




95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Pharmacol. , 27 February 2013
Sec. Pharmacology of Anti-Cancer Drugs
Volume 4 - 2013 | https://doi.org/10.3389/fphar.2013.00018
This article is part of the Research Topic Therapeutic Resistance in Cancer: Mechanisms, Insights and Progress View all 16 articles
The pharmacological inhibitors of poly(ADP-ribose) polymerase (PARP) family of proteins have shown promising results in preclinical studies and clinical trials as a monotherapy or in combination therapy for some cancers. Thus, usage of PARP-inhibitors (PARPi) in cancer therapy is bound to increase with time, but resistance of cancer cells to PARPi is also beginning to be observed. Here we review different known and potential mechanisms by which: (i) PARPi kill cancer cells; and (ii) cancer cells develop resistance to PARPi. Understanding the lethality caused by PARPi and the countermeasures deployed by cancers cells to survive PARPi will help us rationalize the use of this new class of drugs in cancer therapy.
Recent clinical trials with poly(ADP-ribose) polymerase-inhibitors as a monotherapy or in combination therapy have shown promising results against different cancers (Lord and Ashworth, 2012). Therefore, their use in cancer therapy is likely to increase, resulting in the inevitable appearance of PARPi-resistant cancers (Chiarugi, 2012). Here, we first discuss different mechanisms by which PARPi can kill cancer cells and then review several known and potential mechanisms by which cancers can become resistant to PARPi.
There are 18 members of the PARP family of proteins, but therapeutic effect of PARPi on cancer cells is observed only in conjunction with DNA damage; hence DNA damage-responsive PARPs are the most likely mediators of PARPi effect. Among three such PARPs, PARP-1 is the principle responder to DNA damage, as it rapidly reaches the damaged site and mounts a robust catalytic activation response that influences different cellular responses to DNA damage (Javle and Curtin, 2011; Yélamos et al., 2011; Gibson and Kraus, 2012). The activated PARP-1 splits the substrate nicotinamide adenine dinucleotide (NAD+) to release ADP-ribose, nicotinamide, and protons (Affar et al., 2002; Shah et al., 2011). PARP-1 then forms polymers of ADP-ribose (PAR) that post-translationally modify (i.e., PARylate) PARP-1 itself and selected target proteins to control a wide array of cellular processes, such as cell death, transcription, cell division, and DNA repair (Krishnakumar and Kraus, 2010). Among the DNA repair pathways, PARP-1 is widely recognized for its impact on the base excision repair (BER) and single strand break (SSB) repair pathways, but it also influences homologous recombination (HR) and non-homologous end-joining (NHEJ) repair of double strand breaks (DSB; Yélamos et al., 2011; De Vos et al., 2012). In addition, it also plays a role in mismatch repair (Liu et al., 2012) and more recently the nucleotide excision repair pathways (King et al., 2012; Luijsterburg et al., 2012; Pines et al., 2012; Robu et al., 2013).
In contrast to PARP-1, the other two DNA damage-responsive PARPs play a limited role in DNA damage responses. For example, PARP-2, in conjunction with PARP-1, has been shown to affect BER (Schreiber et al., 2002) and restart the stalled replication forks (Bryant et al., 2009). PARP-3 plays a role in NHEJ pathway in conjunction with APLF (Rulten et al., 2011) or PARP-1 (Boehler et al., 2011) and helps activation of PARP-1 (Loseva et al., 2010). In the context of the role of PARPi in inhibiting PARylation activity of PARPs, it is pertinent to note that PARP-2 has a very weak PARylation activity as compared to PARP-1, and many functions of PARP-2 and 3 are associated with PARP-1. Therefore, one could argue that the main target for PARPi is on the role of PARP-1 in DNA repair with possibly some effect on the roles of PARP-2 and 3. Finally, we should not exclude the possibility that the roles of PARP-1 in cell death and transcription are also involved in the therapeutic effect of PARPi.
Most consistent results in clinical trials have been obtained with competitive PARPi, which are analogs of nicotinamide that compete with the substrate NAD+ to bind to the enzyme. Unlike weak inhibitory activity of nicotinamide, its derivatives ranging from the first generation 3-aminobenzamide to the third generation Olaparib and Rucaparib are better inhibitors of PARP-1 and PARP-2 (Table 1). The Iniparib, originally developed as a non-competitive inhibitor of PARP-1, showed early successes in clinical trials, but it is a non-specific and weak inhibitor of PARP-1 (Patel et al., 2012). Hence this review will focus on the results obtained with competitive PARPi.
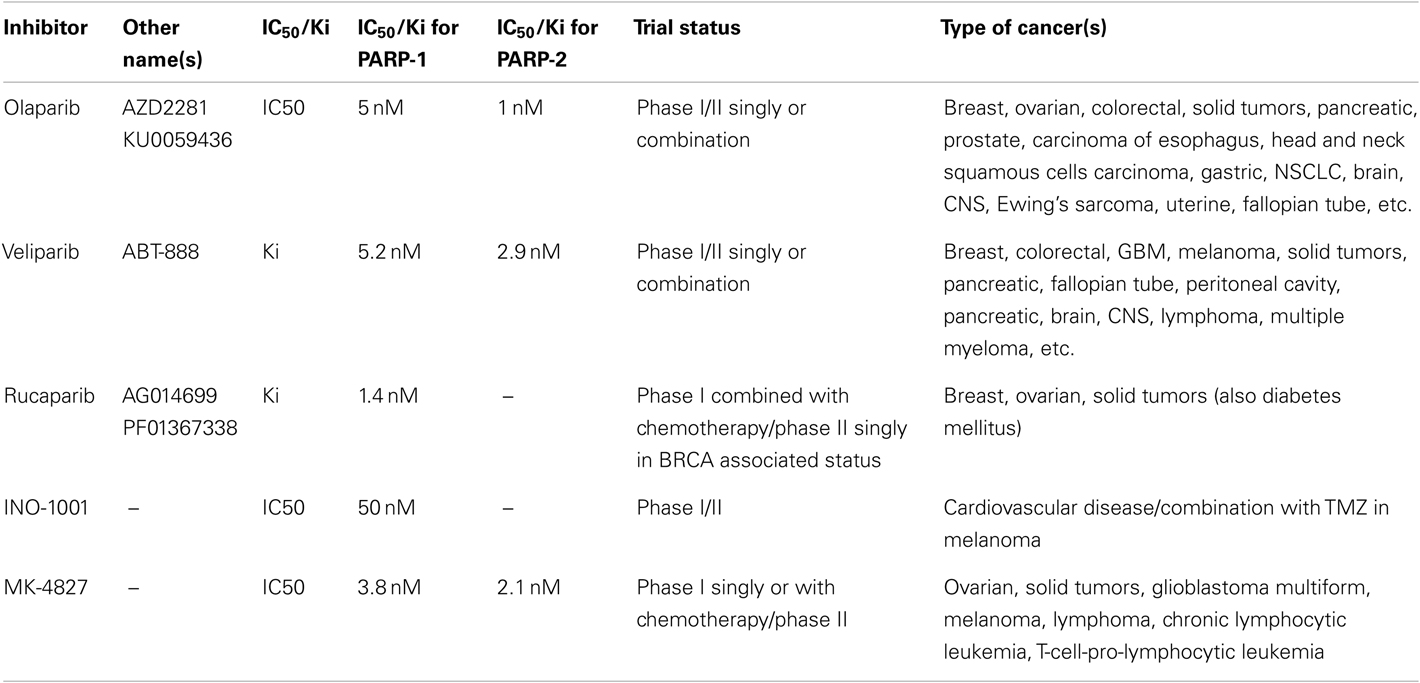
Table 1. Different PARPi currently in clinical trials and their relative inhibitory potential against PARP-1 and PARP-2 (adapted from Davar et al., 2012).
It was suggested that two mutations should be considered synthetic lethal if cells with either mutation are viable but those with both mutations are non-viable (Dobzhansky, 1946). The first success of this approach was observed in 2005, when two groups showed that PARPi, which is non-toxic to normal cells, is lethal to BRCA1/2 cancer cells that are deficient in HR-mediated repair of DSB (Bryant et al., 2005; Farmer et al., 2005; Helleday et al., 2005). Several clinical trials for different cancers have since been launched with PARPi, and a list of current trials is shown in Table 1.
There are different possible mechanisms by which PARPi kill HR-deficient tumor cells (Helleday, 2011). It was initially suggested that constant DNA damage induced by endogenous factors, such as oxidants needs to be repaired by BER in which PARP-1 participates either by binding to SSB or by collaborating with XRCC-1 (Figure 1, steps A and B). Thus, when PARPi block BER, the unrepaired SSB would stall and collapse the replication fork to create DSB (Figure 1, step C). The normal cells would survive by readily repairing these DSB by error-free HR or error-prone NHEJ (Figure 1, steps D or E). However, the DSB would be lethal to HR-deficient BRCA1/2 tumors with an excessive reliance on the error-prone NHEJ repair pathway (Aly and Ganesan, 2011). This scenario is most plausible and is supported by significant evidence, but it does not explain many things, such as lack of accumulation of SSB in PARPi-treated cells or the absence of synthetic lethality by targeting XRCC-1 in BRCA-deficient cells (Helleday, 2011).
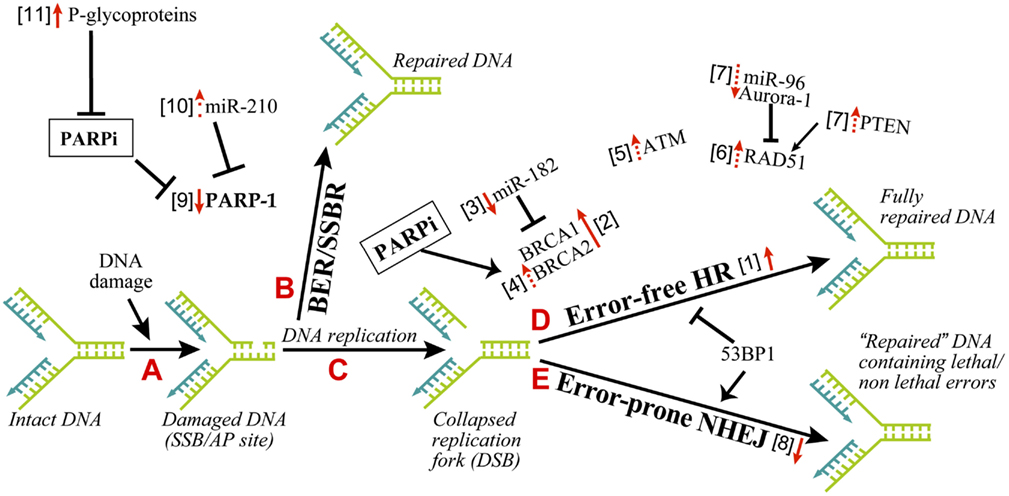
Figure 1. Mechanisms of resistance to PARPi in cancer therapy. The principle explanation for the efficacy of PARPi as synthetic lethal therapy in DNA double strand break (DSB) repair defective cancer cells or as a combination therapy with other agents for other cancers rests on the role of PARP-1 in BER and SSBR. As shown in the series of A–E steps, the inefficient repair of DNA single strand breaks by PARP-1-mediated BER in the presence of PARPi would lead to DSB. An inefficient repair of DSB by HR-deficient cancer cells will kill these cells, whereas normal cells with proper DSB repair capacity will survive. The resistance to PARPi can occur in cancer cells by alteration of various parameters, which influence different steps in this pathway. The changes in these factors, i.e., upregulation or downregulation as pointed by the direction of red arrows, is associated with resistance to PARPi. The solid or dashed arrows indicate known or hypothesized mechanisms of resistance to PARPi, respectively. The numbers within square bracket next to the arrows refer to the explanation in the text for this mechanism of PARPi-resistance.
Therefore, alternative explanations have been offered for synthetic lethality of PARPi in HR-deficient cells. In brief, it has been shown that PARP-1 binds to and is activated by SSB-intermediates formed during BER, which results in release of PARylated PARP-1 from SSB, which are then repaired (Strom et al., 2011). Thus, it is proposed that in the presence of PARPi, SSB bound to PARP-1 would collapse the replication fork and DSB-mediated lethality will occur in HR-deficient cells (Helleday, 2011). It is also possible that the role of PARP-1 in suppressing the error-prone NHEJ is the target for PARPi-induced lethality in HR-deficient cells, because inhibition or downregulation of multiple components of NHEJ, such as Ku80, Artemis, and DNA-PK, made HR-deficient cells resistant to PARPi (Patel et al., 2011). Finally, it has been suggested that since PARP-1 plays a role in reactivating the stalled replication forks, this step could be a target for PARPi-induced lethality in HR-deficient cells (Helleday, 2011).
Cancer cells are known to carry other conditions that create HR-deficiency or BRCAness, which can make them susceptible to synthetic lethality by PARPi. Three such examples are listed here (Figure 1, step D). (i) The protein kinase ataxia telangiectasia mutated (ATM), a key regulator that senses DNA damage, initiates the protein kinase cascade (Wang and Weaver, 2011) and plays a role in HR, is frequently mutated in lymphoid malignancies. Interestingly, PARPi is synthetic lethal to the ATM mutant lymphoid tumor cells (Weston et al., 2010). (ii) Aurora-1 is frequently amplified and overexpressed in breast cancers (Staff et al., 2010). An overexpression of Aurora-1 induces BRCAness in an otherwise HR-competent PIR12 pancreatic tumor cells by causing an impaired recruitment of key HR-protein RAD51, and sensitizes them to synthetic lethality by PARPi (Sourisseau et al., 2010). (iii) PTEN (phosphatase and tensin homolog), which plays a crucial role in regulating PI3K/Akt-1-mTOR signaling pathway, is frequently mutated or decreased in a wide range of human tumors (Salmena et al., 2008). The PTEN-null cancer cells, which are HR-defective due to reduced expression and nuclear localization of RAD51, are sensitive to PARPi (Mendes-Pereira et al., 2009; Dedes et al., 2010; McEllin et al., 2010; Figure 1, step D). Although another study reported that PTEN deficiency in prostate cancer cells is not associated with BRCAness or sensitivity to PARPi (Fraser et al., 2012), suggesting a need for more work in this model.
Finally, PARPi sensitivity has also been reported under circumstances without BRCAness. For example, the depletion of NHEJ components DNA-PK or Ku80 made HR-proficient cells more sensitive to PARPi (Bryant and Helleday, 2006). PARPi sensitivity is also observed in conditions with no apparent defect in any of the DNA repair pathway. The sporadic breast cancer cells overexpressing HER2 (human epidermal growth factor receptor 2) are addicted to overexpression of NF-κB-mediated transcription for survival. Since PARP-1 is a co-activator of NF-κB, the treatment with PARPi abrogates NF-κB-mediated transcription and kills these cancer cells (Nowsheen et al., 2012).
Overall, the ability of PARPi to cause synthetic lethality in cancer cells with BRCAness as well as many other conditions indicates a potential for their use as monotherapy for a wide variety of cancers.
All of the above studies dealing with synthetic lethal effect of PARPi rely on the DNA damage induced by endogenous factors, such as oxidants created during metabolism. Therefore, it is not surprising that PARPi also potentiates lethality of exogenous DNA damaging agents, such as chemotherapeutic agents or ionising radiations (Javle and Curtin, 2011). Such combination therapy has the potential to kill cancer cells with no apparent defect in DNA repair, because chemotherapy induced SSB will be amplified by PARPi to make a large flux of DSB that will overwhelm the normal DSB repair capacity of these tumors and cause death (Figure 1, steps B–E). In the actual clinical conditions for treatment of cancer patients, it is highly likely that PARPi will be used most frequently in combination therapy for DNA repair proficient and even for DNA repair deficient tumors.
There are four categories of known and potential mechanisms of resistance to PARPi in cancer cells, which are described below: (i) increased HR capacity; (ii) altered NHEJ capacity; (iii) decreased levels or activity of PARP-1, and (iv) decreased intracellular availability of PARPi.
Since pre-existing HR defect is the initial lesion that allows PARPi to kill HR-deficient tumors, any of the following conditions that restore HR could result in the resistance to PARPi (Figure 1, step D, arrow #1).
The resistance of BRCA tumors or cells to PARPi was initially identified to be due to reverse mutations in brca1/2 and restoration of HR (Figure 1, step D, arrow #2; Ashworth, 2008; Edwards et al., 2008; Sakai et al., 2008; Swisher et al., 2008; Norquist et al., 2011; Barber et al., 2013). For BRCA2, reverse mutation was in part due to intragenic deletion of the c.6174delT mutation and restoration of the open reading frame (Ashworth, 2008). The genomic instability associated with BRCA loss could be a cause for reverse mutations of brca (Aly and Ganesan, 2011). Certain BRCA1-deficient tumors carry hypomorphic BRCA1 mutations within its population (Drost et al., 2011); hence a selection of cells with restored BRCA function could confer resistance to PARPi.
BRCA1 expression is negatively regulated by the microRNA miR-182; hence miR-182 overexpression sensitizes BRCA1-proficient breast cancer cells to PARPi, whereas its downregulation made them resistant to PARPi (Moskwa et al., 2011; Figure 1, step D, arrow #3). PARP-1 and its activity is a negative modulator of BRCA2, because PARP-1 binds to the silencer-binding region of the brca2 promoter (Wang et al., 2008). Hence PARPi mediated suppression of PARP-1 activity could lead to overexpression of BRCA2 and resistance to PARPi (Figure 1, step D, arrow #4).
53BP1 is a nuclear protein that plays a key role in DNA repair responses and checkpoint control (Bunting et al., 2010). Together, BRCA1 and 53BP1 determine the balance between NHEJ and HR, because the loss of BRCA1 results in a profound defect in HR and increased NHEJ repair, whereas loss of 53BP1 suppresses NHEJ and promotes HR (Figure 1, steps D–E). While cells with defect in BRCA1 alone were susceptible to PARPi, an additional loss of 53BP1 allowed a partial ATM-dependent HR repair (Aly and Ganesan, 2011), making these cells resistant to PARPi (Cao et al., 2009; Bouwman et al., 2010; Bunting et al., 2010; Brandsma and Gent, 2012; Oplustilova et al., 2012). Thus, increased ATM alone could induce resistance to PARPi (Figure 1, steps D–E, arrow #5).
RAD51 is a key HR-protein; therefore any factor that increases RAD51 levels or activity can potentially lead to a resistance to PARPi (Figure 1, step D, arrow #6). The levels of RAD51 are suppressed by miR-96 (Wang et al., 2012) and Aurora-1 (Sourisseau et al., 2010) and increased by PTEN (Dedes et al., 2010). Hence, we hypothesize that decreased miR-96 and Aurora-1 or increased PTEN can increase RAD51 and HR activity leading to the resistance to PARPi (Figure 1, step D, arrows #7). This is indirectly supported by the observation that increased RAD51 levels make colon carcinoma cells resistant to the combined treatment of PARPi and temozolomide (Liu et al., 2009).
One of the causes for synthetic lethality of PARPi in HR-deficient cells is an upregulation of the error-prone NHEJ pathway that is normally suppressed by PARP-1. Hence any decrease in NHEJ capacity in these cells could increase their resistance to PARPi, as shown in BRCA2-deficient cells by inhibition or downregulation of Ku80, Artemis, or DNA-PK (Figure 1, step E, arrow #8; Patel et al., 2011). On the flip side, it has been suggested that normal NHEJ function and the genomic instability mediated by NHEJ could be one of the causes for reversion of the mutation of brca1/2, restoration of partial HR capacities and development of resistance to PARPi in HR-deficient tumors (Chiarugi, 2012; Figure 1, step D, arrow #2). Thus, both increased and decreased NHEJ capacity of cells could lead to resistance to PARPi in different contexts.
The effectiveness of PARPi in anti-cancer therapy requires that its target PARP-1 is available for inhibition; because in PARPi-treated cells, PARP-1 will still bind to DNA strand breaks but will not be activated to form PAR or facilitate DNA repair events. Hence reduced levels of PARP-1 could result in resistance to PARPi (Figure 1, step B, arrow #9). In fact, PARP-1 levels are significantly decreased in the PARPi and temozolomide-resistant clones of colorectal carcinoma HCT116 cells (Liu et al., 2009). Therefore, it will be interesting to see if alterations in PARP-1 levels during different stages in tumor development are also associated with a corresponding change in sensitivity to PARPi. For example, levels of miR-210, which suppresses PARP-1 expression, are initially decreased when normal breast cells are transformed to ductal carcinoma in situ, and they are increased during further transition to the invasive ductal carcinoma stage (Volinia et al., 2012). It will be interesting to test in such a series of samples, whether these changes in miR-210 are inversely associated with alterations in the levels of PARP-1 and directly correlated with the resistance to PARPi (Figure 1, step B, arrow #10). There have been reports of a correlation between the abundance of cytoplasmic PARP-1 and higher sensitivity to chemotherapy in breast cancer samples (Domagala et al., 2011; von Minckwitz et al., 2011; Klauke et al., 2012). However, cytoplasmic PARP-1 was detected at a very low frequency in these tumors, and since we do not know any role for cytoplasmic PARP-1 in DNA damage responses, it is difficult at this moment to rationalize the link between cytoplasmic PARP-1 and resistance to PARPi.
The effectiveness of PARPi is also linked to the catalytic activity of PARP-1. Hence any factor that decreases the activity of PARP-1 could influence the efficacy of PARPi. The cancer cells with normal levels of PARP-1 but decreased enzymatic activity as noted by reduced level of endogenous PARylation are more resistant to PARPi (Oplustilova et al., 2012; Figure 1, step B, arrow #9). As a corollary, HR-deficient tumor cells with higher endogenous PARylation activity are more sensitive to PARPi (Gottipati et al., 2010).
Variant forms of PARP-1 with decreased catalytic activity, such as those created by small nucleotide polymorphism (SNP), could make cancer cells resistant to PARPi. In human cancers, some SNP have indeed been found to some extent, such as V762/A (Lockett et al., 2004; Wang et al., 2007; Zaremba et al., 2009) or M129/T and E251/K (Ogino et al., 2010). However, there is no consensus as to whether V762/A reduces enzyme activity and other mutants do not have significant effect on enzyme function. Thus, it is difficult to predict the effect of SNP on the effectiveness of PARPi.
A cancer cell that can efficiently throw PARPi out of the cell can become relatively resistant to this therapy. The p-glycoproteins (P-gp) also called multi-drug resistance proteins are involved in the efflux of PARPi (Figure 1, step A, arrow #11), because P-gp inhibitors prevent the decrease of PARPi in HCT116 colon cancer cells (Oplustilova et al., 2012) and re-sensitize PARPi-resistant BRCA-1 deficient cells to PARPi (Rottenberg et al., 2008). In the mouse mammary tumor models, PARPi was more effective when P-gp knockout condition was added to BRCA-1 deficient cells (Jaspers et al., 2012). The P-gp belong to ABC transporter family which is inhibited by ADP-ribose, a product of catalytic activity of PARP-1 (Dumitriu et al., 2004). Therefore, it is feasible that PARPi that would prevent formation of ADP-ribose can permit full activity of P-gp to eliminate PARPi from the cells. Nonetheless, more work is needed in this domain because the resistance to drug via upregulation of P-gp has not yet been shown in human tumoral tissues (Borst, 2012).
In cancer treatment with PARPi, the personalization of therapy is important because many factors can influence the efficiency of PARPi, such as HR and NHEJ status, PARP-1 levels or its activity and finally other factors that influence intracellular concentrations of PARPi. Therefore, it would be necessary to assess the status of these controlling factors before beginning the treatment with PARPi (Lord and Ashworth, 2012; Ratner et al., 2012). A thorough understanding of different mechanisms for the resistance to PARPi will permit us to design better PARPi monotherapy as well as combination therapy, and will allow us to identify conditions that can re-sensitize tumor cells to PARPi; and thus treat cancer patients more efficiently.
The authors declare that the research was conducted in the absence of any commercial or financial relationship that could be construed as a potential conflict of interest.
We wish to acknowledge very useful comments made by the reviewers. The scholarship support was received from NSERC and Laval University (Émilie Pouliot and Mihaela Robu). This work was supported by the CIHR operating grant#89964 to Girish M. Shah.
Affar, E. B., Shah, R. G., Dallaire, A.-K., Castonguay, V., and Shah, G. M. (2002). Role of poly(ADP-ribose) polymerase in rapid intracellular acidification induced by alkylating DNA damage. Proc. Natl. Acad. Sci. U.S.A. 99, 245–250.
Aly, A., and Ganesan, S. (2011). BRCA1, PARP, and 53BP1: conditional synthetic lethality and synthetic viability. J. Mol. Cell Biol. 3, 66–74.
Ashworth, A. (2008). A synthetic lethal therapeutic approach: poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J. Clin. Oncol. 26, 3785–3790.
Barber, L. J., Sandhu, S., Chen, L., Campbell, J., Kozarewa, I., Fenwick, K., et al. (2013). Secondary mutations in BRCA2 associated with clinical resistance to a PARP inhibitor. J. Pathol. 229, 422–429.
Boehler, C., Gauthier, L. R., Mortusewicz, O., Biard, D. S., Saliou, J. M., Bresson, A., et al. (2011). Poly(ADP-ribose) polymerase 3 (PARP3), a newcomer in cellular response to DNA damage and mitotic progression. Proc. Natl. Acad. Sci. U.S.A. 108, 2783–2788.
Borst, P. (2012). Cancer drug pan-resistance: pumps, cancer stem cells, quiescence, epithelial to mesenchymal transition, blocked cell death pathways, persisters or what? Open Biol. 2, 120066.
Bouwman, P., Aly, A., Escandell, J. M., Pieterse, M., Bartkova, J., van der Gulden, H., et al. (2010). 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Struct. Mol. Biol. 17, 688–695.
Brandsma, I., and Gent, D. C. (2012). Pathway choice in DNA double strand break repair: observations of a balancing act. Genome Integr. 3, 9.
Bryant, H. E., and Helleday, T. (2006). Inhibition of poly (ADP-ribose) polymerase activates ATM which is required for subsequent homologous recombination repair. Nucleic Acids Res. 34, 1685–1691.
Bryant, H. E., Petermann, E., Schultz, N., Jemth, A. S., Loseva, O., Issaeva, N., et al. (2009). PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J. 28, 2601–2615.
Bryant, H. E., Schultz, N., Thomas, H. D., Parker, K. M., Flower, D., Lopez, E., et al. (2005). Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434, 913–917.
Bunting, S. F., Callen, E., Wong, N., Chen, H. T., Polato, F., Gunn, A., et al. (2010). 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 141, 243–254.
Cao, L., Xu, X., Bunting, S. F., Liu, J., Wang, R. H., Cao, L. L., et al. (2009). A selective requirement for 53BP1 in the biological response to genomic instability induced by Brca1 deficiency. Mol. Cell 35, 534–541.
Chiarugi, A. (2012). A snapshot of chemoresistance to PARP inhibitors. Trends Pharmacol. Sci. 33, 42–48.
Davar, D., Beumer, J. H., Hamieh, L., and Tawbi, H. (2012). Role of PARP inhibitors in cancer biology and therapy. Curr. Med. Chem. 19, 3907–3921.
De Vos, M., Schreiber, V., and Dantzer, F. (2012). The diverse roles and clinical relevance of PARPs in DNA damage repair: current state of the art. Biochem. Pharmacol. 84, 137–146.
Dedes, K. J., Wetterskog, D., Mendes-Pereira, A. M., Natrajan, R., Lambros, M. B., Geyer, F. C., et al. (2010). PTEN deficiency in endometrioid endometrial adenocarcinomas predicts sensitivity to PARP inhibitors. Sci. Transl. Med. 2, 53ra75.
Dobzhansky, T. (1946). Genetics of natural populations. Xiii. Recombination and variability in populations of Drosophila pseudoobscura. Genetics 31, 269–290.
Domagala, P., Huzarski, T., Lubinski, J., Gugala, K., and Domagala, W. (2011). PARP-1 expression in breast cancer including BRCA1-associated, triple negative and basal-like tumors: possible implications for PARP-1 inhibitor therapy. Breast Cancer Res. Treat. 127, 861–869.
Drost, R., Bouwman, P., Rottenberg, S., Boon, U., Schut, E., Klarenbeek, S., et al. (2011). BRCA1 RING function is essential for tumor suppression but dispensable for therapy resistance. Cancer Cell 20, 797–809.
Dumitriu, I. E., Voll, R. E., Kolowos, W., Gaipl, U. S., Heyder, P., Kalden, J. R., et al. (2004). UV irradiation inhibits ABC transporters via generation of ADP-ribose by concerted action of poly(ADP-ribose) polymerase-1 and glycohydrolase. Cell Death Differ. 11, 314–320.
Edwards, S. L., Brough, R., Lord, C. J., Natrajan, R., Vatcheva, R., Levine, D. A., et al. (2008). Resistance to therapy caused by intragenic deletion in BRCA2. Nature 451, 1111–1115.
Farmer, H., McCabe, N., Lord, C. J., Tutt, A. N., Johnson, D. A., Richardson, T. B., et al. (2005). Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921.
Fraser, M., Zhao, H., Luoto, K. R., Lundin, C., Coackley, C., Chan, N., et al. (2012). PTEN deletion in prostate cancer cells does not associate with loss of RAD51 function: implications for radiotherapy and chemotherapy. Clin. Cancer Res. 18, 1015–1027.
Gibson, B. A., and Kraus, W. L. (2012). New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 13, 411–424.
Gottipati, P., Vischioni, B., Schultz, N., Solomons, J., Bryant, H. E., Djureinovic, T., et al. (2010). Poly(ADP-ribose) polymerase is hyperactivated in homologous recombination-defective cells. Cancer Res. 70, 5389–5398.
Helleday, T. (2011). The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Mol. Oncol. 5, 387–393.
Helleday, T., Bryant, H. E., and Schultz, N. (2005). Poly(ADP-ribose) polymerase (PARP-1) in homologous recombination and as a target for cancer therapy. Cell Cycle 4, 1176–1178.
Jaspers, J. E., Kersbergen, A., Boon, U., Sol, W., van Deemter, L., Zander, S. A., et al. (2012). Loss of 53BP1 causes PARP inhibitor resistance in BRCA1-mutated mouse mammary tumors. Cancer Discov. 3, 68–81.
Javle, M., and Curtin, N. J. (2011). The role of PARP in DNA repair and its therapeutic exploitation. Br. J. Cancer 105, 1114–1122.
King, B. S., Cooper, K. L., Liu, K. J., and Hudson, L. G. (2012). Poly(ADP-ribose) contributes to an association between Poly(ADP-ribose)polymerase-1 and xeroderma pigmentosum complementation group A in nucleotide excision repair. J. Biol. Chem. 287, 39824–39833.
Klauke, M. L., Hoogerbrugge, N., Budczies, J., Bult, P., Prinzler, J., Radke, C., et al. (2012). Higher cytoplasmic and nuclear poly(ADP-ribose) polymerase expression in familial than in sporadic breast cancer. Virchows Arch. 461, 425–431.
Krishnakumar, R., and Kraus, W. L. (2010). The PARP side of the nucleus: molecular actions, physiological outcomes, and clinical targets. Mol. Cell 39, 8–24.
Liu, X., Han, E. K., Anderson, M., Shi, Y., Semizarov, D., Wang, G., et al. (2009). Acquired resistance to combination treatment with temozolomide and ABT-888 is mediated by both base excision repair and homologous recombination DNA repair pathways. Mol. Cancer Res. 7, 1686–1692.
Liu, Y., Kadyrov, F. A., and Modrich, P. (2012). PARP-1 enhances the mismatch-dependence of 5′-directed excision in human mismatch repair in vitro. DNA Repair (Amst.) 10, 1145–1153.
Lockett, K. L., Hall, M. C., Xu, J., Zheng, S. L., Berwick, M., Chuang, S. C., et al. (2004). The ADPRT V762A genetic variant contributes to prostate cancer susceptibility and deficient enzyme function. Cancer Res. 64, 6344–6348.
Lord, C. J., and Ashworth, A. (2012). The DNA damage response and cancer therapy. Nature 481, 287–294.
Loseva, O., Jemth, A. S., Bryant, H. E., Schuler, H., Lehtio, L., Karlberg, T., et al. (2010). PARP-3 is a mono-ADP-ribosylase that activates PARP-1 in the absence of DNA. J. Biol. Chem. 285, 8054–8060.
Luijsterburg, M. S., Lindh, M., Acs, K., Vrouwe, M. G., Pines, A., van Attikum, H., et al. (2012). DDB2 promotes chromatin decondensation at UV-induced DNA damage. J. Cell Biol. 197, 267–281.
McEllin, B., Camacho, C. V., Mukherjee, B., Hahm, B., Tomimatsu, N., Bachoo, R. M., et al. (2010). PTEN loss compromises homologous recombination repair in astrocytes: implications for glioblastoma therapy with temozolomide or poly(ADP-ribose) polymerase inhibitors. Cancer Res. 70, 5457–5464.
Mendes-Pereira, A. M., Martin, S. A., Brough, R., McCarthy, A., Taylor, J. R., Kim, J. S., et al. (2009). Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol. Med. 1, 315–322.
Moskwa, P., Buffa, F. M., Pan, Y., Panchakshari, R., Gottipati, P., Muschel, R. J., et al. (2011). miR-182-mediated downregulation of BRCA1 impacts DNA repair and sensitivity to PARP inhibitors. Mol. Cell 41, 210–220.
Norquist, B., Wurz, K. A., Pennil, C. C., Garcia, R., Gross, J., Sakai, W., et al. (2011). Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J. Clin. Oncol. 29, 3008–3015.
Nowsheen, S., Cooper, T., Bonner, J. A., Lobuglio, A. F., and Yang, E. S. (2012). HER2 overexpression renders human breast cancers sensitive to PARP inhibition independently of any defect in homologous recombination DNA repair. Cancer Res. 72, 4796–4806.
Ogino, H., Nakayama, R., Sakamoto, H., Yoshida, T., Sugimura, T., and Masutani, M. (2010). Analysis of poly(ADP-ribose) polymerase-1 (PARP1) gene alteration in human germ cell tumor cell lines. Cancer Genet. Cytogenet. 197, 8–15.
Oplustilova, L., Wolanin, K., Mistrik, M., Korinkova, G., Simkova, D., Bouchal, J., et al. (2012). Evaluation of candidate biomarkers to predict cancer cell sensitivity or resistance to PARP-1 inhibitor treatment. Cell Cycle 11, 3837–3850.
Patel, A. G., De Lorenzo, S. B., Flatten, K. S., Poirier, G. G., and Kaufmann, S. H. (2012). Failure of iniparib to inhibit poly(ADP-Ribose) polymerase in vitro. Clin. Cancer Res. 18, 1655–1662.
Patel, A. G., Sarkaria, J. N., and Kaufmann, S. H. (2011). Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc. Natl. Acad. Sci. U.S.A. 108, 3406–3411.
Pines, A., Vrouwe, M. G., Marteijn, J. A., Typas, D., Luijsterburg, M. S., Cansoy, M., et al. (2012). PARP1 promotes nucleotide excision repair through DDB2 stabilization and recruitment of ALC1. J. Cell Biol. 199, 235–249.
Ratner, E. S., Sartorelli, A. C., and Lin, Z. P. (2012). Poly (ADP-ribose) polymerase inhibitors: on the horizon of tailored and personalized therapies for epithelial ovarian cancer. Curr. Opin. Oncol. 24, 564–571.
Robu, M., Shah, R. G., Petitclerc, N., Brind’Amour, J., Kandan-Kulangara, F., and Shah, G. M. (2013). Role of poly(ADP-ribose) polymerase-1 in the removal of UV-induced DNA lesions by nucleotide excision repair. Proc. Natl. Acad. Sci. U.S.A. 110, 1658–1663.
Rottenberg, S., Jaspers, J. E., Kersbergen, A., van der Burg, E., Nygren, A. O., Zander, S. A., et al. (2008). High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc. Natl. Acad. Sci. U.S.A. 105, 17079–17084.
Rulten, S. L., Fisher, A. E., Robert, I., Zuma, M. C., Rouleau, M., Ju, L., et al. (2011). PARP-3 and APLF function together to accelerate nonhomologous end-joining. Mol. Cell 41, 33–45.
Sakai, W., Swisher, E. M., Karlan, B. Y., Agarwal, M. K., Higgins, J., Friedman, C., et al. (2008). Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 451, 1116–1120.
Salmena, L., Carracedo, A., and Pandolfi, P. P. (2008). Tenets of PTEN tumor suppression. Cell 133, 403–414.
Schreiber, V., Ame, J. C., Dolle, P., Schultz, I., Rinaldi, B., Fraulob, V., et al. (2002). Poly(ADP-ribose) polymerase-2 (PARP-2) is required for efficient base excision DNA repair in association with PARP-1 and XRCC1. J. Biol. Chem. 277, 23028–23036.
Shah, G. M., Kandan-Kulangara, F., Montoni, A., Shah, R. G., Brind’Amour, J., Vodenicharov, M. D., et al. (2011). Approaches to detect PARP-1 activation in vivo, in situ, and in vitro. Methods Mol. Biol. 780, 3–34.
Sourisseau, T., Maniotis, D., McCarthy, A., Tang, C., Lord, C. J., Ashworth, A., et al. (2010). Aurora-A expressing tumour cells are deficient for homology-directed DNA double strand-break repair and sensitive to PARP inhibition. EMBO Mol. Med. 2, 130–142.
Staff, S., Isola, J., Jumppanen, M., and Tanner, M. (2010). Aurora-A gene is frequently amplified in basal-like breast cancer. Oncol. Rep. 23, 307–312.
Strom, C. E., Johansson, F., Uhlen, M., Szigyarto, C. A., Erixon, K., and Helleday, T. (2011). Poly (ADP-ribose) polymerase (PARP) is not involved in base excision repair but PARP inhibition traps a single-strand intermediate. Nucleic Acids Res. 39, 3166–3175.
Swisher, E. M., Sakai, W., Karlan, B. Y., Wurz, K., Urban, N., and Taniguchi, T. (2008). Secondary BRCA1 mutations in BRCA1-mutated ovarian carcinomas with platinum resistance. Cancer Res. 68, 2581–2586.
Volinia, S., Galasso, M., Sana, M. E., Wise, T. F., Palatini, J., Huebner, K., et al. (2012). Breast cancer signatures for invasiveness and prognosis defined by deep sequencing of microRNA. Proc. Natl. Acad. Sci. U.S.A. 109, 3024–3029.
von Minckwitz, G., Muller, B. M., Loibl, S., Budczies, J., Hanusch, C., Darb-Esfahani, S., et al. (2011). Cytoplasmic poly(adenosine diphosphate-ribose) polymerase expression is predictive and prognostic in patients with breast cancer treated with neoadjuvant chemotherapy. J. Clin. Oncol. 29, 2150–2157.
Wang, J., Bian, C., Li, J., Couch, F. J., Wu, K., and Zhao, R. C. (2008). Poly(ADP-ribose) polymerase-1 down-regulates BRCA2 expression through the BRCA2 promoter. J. Biol. Chem. 283, 36249–36256.
Wang, X., and Weaver, D. T. (2011). The ups and downs of DNA repair biomarkers for PARP inhibitor therapies. Am. J. Cancer Res. 1, 301–327.
Wang, X. G., Wang, Z. Q., Tong, W. M., and Shen, Y. (2007). PARP1 Val762Ala polymorphism reduces enzymatic activity. Biochem. Biophys. Res. Commun. 354, 122–126.
Wang, Y., Huang, J. W., Calses, P., Kemp, C. J., and Taniguchi, T. (2012). MiR-96 downregulates REV1 and RAD51 to promote cellular sensitivity to cisplatin and PARP inhibition. Cancer Res. 72, 4037–4046.
Weston, V. J., Oldreive, C. E., Skowronska, A., Oscier, D. G., Pratt, G., Dyer, M. J., et al. (2010). The PARP inhibitor olaparib induces significant killing of ATM-deficient lymphoid tumor cells in vitro and in vivo. Blood 116, 4578–4587.
Yélamos, J., Farrés, J., Llacuna, L., Ampurdanés, C., and Martin-Cabllero, J. (2011). PARP-1 and PARP-2: new players in tumor development. Am. J. Cancer Res. 1, 328–346.
Keywords: poly(ADP-ribose) polymerase, PARP-inhibitors, synthetic lethality, potentiation of anti-cancer therapy, resistance to PARP-inhibitors, DNA damage, DNA repair
Citation: Montoni A, Robu M, Pouliot É and Shah GM (2013) Resistance to PARP-inhibitors in cancer therapy. Front. Pharmacol. 4:18. doi: 10.3389/fphar.2013.00018
Received: 29 December 2012; Paper pending published: 21 January 2013;
Accepted: 05 February 2013; Published online: 27 February 2013.
Edited by:
Gerald Batist, McGill University, CanadaCopyright: © 2013 Montoni, Robu, Pouliot and Shah. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Girish M. Shah, Laboratory for Skin, Cancer Research, (CHU-Q) Hospital, Research Centre of Laval University, Laval University, 2705, Laurier Boulevard, Québec, QC G1V 4G2, Canada. e-mail:Z2lyaXNoLnNoYWhAY3JjaHVsLnVsYXZhbC5jYQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.