 Emma E. Ramsay
Emma E. Ramsay Pierre J. Dilda
Pierre J. Dilda
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 11 August 2014
Sec. Experimental Pharmacology and Drug Discovery
Volume 5 - 2014 | https://doi.org/10.3389/fphar.2014.00181
This article is part of the Research Topic The changing faces of glutathione, a cellular protagonist., Volume I View all 15 articles
Living organisms are continuously exposed to xenobiotics. The major phase of enzymatic detoxification in many species is the conjugation of activated xenobiotics to reduced glutathione (GSH) catalyzed by the glutathione-S-transferase (GST). It has been reported that some compounds, once transformed into glutathione S-conjugates, enter the mercapturic acid pathway whose end products are highly reactive and toxic for the cell responsible for their production. The cytotoxicity of these GSH conjugates depends essentially on GST and gamma-glutamyl transferases (γGT), the enzymes which initiate the mercapturic acid synthesis pathway. Numerous studies support the view that the expression of GST and γGT in cancer cells represents an important factor in the appearance of a more aggressive and resistant phenotype. High levels of tumor GST and γGT expression were employed to selectively target tumor with GST- or γGT-activated drugs. This strategy, explored over the last two decades, has recently been successful using GST-activated nitrogen mustard (TLK286) and γGT-activated arsenic-based (GSAO and Darinaparsin) prodrugs confirming the potential of GSH-conjugates as anticancer drugs.
Glutathione (GSH) plays a myriad of roles in the body. It is a major cellular antioxidant, involved in defense against oxidative stress and redox signaling. GSH also modulates cell proliferation, apoptosis, immune function, and fibrogenesis (Lu, 2013). In cancer cells, GSH and enzymes of the mercapturic acid pathway play a role in resistance to many chemotherapeutic drugs. However, not all xenobiotics (including drugs) are conjugated/inactivated by this pathway (Commandeur et al., 1995); some are in fact activated into cytotoxic compounds. This review explores the importance of the mercapturic acid pathway and the potential of intra-tumor activation of old and recently discovered chemotherapeutics. The expression patterns of participating enzymes of the mercapturic acid pathway could theoretically be employed to drive the activation of such compounds within the tumor.
The mercapturic acid pathway (Figure 1) involves the conjugation of the tripeptide, GSH, to xenobiotics (including drugs) to render them more hydrophilic and facilitate their elimination. Although some spontaneous reactions could occur, the cytosolic glutathione transferases (GST) catalyze the nucleophilic conjugation of GSH with a wide spectrum of electrophiles (Armstrong, 1997). Being the first step in the metabolism and eventual removal of the drug from the body, GST is associated with chemotherapeutic resistance since exposure to these drugs is often associated with induction of GST, especially GST P1-1 (Dahllof et al., 1987; Hamada et al., 1994; Hao et al., 1994). The GSH-conjugated compounds generated are then actively pumped out of the cell by numerous members of the MRP/ABCC family, which appear to have broad and partially overlapping substrate specificity (Ballatori et al., 2009). At the cell surface, the first step in the catabolism of GSH-conjugates is catalyzed by the membrane-bound gamma-glutamyl transferase (γGT) which removes the GSH γ-glutamyl group and transfers it to appropriate acceptors. Similar to what has been described for GST, γGT is also considered to be part of a resistance phenotype. The main reason for this is the role that γGT plays in maintaining appropriate GSH levels in the cells for xenobiotic detoxification by conjugation (Pompella et al., 2006). The newly formed cysteinylglycine S-conjugates are further processed by dipeptidases/aminopeptidases to remove the glycyl group and produce cysteine S-conjugates. These compounds then re-enter the cell via various transporters including organic anion transport polypeptides and cystine/cysteine importers (Hinchman et al., 1998; Budy et al., 2006; Dilda et al., 2008, 2009; Garnier et al., 2014). In the cytosol, N-acetyl transferases create mercapturic acid versions of the xenobiotics which are generally more polar and more water soluble than the parental compound. At this point, the compounds are generally non-toxic and excreted from the body through bile or urine. Alternatively, instead of acetylation, some compounds can be converted by cysteine S-conjugate β-lyase to produce an unstable and highly reactive thiol.
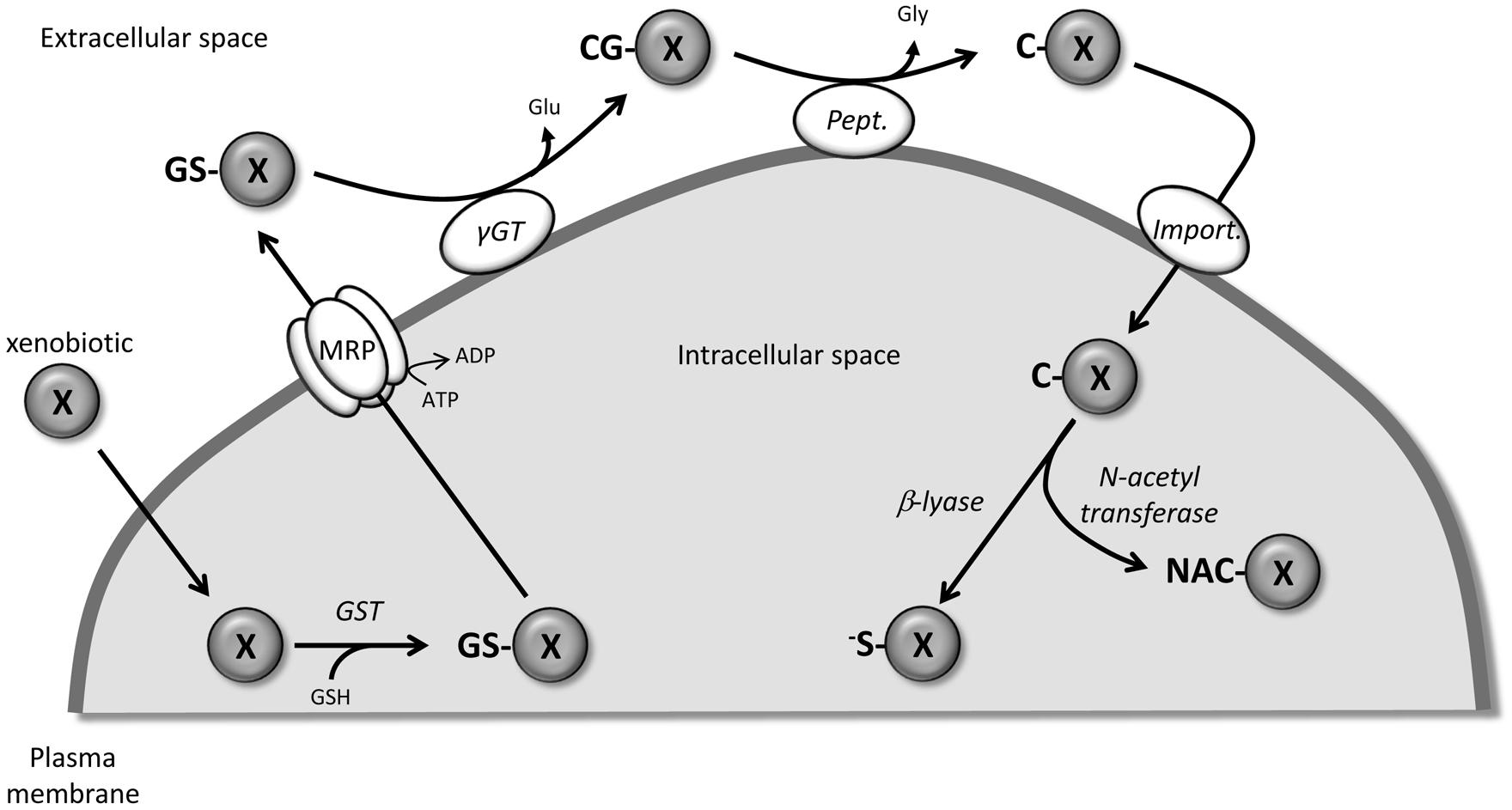
FIGURE 1. Detoxification of xenobiotic through the mercapturic acid synthesis pathway. X, xenobiotic; GS-X, GSH-conjugated version of X; CG-X, cysteinylglycyl-X; C-X, cysteinyl-X; NAC-X, mercapturic acid version of X; -S-X, highly reactive thiol version of X; GST, glutathione S-transferase, γGT, gamma-glutamyl transferase; Pept., peptidase; Import., peptide transporter or cystine/cysteine importer.
There are several examples of drugs becoming toxic/bioactive following their conjugation with GSH. Trichloroethylene is metabolized to form [S-(1,2-dichlorovinyl)GSH], which is the first step in the GSH metabolism pathway. In vitro (Lash et al., 1986; Chen et al., 1990; Cummings and Lash, 2000) and in vivo (Terracini and Parker, 1965; Elfarra et al., 1986), the downstream metabolites of GSH S-dichlorovinyl, such as N-acetyl-S-(1,2-dichlorvinyl)-L-cysteine and S-(1,2-dichlorovinyl)-L-cysteine, induce toxicity towards the cells responsible for their production (Spencer and Schaumburg, 1985; Patel et al., 1993). Consistent with what was observed with trichloroethylene, the GSH-S-conjugate of 4-hydroxynonenal (HNE) causes a loss in cell viability in γGT-expressing cells (Enoiu et al., 2002). This effect was attributed to the cysteinylglycine S-conjugate of HNE (Calonghi et al., 2002; Cerbone et al., 2007; Pettazzoni et al., 2011).
The expression of several enzymes involved in the mercapturic acid pathway (notably GST and γGT) is induced in cancer cells and particularly drug resistant cancer cells (Table 1). The specific expression of these enzymes involved in the local production of cytotoxic/bioactive drugs has been exploited to design and develop various types of anticancer prodrugs. This review describes the enzymes involved in drug activation in the order they participate along the mercapturic acid pathway, GST, γGT, and β-lyase. The potential to exploit the induced expression of these enzymes as predictive markers and the opportunity this presents for drug design is discussed.
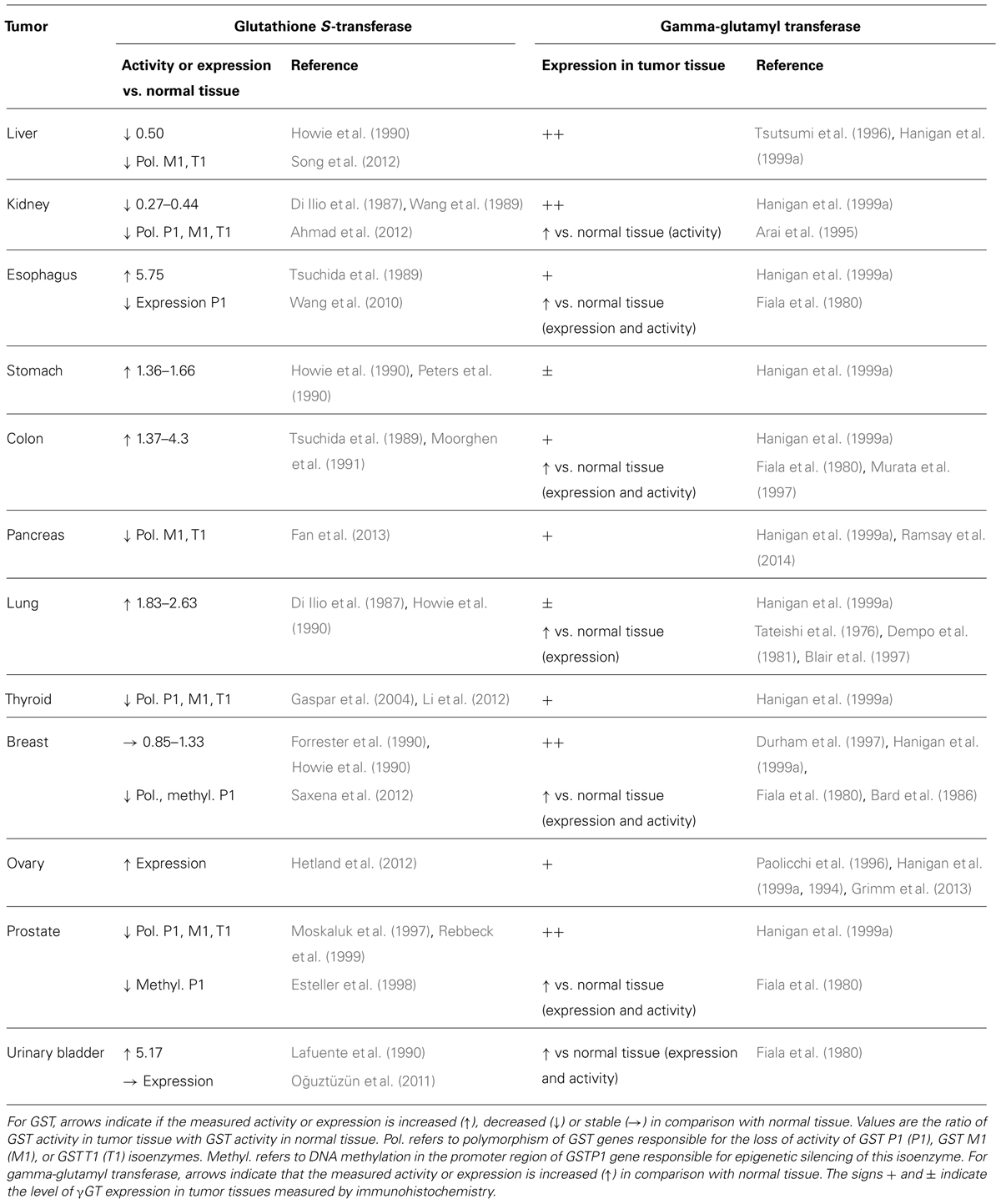
TABLE 1. Activity and expression levels of glutathione-S-transferase and gamma-glutamyl transferase in tumor tissues.
Glutathione transferases (EC 2.5.1.18) are a superfamily of dimeric detoxification enzymes which contribute to the cellular biotransformation of electrophilic compounds (Mannervik et al., 2005). They provide protection against genotoxic and carcinogenic effects of numerous substances of both xenobiotic and endogenous origins. The essential role of GST is to catalyze the conjugation of GSH with a wide variety of compounds, including drugs, resulting in the formation of the corresponding GSH-conjugates and subsequently facilitate their clearance from the body. In humans, various isoforms of GST are present in virtually all tissues with the liver exhibiting the highest cytosolic GST activity level followed by kidney, lung, and intestine (Pacifici et al., 1988).
Although GST detoxifying activity protects cells from endogenous toxic products, it also blunts the effectiveness of certain anticancer drugs (O’Brien and Tew, 1996). GST and GSH are frequently elevated in many tumors relative to surrounding healthy tissue (Howie et al., 1990). Isoenzyme GST P1-1 is notably induced in lung, colon, and stomach cancers and was found to be implicated in cellular resistance to chemotherapeutic agents (Dahllof et al., 1987; Hamada et al., 1994; Hao et al., 1994).
The polymorphism in GSTM1 and GSTT1 genes, leads to complete lack of activity of their corresponding enzymes and is responsible for poor elimination of carcinogenic substances which are potential sources of reactive oxygen species (Rebbeck, 1997; Kumar et al., 2011). This polymorphism has been associated with increased risk of benign prostatic hyperplasia and prostate cancer (Rebbeck et al., 1999) and can be a useful biomarker to identify patients at higher risk for fatal prostate cancer (Agalliu et al., 2006; Liu et al., 2013). In prostate, but also breast, endometrial and hepatocellular carcinomas, another early tumor marker is GSTP1 promoter methylation which is detected at various percentages of clinical samples (Esteller et al., 1998; Zhong et al., 2002; Chan et al., 2005; Lee, 2007; Yoon et al., 2012). The epigenetic silencing of GSTP1 provides a mechanism of resistance which makes tumors with GSTP1 promoter methylation bad candidates for the GST-activated anti-cancer prodrugs presented in this review. However, recent studies demonstrated that it is possible to re-sensitize tumors using a combination of molecules specifically reverting the aberrant DNA methylation in cancer cells (through drugs inhibiting DNA methyltransferase activity) and cytotoxic drugs (Plumb et al., 2000; Cheng et al., 2003; Sabatino et al., 2013).
An approach to solving drug resistance due to over expression of GST is to design specific GST inhibitors (Ruzza et al., 2009). Alternatively, GST overexpression offers the opportunity to target resistant tumors with GST-activated prodrugs. This type of compound undergoes GST-catalyzed breakdown to release locally cytotoxic metabolites and thereby attenuate off target adverse side effects. Amongst these prodrugs, two categories can be distinguished.
The first category consists of prodrugs containing a GSH or GSH-like structure for which a β-elimination reaction, catalyzed by GST, releases a cytotoxic compound. This includes compounds such as nitrogen mustards, which have the property to alkylate DNA after further transformation: TLK286 (canfosfamide, TELCYTATM) and analogs (Lyttle et al., 1994; Satyam et al., 1996; Dourado et al., 2013; Figure 2A). TLK286 showed interesting in vitro and in vivo antiproliferative activity on cells with high GST P1-1 expression as a result of selection for doxorubicin, cyclophosphamide, and platinum resistance (Morgan et al., 1998; Townsend et al., 2002). In murine xenografts, tumor growth inhibition, or regression in response to TLK286 was positively correlated with the level of GST P1-1 expression (Morgan et al., 1998). Several clinical trials demonstrated that TLK286 was active and safe to use in combination treatment regimens with standard chemotherapeutic agents, including platinums, taxanes, and anthracyclines. Noteworthy, clinical efficacy was observed with both relapsed patients with ovarian and non-small cell lung cancers, and in the first-line treatment setting in non-small cell lung cancer patients (Sequist et al., 2009; Vergote et al., 2010).
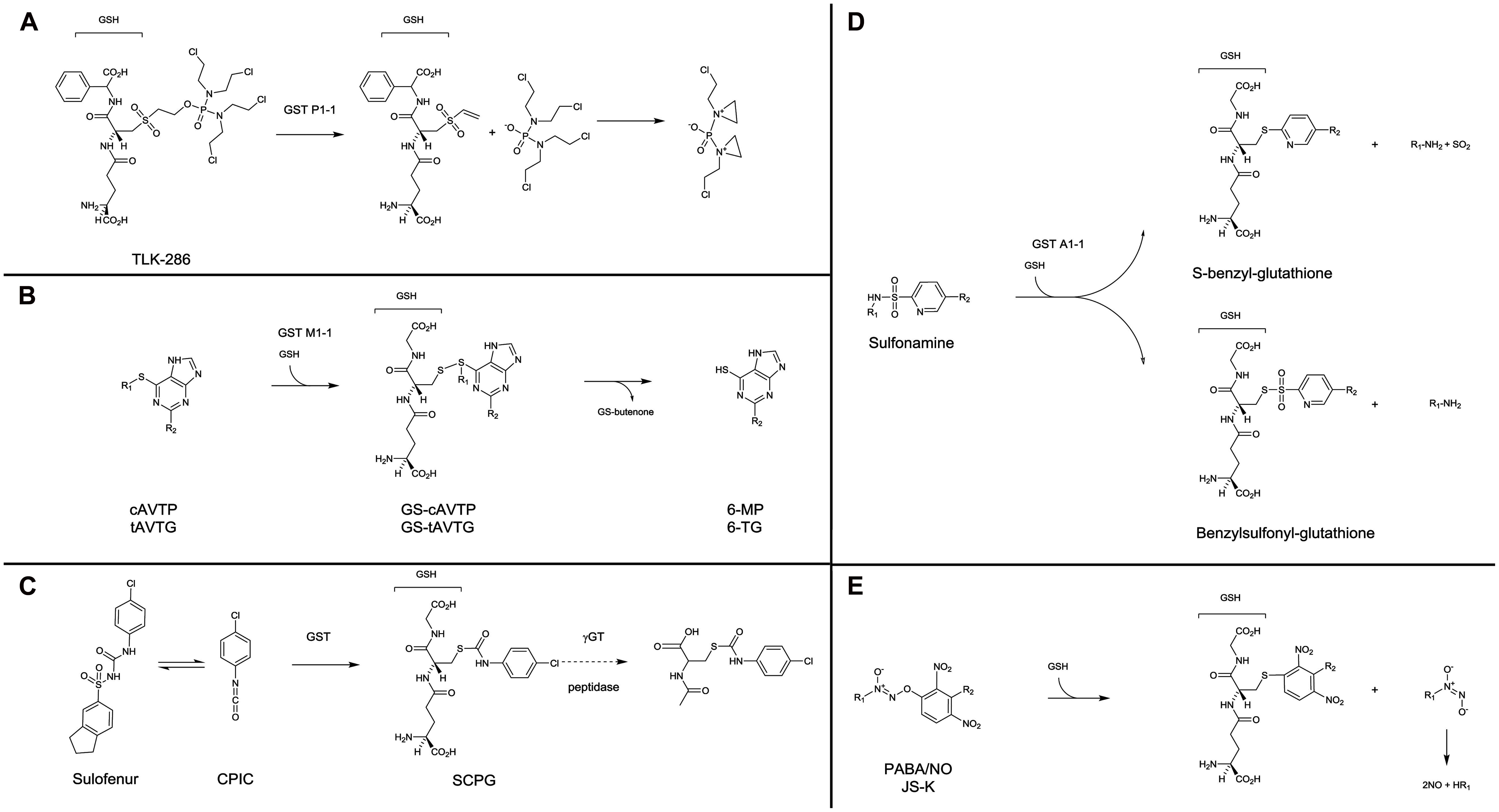
FIGURE 2. Mechanisms of activation of GST prodrugs. TLK286 (A) has a GSH-like moiety in its structure, in contrast, all other presented drugs are conjugated with GSH during their process of activation by GST. (B) for cAVTP, GS-cAVTP, and 6-MP, R1: cis-butenone; R2: H. For tAVTG, GS-tAVTG and 6-TG, R1: trans-butenone; R2: NH2. (D) for sulfonamine compounds, R1: bombesin or aryl derivatives; R2: CN or CF3. (E) for PABA/NO, R1: NMe2; R2: 4-carboxy-N-methylanilinyl. For JS-K, R1: 1-carboxyethyl-4 piperazinyl, R2: H.
The second category regroups prodrugs non-structurally related to GSH. However, their activation by GST involves the production of an intermediate GSH-conjugate.
– In this category, cis-6-(2-acetylvinylthio)-purine (cAVTP) and trans-6-(2-acetylvinylthio)-guanine (tAVTG) contain active thiopurines (6-mercaptopurine and 6-thioguanine, respectively) liberated intracellularly after GSH-conjugation catalyzed by GST M1-1, M2-2, and A4-4, but not GST P1-1 (Gunnarsdottir et al., 2002; Eklund et al., 2007; Figure 2B). Both compounds exhibited a remarkable growth-inhibitory activity towards leukemic and melanoma cells (Gunnarsdottir and Elfarra, 2004). In vivo, on the contrary to what was observed following the administration of bioactive end products (6-mercaptopurine and 6-thioguanine), no reduction of circulating white blood cells was observed upon administration of cAVTP and tAVTG (Gunnarsdottir et al., 2002).
– Using a similar strategy, exocyclic enones displaying antitumor activity were produced by a GSH-dependent reaction catalyzed by GST (Hamilton et al., 2002, 2003). COMC-6 (2-crotonyloxymethyl-2-cyclohexenone) is a potent antitumor agent against both murine and human tumors in culture and in tumor-bearing mice (Aghil et al., 1992).
– Diarylsulfonylureas are a class of antitumor agent with interesting therapeutic efficacy against a wide range of cancers (Howbert et al., 1990; Mohamadi et al., 1992). Sulofenur [(N-(5-indanesulfonyl)-N′-(4-chlorophenyl)urea, LY18664], the prototypic member of this class (Figure 2C), was evaluated in clinical trials on a variety of patients with kidney (Mahjoubi et al., 1993), ovary (O’Brien et al., 1992), breast (Talbot et al., 1993), lung (Munshi et al., 1993), and stomach (Kamthan et al., 1992) cancers. It appeared that sulofenur can undergo metabolic biotransformation to yield the GSH-conjugate of p-chlorophenyl isocyanate [S-(N-p-chlorophenylcarbamoyl) GSH, SCPG; Jochheim et al., 2002]. Whether a direct involvement of GST is necessary or not in the production of this GSH-conjugate remains unclear. However, it was clearly demonstrated that SCPG is further processed through the mercapturic acid pathway to the corresponding N-acetylcysteine conjugate [N-acetyl-S-(p-chlorophenylcarbamoyl)cysteine, NACC], which possesses comparable anticancer activity to the parent compound. The GSH- and cysteine-conjugates produced are susceptible to thiol exchange reactions and may act as carbamoylating agents towards biomacromolecules (Day et al., 1996; Guan et al., 2002). NACC, whose mechanism of action remains unclear, demonstrated selective anticancer activity, and low toxicity, which make NACC and its analogs promising anticancer agents (Chen et al., 2011).
– Another approach was to use the sulphonamidase activity of GST. GST catalyzes the GSH-mediated hydrolysis of sulfonamide bonds, releasing GST competitive inhibitors such as benzylsulfonyl-GSH or S-benzyl-GSH (Koeplinger et al., 1999; Zhao et al., 1999; Axarli et al., 2009; Figure 2D).
– Finally, electrophilic diazenium diolates were designed as prodrugs for spontaneous nitric oxide (NO) release at physiological pH, after a reaction with GSH catalyzed by GST. Essentially two compounds were studied. PABA/NO [(O2-(2,4-dinitro-5-(N-methyl-N-4 carboxyphenylamino)phenyl) 1-N,N-dimethylamino)diazen-1-ium-1,2-diolate; Findlay et al., 2004] and JS-K [O2-(2,4-dinitrophenyl) 1-[(4-ethoxycarbonyl) piperazin-1-yl]diazen-1-ium-1,2-diolate; Shami et al., 2003] were selectively activated by GST M1 and GST A1, respectively, and produced abundant NO in the tumor and contributed to chemotherapy by GSH consumption, DNA synthesis inhibition and inhibition of enzymes responsible for cellular damage repair (Figure 2E). PABA/NO and JS-K demonstrated antitumor activity in mice bearing human ovarian carcinoma (Findlay et al., 2004) and prostate carcinoma (Laschak et al., 2012) or multiple myeloma (Kiziltepe et al., 2007), respectively. Recently, new generations of more stable and potent GST-activated NO prodrugs have been developed (Liu et al., 2012; Fu et al., 2013).
Gamma-glutamyl transferase (EC2.3.2.2) catalyzes the transpeptidation and hydrolysis of the γ-glutamyl group of GSH and related compounds to an acceptor molecule, including water, amino acids, and peptides. It is found on the plasma membrane, facing extracellularly, playing an essential role in the maintenance of intracellular cysteine (Moriarty-Craige and Jones, 2004). Considering GSH’s important role as a cellular antioxidant, γGT has traditionally been considered a component of the cell’s oxidative stress defenses. However, the range of γGT substrates has expanded considerably, now including a range of GSH conjugates, leukotriene C4 (LTC4), S-nitroso-GSH and GSH adducts of xenobiotics, suggesting diverse roles for γGT.
High expression of γGT is commonly found on cells involved in transport, on the luminal surface of secretory and absorptive cells. Its highest expression is on the luminal surface of the proximal tubules in the kidney, whilst the bile ducts, bile canaliculi, and endothelial cells of the nervous system capillaries also have high expression (Shiozawa et al., 1989; Hanigan and Frierson, 1996).
Oxidative stress has been shown to induce γGT expression (Kugelman et al., 1994; Knickelbein et al., 1996; Liu et al., 1998; Borud et al., 2000; Roomi et al., 2006). High γGT expression protects melanoma cells from hydrogen peroxide or ascorbic acid induced oxidative stress (Giommarelli et al., 2008). It also allows the cells to maintain their intracellular GSH levels and subsequently respond to oxidative stress. Conversely, γGT has been demonstrated to have pro-oxidant effects. Combined with metal ions (iron or copper), γGT can induce lipid peroxidation (Stark et al., 1993; Stark and Glass, 1997). γGT has been linked to reactive oxygen species generation in cells (Drozdz et al., 1998; Del Bello et al., 1999; Paolicchi et al., 2002).
Gamma-glutamyl transferases expression has been shown to be increased in numerous cancers. Increased levels have been observed in cancer of the ovary, liver, lung, and breast, and in melanoma and leukemia (Fujisawa et al., 1976; Gerber and Thung, 1980; Dempo et al., 1981; Bard et al., 1986; Corti et al., 2010; Mareš et al., 2012). In many cases the γGT levels are higher in the corresponding primary tumor (Maellaro et al., 2000). An extensive study by Hanigan et al. (1999a) scored the expression of γGT in a variety of tumors. Carcinomas in particular express γGT, with carcinomas of the kidney, liver, and prostate showing strong expression (Hanigan et al., 1999a). Furthermore, some carcinomas of the breast, ovary, uterus, and pancreas were shown to express γGT. On the contrary, non-epithelial malignancies and sarcomas rarely expressed γGT.
Gamma-glutamyl transferases has been considered an early marker of neoplastic transformation. Many early studies have demonstrated in in vivo models the appearance of γGT expression in areas previously negative following exposure to carcinogens (Pompella et al., 2006). The underlying mechanism of this phenomenon remains unclear. A genome-wide analysis of pancreatic cancer implicated GGT1 as playing a role in carcinogenesis (Diergaarde et al., 2010). The proto-oncogene KRAS was shown to be involved in the upregulation of γGT expression. Recently, Moon et al. (2012) have demonstrated that KRAS transformed prostate epithelial cells are more resistant to hydrogen peroxide induced free-radicals than non-transformed cells. They observed an upregulation of GGT2 in the KRAS transfected cells and confirmed its role in resistance to hydrogen peroxide treatment. This has also been observed in colon carcinoma cells where radiation induced γGT activity was mediated through Ras pathway (Pankiv et al., 2006).
The early appearance of γGT in neoplasms suggests the potential for γGT to play a role in tumor progression. γGT has been shown to give cells a growth advantage in vitro and in vivo. High expression of γGT provides cells with greater quantities of cysteine through the breakdown of extracellular GSH (Gerber and Thung, 1980; Hanigan, 1995; Hochwald et al., 1996). This explains the difference in growth rates of cells when moved from the in vitro setting to mice, where extracellular GSH and cysteine is limited in vivo. In clones of melanoma cells, the extent of γGT expression was shown to be proportional to the invasive ability of the clone (Supino et al., 1992).
Beyond its differential expression in cancer, γGT is considered to be part of a resistance phenotype (Pompella et al., 2006). The main reason for this is the role that γGT plays in maintaining GSH levels within the cell. GSH plays an important part in the detoxification of xenobiotics by binding to a range of agents and this allows for GSH mediated expulsion of these compounds from the cell.
The relationship between γGT and chemotherapy resistance is demonstrated by a number of experiments showing that transfection with γGT both in vitro and in vivo leads to resistance to members of the platinum drug family, and in particular to cisplatin (Hanigan et al., 1999b; Daubeuf et al., 2002; Franzini et al., 2006). Further evidence of the relationship comes from biopsies from ovarian adenocarcinoma patients before and after treatment (cisplatin, chlorambucil, and 5FU). Cells grown from biopsies taken before and after treatment, showed a 6.5-fold increase in γGT activity following treatment (Lewis et al., 1988). However, Schäfer et al. (2001) found no direct link between γGT and resistance, despite an evident growth advantage for γGT overexpressing cells.
Another proposed mechanism for platinum drug resistance is the formation of adducts between the platinum drug and the cysteinyl-glycine product of γGT. These complexes have poor ability to cross the cell membrane; as a result the platinum drug rarely reaches its target (DNA). These adducts have been described in the extracellular media of γGT overexpressing cells and the plasma of patients treated with oxaliplatin (Daubeuf et al., 2002, 2003; Paolicchi et al., 2003; Jerremalm et al., 2006;Corti et al., 2010).
Gamma-glutamyl transferases has also been implicated in resistance to radiation therapy. In lymphoid cells, γGT plays a role in maintaining the intracellular GSH levels that are essential for protection against radiation (Jensen and Meister, 1983). Inhibition of γGT in melanoma cells significantly increased the radiosensitivity of a high γGT variant (Prezioso et al., 1994b). In CC531, a colon cancer cell line, γGT was upregulated in a time and dose-dependent manner to irradiation. This increase in γGT activity was attributed to de novo synthesis of the mRNA. It was further demonstrated that signaling through the Ras pathway was responsible for this increase (Pankiv et al., 2006).
The high expression of γGT within aggressive and drug-resistant tumors implies that GSH-conjugated prodrugs, activated at the cell surface by γGT, should be particularly effective against γGT-positive tumors. The metabolism of these compounds by γGT positive tumor cells should produce high local concentrations of bioactive and membrane permeable metabolites that would then block tumor cell proliferation and eventually tumor growth. Increasing the concentration of active metabolites at the tumor site, will subsequently limit the concentration of active drug at other sites, reducing side effects.
GSAO [4-(N-(S-glutathionylacetyl)amino)phenylarsonous acid] and Darinaparsin (ZIO-101; S-dimethylarsino-GSH) are arsenic-based GSH-conjugates with demonstrated antitumor activity evaluated in clinical trials (Don et al., 2003; Dilda et al., 2005b; Diaz et al., 2008; Tsimberidou et al., 2009; Hosein et al., 2012; Horsley et al., 2013). For both drugs, extracellular γGT activity is an essential and limiting step in their activation into membrane permeable compounds (Dilda et al., 2008; Garnier et al., 2014). Indeed, it has recently been demonstrated that tumor γGT could be used for therapeutic delivery (Ramsay et al., 2014).
GSAO. 4-(N-(S-glutathionylacetyl)amino)phenylarsonous acid is a prospective cancer drug and has just completed a phase I dose escalation study in patients with solid tumors refractory to standard therapy (Horsley et al., 2013). Treatment was very well tolerated. Of 34 patients, 20 were evaluated for response (receiving two or more cycles of GSAO). Whilst no patient exhibited an objective response, eight had stable disease, with one patient having stable disease for 18 weeks.
4-(N-(S-glutathionylacetyl)amino)phenylarsonous acid consists of a phenylarsenoxide moiety attached by an N-acetyl linker to the cysteine thiol of reduced GSH (Figure 3). The phenylarsenoxide group is the active moiety, imparting to GSAO its activity by crosslinking closely spaced protein thiols and forming a high affinity ring structure between its arsenic and the thiols (Donoghue et al., 2000). GSAO specifically targets proliferative endothelial cells, which consequently starves the tumor of the nutrients required to support its expanding growth (Don et al., 2003; Dilda et al., 2005b). The GSH moiety contributes to the transport of GSAO in and out of the cell (Dilda et al., 2008, 2005b).
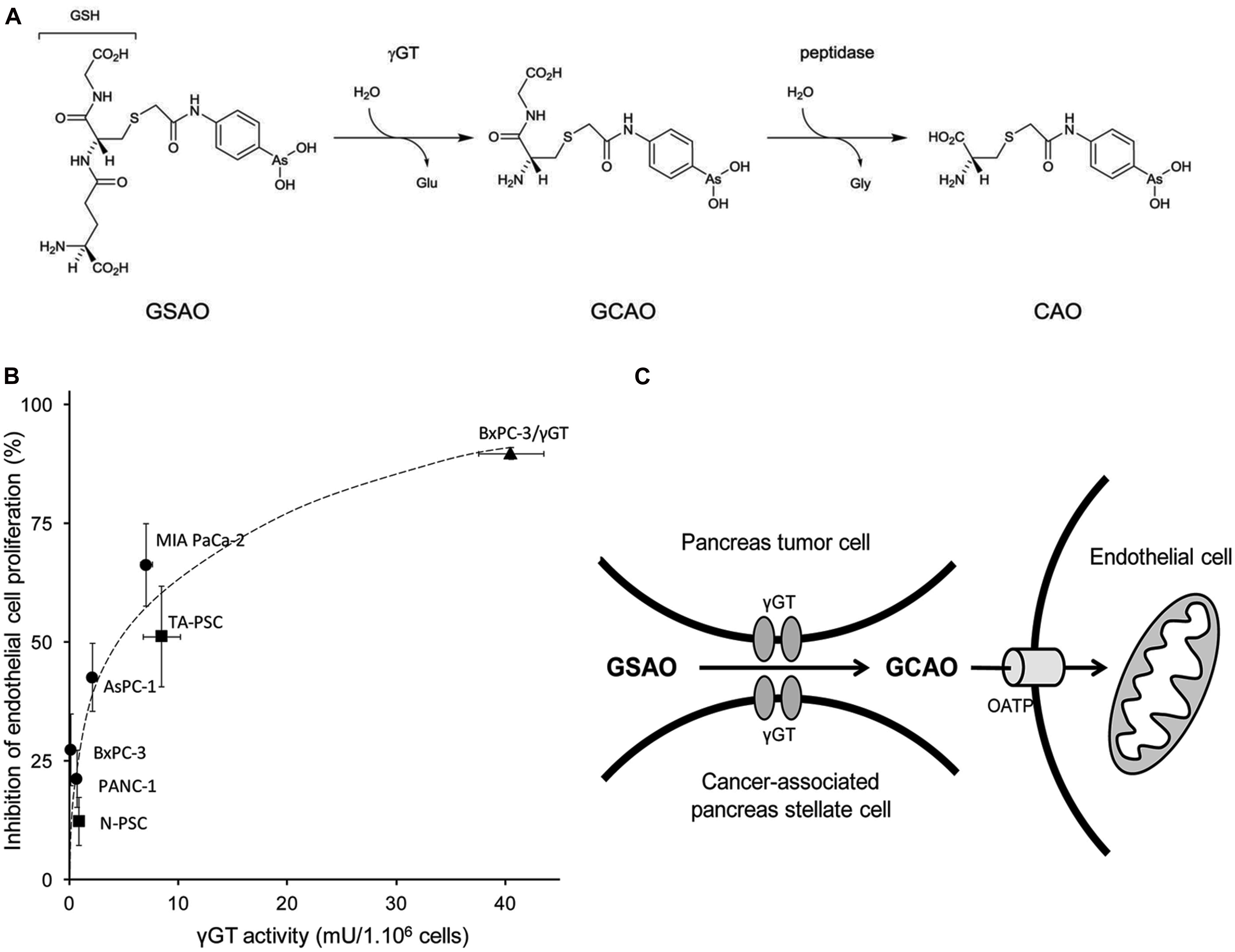
FIGURE 3. Activation process of GSAO and subsequent activity on endothelial cell proliferation. (A) GSAO activation by γGT and peptidase. (B) Pancreatic tumor cell γGT activity positively correlates with GSAO-mediated proliferation arrest of endothelial cells in a transwell model. Pancreatic adenocarcinoma cells are represented by circles and normal and tumor-activated pancreatic stellate cells by squares. BxPC-3 pancreatic tumor cells transfected with γGT are represented as triangles. (C) Model of GSAO activation by tumor γGT (from Ramsay et al., 2014).
4-(N-(S-glutathionylacetyl)amino)phenylarsonous acid in its original form is essentially membrane impermeable. Upon reaching the cell surface the γ-glutamyl group is cleaved by γGT (Dilda et al., 2008), generating the dipeptide form, 4-(N-(S-cysteinylglycylacetyl) amino) phenylarsonous acid (GCAO). GCAO is then able to enter the cell through organic anion transporters (OATPs). Within the cell further processing by dipeptidases likely occurs, resulting in the formation of the single amino acid form of GSAO, 4-(N-(S-cysteinylacetyl)amino)phenylarsonous acid (CAO; Dilda et al., 2008; Figure 3A).
The phenylarsenoxide moiety of CAO has been confirmed to crosslink the cysteine residues 57 and 257 of the adenosine nucleotide translocator (ANT; Park et al., 2012). ANT is the most abundant protein found on the inner mitochondrial membrane and it is responsible for the exchange of matrix ATP for cytosolic ADP across the inner mitochondrial membrane. Disruption of its function has been shown to have major impacts on mitochondrial integrity and cell survival (Halestrap et al., 2002; McStay et al., 2002; Don et al., 2003). The calcium-dependent binding of CAO to ANT induces a conformational change (Halestrap et al., 2002) which results in the opening of the mitochondrial permeability transition pore (MPTP) and allows the equilibration of small solutes and the release of pro-apoptotic proteins from the intermembrane space. The equilibration that results leads to a collapse of the proton-motive force across the membrane and a colloid osmotic pressure that causes massive swelling of the mitochondria (McStay et al., 2002).
There are a number of factors that influence GSAO activity: the level of expression of the enzyme responsible for its activation (γGT); intracellular GSH levels; and the expression of the multi-drug resistance association proteins, ABCC1 (MRP1) and ABCC2 (MRP2). The combination of low intracellular GSH levels and low expression of ABCC1 and ABCC2 in proliferative endothelial cells accounts for GSAO selectivity and subsequently for GSAO anti-angiogenic properties (Dilda et al., 2005a,b; Park and Dilda, 2010).
Pancreatic tumors display the most prominent stromal/desmoplastic reaction of all epithelial tumors (Apte and Wilson, 2012). Knowing that pancreatic tumorigenesis has been associated with expression of γGT by both cancer and tumor-associated stellate cells (Ramsay et al., 2014), Ramsay et al. (2014) have explored the utility of this enzyme in delivering GSAO to pancreatic ductal adenocarcinoma. They demonstrated that human pancreatic tumor and stellate cells activate/process GSAO into its active metabolite and that γGT activity positively correlates with GSAO-mediated proliferation arrest of co-cultured endothelial cells (Figure 3B). Importantly, tumor γGT activity positively correlates with GSAO-mediated inhibition of pancreatic tumor angiogenesis and tumor growth in mice.
4-(N-(S-penicillaminylacetyl)-amino)phenylarsonous acid (PENAO), a cysteine mimetic analog of the cysteine-S-conjugate metabolite of GSAO has been investigated. In this compound, penicillamine replaces the cysteine moiety of CAO. PENAO was shown to have the same molecular target as GSAO (Dilda et al., 2009; Park et al., 2012). By bypassing the cell surface processing of GSAO, PENAO accumulated 85 times faster in cells (Dilda et al., 2009). This corresponds to an increase in anti-proliferative capacity on a variety of endothelial and cancer cell lines. Interestingly, PENAO has a strong anti-proliferative activity against glioblastoma cell lines (Chung et al., 2011) and primary isolates of diffuse intrinsic pontine glioma (Tsoli et al., 2013). In vivo, PENAO demonstrated preclinical activity without signs of toxicity in tumor models of glioblastoma (Chung et al., 2013) and pancreatic carcinoma (Dilda et al., 2009). In the later model, PENAO was approximately 20-fold more efficacious than GSAO (Dilda et al., 2009). Large scale animal toxicity studies demonstrated that PENAO was as well tolerated as GSAO, suggesting an interesting therapeutic window for this compound. PENAO is currently being tested in a clinical Phase I trial in patient with solid tumors refractory to standard chemotherapy. PENAO is another example of a mercapturic acid pathway metabolite with antitumor properties. However, the lack of processing by γGT removes the targeting advantage presented by GSAO.
Darinaparsin. S-dimethylarsino-GSH (Darinaparsin; ZIO-101) is an organic arsenical compound currently in clinical development. It has been tested in Human with hematologic malignancies (Hosein et al., 2012; Nielsen et al., 2013) as well as solid tumors (Tsimberidou et al., 2009; Wu et al., 2010). Significantly more potent and better tolerated than arsenic trioxide, Darinaparsin showed encouraging responses in T-cell lymphoma (Hosein et al., 2012) and AML (Nielsen et al., 2013) patients. However, in a phase II evaluation of the compound in hepatocellular carcinoma, no objective response was shown (Wu et al., 2010).
Darinaparsin belongs to the class of organic arsenical compounds that are generally considered less toxic than inorganic ones (Waxman and Anderson, 2001; Dilda and Hogg, 2007). It was synthesized by conjugating GSH to dimethyl arsenic (Figure 4). Darinaparsin shares some characteristics with other arsenicals but has also unique properties. As do other arsenicals, it induces G2/M cell cycle arrest and triggers apoptosis through disruption of mitochondrial functions and JNK activation and is responsible for reactive oxygen species production (Diaz et al., 2008). However, unlike arsenic trioxide, GSAO (Dilda et al., 2005b) or PENAO (Dilda et al., 2009), Darinaparsin activity is unaffected by the expression of ABCC1 (MRP-1), modulation of GSH levels (Diaz et al., 2008) and heme-oxygenase inhibition (Garnier et al., 2013).
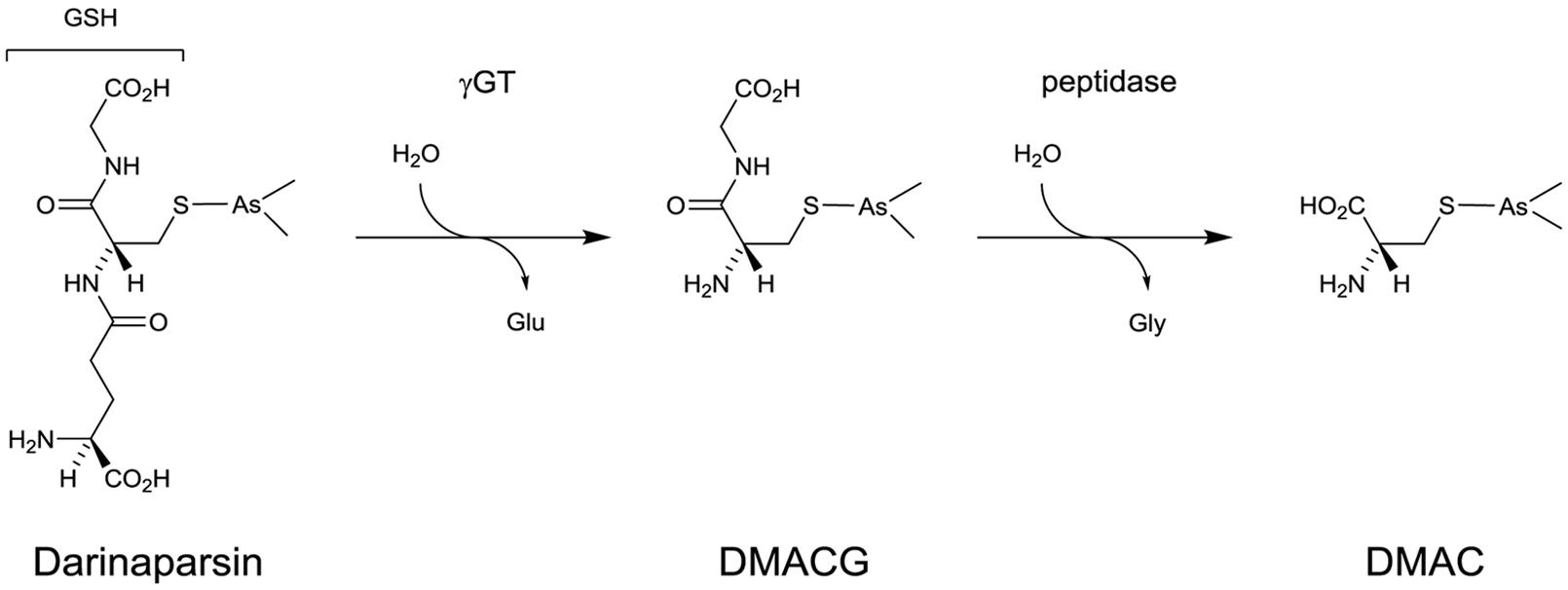
FIGURE 4. Activation process of Darinaparsin.
Darinaparsin was characterized in vitro as a potent inducer of growth arrest and apoptosis in a range of hematologic malignancies such as acute promyelocytic leukemia (APL), acute lymphoblastic leukemia, B cell lymphoma, and multiple myeloma (Diaz et al., 2008; Matulis et al., 2009). In contrast to arsenic trioxide (Lallemand-Breitenbach et al., 2012) and like PENAO and melarsoprol (Chen et al., 2003), Darinaparsin, at concentrations that induce apoptosis, does not induce PML/RAR degradation in APL cells. In the context of solid cancer, Darinaparsin was found to have preferential cytotoxic and radiosensitizing effects as compared with normal cells both in vitro and in a clinically relevant model (Tian et al., 2012, 2013).
It was recently demonstrated that Darinaparsin, which is essentially a GSH S-conjugate of dimethylarsenic, needs to be processed at the cell surface before exerting its activity on various cancer cells. The GSH moiety of the drug has to be processed by γGT before further processing by dipeptidases to generate dimethylarsino-cysteine (DMAC), a cysteine S-conjugate which is imported via cystine transporters (Garnier et al., 2014; Figure 4). Whether DMAC could be subsequently transformed intracellularly into (i) a highly reactive and cytotoxic thiol through β-lyase activity or (ii) dimethylarsenite/dimethylarsenate is not known. Similarly to what was observed in the case of GSAO and CAO (Dilda et al., 2008), Garnier et al. (2014) demonstrated that DMAC recapitulates the effects of Darinaparsin and that γGT is the first and limiting step in the activation of the drug. They also linked Darinaparsin efficacy to the level of expression of cystine/cysteine importing systems in cancer cells.
S-nitrosoglutathione (GSNO) is the S-nitrosated derivative of GSH and is thought to be a critical mediator of the downstream signaling effects of NO (Broniowska et al., 2013). GSNO plays roles as a carrier for NO and in protein S-nitrosation or S-glutathiolation (Broniowska et al., 2013). GSNO can be cleaved by γGT (Hogg et al., 1997; Angeli et al., 2009; Bramanti et al., 2009), generating S-nitrosocysteinylglycine and releasing NO in vitro. GSNO has been tested in a number of clinical trials, the majority focusing on the cardiovascular effects of GSNO as a NO donor (Broniowska et al., 2013). As do other NO-donating compounds, GSNO induces oxidative stress and initiates apoptosis (Turchi, 2006). GSNO promotes NO-induced apoptosis in colon carcinoma cell lines through depletion of intracellular GSH and the release of NO, (Ho et al., 1999; Liu et al., 2003). In a variety of cell lines, GSNO treatment combined with a MEK inhibitor was shown to inhibit the proliferation and invasive phenotype of the cells (Furuhashi et al., 2012). In T and B lymphocytes the anti-proliferative effect of GSNO was dependent on γGT activity (Henson et al., 1999). The anti-proliferative activity of GSNO, combined with the overexpression of γGT in tumor cells, suggests the possibility of its utility in cancer therapy. This requires further understanding of the roles that NO plays in cancer.
In the light of the promising results obtained, notably with the arsenical-based GSH-conjugates, it appears that tumor γGT could potentially be employed in drug targeting and delivery. In addition to cleavage of the γ-glutamyl from a GSH moiety, γGT can also catalyze the cleavage of γ-glutamyl moieties from a variety of compounds. This property along with tissue/tumor expression of γGT was used to develop γ-glutamyl prodrugs.
Due to the tissue distribution of γGT, γ-glutamyl prodrugs have been mostly developed for kidney applications. An anti-nociceptive prodrug, γ-glutamyl-dermorphin, was explored by comparing the pain threshold of mice (Misicka et al., 1996). More extensively investigated was the γ-glutamyl conjugate of L-DOPA (dihydroxyphenylalanine). This prodrug provides the precursor of dopamine, DOPA, upon activation and was explored as a potential renal vasodilator (Wilk et al., 1978; Worth et al., 1985; Sadiq et al., 2000). Clinical testing of the prodrug determined that despite reasonable kidney specificity it had low bioavailability (Lee, 1990). A number of other compounds were conjugated to a glutamyl group and tested; however none were approved for use (Huttunen and Rautio, 2011).
In terms of potential antitumor activity, γ-glutamyl-protected N-hydroxyguanidines (NHGs) have been developed to explore the ability to deliver NO to the kidney (Figure 5A). The NHGs include Nw-hydroxy-L-arginine, which is an intermediate in the NO synthase synthesis of NO (Zhang et al., 2013b). Whilst promising results were seen, there was a propensity for the potential compounds to cyclize (Zhang et al., 2013a,b) suggesting that NHG conjugation to GSH may be more stable. (Zhang et al., 2013a). As described above for PABA/NO, JS-K, and GSNO, the NHGs, in accordance with γGT expression patterns, contribute to chemotherapy by producing locally high levels of NO which can rapidly react with O2⋅- to generate the potent oxidant peroxynitrite that, in turn causes extensive cellular damage, including the nitration of protein tyrosine residues (Kirsch et al., 2001).
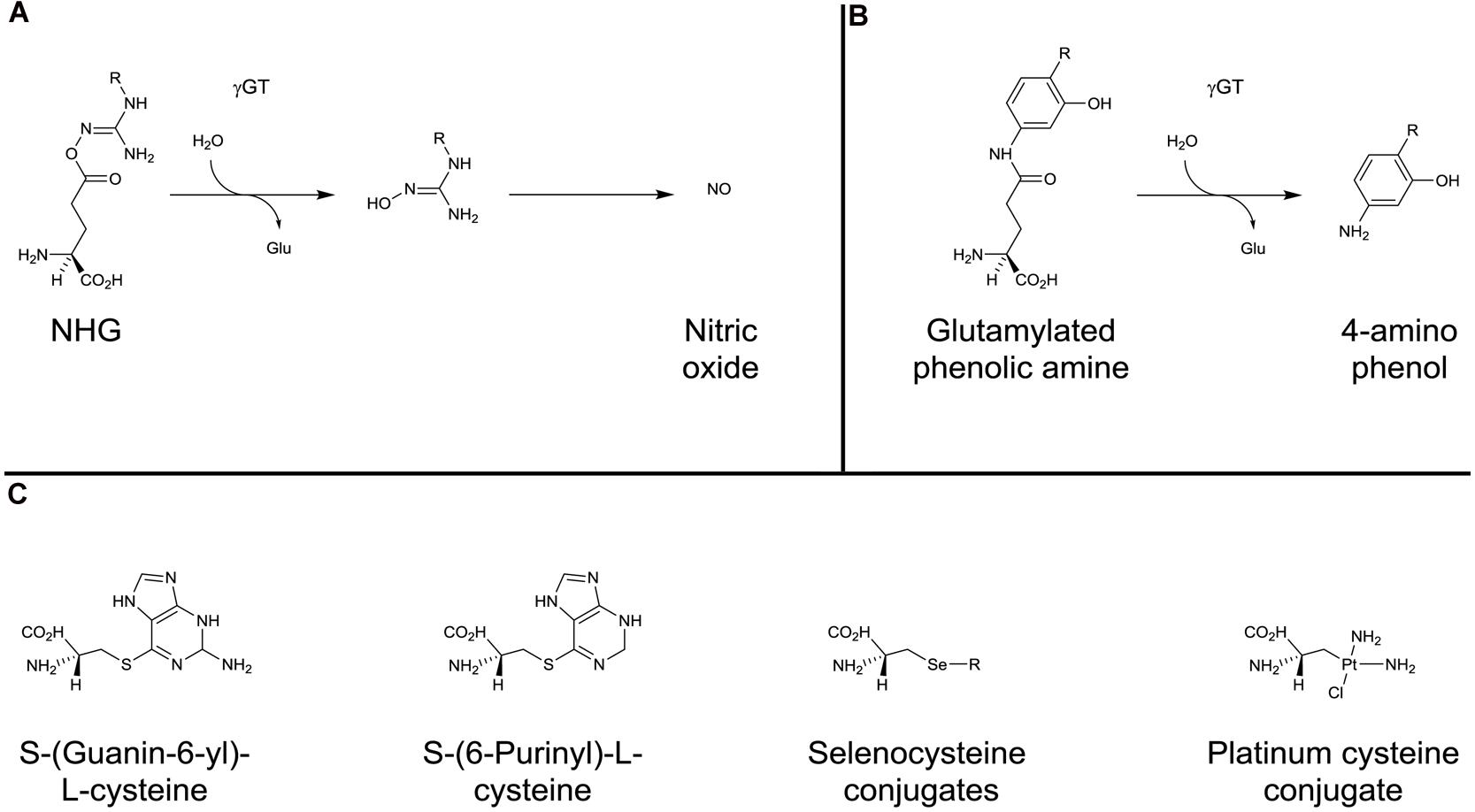
FIGURE 5. γ-glutamyl conjugates and β-lyase substrates. Activation of N-hydroxyguanidines (A) and glutamylated phenolic amines (B) by γGT. (C) examples of cysteine- and selenocysteine-conjugates activated by β-elimination catalyzed by β-lyase.
Glutamylated phenolic amine compounds have been shown to be activated by γGT (Figure 5B). γ-L-glutaminyl-4-hydroxybenzene and γ-L-glutaminyl-4-iodobenzene have been demonstrated to be activated by tyrosinase (Prezioso et al., 1993) and γGT (Prezioso et al., 1994a). Both compounds were shown to have greater cytotoxicity in melanoma cell lines when γGT activity was not inhibited (Prezioso et al., 1994a).
Gamma-glutamyl transferase being the initial and limiting step in γGT-activated prodrugs processing and because γGT is highly expressed on the luminal surface of the proximal tubules in the kidney, potential nephrotoxicity could have been a limitation in the usage of this class of compound when administered systematically. In the case of GSAO, a reversible renal toxicity described in preclinical studies at the maximal tolerated dose was not observed in human during clinical Phase I (Horsley et al., 2013). Similarly, Darinaparsin, when employed in patients (Tsimberidou et al., 2009; Wu et al., 2010; Hosein et al., 2012) didn’t display any toxicity profile related to renal dysfunction at dose-limiting toxicity (DLT). Taken together, these clinical studies indicate that the γGT-activated prodrugs have not necessarily nephrotoxic properties. This type of adverse effects is most likely dependent on the nature of the metabolites produced by the mercapturic acid pathway and particularly if the end products are substrates for cysteine conjugate β-lyases. This enzyme is notably responsible for the nephrotoxicity of cisplatin (see below). However, recent studies demonstrated that the modulation of renal GSH content could prevent such toxicity (Firdous and Kuttan, 2012; Sener et al., 2012; Ghosh et al., 2013).
Cysteine conjugates produced by dipeptidases or amino peptidases following the GST and γGT steps, are transformed by cysteine conjugate β-lyases present in the cytosol and mitochondria. The β-lyases catalyze the β-elimination of various L-cysteine S-conjugates to the corresponding thiols (Cooper, 1998). The thiols released can be either toxic or pharmacologically active for the cells responsible for their production (Commandeur et al., 1995). Due to the high expression of cysteine conjugate β-lyase in the proximal tubule of the kidney, administration of cysteine-S-conjugates of haloalkenes to rodents results in selective nephrotoxicity (Stevens et al., 1989; Hayden and Stevens, 1990).
The specific localization along with intense expression of these enzymes in renal carcinoma has led to the design of kidney-selective prodrugs that bypass earlier steps of the mercapturic acid pathway (GST, γGT, and peptidase): these compounds include S-(guanin-6-yl)-L-cysteine (Elfarra et al., 1995), S-(6-Purinyl)-L-cysteine (Hwang and Elfarra, 1991; Lash et al., 1997) and selenocysteine-conjugates (Commandeur et al., 2000; Figure 5C). The potential of these types of compounds as kidney-selective antitumor prodrugs has been demonstrated in tissue distribution experiments. However, because of a lower ratio of kidney/liver β-lyase activity in human than rodents, kidney selectivity remains to be confirmed.
Cisplatin is one of the most effective anticancer agents in the treatment of solid tumors, including, breast, testicular, and ovarian cancer. The drug binds DNA, which is toxic to dividing tumor cells. However, therapy with cisplatin is notably limited by a toxicity mechanism in the non-dividing proximal tubule cells of the kidney that is distinct from DNA cross-linking. It was demonstrated that cysteine S-conjugate β-lyase highly expressed in the kidney was responsible for the observed nephrotoxicity (Zhang and Hanigan, 2003; Townsend et al., 2003). Even if cysteine S-conjugate β-lyase is responsible for the synthesis of the highly reactive and cytotoxic thiol version of cisplatin (Figure 1), the activation of cisplatin into a nephrotoxin relies on the three previous steps of the classical mercapturic acid pathway involving GSTs (GSH conjugation), γGT (production of CisPt-cys-glu) and dipeptidase (production of CisPt-cys; Figure 5C). Indeed, the inhibition of γGT or cysteine S-conjugate β-lyase (both highly expressed in proximal tubule cells of the kidney) blocks the nephrotoxicity of cisplatin in mice (Townsend et al., 2003). In this process, the action of γGT is a key and limiting step as it transformed the essentially membrane impermeable CisPt-GSH into a membrane permeable CisPt-cys-glu that can be taken up by the cells.
The traditional approach to identify potential cancer therapies has been to identify a drug that helps the greatest number of patients. In doing this, the potential of therapies that have a significant impact on a small subset is lost within the larger sample population. The current trend to target specific abnormalities in cancer allows for the development of predictive markers and companion tests to identify which patients have a particular tumor characteristic. From this stems the ability to identify the patient population that will (or will not) respond to the drug. Developing new therapies with the target population in mind will facilitate the development of drugs that will induce a greater tumor response for individual patients. For this to occur, a marker that predicts patient response to the drug is essential.
This review recapitulates the different drug strategies involving GSH-conjugates, and the enzymes of the mercapturic acid pathway, employed over the last twenty years. Amongst the long list of compounds investigated, a limited number of GSH-conjugates have recently progressed towards clinical trials for the treatment of cancer patients. Both GST-activated nitrogen mustard (TLK286) and γGT-activated arsenic-based (GSAO and Darinaparsin) prodrugs have displayed promising activities along with safe profiles.
Given the complexity of mechanisms involved in drug resistance and survival pathways of tumor cells, combination strategies of GSH-conjugates with conventional chemotherapy or targeted drugs should provide very interesting perspectives for the treatment of drug resistant tumors. This approach has already been explored successfully with TLK286 when combined with pegylated liposomal doxorubicin (Kavanagh et al., 2010; Vergote et al., 2010) or with carboplatin plus paclitaxel (Sequist et al., 2009). Moreover, knowing that the combinations of arsenic trioxide with targeted drugs such as mTOR (Guilbert et al., 2013; Iwanami et al., 2013), EGFR (Noh et al., 2010; Kryeziu et al., 2013), or proteasome inhibitors (Jung et al., 2012) demonstrate potent profiles on a variety of malignancies, it would be of great interest to investigate the antitumor properties of such inhibitors in combination with the arsenical-based GSH-conjugates GSAO and Darinaparsin.
Increased GST and/or γGT tumor expression, generally considered as a bad prognosis marker (associated with drug resistance and disease progression), and could potentially be used as a predictive marker, allowing doctors, after biopsy, to identify which patients will respond best to GST or γGT-activated prodrugs. Predictive biomarkers provide a number of benefits, including: reduced unnecessary treatment; reduced quantity of adverse events; reduced drug attrition rates; improved therapeutic benefit; and better control of medical costs (La Thangue and Kerr, 2011). Whilst taking the time to test for GST and/or γGT tumor expression may slightly delay treatment, it will ensure that GSH-conjugates anticancer prodrugs will be administered to the patients that are more likely to respond.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to thank Prof. Philip J. Hogg and Dr. Stéphanie Decollogne for critical reading of the manuscript and Prof. Stanley B. Wild for his help with chemical nomenclature. Emma E. Ramsay is a recipient of scholarships from the National Health and Medical Research Council of Australia, the Cancer Institute New South Wales, and the Translational Cancer Research Network.
6-MP, 6-mercaptopurine; 6-TG, 6-thioguanine (6-mercaptopurine and 6-thioguanine); CAO, 4-[N-(S-cysteinylacetyl)amino]phenylarsonous acid; cAVTP, cis-6-(2-acetylvinylthio)-purine; CPIC, p-chlorophenyl isocyanate; DMAC, dimethylarsino-cysteine; DMACG, dimethylarsino-cysteinylglycine; EGFR, epidermal growth factor receptor; GSH, glutathione; GCAO, 4-[N-(S-cysteinylglycylacetyl) amino] phenylarsonous acid; γGT, gamma-glutamyl transferase; GSAO, [4-(N-(S-glutathionylacetyl) amino)phenylarsonous acid]; GS-cAVTP, glutathione s-cis-6-(2-acetylvinylthio)-purine; Glu, glutamate; Gly, glycine; GS-tAVTG, glutathione s-trans-6-(2-acetylvinylthio)-guanine; PENAO, 4-(N-(S-penicillaminylacetyl)-amino)phenylarsonous acid; SCPG, S-(N-pchlorophenylcarbamoyl) glutathione; tAVTG, trans-6-(2-acetylvinylthio)-guanine.
Agalliu, I., Langeberg, W. J., Lampe, J. W., Salinas, C. A., and Stanford, J. L. (2006). Glutathione S-transferase M1, T1, and P1 polymorphisms and prostate cancer risk in middle-aged men. Prostate 66, 146–156. doi: 10.1002/pros.20305
Aghil, O., Bibby, M. C., Carrington, S. J., Double, J., Douglas, K. T., Phillips, R. M.,et al. (1992). Synthesis and cytotoxicity of shikimate analogues. Structure:activity studies based on 1-crotonyloxymethyl-3R,4R,5R-trihydroxycyclohex-2-enone. Anticancer Drug Des. 7, 67–82.
Ahmad, S. T., Arjumand, W., Seth, A., Saini, A. K., and Sultana, S. (2012). Impact of glutathione transferase M1, T1, and P1 gene polymorphisms in the genetic susceptibility of North Indian population to renal cell carcinoma. DNA Cell Biol. 31, 636–643. doi: 10.1089/dna.2011.1392
Angeli, V., Tacito, A., Paolicchi, A., Barsacchi, R., Franzini, M., Baldassini, R.,et al. (2009). A kinetic study of γ-glutamyltransferase (GGT)-mediated S-nitrosoglutathione catabolism. Arch. Biochem. Biophys. 481, 191–196. doi: 10.1016/j.abb.2008.10.027
Apte, M. V., and Wilson, J. S. (2012). Dangerous liaisons: pancreatic stellate cells and pancreatic cancer cells. J. Gastroenterol. Hepatol. 27(Suppl. 2) 69–74. doi: 10.1111/j.1440-1746.2011.07000.x
Arai, K., Sumi, S. H., Yoshida, K. I., and Komoda, T. (1995). A precursor form of human kidney γ-glutamyl transferase in normal and cancerous tissues, and its possible post-translational modification. Biochim. Biophys. Acta. 1253, 33–38. doi: 10.1016/0167-4838(95)00141-G
Armstrong, R. N. (1997). Structure, catalytic mechanism, and evolution of the glutathione transferases. Chem. Res. Toxicol. 10, 2–18. doi: 10.1021/tx960072x
Axarli, I., Labrou, N. E., Petrou, C., Rassias, N., Cordopatis, P., and Clonis, Y. D. (2009). Sulphonamide-based bombesin prodrug analogues for glutathione transferase, useful in targeted cancer chemotherapy. Eur. J. Med. Chem. 44, 2009–2016. doi: 10.1016/j.ejmech.2008.10.009
Ballatori, N., Krance, S. M., Marchan, R., and Hammond, C. L. (2009). Plasma membrane glutathione transporters and their roles in cell physiology and pathophysiology. Mol. Aspects Med. 30, 13–28. doi: 10.1016/j.mam.2008.08.004
Bard, S., Noel, P., Chauvin, F., and Quash, G. (1986). γ-glutamyltranspeptidase activity in human breast lesions: an unfavourable prognostic sign. Br. J. Cancer 53, 637–642. doi: 10.1038/bjc.1986.107
Blair, S. L., Heerdt, P., Sachar, S., Abolhoda, A., Hochwald, S., Cheng, H.,et al. (1997). Glutathione metabolism in patients with non-small cell lung cancers. Cancer Res. 57, 152–155.
Borud, O., Mortensen, B., Mikkelsen, I. M., Leroy, P., Wellman, M., and Huseby, N. E. (2000). Regulation of γ-glutamyltransferase in cisplatin-resistant and – sensitive colon carcinoma cells after acute cisplatin and oxidative stress exposures. Int. J. Cancer 88, 464–468. doi: 10.1002/1097-0215(20001101)88:3<464::AID-IJC20>3.0.CO;2-F
Bramanti, E., Angeli, V., Franzini, M., Vecoli, C., Baldassini, R., Paolicchi, A.,et al. (2009). Exogenous vs. endogenous γ-glutamyltransferase activity: implications for the specific determination of S-nitrosoglutathione in biological samples. Arch. Biochem. Biophys. 487, 146–152. doi: 10.1016/j.abb.2009.05.012
Broniowska, K. A., Diers, A. R., and Hogg, N. (2013). S-Nitrosoglutathione. Biochim. Biophys. Acta 1830, 3173–3181. doi: 10.1016/j.bbagen.2013.02.004
Budy, B., O’neill, R., Dibello, P. M., Sengupta, S., and Jacobsen, D. W. (2006). Homocysteine transport by human aortic endothelial cells: identification and properties of import systems. Arch. Biochem. Biophys. 446, 119–130. doi: 10.1016/j.abb.2005.12.014
Calonghi, N., Boga, C., Cappadone, C., Pagnotta, E., Bertucci, C., Fiori, J.,et al. (2002). Cytotoxic and cytostatic effects induced by 4-hydroxynonenal in human osteosarcoma cells. Biochem. Biophys. Res. Commun. 293, 1502–1507. doi: 10.1016/S0006-291X(02)00397-2
Cerbone, A., Toaldo, C., Laurora, S., Briatore, F., Pizzimenti, S., Dianzani, M. U.,et al. (2007). 4-Hydroxynonenal and Pparγ ligands affect proliferation, differentiation, and apoptosis in colon cancer cells. Free Radic. Biol. Med. 42, 1661–1670. doi: 10.1016/j.freeradbiomed.2007.02.009
Chan, Q. K. Y., Khoo, U.-S., Chan, K. Y. K., Ngan, H. Y. S., Li, S.-S., Chiu, P.-M.,et al. (2005). Promoter methylation and differential expression of π-class glutathione S-transferase in endometrial carcinoma. J. Mol. Diagn. 7, 8–16. doi: 10.1016/S1525-1578(10)60003-7
Chen, G. Q., Zhou, L., Styblo, M., Walton, F., Jing, Y., Weinberg, R.,et al. (2003). Methylated metabolites of arsenic trioxide are more potent than arsenic trioxide as apoptotic but not differentiation inducers in leukemia and lymphoma cells. Cancer Res. 63, 1853–1859.
Chen, J. C., Stevens, J. L., Trifillis, A. L., and Jones, T. W. (1990). Renal cysteine conjugate β-lyase-mediated toxicity studied with primary cultures of human proximal tubular cells. Toxicol. Appl. Pharmacol. 103, 463–473. doi: 10.1016/0041-008X(90)90319-P
Chen, W., Seefeldt, T., Young, A., Zhang, X., and Guan, X. (2011). Design, synthesis, and biological evaluation of N-acetyl-S-(p-chlorophenylcarbamoyl)cysteine and its analogs as a novel class of anticancer agents. Bioorg. Med. Chem. 19, 287–294. doi: 10.1016/j.bmc.2010.11.026
Cheng, J. C., Matsen, C. B., Gonzales, F. A., Ye, W., Greer, S., Marquez, V. E.,et al. (2003). Inhibition of Dna methylation and reactivation of silenced genes by zebularine. J. Natl. Cancer Inst. 95, 399–409. doi: 10.1093/jnci/95.5.399
Chung, S., Decollogne, S., Luk, P., Shen, H., Ha, W., Day, B.,et al. (2013). “Preclinical evaluation of Penao: a potent mitochondrial specific, arsenical-based inhibitor for Glioblastoma,” in Proceedings of the 4th Quadrennial Meeting of the World Federation of Neuro-Oncology, San Francisco, 15, iii40.
Chung, S., Mc Donald, K. L., Shen, H., Day, B. W., Stringer, B. W., John, T.,et al. (2011). “Targeting glioblastoma metabolism with a novel arsenic-based metabolic inhibitor, Penao,” in Proceedings of the SNO Scientific Meeting on Neuro-Oncology, Orange County, CA, 13, 118.
Commandeur, J. N., Andreadou, I., Rooseboom, M., Out, M., De Leur, L. J., Groot, E.,et al. (2000). Bioactivation of selenocysteine Se-conjugates by a highly purified rat renal cysteine conjugate β-lyase/glutamine transaminase K. J. Pharmacol. Exp. Ther. 294, 753–761.
Commandeur, J. N., Stijntjes, G. J., and Vermeulen, N. P. (1995). Enzymes and transport systems involved in the formation and disposition of glutathione S-conjugates. Role in bioactivation and detoxication mechanisms of xenobiotics. Pharmacol. Rev. 47, 271–330.
Cooper, A. J. (1998). Mechanisms of cysteine S-conjugate β-lyases. Adv. Enzymol. Relat. Areas Mol. Biol. 72, 199–238.
Corti, A., Franzini, M., Paolicchi, A., and Pompella, A. (2010). γ-glutamyltransferase of cancer cells at the crossroads of tumor progression, drug resistance and drug targeting. Anticancer Res. 30, 1169–1181.
Cummings, B. S., and Lash, L. H. (2000). Metabolism and toxicity of trichloroethylene and S-(1,2-dichlorovinyl)-L-cysteine in freshly isolated human proximal tubular cells. Toxicol. Sci. 53, 458–466. doi: 10.1093/toxsci/53.2.458
Dahllof, B., Martinsson, T., Mannervik, B., Jensson, H., and Levan, G. (1987). Characterization of multidrug resistance in Sewa mouse tumor cells: increased glutathione transferase activity and reversal of resistance with verapamil. Anticancer Res. 7, 65–69.
Daubeuf, S., Balin, D., Leroy, P., and Visvikis, A. (2003). Different mechanisms for γ-glutamyltransferase-dependent resistance to carboplatin and cisplatin. Biochem. Pharmacol. 66, 595–604. doi: 10.1016/S0006-2952(03)00343-5
Daubeuf, S., Leroy, P., Paolicchi, A., Pompella, A., Wellman, M., Galteau, M. M.,et al. (2002). Enhanced resistance of HeLa cells to cisplatin by overexpression of γ-glutamyltransferase. Biochem. Pharmacol. 64, 207–216. doi: 10.1016/S0006-2952(02)01118-8
Day, B. W., Jin, R., and Karol, M. H. (1996). In vivo and in vitro reactions of toluene diisocyanate isomers with guinea pig hemoglobin. Chem. Res. Toxicol. 9, 568-73. doi: 10.1021/tx9501703
Del Bello, B., Paolicchi, A., Comporti, M., Pompella, A., and Maellaro, E. (1999). Hydrogen peroxide produced during γ-glutamyl transpeptidase activity is involved in prevention of apoptosis and maintenance of proliferation in U937 cells. FASEB J. 13, 69–79.
Dempo, K., Elliott, K. A. C., Desmond, W., and Fishman, W. H. (1981). Demonstration of γ-glutamyl transferase, alkaline phosphatase, Cea and Hcg in human lung cancer. Oncodev. Biol. Med. 2, 21–37.
Di Ilio, C., Del Boccio, G., Aceto, A., and Federici, G. (1987). Alteration of glutathione transferase isoenzyme concentrations in human renal carcinoma. Carcinogenesis 8, 861–864. doi: 10.1093/carcin/8.6.861
Diaz, Z., Mann, K. K., Marcoux, S., Kourelis, M., Colombo, M., Komarnitsky, P. B.,et al. (2008). A novel arsenical has antitumor activity toward As2O3-resistant and MRP1/ABCC1-overexpressing cell lines. Leukemia 22, 1853–1863. doi: 10.1038/leu.2008.194
Diergaarde, B., Brand, R., Lamb, J., Cheong, S. Y., Stello, K., Barmada, M. M.,et al. (2010). Pooling-based genome-wide association study implicates γ-glutamyltransferase 1 (GGT1) gene in pancreatic carcinogenesis. Pancreatology 10, 194–200. doi: 10.1159/000236023
Dilda, P. J., Decollogne, S., Rossiter-Thornton, M., and Hogg, P. J. (2005a). Para to ortho repositioning of the arsenical moiety of the angiogenesis inhibitor 4-(N-(S-glutathionylacetyl)amino)phenylarsenoxide results in a markedly increased cellular accumulation and antiproliferative activity. Cancer Res. 65, 11729–11734. doi: 10.1158/0008-5472.CAN-05-2797
Dilda, P. J., Don, A. S., Tanabe, K. M., Higgins, V. J., Allen, J. D., Dawes, I. W.,et al. (2005b). Mechanism of selectivity of an angiogenesis inhibitor from screening a genome-wide set of Saccharomyces cerevisiae deletion strains. J. Natl. Cancer Inst. 97, 1539–1547. doi: 10.1093/jnci/dji316
Dilda, P. J., Decollogne, S., Weerakoon, L., Norris, M. D., Haber, M., Allen, J. D.,et al. (2009). Optimization of the antitumor efficacy of a synthetic mitochondrial toxin by increasing the residence time in the cytosol. J. Med. Chem. 52, 6209–6216. doi: 10.1021/jm9008339
Dilda, P. J., and Hogg, P. J. (2007). Arsenical-based cancer drugs. Cancer Treat. Rev. 33, 542–564. doi: 10.1016/j.ctrv.2007.05.001
Dilda, P. J., Ramsay, E. E., Corti, A., Pompella, A., and Hogg, P. J. (2008). Metabolism of the tumor angiogenesis inhibitor 4-(N-(S-Glutathionylacetyl)amino)phenylarsonous acid. J. Biol. Chem. 283, 35428–35434. doi: 10.1074/jbc.M804470200
Don, A. S., Kisker, O., Dilda, P., Donoghue, N., Zhao, X., Decollogne, S.,et al. (2003). A peptide trivalent arsenical inhibits tumor angiogenesis by perturbing mitochondrial function in angiogenic endothelial cells. Cancer Cell 3, 497–509. doi: 10.1016/S1535-6108(03)00109-0
Donoghue, N., Yam, P. T. W., Jiang, X. M., and Hogg, P. J. (2000). Presence of closely spaced protein thiols on the surface of mammalian cells. Protein Sci. 9, 2436–2445. doi: 10.1110/ps.9.12.2436
Dourado, D. F. A. R., Fernandes, P. A., Ramos, M. J., and Mannervik, B. (2013). Mechanism of glutathione transferase P1-1-catalyzed activation of the prodrug canfosfamide (TLK286, TELCYTA.) Biochemistry 52, 8069–8078. doi: 10.1021/bi4005705
Drozdz, R., Parmentier, C., Hachad, H., Leroy, P., Siest, G., and Wellman, M. (1998). γ-glutamyltransferase dependent generation of reactive oxygen species from a glutathione/transferrin system. Free Radic. Biol. Med. 25, 786–792. doi: 10.1016/S0891-5849(98)00127-0
Durham, J. R., Frierson H. F. Jr., and Hanigan, M. H. (1997). Gamma-glutamyl transpeptidase immunoreactivity in benign and malignant breast tissue. Breast Cancer Res. Treat. 45, 55–62. doi: 10.1023/A:1005889006557
Eklund, B. I., Gunnarsdottir, S., Elfarra, A. A., and Mannervik, B. (2007). Human glutathione transferases catalyzing the bioactivation of anticancer thiopurine prodrugs. Biochem. Pharmacol. 73, 1829–1841. doi: 10.1016/j.bcp.2007.02.002
Elfarra, A. A., Duescher, R. J., Hwang, I. Y., Sicuri, A. R., and Nelson, J. A. (1995). Targeting 6-thioguanine to the kidney with S-(guanin-6-yl)-L-cysteine. J. Pharmacol. Exp. Ther. 274, 1298–1304.
Elfarra, A. A., Jakobson, I., and Anders, M. W. (1986). Mechanism of S-(1,2-dichlorovinyl)glutathione-induced nephrotoxicity. Biochem. Pharmacol. 35, 283–288. doi: 10.1016/0006-2952(86)90527-7
Enoiu, M., Herber, R., Wennig, R., Marson, C., Bodaud, H., Leroy, P.,et al. (2002). γ-glutamyltranspeptidase-dependent metabolism of 4-hydroxynonenal-glutathione conjugate. Arch. Biochem. Biophys. 397, 18–27. doi: 10.1006/abbi.2001.2633
Esteller, M., Corn, P. G., Urena, J. M., Gabrielson, E., Baylin, S. B., and Herman, J. G. (1998). Inactivation of glutathione S-transferase P1 gene by promoter hypermethylation in human neoplasia. Cancer Res. 58, 4515–4518.
Fan, Y., Zhang, W., Shi, C. Y., and Cai, D. F. (2013). Associations of GSTM1 and GSTT1 polymorphisms with pancreatic cancer risk: evidence from a meta-analysis. Tumor Biol. 34, 705–712. doi: 10.1007/s13277-012-0598-6
Fiala, S., Trout, E. C. Jr., Teague, C. A., and Fiala, E. S. (1980). γ-Glutamyltransferase, a common marker of human epithelial tumors? Cancer Detect. Prev. 3, 471–485.
Findlay, V. J., Townsend, D. M., Saavedra, J. E., Buzard, G. S., Citro, M. L., Keefer, L. K.,et al. (2004). Tumor cell responses to a novel glutathione S-transferase-activated nitric oxide-releasing prodrug. Mol. Pharmacol. 65, 1070–1079. doi: 10.1124/mol.65.5.1070
Firdous, A. P., and Kuttan, R. (2012). Amelioration of cisplatin-induced toxicity in mice by carotenoid meso-zeaxanthin. Hum. Exp. Toxicol. 31, 710–717. doi: 10.1177/0960327111431707
Forrester, L. M., Hayes, J. D., Millis, R., Barnes, D., Harris, A. L., Schlager, J. J.,et al. (1990). Expression of glutathione S-transferases and cytochrome P450 in normal and tumor breast tissue. Carcinogenesis 11, 2163–2170. doi: 10.1093/carcin/11.12.2163
Franzini, M., Corti, A., Lorenzini, E., Paolicchi, A., Pompella, A., De Cesare, M.,et al. (2006). Modulation of cell growth and cisplatin sensitivity by membrane γ-glutamyltransferase in melanoma cells. Eur. J. Cancer 42, 2623–2630. doi: 10.1016/j.ejca.2006.04.016
Fu, J., Liu, L., Huang, Z., Lai, Y., Ji, H., Peng, S.,et al. (2013). Hybrid molecule from O2-(2,4-dinitrophenyl)diazeniumdiolate and oleanolic acid: a glutathione S-transferaseπ-activated nitric oxide prodrug with selective anti-human hepatocellular carcinoma activity and improved stability. J. Med. Chem. 56, 4641–4655. doi: 10.1021/jm400393u
Fujisawa, K., Kurihara, N., Nishikawa, H., Kimura, A., and Kojima, M. (1976). Carcinoembryonic character of γ glutamyltranspeptidase in primary hepatocellular carcinoma. Gastroenterol. Jpn. 11, 380–386. doi: 10.1007/BF02777380
Furuhashi, S., Sugita, H., Takamori, H., Horino, K., Nakahara, O., Okabe, H.,et al. (2012). No donor and Mek inhibitor synergistically inhibit proliferation and invasion of cancer cells. Int. J. Oncol. 40, 807–815. doi: 10.3892/ijo.2011.1243
Garnier, N., Petruccelli, L. A., Molina, M. F., Kourelis, M., Kwan, S., Diaz, Z.,et al. (2013). The novel arsenical Darinaparsin circumvents Brg1-dependent, Ho-1-mediated cytoprotection in leukemic cells. Leukemia 27, 2220–2228. doi: 10.1038/leu.2013.54
Garnier, N., Redstone, G. G., Dahabieh, M. S., Nichol, J. N., Del Rincon, S. V., Gu, Y.,et al. (2014). The novel arsenical darinaparsin is transported by cystine importing systems. Mol. Pharmacol. 85, 576–585. doi: 10.1124/mol.113.089433
Gaspar, J., Rodrigues, S., Gil, O. M., Manita, I., Ferreira, T. C., Limbert, E.,et al. (2004). Combined effects of glutathione S-transferase polymorphisms and thyroid cancer risk. Cancer Genet. Cytogenet. 151, 60–67. doi: 10.1016/j.cancergencyto.2003.09.018
Gerber, M. A., and Thung, S. N. (1980). Enzyme patterns in human hepatocellular carcinoma. Am. J. Pathol. 98, 395–400.
Ghosh, P., Roy, S. S., Chakraborty, P., Ghosh, S., and Bhattacharya, S. (2013). Effects of organoselenium compound 2-(5-selenocyanatopentyl)- benzo[de]isoquinoline 1,3-dione on cisplatin induced nephrotoxicity and genotoxicity: an investigation of the influence of the compound on oxidative stress and antioxidant enzyme system. Biometals 26, 61–73. doi: 10.1007/s10534-012-9594-y
Giommarelli, C., Corti, A., Supino, R., Favini, E., Paolicchi, A., Pompella, A.,et al. (2008). Cellular response to oxidative stress and ascorbic acid in melanoma cells overexpressing γ-glutamyltransferase. Eur. J. Cancer 44, 750–759. doi: 10.1016/j.ejca.2008.02.010
Grimm, C., Hofstetter, G., Aust, S., Mutz-Dehbalaie, I., Bruch, M., Heinze, G.,et al. (2013). Association of γ-glutamyltransferase with severity of disease at diagnosis and prognosis of ovarian cancer. Br. J. Cancer 109, 610–614. doi: 10.1038/bjc.2013.323
Guan, X., Hoffman, B. N., Mcfarland, D. C., Gilkerson, K. K., Dwivedi, C., Erickson, A. K.,et al. (2002). Glutathione and mercapturic acid conjugates of sulofenur and their activity against a human colon cancer cell line. Drug Metab. Dispos. 30, 331–335. doi: 10.1124/dmd.30.3.331
Guilbert, C., Annis, M. G., Dong, Z., Siegel, P. M., Miller, W. H. Jr., and Mann, K. K. (2013). Arsenic trioxide overcomes rapamycin-induced feedback activation of akt and erk signaling to enhance the anti-tumor effects in breast cancer. PLoS ONE 8:e85995. doi: 10.1371/journal.pone.0085995
Gunnarsdottir, S., and Elfarra, A. A. (2004). Cytotoxicity of the novel glutathione-activated thiopurine prodrugs cis-AVTP [cis-6-(2-acetylvinylthio)purine] and trans-AVTG [trans-6-(2-acetylvinylthio)guanine] results from the National Cancer Institute’s anticancer drug screen. Drug Metab. Dispos. 32, 321–327. doi: 10.1124/dmd.32.3.321
Gunnarsdottir, S., Rucki, M., and Elfarra, A. A. (2002). Novel glutathione-dependent thiopurine prodrugs: evidence for enhanced cytotoxicity in tumor cells and for decreased bone marrow toxicity in mice. J. Pharmacol. Exp. Ther. 301, 77–86. doi: 10.1124/jpet.301.1.77
Halestrap, A. P., Mcstay, G. P., and Clarke, S. J. (2002). The permeability transition pore complex: another view. Biochimie 84, 153–166. doi: 10.1016/S0300-9084(02)01375-5
Hamada, S., Kamada, M., Furumoto, H., Hirao, T., and Aono, T. (1994). Expression of glutathione S-transferase-pi in human ovarian cancer as an indicator of resistance to chemotherapy. Gynecol. Oncol. 52, 313–319. doi: 10.1006/gyno.1994.1055
Hamilton, D. S., Ding, Z., Ganem, B., and Creighton, D. J. (2002). Glutathionyl transferase catalyzed addition of glutathione to Comc: a new hypothesis for antitumor activity. Org. Lett. 4, 1209–1212. doi: 10.1021/ol025650h
Hamilton, D. S., Zhang, X., Ding, Z., Hubatsch, I., Mannervik, B., Houk, K. N.,et al. (2003). Mechanism of the glutathione transferase-catalyzed conversion of antitumor 2-crotonyloxymethyl-2-cycloalkenones to Gsh adducts. J. Am. Chem. Soc. 125, 15049–15058. doi: 10.1021/ja030396p
Hanigan, M. H. (1995). Expression of γ-glutamyl transpeptidase provides tumor cells with a selective growth advantage at physiologic concentrations of cyst(e)ine. Carcinogenesis 16, 181–185. doi: 10.1093/carcin/16.2.181
Hanigan, M. H., and Frierson, H. F. Jr. (1996). Immunohistochemical detection of γ-glutamyl transpeptidase in normal human tissue. J. Histochem. Cytochem. 44, 1101–1108. doi: 10.1177/44.10.8813074
Hanigan, M. H., Frierson, H. F. Jr., Brown, J. E., Lovell, M. A., and Taylor, P. T. (1994). Human ovarian tumors express γ-glutamyl transpeptidase. Cancer Res. 54, 286–290.
Hanigan, M. H., Frierson, H. F. Jr., Swanson, P. E., and De Young, B. R. (1999a). Altered expression of γ-glutamyl transpeptidase in human tumors. Hum. Pathol. 30, 300–305. doi: 10.1016/S0046-8177(99)90009-6
Hanigan, M. H., Gallagher, B. C., Townsend, D. M., and Gabarra, V. (1999b). γ-glutamyl transpeptidase accelerates tumor growth and increases the resistance of tumors to cisplatin in vivo. Carcinogenesis 20, 553–559. doi: 10.1093/carcin/20.4.553
Hao, X. Y., Widersten, M., Ridderstrom, M., Hellman, U., and Mannervik, B. (1994). Co-variation of glutathione transferase expression and cytostatic drug resistance in HeLa cells: establishment of class Mu glutathione transferase M3-3 as the dominating isoenzyme. Biochem. J. 297(Pt 1), 59–67.
Hayden, P. J., and Stevens, J. L. (1990). Cysteine conjugate toxicity, metabolism, and binding to macromolecules in isolated rat kidney mitochondria. Mol. Pharmacol. 37, 468–476.
Henson, S. E., Nichols, T. C., Holers, V. M., and Karp, D. R. (1999). The ectoenzyme γ-glutamyl transpeptidase regulates antiproliferative effects of S-nitrosoglutathione on human T and B lymphocytes. J. Immunol. 163, 1845–1852.
Hetland, T. E., Nymoen, D. A., Emilsen, E., Kærn, J., Tropé, C. G., Florenes, V. A.,et al. (2012). MGST1 expression in serous ovarian carcinoma differs at various anatomic sites, but is unrelated to chemoresistance or survival. Gynecol. Oncol. 126, 460–465. doi: 10.1016/j.ygyno.2012.05.029
Hinchman, C. A., Rebbeor, J. F., and Ballatori, N. (1998). Efficient hepatic uptake and concentrative biliary excretion of a mercapturic acid. Am. J. Physiol. 275, G612–G619.
Ho, Y. S., Liu, H. L., Duh, J. S., Chen, R. J., Ho, W. L., Jeng, J. H.,et al. (1999). Induction of apoptosis by S-nitrosoglutathione and Cu2+ or Ni2+ ion through modulation of bax, bad, and bcl-2 proteins in human colon adenocarcinoma cells. Mol. Carcinog. 26, 201–211. doi: 10.1002/(SICI)1098-2744(199911)26:3<201::AID-MC9>3.0.CO;2-K
Hochwald, S. N., Harrison, L. E., Rose, D. M., Anderson, M., and Burt, M. E. (1996). γ-glutamyl transpeptidase mediation of tumor glutathione utilization in vivo. J. Natl. Cancer Inst. 88, 193–197. doi: 10.1093/jnci/88.3-4.193
Hogg, N., Singh, R. J., Konorev, E., Joseph, J., and Kalyanaraman, B. (1997). S-nitrosoglutathione as a substrate for γ-glutamyl transpeptidase. Biochem. J. 323, 477–481.
Horsley, L., Cummings, J., Middleton, M., Ward, T., Backen, A., Clamp, A.,et al. (2013). A phase 1 trial of intravenous 4-(N-(S-glutathionylacetyl)amino) phenylarsenoxide (GSAO) in patients with advanced solid tumours. Cancer Chemother. Pharmacol. 72, 1343–1352. doi: 10.1007/s00280-013-2320-9
Hosein, P. J., Craig, M. D., Tallman, M. S., Boccia, R. V., Hamilton, B. L., Lewis, J. J.,et al. (2012). A multicenter phase II study of darinaparsin in relapsed or refractory Hodgkin’s and non-Hodgkin’s lymphoma. Am. J. Hematol. 87, 111–114. doi: 10.1002/ajh.22232
Howbert, J. J., Grossman, C. S., Crowell, T. A., Rieder, B. J., Harper, R. W., Kramer, K. E.,et al. (1990). Novel agents effective against solid tumors: the diarylsulfonylureas. Synthesis, activities, and analysis of quantitative structure-activity relationships. J. Med. Chem. 33, 2393–2407. doi: 10.1021/jm00171a013
Howie, A. F., Forrester, L. M., Glancey, M. J., Schlager, J. J., Powis, G., Beckett, G. J.,et al. (1990). Glutathione S-transferase and glutathione peroxidase expression in normal and tumour human tissues. Carcinogenesis 11, 451–458. doi: 10.1093/carcin/11.3.451
Huttunen, K. M., and Rautio, J. (2011). Prodrugs - An efficient way to breach delivery and targeting barriers. Curr. Top. Med. Chem. 11, 2265–2287. doi: 10.2174/156802611797183230
Hwang, I. Y., and Elfarra, A. A. (1991). Kidney-selective prodrugs of 6-mercaptopurine: biochemical basis of the kidney selectivity of S-(6-purinyl)-L-cysteine and metabolism of new analogs in rats. J. Pharmacol. Exp. Ther. 258, 171–177.
Iwanami, A., Cloughesy, T. F., Cavenee, W. K., and Mischel, P. S. (2013). Arsenic reverses glioblastoma resistance to mtor-targeted therapies. Cell Cycle 12, 1473–1474. doi: 10.4161/cc.24747
Jensen, G. L., and Meister, A. (1983). Radioprotection of human lymphoid cells by exogenously supplied glutathione is mediated by γ-glutamyl transpeptidase. Proc. Natl. Acad. Sci. U.S.A. 80, 4714–4717. doi: 10.1073/pnas.80.15.4714
Jerremalm, E., Wallin, I., Yachnin, J., and Ehrsson, H. (2006). Oxaliplatin degradation in the presence of important biological sulphur-containing compounds and plasma ultrafiltrate. Eur. J. Pharm. Sci. 28, 278–283. doi: 10.1016/j.ejps.2006.03.001
Jochheim, C. M., Davis, M. R., Baillie, K. M., Ehlhardt, W. J., and Baillie, T. A. (2002). Glutathione-dependent metabolism of the antitumor agent sulofenur. Evidence for the formation of p-chlorophenyl isocyanate as a reactive intermediate. Chem. Res. Toxicol. 15, 240–248. doi: 10.1021/tx0155698
Jung, H. J., Chen, Z., and Mccarty, N. (2012). Synergistic anticancer effects of arsenic trioxide with bortezomib in mantle cell lymphoma. Am. J. Hematol. 87, 1057–1064. doi: 10.1002/ajh.23317
Kamthan, A., Scarffe, J. H., Walling, J., Hatty, S., Peters, B., Coleman, R.,et al. (1992). A phase II study of sulofenur (Ly186641) in gastric cancer. Anticancer Drugs 3, 331–335. doi: 10.1097/00001813-199208000-00003
Kavanagh, J. J., Levenback, C. F., Ramirez, P. T., Wolf, J. L., Moore, C. L., Jones, M. R.,et al. (2010). Phase 2 study of canfosfamide in combination with pegylated liposomal doxorubicin in platinum and paclitaxel refractory or resistant epithelial ovarian cancer. J. Hematol. Oncol. 3, 9. doi: 10.1186/1756-8722-3-9
Kirsch, M., Lehnig, M., Korth, H. G., Sustmann, R., and De Groot, H. (2001). Inhibition of peroxynitrite-induced nitration of tyrosine by glutathione in the presence of carbon dioxide through both radical repair and peroxynitrate formation. Chemistry 7, 3313–3320. doi: 10.1002/1521-3765(20010803)7:15<3313::AID-CHEM3313>3.0.CO;2-7
Kiziltepe, T., Hideshima, T., Ishitsuka, K., Ocio, E. M., Raje, N., Catley, L.,et al. (2007). JS-K, a GST-activated nitric oxide generator, induces DNA double-strand breaks, activates DNA damage response pathways, and induces apoptosis in vitro and in vivo in human multiple myeloma cells. Blood 110, 709–718. doi: 10.1182/blood-2006-10-052845
Knickelbein, R. G., Ingbar, D. H., Seres, T., Snow, K., Johnston R. B. Jr., Fayemi, O.,et al. (1996). Hyperoxia enhances expression of γ-glutamyl transpeptidase and increases protein S-glutathiolation in rat lung. Am. J. Physiol. 270, L115–L122.
Koeplinger, K. A., Zhao, Z., Peterson, T., Leone, J. W., Schwende, F. S., Heinrikson, R. L.,et al. (1999). Activated sulfonamides are cleaved by glutathione-S-transferases. Drug Metab. Dispos. 27, 986–991.
Kryeziu, K., Jungwirth, U., Hoda, M. A., Ferk, F., Knasmüller, S., Karnthaler-Benbakka, C.,et al. (2013). Synergistic anticancer activity of arsenic trioxide with erlotinib is based on inhibition of EGFR-mediated DNA double-strand break repair. Mol. Cancer Ther. 12, 1073–1084. doi: 10.1158/1535-7163.MCT-13-0065
Kugelman, A., Choy, H. A., Liu, R., Shi, M. M., Gozal, E., and Forman, H. J. (1994). γ-Glutamyl transpeptidase is increased by oxidative stress in rat alveolar L2 epithelial cells. Am. J. Respir. Cell Mol. Biol. 11, 586–592. doi: 10.1165/ajrcmb.11.5.7946387
Kumar, V., Yadav, C. S., Datta, S. K., Singh, S., Ahmed, R. S., Goel, S.,et al. (2011). Association of GSTM1 and GSTT1 polymorphism with lipid peroxidation in benign prostate hyperplasia and prostate cancer: a pilot study. Dis. Markers 30, 163–169. doi: 10.1155/2011/624961
La Thangue, N. B., and Kerr, D. J. (2011). Predictive biomarkers: a paradigm shift towards personalized cancer medicine. Nat. Rev. Clin. Oncol. 8, 587–596. doi: 10.1038/nrclinonc.2011.121
Lafuente, A., Giralt, M., Cervello, I., Pujol, F., and Mallol, J. (1990). Glutathione-S-transferase activity in human superficial transitional cell carcinoma of the bladder comparison with healthy controls. Cancer 65, 2064–2068. doi: 10.1002/1097-0142(19900501)65:9<2064::AID-CNCR2820650929>3.0.CO;2-2
Lallemand-Breitenbach, V., Zhu, J., Chen, Z., and De The, H. (2012). Curing Apl through Pml/Rara degradation by As2O3. Trends Mol. Med. 18, 36–42. doi: 10.1016/j.molmed.2011.10.001
Laschak, M., Spindler, K. D., Schrader, A. J., Hessenauer, A., Streicher, W., Schrader, M.,et al. (2012). JS-K, a glutathione/glutathione S-transferase-activated nitric oxide releasing prodrug inhibits androgen receptor and WNT-signaling in prostate cancer cells. BMC Cancer 12:130. doi: 10.1186/1471-2407-12-130
Lash, L. H., Elfarra, A. A., and Anders, M. W. (1986). Renal cysteine conjugate β-lyase. Bioactivation of nephrotoxic cysteine S-conjugates in mitochondrial outer membrane. J. Biol. Chem. 261, 5930–5935.
Lash, L. H., Shivnani, A., Mai, J., Chinnaiyan, P., Krause, R. J., and Elfarra, A. A. (1997). Renal cellular transport, metabolism, and cytotoxicity of S-(6-purinyl)glutathione, a prodrug of 6-mercaptopurine, and analogues. Biochem. Pharmacol. 54, 1341–1349. doi: 10.1016/S0006-2952(97)00401-2
Lee, J. S. (2007). GSTP1 promoter hypermethylation is an early event in breast carcinogenesis. Virchows Archiv. 450, 637–642. doi: 10.1007/s00428-007-0421-8
Lee, M. R. (1990). Five years’ experience with γ-L-glutamyl-L-dopa: a relatively renally specific dopaminergic prodrug in man. J. Auton. Pharmacol. 10, s103–s108. doi: 10.1111/j.1474-8673.1990.tb00235.x
Lewis, A. D., Hayes, J. D., and Wolf, C. R. (1988). Glutathione and glutathione-dependent enzymes in ovarian adenocarcinoma cell lines derived from a patient before and after the onset of drug resistance: intrinsic differences and cell cycle effects. Carcinogenesis 9, 1283–1287. doi: 10.1093/carcin/9.7.1283
Li, J., Long, J., Hu, Y., Tan, A., Guo, X., and Zhang, S. (2012). Glutathione S-transferase M1, T1, and P1 polymorphisms and thyroid cancer risk: a meta-analysis. Cancer Epidemiol. 36, e333–e340. doi: 10.1016/j.canep.2012.06.002
Liu, D., Liu, Y., Ran, L., Shang, H., and Li, D. (2013). Gstt1 and Gstm1 polymorphisms and prostate cancer risk in Asians: a systematic review and meta-analysis. Tumor Biol. 34, 2539–2544. doi: 10.1007/s13277-013-0778-z
Liu, L., Fu, J., Li, T., Cui, R., Ling, J., Yu, X.,et al. (2012). Ng, a novel PABA/NO-based oleanolic acid derivative, induces Human hepatoma cell apoptosis via a ROS/MAPK-dependent mitochondrial pathway. Eur. J. Pharmacol 691, 61–68. doi: 10.1016/j.ejphar.2012.07.031
Liu, Q., Chan, S. T. F., and Mahendran, R. (2003). Nitric oxide induces cyclooxygenase expression and inhibits cell growth in colon cancer cell lines. Carcinogenesis 24, 637–642. doi: 10.1093/carcin/bgg014
Liu, R. M., Shi, M. M., Giulivi, C., and Forman, H. J. (1998). Quinones increase γ-glutamyl transpeptidase expression by multiple mechanisms in rat lung epithelial cells. Am. J. Physiol. 274, L330–L336.
Lu, S. C. (2013). Glutathione synthesis. Biochim. Biophys. Acta 1830, 3143–3153. doi: 10.1016/j.bbagen.2012.09.008
Lyttle, M. H., Satyam, A., Hocker, M. D., Bauer, K. E., Caldwell, C. G., Hui, H. C.,et al. (1994). Glutathione-S-transferase activates novel alkylating agents. J. Med. Chem. 37, 1501–1507. doi: 10.1021/jm00036a016
Maellaro, E., Dominici, S., Del Bello, B., Valentini, M. A., Pieri, L., Perego, P.,et al. (2000). Membrane γ-glutamyl transpeptidase activity of melanoma cells: effects on cellular H2O2 production, cell surface protein thiol oxidation and NF-κB activation status. J. Cell Sci. 113, 2671–2678.
Mahjoubi, M., Kattan, J., Bonnay, M., Schmitt, H., and Droz, J. P. (1993). Phase II trial of LY 186641 in advanced renal cancer. Invest. New Drugs 11, 323–328. doi: 10.1007/BF00874431
Mannervik, B., Board, P. G., Hayes, J. D., Listowsky, I., and Pearson, W. R. (2005). Nomenclature for mammalian soluble glutathione transferases. Methods Enzymol. 401, 1–8. doi: 10.1016/S0076-6879(05)01001-3
Mareš, V., Stremeòová, J., Lisá, V., Kozáková, H., Marek, J., Syrùèek, M.,et al. (2012). Compartment- and malignance-dependent up-regulation of γ-glutamyltranspeptidase and dipetidylpeptidase-IV activity in human brain gliomas. Histol. Histopathol. 27, 931–940.
Matulis, S. M., Morales, A. A., Yehiayan, L., Croutch, C., Gutman, D., Cai, Y.,et al. (2009). Darinaparsin induces a unique cellular response and is active in an arsenic trioxide-resistant myeloma cell line. Mol. Cancer Ther. 8, 1197–1206. doi: 10.1158/1535-7163.MCT-08-1072
McStay, G. P., Clarke, S. J., and Halestrap, A. P. (2002). Role of critical thiol groups on the matrix surface of the adenine nucleotide translocase in the mechanism of the mitochondrial permeability transition pore. Biochem. J. 367, 541–548. doi: 10.1042/BJ20011672
Misicka, A., Maszczynska, I., Lipkowski, A. W., Stropova, D., Yamamura, H. I., and Hruby, V. J. (1996). Synthesis and biological properties of γ-glutamyl-dermorphin, a prodrug. Life Sci. 58, 905–911. doi: 10.1016/0024-3205(96)00033-1
Mohamadi, F., Spees, M. M., and Grindey, G. B. (1992). Sulfonylureas: a new class of cancer chemotherapeutic agents. J. Med. Chem. 35, 3012–3016. doi: 10.1021/jm00094a013
Moon, D. O., Kim, B. Y., Jang, J. H., Kim, M. O., Jayasooriya, R. G. P. T., Kang, C. H.,et al. (2012). K-Ras transformation in prostate epithelial cell overcomes H2O2-induced apoptosis via upregulation of γ-glutamyltransferase-2. Toxicol. In Vitro 26, 429–434. doi: 10.1016/j.tiv.2012.01.013
Moorghen, M., Cairns, J., Forrester, L. M., Hayes, J. D., Hall, A., Cattan, A. R.,et al. (1991). Enhanced expression of glutathione S-transferases in colorectal carcinoma compared to non-neoplastic mucosa. Carcinogenesis 12, 13–17. doi: 10.1093/carcin/12.1.13
Morgan, A. S., Sanderson, P. E., Borch, R. F., Tew, K. D., Niitsu, Y., Takayama, T.,et al. (1998). Tumor efficacy and bone marrow-sparing properties of Ter286, a cytotoxin activated by glutathione S-transferase. Cancer Res. 58, 2568–2575.
Moriarty-Craige, S. E., and Jones, D. P. (2004). Extracellular thiols and thiol/disulfide redox in metabolism. Annu. Rev. Nutr. 24, 481–509. doi: 10.1146/annurev.nutr.24.012003.132208
Moskaluk, C. A., Duray, P. H., Cowan, K. H., Linehan, M., and Merino, M. J. (1997). Immunohistochemical expression of π-class glutathione S-transferase is down-regulated in adenocarcinoma of the prostate. Cancer 79, 1595–1599. doi: 10.1002/(SICI)1097-0142(19970415)79:8<1595::AID-CNCR23>3.0.CO;2-S
Munshi, N. C., Seitz, D. E., Fossella, F., Lippman, S. M., and Einhorn, L. H. (1993). Phase II study of sulofenur (LY 186641). A novel antineoplastic agent in advanced non-small cell lung cancer. Invest New Drugs 11, 87–90. doi: 10.1007/BF00873919
Murata, J. I., Ricciardi-Castagnoli, P., Mange, P. D. L., Martin, F., and Juillerat-Jeanneret, L. (1997). Microglial cells induce cytotoxic effects toward colon carcinoma cells: measurement of tumor cytotoxicity with a γ-glutamyl transpeptidase assay. Int. J. Cancer 70, 169–174. doi: 10.1002/(SICI)1097-0215(19970117)70:2<169::AID-IJC6>3.0.CO;2-V
Nielsen, T. H., Johnson, N., Garnier, N., Kwan, S., Yao, L., Cocolakis, E.,et al. (2013). Monitoring response and resistance to the novel arsenical Darinaparsin in an AML patient. Front. Pharmacol. 4:9. doi: 10.3389/fphar.2013.00009
Noh, E. K., Kim, H., Park, M. J., Baek, J. H., Park, J. H., Cha, S. J.,et al. (2010). Gefitinib enhances arsenic trioxide (AS2O3)-induced differentiation of acute promyelocytic leukemia cell line. Leuk. Res. 34, 1501–1505. doi: 10.1016/j.leukres.2010.02.016
O’Brien, M. E., Hardy, J., Tan, S., Walling, J., Peters, B., Hatty, S.,et al. (1992). A phase II study of sulofenur, a novel sulfonylurea, in recurrent epithelial ovarian cancer. Cancer Chemother. Pharmacol. 30, 245–248. doi: 10.1007/BF00686324
O’Brien, M. L., and Tew, K. D. (1996). Glutathione and related enzymes in multidrug resistance. Eur. J. Cancer 32A, 967–978. doi: 10.1016/0959-8049(96)00051-2
Oğuztüzün, S., Sezgin, Y., Yazιcι, S., Fιrat, P., Özhavzalι, M., and Özen, H. (2011). Expression of glutathione-S-transferases isoenzymes and p53 in exfoliated human bladder cancer cells. Urol. Oncol. 29, 538–544. doi: 10.1016/j.urolonc.2009.08.001
Pacifici, G. M., Franchi, M., Bencini, C., Repetti, F., Di Lascio, N., and Muraro, G. B. (1988). Tissue distribution of drug-metabolizing enzymes in humans. Xenobiotica 18, 849–856. doi: 10.3109/00498258809041723
Pankiv, S., Møller, S., Bjørkøy, G., Moens, U., and Huseby, N. E. (2006). Radiation-induced upregulation of γ-glutamyltransferase in colon carcinoma cells is mediated through the Ras signal transduction pathway. Biochim. Biophys. Acta 1760, 151–157. doi: 10.1016/j.bbagen.2005.11.006
Paolicchi, A., Dominici, S., Pieri, L., Maellaro, E., and Pompella, A. (2002). Glutathione catabolism as a signaling mechanism. Biochem. Pharmacol. 64, 1027–1035. doi: 10.1016/S0006-2952(02)01173-5
Paolicchi, A., Pompella, A., Tonarelli, P., Gadducci, A., Genazzani, A. R., Zunino, F.,et al. (1996). Gamma-glutamyltranspeptidase activity in human ovarian carcinoma. Anticancer Res. 16, 3053–3058.
Paolicchi, A., Sotiropuolou, M., Perego, P., Daubeuf, S., Visvikis, A., Lorenzini, E.,et al. (2003). γ-Glutamyl transpeptidase catalyses the extracellular detoxification of cisplatin in a human cell line derived from the proximal convoluted tubule of the kidney. Eur. J. Cancer 39, 996–1003. doi: 10.1016/S0959-8049(03)00067-4
Park, D., Chiu, J., Perrone, G. G., Dilda, P. J., and Hogg, P. J. (2012). The tumour metabolism inhibitors GSAO and PENAO react with cysteines 57 and 257 of mitochondrial adenine nucleotide translocase. Cancer Cell Int. 12, 11. doi: 10.1186/1475-2867-12-11
Park, D., and Dilda, P. J. (2010). Mitochondria as targets in angiogenesis inhibition. Mol. Aspects Med. 31, 113–131. doi: 10.1016/j.mam.2009.12.005
Patel, N. J., Fullone, J. S., and Anders, M. W. (1993). Brain uptake of S-(1,2-dichlorovinyl)glutathione and S-(1,2-dichlorovinyl)-L-cysteine, the glutathione and cysteine S-conjugates of the neurotoxin dichloroacetylene. Mol. Brain Res. 17, 53–58. doi: 10.1016/0169-328X(93)90072-W
Peters, W. H. M., Wormskamp, N. G. M., and Thies, E. (1990). Expression of glutathione S-transferases in normal gastric mucosa and in gastric tumors. Carcinogenesis 11, 1593–1596. doi: 10.1093/carcin/11.9.1593
Pettazzoni, P., Pizzimenti, S., Toaldo, C., Sotomayor, P., Tagliavacca, L., Liu, S.,et al. (2011). Induction of cell cycle arrest and DNA damage by the Hdac inhibitor panobinostat (LBH589) and the lipid peroxidation end product 4-hydroxynonenal in prostate cancer cells. Free Radic. Biol. Med. 50, 313–322. doi: 10.1016/j.freeradbiomed.2010.11.011
Plumb, J. A., Strathdee, G., Sludden, J., Kaye, S. B., and Brown, R. (2000). Reversal of drug resistance in human tumor xenografts by 2′-deoxy-5-azacytidine-induced demethylation of the HMLH1 gene promoter. Cancer Res. 60, 6039–6044.
Pompella, A., De Tata, V., Paolicchi, A., and Zunino, F. (2006). Expression of γ-glutamyltransferase in cancer cells and its significance in drug resistance. Biochem. Pharmacol. 71, 231–238. doi: 10.1016/j.bcp.2005.10.005
Prezioso, J. A., Damodaran, K. M., Wang, N., and Bloomer, W. D. (1993). Mechanism(s) regulating inhibition of thymidylate synthase and growth by γ-L-glutaminyl-4-hydroxy-3-iodobenzene, a novel melanin precursor, in melanogenic melanoma cells. Biochem. Pharmacol. 45, 473–481. doi: 10.1016/0006-2952(93)90085-B
Prezioso, J. A., Hughey, R. P., Wang, N., Damodaran, K. M., and Bloomer, W. D. (1994a). γ-Glutamyltranspeptidase expression regulates the growth-inhibitory activity of the anti-tumor prodrug γ-L-glutaminyl-4-hydroxy-3-iodobenzene. Int. J. Cancer 56, 874–879. doi: 10.1002/ijc.2910560620
Prezioso, J. A., Shields, D., Wang, N., and Rosenstein, M. (1994b). Role of γ-glutamyltranspeptidase-mediated glutathione transport on the radiosensitivity of B16 melanoma variant cell lines. Int. J. Radiat. Oncol. Biol. Phys. 30, 373–381. doi: 10.1016/0360-3016(94)90017-5
Ramsay, E. E., Decollogne, S., Joshi, S., Corti, A., Apte, M., Pompella, A.,et al. (2014). Employing pancreatic tumor γ-glutamyltransferase for therapeutic delivery. Mol. Pharm. 11, 1500–1511. doi: 10.1021/mp400664t
Rebbeck, T. R. (1997). Molecular epidemiology of the human glutathione S-transferase genotypes GSTM1 and GSTT1 in cancer susceptibility. Cancer Epidemiol. Biomarkers Prev. 6, 733–743.
Rebbeck, T. R., Walker, A. H., Jaffe, J. M., White, D. L., Wein, A. J., and Malkowicz, S. B. (1999). Glutathione S-transferase-μ (Gstm1) and -𝜃 (Gstt1) genotypes in the etiology of prostate cancer. Cancer Epidemiol. Biomarkers Prev. 8, 283–287.
Roomi, M. W., Gaal, K., Yuan, Q. X., French, B. A., Fu, P., Bardag-Gorce, F.,et al. (2006). Preneoplastic liver cell foci expansion induced by thioacetamide toxicity in drug-primed mice. Exp. Mol. Pathol. 81, 8–14. doi: 10.1016/j.yexmp.2006.02.006
Ruzza, P., Rosato, A., Rossi, C. R., Floreani, M., and Quintieri, L. (2009). Glutathione transferases as targets for cancer therapy. Anticancer Agents Med. Chem. 9, 763–777. doi: 10.2174/187152009789056895
Sabatino, M. A., Geroni, C., Ganzinelli, M., Ceruti, R., and Broggini, M. (2013). Zebularine partially reverses GST methylation in prostate cancer cells and restores sensitivity to the DNA minor groove binder brostallicin. Epigenetics 8. doi: 10.4161/epi.24916
Sadiq, S., Berndt, T. J., Nath, K. A., and Knox, F. G. (2000). Effect of γ-L-glutamyl-L-DOPA on phosphate excretion. J. Lab. Clin. Med. 135, 52–56. doi: 10.1016/S0022-2143(00)70020-5
Satyam, A., Hocker, M. D., Kane-Maguire, K. A., Morgan, A. S., Villar, H. O., and Lyttle, M. H. (1996). Design, synthesis, and evaluation of latent alkylating agents activated by glutathione S-transferase. J. Med. Chem. 39, 1736–1747. doi: 10.1021/jm950005k
Saxena, A., Dhillon, V. S., Shahid, M., Khalil, H. S., Rani, M., Das, T. P.,et al. (2012). GSTP1 methylation and polymorphism increase the risk of breast cancer and the effects of diet and lifestyle in breast cancer patients. Exp. Ther. Med. 4, 1097–1103. doi: 10.3892/etm.2012.710
Schäfer, C., Fels, C., Brucke, M., Holzhausen, H. J., Bahn, H., Wellman, M.,et al. (2001). Gamma-glutamyl transferase expression in higher-grade astrocytic glioma. Acta Oncol. 40, 529–535. doi: 10.1080/028418601750288271
Sener, M. T., Sener, E., Tok, A., Polat, B., Cinar, I., Polat, H.,et al. (2012). Biochemical and histologic study of lethal cisplatin nephrotoxicity prevention by mirtazapine. Pharmacol. Rep. 64, 594–602. doi: 10.1016/S1734-1140(12)70855-1
Sequist, L. V., Fidias, P. M., Temel, J. S., Kolevska, T., Rabin, M. S., Boccia, R. V.,et al. (2009). Phase 1-2a multicenter dose-ranging study of canfosfamide in combination with carboplatin and paclitaxel as first-line therapy for patients with advanced non-small cell lung cancer. J. Thorac. Oncol. 4, 1389–1396. doi: 10.1097/JTO.0b013e3181b6b84b
Shami, P. J., Saavedra, J. E., Wang, L. Y., Bonifant, C. L., Diwan, B. A., Singh, S. V.,et al. (2003). JS-K, a glutathione/glutathione S-transferase-activated nitric oxide donor of the diazeniumdiolate class with potent antineoplastic activity. Mol. Cancer Ther. 2, 409–417.
Shiozawa, M., Yamashita, S., Aiso, S., and Yasuda, K. (1989). A monoclonal antibody against human kidney γ-glutamyl transpeptidase: preparation, immunochemical, and immunohistochemical characterization. J. Histochem. Cytochem. 37, 1053–1061. doi: 10.1177/37.7.2567299
Song, K., Yi, J., Shen, X., and Cai, Y. (2012). Genetic polymorphisms of glutathione S-transferase genes GSTM1, GSTT1 and risk of hepatocellular carcinoma. PLoS ONE 7:e48924. doi: 10.1371/journal.pone.0048924
Spencer, P. S., and Schaumburg, H. H. (1985). Organic solvent neurotoxicity: facts and research needs. Scand. J. Work Environ. Health 11, 53–60.
Stark, A. A., and Glass, G. A. (1997). Role of copper and ceruloplasmin in oxidative mutogenesis induced by the glutathione-γ-glutamyl transpeptidase system and by other thiols. Environ. Mol. Mutagen. 29, 63–72. doi: 10.1002/(SICI)1098-2280 (1997)29:1<63::AID-EM9>3.0.CO;2-E
Stevens, J. L., Hatzinger, P. B., and Hayden, P. J. (1989). Quantitation of multiple pathways for the metabolism of nephrotoxic cysteine conjugates using selective inhibitors of L-α-hydroxy acid oxidase (L-amino acid oxidase) and cysteine conjugate β-lyase. Drug Metab. Dispos. 17, 297–303.
Supino, R., Mapelli, E., Sanfilippo, O., and Silvestro, L. (1992). Biological and enzymatic features of human melanoma clones with different invasive potential. Melanoma Res. 2, 377–384. doi: 10.1097/00008390-199212000-00012
Talbot, D. C., Smith, I. E., Nicolson, M. C., Powles, T. J., Button, D., and Walling, J. (1993). Phase II trial of the novel sulphonylurea sulofenur in advanced breast cancer. Cancer Chemother. Pharmacol. 31, 419–422. doi: 10.1007/BF00686159
Tateishi, N., Higashi, T., Nomura, T., Naruse, A., and Nakashima, K. (1976). Higher transpeptidation activity and broad acceptor specificity of γ glutamyltransferases of tumors. Gann 67, 215–222.
Terracini, B., and Parker, V. H. (1965). A pathological study on the toxicity of S-dichlorovinyl-L-cysteine. Food Cosmet. Toxicol. 3, 67–74. doi: 10.1016/S0015-6264(65)80010-4
Tian, J., Doi, H., Saar, M., Santos, J., Li, X., Peehl, D. M.,et al. (2013). Radioprotection and cell cycle arrest of intestinal epithelial cells by darinaparsin, a tumor radiosensitizer. Int. J. Radiat. Oncol. Biol. Phys. 87, 1179–1185. doi: 10.1016/j.ijrobp.2013.08.051
Tian, J., Zhao, H., Nolley, R., Reese, S. W., Young, S. R., Li, X.,et al. (2012). Darinaparsin: solid tumor hypoxic cytotoxin and radiosensitizer. Clin. Cancer Res 18, 3366–3376. doi: 10.1158/1078-0432.CCR-11-3179
Townsend, D. M., Deng, M., Zhang, L., Lapus, M. G., and Hanigan, M. H. (2003). Metabolism of Cisplatin to a nephrotoxin in proximal tubule cells. J. Am. Soc. Nephrol. 14, 1–10. doi: 10.1097/01.ASN.0000042803.28024.92
Townsend, D. M., Shen, H., Staros, A. L., Gaté, L., and Tew, K. D. (2002). Efficacy of a glutathione S-transferase π-activated prodrug in platinum-resistant ovarian cancer cells. Mol. Cancer Ther. 1, 1089–1095.
Tsimberidou, A. M., Camacho, L. H., Verstovsek, S., Ng, C., Hong, D. S., Uehara, C. K.,et al. (2009). A phase I clinical trial of darinaparsin in patients with refractory solid tumors. Clin. Cancer Res 15, 4769–4776. doi: 10.1158/1078-0432.CCR-08-2984
Tsoli, M., Luk, P., Dilda, P. J., Haber, M. & Ziegler, D. (2013). “Targeting mitochondria and metabolism as a novel therapeutic approach in the treatment of diffuse intrinsic pontine glioma,” in Proceedings of the 2nd Annual Pediatric Neuro-Oncology Basic and Translational Research Conference, Neuro-Oncology, Fort Lauderdale, FL, 15, 21.
Tsuchida, S., Sekine, Y., Shineha, R., Nishihira, T., and Sato, K. (1989). Elevation of the placental glutathione S-transferase form (Gst-π) in tumor tissues and the levels in sera of patients with cancer. Cancer Res. 49, 5225–5229.
Tsutsumi, M., Sakamuro, D., Takada, A., Zang, S. C., Furukawa, T., and Taniguchi, N. (1996). Detection of a unique γ-glutamyl transpeptidase messenger Rna species closely related to the development of hepatocellular carcinoma in humans: a new candidate for early diagnosis of hepatocellular carcinoma. Hepatology 23, 1093–1097.
Turchi, J. J. (2006). Nitric oxide and cisplatin resistance: NO easy answers. Proc. Natl. Acad. Sci. U.S.A. 103, 4337–4338. doi: 10.1073/pnas.0601001103
Vergote, I., Finkler, N. J., Hall, J. B., Melnyk, O., Edwards, R. P., Jones, M.,et al. (2010). Randomized phase III study of canfosfamide in combination with pegylated liposomal doxorubicin compared with pegylated liposomal doxorubicin alone in platinum-resistant ovarian cancer. Int. J. Gynecol. Cancer 20, 772–780. doi: 10.1111/IGC.0b013e3181daaf59
Wang, Y., Teicher, B. A., Shea, T. C., Holden, S. A., Rosbe, K. W., Al-Achi, A.,et al. (1989). Cross-resistance and glutathione-S-transferase-π levels among four human melanoma cell lines selected for alkylating agent resistance. Cancer Res. 49, 6185–6192.
Wang, Z., He, W., Yang, G., Wang, J., Wang, Z., Nesland, J. M.,et al. (2010). Decreased expression of GST pi is correlated with a poor prognosis in human esophageal squamous carcinoma. BMC Cancer 10:352. doi: 10.1186/1471-2407-10-352
Waxman, S., and Anderson, K. C. (2001). History of the development of arsenic derivatives in cancer therapy. Oncologist 6(Suppl. 2), 3–10. doi: 10.1634/theoncologist.6-suppl_2-3
Wilk, S., Mizoguchi, H., and Orlowski, M. (1978). γ-glutamyl dopa: a kidney-specific dopamine precursor. J. Pharmacol. Exp. Ther. 206, 227–232.
Worth, D. P., Harvey, J. N., Brown, J., and Lee, M. R. (1985). γ-L-glutamyl-L-dopa is a dopamine pro-drug, relatively specific for the kidney in normal subjects. Clin. Sci. 69, 207–214.
Wu, J., Henderson, C., Feun, L., Van Veldhuizen, P., Gold, P., Zheng, H.,et al. (2010). Phase II study of darinaparsin in patients with advanced hepatocellular carcinoma. Invest. New Drugs 28, 670–676. doi: 10.1007/s10637-009-9286-9
Yoon, H. Y., Kim, Y. W., Kang, H. W., Kim, W. T., Yun, S. J., Lee, S. C.,et al. (2012). DNA methylation of GSTP1 in human prostate tissues: pyrosequencing analysis. Korean J. Urol. 53, 200–205. doi: 10.4111/kju.2012.53.3.200
Zhang, L., and Hanigan, M. H. (2003). Role of cysteine S-conjugate β-lyase in the metabolism of cisplatin. J. Pharmacol. Exp. Ther. 306, 988–994. doi: 10.1124/jpet.103.052225
Zhang, Q., Kulczynska, A., Webb, D. J., Megson, I. L., and Botting, N. P. (2013a). A new class of NO-donor pro-drugs triggered by γ-glutamyl transpeptidase with potential for reno-selective vasodilatation. Chem. Commun. 49, 1389–1391. doi: 10.1039/c2cc38382a
Zhang, Q., Milliken, P., Kulczynska, A., Slawin, A. M. Z., Gordon, A., Kirkby, N. S.,et al. (2013b). Development and characterization of glutamyl-protected N-hydroxyguanidines as reno-active nitric oxide donor drugs with therapeutic potential in acute renal failure. J. Med. Chem. 56, 5321–5334. doi: 10.1021/jm400146r
Zhao, Z., Koeplinger, K. A., Peterson, T., Conradi, R. A., Burton, P. S., Suarato, A.,et al. (1999). Mechanism, structure-activity studies, and potential applications of glutathione S-transferase-catalyzed cleavage of sulfonamides. Drug Metab. Dispos. 27, 992–998.
Keywords: glutathione S-conjugate, prodrug, cancer, drug resistance, glutathione transferase, gamma-glutamyl transferase
Citation: Ramsay EE and Dilda PJ (2014) Glutathione S-conjugates as prodrugs to target drug-resistant tumors. Front. Pharmacol. 5:181. doi: 10.3389/fphar.2014.00181
Received: 31 May 2014; Paper pending published: 10 June 2014;
Accepted: 16 July 2014; Published online: 11 August 2014.
Edited by:
Alfonso Pompella, Università di Pisa, ItalyReviewed by:
Marc Diederich, Laboratoire de Biologie Moléculaire et Cellulaire du Cancer, Hôpital Kirchberg, LuxembourgCopyright © 2014 Ramsay and Dilda. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pierre J. Dilda, Tumour Metabolism Group, Adult Cancer Program, Lowy Cancer Research Centre and Prince of Wales Clinical School, Faculty of Medicine, University of New South Wales, Sydney, NSW 2052, Australia e-mail:cC5kaWxkYUB1bnN3LmVkdS5hdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.