 Alicia Mattiazzi
Alicia Mattiazzi Evangelia G. Kranias
Evangelia G. Kranias- 1Facultad de Medicina, Centro de Investigaciones Cardiovasculares, Conicet La Plata-Universidad Nacional de La Plata, La Plata, Argentina
- 2Department of Pharmacology and Cell Biophysics, College of Medicine, University of Cincinnati, Cincinnati, OH, USA
Phospholamban (PLN) is a phosphoprotein in cardiac sarcoplasmic reticulum (SR) that is a reversible regulator of the Ca2+-ATPase (SERCA2a) activity and cardiac contractility. Dephosphorylated PLN inhibits SERCA2a and PLN phosphorylation, at either Ser16 by PKA or Thr17 by Ca2+-calmodulin-dependent protein kinase (CaMKII), reverses this inhibition. Through this mechanism, PLN is a key modulator of SR Ca2+ uptake, Ca2+ load, contractility, and relaxation. PLN phosphorylation is also the main determinant of β1-adrenergic responses in the heart. Although phosphorylation of Thr17 by CaMKII contributes to this effect, its role is subordinate to the PKA-dependent increase in cytosolic Ca2+, necessary to activate CaMKII. Furthermore, the effects of PLN and its phosphorylation on cardiac function are subject to additional regulation by its interacting partners, the anti-apoptotic HAX-1 protein and Gm or the anchoring unit of protein phosphatase 1. Regulation of PLN activity by this multimeric complex becomes even more important in pathological conditions, characterized by aberrant Ca2+-cycling. In this scenario, CaMKII-dependent PLN phosphorylation has been associated with protective effects in both acidosis and ischemia/reperfusion. However, the beneficial effects of increasing SR Ca2+ uptake through PLN phosphorylation may be lost or even become deleterious, when these occur in association with alterations in SR Ca2+ leak. Moreover, a major characteristic in human and experimental heart failure (HF) is depressed SR Ca2+ uptake, associated with decreased SERCA2a levels and dephosphorylation of PLN, leading to decreased SR Ca2+ load and impaired contractility. Thus, the strategy of altering SERCA2a and/or PLN levels or activity to restore perturbed SR Ca2+ uptake is a potential therapeutic tool for HF treatment. We will review here the role of CaMKII-dependent phosphorylation of PLN at Thr17 on cardiac function under physiological and pathological conditions.
Introduction
A major characteristic of human and experimental heart failure (HF) is altered Ca2+ cycling, associated with decreased contractility, which partially reflects the impaired function of the sarcoplasmic reticulum (SR) membrane. During a normal excitation-contraction-coupling cycle (ECC), Ca2+ enters the cell through the L-type Ca2+ channels leading to activation of the ryanodine receptors (RyR2) in the SR and release of Ca2+ from this membrane system. This Ca2+-induced-Ca2+-release mechanism (Fabiato and Fabiato, 1977) underlies a fine-tuned synchronization of Ca2+ cycling in the heart, coordinating contraction and relaxation. Relaxation is mediated mainly by the activity of the SR Ca2+-ATPase (SERCA2a) and to a lesser extent by the Na+/Ca2+ exchanger (NCX). Thus, the SR is the major regulator of Ca2+-handling during the cardiac excitation-contraction-relaxation cycle (Bers, 2001).
The activity of SERCA2a is under the reversible control of phospholamban (PLN), an SR associated protein (Tada et al., 1975). PLN is a 52 amino acid phosphoprotein, which, in the dephosphorylated state, inhibits the apparent Ca2+-affinity of SERCA2a (James et al., 1989; Kim et al., 1990). PLN can be phosphorylated at three distinct sites in vitro: Ser16 by cyclic AMP (cAMP)- and cGMP-dependent protein kinases, Thr17 by Ca2+-calmodulin-dependent protein kinase II (CaMKII), and Ser16 by protein kinase C (Movsesian et al., 1984; Simmerman et al., 1986; Huggins et al., 1989). Phosphorylation of these sites in vitro relieves the inhibition of PLN on SERCA2a and increases SR Ca2+ uptake. Whereas Ser10 phosphorylation by PKC does not occur in intact hearts (Edes and Kranias, 1990), cGMP phosphorylation of PLN has been described in isolated myocytes (Bartel et al., 1995). However, the physiological significance of this pathway is still unclear. In contrast, phosphorylation of Ser16 and Thr17 by PKA and CaMKII has been shown to be a key mediator of the positive inotropic and relaxant effects of ß1-adrenergic stimulation in the heart. The increase in SERCA2a activity and Ca2+ uptake rate elicited by the phosphorylation of these sites, leads to an increase in the velocity of relaxation, SR Ca2+ load and SR Ca2+ release which, in association with L-type Ca2+ channel and RyR2 phosphorylation, mediate the enhanced contractility produced by ß1-stimulation (Lindemann et al., 1983; Lindemann and Watanabe, 1985; Vittone et al., 1990; Napolitano et al., 1992; Mundiña-Weilenmann et al., 1996; Kuschel et al., 1999). Dephosphorylation of PLN, occurring by a SR-associated type 1 phosphatase (PP1; MacDougall et al., 1991), reverses the activation of SERCA2a and the stimulatory effects of β1-agonists. This article will discuss the role of Thr17 phosphorylation of PLN and address its significance under physiological and pathological processes.
The Phospholamban Regulatome
Phospholamban was first described as a cAMP-dependent protein kinase substrate in the early 1970s. The phosphorylated amino acid was shown to be Ser16 and phosphorylation enhanced SERCA2a activity and Ca2+-uptake (Kirchberger et al., 1972). Subsequently, PLN was shown to be also phosphorylated by a SR-associated Ca2+-CaM-kinase (CaMKII) at Thr17 and this phosphorylation occurred independently of its PKA-phosphorylation (Bilezikjian et al., 1981; Davis et al., 1990). Phosphorylation by CaMKII also enhances SR Ca2+-transport through an increase in the apparent affinity of the SERCA2a for Ca2+ (KCa). Thus, it was initially proposed that phosphorylated PLN acts as a stimulator of cardiac SERCA2a activity. However, in the late 1980s and early 1990s, there were two significant breakthroughs: (a) in vitro studies of reconstituted SR membrane systems (James et al., 1989; Kim et al., 1990); and (b) in vivo studies in mouse models with ablation or overexpression of PLN (Luo et al., 1994, 1996; Kadambi et al., 1996), which demonstrated that dephosphorylated PLN is actually an inhibitor of SERCA2 and phosphorylation relieves this inhibition, giving the appearance of phosphorylation-induced stimulation. These findings, together with the characterization and identification of a cardiac SR–associated protein phosphatase that can dephosphorylate PLN (Kranias, 1985), has led to our current understanding of PLN as a reversible inhibitor of cardiac SR Ca2+-ATPase activity.
Furthermore, recent studies showed that the activity of PLN can itself be regulated by the HS-1 associated protein X-1 (HAX-1), which is ubiquitously expressed in mitochondria and SR. HAX-1 physically interacts with PLN and the binding region of PLN includes amino acids 16–22 with both Ser16 and Thr17 phosphorylation sites. Interestingly, phosphorylation of PLN diminishes its binding to HAX-1, indicating that this interaction may be physiologically relevant in the heart (Vafiadaki et al., 2007). Indeed, HAX-1 has been found to increase PLN inhibition of SR Ca2+ cycling and cardiac contractility in vivo, whereas β1-adrenergic stimulation relieves this inhibition (Zhao et al., 2009; Lam et al., 2013).
Besides HAX-1, other regulatory proteins such as PKA, CAMKII and PP1 are also associated with PLN, achieving an efficient and compartmentalized complex that regulates SR Ca2+-cycling and cardiac function. PP1 is a negative regulator of PLN activity through its dephosphorylation and increased inhibition of SERCA2a. Interestingly, the type 1 enzyme is modulated by its endogenous inhibitors, Inhibitor-1 (I-1) and Inhibitor-2 (I-2). Inhibitor-1 gets activated upon its PKA phosphorylation at Thr-35 resulting in potent inhibition of PP1 activity and amplification of the β1-adrenergic receptor stimulatory effects (Iyer et al., 1988; Neumann et al., 1991; Gupta et al., 1996). More recently, the small heat shock protein 20 (Hsp20) was also identified as a novel interacting partner of PP1 and inhibitor of its enzymatic activity, resulting in diminished PLN inhibition and enhanced cardiac function (Qian et al., 2011). Thus, there is a multimeric functionally coupled signaling complex, which reversibly regulates SR Ca2+ cycling in the cell, composed of SERCA, PLN, HAX-1, PKA, CAMKII, PP1, I-1, and Hsp20 (Figure 1).
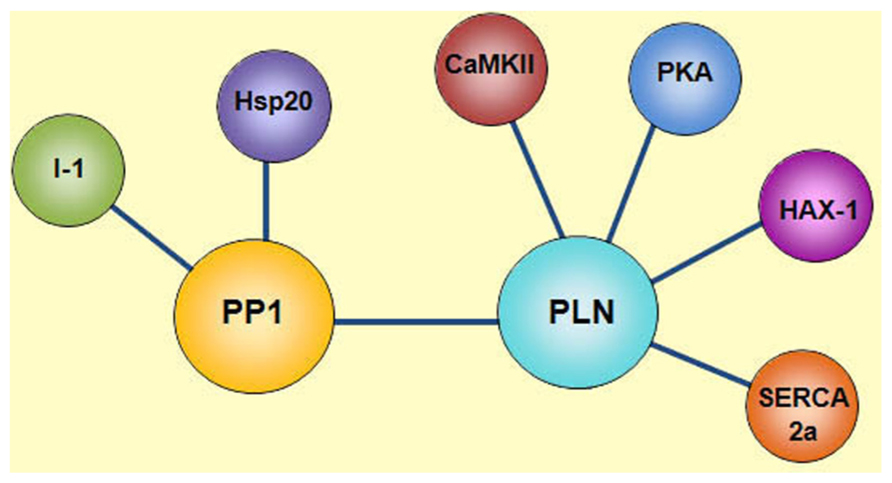
FIGURE 1. Phospholamban regulatome. Scheme of the multimeric protein complex constituted by SERCA2a, PLN, HAX-1, PKA, CAMKII, PP1, Inhibitor-1 (I-1), and Hsp20, which reversibly regulates SR Ca2+ transport in the cell. SERCA2a activity is regulated by its reversible inhibitor PLN and the histidine rich Ca2+-binding protein (HRC). Phosphorylation of PLN is mediated by cAMP-dependent protein kinase (PKA) at Ser16 site and Ca2+-calmodulin-dependent protein kinase (CaMKII) at Thr17 site. Dephosphorylation of these sites occurs by protein phosphatase 1 (PP1). The activity of PP1 is regulated by inhibitor-1 (I-1) and Hsp20.
Phosphorylation of PLN by CaMKII Under Physiological Conditions
ß1-Adrenergic Stimulation
It is well established that stimulation by β1-agonists at the cell membrane, initiates a signal-transduction pathway that involves the Gs proteins to stimulate cAMP formation by adenylate cyclase, followed by PKA activation (Figure 2). PKA then phosphorylates several proteins in the cardiac myocytes to induce positive chronotropic, inotropic, and relaxant effects, the so-called “fight or flight response,” which is considered the most effective mechanism to acutely increase cardiac output. The underlying phosphoproteins include PLN and RyR2 at the SR level, the L-type Ca2+ channel and phospholemman, at the sarcolemma level, and troponin I (TnI), C protein, and myosin light chain, at the level of the myofibrils (Bers, 2001).
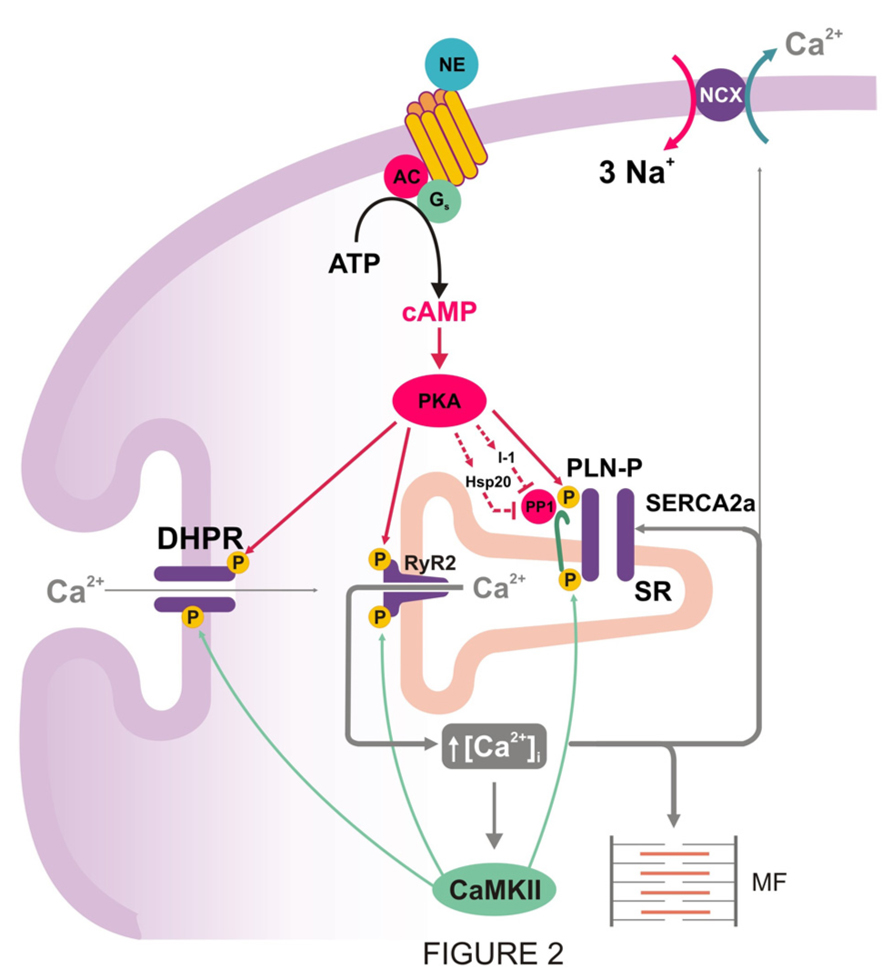
FIGURE 2. PKA mediated increase in cytosolic Ca2+ and inhibition of PP1: two prerequisites for CaMKII-dependent phosphorylation of PLN during β1-adrenergic stimulation. PKA-dependent phosphorylation of Ca2+ handling proteins, particularly L-type Ca2+ channel and PLN, produces an increase in cytosolic Ca2+ that is necessary to activate CaMKII and produce CaMKII-dependent phosphorylation. PKA also increases inhibitor-1 and Hsp20 phosphorylation, amplifying the stimulatory effects of β1–adrenergic stimulation on SR Ca2+-transport, relaxation, and contractility.
The role of PLN phosphorylation vs. the phosphorylation of other proteins, which are also involved in ECC, was demonstrated by the generation and characterization of gene knockout and transgenic models with ablation, reduction or overexpression of PLN in the heart. Ablation of PLN was associated with enhanced affinity of SERCA2a for Ca2+, contractility and relaxation (Luo et al., 1994, 1996). In contrast, overexpression of PLN decreased Ca2+affinity of SERCA2a and diminished SR Ca2+-load and cardiac contractility (Kadambi et al., 1996). Importantly, studies at the cardiomyocyte, organ and intact animal levels from PLN-deficient mice, indicated a significant attenuation of the inotropic and lusitropic effects of isoproterenol, compared with wild types (Luo et al., 1994; Hoit et al., 1995), and revealed that PLN is a major mediator of the β1-adrenergic response in the mammalian heart. However, as noted above, PLN is not only phosphorylated by PKA, at Ser16, but also by CaMKII at Thr17, during β1-adrenergic stimulation. These phosphorylations appear to occur independently of each other in vitro (Bilezikjian et al., 1981; Davis et al., 1990). However, in vivo attempts to phosphorylate PLN by CaMKII indicated that an increase in cAMP levels was a requirement for CaMKII activation (Lindemann et al., 1983; Lindemann and Watanabe, 1985; Vittone et al., 1990; Napolitano et al., 1992; Mundiña-Weilenmann et al., 1996; Kuschel et al., 1999). This may be due to required PKA-phosphorylation of PLN, as well as L-type Ca2+ channels and RyR2, to enhance cytosolic Ca2+, necessary to activate CaMKII. Indeed the availability of transgenic models, expressing either wild type PLN (PLN-WT), Ser16 → Ala mutant PLN (PLN-S16A) or Thr17 → Ala mutant PLN (PLN-T17A) in the cardiac compartment of PLN knockout mice, indicated that the phosphorylation of Ser16 of PLN is a prerequisite for the phosphorylation of Thr17 (Luo et al., 1998). In addition, these studies showed that Ser16 can be phosphorylated independently of Thr17 in vivo and that phosphorylation of Ser16 was sufficient for mediating the maximal cardiac responses to β1-adrenergic stimulation (Chu et al., 2000).
The role of CaMKII-phosphorylation of PLN was also addressed in a model with expression of a CaMKII inhibitory peptide targeted to the longitudinal SR (AIP4-LSR TG; Ji et al., 2006). The results indicated that Thr17 PLN-phosphorylation as well as SR Ca2+-uptake and contractile parameters were decreased. However, the response to isoproterenol remained unaltered. Similarly, transgenic mice with CaMKII inhibition (AC3-I mice), decreased SR Ca2+-content without changes in the myocyte response to isoproterenol (Zhang et al., 2005). These findings suggested a predominant role of Ser16 phosphorylation over that of Thr17 in the β1-adrenergic response. Furthermore, experiments in perfused rat hearts using the PKA inhibitor H-89, confirmed that PKA activation is required for β1-adreniceptor mediated phosphorylation of the Thr17 site in PLN (Said et al., 2002). It was further demonstrated that, when both PLN phosphorylation sites are present, the CaMKII site contributes to PLN phosphorylation and enhanced mechanical effects only at relatively high levels of β1-adrenergic stimulation, i.e., isoproterenol concentrations ≥10 nM. The lack of contribution of Thr17 site to PLN phosphorylation at lower isoproterenol concentrations was attributed to a moderate increase in PKA activity, which would raise intracellular Ca2+ to a level not sufficient to activate CaMKII and phosphorylate Thr17 site (Mundiña-Weilenmann et al., 1996; Said et al., 2002). Taken together, these findings support the notion that CaMKII is a contributor in the stimulatory effects of β1-adrenergic receptor in the heart. However, PKA activation is required to create the necessary conditions for CaMKII activation and Thr17 phosphorylation (Figure 2). A similar conclusion should hold true for the different Ca2+ handling proteins which are phosphorylated by both kinases, like L-type Ca2+ channels or RyR2. Interestingly, sustained β1-adrenergic receptor stimulation enhanced cell contraction and Ca2+ transients by a mechanism which is largely PKA-independent but sensitive to CaMKII-inhibitors. In these studies, a shift from Ser16 to Thr17 phosphorylation pathway was observed, underscoring the role of CaMKII during prolonged β1-adrenergic stimulation (Wang et al., 2004). In addition, β1-adrenoceptors activate the guanine nucleotide exchange protein that is directly activated by cAMP (Epac), independently of, and in parallel with, PKA. Indeed, Oestreich et al. (2009) identified RyR2 and PLN as two effector targets of a pathway mediated by Epac-PLCє-PKCє-CaMKII. These authors described an increase in Ca2+ transient mainly attributed to an increase in RyR2 sensitivity by Ca2+ influx activation. Although the specific role of Thr17 phosphorylation of PLN in these effects was not directly tested, they showed that β-adrenergic stimulation-mediated enhancement of SR Ca2+ load and myoplasmic Ca2+ clearance were not significantly altered by PLCє ablation, suggesting a poor role of the pathway described, on SR Ca2+ uptake (Oestreich et al., 2007). Moreover, other results showed that Epac activation decreases the amplitude of evoked Ca2+ transient due to Epac-induced SR Ca2+ leak by CaMKII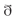 -phosphorylation of RyR2 and SR depletion (Pereira et al., 2007, 2013). The different outcomes of the effects of Epac on Ca2+ transient amplitude may be due to different experimental protocols, since Epac activation produces an initial increase in Ca2+ transients before reaching a steady state, in which Ca2+ transients are decreased. Yet, both results are consistent with an increase in RyR2 activation produced by Epac. The more recent study by Pereira et al. (2013) further showed that inhibition of PKA-dependent effects of isoproterenol by H-89 pretreatment blocked the isoproterenol-induced increase of Ca2+ transient amplitude, speed of relaxation and SR Ca2+ load. In contrast, isoproterenol still greatly increased SR Ca2+ spark frequency and decreased Ca2+ transient amplitude. Both of these effects were similar to the steady state responses produced by Epac activation. These results would imply that: (1) most of the isoproterenol-induced PKA-independent Ca2+ leak enhancement is mediated by Epac; and (2) the contribution of Epac to isoproterenol–induced SR Ca2+ reuptake through PLN phosphorylation, is very modest, if any, since no relaxant effects of isoproterenol could be detected after PKA inhibition.
-phosphorylation of RyR2 and SR depletion (Pereira et al., 2007, 2013). The different outcomes of the effects of Epac on Ca2+ transient amplitude may be due to different experimental protocols, since Epac activation produces an initial increase in Ca2+ transients before reaching a steady state, in which Ca2+ transients are decreased. Yet, both results are consistent with an increase in RyR2 activation produced by Epac. The more recent study by Pereira et al. (2013) further showed that inhibition of PKA-dependent effects of isoproterenol by H-89 pretreatment blocked the isoproterenol-induced increase of Ca2+ transient amplitude, speed of relaxation and SR Ca2+ load. In contrast, isoproterenol still greatly increased SR Ca2+ spark frequency and decreased Ca2+ transient amplitude. Both of these effects were similar to the steady state responses produced by Epac activation. These results would imply that: (1) most of the isoproterenol-induced PKA-independent Ca2+ leak enhancement is mediated by Epac; and (2) the contribution of Epac to isoproterenol–induced SR Ca2+ reuptake through PLN phosphorylation, is very modest, if any, since no relaxant effects of isoproterenol could be detected after PKA inhibition.
Phosphorylation of Thr17 of PLN in the Absence of β1-Adrenergic Stimulation
As indicated above, several studies showed that CaMKII-dependent PLN phosphorylation can only occur in the intact beating heart in the presence of ß1-adrenergic stimulation, while it occurs independently of cAMP-PKA activation in vitro. To address this apparent discrepancy, the phosphatase inhibitor okadaic acid was used in the presence of high extracellular Ca2+. Under phosphatase inhibition, increasing Ca2+, increased contractility, relaxation and phosphorylation of Thr17 of PLN, without significantly changing either cAMP or Ser16 phosphorylation (Mundiña-Weilenmann et al., 1996). These findings indicated that Thr17 can be phosphorylated independently of Ser16 of PLN in the intact heart, in accordance with the in vitro studies. Thus, the relative balance of protein kinase (PKA and CaMKII) and phosphatase activities appears to regulate phosphorylation of Thr17 and Ser16 in PLN.
Stimulation frequency (SF), a fundamental physiological modulator of myocardial performance, is another example in which Thr17 phosphorylation of PLN can occur in the absence of prior Ser16 phosphorylation (Hagemann et al., 2000; Zhao et al., 2004; Valverde et al., 2005). These findings are in concert with the fact that CaMKII can decode the frequency of Ca2+ spikes into distinct amounts of kinase activity (De Koninck and Schulman, 1998), and indicate that SF can produce a sustained increase in CaMKII, which leads to the phosphorylation of Thr17 in PLN, without the requirement of phosphatase inhibition. Moreover, these results prompted the link between the observed Thr17 phosphorylation and the relaxant effect of increasing SF (frequency-dependent acceleration of relaxation or FDAR, Bers, 2001; Hagemann et al., 2000). Indeed, FDAR was inhibited in the presence of CaMKII-inhibitors and in cardiomyocytes expressing the mutant T17A-PLN (Zhao et al., 2004). However, although the involvement of SR and CaMKII in FDAR was supported by several studies (Bassani et al., 1995; DeSantiago et al., 2002; Picht et al., 2007; Wu et al., 2012), a recent report challenged these previous findings by showing that FDAR was still present in CaMKIIδ-KO mice (Neef et al., 2013). These results would suggest that either a CaMKIIδ-independent mechanism or another CaMKII isoform, like CaMKIIγ, is playing a role in FDAR.
The role of Thr17 phosphorylation of PLN on FDAR was also questioned on the basis of three main findings: (1) FDAR precedes the phosphorylation of Thr17 site of PLN (Valverde et al., 2005; Huke and Bers, 2007); (2) Most studies concur that the main regulatory effect of PLN phosphorylation is to increase the apparent Ca2+ affinity of SERCA2a (Simmerman and Jones, 1998), while FDAR is associated with an increase in the maximal velocity of SR Ca2+ uptake (Picht et al., 2007); and (3) FDAR has been also detected in PLNKO mice in one study (DeSantiago et al., 2002), although this finding was not observed in other studies (Bluhm et al., 2000; Wu et al., 2012).
Taken together, the underlying molecular steps that encompass the FDAR process are currently unclear. Although most of the experimental evidence indicates that CaMKII is involved in FDAR, some studies have challenged this possibility and the participation of PLN in FDAR. Thus, it is likely that several rather than a single mechanism, are associated with this phenomenon.
Phosphorylation of PLN by CaMKII Under Pathological Conditions
Acidosis
Mechanical recovery during acidosis
An understanding of how pH changes alter cardiac function is important for a better comprehension of some cardiac pathological situations, which are important in the clinical setting. Myocardial ischemia is particularly relevant along these lines: in human, acidosis can be detected 15 s after the occlusion of the coronary artery and is a major mechanism for the loss of contractility during ischemia (Poole-Wilson, 1989). Substantial changes in intracellular pH may also occur in disorders of different origins which affect cardiac function, like sleep apnea/hypopnea syndrome, diabetic ketoacidosis or in patients on dialysis.
Acidosis produces a rapid decrease in the strength of contraction (Cingolani et al., 1970; Allen and Orchard, 1983), which is largely due to a decrease in myofilament Ca2+ responsiveness (Fabiato and Fabiato, 1978). This decrease displaces Ca2+ from troponin C, and would be the main mechanism responsible for the early increase in diastolic Ca2+ during acidosis. The initial fall in contractility is followed by an increase in the amplitude of intracellular Ca2+ transients and contractile force (Mattiazzi and Cingolani, 1977a, b; Allen and Orchard, 1983, the mechanism of which is not intuitively obvious, because acidosis inhibits most of the steps of excitation-contraction-coupling (Orchard and Kentish, 1990). Earlier experiments suggested that acidosis-induced activation of Na+-H+ exchanger (NHE), by increasing cytosolic Na+ and then Ca2+ through the NCX, was sufficient to overcome the inhibitory effect of acidosis on SERCA2a, increasing SR Ca2+ and intracellular Ca2+ transients (Harrison et al., 1992). However, inhibition of NHE does not always prevent intracellular Ca2+ and mechanical recovery (Choi et al., 2000; DeSantiago et al., 2004), indicating that additional mechanisms may play a role (Figure 3A). A major clue supporting this possibility was given by experiments showing that Ca2+ and contractile recovery during acidosis require an intact SR and CaMKII activity, suggesting that CaMKII-dependent phosphorylation at the SR level is involved in the recovery mechanism (Pérez et al., 1995; Komukai et al., 2001; Nomura et al., 2002; DeSantiago et al., 2004; Mundiña-Weilenmann et al., 2005; Neef et al., 2013). Indeed, it was shown that phosphorylation of the Thr17 site of PLN transiently increased at the onset of acidosis, possibly favored by the increase in intracellular (diastolic) Ca2+ and the inhibition of phosphatase induced by acidosis (Allen and Orchard, 1983; Vittone et al., 1998). This phosphorylation was associated with the initial and most significant portion of the contractile/relaxation recovery, and both were blunted by CaMKII-inhibition (Mundiña-Weilenmann et al., 2005). Thus, CaMKII-dependent PLN phosphorylation provides a mechanism to overcome the depressant effect of acidosis on SERCA2a (Mandel et al., 1982). These increases in SR Ca2+ content and release also counteract the effect of acidosis on contractile proteins, thereby helping to maintain contractile force. More recent experiments showed that CaMKII also activates NHE which may add to the direct activation of the exchanger induced by acidosis (Vila-Petroff et al., 2010; Figure 3A). Interestingly, experiments by DeSantiago et al. (2004) showed absence of mechanical recovery in myocytes lacking PLN (PLNKO). This finding may be taken to indicate that PLN is essential for SR Ca2+ and mechanical recovery during acidosis. However, the effects of PLN ablation mimic maximal PLN phosphorylation. Therefore the results of DeSantiago et al. (2004) actually raise the question of whether accelerating SR Ca2+ reuptake during acidosis is beneficial, favoring mechanical recovery as discussed above, or harmful, hindering it. An explanation to these apparent contradictory results may lie on the fact that intracellular Ca2+ and mechanical recovery during acidosis require an increase in SR Ca2+ uptake above steady state, a condition that cannot be accomplished in PLNKO mice in which basal SR Ca2+ uptake is already at maximal levels. In line with DeSantiago’s results, Nomura et al. (2002) showed that the mechanical recovery from acidosis did not occur in highly phosphorylated myocytes treated with isoproterenol and a phosphatase inhibitor.
FIGURE 3. (A) Intracellular mechanisms that may contribute to the mechanical recovery during acidosis. Acidosis produces a decrease in myofilament Ca2+ responsiveness which increases diastolic Ca2+ ([Ca2+]d). Activation of NHE and direct acidosis inhibition of Na+-Ca2+ exchanger (NCX), would contribute to the increase in cytosolic Ca2+. Acidosis also inhibits PP1. The simultaneous increase in cytosolic Ca2+ and inhibition of PP1 activates CaMKII and enhances PLN phosphorylation at Thr17 site. As a consequence, there is an increase in SR Ca2+ uptake, able to offset the direct acidosis-induced inhibition of SERCA2a activity. This would lead to enhanced SR Ca2+ release and Ca2+ transients, which counteract the negative effect of acidosis on contractile proteins and supply the substrate for the slow mechanical recovery during acidosis. (B). Putative intracellular mechanisms of post-acidosis induced-arrhythmias. Upon returning to control pH, the inhibitory effects of acidosis are rapidly removed, favoring the increase in Ca2+ cycling and the contractile recovery towards control levels. However, the relief of RyR2 from the previous constrain produced by acidosis, evokes also an increase in diastolic Ca2+ leak from the (Ca2+-loaded) SR. Such release may activate inward currents through the NCX, also relieved from the inhibition evoked by acidosis. The inward Na+ current, if large enough, can trigger arrhythmias.
Acidosis and post-acidosis arrhythmias
The increase in SR Ca2+ load during acidosis, responsible for the mechanical recovery, may also increase spontaneous SR Ca2+ release and produce extra-systoles (Orchard et al., 1987). Moreover, returning to normal pH after acidosis is also arrhythmogenic: recovery of pH induces an increase in SR Ca2+ leak. This effect was attributed to the increase in the opening probability of RyR2 due to the pH increase after acidosis and the acidosis-induced increase in SR Ca2+ content, still present at the beginning of post-acidosis (Said et al., 2008). The return to normal pH also leads to recovery of the previous acidosis-induced inhibition of NCX (Philipson et al., 1982), favoring Ca2+ extrusion and Na+ gain into the cell, membrane depolarization and eventually triggered arrhythmias (Said et al., 2008; Figure 3B). Together, these results indicate that post-acidosis CaMKII-dependent DADs are triggered by two concurrent factors: (1) acidosis-induced increase in SR Ca2+ content; and (2) relief of RyR2 and NCX, previously inhibited by acidosis.
Ischemia/Reperfusion (I/R)
Stunning
The role of CaMKII in I/R will be addressed in detail elsewhere in this issue. We will briefly refer here to the role of CaMKII-dependent PLN phosphorylation in this pathological situation. In the last few years, a dual effect of CaMKII-dependent protein phosphorylation (beneficial and detrimental) has been described in the scenario of I/R in the intact heart. The beneficial effect of CaMKII usually refers to the intracellular Ca2+ and contractile recovery that occurs during stunning, a fully reversible post-ischemic dysfunction (Braunwald and Kloner, 1982). Initially, this beneficial effect was associated with an increase in the phosphorylation of Thr17 site in PLN at the onset of reperfusion (Vittone et al., 2002). Further experiments in transgenic mice in which Thr17 and/or Ser16 sites of PLN were mutated to Ala and direct measurements of intracellular Ca2+, demonstrated that Thr17 phosphorylation was essential for the recovery of Ca2+ transients and contractility in the stunned heart (Said et al., 2003; Valverde et al., 2006). These findings confirmed that the increase in Thr17 phosphorylation of PLN upon reperfusion, although transient, offers a mechanism that helps to limit cytosolic Ca2+ overload, by accelerating SR Ca2+ reuptake and thereby ameliorating intracellular Ca2+ handling (Figure 4). In contrast, when SR Ca2+ reuptake is highly enhanced by ablation of PLN, post-ischemic recovery of contractile function was negligible (Cross et al., 2003). A possible explanation for this apparent paradox is the higher ATP consumption of PLNKO hyperactive hearts relative to WTs, which may greatly influence contractile recovery. A second possibility that does not exclude the first one, is that under conditions in which RyR2 are altered, a persistent and exacerbated SR Ca2+ uptake, would greatly elevate SR Ca2+ content and enhance the propensity for SR Ca2+ leak, which may conspire against contractile recovery and favor reperfusion arrhythmias. Indeed, a rise in CaMKII phosphorylation of Ser2814 in RyR2 and an abrupt increase in SR Ca2+ release at the onset of reflow were recently associated with early reperfusion arrhythmias. This occurs in spite of the fact that Thr17 site of PLN was also phosphorylated (Said et al., 2011; Valverde et al., 2010). These results strongly suggest that the beneficial effects of increasing SR Ca2+ uptake in I/R, may turn to be deleterious under conditions in which the balance between SR Ca2+ uptake and leak is lost (Figure 4).
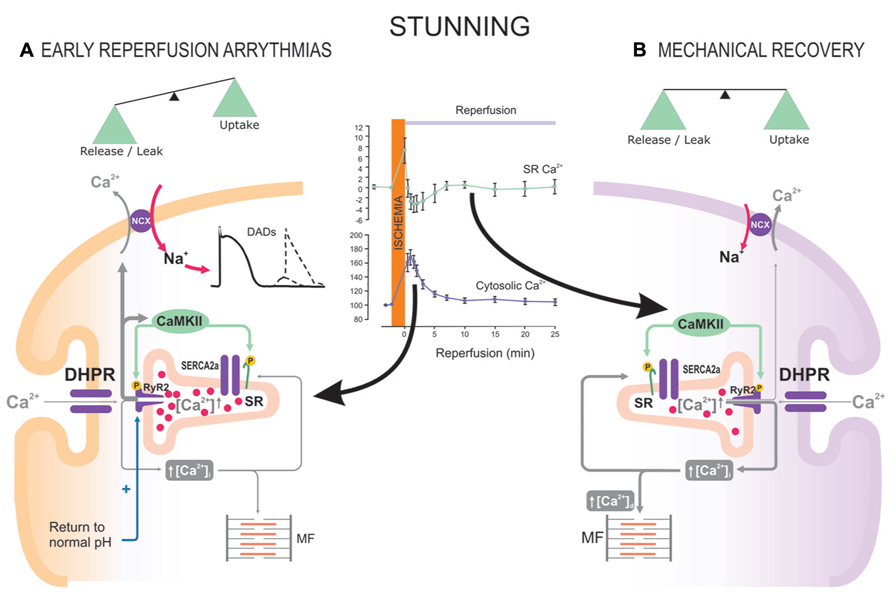
FIGURE 4. The beneficial or detrimental effects of CaMKII activation and PLN phosphorylation in I/R depend on a tight balance between SR Ca2+ reuptake and leak. Reperfusion after a short ischemic period (stunning) is associated with an increase in CaMKII-dependent PLN and RyR2 phosphorylation. During early reperfusion (A), there is: abrupt release of SR Ca2+ (inset of the Figure, Valverde et al., 2010), possibly favored by the ischemia-induced increase in SR Ca2+ content; relief of RyR2 inhibition exerted by the ischemic acidosis; and increase in Ser2814 phosphorylation of RyR2 (Said et al., 2011). CaMKII-dependent phosphorylation of PLN does not counteract SR Ca2+ release, which is partially responsible for early reperfusion arrhythmias. (B) After the first minutes of reperfusion, the increase in Thr17 phosphorylation of PLN appears to successfully counteract SR Ca2+ leak, leading to Ca2+ transients and mechanical recovery.
As will be discussed below, this unbalance may constitute a major cause of the detrimental effect of CaMKII activation in the irreversible I/R.
Irreversible ischemia/reperfusion
After a prolonged ischemic period, reperfusion evokes irreversible cardiac injury. Under these conditions, myocytes die by apoptosis, autophagy and necrosis. The rise in Ca2+ during ischemia and reperfusion leads to mitochondrial Ca2+ accumulation, which is greatly favored by the close association between mitochondria and SR and constitutes a main event in the initiation of cell death (Rizzuto and Pozzan, 2006).
Experimental evidence consistently indicates that CaMKII-inhibition is protective in the irreversible I/R injury (Zhang et al., 2005; Vila-Petroff et al., 2007; Salas et al., 2010). Although the mechanisms for myocardial protection by CaMKII inhibition are still unclear, the CaMKII deleterious pathway in I/R certainly involves the SR and the mitochondria (Vila-Petroff et al., 2007; Salas et al., 2010; Joiner et al., 2012; Valverde et al., 2013). Phosphorylation of Thr17, the CaMKII site of PLN, has been shown to increase at the onset of reperfusion (Vila-Petroff et al., 2007; Salas et al., 2010). This finding may suggest either that the effect of PLN phosphorylation is part of the deleterious cascade of CaMKII activation, or that this phosphorylation is beneficial, although insufficient to counteract the effect of simultaneous detrimental mechanisms. The experimental outcome of testing these possibilities has remained controversial. Referent to the first one, Yang et al. (2006) demonstrated that the protective effect of chronic CaMKII inhibition in AC3-I mice was lost, when they were interbred with PLNKO mice and submitted to myocardial infarction, supporting a detrimental effect of enhancing of SR Ca2+ uptake. Referent to the second possibility, several studies demonstrated that accelerating SR Ca2+ uptake by different means (i.e., overexpressing SERCA1a, with higher kinetics than SERCA2a, or expressing a repressor of PLN activity, PP1 inhibitor-1), alleviated post-ischemic cardiac injury (Talukder et al., 2007, 2008; Nicolaou et al., 2009), supporting a beneficial effect of accelerating SR Ca2+ uptake. A possible clue to explain these controversial findings is given by results showing that proteins, different from PLN, may be involved in the cascade by which CaMKII activity is deleterious in I/R. A decrease in the expression of RyR2 has been described in I/R (Salas et al., 2010), compatible with a degradation/damage of these channels by the concerted action of calpains and proteasomes (Pedrozo et al., 2010), that would lead to an increase in the opening probability of RyR2 (Domenech et al., 2003). Moreover, redox alterations or CaMKII-dependent phosphorylation might also influence the activity of RyR2 and SR Ca2+ leak in I/R (Hidalgo et al., 2004; Said et al., 2011; Valverde et al., 2013). These alterations would favor the unbalance between SR Ca2+uptake and leak, promoting mitochondrial Ca2+ overload and cell death. This cascade would be further stimulated by the recently described CaMKII-dependent phosphorylation of mitochondrial Ca2+ uniporter (Joiner et al., 2012). Taken together, these findings suggest that the progression toward a beneficial or detrimental effect of CaMKII activation and PLN phosphorylation in I/R would critically depend on the balance between the extent of SR Ca2+ reuptake and SR Ca2+ leak, largely given by the status/characteristics of other proteins, also involved in SR Ca2+ handling, like RyR2 (Figure 4).
Heart Failure
Heart failure develops when the heart is unable to provide an adequate cardiac output to meet the metabolic needs of the organism. Mechanical dysfunction and arrhythmias are hallmark features of HF, being aberrant Ca2+ handling a main cause of these two characteristic alterations. Indeed, there is evidence supporting a decrease in intracellular Ca2+-transient and diminished SR Ca2+ content, an outcome that constitutes the major origin of the altered contractility in HF (O’Rourke et al., 1999; Piacentino et al., 2003), and that can be attributed to alterations in the expression/activity of different Ca2+ regulatory proteins. In particular, a decrease in SERCA2a and an increase in NCX expressions have been described in different HF models and species, including human (Hasenfuss, 1998). An enhanced SR Ca2+ leak, through hyperphosphorylated RyR2, would also contribute to the decrease in SR Ca2+ content and Ca2+ release, typical of HF (Ai et al., 2005; Shan et al., 2010; Respress et al., 2012).
The decrease in SERCA2a expression is not associated with a parallel decrease in PLN, which would produce an increase in the functional stoichiometry PLN/SERCA, with a decrease in SERCA2a Ca2+ affinity and SR Ca2+ uptake rate and a prolongation of relaxation times (Meyer et al., 1995). Moreover, phosphorylation of PLN has been found to be decreased, either at Ser16 (Schwinger et al., 1999; Sande et al., 2002), Thr17 (Netticadan et al., 2000), or both (Huang et al., 1999; Mishra et al., 2003), accounting for increased inhibition of SERCA2a. These findings may be due to the attenuation of β1-adrenergic cascade, due to receptor desensitization, down-regulation and uncoupling, typical of the disease progression (Bristow et al., 1982; Dash et al., 2001; Port and Bristow, 2001), and/or the increase in PP1 activity, described in HF (Bibb et al., 2001; Carr et al., 2002; Gupta et al., 2003). Indeed, in human failing myocardium, phosphorylation of Ser16 in PLN decreased because of increases in PP1 activity (Schwinger et al., 1999), whereas phosphorylation of Thr17 decreased due to increased activity of PP2B (calcineurin; Münch et al., 2002). Interestingly, this decrease occurred despite an increase in CaMKII activity characteristic of HF. Taken together, these results indicate that the increase in SERCA2a/PLN ratio and the diminished phosphorylation of PLN, are key determinants of the depressed SR Ca2+ uptake in HF, leading to an increase in diastolic Ca2+, a decrease in SR Ca2+ stores and therefore in Ca2+ available for contraction. This results in reduced contractile force, impaired relaxation and altered force-frequency relationship.
Given this central role of SERCA2a and PLN in the defective Ca2+ handling typical of HF, the strategy of altering SERCA2a and/or PLN levels or activity to restore perturbed Ca2+ uptake into the SR are potential therapeutic strategies for HF treatment (del Monte and Hajjar, 2003). Indeed, overexpression of SERCA2a can restore Ca2+ handling and contractile function in animal models (Cutler et al., 2012) and in human HF (del Monte et al., 1999; Jaski et al., 2009), suggesting that repairing SERCA2a expression may be a viable therapy. Moreover PLN ablation prevented HF in a mouse model of dilated cardiomyopathy caused by deficiency of the muscle-specific LIM protein (Arber et al., 1997; Minamisawa et al., 1999). In isolated human HF myocytes, gene therapy with antisense against PLN improved contractile and diastolic function (del Monte et al., 2002). In contrast, PLN ablation increased SR Ca2+ filling and contractility in mice with cardiomyopathy attributable to overexpression of CaMKII. This led to premature death and mitochondrial Ca2+ overload, suggesting that accelerating SR Ca2+ uptake and increasing SR Ca2+ load, is disadvantageous at least in the presence of excessive CaMKII activity (Zhang et al., 2010). These findings are consistent with the idea already discussed for I/R: in the face of phosphorylated RyR2 channels, as is the case of CaMKII overexpressing mice, repletion of Ca2+ stores through PLN ablation could further worsen overall heart function, via mitochondrial Ca2+ loading, cell death, and arrhythmias.
Concluding Remarks
We have described that PLN and its CaMKII-dependent phosphorylation are part of a multimeric functionally coupled signaling complex, composed of SERCA, PLN, HAX-1, PKA, CaMKII, PP1, I-1, and Hsp20, which reversibly regulates SR Ca2+ cycling. Although CaMKII-dependent PLN phosphorylation contributes to β1-adrenergic mechanical response, its role is subordinate to the PKA-dependent increase in cytosolic Ca2+ and inhibition of phosphatase, necessary to activate CaMKII and phosphorylate Thr17 of PLN. These requirements are also achieved under different pathological situations, like acidosis and I/R, independent of PKA activation. Under these conditions, CaMKII-dependent PLN phosphorylation may paradoxically produce either favorable or harmful cardiac effects. The findings summarized in this review also suggest that the beneficial or detrimental effects associated with CaMKII activation and PLN phosphorylation depend on a tight balance between SR Ca2+ reuptake and leak, determined by the status/characteristics of other SR proteins, among which the RyR2 is a main candidate. A moderate or even high increase in SR Ca2+ uptake (and content) due to PLN phosphorylation, would enhance RyR2 opening due to the regulatory effect of intra-SR Ca2+. However, in the absence of additional RyR2 modifications, the increased SERCA2a activity, produced by PLN phosphorylation, may cope with the enhanced diastolic SR Ca2+ release/leak. In contrast, even moderate increases in SR Ca2+ may increase diastolic SR Ca2+ release under conditions where RyR2 activity is altered independently of intra-SR Ca2+-induced modifications, enhancing the propensity to arrhythmias and leading to mitochondrial Ca2+ overload, which favors apoptosis and necrosis. Thus, increasing SERCA2a activity by PLN phosphorylation seems to have the potential of producing salutary effects in a number of diseases, as long as these effects are achieved under conditions in which diastolic Ca2+ release is satisfactorily controlled. Future research in this area is needed to parse the contribution of different players involved in the balance/interaction between SR Ca2+ reuptake and leak, including the RyR2 regulators or the L-type Ca2+ channels. More specifically to the subject of this review, the recently described multimeric SERCA/PLN-ensemble may represent a nodal point in the interaction of several protein partners, regulating and modifying the fine-tuned control of Ca2+ cycling achieved by the duo SERCA-PLN. An intensive scrutiny of the various proteins of this new pathway will give new insights into their role in SR Ca2+ uptake control and may provide novel therapeutic avenues which can contribute to solve the abnormalities in Ca2+ handling underlying different pathological process.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by PICT 1903 (FONCyT) and PIP 02139 (CONICET) to Alicia Mattiazzi; and NIH HL26057 and HL64018 to Evangelia G. Kranias. The authors are grateful to Mrs. Inés Vera for her assistance in preparing the figures of the manuscript and Ms. Erica Vanderbilt for secretarial assistance.
References
Ai, X., Curran, J. W., Shannon, T. R., Bers, D. M., and Pogwizd, S. M. (2005). Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ. Res. 97, 1314–1322. doi: 10.1161/01.RES.0000194329.41863.89
Allen, D. G., and Orchard, C. H. (1983). The effects of changes of pH on intracellular calcium transients in mammalian cardiac muscle. J. Physiol. (Lond.) 335, 555–567.
Arber, S., Hunter, J. J., Ross, J. Jr., Hongo, M., Sansig, G., Borg, J., et al. (1997). MLP-deficient mice exhibit a disruption of cardiac cytoarchitectural organization, dilated cardiomyopathy, and heart failure. Cell 88, 393–403. doi: 10.1016/S0092-8674(00)81878-4
Bartel, S., Willenbrock, R., Haase, H., Karczewski, P., Wallukat, G., Dietz, R., et al. (1995). Cyclic GMP-mediated phospholamban phosphorylation in intact cardiomyocytes. Biochem. Biophys. Res. Commun. 214, 75–80. doi: 10.1006/bbrc.1995.2258
Bassani, R., Mattiazzi, A., and Bers, D. M. (1995). CaMK-II is responsible for activity-dependent acceleration of relaxation in intact rat ventricular myocytes. Am. J. Physiol. 268, H703–H712.
Bers, D. M. (2001). Excitation-Contraction Coupling and Cardiac Contractile Force, 2nd Edn. Dordrecht: Kluwer Academic Publishers. doi: 10.1007/978-94-010-0658-3
Bibb, J. A., Nishi, A., O’Callaghan, J. P., Ule, J., Lan, M., Snyder, G. L., et al. (2001). Phosphorylation of protein phosphatase inhibitor-1 by Cdk5. J. Biol. Chem. 276, 14490–14497.
Bilezikjian, L. M., Kranias, E. G., Potter, J. D., and Schwartz, A. (1981). Studies on phosphorylation of canine cardiac sarcoplasmic reticulum by calmodulin-dependent protein kinase. Circ. Res. 49, 1356–1362. doi: 10.1161/01.RES.49.6.1356
Bluhm, W. F., Kranias, E. G., Dillmann, W. H., and Meyer, M. (2000). Phospholamban: a major determinant of the cardiac force–frequency relationship. Am. J. Physiol. 278, H249–H255.
Braunwald, E., and Kloner, R. A. (1982). The stunned myocardium: prolonged postischemic ventricular dysfunction. Circulation 66, 1146–1149. doi: 10.1161/01.CIR.66.6.1146
Bristow, M. R., Ginsburg, R., Minobe, W., Cubicciotti, R. S., Sageman, W. S., Lurie, K., et al. (1982). Decreased catecholamine sensitivity and beta-adrenergic-receptor density in failing human hearts. N. Engl. J. Med. 307, 205–211. doi: 10.1056/NEJM198207223070401
Carr, A. N., Schmidt, A. G., Suzuki, Y., del Monte, F., Sato, Y., Lanner, C., et al. (2002). Type 1 phosphatase, a negative regulator of cardiac function. Mol. Cell. Biol. 22, 4124–4135. doi: 10.1128/MCB.22.12.4124-4135.2002
Choi, H. S., Trafford A. W., Orchard, C. H., and Eisner, D. A. (2000). The effect of acidosis on systolic Ca2+ and sarcoplasmic reticulum calcium content in isolated rat ventricular myocytes. J. Physiol. (Lond.) 529, 661–668. doi: 10.1111/j.1469-7793.2000.00661.x
Chu, G., Lester, J. W., Young, K. B., Luo, W., Zhai, J., and Kranias, E. G. (2000). A single site (Ser16) phosphorylation in phospholamban is sufficient in mediating its maximal cardiac responses to β-agonists. J. Biol. Chem. 275, 38938–38943. doi: 10.1074/jbc.M004079200
Cingolani, H. E., Mattiazzi, A. R., Blesa, E. S., and González, N. C. (1970). Contractility in isolated mammalian heart muscle after acid base changes. Circ. Res. 26, 269–278. doi: 10.1161/01.RES.26.3.269
Cross, H. R., Kranias, E. G., Murphy, E., and Steenbergen, C. (2003). Ablation of PLB exacerbates ischemic injury to a lesser extent in female than male mice: protective role of NO. Am. J. Physiol. 284, H683–H690.
Cutler, M. J., Wan, X., Plummer, B. N., Liu, H., Deschenes, I., Laurita, K. R., et al. (2012). Targeted sarcoplasmic reticulum Ca2+ATPase 2a gene delivery to restore electrical stability in the failing heart. Circulation 126, 2095–2104. doi: 10.1161/CIRCULATIONAHA.111.071480
Dash, R., Kadambi, V., Schmidt, A. G., Tepe, N. M., Biniakiewicz, D., Gerst, M. J., et al. (2001). Interactions between phospholamban and beta-adrenergic drive may lead to cardiomyopathy and early mortality. Circulation 103, 889–896. doi: 10.1161/01.CIR.103.6.889
Davis, B. A., Edes, I., Gupta, R. C., Young, E. F., Kim, H. W., Steenaart, N. A., et al. (1990). The role of phospholamban in the regulation of calcium transport by cardiac sarcoplasmic reticulum. Mol. Cell. Biochem. 99, 83–88. doi: 10.1007/BF00230337
De Koninck, P., and Schulman, H. (1998). Sensitivity of CaM kinase II to the frequency of Ca2+ oscillations. Science 279, 227–230. doi: 10.1126/science.279.5348.227
del Monte, F., and Hajjar, R. J. (2003). Targeting calcium cycling proteins in heart failure through gene transfer. J. Physiol. (Lond.) 546, 49–61. doi: 10.1113/jphysiol.2002.026732
del Monte, F., Harding, S. E., Dec, G. W., Gwathmey, J. K., and Hajjar, R. J. (2002). Targeting phospholamban by gene transfer in human heart failure. Circulation 105, 904–907. doi: 10.1161/hc0802.105564
del Monte, F., Harding, S. E., Schmidt, U., Matsui, T., Kang, Z. B., Dec, G. W., et al. (1999). Restoration of contractile function in isolated cardiomyocytes from failing human hearts by gene transfer of SERCA2a. Circulation 100, 2308–2311. doi: 10.1161/01.CIR.100.23.2308
DeSantiago, J., Maier, L. S., and Bers, D. M. (2002). Frequency-dependent acceleration of relaxation in the heart depends on CaMKII, but not phospholamban. J. Mol. Cell Cardiol. 34, 975–984. doi: 10.1006/jmcc.2002.2034
DeSantiago, J., Maier, L. S., and Bers, D. M. (2004). Phospholamban is required for CaMKII-dependent recovery of Ca transients and SR Ca reuptake during acidosis in cardiac myocytes. J. Mol. Cell Cardiol. 36, 67–74. doi: 10.1016/j.yjmcc.2003.10.012
Domenech, R. J., Sánchez, G., Donoso, P., Parra, V., and Macho, P. (2003). Effect of tachycardia on myocardial sarcoplasmic reticulum and Ca2+dynamics: a mechanism for preconditioning? J. Mol. Cell Cardiol. 35, 1429–1437. doi: 10.1016/j.yjmcc.2003.09.006
Edes, I., and Kranias, E. G. (1990). Phospholamban and troponin I are substrates for protein kinase C in vitro but not in intact beating guinea pig hearts. Circ. Res. 67, 394–400. doi: 10.1161/01.RES.67.2.394
Fabiato, A., and Fabiato, F. (1977). Calcium release from the sarcoplasmic reticulum. Circ. Res. 40, 119–129. doi: 10.1161/01.RES.40.2.119
Fabiato, A., and Fabiato, F. (1978). Myofilament-generated tension oscillations during partial calcium activation and activation dependence of the sarcomere length–tension relation of skinned cardiac cells. J. Gen. Physiol. 72, 667–6 99.
Gupta, R. C., Mishra, S., Rastogi, S., Imai, M., Habib, O., and Sabbah, H. N. (2003). Cardiac SR-coupled PP1 and expression are increased and inhibitor-1 protein expression is decreased in failing hearts. Am. J. Physiol. 285, H2373–H2381.
Gupta, R. C., Neumann, J., Watanabe, A. M., Lesch, M., and Sabbah, H. N. (1996). Evidence for presence and hormonal regulation of protein phosphatase inhibitor-1 in ventricular cardiomyocyte. Am. J. Physiol. 270, H1159–H1164.
Hagemann, D., Kuschel, M., Kuramochi, T., Zhu, W., Cheng, H., and Xiao, R.-P. (2000). Frequency-encoding Thr17 phospholamban phosphorylation is independent of Ser16 phosphorylation in cardiac myocytes. J. Biol. Chem. 275, 22532–22536. doi: 10.1074/jbc.C000253200
Harrison, S. M., Frampton, J. E., McCall, E., Boyett, M. R., and Orchard, C. H. (1992). Contraction and intracellular Ca2+, Na+ and H+ during acidosis in rat ventricular myocytes. Am. J. Physiol. 262, C348–C357.
Hasenfuss, G. (1998). Alterations in calcium regulatory proteins in heart failure. Cardiovasc. Res. 37, 279–289. doi: 10.1016/S0008-6363(97)00277-0
Hidalgo, C., Bull, R., Behrens, M. I., and Donoso, P. (2004). Redox regulation of RyR-mediated Ca2+ release in muscle and neurons. Biol. Res. 37, 539–552. doi: 10.4067/S0716-97602004000400007
Hoit, B. D., Khoury, S. F., Kranias, E. G., Ball, N., and Walsh, R. A. (1995). In vivo echocardiographic detection of enhanced left ventricular function in gene targeted mice with phospholamban deficiency. Circ Res. 77, 632–637. doi: 10.1161/01.RES.77.3.632
Huang, B., Wang, S., Qin, D., Boutjdir, M., and El-Sherif, N. (1999). Dimished basal phosphorylation level of phospholamban in the postinfarctium remodeled rat ventricle. Role of beta adrenergic pathway, Gi protein, phosphodiesterase and phosphatases. Circ. Res. 85, 848–855. doi: 10.1161/01.RES.85.9.848
Huggins, J. P., Cook, E. A., Piggott, J. R., Mattinsley, T. J., and England, P. J. (1989). Phospholamban is a good substrate for cyclic GMP-dependent protein kinase in vitro, but not in intact cardiac or smooth muscle. Biochem. J. 260, 829–835.
Huke, S., and Bers, D. M. (2007). Temporal dissociation of frequency-dependent acceleration of relaxation and protein phosphorylation by CaMKII. J. Mol. Cell Cardiol. 42, 590–599. doi: 10.1016/j.yjmcc.2006.12.007
Iyer, R. B., Koritz, S. B., and Kirchberger, M. A. (1988). A regulation of the level of phosphorylated phospholamban by inhibitor-1 in rat heart preparations in vitro. Mol. Cell. Endocrinol. 55, 1–6. doi: 10.1016/0303-7207(88)90084-6
James, P., Inui, M., Tada, M., Chiesi, M., and Carafoli, E. (1989). Nature and site of phospholamban regulation of the Ca2+ pump of sarcoplasmic reticulum. Nature 342, 90–92. doi: 10.1038/342090a0
Jaski, B. E., Jessup, M. L., Mancini, D. M., Cappola, T. P., Pauly, D. F., Greenberg, B., et al. (2009). Calcium up-regulation by percutaneous administration of gene therapy in cardiac disease (CUPID) trial investigators. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID Trial), a first-inhuman phase 1/2 clinical trial. J. Card. Fail. 15, 171–181. doi: 10.1016/j.cardfail.2009.01.013
Ji, J., Zhao, W., Li, B., Desantiago, J., Picht, E., Kaetzel, M., et al. (2006). Targeted inhibition of sarcoplasmic reticulum CaMKII activity results in alterations of Ca2+ homeostasis and cardiac contractility. Am. J. Physiol. 290, H599–H606.
Joiner, M. A., Kovall, O. M., Li, J., He, B. J., Allamargot, C., Gao, Z., et al. (2012). CaMKII determines mitochondrial stress responses in heart. Nature 491, 269–274. doi: 10.1038/nature11444
Kadambi, V. J., Ponniah, S., Harrer, J., Hoit, B., Dorn, G. W., Walsh, R. A., et al. (1996). Cardiac-specific overexpression of phospholamban alters calcium kinetics and resultant cardiomyocyte mechanics in transgenic mice. J. Clin. Invest. 97, 533–539. doi: 10.1172/JCI118446
Kim, H. W., Steenaart, N. A., Ferguson, D. G., and Kranias, E. G. (1990). Functional reconstitution of the cardiac sarcoplasmic reticulum Ca2+-ATPase with phospholamban in phospholipid vesicles. J. Biol. Chem. 265, 1702–1709.
Kirchberger, M. A., Tada, M., Repke, D. I., and Katz, A. M. (1972). Cyclic adenosine 3’, 5’-monophosphate dependent protein kinase stimulation of calcium uptake by canine cardiac microsomes. J. Mol. Cell Cardiol. 4, 673–680. doi: 10.1016/0022-2828(72)90120-4
Komukai, K., Pascarel, C., and Orchard, C. H. (2001). Compensatory role of CaMKII on ICa and SR function during acidosis in rat ventricular myocytes. Pflügers Arch. 442, 353–361. doi: 10.1007/s004240100549
Kranias, E. G. (1985). Regulation of calcium transport by protein phosphatase activity associated with cardiac sarcoplasmic reticulum. J. Biol. Chem. 260, 11006–11010.
Kuschel, M., Karczewski, P., Hempel, P., Schlegel, W. P., Krause, E. G., and Bartel, S. (1999). Ser16 prevails over Thr17 phospholamban phosphorylation in the beta-adrenergic regulation of cardiac relaxation. Am. J. Physiol. 276, H1625–H1633.
Lam, C. K., Zhao, W., Cai, W., Vafiadaki, E., Florea, S. M., Ren, X., et al. (2013). Novel role of HAX-1 in ischemic injury protection involvement of heat shock protein 90. Circ. Res. 112, 79–89. doi: 10.1161/CIRCRESAHA.112.279935
Lindemann, J. P., and Watanabe, A. M. (1985). Phosphorylation of phospholamban in intact myocardium. Role of Ca2+-calmodulin-dependent mechanisms. J. Biol. Chem. 260, 4516–4525.
Lindemann, J. P., Jones, L. R., Hathaway, D. R., Henry, B. G., and Watanabe, A. M. (1983). β-adrenergic simulation of phospholamban phosphorylation and Ca2+-ATPase activity in guine pig ventricles. J. Biol. Chem. 258, 464–471.
Luo, W., Chu, G., Sato, Y., Zhou, Z., Kadambi, V. J., and Kranias, E. G. (1998). Transgenic approaches to define the functional role of dual site phospholamban phosphorylation. J. Biol. Chem. 273, 4734–4739. doi: 10.1074/jbc.273.8.4734
Luo, W., Grupp, I. L., Harrer, J., Ponniah, S., Grupp, G., Duffy, J. J., et al. (1994). Targeted ablation of the phospholamban gene is associatied with markedly enhanced myocardial contractility and loss of β-agonist simulation. Circ. Res. 75, 401–409. doi: 10.1161/01.RES.75.3.401
Luo, W., Wolska, B. M., Grupp, I. L., Harrer, J. M., Haghighi, K., Ferguson, D. G., et al. (1996). Phospholamban gene dosage effects in the mammalian heart. Circ. Res. 78, 839–847. doi: 10.1161/01.RES.78.5.839
MacDougall, L. K., Jones, L. R., and Cohen, P. (1991). Identification of the major protein phosphatases in mammalian cardiac muscle which dephosphorylate phospholamban. Eur. J. Biochem. 196, 725–734. doi: 10.1111/j.1432-1033.1991.tb15871.x
Mandel, F., Kranias, E. G., de Gende, A. G., Sumida, M., and Schwart, A. (1982). The effect of pH on the transient-state kinetics of Ca2+–Mg2+-ATPase of cardiac sarcoplasmic reticulum. Circ. Res. 50, 310–317. doi: 10.1161/01.RES.50.2.310
Mattiazzi, A., and Cingolani, H. E. (1977a). Biphasic effect of hypercapnia on myocardial contractility. Arch. Int. Physiol. Biochim. 85, 11–25.
Mattiazzi, A., and Cingolani, H. E. (1977b). Paradoxical effect of hypercapnia on toad ventricular muscle. Circ. Res. 41, 117–122. doi: 10.1161/01.RES.41.1.117
Meyer, M., Schillinger, W., Pieske, B, Holubarsch, C., Heilmann, C., Posival, H., et al. (1995). Alterations of sarcoplasmic reticulum proteins in failing human dilated cardiomyopathy. Circulation 92, 778–784. doi: 10.1161/01.CIR.92.4.778
Minamisawa, S., Hoshijima, M., Chu, G., Ward, C. A., Frank, K., Gu, Y., et al. (1999). Chronic phospholamban-sarcoplasmic reticulum calcium ATPase interaction is the critical calcium cycling defect in dilated cardiomyopathy. Cell 99, 313–322. doi: 10.1016/S0092-8674(00)81662-1
Mishra, S., Sabbah, H. N., Jain, J. C., and Gupta, R. C. (2003). Reduced Ca2+-calmodulin-dependent protein kinase activity and expression of LV myocardium of dogs with heart failure. Am. J. Physiol. 284, H876–H883.
Movsesian, M. A., Nishikawa, M., and Adelstein, R. S. (1984). Phosphorylation of phospholamban by calcium-activated, phospholipid-dependent protein kinase. Stimulation of cardiac sarcoplasmic reticulum calcium uptake. J. Biol. Chem. 259, 8029–8032.
Münch, G., Bölck, B., Karczewski, P., and Schwinger, R. H. (2002). Evidence for calcineurin-mediated regulation of SERCA2a activity in human myocardium. J. Mol. Cell Cardiol. 34, 321–334. doi: 10.1006/jmcc.2001.1515
Mundiña-Weilenmann, C., Ferrero, P., Said, M., Vittone, L., Kranias, E. G., and Mattiazzi, A. (2005). Role of phosphorylation of Thr17 residue of phospholambanin mechanical recovery during hypercapnic acidosis. Cardiovasc. Res. 66, 114–122. doi: 10.1016/j.cardiores.2004.12.028
Mundiña-Weilenmann, C., Vittone, L., Ortale, M., Chiappe de Cingolani, G., and Mattiazzi, A. (1996). Immunodetection of phosphorylation sites gives new insights into the mechanisms underlying phospholamban phosphorylation in the intact heart. J. Biol. Chem. 271, 33561–33567. doi: 10.1074/jbc.271.52.33561
Napolitano, R., Vittone, L., Mundiña Weilenmann, C., Chiappe de Cingolani, G., and Mattiazzi, A. (1992). Phosphorylation of phospholamban in the intact heart. A study on the physiological role of the Ca-calmodulin-dependent protein kinase system. J. Mol. Cell Cardiol. 24, 387–396. doi: 10.1016/0022-2828(92)93193-N
Neef, S., Sag, C. M., Daut, M., Bäumer, H., Grefe, C., El-Armouche A, et al. (2013). While systolic cardiomyocyte function is preserved, diastolic myocyte function and recovery from acidosis are impaired in CaMKIIδ-KO mice. J. Mol. Cell Cardiol. 59, 107–116. doi: 10.1016/j.yjmcc.2013.02.014
Netticadan, T., Temsah, R., Kawabata, K., and Dallha, NS. (2000). Sarcoplasmic reticulum Ca2+/calmodulin-dependent kinase is altered in heart failure. Circ. Res. 86, 596–605. doi: 10.1161/01.RES.86.5.596
Neumann, J., Gupta, R. C., Schmitz, W., Scholz, H., Nairn, A. C., and Watanabe, A. M. (1991). Evidence for isoproterenol-induced phosphorylation of phosphatase inhibitor-1 in the intact heart. Circ. Res. 69, 1450–1457. doi: 10.1161/01.RES.69.6.1450
Nicolaou, P., Rodriguez, P., Ren, X., Zhou, X., Qian, J., Sadayappan, S., et al. (2009). Inducible expression of active protein phosphatase-1 inhibitor-1 enhances basal cardiac function and protects against ischemia/reperfusion injury. Circ. Res. 104, 1012–1020. doi: 10.1161/CIRCRESAHA.108.189811
Nomura, N., Satoh, H., Terada, H., Matsunaga, M., Watanabe, H., and Hayashi, H. (2002). CaMKII-dependent reactivation of SR Ca2+ uptake and contractile recovery during intracellular acidosis. Am. J. Physiol. 283, H193–H203.
O’Rourke, B., Kass, D. A., Tomaselli, G. F., Kääb, S., Tunin, R., and Marbán, E. (1999). Mechanisms of altered excitation-contraction coupling in canine tachicardia-induced heart failure. I. Experimental studies. Circ. Res. 84, 562–570. doi: 10.1161/01.RES.84.5.562
Oestreich, E. A., Malik, S., Goonasekera, S. A., Blaxall, B. C., Kelley, G. G., Dirksen, R. T., et al. (2009). Epac and phospholipase Cε regulate Ca2+ release in the heart by activation of protein kinase Cε and calcium-calmodulin kinase II. J. Biol. Chem. 284, 1514–1522. doi: 10.1074/jbc.M806994200
Oestreich, E. A., Wang, H., Malik, S., Kaproth-Joslin, K. A., Blaxall, B. C., Kelley, G. G., et al. (2007). Epac-mediated activation of phospholipase Cε plays a critical role in β-adrenergic receptor-dependent enhancement of Ca2+ mobilization in cardiac myocytes. J. Biol. Chem. 282, 5488–5495. doi: 10.1074/jbc.M608495200
Orchard, C. H., Houser, S. R., Kort, A. A., Bahinski, A., Capogrossi, M. C., and Lakatta, E. G. (1987). Acidosis facilitates spontaneous sarcoplasmic reticulum Ca2+ release in rat myocardium. J. Genet. Physiol. 90, 145–165. doi: 10.1085/jgp.90.1.145
Orchard, C. H., and Kentish, J. C. (1990). Effects of changes of pH on the contractile function of cardiac muscle. Am. J. Physiol. 258, C967–C981.
Pedrozo, Z., Sánchez, G., Torrealba, N., Valenzuela, R., Fernández, C., Hidalgo, C., et al. (2010). Calpains and proteasomes mediate degradation of ryanodine receptors in a model of cardiac ischemic reperfusion. Biochim. Biophys. Acta 1802, 356–362. doi: 10.1016/j.bbadis.2009.12.005
Pereira, L., Cheng, H., Lao, D. H., Na, L., van Oort, R. J., Brown, J. H., et al. (2013). Epac2 mediates cardiac β1-adrenergic-dependent sarcoplasmic reticulum Ca2+ leak and arrhythmia. Circulation 127, 913–922. doi: 10.1161/CIRCULATIONAHA.12.148619
Pereira, L., Metrich, M., Fernández-Velasco, M., Lucas, A., Leroy, J., Perrier, R., et al. (2007). The cAMP binding protein Epac modulates Ca2+ sparks by a Ca2+/calmodulin kinase signalling pathway in rat cardiac myocytes. J. Physiol. 583, 685–694. doi: 10.1113/jphysiol.2007.133066
Pérez, N. G., Mattiazzi, A. Camilion de Hurtado, M. C., and Cingolani, H. E. (1995). Myocardial contractility recovery during hypercapnic acidosis: its dissociation from recovery in pHi by ryanodine. Can. J. Cardiol. 11, 553–560.
Philipson, K. D., Bersohn, M. M., and Nishimoto, A. Y. (1982). Effects of pH on Na+–Ca2+ exchange in canine sarcolemmal vesicles. Circ. Res. 50, 287–293. doi: 10.1161/01.RES.50.2.287
Piacentino, V. III, Weber, C. R., Chen, X., Weisser-Thomas, J., Margulies, K. B., Bers, D. M., et al. (2003). Cellular basis of abnormal calcium transients of failing human ventricular myocytes. Circ. Res. 92, 651–658. doi: 10.1161/01.RES.0000062469.83985.9B
Picht, E., DeSantiago, J., Huke, S., Kaetzel, M. A., Dedman, J. R., and Bers, D. M. (2007). CaMKII inhibition targeted to the sarcoplasmic reticulum inhibits frequency-dependent acceleration of relaxation and Ca2+ current facilitation. J. Mol. Cell Cardiol. 42, 196–205. doi: 10.1016/j.yjmcc.2006.09.007
Poole-Wilson, P. A. (1989). Regulation of intracellular pH in the myocardium; relevance to pathology. Mol. Cell. Biochem. 89, 151–155. doi: 10.1007/BF00220768
Port, J. D., and Bristow, M. R. (2001). Altered beta-adrenergic receptor gene regulation and signaling in chronic heart failure. J. Mol. Cell Cardiol. 33, 887–905. doi: 10.1006/jmcc.2001.1358
Qian, J., Vafiadaki, E., Florea, S., Singh, V., Song, W., Lam, C. K., et al. (2011). Small heat shock protein 20 interacts with protein phosphatise-1 and enhances sarcoplasmic reticulum calcium cycling. Circ. Res. 108, 1429–1438. doi: 10.1161/CIRCRESAHA.110.237644
Respress, J. L., van Oort, R. J., Li, N., Rolim, N., Dixit, S. S, deAlmeida, A., et al. (2012). Role of RyR2 phosphorylation at S2814 during heart failure progression. Circ. Res. 110, 1474–1483. doi: 10.1161/CIRCRESAHA.112.268094
Rizzuto, R., and Pozzan, T. (2006). Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol. Rev. 86, 369–408. doi: 10.1152/physrev.00004.2005
Said, M., Becerra, R., Palomeque, J., Rinaldi, G., M. Kaetzel, M. A., Diaz-Sylvester, P. L., et al. (2008). Increased intracellular Ca2+ and SR Ca2+ load contribute to arrhythmias after acidosis in rat heart. Role of Ca2+/calmodulin-dependent protein kinase II. Am. J. Physiol. 295, H1669–H1683. doi: 10.1152/ajpheart.00010.2008
Said, M., Becerra, R., Valverde, C. A., Kaetzel, M. A., Dedman, J. R., Mundiña-Weilenmann, C., et al. (2011). Calcium-calmodulin dependent protein kinase II (CaMKII): a main signal responsible for early reperfusion arrhythmias. J. Mol. Cell Cardiol. 51, 936–944. doi: 10.1016/j.yjmcc.2011.08.010
Said, M., Mundiña-Weilenmann, C., Vittone, L., and Mattiazzi, A. (2002). The relative relevance of phosphorylation of the Thr17 residue of phospholamban is different at different levels of β-adrenergic stimulation. Pflügers Arch. 444, 801–809. doi: 10.1007/s00424-002-0885-y
Said, M., Vittone, L., Mundiña-Weilenmann, C., Ferrero, P., Kranias, E. G., and Mattiazzi, A. (2003). Role of dual-site phospholamban phosphorylation in the stunned heart: insights from phospholamban site-specific mutants. Am. J. Physiol. 285, H1198–H1205. doi: 10.1152/ajpheart.00209.2003
Salas, M. A., Valverde, C. A., Sánchez, G., Said, M., Rodriguez, J. S., Portiansky, E. L., et al. (2010). The signaling pathway of CaMKII-mediated apoptosis and necrosis in the ischemia/reperfusion injury. J. Mol. Cell Cardiol. 48, 1298–1306. doi: 10.1016/j.yjmcc.2009.12.015
Sande, J. B., Sjaastad, I., Hoen, I. B., Bokenes, J., Tonnessen, T., Holt, E., et al. (2002). Reduced level of serine16 phosphorylated phospholamban in the failing rat myocardium: a major contributor to reduced SERCA2 activity. Cardiovasc. Res. 53, 383–391. doi: 10.1016/S0008-6363(01)00489-8
Schwinger, R. H., Munch, G., Bolk, B., Karczewski, P., Krause, E-G., and Erdmann, E. (1999). Reduced Ca2+-sensitivity of SERCA2a in failing human myocardium due to reduced Serin-16 phospholamban phosphorylation. J. Mol. Cell Cardiol. 31, 479–491. doi: 10.1006/jmcc.1998.0897
Shan, J., Betzenhauser, M. J., Kushnir, A., Reiken, S., Meli, A. C., Wronska, A., et al. (2010). Role of chronic ryanodine receptor phosphorylation in heart failure and β-adrenergic receptor blockade in mice. J. Clin. Invest. 120, 4375–4387. doi: 10.1172/JCI37649
Simmerman, H. K. B., and Jones, L. R. (1998). Phospholamban: protein structure, mechanism of action and role in cardiac function. Physiol. Rev. 78, 921–947.
Simmerman, H. K., Collins, J. H., Theibert, J. L., Wegener, A. D., and Jones, L. R. (1986). Sequence analysis of phospholamban. Identification of phosphorylation sites and two major structural domains. J Biol Chem. 61, 13333–13341.
Tada, M., Kirchberger, M. A., and Katz, A. M. (1975). Phosphorylation of a 22,000-dalton component of the cardiac sarcoplasmic reticulum by adenosine 3’,5’-monophosphate-dependet protein kinase. J. Biol. Chem. 250, 2640–2647.
Talukder, M. A., Kalyanasundaram, A., Zhao, X., Zuo, L., Bhupathy, P., Babu, G. J., et al. (2007). Expression of SERCA isoform with faster Ca2+-transport properties improves post-ischemic cardiac function and Ca2+-handling and decreases myocardial infarction. Am. J. Physiol. 293, H2418–H2428.
Talukder, M. A., Kalyanasundaram, A., Zuo, L., Velayutham, M., Nishijima, Y., Periasamy, M., et al. (2008). Is reduced SERCA2a expression detrimental or beneficial to postischemic cardiac function and injury? Evidence from heterozygous SERCA2a knockout mice. Am. J. Physiol. 294, H1426–H1434. doi: 10.1152/ajpheart.01016.2007
Vafiadaki, E., Sanoudou, D., Arvanitis, D. A., Catino, D. H., Kranias, E. G., and Kontrogianni-Konstantopoulos, A. (2007). Phospholamban interacts with HAX-1, a mitochondrial protein with anti-apoptotic function. J. Mol. Biol. 367, 65–79. doi: 10.1016/j.jmb.2006.10.057
Valverde, C. A., Di Carlo, M. N., Said, M., Wehrens, X. H., Salas, M., and Mattiazzi, A. (2013). Phosphorylation of ryanodine receptors (RyR2) is a key player in the ischemia/reperfusion-induced infarct size and decrease in cardiac function. J. Mol. Cell Cardiol. 65(Suppl.), S109.
Valverde, C. A., Kornyeyev, D., Ferreiro, M., Petrosky, A. D., Mattiazzi, A., and Escobar, A. L. (2010). Transient Ca2+ depletion of the sarcoplasmic reticulum at the onset of reperfusion. Cardiovasc. Res. 85, 671–680. doi: 10.1093/cvr/cvp371
Valverde, C. A., Mundiña-Weilenmann, C., Reyes, M., Kranias, E. G., Escobar, A. L., and Mattiazzi, A. (2006). Phospholamban phosphorylation sites enhance the recovery of intracellular Ca2+ after perfusion arrest in isolated, perfused mouse heart. Cardiovasc. Res. 70, 335–345. doi: 10.1016/j.cardiores.2006.01.018
Valverde, C., Mundiña-Weilenmann, C., Said, M., Ferrero, P., Vittone, L., Salas, M., et al. (2005). Frequency-dependent acceleration of relaxation in mammalian heart: a property not relying on phospholamban and SERCA2a phosphorylation. J. Physiol. (Lond.) 562, 801–813. doi: 10.1113/jphysiol.2004.075432
Vila-Petroff, M., Mundiña-Weilenmann, C., Lezcano, N., Snabaitis, A. K., Huergo, M. A., Valverde, C. A., et al. (2010). Ca(2+)/calmodulin-dependent protein kinase II contributes to intracellular pH recovery from acidosis via Na(+)/H(+) exchanger activation. J. Mol. Cell Cardiol. 49, 106–112. doi: 10.1016/j.yjmcc.2009.12.007
Vila-Petroff, M., Salas, M., Said, M., Valverde, C. A., Sapia, L., Portiansky, E., et al. (2007). CaMKII-Inhibition Protects against Necrosis and Apoptosis in the Irreversible Ischemia-Reperfusion Injury. Cardiovasc. Res. 73, 689–698. doi: 10.1016/j.cardiores.2006.12.003
Vittone, L., Mundiña-Weilenmann, C., Chiappe de Cingolani, G., and Mattiazzi, A. (1990). cAMP and calcium dependent mechanisms of phospholamban phosphorylation in intact hearts. Am. J. Physiol. 258, H318–H325.
Vittone, L., Mundiña-Weilenmann, C., Said, M., and Mattiazzi, A. (1998). Mechanisms involved in the acidosis enhancement of the isoproterenol-induced phosphorylation of phospholamban in the intact heart. J. Biol. Chem. 273, 9804–9811. doi: 10.1074/jbc.273.16.9804
Vittone, L., Mundiña-Weilenmann, C., Said, M., Ferrero, P., and Mattiazzi, A. (2002). Time course and mechanisms of phosphorylation of phospholamban residues in ischemia reperfused rat hearts. Dissociation of phospholamban phosphorylation pathways. J. Mol. Cell Cardiol. 34, 39–50. doi: 10.1006/jmcc.2001.1488
Wang, W., Zhu, W., Wang, S., Yang, D., Crow, M. T., Xiao, R.-P., and Cheng, H. (2004). Sustained β1-adrenergic stimulation modulates cardiac contractility by Ca2+/calmodulin kinase signaling pathway. Circ. Res. 95, 798–806. doi: 10.1161/01.RES.0000145361.50017.aa
Wu, Y., Luczak, E. D., Lee, E.-J., Hidalgo, C., Yang, J., Gao, Z., et al. (2012). CaMKII effects on inotropic but not lusitropic force frequency responses require phospholamban. J. Mol. Cell Cardiol. 53, 429–436. doi: 10.1016/j.yjmcc.2012.06.019
Yang. Y., Zhu, W. Z., Joiner, M. L., Zhang, R., Oddis, C. V., Hou, Y., et al. (2006). Calmodulin kinase II inhibition protects against myocardial cell apoptosis in vivo. Am. J. Physiol. 291, H3065–H3075.
Zhang, R., Khoo, M. S., Wu, Y., Yang, Y., Grueter, C. E., Ni, G., et al. (2005). Calmodulin kinase II inhibition protects against structural heart disease. Nat. Med. 11, 409–417. doi: 10.1038/nm1215
Zhang, T., Guo, T., Mishra, S., Dalton, N. D., Kranias, E. G., Peterson, K. L., et al. (2010). Phospholamban ablation rescues sarcoplasmic reticulum Ca(2+) handling but exacerbates cardiac dysfunction in CaMKIIdeltaC transgenic mice. Circ. Res. 106, 354–362. doi: 10.1161/CIRCRESAHA.109.207423
Zhao, W., Uehara, Y., Chu, G., Song, Q., Qian, J., Young, K., et al. (2004). Threonine-17 phosphorylation of phospholamban: a key determinant of frequency-dependent increase of cardiac contractility. J. Mol. Cell Cardiol. 37, 607–612. doi: 10.1016/j.yjmcc.2004.05.013
Keywords: myocardium, CaMKII, PLN regulation, acidosis, ischemia/reperfusion injury, heart failure
Citation: Mattiazzi A and Kranias EG (2014) The role of CaMKII regulation of phospholamban activity in heart disease. Front. Pharmacol. 5:5. doi: 10.3389/fphar.2014.00005
Received: 22 November 2013; Paper pending published: 20 December 2013;
Accepted: 07 January 2014; Published online: 27 January 2014.
Edited by:
Donald M. Bers, University of California at Davis, USAReviewed by:
Mohamed Trebak, State University of New York, USAClemens Möller, Albstadt-Sigmaringen University, Germany
Copyright © 2014 Mattiazzi and Kranias. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Evangelia G. Kranias, Department of Pharmacology and Cell Biophysics, College of Medicine, University of Cincinnati, 231 Albert Sabin Way, Cincinnati, OH, USA e-mail:a3JhbmlhZWdAdWNtYWlsLnVjLmVkdQ==