
- 1 Division of Neonatology, Children’s Hospital of Philadelphia, University of Pennsylvania School of Medicine, Philadelphia, PA, USA
- 2 Division of Neonatology, Department of Pediatrics, University of Pennsylvania, Philadelphia, PA, USA
The heme degradation pathway has been conserved throughout phylogeny and allows for the removal of a pro-oxidant and the generation of unique molecules including bile pigments with important cellular functions. The impact of bile pigments on health and disease are reviewed, as is the special circumstance of neonatal hyperbilirubinemia. In addition, the importance of promoter polymorphisms in the UDP-glucuronosyl transferase gene (UGTA1), which is key to the elimination of excess bilirubin and to the prevention of its toxicity, are discussed. Overall, the duality of bile pigments as either cytoprotective or toxic molecules is highlighted.
Introduction
The generation of bile pigments occurs through a unique pathway for the degradation of heme, limited by the enzyme hemeoxygenase (HO). This enzymatic reaction requires molecular oxygen (O2) and NADPH as a reducing equivalent and results in the formation of biliverdin and the release of carbon monoxide (CO) in an equimolar ratio. In addition, reduction of heme iron from Fe2+–Fe3+ occurs with the transfer of electrons from oxygen with NADPH as a source of reducing equivalents. Also, oxidative cleavage of the α-methene carbon bridge in the heme molecule forms biliverdin, releases carbon monoxide and iron (Figure 1). In most circumstances, heme does not accumulate freely but rather, it is bound to hemo proteins that are essential for cellular metabolism. An intricate enzymatic cascade regulates the production of heme (Ryter and Tyrrell, 2000). In pathological conditions, free heme is released from hemoglobin and can deposit in tissues because it is lipophilic and can lead to the formation of oxygen radicals therefore, the enzymatic reaction of HO is essential to preventing this since HO-1 is highly inducible by the substrate heme or by oxidative stress (Chang et al., 2005). The constitutive isoenzyme HO-2 can also catalyze the degradation of heme and is found in high abundance in the brain and testes (Maines et al., 1986; Maines, 1988). In the last steps of the reaction, biliverdin reductase (BVR), a microsomal enzyme, converts biliverdin into bilirubin in a non-rate-limiting fashion (Figure 1). Bilirubin, unlike biliverdin is not water soluble, but rather lipophilic, and can penetrate cellular membranes. To be made water soluble and therefore excretable in the gastrointestinal tract, it must be conjugated. The latter is regulated by UDP-glucuronosyl transferase 1A1 (UGT1A1), an enzyme that adds two glucuronide residues to bilirubin to render it water soluble (Figure 1).
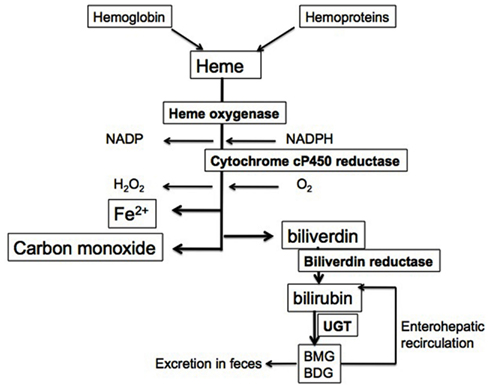
Figure 1. Heme degradation pathway. Heme from hemoglobin and cellular hemo proteins is metabolized in a rate-limiting step by HO. The reaction utilizes molecular oxygen and NADPH as a reducing equivalent. This leads to the release of iron, water, and CO as well as biliverdin. The latter is converted in a non-rate-limiting fashion to bilirubin by BVR. Since bilirubin is not water soluble, it is conjugated to monoglucuronide and diglucuronide forms, which can then be excreted. These glucuronide residues can be removed by intestinal bacteria and allow unconjugated bilirubin to re-enter the circulation.
The byproducts of HO-mediated heme degradation can have both positive and negative effects on cellular function. These will be outlined in this review.
Bilirubin
Effects of Bilirubin in the Neonate
Newborns have elevated numbers of red blood cells with a shortened life-span. When these cells lyse, heme is released from hemoglobin. In addition, due to a reduced ability to conjugate bilirubin formed during the degradation of heme, this pigment can accumulate in the serum in the first days of life leading to a transient hyperbilirubinemia, which typically resolves within the first weeks of life. In fact, the average full-term newborn infant has a peak serum bilirubin concentration of 5–6 mg/dL (86–103 μmol/L). This level is referred to as physiologic jaundice. However, in some circumstances, such as increased accumulation of heme (i.e., birth trauma, bruising, hemolysis), serum bilirubin levels can increase beyond the physiologic range. If serum bilirubin values are between 7 and 17 mg/dL (104–291 μmol/L), this is then referred to as exaggerated physiologic jaundice. However, this must be judged according to the infant’s age in hours on the bilirubin nomogram, as serum bilirubin levels change rapidly during the first week of life. Serum bilirubin concentrations higher than 17 mg/dL in full-term infants are considered pathologic and can be associated with adverse sequelae. Fortunately, most infants will not be affected until their bilirubin levels are significantly higher than 17 mg/dL because the toxicity of bilirubin is dictated by many factors including age (the younger, the more vulnerable), maturity (prematures are more vulnerable), and associated illnesses (hemolysis, sepsis, acidosis worsen bilirubin toxicity) amongst other factors. The most severe manifestation of bilirubin toxicity is kernicterus, a rare but devastating condition with acute neurological abnormalities including seizures, opisthotonus, and hypertonia and long lasting sequelae including sensorineural deafness, athetoid cerebral palsy, and delayed motor skills (Dennery et al., 2001).
Prior to the late 1980s, physicians had a very aggressive approach to the management of hyperbilirubinemia in neonates, which included institution of phototherapy at low levels of bilirubin and exchange transfusion if the level exceeded 20 mg/dL in all cases. This approach changed radically since kernicterus was such a rare condition and, with the advent of Rhogam prophylaxis for Rh-negative pregnant mothers, kernicterus was thought by some to be nearly eradicated in developed nations (Watchko and Oski, 1983). In addition, reports of the beneficial, antioxidant effects of bilirubin (Stocker et al., 1987, 1990; McDonagh, 1990) made this aggressive approach seem even more unwarranted. Alas, with this change in attitude, pediatricians saw a resurgence of bilirubin neurotoxicity, and were faced with litigation for the negligent practice of not having taken simple measures to prevent the devastating consequences of hyperbilirubinemia (Maisels and Newman, 2007; Bhutani et al., 1999). In the present day, we have adopted a more thoughtful approach to this preventable problem by using strict guidelines for universal screening and by instituting therapy guided by age-based serum bilirubin thresholds (Bhutani et al., 1999; Maisels and Newman, 2007).
It is not clear why bilirubin accumulates in the first days of life and what is its value. Physiologic hyperbilirubinemia is a phenomenon only seen in mammals. Many have speculated a teleological benefit to this reaction (McDonagh, 1990) and laboratory investigations do confirm that bilirubin has significant antioxidant properties both in vitro (Stocker et al., 1987; Mireles et al., 1999; Granato et al., 2003) and in vivo (Dennery et al., 1995; Mayer, 2000). Perhaps this may be useful in the transition from the relatively hypoxic environment of the wound to ambient air. This is not yet clear. Also, the exact dose at which bilirubin is toxic to cells vs. beneficial is also not yet known.
In infants with Rh hemolytic disease, peak serum bilirubin concentrations above 20 mg/dL predict poor outcome however, many infants without an obvious hemolytic etiology for their jaundice are normal at serum bilirubin concentrations of 25 mg/dL or higher. Looking at this more closely, 8% of infants with Rh-associated hemolysis and serum bilirubin concentrations of 19–24 mg/dL had kernicterus whereas this condition was observed in 73% of infants with concentrations of 30–40 mg/dL demonstrating the dose effect of bilirubin, at least in hemolytic children. Bilirubin enters the brain and targets the basal ganglia (Johnston and Hoon, 2000) and the auditory nerve preferentially (Shapiro and Nakamura, 2001). This occurs if bilirubin is not bound to albumin or is unconjugated or if there has been damage to the blood–brain barrier. As an example, a newborn infant with a serum albumin concentration of 3 g/dL can bind 25 mg/dL of bilirubin. In current clinical practice, the bilirubin albumin ratio is taken into consideration to determine levels at which physicians should proceed to more aggressive management of hyperbilirubinemia (i.e., exchange transfusion). If the serum albumin concentration is low, the risk of kernicterus increases because free bilirubin enters tissues and causes its toxic effects (Ahlfors et al., 2009). Conditions that alter the blood–brain barrier, such as infection, acidosis, hyperoxia, sepsis, prematurity, and hyperosmolarity, may also affect the entry of bilirubin into the brain (Dennery et al., 2001).
Mechanisms of Toxicity of Bilirubin
Bilirubin has high affinity for membrane phospholipids thereby entering cells and inhibiting mitochondrial enzymes (Chuniaud et al., 1996; Rodrigues et al., 2002a), interfering with DNA synthesis, inducing DNA-strand breaks (Rosenstein et al., 1983), and inhibiting protein synthesis and phosphorylation. In the brain, bilirubin inhibits the uptake of tyrosine, reduces synaptic transmission, and inhibits N-methyl-D-aspartate-receptor ion channels. Overall, bilirubin can interfere with neuro excitatory signals and impair nerve conduction particularly in the auditory nerve (Bratlid, 1990). Unconjugated bilirubin directly interacts with mitochondria influencing membrane lipid and protein properties, redox status, and cytochrome c content (Rodrigues et al., 2002b). It can work in concert with amyloid β peptide to activate apoptosis in neural cells (Rodrigues et al., 2000). Interestingly, younger animals are less susceptible to bilirubin-related mitochondrial injury (Rodrigues et al., 2002c) and the toxicity of bilirubin is not restricted to neonates. In patients with Crigler–Najjar I syndrome and absent activity of the UGTA1, therefore a complete lack of ability to conjugate bilirubin, the risk of kernicterus is quite high especially in the face of intercurrent illnesses and these patients require life-long phototherapy and/or liver transplantation (Strauss et al., 2006).
In the nervous system, the susceptibility of bilirubin varies with cell type (Ngai et al., 2000). In brain endothelial cells, bilirubin resulted in apoptosis in a time-dependent manner (Akin et al., 2002). Unconjugated bilirubin induced protein oxidation and lipid peroxidation and reduced antioxidant defenses in neuronal cells in culture (Brito et al., 2008). Although toxic to cultured neuroblastoma cells, exposure to unconjugated bilirubin induced genes involved in the endoplasmic reticulum stress response in surviving cells thereby enhancing cellular homeostasis (Calligaris et al., 2009). In astrocytes, unconjugated bilirubin up-regulated the multidrug resistance-associated protein-1 and increased its trafficking to the plasma membrane, thus reducing its cytotoxicity by preventing its intracellular accumulation (Gennuso et al., 2004).
The toxic effects of bilirubin are not limited to the brain. Unconjugated bilirubin can mediate apoptosis in cultured hepatocytes by increasing oxidative stress and enhancing caspase-9 activity (Oakes and Bend, 2005). In erythrocytes, high bilirubin concentrations can induce hemolysis and lead to membrane disruption, which could theoretically worsen hemolytic anemia (Brites et al., 1997). We observed that in erythrocytes derived from cord blood, concentrations of bilirubin equal to or exceeding 30 mg/dL were associated with increased protein oxidation, decreased erythrocyte glucose-6 phosphate dehydrogenase and adenosine triphosphatase activity as well as altered cell membrane integrity (Mireles et al., 1999). There was a correlation between the release of unconjugated bilirubin and hepatotoxicity after TNF-a administration, in mice and this was resolved with HO inhibitors (Van Molle and Libert, 2003). Another important cytotoxicity of bilirubin involves its effects on complement activation, a key element of immune defense. Unconjugated bilirubin interferes with the interaction between C1q and immunoglobulins, which results in decreased complement activation via the classical pathway (Basiglio et al., 2010).
Antioxidant Benefits of Bilirubin
Despite the fact that bilirubin may be toxic at higher concentrations, there is still significant evidence that it is a potent antioxidant at micromolar concentrations in vitro and in vivo. In fact, bilirubin is the most abundant cellular antioxidant. In vitro, bilirubin is a chain-breaking molecule that can scavenge the hydroxyl radical better than a-tocopherol, a well-known antioxidant against lipid peroxidation (Stocker et al., 1987; Mireles et al., 1999). Although incubation with bilirubin and albumin at concentrations greater than 30 mg/dL was associated with dose-dependent injury in erythrocytes derived from cord blood, protection against lipid peroxidation was seen at lower concentrations (Mireles et al., 1999), indicating the duality of bilirubin as a cytotoxic and cytoprotective molecule. Similarly, hemeoxygenase is both detrimental and beneficial based on levels of activity. Nevertheless, in that study, no change in bilirubin levels could be detected to explain the toxicity of hemeoxygenase (Suttner and Dennery, 1999).
Similarly, in vivo, there are several examples of the beneficial effects of hyperbilirubinemia. In 50 patients older than 40 years with Gilbert syndrome, a relatively benign condition leading to mild to moderate unconjugated hyperbilirubinemia because of impaired glucuronidation, occurrence of ischemic heart disease was compared to that of a large cohort of patients without the disease. Ischemic heart disease occurred in only 2% of the Gilbert patients compared to 12.1% of the controls and, interestingly, hyperbilirubinemia rather than elevation of HDL cholesterol levels seemed to be more important in protection from ischemic heart disease (Vitek et al., 2002). In a case report, resolution of corticosteroid- and cyclophosphamide-resistant pulmonary fibrosis occurred with onset of hyperbilirubinemia due to biliary obstruction in a patient who developed elevated conjugated bilirubin levels (Ohrui et al., 2001), suggesting that higher serum bilirubin levels could reverse pulmonary fibrosis. The mechanisms by which this could occur are not yet explored. In organ transplantation, bilirubin can be protective against graft rejection (Ollinger et al., 2007). Injection of bilirubin in mouse organ recipients prolonged islet allograft survival and induced tolerance induction and graft acceptance via a regulatory T cell-dependent mechanism involving CD4(+) and CD25(+) cells. In fact, bilirubin enhanced de novo generation of regulatory T cells in the recipients thereby preventing rejection (Rocuts et al., 2010). Another novel mechanisms by which bilirubin may be protective is by the regulation of rapid eye movement sleep and by mediating some of the antidepressant effects of ambient light (Oren, 1997). Whether the antioxidant effects mediate these benefits is not yet clear.
Overall, the beneficial effects of bilirubin have been demonstrated in various models but beyond a certain threshold, bilirubin is clearly toxic.
Epidemiologic Evidence of Bilirubin as a Cytoprotective Molecule
To further understand whether bilirubin is cytoprotective in humans, epidemiologic studies can provide a clue. Regulation of bilirubin conjugation is key in the accumulation of bilirubin and its potential benefits or toxicity, therefore, studies comparing patients with differences in the ability to conjugate bilirubin may provide clues. The promoter of the UGT1A1 gene has regions of TA repeats, which regulate its transcriptional efficiency. In Caucasian populations, an additional TA repeat (TA7 vs. TA6) is necessary but not sufficient to cause Gilbert syndrome (Bartlett and Gourley, 2011). Strong associations between polymorphisms in the UGT1A1 gene and human disease have been shown. In particular, there have been associations with altered bilirubin conjugation and the occurrence of various cancers. The common UGT1A1*28 allele results in elevated plasma bilirubin levels and is strongly associated with Gilbert syndrome in Caucasians. Low serum bilirubin levels observed in a Caucasian cohort with predicted high activity of UGT1A1 were associated with an increased risk of esophageal cancer. Interestingly, the UGT1A8 and UGT2B4 genotypes, associated with decreased UGT enzyme activity and increased unconjugated bilirubin levels, were also significantly associated with increased risk of esophageal cancer (Dura et al., 2012). In another study, the UGT1A gene cluster on chromosome 2q37.1 was identified in a cohort of patients with bladder cancer suggesting that enhanced UGT1A may protect from bladder cancer by increasing the removal of carcinogens from bladder epithelium (Tang et al., 2012). In a meta-analysis of 21 case-control studies cancer risk was associated with intermediate, and low activity of UGT1A7 genotypes, found predominantly in Asians (Lu et al., 2011). In contrast to the other studies, the TA repeat polymorphism of UGT1A1 gene did not alter prostate cancer risk susceptibility in Caucasian men (Karatzas et al., 2010). Overall, these studies suggest that the concentration of bilirubin in the serum determines whether it is beneficial or detrimental.
Not only does the UGT1 gene play a role in cancer, it appears to have important effects in other diseases. For example, the homozygous state associated with higher serum bilirubin levels appeared to be protective against Crohn’s disease (de Vries et al., 2012).
Serum bilirubin, independent of variation in UGT promoter activity, is also associated with diseases in large populations, in particular in cardiovascular disease. In a Swedish cohort, plasma bilirubin was lower in 231 cases of ischemic stroke than in 462 matched controls but the difference reached significance only in women (Ekblom et al., 2010). In males with coronary artery disease, there was inverse association between serum total bilirubin and coronary artery calcification score. Additionally, bilirubin was associated with reduced c-reactive protein levels, which could explain the lower calcification scores (Zhang et al., 2012). In another study, bilirubin levels were also inversely associated with the presence of coronary heart disease. Interestingly, bilirubin levels were significantly raised after treatment with 80 mg simvastatin independent of changes in liver enzymes (Nolting et al., 2011).
Despite the beneficial effects of bilirubin, the biggest challenge remains determining a specific threshold at which bilirubin is toxic vs. beneficial. It seems paradoxical that early events in bilirubin toxicity may involve increased oxidative stress and changes in redox status (Tell and Gustincich, 2009) yet conversely, bilirubin alleviates oxidative stress.
Antioxidant Properties of Biliverdin
Although biliverdin does not accumulate in mammals, since it is rapidly converted to bilirubin through the action of biliverdin reductase (BVR), it may have important signaling effects. In macrophages, biliverdin activates endothelial nitric oxide, resulting in NO-dependent S-nitrosylation of BVR. The mechanisms by which biliverdin mediates this effect was via the repression of Toll-like receptor-4 (Wegiel et al., 2011). In another study, rats injected intraperitoneally with biliverdin before undergoing lung transplantation had less evidence of inflammation, oxidative injury, and apoptosis suggesting that biliverdin has anti-inflammatory and anti-apoptotic effects (Wang et al., 2010). Despite these data, the most plausible effect of biliverdin is to serve as a signaling molecule that may regulate BVR (Lerner-Marmarosh et al., 2008). The properties of this enzyme have been reviewed at length in a previous issue of this journal (Gibbs et al., 2012).
Other Possible Mediators of HO-related Cytoprotective Effects
Although the byproducts of the HO-1 reaction are important cytoprotective molecules, a likely factor that influences the beneficial effects of the HO reaction is the degradation of a potent oxidant, heme. In one example, induction of HO-1 prevented photodynamic therapy-induced tumor necrosis, but neither bilirubin, biliverdin nor CO was responsible for this cytoprotection. In fact, the iron chelator desferrioxamine enhanced the cytotoxic effects of photodynamic therapy suggesting that heme was key important to enhancing the tumor killing effects of this therapy (Nowis et al., 2006).
Summary
Overall, the byproducts of the HO reaction are important cytoprotective molecules that have clinically significant effects in various diseases. Nevertheless, in most cases, these molecules can also be cytotoxic under specific circumstances and/or at high concentrations. Potential therapeutic interventions will need to balance the potential benefits with the risk of toxicity to be most effective.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ahlfors, C. E., Amin, S. B., and Parker, A. E. (2009). Unbound bilirubin predicts abnormal automated auditory brainstem response in a diverse newborn population. J. Perinatol. 29, 305–309.
Akin, E., Clower, B., Tibbs, R., Tang, J., and Zhang, J. (2002). Bilirubin produces apoptosis in cultured bovine brain endothelial cells. Brain Res. 931, 168–175.
Bartlett, M. G., and Gourley, G. R. (2011). Assessment of UGT polymorphisms and neonatal jaundice. Semin. Perinatol. 35, 127–133.
Basiglio, C. L., Arriaga, S. M., Pelusa, F., Almara, A. M., Kapitulnik, J., and Mottino, A. D. (2010). Complement activation and disease: protective effects of hyperbilirubinaemia. Clin. Sci. 118, 99–113.
Bhutani, V. K., Johnson, L. H., and Sivieri, E. M. (1999). Predictive ability of a predischarge hour-specific serum bilirubin for subsequent significant hyperbilirubinemia in healthy term and near-term newborns. Pediatrics 103, 6–14.
Brites, D., Silva, R., and Brito, A. (1997). Effect of bilirubin on erythrocyte shape and haemolysis, under hypotonic, aggregating or non-aggregating conditions, and correlation with cell age. Scand. J. Clin. Lab. Invest. 57, 337–349.
Brito, M. A., Lima, S., Fernandes, A., Falcao, A. S., Silva, R. F., Butterfield, D. A., and Brites, D. (2008). Bilirubin injury to neurons: contribution of oxidative stress and rescue by glycoursodeoxycholic acid. Neurotoxicology 29, 259–269.
Calligaris, R., Bellarosa, C., Foti, R., Roncaglia, P., Giraudi, P., Krmac, H., Tiribelli, C., and Gustincich, S. (2009). A transcriptome analysis identifies molecular effectors of unconjugated bilirubin in human neuroblastoma SH-SY5Y cells. BMC Genomics 10, 543. doi:10.1186/1471-2164-10-543
Chang, E. F., Claus, C. P., Vreman, H. J., Wong, R. J., and Noble-Haeusslein, L. J. (2005). Heme regulation in traumatic brain injury: relevance to the adult and developing brain. J. Cereb. Blood Flow Metab. 25, 1401–1417.
Chuniaud, L., Dessante, M., Chantoux, F., Blondeau, J. P., Francon, J., and Trivin, F. (1996). Cytotoxicity of bilirubin for human fibroblasts and rat astrocytes in culture. Effect of the ratio of bilirubin to serum albumin. Clin. Chim. Acta 256, 103–114.
de Vries, H. S., Te Morsche, R. H., Jenniskens, K., Peters, W. H., and De Jong, D. J. (2012). A functional polymorphism in UGT1A1 related to hyperbilirubinemia is associated with a decreased risk for Crohn’s disease. J. Crohns Colitis. 6, 597–602.
Dennery, P. A., McDonagh, A. F., Spitz, D. R., Rodgers, P. A., and Stevenson, D. K. (1995). Hyperbilirubinemia results in reduced oxidative injury in neonatal Gunn rats exposed to hyperoxia. Free Radic. Biol. Med. 19, 395–404.
Dennery, P. A., Seidman, D. S., and Stevenson, D. K. (2001). Neonatal hyperbilirubinemia. N. Engl. J. Med. 344, 581–590.
Dura, P., Salomon, J., Te Morsche, R. H., Roelofs, H. M., Kristinsson, J. O., Wobbes, T., Witteman, B. J., Tan, A. C., Drenth, J. P., and Peters, W. H. (2012). High enzyme activity UGT1A1 or low activity UGT1A8 and UGT2B4 genotypes increase esophageal cancer risk. Int. J. Oncol. 40, 1789–1796.
Ekblom, K., Marklund, S. L., Johansson, L., Osterman, P., Hallmans, G., Weinehall, L., Wiklund, P. G., and Hultdin, J. (2010). Bilirubin and UGT1A1*28 are not associated with lower risk for ischemic stroke in a prospective nested case-referent setting. Cerebrovasc. Dis. 30, 590–596.
Gennuso, F., Fernetti, C., Tirolo, C., Testa, N., L’Episcopo, F., Caniglia, S., Morale, M. C., Ostrow, J. D., Pascolo, L., Tiribelli, C., and Marchetti, B. (2004). Bilirubin protects astrocytes from its own toxicity by inducing up-regulation and translocation of multidrug resistance-associated protein 1 (Mrp1). Proc. Natl. Acad. Sci. U.S.A. 101, 2470–2475.
Gibbs, P. E., Tudor, C., and Maines, M. D. (2012). Biliverdin reductase: more than a namesake – the reductase, its Peptide fragments, and biliverdin regulate activity of the three classes of protein kinase C. Front. Pharmacol. 3:31. doi:10.3389/fphar.2012.00031
Granato, A., Gores, G., Vilei, M. T., Tolando, R., Ferraresso, C., and Muraca, M. (2003). Bilirubin inhibits bile acid induced apoptosis in rat hepatocytes. Gut 52, 1774–1778.
Johnston, M. V., and Hoon, A. H. Jr. (2000). Possible mechanisms in infants for selective basal ganglia damage from asphyxia, kernicterus, or mitochondrial encephalopathies. J. Child Neurol. 15, 588–591.
Karatzas, A., Giannatou, E., Tzortzis, V., Gravas, S., Aravantinos, E., Moutzouris, G., Melekos, M., and Tsezou, A. (2010). Genetic polymorphisms in the UDP-glucuronosyltransferase 1A1 (UGT1A1) gene and prostate cancer risk in Caucasian men. Cancer Epidemiol. 34, 345–349.
Lerner-Marmarosh, N., Miralem, T., Gibbs, P. E., and Maines, M. D. (2008). Human biliverdin reductase is an ERK activator; hBVR is an ERK nuclear transporter and is required for MAPK signaling. Proc. Natl. Acad. Sci. U.S.A. 105, 6870–6875.
Lu, P. H., Chen, M. B., Wu, X. Y., Gu, J. H., Liu, Y., and Gu, R. M. (2011). Genetic polymorphisms of UGT1A7 and cancer risk: evidence from 21 case-control studies. Cancer Invest. 29, 645–654.
Maines, M. D. (1988). Heme oxygenase: function, multiplicity, regulatory mechanisms, and clinical applications. FASEB J. 2, 2557–2568.
Maines, M. D., Trakshel, G. M., and Kutty, R. K. (1986). Characterization of two constitutive forms of rat liver microsomal heme oxygenase. Only one molecular species of the enzyme is inducible. J. Biol. Chem. 261, 411–419.
Maisels, M. J., and Newman, T. B. (2007). Kernicterus, the Daubert decision, and evidence-based medicine. Pediatrics 119, 1038; author reply 1038–1039.
Mayer, M. (2000). Association of serum bilirubin concentration with risk of coronary artery disease. Clin. Chem. 46, 1723–1727.
Mireles, L. C., Lum, M. A., and Dennery, P. A. (1999). Antioxidant and cytotoxic effects of bilirubin on neonatal erythrocytes. Pediatr. Res. 45, 355–362.
Ngai, K. C., Yeung, C. Y., and Leung, C. S. (2000). Difference in susceptibilities of different cell lines to bilirubin damage. J. Paediatr. Child Health 36, 51–55.
Nolting, P. R., Kusters, D. M., Hutten, B. A., and Kastelein, J. J. (2011). Serum bilirubin levels in familial hypercholesterolemia: a new risk marker for cardiovascular disease? J. Lipid Res. 52, 1755–1759.
Nowis, D., Legat, M., Grzela, T., Niderla, J., Wilczek, E., Wilczynski, G. M., Glodkowska, E., Mrowka, P., Issat, T., Dulak, J., Jozkowicz, A., Was, H., Adamek, M., Wrzosek, A., Nazarewski, S., Makowski, M., Stoklosa, T., Jakobisiak, M., and Golab, J. (2006). Heme oxygenase-1 protects tumor cells against photodynamic therapy-mediated cytotoxicity. Oncogene 25, 3365–3374.
Oakes, G. H., and Bend, J. R. (2005). Early steps in bilirubin-mediated apoptosis in murine hepatoma (Hepa 1c1c7) cells are characterized by aryl hydrocarbon receptor-independent oxidative stress and activation of the mitochondrial pathway. J. Biochem. Mol. Toxicol. 19, 244–255.
Ohrui, T., Higuchi, M., Kanda, A., Matsui, T., Sato, E., and Sasaki, H. (2001). A patient with exacerbation of idiopathic pulmonary fibrosis which was resolved probably due to the coexisting hyperbilirubinemia? Tohoku J. Exp. Med. 193, 245–249.
Ollinger, R., Wang, H., Yamashita, K., Wegiel, B., Thomas, M., Margreiter, R., and Bach, F. H. (2007). Therapeutic applications of bilirubin and biliverdin in transplantation. Antioxid. Redox Signal. 9, 2175–2185.
Oren, D. A. (1997). Bilirubin, REM sleep, and phototransduction of environmental time cues. A hypothesis. Chronobiol. Int. 14, 319–329.
Rocuts, F., Zhang, X., Yan, J., Yue, Y., Thomas, M., Bach, F. H., Czismadia, E., and Wang, H. (2010). Bilirubin promotes de novo generation of T regulatory cells. Cell Transplant. 19, 443–451.
Rodrigues, C. M., Sola, S., and Brites, D. (2002a). Bilirubin induces apoptosis via the mitochondrial pathway in developing rat brain neurons. Hepatology 35, 1186–1195.
Rodrigues, C. M., Sola, S., Brito, M. A., Brites, D., and Moura, J. J. (2002b). Bilirubin directly disrupts membrane lipid polarity and fluidity, protein order, and redox status in rat mitochondria. J. Hepatol. 36, 335–341.
Rodrigues, C. M., Sola, S., Silva, R. F., and Brites, D. (2002c). Aging confers different sensitivity to the neurotoxic properties of unconjugated bilirubin. Pediatr. Res. 51, 112–118.
Rodrigues, C. M., Sola, S., Silva, R., and Brites, D. (2000). Bilirubin and amyloid-beta peptide induce cytochrome c release through mitochondrial membrane permeabilization. Mol. Med. 6, 936–946.
Rosenstein, B. S., Ducore, J. M., and Cummings, S. W. (1983). The mechanism of bilirubin-photosensitized DNA strand breakage in human cells exposed to phototherapy light. Mutat. Res. 112, 397–406.
Ryter, S. W., and Tyrrell, R. M. (2000). The heme synthesis and degradation pathways: role in oxidant sensitivity. Heme oxygenase has both pro- and antioxidant properties. Free Radic. Biol. Med. 28, 289–309.
Shapiro, S. M., and Nakamura, H. (2001). Bilirubin and the auditory system. J. Perinatol. 21(Suppl. 1), S52–S55; discussion S59–S62.
Stocker, R., McDonagh, A. F., Glazer, A. N., and Ames, B. N. (1990). Antioxidant activities of bile pigments: biliverdin and bilirubin. Meth. Enzymol. 186, 301–309.
Stocker, R., Yamamoto, Y., McDonagh, A. F., Glazer, A. N., and Ames, B. N. (1987). Bilirubin is an antioxidant of possible physiological importance. Science 235, 1043–1046.
Strauss, K. A., Robinson, D. L., Vreman, H. J., Puffenberger, E. G., Hart, G., and Morton, D. H. (2006). Management of hyperbilirubinemia and prevention of kernicterus in 20 patients with Crigler-Najjar disease. Eur. J. Pediatr. 165, 306–319.
Suttner, D. M., and Dennery, P. A. (1999). Reversal of HO-1 related cytoprotection with increased expression is due to reactive iron. FASEB J. 13, 1800–1809.
Tang, W., Fu, Y. P., Figueroa, J. D., Malats, N., Garcia-Closas, M., Chatterjee, N., Kogevinas, M., Baris, D., Thun, M., Hall, J. L., De Vivo, I., Albanes, D., Porter-Gill, P., Purdue, M. P., Burdett, L., Liu, L., Hutchinson, A., Myers, T., Tardon, A., Serra, C., Carrato, A., Garcia-Closas, R., Lloreta, J., Johnson, A., Schwenn, M., Karagas, M. R., Schned, A., Black, A., Jacobs, E. J., Diver, W. R., Gapstur, S. M., Virtamo, J., Hunter, D. J., Fraumeni, J. F. Jr., Chanock, S. J., Silverman, D. T., Rothman, N., and Prokunina-Olsson, L. (2012). Mapping of the UGT1A locus identifies an uncommon coding variant that affects mRNA expression and protects from bladder cancer. Hum. Mol. Genet. 21, 1918–1930.
Tell, G., and Gustincich, S. (2009). Redox state, oxidative stress, and molecular mechanisms of protective and toxic effects of bilirubin on cells. Curr. Pharm. Des. 15, 2908–2914.
Van Molle, W., and Libert, C. (2003). Bilirubin release induced by tumor necrosis factor in combination with galactosamine is toxic to mice. Cytokine 23, 94–100.
Vitek, L., Jirsa, M., Brodanova, M., Kalab, M., Marecek, Z., Danzig, V., Novotny, L., and Kotal, P. (2002). Gilbert syndrome and ischemic heart disease: a protective effect of elevated bilirubin levels. Atherosclerosis 160, 449–456.
Wang, J., Zhou, H. C., Pan, P., Zhang, N., and Li, W. Z. (2010). Exogenous biliverdin improves the function of lung grafts from brain dead donors in rats. Transplant. Proc. 42, 1602–1609.
Wegiel, B., Gallo, D., Csizmadia, E., Roger, T., Kaczmarek, E., Harris, C., Zuckerbraun, B. S., and Otterbein, L. E. (2011). Biliverdin inhibits Toll-like receptor-4 (TLR4) expression through nitric oxide-dependent nuclear translocation of biliverdin reductase. Proc. Natl. Acad. Sci. U.S.A. 108, 18849–18854.
Keywords: neonatal jaundice, kernicterus, UDP-glucuronyltransferase, antioxidant, polymorphisms
Citation: Dennery PA (2012) Evaluating the beneficial and detrimental effects of bile pigments in early and later life. Front. Pharmacol. 3:115. doi: 10.3389/fphar.2012.00115
Received: 10 April 2012; Paper pending published: 23 April 2012;
Accepted: 29 May 2012; Published online: 22 June 2012.
Edited by:
Jaime Kapitulnik, The Hebrew University of Jerusalem, IsraelReviewed by:
David K. Stevenson, Stanford University School of Medicine, USAMichael Kaplan, Shaare Zedek Medical Center, Israel
Copyright: © 2012 Dennery. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Phyllis A. Dennery, Division of Neonatology, Department of Pediatrics, University of Pennsylvania, 34th and Civic Center Boulevard, Philadelphia, PA 19104-4318, USA. e-mail:ZGVubmVyeUBlbWFpbC5jaG9wLmVkdQ==