeting disease networks associated adaptive processes induced by drug and/or disease, and (iv) incorporating cellular and animal models with greater predictive validity in drug screens.
Multi-Center Genetic Studies and Use of hiPSCs to Derive Insights into Disease Biology
Tracking with the great advances in cancer drug development that have benefitted from the genomics revolution, forward genetics strategies to elucidate the complexity of human disease have been accelerated as the cost of assaying nucleic acid sequences continues to drop exponentially and ever bigger cohorts of diseased individuals are assembled. Whether performing a genome-wide association study or whole exome/genome sequencing studies in case/control cohorts or families segregating diseases of interest, the quest has been to identify specific genes, pathways and networks that are critical for disease onset, progression and severity and thereby rationalizing the selection or prioritization of molecular targets or pathways for drug discovery.
A genetics case study for schizophrenia
Schizophrenia is a complex and heterogeneous disorder with an estimated heritability of about 80% (Sullivan et al., 2003). Much like cancer, many types of DNA variations, [single nucleotide polymorphisms (SNPs), copy number variations (CNVs), and small exonic missense and nonsense mutations) as well as epigenetic and/or environmental factors contribute to the risk of SZ. The genetic risk factors for SZ include both rare variants conferring large relative risks (e.g., CNVs) as well as common SNP variants, the latter (Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014) with modest individual effect sizes (Purcell et al., 2009).
Copy number variations are ubiquitous in the population (McCarroll et al., 2008); it is now widely held that in addition to a number of fairly uncommon syndromes, they also contribute to more common disorders such as SZ. In fact, though linkage studies have been unsuccessful in identifying highly penetrant genes (Ng et al., 2009), a large body of work (reviewed Malhotra and Sebat, 2012) defined the following principles across >10,000 SZ samples: (1) genome-wide rates of large (>100 kb), rare (<1%) CNVs are elevated, (2) rates of de novo CNVs are elevated 2–50 fold, (3) CNVs generally contain many genes and confer large relative risks (2–50), (4) specific sites of CNVs are often also found in multiple additional neurological diseases, and (5) CNVs are enriched in neuronal functions, particularly those that are involved in synaptic activity and neurodevelopmental processes. CNVs represent a polygenic burden of rare disruptive mutations, one that is particularly enriched in gene sets including the voltage-gated calcium ion channel and the post-synaptic density (Purcell et al., 2014).
While individually penetrant CNVs are only found in a minority of patients (perhaps 5–10%), common DNA variants (minor allele frequency >5%) are significant contributors to the heritability of SZ, accounting for ∼30% of the variance in liability (Lee et al., 2012). It is now widely held that SZ risk also involves 1000s of common alleles of very small effect (Purcell et al., 2009). The earliest convincing evidence for a contribution to common variants in SZ included the major histocompatibility complex (MHC; Purcell et al., 2009; Shi et al., 2009; Stefansson et al., 2009), subsequent work also implicated the microRNA (miR)-137 as well as four of its targets (Ripke et al., 2011). Recent GWAS of ∼38,000 SZ patients and ∼115,000 controls by the Psychiatric Genomics Consortium Schizophrenia Group have now identified 108 genome-wide significant loci, most of which are novel, but individually have small effects (relative risk ranging from 1.09–1.17; Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014). In fact, it was recently estimated that more than 6300 common SNPs collectively account for at least 32% of the genetic risk for SZ (Ripke et al., 2013). Interestingly, these loci have begun to implicate pathways: in addition to at least one target of current neuropharmacology (the dopamine receptor DRD2), for the first time, critical glutamatergic genes such as GRM3 and GRIN2A, and calcium channel subunits (CACNA1C and CACNA1l) have been associated with SZ.
Studying de novo point mutations is also a powerful tool; it was recently shown that small de novo mutations affecting one or a few nucleotides are overrepresented among glutamatergic post-synaptic proteins (Fromer et al., 2014). The location of rare disruptive loss-of-function mutations, enriched in glutamatergic and calcium signaling, have been shown to overlap with SZ-associated CNV (Purcell et al., 2014). Similarly, common and rare variants can overlap: a novel variant at 16p11.2 (rs4583255[T]; odds ratio = 1.08) substantially increases risk of psychosis (Steinberg et al., 2014).
However, genetic data on their own are not sufficient to garner insights into disease biology for several reasons. Although disease-associated loci are identified, the causal genes are not always known. Even when the genes implicated are known, an understanding of the functional relevance of the genetic variant, whether it is an activating or inhibitory variant, has to be experimentally derived to design drug discovery strategy. For SZ, the functional implications of the DNA variants in glutamatergic genes or calcium channels remain to be elucidated. Both animal model and clinical studies indicate that SZ is associated with hyper-glutamatergic neurotransmission, at least early in the disease (reviewed by Poels et al., 2014). It is critical to know whether disease-associated GRM3 or GRIN2A SNPs predispose to SZ by increasing glutamatergic neurotransmission at a vulnerable developmental stage. The importance of such functional data is exemplified by the recent failure of two glutamate targeting ligands in Ph3 trials. First, a GRM2/3 agonist, LY2140023, from Eli Lilly and Company, which is predicted mechanistically to reduce glutamatergic neurotransmission. Although, in a Phase 2 study, this compound significantly reduced the symptoms of SZ (Patil et al., 2007), it failed to do so in subsequent larger Phase 3 studies (Adams et al., 2013, 2014). Similarly, on January 21, 2014, Roche announced that the GlyT1 inhibitor, bitopertin, failed to meet its primary end-points in two Phase 3 trials in SZ. What then is the significance of glutamatergic pathway replicably associated to SZ by genetic studies? From the clinical studies we cannot conclude whether the targets were wrong or the patient population was wrong. One key factor to consider is patient heterogeneity. Thus only a minority of patients carry DNA variants in glutamatergic genes (Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014) but the drug studies did not stratify patients on the basis of gene variants or glutamatergic imaging or physiological markers. Using the cancer example, two paradigms need to be adopted to leverage the genomic data. First, to refine the taxonomy of SZ and other CNS disorders on the basis of molecular markers. This will allow identification of patient populations most likely to respond to a drug mechanism. Second, use model systems to understand functional biological implications of gene variants, as detailed below.
hiPSC as a model system to translate genetic findings into functional insights
In order to understand the complex network interactions contributing to the entire genetic risk in any given patient, and between patients, one must be able to study the full genetic background, even without knowing all the risk alleles contributing to the disease. Today, hiPSC-based models for many CNS disorders have been established, by reprogramming patient somatic cells into hiPSCs, and subsequently differentiating these stem cells into different types of neurons (Dimos et al., 2008; Park et al., 2008; Baek et al., 2009; Ebert et al., 2009; Hotta et al., 2009; Lee et al., 2009; Soldner et al., 2009; Marchetto et al., 2010; Nguyen et al., 2011; Pasca et al., 2011). This type of technology has made it possible to connect genetic data to biological insights, elucidating molecular and physiological changes in different neural cell types, something that was incredibly difficult or even impossible prior to hiPSCs.
A number studies of psychiatric disorders have reproducibly demonstrated that even small patient hiPSC cohorts can reveal robust and repeatable neural phenotypes, meriting further investigation. For example, many groups have generated hiPSCs from Rett Syndrome patients, and consistent with post-mortem patient studies, all have reported that neuronal soma size is reduced compared with controls. Additionally, other disease-relevant phenotypes such as reduced spine density, decreased neuronal spontaneous calcium signaling and decreased spontaneous excitatory and inhibitory post-synaptic currents have been reported (Marchetto et al., 2010; Ananiev et al., 2011; Cheung et al., 2011a). Timothy syndrome (TS) is caused by a mutation in the L-type calcium channel Ca(v)1.2 and associated with heart arrhythmias and ASD. TS hiPSC derived cortical neural progenitor cells (NPCs) and neurons show aberrant calcium signaling, (Pasca et al., 2011) ameliorated by treatment with roscovitine, a cyclin-dependent kinase inhibitor and atypical L-type-channel blocker (Pasca et al., 2011). Though early proof-of-concept studies of hiPSC neuronal pathology focused on diseases characterized by both the loss of function of a single gene product and rapid disease progression in early childhood (Ebert et al., 2009; Lee et al., 2009; Marchetto et al., 2010), many groups have recently extended these studies to complex genetic psychiatric disorders.
To date, SZ has lacked a human cell-based platform that incorporates the heterogeneity of this complex genetic disorder with which potential therapeutic compounds might be identified by high throughput screening.
hiPSC-based studies of schizophrenia
For a discovery made in 2006, that transient expression of just four factors (OCT3/4, KLF4, SOX2, and c-MYC) is sufficient to directly reprogram adult somatic cells into an induced pluripotent stem cell (iPSC) state (Takahashi and Yamanaka, 2006; Takahashi et al., 2007; Yu et al., 2007), Shinya Yamanaka was awarded the 2012 Nobel Prize in Medicine. With this revolutionary advance, hiPSCs are now be routinely generated from patient skin or blood cells, owing to the relative ease of tissue access, and are believed to be capable of differentiating into every cell type found in the adult (Maherali et al., 2007; Meissner et al., 2007; Takahashi et al., 2007; Wernig et al., 2007; Yu et al., 2007). Because hiPSCs can be derived from adult humans, after the development of disease, hiPSCs represent a potentially limitless source of human cells with which to study the onset and progression of neurological disease, even without knowing which genes are interacting to produce the disease state in an individual patient.
In previous publications, we directly reprogrammed fibroblasts from four SZ patients into hiPSCs and differentiated these disorder-specific hiPSCs into forebrain NPCs (Brennand et al., 2014) and neurons (Brennand et al., 2011). Gene expression comparisons of our hiPSC-derived NPCs and 6-week-old neurons to the Allen Brain Atlas indicate that our hiPSC neural cells, from controls and patients with SZ, resemble fetal rather than adult brain tissue (Brennand et al., 2014), indicating that hiPSC-based models may not yet be suited for the study of the late features of this disorder. SZ hiPSC NPCs show evidence of aberrant migration and increased oxidative stress (Brennand et al., 2014), while SZ hiPSC neurons showed diminished neuronal connectivity in conjunction with decreased neurite number, PSD-95 and glutamate receptor expression. Key cellular and molecular elements of the SZ phenotype were ameliorated following treatment of SZ hiPSC neurons with the antipsychotic loxapine (Brennand et al., 2011). Others have also reported that SZ hiPSC neural cells show increased oxidative stress (Paulsen et al., 2011; Robicsek et al., 2013), aberrant responses to environmental stresses (Hashimoto-Torii et al., 2014) and have reduced synaptic maturation (Robicsek et al., 2013; Wen et al., 2014; Yu et al., 2014; Zhang et al., 2014b).
Until recently, functional differences in SZ hiPSC neurons had not been identified, likely owing to the heterogeneity in hiPSC neuronal culture. Now, the first phenotypic characterization of a single neuronal subtype (hippocampal dentate gyrus granule neurons) shows reduced neuronal activity and spontaneous neurotransmitter release in SZ hiPSC-derived neurons (Yu et al., 2014). This demonstration that functional deficits can be detected in live human neurons in vitro convincing shows that phenotypic assays (if not molecular comparisons) must be conducted in specific and defined neuronal subpopulations.
A growing body of evidence links SZ with abnormal functioning of dopaminergic, GABAergic and glutamatergic neurons. Although pharmacological modulation of dopamine transmission helps manage the positive symptoms of SZ for some patients (Weinberger, 1987; Kessler et al., 2009), emerging evidence indicates that aberrant dopamine transmission is most likely downstream from dysfunctional GABAergic and glutamatergic neurons of the prefrontal cortex (Wen et al., 2010; Demjaha et al., 2013). hiPSCs can now be differentiated to cortical pyramidal (Espuny-Camacho et al., 2013), interneuron (Maroof et al., 2013; Nicholas et al., 2013) and midbrain dopaminergic fate (Chambers et al., 2009; Kriks et al., 2011), providing multiple avenues for studying SZ in precisely defined subpopulations of neurons. Efficient protocols to differentiate hiPSCs into dopamine neurons have been systematically optimized and yields now exceed >80% (Chambers et al., 2009; Kriks et al., 2011). After neural induction, DA specification occurs recapitulating the activation of Sonic hedgehog (SHH) and Wnt/β-catenin signaling that patterns dopaminergic neurons in the floor plate region of the ventral midline. Recently published methods to generate GABAergic neurons are similar, reiterating embryonic development of the ventral telencephalon via the inhibition of WNT signals and timed exposure to SHH signals (15,16). Differentiation to glutamatergic fate occurs in the absence of bone morphogenetic protein (BMP), Wnt/β-catenin and TGF-β/activin/nodal pathways (Mariani et al., 2012; Shi et al., 2012). Protracted cortical differentiation (50–70 days) seems to mimic human developmental temporal patterning, resulting in sequential specification of cortical layer identity (Shi et al., 2012; Espuny-Camacho et al., 2013).
The ability to rapidly induce neurons, rather than rely on protracted differentiation protocols, would clearly be advantageous when considering systematic comparisons of 100s of SZ patients or high throughput screening of 1000s of potential therapeutics. Mouse (Vierbuchen et al., 2010) and human (Pang et al., 2011) fibroblasts can be induced in less than 6 days into iNeurons, via lentiviral (LV) overexpression of just BRN2, ASCL1, and MYTL1 – with the addition of NEUROD1 in human cells. Though rapid, the process is inefficient, occurring in just 2–4% of the original fibroblasts, and generates relatively immature neurons unable to form synapses on their own. The addition of key microRNAs improves the process, resulting in mature neurons capable of forming fully functional synapses in pure cultures (Ambasudhan et al., 2011; Yoo et al., 2011). Yields remain at approximately 10%, with substantial variability between fibroblast lines, and the temporal and spatial identity of iNs, relative to the human brain, is unresolved. The ability to generate neuronal populations of a specific sub-type would be ideal for cell-based studies. Already, using pools of cell type specific transcription factors, human fibroblasts can be induced into midbrain dopaminergic neurons (Caiazzo et al., 2011). Though faster than hiPSC reprogramming and subsequent neuronal differentiation, of primary consideration is that this methodology transforms precious primary patient-derived cells into terminally differentiated neurons, limiting the cellular material available for studies.
In comparison to methods of growth factor-directed differentiation, or fibroblast derived iNeurons, inducible LV overexpression of NGN2 in hiPSCs rapidly induces pure populations of functional excitatory neurons, with a transcript profile indicative of cortical layer II/III neurons, in as little as in 21 days (Zhang et al., 2013b). We predict that similar methods for rapid and directed induction of a variety of pure neuronal subpopulations will soon be ubiquitous. While one might fear that this progression toward faster and more defined neuronal induction will bypass normal neural development, potentially limiting the ability to observe early phenotypes such as neural migration, specification or maturation, we note recent evidence that iNeurons derived from patients with an autism-associated neuroligin-3 (NLGN3) mutation perfectly recapitulated the molecular and synaptic defects observed in the Nlgn3 mouse model (Chanda et al., 2013).
To date, most hiPSC studies have been conducted on cells derived from a handful of cases and controls, typically around 3–6 patient lines. However, efforts are underway to make the process of converting somatic cells into stem cells more uniform, efficient, and cost-effective. Thus one can anticipate a time in the near future where 100s of cell lines representing 100s of individual patients may be studied to understand disease biology for common disorders such as SZ. In concert with functional genomics, high throughput electrophysiology, imaging and integrative systems biology approaches, this platform could provide insights into common and unique mechanisms of syndromic diseases upon which new drug discovery paradigms may be founded. Thus analogous to the oncology field, hiPSCs is poised to provide a cellular model platform that could enable personalized medicine for psychiatric indications. Additionally, these cell lines and associated phenotypes will form a powerful platform for drug-screening assays with direct relevance to disease biology.
Multiscale Biology Approach to Understanding Disease Biology and Identifying Therapeutic Targets
Given the enormous amount of panomic data that have been generated to characterize common human diseases, this data can be integrated in order to build predictive network models of normal and disease states, which can elucidate the key biological drivers of the disease state. To fully understand complex neurological diseases, we must link molecular biology to physiology (Schadt, 2009; Schadt et al., 2009; Califano et al., 2012). Multimodal models can be used to identify disease signatures, by using the networks to organize the signatures according to the sub-networks (and the biological processes that they define), which are associated with disease (Schadt et al., 2005, 2008; Chen et al., 2008; Emilsson et al., 2008; Yang et al., 2009, 2010; Zhong et al., 2010a,b; Zhu et al., 2010; Ambasudhan et al., 2011; Greenawalt et al., 2011; Wang et al., 2012). Ultimately, integrating diverse, large-scale data provides a path to predict which drug effects might best counteract the molecular networks underlying disease (Figure 2).
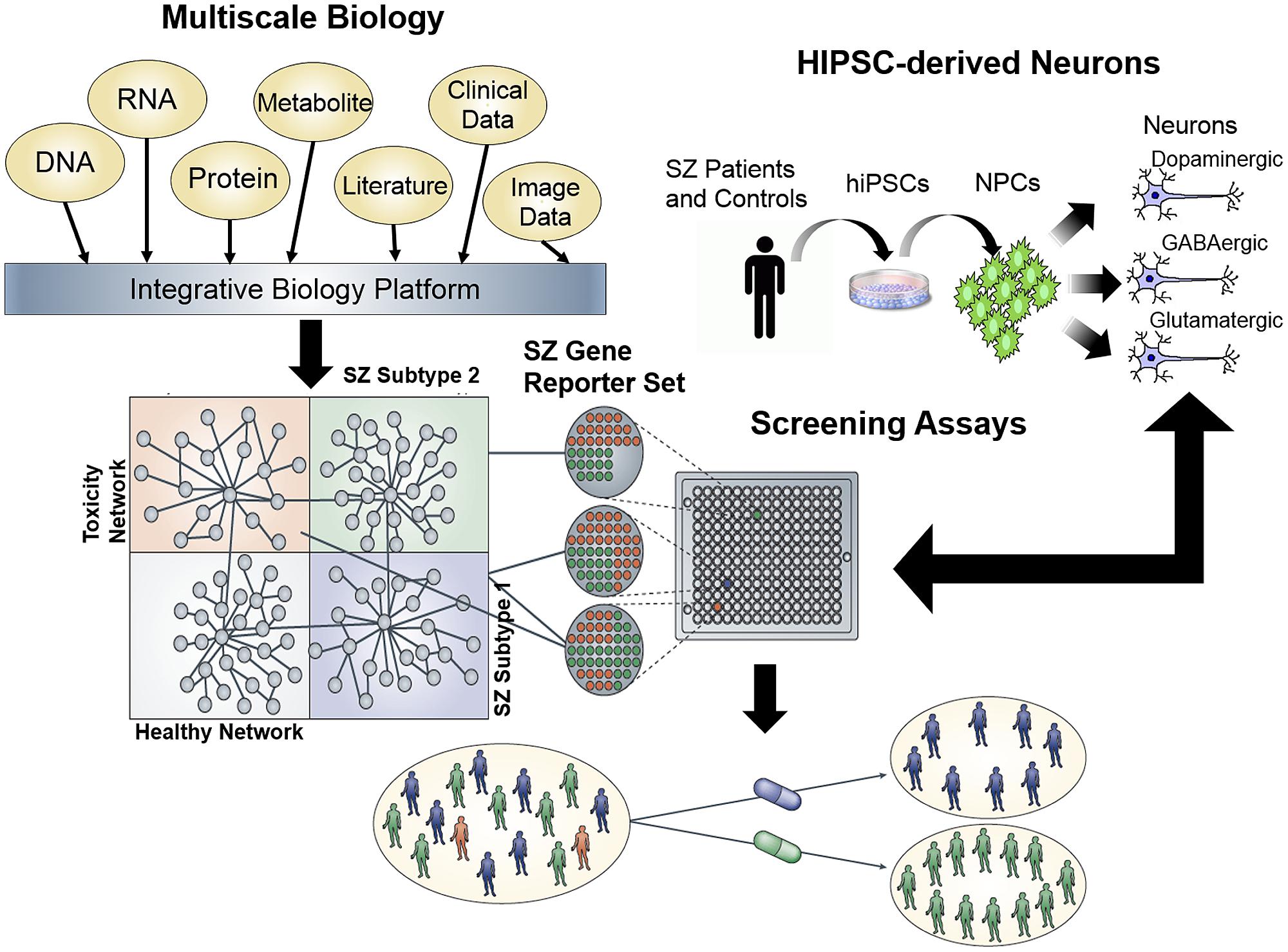
FIGURE 2. Schematic for a network screening approach to drug discovery. First, integration of multiscale data sets can be reduced to a minimal gene reporter set specific to a disease of interest, in this case, schizophrenia. Second, expression of this gene reporter set can be used as a platform for high throughput screening of hiPSC neurons generated from schizophrenia patients, in order to identify compounds capable of ameliorating the gene expression signature in patient-derived neurons.
Building and applying multiscale network models
Integrative network models utilize panomic data to derive causal relationships among 1000s of intermediate molecular traits and between molecular and higher order physiological traits associated with disease (Barabasi and Oltvai, 2004; Zhu et al., 2012). In this context, networks are represented graphically as nodes and edges, where nodes represent individual molecular and clinical features (gene expression levels, metabolite levels, protein states, methylation levels, biochemical measures, and so on) and edges represent the interactions among these variables. How molecular traits and disease traits causally relate to each other can be modeled using pairwise causality tests (Schadt et al., 2005; Millstein et al., 2009) or probabilistic graphical models, such as RIMBANet (Zhu et al., 2004, 2007, 2008, 2010, 2012), in which all available traits are considered simultaneously. A number of studies performed by us and others, in a variety of species, have demonstrated that predictive networks like Bayesian networks can capture fundamental properties of complex systems in states that give rise to complex phenotypes (Jansen et al., 2003; Lee et al., 2004; Zhu et al., 2004, 2007, 2008, 2012; Schadt et al., 2008; Zhang et al., 2013a). The available molecular data that informs on disease, derived from different tissues in different states, providing the necessary ingredients to reconstruct causal network models of disease (Figure 1).
High dimensional panomic data will increasingly be generated in hiPSC derived neurons, with 100s or even 1000s of samples generated from disease cohorts now possible for (relatively) low costs. This type of panomic data permits the construction of interaction and differential connectivity networks, which characterize the connectivity patterns of the molecular networks in disease relevant cell types between those with and without disease. Interaction networks and differential connectivity measures, such as the module-centric differential co-regulation (MDC) measure, provide for deeper insights into the molecular processes involved in disease. For example, by applying MDC to the molecular interaction networks generated from late-onset AD brain regions compared to these same brain regions in non-demented controls (Zhang et al., 2013a), we determine that one module with a significant gain of connectivity in AD patients was enriched for immune function and microglia. This type of analysis, comparing disease cases to controls, can now be carried out on hiPSC-derived cell types in order to generate sets of genes from co-expression modules that are differentially connected.
The network constructs discussed above provide a convenient framework for understanding the core biological processes involved in a given disease of interest, as well as for elucidating the master regulators of disease. Individual signatures of disease, or a given perturbation, can be projected onto the network models, in order to identify the sub-networks that best organize the signatures according to biological processes. For example, gene expression traits monitored in hiPSC-derived neurons can be identified as changed or not, in response to treatment with a given small molecule compound. This signature would represent a complex mixture of changes that reflect the proteins specifically targeted by the compound, the primary response of genes to those specific targets, downstream changes that result from changes in these molecular states, changes induced by unintended targets, and so on. By projecting this complex signature onto a more comprehensive multiscale network model, the signature can be broken up into different coherent components that reflect different biological processes and molecular functions associated with the action of the drug on the cell system under study (Figure 3). The identified components represent sub-networks that can in turn elucidate potential disease mechanisms defined by them. The master regulators of these sub-networks can in turn be identified using key driver analysis methods (Zhu et al., 2008, 2012; Tran et al., 2011; Zhang et al., 2013a) that involve finding the largest connected graphs containing the sub-networks, and then perturbing each node (or combination of nodes) in this expanded sub-network in silico to predict the network response. Those nodes that significantly alter the state of the network are declared as key drivers or master regulators of the sub-network. We have previously demonstrated this type of key driver analysis to identify networks and their corresponding key drivers associated with inflammatory bowel disease (IBD), AD, and other such common human diseases (Jostins et al., 2012; Wang et al., 2012; Zhang et al., 2013a).
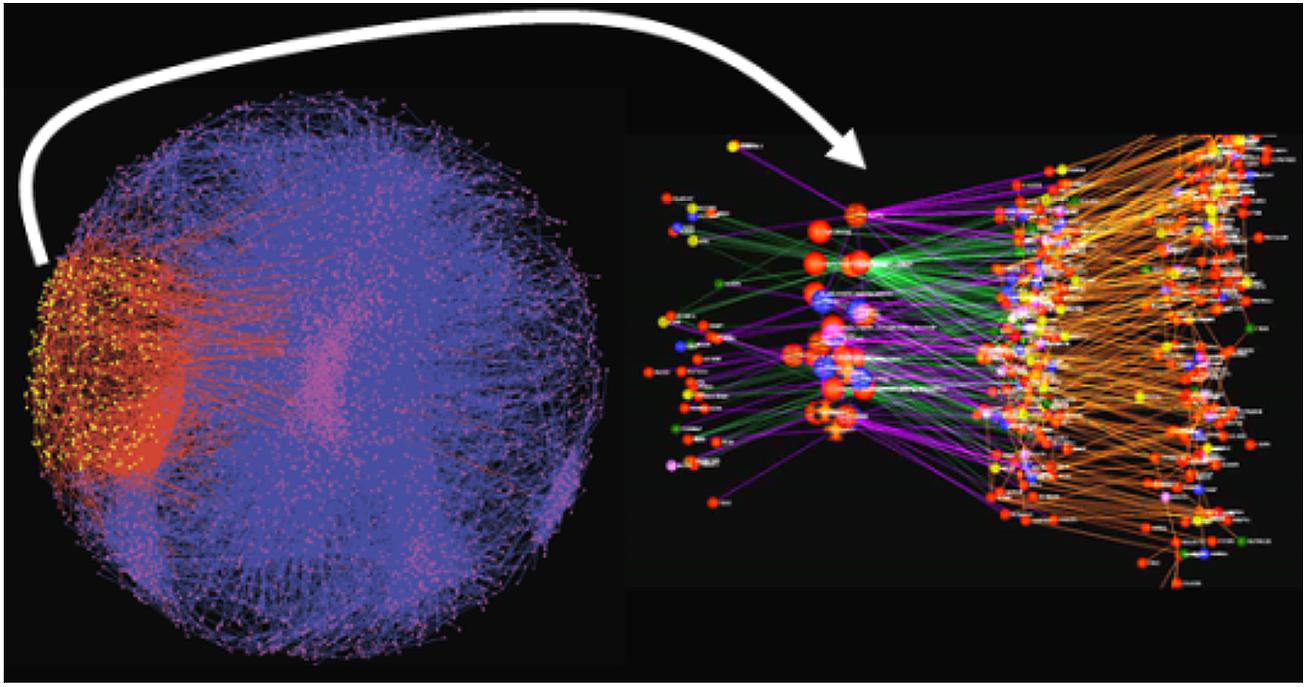
FIGURE 3. Inflammatory bowel disease-associated gene set projected onto a predictive network model. The yellow nodes and red edges indicate the sub-network most significantly enriched for the signature gene set. The right panel is an expanded view of the impacted sub-network. The larger nodes represent key drivers that are >10-fold enriched for genes associated with IBD, whereas the sub-network itself is roughly 4-fold enriched.
Application of multiscale networks to high throughput screening
Multiscale models defining networks for a given neurological disease can be used to construct gene expression assays for high throughput screening (Figure 2). The effect of any given perturbagen, whether a small molecule compound, natural product, RNAi-based construct and so on, on a specific network of interest can be assayed directly in cell-based systems (such as hiPSC-derived neurons derived from SZ or AD patients), which more accurately reflect the states of networks underlying disease. Complementing the network-based screens that use molecular network state as a readout, are cellular phenotyping assays that also aid in the linking of molecular states of disease to pathophysiological states. Screening carried out in this way can lead to the rapid identification of compounds that affect disease networks in favorable ways, while simultaneously identifying compounds that hit networks associated with toxicity or other adverse events (Figure 2). In this way, compounds can be identified that target specific subtypes of disease without targeting networks that can lead to toxicity or adverse events.
Network constructs can be used to inform on molecular responses to perturbations with small molecules or other perturbagens, where the networks enable a direct link between molecular biology and pathophysiology. In a high throughput screening context, where transcription or other molecular features are the readout, in addition to cell-based phenotypes, the aim is to identify molecular responses to the perturbagens that are predicted to associate with physiological changes in favorable directions, while simultaneously being predicted to have a minimal adverse event profile. Networks can be integrated with molecular screening data to identify those perturbagens from the screen that have similar mechanisms of action, that impact key disease related processes, or that impact key driver genes of diseases of interest. We and others have previously made use of network models to inform on perturbagen-induced molecular signatures as a means of predicting and validating the impact a given gene or genes had on molecular states and the pathophysiology of disease-associated with those states (Mehrabian et al., 2005; Schadt et al., 2005; Chen et al., 2008; Zhu et al., 2008, 2012).
This type of approach has been more generally applied to repurpose existing drugs for novel indications. For example, IBD signatures were derived from surgical specimens and intersected with Connectivity Map data representing transcriptional readouts across a number of cell lines in response to treatment with many 100s of drugs using a novel pattern-matching algorithm (Dudley et al., 2011). From this search the anticonvulsant drug topiramate was identified and experimentally validated as a novel treatment for IBD (Dudley et al., 2011). Topiramate has primary indications for seizure disorders and no history of efficacious use for IBD or other inflammatory diseases. Using a chemically induced (2,4,6-trinitrobenzenesulfonic acid) rodent model of IBD to evaluate the activity of topiramate administered in the presence of an IBD phenotype, a statistically significant reduction in gross pathophysiological and histopathological measures of severity of the induced IBD phenotype in the population of animals receiving topiramate compared to untreated vehicle controls was observed.
Returning one last time to novel successes in cancer therapeutics, in a separate study, this same computational drug repurposing strategy was applied to transcriptional profiles of small cell lung cancer (SCLC; Jahchan et al., 2013). The system identified imipramine (a tricyclic antidepressant), bepridil (a calcium channel blocker), and promethazine (a phenothiazine antihistamine) as having anti-SCLC activity. These predictions were experimentally validated, demonstrating anti-SCLC activity across a number of in vitro and in vivo experiments using human and animal model systems. The same approach was used to identify and validate anti-neoplastic activity of the anti-ulcer drug cimetidine against NSCLC (Sirota et al., 2011).
While low cost sequencing assays have provided an unprecedented amount of data on genetic loci and variants associated with common syndromic disorders, these loci by themselves are not sufficient to garner the most informative insights into disease mechanisms upon which new drug discovery efforts may be founded. Again, analogous to the oncology field, multiscale predictive models now provide a computational platform that has the potential to significantly improve the success rate of neurological drug discovery if integrated appropriately.
Conclusion
After considering the frequent clinical failures for novel drugs together with the novel cancer research paradigms that have led to improved drug discover, here we have discussed two transformational technologies that have and will continue to provide unprecedented insights into molecular mechanisms associated with complex diseases of the brain, and thereby de-risk drug discovery. First is the ability to generate on a large-scale hiPSCs derived neurons; this cellular model recapitulates disease mechanisms in vitro, thereby enabling studies of: disease mechanisms, genotype-phenotype relationships, and causative or risk factors. Furthermore, hiPSC derived neurons offer the ability to engineer assays that have direct relevance to disease biology, analogous to proliferation assays on cancer cell lines. Second, advances in biotechnology have enabled very low cost sequencing of nucleic acids, leading to big data and identification of large ensembles of gene loci and variants, which necessitated a revolution in computing and big data analytics to more comprehensively integrate very large-scale data and infer predictive models from it. Using these methods, we have already identified novel insights into disease mechanisms in AD, which are now the focus of drug discovery in small and large pharmaceutical companies; we expect that a similar approach will yield a better understanding into the mechanisms and treatment of psychiatric disorders, such as SZ. Together, these technologies can spawn a new generation of drug screening paradigms where screening assays (either in vitro or in silico) that capture far more of the relevant biology for common human diseases may be performed. The prediction is that such screens will significantly improve discovery of targets and drugs, as well as diagnostic tests, to make personalized therapies a reality for CNS disorders.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The Schadt Laboratory is supported in part by NIH grants R01 MH097276 (NIMH) and RO1 AG046170 (NIA) Kristen J. Brennand is a New York Stem Cell Robertson Investigator. The Brennand Laboratory is supported by a Brain and Behavior Young Investigator Grant, NIH grant R01 MH101454 (NIMH) and New York Stem Cell Foundation.
References
Adams, D. H., Kinon, B. J., Baygani, S., Millen, B. A., Velona, I., Kollack-Walker, S.,et al. (2013). A long-term, phase 2, multicenter, randomized, open-label, comparative safety study of pomaglumetad methionil (LY2140023 monohydrate) versus atypical antipsychotic standard of care in patients with schizophrenia. BMC Psychiatry 13:143. doi: 10.1186/1471-244X-13-143
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Adams, D. H., Zhang, L., Millen, B. A., Kinon, B. J., and Gomez, J. C. (2014). Pomaglumetad methionil (LY2140023 monohydrate) and aripiprazole in patients with schizophrenia: a phase 3, multicenter, double-blind comparison. Schizophr. Res. Treatment 2014:758212. doi: 10.1155/2014/758212
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Alexandrov, L. B., Nik-Zainal, S., Wedge, D. C., Aparicio, S. A., Behjati, S., Biankin, A. V.,et al. (2013). Signatures of mutational processes in human cancer. Nature 500, 415–421. doi: 10.1038/nature12477
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Alzheimer’s Association, A. S. (2014). Changing the Trajectory of Alzheimer’s Disease: A National Imperative. Available at: http://www.alz.org/documents_custom/trajectory.pdf
Ambasudhan, R., Talantova, M., Coleman, R., Yuan, X., Zhu, S., Lipton, S. A.,et al. (2011). Direct reprogramming of adult human fibroblasts to functional neurons under defined conditions. Cell Stem Cell 9, 113–118. doi: 10.1016/j.stem.2011.07.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ananiev, G., Williams, E. C., Li, H., and Chang, Q. (2011). Isogenic pairs of wild type and mutant Induced Pluripotent Stem Cell (iPSC) lines from Rett Syndrome patients as in vitro disease model. PLoS ONE 6:e25255. doi: 10.1371/journal.pone.0025255
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Astsaturov, I., Ratushny, V., Sukhanova, A., Einarson, M. B., Bagnyukova, T., Zhou, Y.,et al. (2010). Synthetic lethal screen of an EGFR-centered network to improve targeted therapies. Sci. Signal. 3:ra67. doi: 10.1126/scisignal.2001083
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Baek, K. H., Zaslavsky, A., Lynch, R. C., Britt, C., Okada, Y., Siarey, R. J.,et al. (2009). Down’s syndrome suppression of tumour growth and the role of the calcineurin inhibitor DSCR1. Nature 459, 1126–1130. doi: 10.1038/nature08062
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bajrami, I., Kigozi, A., Van Weverwijk, A., Brough, R., Frankum, J., Lord, C. J.,et al. (2012). Synthetic lethality of PARP and NAMPT inhibition in triple-negative breast cancer cells. EMBO Mol. Med. 4, 1087–1096. doi: 10.1002/emmm.201201250
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Baoi, J. (2014). Prevalence of autism spectrum disorder among children aged 8 years. MMWR Morb. Mortal. Wkly. Rep. 63, 1–21.
Barabasi, A. L., and Oltvai, Z. N. (2004). Network biology: understanding the cell’s functional organization. Nat. Rev. Genet. 5, 101–113. doi: 10.1038/nrg1272
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Barber, L. J., Sandhu, S., Chen, L., Campbell, J., Kozarewa, I., Fenwick, K.,et al. (2013). Secondary mutations in BRCA2 associated with clinical resistance to a PARP inhibitor. J. Pathol. 229, 422–429. doi: 10.1002/path.4140
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Barretina, J., Caponigro, G., Stransky, N., Venkatesan, K., Margolin, A. A., Kim, S.,et al. (2012). The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607. doi: 10.1038/nature11003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Berman, D. M., Karhadkar, S. S., Hallahan, A. R., Pritchard, J. I., Eberhart, C. G., Watkins, D. N.,et al. (2002). Medulloblastoma growth inhibition by hedgehog pathway blockade. Science 297, 1559–1561. doi: 10.1126/science.1073733
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brennand, K. J., Silvas, J., Kim, Y., Tran, N., Simone, A., Ladran, I.,et al. (2014). Phenotypic differences in hiPSC NPCs derived from patients with schizophrenia. Mol. Psychiatry doi: 10.1038/mp.2014.22 [Epub ahead of print].
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brennand, K. J., Simone, A., Jou, J., Gelboin-Burkhart, C., Tran, N., Sangar, S.,et al. (2011). Modelling schizophrenia using human induced pluripotent stem cells. Nature 473, 221–225. doi: 10.1038/nature09915
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bystrykh, L., Weersing, E., Dontje, B., Sutton, S., Pletcher, M. T., Wiltshire, T.,et al. (2005). Uncovering regulatory pathways that affect hematopoietic stem cell function using ‘genetical genomics.’ Nat. Genet. 37, 225–232. doi: 10.1038/ng1497
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cagan, R. (2013). Bench to bedside with fruit flies: an interview with Ross Cagan. Dis. Model. Mech. 6, 567–569. doi: 10.1242/dmm.012443
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Caiazzo, M., Dell’Anno, M. T., Dvoretskova, E., Lazarevic, D., Taverna, S., Leo, D.,et al. (2011). Direct generation of functional dopaminergic neurons from mouse and human fibroblasts. Nature 476, 224–227. doi: 10.1038/nature10284
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Califano, A., Butte, A. J., Friend, S., Ideker, T., and Schadt, E. (2012). Leveraging models of cell regulation and GWAS data in integrative network-based association studies. Nat. Genet. 44, 841–847. doi: 10.1038/ng.2355
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cancer Genome Atlas Research Network. (2013). Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 499, 43–49. doi: 10.1038/nature12222
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cancer Genome Atlas Research Network. (2014). Comprehensive molecular characterization of gastric adenocarcinoma. Nature 513, 202–209. doi: 10.1038/nature13480
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cancer Genome Atlas Research Network, Kandoth, C., Schultz, N., Cherniack, A. D., Akbani, R., Liu, Y.,et al. (2013a). Integrated genomic characterization of endometrial carcinoma. Nature 497, 67–73. doi: 10.1038/nature12113
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cancer Genome Atlas Research Network, Weinstein, J. N., Collisson, E. A., Mills, G. B., Shaw, K. R., Ozenberger, B. A.,et al. (2013b). The cancer genome atlas pan-cancer analysis project. Nat. Genet. 45, 1113–1120. doi: 10.1038/ng.2764
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chambers, S. M., Fasano, C. A., Papapetrou, E. P., Tomishima, M., Sadelain, M., and Studer, L. (2009). Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 27, 275–280. doi: 10.1038/nbt.1529
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chanda, S., Marro, S., Wernig, M., and Sudhof, T. C. (2013). Neurons generated by direct conversion of fibroblasts reproduce synaptic phenotype caused by autism-associated neuroligin-3 mutation. Proc. Natl. Acad. Sci. U.S.A. 110, 16622–16627. doi: 10.1073/pnas.1316240110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, Y., Zhu, J., Lum, P. Y., Yang, X., Pinto, S., MacNeil, D. J.,et al. (2008). Variations in DNA elucidate molecular networks that cause disease. Nature 452, 429–435. doi: 10.1038/nature06757
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cheung, A. Y., Horvath, L. M., Grafodatskaya, D., Pasceri, P., Weksberg, R., Hotta, A.,et al. (2011a). Isolation of MECP2-null Rett Syndrome patient hiPS cells and isogenic controls through X-chromosome inactivation. Hum. Mol. Genet. 20, 2103–2115. doi: 10.1093/hmg/ddr093
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cheung, H. W., Cowley, G. S., Weir, B. A., Boehm, J. S., Rusin, S., Scott, J. A.,et al. (2011b). Systematic investigation of genetic vulnerabilities across cancer cell lines reveals lineage-specific dependencies in ovarian cancer. Proc. Natl. Acad. Sci. U.S.A. 108, 12372–12377. doi: 10.1073/pnas.1109363108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cobleigh, M. A., Vogel, C. L., Tripathy, D., Robert, N. J., Scholl, S., Fehrenbacher, L.,et al. (1999). Multinational study of the efficacy and safety of humanized anti-HER2 monoclonal antibody in women who have HER2-overexpressing metastatic breast cancer that has progressed after chemotherapy for metastatic disease. J. Clin. Oncol. 17, 2639–2648.
Corcoran, R. B., Ebi, H., Turke, A. B., Coffee, E. M., Nishino, M., Cogdill, A. P.,et al. (2012). EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2, 227–235. doi: 10.1158/2159-8290.CD-11-0341
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cortes, J. E., Kantarjian, H., Shah, N. P., Bixby, D., Mauro, M. J., Flinn, I.,et al. (2012). Ponatinib in refractory Philadelphia chromosome-positive leukemias. N. Engl. J. Med. 367, 2075–2088. doi: 10.1056/NEJMoa1205127
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cummings, J. L., Morstorf, T., and Zhong, K. (2014). Alzheimer’s disease drug-development pipeline: few candidates, frequent failures. Alzheimers Res. Ther. 6, 37. doi: 10.1186/alzrt269
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dar, A. C., Das, T. K., Shokat, K. M., and Cagan, R. L. (2012). Chemical genetic discovery of targets and anti-targets for cancer polypharmacology. Nature 486, 80–84. doi: 10.1038/nature11127
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Das, T. K., and Cagan, R. L. (2013). A Drosophila approach to thyroid cancer therapeutics. Drug Discov. Today Technol. 10, e65–e71. doi: 10.1016/j.ddtec.2012.09.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Das Thakur, M., and Stuart, D. D. (2013). The evolution of melanoma resistance reveals therapeutic opportunities. Cancer Res. 73, 6106–6110. doi: 10.1158/0008-5472.CAN-13-1633
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Davare, M. A., Saborowski, A., Eide, C. A., Tognon, C., Smith, R. L., Elferich, J.,et al. (2013). Foretinib is a potent inhibitor of oncogenic ROS1 fusion proteins. Proc. Natl. Acad. Sci. U.S.A. 110, 19519–19524. doi: 10.1073/pnas.1319583110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Demetri, G. D., von Mehren, M., Blanke, C. D., Van den Abbeele, A. D., Eisenberg, B., Roberts, P. J.,et al. (2002). Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N. Engl. J. Med. 347, 472–480. doi: 10.1056/NEJMoa020461
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Demjaha, A., Egerton, A., Murray, R. M., Kapur, S., Howes, O. D., Stone, J. M.,et al. (2013). Antipsychotic treatment resistance in schizophrenia associated with elevated glutamate levels but normal dopamine function. Biol. Psychiatry 75, e11–e13. doi: 10.1016/j.biopsych.2013.06.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dermime, S., Grignani, F., Clerici, M., Nervi, C., Sozzi, G., Talamo, G. P.,et al. (1993). Occurrence of resistance to retinoic acid in the acute promyelocytic leukemia cell line NB4 is associated with altered expression of the pml/RAR alpha protein. Blood 82, 1573–1577.
Dimos, J. T., Rodolfa, K. T., Niakan, K. K., Weisenthal, L. M., Mitsumoto, H., Chung, W.,et al. (2008). Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science 321, 1218–1221. doi: 10.1126/science.1158799
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Druker, B. J. (2008). Translation of the Philadelphia chromosome into therapy for CML. Blood 112, 4808–4817. doi: 10.1182/blood-2008-07-077958
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dudley, J. T., Sirota, M., Shenoy, M., Pai, R. K., Roedder, S., Chiang, A. P.,et al. (2011). Computational repositioning of the anticonvulsant topiramate for inflammatory bowel disease. Sci. Transl. Med. 3:96ra76. doi: 10.1126/scitranslmed.3002648
Ebert, A. D., Yu, J., Rose, F. F. Jr., Mattis, V. B., Lorson, C. L., Thomson, J. A.,et al. (2009). Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature 457, 277–280. doi: 10.1038/nature07677
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Eifert, C., and Powers, R. S. (2012). From cancer genomes to oncogenic drivers, tumour dependencies and therapeutic targets. Nat. Rev. Cancer 12, 572–578. doi: 10.1038/nrc3299
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Emilsson, V., Thorleifsson, G., Zhang, B., Leonardson, A. S., Zink, F., Zhu, J.,et al. (2008). Genetics of gene expression and its effect on disease. Nature 452, 423–428. doi: 10.1038/nature06758
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Espuny-Camacho, I., Michelsen, K. A., Gall, D., Linaro, D., Hasche, A., Bonnefont, J.,et al. (2013). Pyramidal neurons derived from human pluripotent stem cells integrate efficiently into mouse brain circuits in vivo. Neuron 77, 440–456. doi: 10.1016/j.neuron.2012.12.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Farmer, H., McCabe, N., Lord, C. J., Tutt, A. N., Johnson, D. A., Richardson, T. B.,et al. (2005). Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921. doi: 10.1038/nature03445
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fong, P. C., Boss, D. S., Yap, T. A., Tutt, A., Wu, P., Mergui-Roelvink, M.,et al. (2009). Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 361, 123–134. doi: 10.1056/NEJMoa0900212
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Friboulet, L., Li, N., Katayama, R., Lee, C. C., Gainor, J. F., Crystal, A. S.,et al. (2014). The ALK inhibitor ceritinib overcomes crizotinib resistance in non-small cell lung cancer. Cancer Discov. 4, 662–673. doi: 10.1158/2159-8290.CD-13-0846
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fromer, M., Pocklington, A., Kavanagh, D., Williams, H., Dwyer, S., Gormley, P.,et al. (2014). De novo mutations in schizophrenia implicate synaptic networks. Nature 506, 179–184. doi: 10.1038/nature12929
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Garnett, M. J., Edelman, E. J., Heidorn, S. J., Greenman, C. D., Dastur, A., Lau, K. W.,et al. (2012). Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 483, 570–575. doi: 10.1038/nature11005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ghazalpour, A., Doss, S., Sheth, S. S., Ingram-Drake, L. A., Schadt, E. E., Lusis, A. J.,et al. (2005). Genomic analysis of metabolic pathway gene expression in mice. Genome Biol. 6:R59. doi: 10.1186/gb-2005-6-7-r59
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Greenawalt, D. M., Dobrin, R., Chudin, E., Hatoum, I. J., Suver, C., Beaulaurier, J.,et al. (2011). A survey of the genetics of stomach, liver, and adipose gene expression from a morbidly obese cohort. Genome Res. 21, 1008–1016. doi: 10.1101/gr.112821.110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hall-Jackson, C. A., Eyers, P. A., Cohen, P., Goedert, M., Boyle, F. T., Hewitt, N.,et al. (1999). Paradoxical activation of Raf by a novel Raf inhibitor. Chem. Biol. 6, 559–568. doi: 10.1016/S1074-5521(99)80088-X
Hashimoto-Torii, K., Torii, M., Fujimoto, M., Nakai, A., El Fatimy, R., Mezger, V.,et al. (2014). Roles of heat shock factor 1 in neuronal response to fetal environmental risks and its relevance to brain disorders. Neuron 82, 560–572. doi: 10.1016/j.neuron.2014.03.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hata, A. N., Yeo, A., Faber, A. C., Lifshits, E., Chen, Z., Cheng, K. A.,et al. (2014). Failure to induce apoptosis via BCL-2 family proteins underlies lack of efficacy of combined MEK and PI3K inhibitors for KRAS mutant lung cancers. Cancer Res. 74, 3146–3156. doi: 10.1158/0008-5472.CAN-13-3728
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hatzivassiliou, G., Song, K., Yen, I., Brandhuber, B. J., Anderson, D. J., Alvarado, R.,et al. (2010). RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 464, 431–435. doi: 10.1038/nature08833
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hebert, L. E., Weuve, J., Scherr, P. A., and Evans, D. A. (2013). Alzheimer disease in the United States (2010-2050) estimated using the 2010 census. Neurology 80, 1778–1783. doi: 10.1212/WNL.0b013e31828726f5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Heidorn, S. J., Milagre, C., Whittaker, S., Nourry, A., Niculescu-Duvas, I., Dhomen, N.,et al. (2010). Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell 140, 209–221. doi: 10.1016/j.cell.2009.12.040
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hirabayashi, S., Baranski, T. J., and Cagan, R. L. (2013). Transformed Drosophila cells evade diet-mediated insulin resistance through wingless signaling. Cell 154, 664–675. doi: 10.1016/j.cell.2013.06.030
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hotta, A., Cheung, A. Y., Farra, N., Garcha, K., Chang, W. Y., Pasceri, P.,et al. (2009). EOS lentiviral vector selection system for human induced pluripotent stem cells. Nat. Protoc. 4, 1828–1844. doi: 10.1038/nprot.2009.201
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Huang, S., Holzel, M., Knijnenburg, T., Schlicker, A., Roepman, P., McDermott, U.,et al. (2012). MED12 controls the response to multiple cancer drugs through regulation of TGF-beta receptor signaling. Cell 151, 937–950. doi: 10.1016/j.cell.2012.10.035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ideker, T., Ozier, O., Schwikowski, B., and Siegel, A. F. (2002). Discovering regulatory and signalling circuits in molecular interaction networks. Bioinformatics 18(Suppl. 1), S233–S240. doi: 10.1093/bioinformatics/18.suppl_1.S233
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jahchan, N. S., Dudley, J. T., Mazur, P. K., Flores, N., Yang, D., Palmerton, A.,et al. (2013). A drug repositioning approach identifies tricyclic antidepressants as inhibitors of small cell lung cancer and other neuroendocrine tumors. Cancer Discov. 3, 1364–1377. doi: 10.1158/2159-8290.CD-13-0183
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jansen, R., Yu, H., Greenbaum, D., Kluger, Y., Krogan, N. J., Chung, S.,et al. (2003). A Bayesian networks approach for predicting protein-protein interactions from genomic data. Science 302, 449–453. doi: 10.1126/science.1087361
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Joseph, E. W., Pratilas, C. A., Poulikakos, P. I., Tadi, M., Wang, W., Taylor, B. S.,et al. (2010). The RAF inhibitor PLX4032 inhibits ERK signaling and tumor cell proliferation in a V600E BRAF-selective manner. Proc. Natl. Acad. Sci. U.S.A. 107, 14903–14908. doi: 10.1073/pnas.1008990107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jostins, L., Ripke, S., Weersma, R. K., Duerr, R. H., McGovern, D. P., Hui, K. Y.,et al. (2012). Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491, 119–124. doi: 10.1038/nature11582
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kandoth, C., McLellan, M. D., Vandin, F., Ye, K., Niu, B., Lu, C.,et al. (2013). Mutational landscape and significance across 12 major cancer types. Nature 502, 333–339. doi: 10.1038/nature12634
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kessler, R. M., Woodward, N. D., Riccardi, P., Li, R., Ansari, M. S., Anderson, S.,et al. (2009). Dopamine D2 receptor levels in striatum, thalamus, substantia nigra, limbic regions, and cortex in schizophrenic subjects. Biol. Psychiatry 65, 1024–1031. doi: 10.1016/j.biopsych.2008.12.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kidd, B. A., Peters, L. A., Schadt, E. E., and Dudley, J. T. (2014). Unifying immunology with informatics and multiscale biology. Nat. Immunol. 15, 118–127. doi: 10.1038/ni.2787
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kola, I., and Landis, J. (2004). Can the pharmaceutical industry reduce attrition rates? Nat. Rev. Drug Discov. 3, 711–715. doi: 10.1038/nrd1470
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Konieczkowski, D. J., Johannessen, C. M., Abudayyeh, O., Kim, J. W., Cooper, Z. A., Piris, A.,et al. (2014). A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer Discov. 4, 745–748. doi: 10.1158/2159-8290.CD-13-0424
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kriks, S., Shim, J. W., Piao, J., Ganat, Y. M., Wakeman, D. R., Xie, Z.,et al. (2011). Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature 480, 547–551. doi: 10.1038/nature10648
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kumar, M. S., Hancock, D. C., Molina-Arcas, M., Steckel, M., East, P., Diefenbacher, M.,et al. (2012). The GATA2 transcriptional network is requisite for RAS oncogene-driven non-small cell lung cancer. Cell 149, 642–655. doi: 10.1016/j.cell.2012.02.059
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lamba, S., Russo, M., Sun, C., Lazzari, L., Cancelliere, C., Grernrum, W.,et al. (2014). RAF suppression synergizes with MEK Inhibition in KRAS Mutant Cancer Cells. Cell Rep. 8, 1475–1483. doi: 10.1016/j.celrep.2014.07.033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, G., Papapetrou, E. P., Kim, H., Chambers, S. M., Tomishima, M. J., Fasano, C. A.,et al. (2009). Modelling pathogenesis and treatment of familial dysautonomia using patient-specific iPSCs. Nature 461, 402–406. doi: 10.1038/nature08320
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, I., Date, S. V., Adai, A. T., and Marcotte, E. M. (2004). A probabilistic functional network of yeast genes. Science 306, 1555–1558. doi: 10.1126/science.1099511
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, S. H., DeCandia, T. R., Ripke, S., Yang, J., Sullivan, P. F., Goddard, M. E.,et al. (2012). Estimating the proportion of variation in susceptibility to schizophrenia captured by common SNPs. Nat. Genet. 44, 247–250. doi: 10.1038/ng.1108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lito, P., Pratilas, C. A., Joseph, E. W., Tadi, M., Halilovic, E., Zubrowski, M.,et al. (2012). Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell 22, 668–682. doi: 10.1016/j.ccr.2012.10.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lito, P., Saborowski, A., Yue, J., Solomon, M., Joseph, E., Gadal, S.,et al. (2014). Disruption of CRAF-mediated MEK activation is required for effective MEK inhibition in KRAS mutant tumors. Cancer Cell 25, 697–710. doi: 10.1016/j.ccr.2014.03.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lord, C. J., and Ashworth, A. (2013). Mechanisms of resistance to therapies targeting BRCA-mutant cancers. Nat. Med. 19, 1381–1388. doi: 10.1038/nm.3369
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lum, P. Y., Chen, Y., Zhu, J., Lamb, J., Melmed, S., Wang, S.,et al. (2006). Elucidating the murine brain transcriptional network in a segregating mouse population to identify core functional modules for obesity and diabetes. J. Neurochem. 97(Suppl. 1), 50–62. doi: 10.1111/j.1471-4159.2006.03661.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Luo, J., Emanuele, M. J., Li, D., Creighton, C. J., Schlabach, M. R., Westbrook, T. F.,et al. (2009a). A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell 137, 835–848. doi: 10.1016/j.cell.2009.05.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Luo, J., Solimini, N. L., and Elledge, S. J. (2009b). Principles of cancer therapy: oncogene and non-oncogene addiction. Cell 136, 823–837. doi: 10.1016/j.cell.2009.02.024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Maherali, N., Sridharan, R., Xie, W., Utikal, J., Eminli, S., Arnold, K.,et al. (2007). Directly reprogrammed fibroblasts show global epigenetic remodeling and widespread tissue contribution. Cell Stem Cell 1, 55–70. doi: 10.1016/j.stem.2007.05.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mair, B., Kubicek, S., and Nijman, S. M. (2014). Exploiting epigenetic vulnerabilities for cancer therapeutics. Trends Pharmacol. Sci. 35, 136–145. doi: 10.1016/j.tips.2014.01.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Malhotra, D., and Sebat, J. (2012). CNVs: harbingers of a rare variant revolution in psychiatric genetics. Cell 148, 1223–1241. doi: 10.1016/j.cell.2012.02.039
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Marchetto, M. C., Carromeu, C., Acab, A., Yu, D., Yeo, G. W., Mu, Y.,et al. (2010). A model for neural development and treatment of rett syndrome using human induced pluripotent stem cells. Cell 143, 527–539. doi: 10.1016/j.cell.2010.10.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mariani, J., Simonini, M. V., Palejev, D., Tomasini, L., Coppola, G., Szekely, A. M.,et al. (2012). Modeling human cortical development in vitro using induced pluripotent stem cells. Proc. Natl. Acad. Sci. U.S.A. 109, 12770–12775. doi: 10.1073/pnas.1202944109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Maroof, A. M., Keros, S., Tyson, J. A., Ying, S. W., Ganat, Y. M., Merkle, F. T.,et al. (2013). Directed differentiation and functional maturation of cortical interneurons from human embryonic stem cells. Cell Stem Cell 12, 559–572. doi: 10.1016/j.stem.2013.04.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McCabe, N., Lord, C. J., Tutt, A. N., Martin, N. M., Smith, G. C., and Ashworth, A. (2005). BRCA2-deficient CAPAN-1 cells are extremely sensitive to the inhibition of Poly (ADP-Ribose) polymerase: an issue of potency. Cancer Biol. Ther. 4, 934–936. doi: 10.4161/cbt.4.9.2141
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McCarroll, S. A., Kuruvilla, F. G., Korn, J. M., Cawley, S., Nemesh, J., Wysoker, A.,et al. (2008). Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat. Genet. 40, 1166–1174. doi: 10.1038/ng.238
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McDermott, U., Iafrate, A. J., Gray, N. S., Shioda, T., Classon, M., Maheswaran, S.,et al. (2008). Genomic alterations of anaplastic lymphoma kinase may sensitize tumors to anaplastic lymphoma kinase inhibitors. Cancer Res. 68, 3389–3395. doi: 10.1158/0008-5472.CAN-07-6186
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mehrabian, M., Allayee, H., Stockton, J., Lum, P. Y., Drake, T. A., Castellani, L. W.,et al. (2005). Integrating genotypic and expression data in a segregating mouse population to identify 5-lipoxygenase as a susceptibility gene for obesity and bone traits. Nat. Genet. 37, 1224–1233. doi: 10.1038/ng1619
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Meissner, A., Wernig, M., and Jaenisch, R. (2007). Direct reprogramming of genetically unmodified fibroblasts into pluripotent stem cells. Nat. Biotechnol. 25, 1177–1181. doi: 10.1038/nbt1335
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Millstein, J., Zhang, B., Zhu, J., and Schadt, E. E. (2009). Disentangling molecular relationships with a causal inference test. BMC Genet. 10:23. doi: 10.1186/1471-2156-10-23
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Muellner, M. K., Uras, I. Z., Gapp, B. V., Kerzendorfer, C., Smida, M., Lechtermann, H.,et al. (2011). A chemical-genetic screen reveals a mechanism of resistance to PI3K inhibitors in cancer. Nat. Chem. Biol. 7, 787–793. doi: 10.1038/nchembio.695
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Muller, F. L., Colla, S., Aquilanti, E., Manzo, V. E., Genovese, G., Lee, J.,et al. (2012). Passenger deletions generate therapeutic vulnerabilities in cancer. Nature 488, 337–342. doi: 10.1038/nature11331
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
National Research Council (US) Committee on A Framework for Developing a New Taxonomy of Disease. (2011). Toward Precision Medicine : Building a Knowledge Network for Biomedical Research and a New Taxonomy of Disease. Washington, DC, National Academies Press.
Ng, M. Y., Levinson, D. F., Faraone, S. V., Suarez, B. K., DeLisi, L. E., Arinami, T.,et al. (2009). Meta-analysis of 32 genome-wide linkage studies of schizophrenia. Mol. Psychiatry 14, 774–785. doi: 10.1038/mp.2008.135
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nguyen, H. N., Byers, B., Cord, B., Shcheglovitov, A., Byrne, J., Gujar, P.,et al. (2011). LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell 8, 267–280. doi: 10.1016/j.stem.2011.01.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nicholas, C. R., Chen, J., Tang, Y., Southwell, D. G., Chalmers, N., Vogt, D.,et al. (2013). Functional maturation of hPSC-derived forebrain interneurons requires an extended timeline and mimics human neural development. Cell Stem Cell 12, 573–586. doi: 10.1016/j.stem.2013.04.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Niederst, M. J., and Engelman, J. A. (2013). Bypass mechanisms of resistance to receptor tyrosine kinase inhibition in lung cancer. Sci. Signal. 6:re6. doi: 10.1126/scisignal.2004652
O’Hare, T., Deininger, M. W., Eide, C. A., Clackson, T., and Druker, B. J. (2011). Targeting the BCR-ABL signaling pathway in therapy-resistant Philadelphia chromosome-positive leukemia. Clin. Cancer Res. 17, 212–221. doi: 10.1158/1078-0432.CCR-09-3314
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Paez, J. G., Janne, P. A., Lee, J. C., Tracy, S., Greulich, H., Gabriel, S.,et al. (2004). EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 304, 1497–1500. doi: 10.1126/science.1099314
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pang, Z. P., Yang, N., Vierbuchen, T., Ostermeier, A., Fuentes, D. R., Yang, T. Q.,et al. (2011). Induction of human neuronal cells by defined transcription factors. Nature 476, 220–223. doi: 10.1038/nature10202
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Park, I. H., Arora, N., Huo, H., Maherali, N., Ahfeldt, T., Shimamura, A.,et al. (2008). Disease-specific induced pluripotent stem cells. Cell 134, 877–886. doi: 10.1016/j.cell.2008.07.041
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pasca, S. P., Portmann, T., Voineagu, I., Yazawa, M., Shcheglovitov, A., Pasca, A. M.,et al. (2011). Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat. Med. 17, 1657–1662. doi: 10.1038/nm.2576
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Patil, S. T., Zhang, L., Martenyi, F., Lowe, S. L., Jackson, K. A., Andreev, B. V.,et al. (2007). Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: a randomized Phase 2 clinical trial. Nat. Med. 13, 1102–1107. doi: 10.1038/nm1632
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Paulsen, B. D., Maciel, R. D., Galina, A., da Silveira, M. S., Souza, C. D., Drummond, H.,et al. (2011). Altered oxygen metabolism associated to neurogenesis of induced pluripotent stem cells derived from a schizophrenic patient. Cell Transplant. 21, 1547–1559. doi: 10.3727/096368911X600957
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Poels, E. M., Kegeles, L. S., Kantrowitz, J. T., Slifstein, M., Javitt, D. C., Lieberman, J. A.,et al. (2014). Imaging glutamate in schizophrenia: review of findings and implications for drug discovery. Mol. Psychiatry 19, 20–29. doi: 10.1038/mp.2013.136
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Poulikakos, P. I., Zhang, C., Bollag, G., Shokat, K. M., and Rosen, N. (2010). RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 464, 427–430. doi: 10.1038/nature08902
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Prahallad, A., Sun, C., Huang, S., Di Nicolantonio, F., Salazar, R., Zecchin, D.,et al. (2012). Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 483, 100–103. doi: 10.1038/nature10868
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Purcell, S., Moran, J., Fromer, M., Ruderfer, D., Solovieff, N., Roussos, P.,et al. (2014). A polygenic burden of rare disruptive mutations in schizophrenia. Nature 506, 185–190. doi: 10.1038/nature12975
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Purcell, S. M., Wray, N. R., Stone, J. L., Visscher, P. M., O’Donovan, M. C., Sullivan, P. F.,et al. (2009). Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 460, 748–752. doi: 10.1038/nature08185
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rehman, F. L., Lord, C. J., and Ashworth, A. (2010). Synthetic lethal approaches to breast cancer therapy. Nat. Rev. Clin. Oncol. 7, 718–724. doi: 10.1038/nrclinonc.2010.172
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Riabinska, A., Daheim, M., Herter-Sprie, G. S., Winkler, J., Fritz, C., Hallek, M.,et al. (2013). Therapeutic targeting of a robust non-oncogene addiction to PRKDC in ATM-defective tumors. Sci. Transl. Med. 5:189ra178. doi: 10.1126/scitranslmed.3005814
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rikova, K., Guo, A., Zeng, Q., Possemato, A., Yu, J., Haack, H.,et al. (2007). Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 131, 1190–1203. doi: 10.1016/j.cell.2007.11.025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ripke, S., O’Dushlaine, C., Chambert, K., Moran, J. L., Kahler, A. K., Akterin, S.,et al. (2013). Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat. Genet. 45, 1150–1159. doi: 10.1038/ng.2742
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ripke, S., Sanders, A. R., Kendler, K. S., Levinson, D. F., Sklar, P., Holmans, P. A.,et al. (2011). Genome-wide association study identifies five new schizophrenia loci. Nat. Genet. 43, 969–976. doi: 10.1038/ng.940
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Robert, C., Thomas, L., Bondarenko, I., O’Day, S., Jeffrey Weber, M. D., Garbe, C.,et al. (2011). Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N. Engl. J. Med. 364, 2517–2526. doi: 10.1056/NEJMoa1104621
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Robicsek, O., Karry, R., Petit, I., Salman-Kesner, N., Muller, F. J., Klein, E.,et al. (2013). Abnormal neuronal differentiation and mitochondrial dysfunction in hair follicle-derived induced pluripotent stem cells of schizophrenia patients. Mol. Psychiatry 18, 1067–1076. doi: 10.1038/mp.2013.67
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Roller, D. G., Axelrod, M., Capaldo, B. J., Jensen, K., Mackey, A., Weber, M. J.,et al. (2012). Synthetic lethal screening with small-molecule inhibitors provides a pathway to rational combination therapies for melanoma. Mol. Cancer Ther. 11, 2505–2515. doi: 10.1158/1535-7163.MCT-12-0461
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rudin, C. M., Hann, C. L., Laterra, J., Yauch, R. L., Callahan, C. A., Fu, L.,et al. (2009). Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N. Engl. J. Med. 361, 1173–1178. doi: 10.1056/NEJMoa0902903
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schadt, E. E. (2005). Exploiting naturally occurring DNA variation and molecular profiling data to dissect disease and drug response traits. Curr. Opin. Biotechnol. 16, 647–654. doi: 10.1016/j.copbio.2005.10.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schadt, E. E. (2009). Molecular networks as sensors and drivers of common human diseases. Nature 461, 218–223. doi: 10.1038/nature08454
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schadt, E. E., Friend, S. H., and Shaywitz, D. A. (2009). A network view of disease and compound screening. Nat. Rev. Drug Discov. 8, 286–295. doi: 10.1038/nrd2826
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schadt, E. E., Lamb, J., Yang, X., Zhu, J., Edwards, S., Guhathakurta, D.,et al. (2005). An integrative genomics approach to infer causal associations between gene expression and disease. Nat. Genet. 37, 710–717. doi: 10.1038/ng1589
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schadt, E. E., Molony, C., Chudin, E., Hao, K., Yang, X., Lum, P. Y.,et al. (2008). Mapping the genetic architecture of gene expression in human liver. PLoS Biol. 6:e107. doi: 10.1371/journal.pbio.0060107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schadt, E. E., Monks, S. A., Drake, T. A., Lusis, A. J., Che, N., Colinayo, V.,et al. (2003). Genetics of gene expression surveyed in maize, mouse and man. Nature 422, 297–302. doi: 10.1038/nature01434
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schizophrenia Working Group of the Psychiatric Genomics Consortium. (2014). Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427. doi: 10.1038/nature13595
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schmit, F., Utermark, T., Zhang, S., Wang, Q., Von, T., Roberts, T. M.,et al. (2014). PI3K isoform dependence of PTEN-deficient tumors can be altered by the genetic context. Proc. Natl. Acad. Sci. U.S.A. 111, 6395–6400. doi: 10.1073/pnas.1323004111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shi, J., Levinson, D. F., Duan, J., Sanders, A. R., Zheng, Y., Pe’er, I.,et al. (2009). Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature 460, 753–757. doi: 10.1038/nature08192
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shi, Y., Kirwan, P., Smith, J., Robinson, H. P., and Livesey, F. J. (2012). Human cerebral cortex development from pluripotent stem cells to functional excitatory synapses. Nat. Neurosci. 15, 477–486. doi: 10.1038/nn.3041
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sirota, M., Dudley, J. T., Kim, J., Chiang, A. P., Morgan, A. A., Sweet-Cordero, A.,et al. (2011). Discovery and preclinical validation of drug indications using compendia of public gene expression data. Sci. Transl. Med. 3:96ra77. doi: 10.1126/scitranslmed.3001318
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Soda, M., Choi, Y. L., Enomoto, M., Takada, S., Yamashita, Y., Ishikawa, S.,et al. (2007). Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 448, 561–566. doi: 10.1038/nature05945
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Soldner, F., Hockemeyer, D., Beard, C., Gao, Q., Bell, G. W., Cook, E. G.,et al. (2009). Parkinson’s disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell 136, 964–977. doi: 10.1016/j.cell.2009.02.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Solit, D. B., Garraway, L. A., Pratilas, C. A., Sawai, A., Getz, G., Basso, A.,et al. (2006). BRAF mutation predicts sensitivity to MEK inhibition. Nature 439, 358–362. doi: 10.1038/nature04304
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stefansson, H., Ophoff, R. A., Steinberg, S., Andreassen, O. A., Cichon, S., Rujescu, D.,et al. (2009). Common variants conferring risk of schizophrenia. Nature 460, 744–747. doi: 10.1038/nature08186
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Steinberg, S., de Jong, S., Mattheisen, M., Costas, J., Demontis, D., Jamain, S.,et al. (2014). Common variant at 16p11.2 conferring risk of psychosis. Mol. Psychiatry 19, 108–114. doi: 10.1038/mp.2012.157
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stratton, M. R. (2011). Exploring the genomes of cancer cells: progress and promise. Science 331, 1553–1558. doi: 10.1126/science.1204040
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sullivan, P. F., Kendler, K. S., and Neale, M. C. (2003). Schizophrenia as a complex trait: evidence from a meta-analysis of twin studies. Arch. Gen. Psychiatry 60, 1187–1192. doi: 10.1001/archpsyc.60.12.1187
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sun, C., Hobor, S., Bertotti, A., Zecchin, D., Huang, S., Galimi, F.,et al. (2014). Intrinsic resistance to MEK inhibition in KRAS mutant lung and colon cancer through transcriptional induction of ERBB3. Cell Rep. 7, 86–93. doi: 10.1016/j.celrep.2014.02.045
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Takahashi, K., Tanabe, K., Ohnuki, M., Narita, M., Ichisaka, T., Tomoda, K.,et al. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872. doi: 10.1016/j.cell.2007.11.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Takahashi, K., and Yamanaka, S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676. doi: 10.1016/j.cell.2006.07.024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Topalian, S. L., Sznol, M., McDermott, D. F., Kluger, H. M., Carvajal, R. D., Sharfman, W. H.,et al. (2014). Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J. Clin. Oncol. 32, 1020–1030. doi: 10.1200/JCO.2013.53.0105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Toyoshima, M., Howie, H. L., Imakura, M., Walsh, R. M., Annis, J. E., Chang, A. N.,et al. (2012). Functional genomics identifies therapeutic targets for MYC-driven cancer. Proc. Natl. Acad. Sci. U.S.A. 109, 9545–9550. doi: 10.1073/pnas.1121119109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Traer, E., Javidi-Sharifi, N., Agarwal, A., Dunlap, J., English, I., Martinez, J.,et al. (2014). Ponatinib overcomes FGF2-mediated resistance in CML patients without kinase domain mutations. Blood 123, 1516–1524. doi: 10.1182/blood-2013-07-518381
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tran, L. M., Zhang, B., Zhang, Z., Zhang, C., Xie, T., Lamb, J. R.,et al. (2011). Inferring causal genomic alterations in breast cancer using gene expression data. BMC Syst. Biol. 5:121. doi: 10.1186/1752-0509-5-121
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Turner, N. C., Lord, C. J., Iorns, E., Brough, R., Swift, S., Elliott, R.,et al. (2008). A synthetic lethal siRNA screen identifying genes mediating sensitivity to a PARP inhibitor. EMBO J. 27, 1368–1377. doi: 10.1038/emboj.2008.61
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
van’t Veer, L. J., Dai, H., van de Vijver, M. J., He, Y. D., Hart, A. A., Mao, M.,et al. (2002). Gene expression profiling predicts clinical outcome of breast cancer. Nature 415, 530–536. doi: 10.1038/415530a
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vierbuchen, T., Ostermeier, A., Pang, Z. P., Kokubu, Y., Sudhof, T. C., and Wernig, M. (2010). Direct conversion of fibroblasts to functional neurons by defined factors. Nature 463, 1035–1041. doi: 10.1038/nature08797
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Von Hoff, D. D., LoRusso, P. M., Rudin, C. M., Reddy, J. C., Yauch, R. L., Tibes, R.,et al. (2009). Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N. Engl. J. Med. 361, 1164–1172. doi: 10.1056/NEJMoa0905360
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, I. M., Zhang, B., Yang, X., Zhu, J., Stepaniants, S., Zhang, C.,et al. (2012). Systems analysis of eleven rodent disease models reveals an inflammatome signature and key drivers. Mol. Syst. Biol. 8, 594. doi: 10.1038/msb.2012.24
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, S., Yehya, N., Schadt, E. E., Wang, H., Drake, T. A., and Lusis, A. J. (2006). Genetic and genomic analysis of a fat mass trait with complex inheritance reveals marked sex specificity. PLoS Genet. 2:e15. doi: 10.1371/journal.pgen.0020015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Weinberger, D. R. (1987). Implications of normal brain development for the pathogenesis of schizophrenia. Arch. Gen. Psychiatry 44, 660–669. doi: 10.1001/archpsyc.1987.01800190080012
Wells, S. A. Jr., Gosnell, J. E., Gagel, R. F., Moley, J., Pfister, D., Sosa, J. A.,et al. (2010). Vandetanib for the treatment of patients with locally advanced or metastatic hereditary medullary thyroid cancer. J. Clin. Oncol. 28, 767–772. doi: 10.1200/JCO.2009.23.6604
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wen, L., Lu, Y. S., Zhu, X. H., Li, X. M., Woo, R. S., Chen, Y. J.,et al. (2010). Neuregulin 1 regulates pyramidal neuron activity via ErbB4 in parvalbumin-positive interneurons. Proc. Natl. Acad. Sci. U.S.A. 107, 1211–1216. doi: 10.1073/pnas.0910302107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wen, Z., Nguyen, H. N., Guo, Z., Lalli, M. A., Wang, X., Su, Y.,et al. (2014). Synaptic dysregulation in a human iPS cell model of mental disorders. Nature doi: 10.1038/nature13716 [Epub ahead of print].
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wernig, M., Meissner, A., Foreman, R., Brambrink, T., Ku, M., Hochedlinger, K.,et al. (2007). In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature 448, 318–324. doi: 10.1038/nature05944
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Whitehurst, A. W., Bodemann, B. O., Cardenas, J., Ferguson, D., Girard, L., Peyton, M.,et al. (2007). Synthetic lethal screen identification of chemosensitizer loci in cancer cells. Nature 446, 815–819. doi: 10.1038/nature05697
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yadav, V., Zhang, X., Liu, J., Estrem, S., Li, S., Gong, X. Q.,et al. (2012). Reactivation of mitogen-activated protein kinase (MAPK) pathway by FGF receptor 3 (FGFR3)/Ras mediates resistance to vemurafenib in human B-RAF V600E mutant melanoma. J. Biol. Chem. 287, 28087–28098. doi: 10.1074/jbc.M112.377218
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yang, X., Deignan, J. L., Qi, H., Zhu, J., Qian, S., Zhong, J.,et al. (2009). Validation of candidate causal genes for obesity that affect shared metabolic pathways and networks. Nat. Genet. 41, 415–423. doi: 10.1038/ng.325
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yang, X., Peterson, L., Thieringer, R., Deignan, J. L., Wang, X., Zhu, J.,et al. (2010). Identification and validation of genes affecting aortic lesions in mice. J. Clin. Invest. 120, 2414–2422. doi: 10.1172/JCI42742
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yauch, R. L., Dijkgraaf, G. J., Alicke, B., Januario, T., Ahn, C. P., Holcomb, T.,et al. (2009). Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science 326, 572–574. doi: 10.1126/science.1179386
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yoo, A. S., Sun, A. X., Li, L., Shcheglovitov, A., Portmann, T., Li, Y.,et al. (2011). MicroRNA-mediated conversion of human fibroblasts to neurons. Nature 476, 228–231. doi: 10.1038/nature10323
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yu, D. X., Di Giorgio, F. P., Yao, J., Marchetto, M. C., Brennand, K., Wright, R.,et al. (2014). Modeling hippocampal neurogenesis using human pluripotent stem cells. Stem Cell Reports 2, 295–310. doi: 10.1016/j.stemcr.2014.01.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yu, J., Vodyanik, M. A., Smuga-Otto, K., Antosiewicz-Bourget, J., Frane, J. L., Tian, S.,et al. (2007). Induced pluripotent stem cell lines derived from human somatic cells. Science 318, 1917–1920. doi: 10.1126/science.1151526
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, B., Gaiteri, C., Bodea, L. G., Wang, Z., McElwee, J., Podtelezhnikov, A. A.,et al. (2013a). Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 153, 707–720. doi: 10.1016/j.cell.2013.03.030
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, Y., Pak, C., Han, Y., Ahlenius, H., Zhang, Z., Chanda, S.,et al. (2013b). Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 78, 785–798. doi: 10.1016/j.neuron.2013.05.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, J., Fujimoto, J., Zhang, J., Wedge, D. C., Song, X., Zhang, J.,et al. (2014a). Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science 346, 256–259. doi: 10.1126/science.1256930
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, L., Song, X., Mohri, Y., and Qiao, L. (2014b). Role pf inflammation and tumor microenvironment in the development of gastrointestinal cancers: what induced pluripotent stem cells can do? Curr. Stem Cell Res. Ther. doi: 10.2174/1574888X09666140812112305 [Epub ahead of print].
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhong, H., Beaulaurier, J., Lum, P. Y., Molony, C., Yang, X., Macneil, D. J.,et al. (2010a). Liver and adipose expression associated SNPs are enriched for association to type 2 diabetes. PLoS Genet. 6:e1000932. doi: 10.1371/journal.pgen.1000932
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhong, H., Yang, X., Kaplan, L. M., Molony, C., and Schadt, E. E. (2010b). Integrating pathway analysis and genetics of gene expression for genome-wide association studies. Am. J. Hum. Genet. 86, 581–591. doi: 10.1016/j.ajhg.2010.02.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhou, P., Shaffer, D. R., Alvarez Arias, D. A., Nakazaki, Y., Pos, W., Torres, A. J.,et al. (2014). In vivo discovery of immunotherapy targets in the tumour microenvironment. Nature 506, 52–57. doi: 10.1038/nature12988
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhu, J., Chen, Y., Leonardson, A. S., Wang, K., Lamb, J. R., Emilsson, V.,et al. (2010). Characterizing dynamic changes in the human blood transcriptional network. PLoS Comput. Biol. 6:e1000671. doi: 10.1371/journal.pcbi.1000671
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhu, J., Lum, P. Y., Lamb, J., GuhaThakurta, D., Edwards, S. W., Thieringer, R.,et al. (2004). An integrative genomics approach to the reconstruction of gene networks in segregating populations. Cytogenet. Genome Res. 105, 363–374. doi: 10.1159/000078209
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhu, J., Sova, P., Xu, Q., Dombek, K. M., Xu, E. Y., Vu, H.,et al. (2012). Stitching together multiple data dimensions reveals interacting metabolomic and transcriptomic networks that modulate cell regulation. PLoS Biol. 10:e1001301. doi: 10.1371/journal.pbio.1001301
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhu, J., Wiener, M. C., Zhang, C., Fridman, A., Minch, E., Lum, P. Y.,et al. (2007). Increasing the power to detect causal associations by combining genotypic and expression data in segregating populations. PLoS Comput. Biol. 3:e69. doi: 10.1371/journal.pcbi.0030069
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhu, J., Zhang, B., Smith, E. N., Drees, B., Brem, R. B., Kruglyak, L.,et al. (2008). Integrating large-scale functional genomic data to dissect the complexity of yeast regulatory networks. Nat. Genet. 40, 854–861. doi: 10.1038/ng.167
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: stem cell-based screening, systems biology and network biology, drug discovery screening, complex disease mechanism, high throughput biology
Citation: Schadt EE, Buchanan S, Brennand KJ and Merchant KM (2014) Evolving toward a human-cell based and multiscale approach to drug discovery for CNS disorders. Front. Pharmacol. 5:252. doi: 10.3389/fphar.2014.00252
Received: 08 August 2014; Accepted: 30 October 2014;
Published online: 02 December 2014.
Edited by:
Birgit T. Priest, Eli Lilly and Company, USAReviewed by:
Douglas Auld, Novartis Institutes for BioMedical Research, USAJonathan M. Solomon, Novartis Institutes for BioMedical Research, USA
Copyright © 2014 Schadt, Buchanan, Brennand and Merchant. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eric E. Schadt, Icahn Institute for Genomics and Multiscale Biology, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA e-mail: eric.schadt@mssm.edu
† Present address: Kalpana M. Merchant, Trans Thera Consulting Co., 6702 South 850 East, Zionsville, IN 46077, USA