




95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 03 May 2012
Sec. Experimental Pharmacology and Drug Discovery
Volume 3 - 2012 | https://doi.org/10.3389/fphar.2012.00082
This article is part of the Research Topic Sirtuins in Biology and Disease View all 10 articles
Sirtuin proteins are conserved regulators of aging that have recently emerged as important modifiers of several diseases which commonly occur later in life such as cancer, diabetes, cardiovascular, and neurodegenerative diseases. In mammals, there are seven sirtuins (SIRT1-7), which display diversity in subcellular localization and function. SIRT1 has received much of attention due to its possible impact on longevity, while important biological and therapeutic roles of other sirtuins have been underestimated and just recently recognized. Here we focus on SIRT2, a member of the sirtuin family, and discuss its role in cellular and tissue-specific functions. This review summarizes the main scientific advances on SIRT2 protein biology and explores its potential as a therapeutic target for treatment of age-related disorders.
Silent information regulator 2 (SIR2) is the eponymous gene of a whole family of conserved genes named sirtuins, which are present in organisms ranging from bacteria to plants, and animals (Haigis and Sinclair, 2010). Original studies of aging in S. cerevisiae led to the discovery that extra copies of the SIR2 gene extended lifespan by 50%, whereas SIR2 deletion reduced longevity (Kaeberlein et al., 1999). In yeast, SIR2 promotes longevity by suppressing the formation of toxic extrachromosomal rDNA circles (Sinclair and Guarente, 1997). Remarkably, the fly and worm SIR2 orthologs also play a role in the regulation of lifespan (Tissenbaum and Guarente, 2001; Rogina and Helfand, 2004). However, the increase of longevity due to Sir2 overexpression in fly and nematode models has recently been challenged, and is now considered to be about 15% in nematodes (Burnett et al., 2011; Viswanathan and Guarente, 2011). Nevertheless, several studies in yeast and fly models suggested that SIR2 could be a critical mediator of the beneficial effects of caloric restriction (CR) (Chen and Guarente, 2007). CR is a dietary regimen that reduces caloric intake by a total of 30% without malnutrition. CR has been shown to slow down the aging process and increase lifespan in all laboratory models tested (Koubova and Guarente, 2003). In mammals, several studies found that CR leads to an increase of sirtuins protein expression in a variety of tissues (Cohen et al., 2004; Shi et al., 2005). Importantly, SIRT1 was found to be required for the increase of physical activity induced by CR (Chen et al., 2005). These studies suggest that mammalian sirtuins are involved in the activation of biological responses during CR required for the increased lifespan.
The discovery that SIR2 extended lifespan in yeast, worms, and flies incited scientists to further study mammalian sirtuins, since questions such as whether sirtuins promote health and protect against aging-associated disorders are of great scientific and social/economic interest. In mammals there are seven sirtuins, SIRT1-7, all possessing a highly conserved central NAD+- binding site and common catalytic domain (Frye, 2000; Landry et al., 2000; Smith et al., 2000; Tanner et al., 2000). Sirtuins are NAD+-dependent deacetylases, linking their enzymatic activity to the energy state of the cell (Nakagawa et al., 2009). Therefore it is crucial to study the effect of nicotinamide phosphoribosyltransferase (NAMPT) on SIRT2 activity, a key enzyme that regulates NAD biosynthesis. The sirtuin-mediated deacetylation reaction couples lysine deacetylation to NAD+-hydrolysis. This hydrolysis yields O-acetyl-ADP-ribose, the deacetylated substrate and nicotinamide, which is an inhibitor of sirtuins activity (Tanner et al., 2000). During glycolysis and the citric acid cycle NAD+ is reduced to NADH, hence the ratio of NAD+ to NADH is inversely proportional to energy availability. Interestingly, in yeast cells NADH can competitively inhibit the deacetylase reaction catalyzed by sirtuins and NADH levels are decreased by CR (Lin et al., 2004). Thus it is tempting to speculate that mammalian sirtuins should have augmented enzymatic activity under conditions of low energy, high NAD+ to NADH ratio, and therefore they function as sensors for the cellular energy status. Although the catalytic domain of mammalian sirtuins is conserved, they are structurally different with respect to their N- and C-termini, to their localization within the cell, and in that they utilize different substrates and protein binding partners (Blander and Guarente, 2004). SIRT1 is mainly nuclear, although it can transiently be found in the cytoplasm (Haigis and Guarente, 2006). While SIRT2 is primarily a cytoplasmic protein, it can transiently shuttle into the nucleus in a cell cycle-dependent manner (North et al., 2003; North and Verdin, 2007a). SIRT3, SIRT4, and SIRT5 are mitochondrial proteins. SIRT6 is a nuclear protein and SIRT7 is nucleolar (Liszt et al., 2005; Ford et al., 2006; Figure 1). Despite the fact that some sirtuins can have redundant functions, different biological roles may be determined by intracellular compartmentalization, and by different tissue expression pattern.
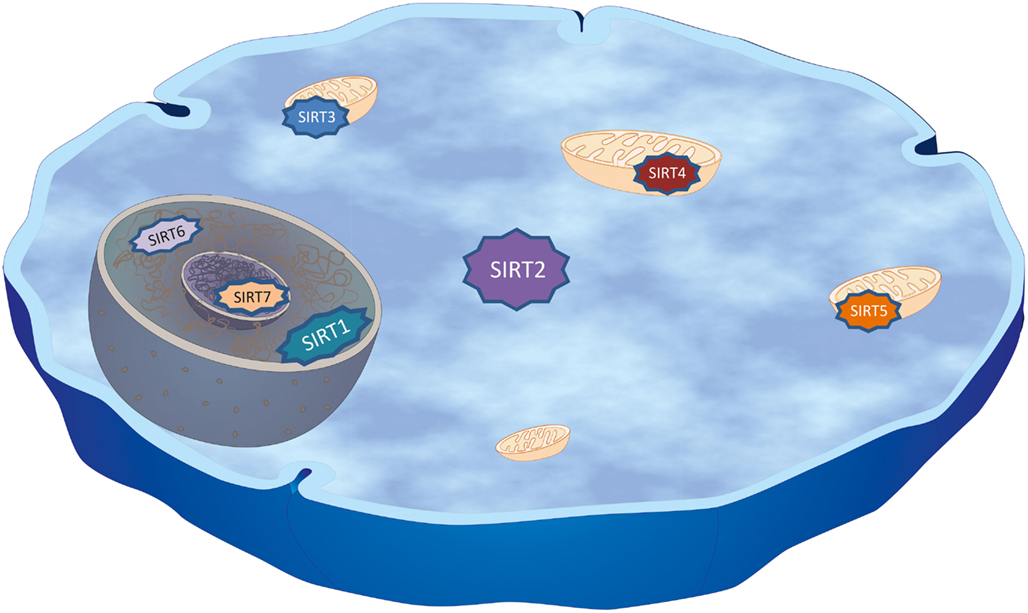
Figure 1. Cellular distribution of mammalian sirtuins. Mammals possess seven sirtuins, SIRT1–7, that have a NAD+-dependent catalytic core domain in common. Additional variable N-terminal and C-terminal sequences flank this core domain leading to diverse subcellular localizations that may account for differences in their biological functions as well as different substrates and binding partners within the cell. SIRT1 is mainly nuclear, modulating, amongst others, chromatin structure by deacetylating specific lysine residues in histones H1, H3, and H4, although it can transiently be found in the cytoplasm. SIRT2, the closest homolog of SIRT1, is primarily a cytoplasmic protein, but can transiently shuttle into the nucleus during mitosis, where it deacetylates Lys-56 of histone H3 and Lys-16 of histone H4. In the cytoplasm, SIRT2 deacetylates microtubules, in particular α-tubulin at Lys-40 and a variety of other substrates (Table 1). SIRT3, SIRT4, and SIRT5 were found to co-localize with mitochondria indicating key regulative roles in metabolism and energy usage of the cell. SIRT6 is a nuclear protein found to regulate DNA repair, while SIRT7 co-localizes with the nucleolus and was shown to be involved in rRNA transcription.
Among all mammalian sirtuins, SIRT1 has been the most extensively studied. Nevertheless, studies of the other mammalian sirtuins have uncovered a variety of substrates, interacting partners and biological relevance in diverse cellular processes. Here we focus on the biological role of SIRT2, the only cytoplasmic member of the family, explore its various roles in age-associated disorders and discuss possible therapeutic applications.
SIRT2 is the mammalian ortholog of yeast HST2 (Perrod et al., 2001). Similar to SIR2, HST2 is upregulated by CR as well as oxidative stress and extends lifespan by a SIR2-independent mechanism (Lamming et al., 2005; Zhu et al., 2012). In future experiments, it will be important to determine if mice overexpressing SIRT2 mice show an altered lifespan. It would be equally important to determine whether SIRT2 is required for any of the protective phenotypes mediated by CR.
SIRT2 is found primarily in the cytoplasm, co-localizes with microtubules and deacetylates the major component of microtubules, α-tubulin at lysine 40 (North et al., 2003). In the same study the authors identified the microtubule deacetylase HDAC6 as a binding partner of SIRT2. It is unclear whether SIRT2 deacetylates soluble or polymerized α-tubulin, and thus could be formally considered as a microtubule deacetylase.
SIRT2 transiently migrates to the nuclei during G2/M transition and deacetylates histone H4 at lysine 16, thereby modulating chromatin condensation during metaphase (Vaquero et al., 2006). Interestingly, SIRT1 also deacetylates lysine 16 of histone H4, indicating a possible synergistic relationship or functional redundancy between these sirtuins (Imai et al., 2000; Vaquero et al., 2004). When SIRT2 is overexpressed it delays cell cycle progression (Dryden et al., 2003). Moreover, SIRT2 protein levels vary accordingly to the cell cycle phase, increasing during mitosis. Altogether, these observations implicate SIRT2 in the regulation of the cell cycle (Inoue et al., 2007).
In addition to α-tubulin and histone H4 substrates, SIRT2 deacetylates forkhead transcription factors of class O, FOXO1, and FOXO3 (Li et al., 2007; Wang and Tong, 2009; Zhu et al., 2012). Since FOXO transcription factors are involved in multiple cellular processes, such as DNA repair, cell cycle, apoptosis, metabolism, and aging, SIRT2 is therefore also connected with these diverse pathways (Calnan and Brunet, 2008).
SIRT2 deacetylates lysine residues in the catalytic domain of p300, a histone acetyltransferase, which maintains its active form by autoacetylation (Black et al., 2008). The known consequence of SIRT2-dependent deacetylation of p300 is the de-repression of p53 transcriptional activity (Han et al., 2008). Notably, p300 is capable of inhibiting SIRT2 activity by direct acetylation, thus demonstrating a complex relationship and regulation of acetylase and deacetylase activities in cells. Interestingly, posttranslational phosphorylation also negatively modulates SIRT2 activity. Phosphorylation at serine 331 inhibits SIRT2 catalytic activity (Pandithage et al., 2008). SIRT2 is phosphorylated by various cycline-CDK complexes at serine 331 and is dephosphorylated by CDC14B phosphatase (Dryden et al., 2003; Southwood et al., 2007; Pandithage et al., 2008).
In addition, several proteins such as 14-3-3 β/γ and homeobox transcription factor 10 are binding partners but not deacetylation substrates of SIRT2 (Bae et al., 2004; Jin et al., 2008). The currently known SIRT2 substrates/binding partners suggest a complex and apparently diverse function for this sirtuin in the cell (Table 1).
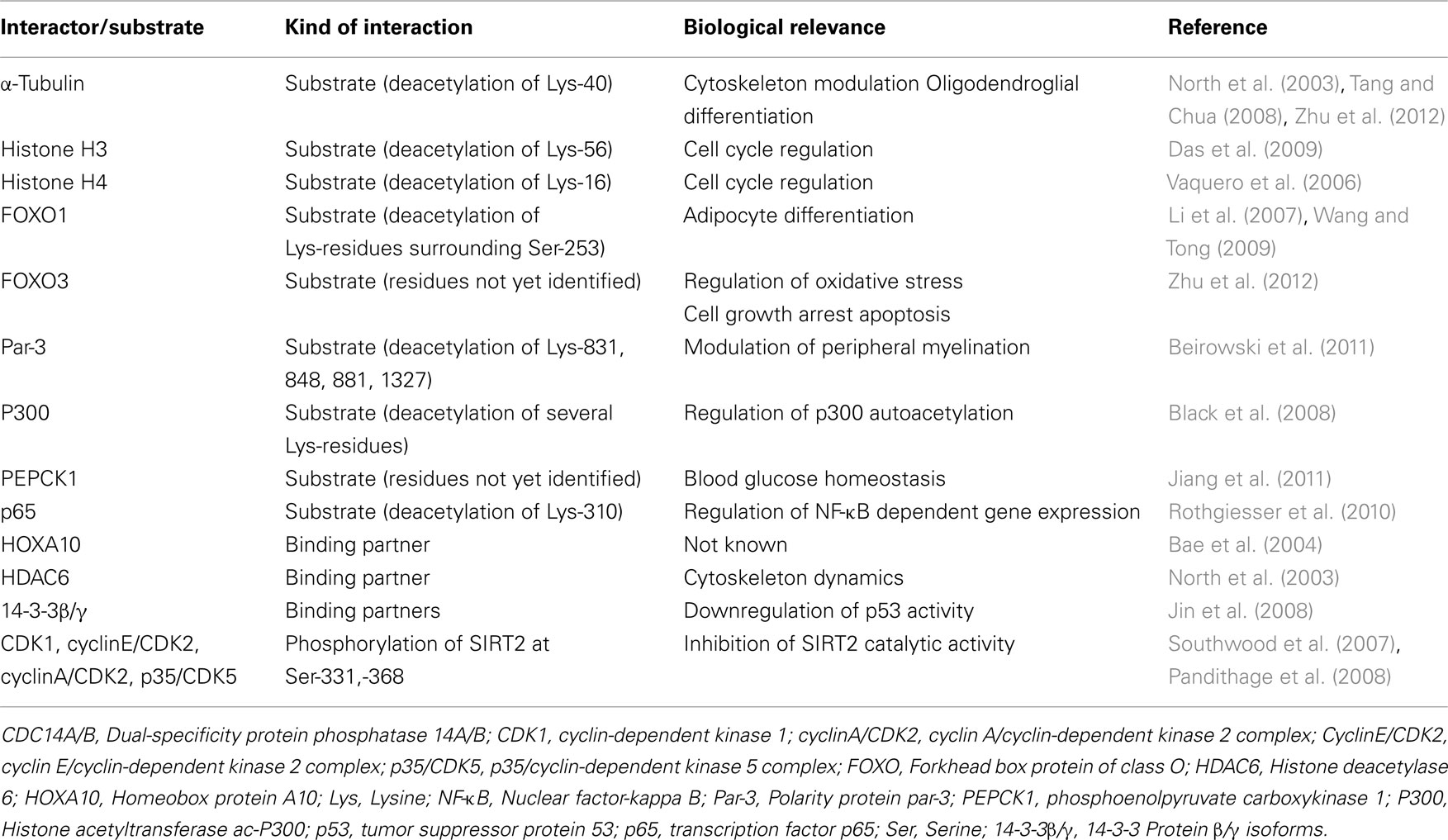
Table 1. SIRT2 substrates and interactors and their biological relevance.
In western societies high fat and low fibber diets, together with a sedentary life style, are associated with a high prevalence of metabolic syndrome, that increases with age (Ford et al., 2002). Metabolic syndrome is the condition brought about by: obesity; insulin resistance; hypertension; and elevated lipid content in the blood. Metabolic syndrome increases the risk of serious health problems (Moller and Kaufman, 2005). In the obese it is the proportion of body fat which is significant, not purely weight per se. Dramatically, obesity is associated with a decrease in life expectancy (Haslam and James, 2005).
Among all the mammalian sirtuins, the SIRT2 transcript is the most abundant in adipocytes (Li et al., 2007); this alone may indicate a role in the regulation of adipose tissue functionality. Together with the observation that SIRT2 decreases during preadipocyte differentiation and regulates adipocyte differentiation in a negative manner by deacetylating FOXO1, it raises the exciting hypothesis that this protein has a pivotal role in the regulation of fat abundance (Jing et al., 2007; Wang and Tong, 2009). Thus, it can be speculated that modulating SIRT2 activity may ameliorate, at least in part, the metabolic disturbances in the obese resulting from increased fat mass. In support of this hypothesis, retroviral expression of SIRT2 in adipocytes was found to promote lipolysis (Wang and Tong, 2009). Thus, SIRT2 activators could be used as a novel therapeutic approach to obesity and in a more general way to metabolic syndrome.
SIRT2 expression is elevated in white adipose tissue and kidney of caloric-restricted mice (Zhu et al., 2012). It is interesting to note that CR reverses the modifications to some physiological parameters caused by metabolic syndrome. Moreover CR protects against the diseases to which metabolic syndrome is a risk factor (Guarente, 2006). Another exciting study raised the possibility that SIRT2 plays a role in blood glucose homeostasis by deacetylating and stabilizing phosphoenolpyruvate carboxykinase 1 (PEPCK1), an important enzyme in gluconeogenesis (Jiang et al., 2011). When SIRT2 is activated by low glucose conditions, PEPCK1 is stabilized and it shifts the equilibrium toward the generation of glucose from non-carbohydrate carbon sources, mimicking a fast or exercise state in the organisms. Conversely, in the presence of high glucose SIRT2 expression is suppressed, leading to PEPCK1 degradation by the ubiquitin proteasome system (Jiang et al., 2011). This study raises the question of how glucose availability regulates SIRT2 transcription. Since other sirtuins are also linked to metabolic homeostasis, another question arises: how does SIRT2 communicate with other family members to promote metabolic homeostasis? Further insights into the intricate regulation of the metabolic network will likely emerge when we identify novel SIRT2 targets in organs such as liver, fat, pancreas and muscle. The study of tissue-specific knockouts will also contribute to a better understanding of the regulatory pathways that control metabolism in health and disease and assess a possible therapeutic role of SIRT2 modulation in metabolic syndromes. Genetic studies should also address the safety of targeting SIRT2 activity pharmacologically and the extent of the possible benefits.
Cancer cells have the ability to divide and grow in an uncontrolled manner. A common event in various cancers is the presence of mutant proteins, namely the machinery responsible for cell cycle control and differentiation (Vogelstein and Kinzler, 2004). Several studies are in favor of SIRT2 working as a mitotic check-point, thus suggesting a role in tumorigenesis (Inoue et al., 2007). SIRT2 protein and RNA levels are decreased in gliomas, melanomas, and gastric carcinomas (Hiratsuka et al., 2003; Peters et al., 2010). We can speculate that in human gliomas this decrease may be associated with the deletion of the SIRT2 gene, since SIRT2 is located at 19q13.2, a region frequently deleted in this kind of carcinoma (von Deimling et al., 1994; Rasheed et al., 1999; Hiratsuka et al., 2003). Moreover, in a glioma cell line SIRT2 inhibits colony formation (Hiratsuka et al., 2003). In melanomas SIRT2 is mutated in the catalytic domain, eliminating its enzymatic activity (Lennerz et al., 2005). Another important observation is that when the serine 368 SIRT2 mutant (phosphorylation site) is overexpressed in a glioblastoma cell line, it leads to a reduction of hyperploid cells under mitotic stress exposure (North and Verdin, 2007b). These two studies indicate that the enzymatic function of SIRT2 may play a role in cancer, at least in the case of melanoma and glioblastoma. It remains to be determined whether SIRT2 enzymatic activity is also important in other kinds of cancers. In a recent report, SIRT2 deficiency in mice led to augmented levels of Aurora-A and -B, known to direct centrosome amplification, aneuploidy, and mitotic cell death. Surprisingly, gender-specific tumorigenesis was observed in the SIRT2-deficient mice, where females primarily developed mammary tumors, while males developed more hepatocellular carcinoma (Kim et al., 2011). The same study showed SIRT2-dependent regulation of the anaphase-promoting complex/cyclosome activity through deacetylation of its coactivators, APC (CDH1) and CDC20. The previous data combined with other study showing that lysine 53 of histone H3, which is deacetylated by SIRT2 and SIRT1, was hyperacetylated in cancer cells, implies that SIRT2 may act as a tumor suppressor gene (Das et al., 2009). Downregulation of SIRT2 renders cancer cells to apoptosis. For example, SIRT2 downregulation caused apoptosis in a cancer cell line such as HeLa but not in normal cells, suggesting a possible therapeutic avenue for intervention (Li et al., 2011). In addition, SIRT2 regulates the expression of the pro-apoptotic protein BIM due to its ability to deacetylate FOXO3 (Wang et al., 2012). Overall, SIRT2 overexpression is found to reduce cell proliferation and to regulate cell death in response to DNA damage stress. These and previously published results with cancer cell lines suggest SIRT2 as a tumor suppressor gene (Das et al., 2009). The dual role of sirtuins in cancer has recently been reviewed and although most evidences point to SIRT2 as a tumor suppressor gene. Further studies will be important to elucidate the role of SIRT2 activation in specific human cancers (Bosch-Presegue and Vaquero, 2011). These critical studies will illuminate potential and feasible therapeutic interventions for cancer treatment.
All seven members of the mammalian SIR2 family are expressed in the brain. SIRT2 is the most abundant, although not much is known about its role in this organ (Zhu et al., 2012). SIRT2 is expressed in nearly all brain cells including olfactory and hippocampal neurons, while primarily found in the myelin producing cells of the central nervous system (CNS): oligodendrocytes (OL; Pandithage et al., 2008; Zhu et al., 2012). There are three known alternatively spliced SIRT2 isoforms in mammals. The full-length SIRT2 protein (isoform SIRT2.1) showed only moderate expression in the CNS, comparable to that seen in peripheral tissues. In sharp contrast, a significant increase of the second isoform, SIRT2.2, was observed during postnatal mouse development. SIRT2.2 expression levels remained remarkably high in the adult CNS, with maximal accumulation in aged brains (Maxwell et al., 2011).
SIRT2 expression in OL plays a role in myelinogenesis, while in neurons its function is essentially unknown. In OLs, SIRT2 is rather localized to the myelin sheath than to the cell body (Werner et al., 2007). SIRT2 appears to decelerate cell differentiation of OL precursor cells through deacetylating α-tubulin (Li et al., 2007). In accordance with the suggested role for SIRT2 as an OL differentiation inhibitor, gene knockdown causes upregulation of myelin basic protein and promotes OL differentiation. Moreover, it appears that SIRT2 functions in myelin sheaths at sites of microtubule remodeling (Southwood et al., 2007; Harting and Knoll, 2010). Based on a recent report, SIRT2 is highly abundant during active myelination, and protein levels are regulated by the QKI-dependent pathway and mediated through selective regulation of proteolipid protein PLP (Zhu et al., 2012). Consistent with this mechanism, the presence of the SIRT2.2 isoform is severely reduced in brain of Plp1 knockout mice. Recently, mice with tissue-specific knockout of SIRT2 in Schwann cells were found to display a transient delay in myelination (Beirowski et al., 2011). It appears that in Schwann cells SIRT2 deacetylates an important regulator of cell polarity Par-3. SIRT2 deacetylates Par-3 and leads to a decrease in the activity of the polarity complex signaling component aPKC, thus regulating myelin formation. Notably, SIRT2 knockout did not lead to an increase of α-tubulin acetylation in Schwann cells. It remains to be tested whether pharmacological stimulation of SIRT2 can facilitate re-myelination in neuronal injuries or to be therapeutically beneficial in human conditions such as multiple sclerosis, characterized by progressive loss of myelin.
In neurons, SIRT2 is evenly distributed within the cytoplasm, neurites, and their growth cones (Pandithage et al., 2008). One important feature regulated by SIRT2 is neuronal motility, which is strongly dependent on the dynamic properties of the cytoskeleton, particularly those of actin filaments and microtubules. The obvious connection is the ability of SIRT2 to deacetylate α-tubulin, which is the main component of microtubules, together with β-tubulin. SIRT2 inhibits neurite outgrowth and growth cone collapse in postmitotic hippocampal neurons (Pandithage et al., 2008). Interestingly, this effect was antagonized by CDK-dependent phosphorylation of SIRT2, raising the important question of how posttranslational modifications, namely SIRT2 phosphorylation regulates its activity (Pandithage et al., 2008). Recent studies reveal that focal areas of endogenous SIRT2 expression correlate with reduced α-tubulin acetylation in primary mouse cortical neurons, further suggesting that SIRT2 may function as a microtubule deacetylase in mature neurons (Maxwell et al., 2011). Noteworthy, there are no direct evidences convincingly demonstrating that SIRT2 deacetylates microtubules in neurons. Overexpression of SIRT2 leads to inhibition of neurite elongation and impairment of migration in primary hippocampal neurons (Pandithage et al., 2008). However, no studies have been published so far indicating a direct role for SIRT2 in neuronal differentiation. The fact that SIRT2 deacetylates FOXO3 and thereby regulates its transcriptional activity, suggests a role for SIRT2 in neural stem cells homeostasis; because FOXO3 regulates the neuronal stem cell pool by maintaining quiescence (Renault et al., 2009). Besides, SIRT1 modulates neuroblastoma cell differentiation by deacetylating FOXO3 (Kim et al., 2009). Thus, it is likely to assume that SIRT2 might also play a role in neuronal differentiation. The tumor suppressor p53, has recently been ascribed a role in the regulation of neuronal cell migration, neurite outgrowth, growth cone motility, and axonal regeneration (Di Giovanni et al., 2006; Tedeschi and Di Giovanni, 2009). Since SIRT2 was shown to deacetylate p53 in vitro, we cannot rule out the possibility of p53 being an in vivo substrate for SIRT2 (Nahhas et al., 2007). Whether SIRT2 mediates these p53 dependent processes, is an intriguing question. Clearly more studies are needed to assess the precise role of SIRT2 in neuronal networks.
Neurodegenerative disorders affect mostly the elderly population and strongly contribute to a tremendous increase in health expenditures due to the augmentation in life expectancy. A common hallmark to several neurodegenerative disorders is the presence of abnormal protein inclusions in the brain containing specific misfolded proteins (Mattson and Magnus, 2006). However, the precise functions of those proteins under physiological and pathological conditions remain unclear. Nevertheless, SIRT2 was associated with the aggregation of proteins such as α-synuclein and huntingtin, involved in Parkinson’s and Huntington’s disease (HD), respectively (Outeiro et al., 2007; Pallos et al., 2008; Luthi-Carter et al., 2010). In addition, SIRT2 was indirectly associated with cellular processes implicated in the pathophysiology of neurodegenerative disorders, namely autophagy, oxidative stress, and inflammation.
Modifications in autophagy pathways were described in neurodegenerative diseases and in the normal aging brain (Wong and Cuervo, 2010). Importantly, activation of autophagy may mitigate or even prevent these disorders. Autophagy is a degradation mechanism by which cells clear out organelles, proteins, and protein aggregates that may be too large to be degraded by the ubiquitin proteasome system (Kraft et al., 2010). Several stimuli can activate autophagy and, notably, one of the main triggers is nutrient deprivation. In response to oxidative stress or serum deprivation SIRT2 releases FOXO1, which is then acetylated and binds to ATG7 and thus induce autophagy in the context of cancer (Zhao et al., 2010a,b). Accordingly, it would be interesting to test whether SIRT2 also mediates autophagy through deacetylation of FOXO1 in the context of neurodegeneration. This hypothesis seems plausible concerning the stimulus for SIRT2 to release FOXO1 and making it available to activate autophagy. Interestingly, SIRT1 also plays a role in the regulation of autophagy through deacetylation of ATG5, 7, and 8 (Lee et al., 2008). It is widely accepted that oxidative stress is implicated in the pathogenesis of neurodegenerative diseases. SIRT2 elevates the expression of the antioxidant mitochondrial superoxide dismutase (MnSOD) due to its ability to deacetylate FOXO3 and consequent increase of FOXO3 DNA-binding activity (Wang et al., 2007). Interestingly, MnSOD enzymatic activity is regulated by SIRT3 deacetylation in response to stress (Tao et al., 2010).
Another interesting link between SIRT2 and neurodegeneration is the nuclear factor-kappa B (NF-κB). NF-κB plays a pivotal role in regulating gene expression programs related to aging and inflammation, namely by inducing the expression of pro-inflammatory cytokines. New emerging data point to an association between chronic neuroinflammation and the exacerbation of several neurodegenerative diseases (Salminen and Kaarniranta, 2009). For instance, as the organism ages NF-κB transcription is activated and its incorrect regulation may elicit neurodegeneration. SIRT2 has been reported to interact with p65, an NF-κB family member, in the cytoplasm and to deacetylate it at lysine 310 after stimulation with TNF-α (Rothgiesser et al., 2010). Interestingly, deacetylation of p65 by SIRT1 antagonizes NF-κB activity (Yeung et al., 2004).
SIRT2 in Parkinson’s disease. Parkinson’s disease (PD) is the second most common neurodegenerative disorder, after Alzheimer’s disease affecting around 4 million people worldwide. The disease is characterized by the dysfunction and degeneration of dopaminergic neurons in the substantia nigra, which alters neurotransmitter balance in the striatum resulting in the progressive loss of movement control (Gelb et al., 1999; Forman et al., 2005). The vast majority of PD cases are sporadic and aging is the main risk factor, although inherited forms account for 5–10% of the cases. The pathological hallmark of PD and other synucleinopathies is the accumulation of protein aggregates called Lewy bodies (LB), which consist mainly of α-synuclein (Spillantini et al., 1997). LBs occur in the central, peripheral, and autonomic nervous system. The biological significance of LBs, their impact on neurodegeneration, and whether they are detrimental, and interfere with normal cell function, or are a structural manifestation of a cytoprotective response to confine and eliminate cytotoxic proteins is currently unclear.
SIRT2 regulates α-synuclein inclusion number, size, and cytotoxicity. Inhibition of SIRT2 function, either pharmacologically or genetically, led to a rescue of α-synuclein toxicity in models of PD, namely dopaminergic neurons and in an in vivo fly model (Outeiro et al., 2007). Inhibition of SIRT2 promoted the development of fewer and bigger aggregates. Interestingly, a smaller quantity of LB-like inclusions which are bigger in size was ascribed a protective role in neuronal cell death in vitro (Outeiro et al., 2007). However, the exact molecularSIRT2 inhibition-dependent neuroprotective mechanism is still elusive. It is tempting to speculate that SIRT2 inhibition increases microtubule-dependent transport of putative neurotoxic α-synuclein oligomers to the nucleation aggregation site, which facilitates formation of large benign inclusions. Consistent with this idea, an interaction between microtubules and α-synuclein was observed by several groups (Iseki et al., 2000; Payton et al., 2001; Alim et al., 2004). Speculatively, the affinity of interaction between oligomeric α-synuclein and acetylated microtubules may be enhanced by inhibition of microtubule deacetylase SIRT2 or/and HDAC6. We envision that genetic crosses between PD and SIRT2 brain-specific knockout mice will provide valuable clues on neuroprotective mechanism(s) and further report on therapeutic potentials of SIRT2.
SIRT2 in Huntington’s disease. Huntington’s disease is an autosomal dominant neurodegenerative disorder caused by mutant huntingtin protein containing pathologically extended polyglutamine repeats (Bates, 2003). The disease is characterized by a gradual and progressive loss of neurons, predominantly in the cortex and striatum leading to impairment in muscle coordination, cognitive decline, and dementia. Currently, there is no cure for HD and treatments can only mildly relieve some of its symptoms. Pharmacological inhibition of SIRT2 achieved neuroprotection in cellular and invertebrate models of HD through a negative regulation of sterol biosynthesis (Luthi-Carter et al., 2010). In primary striatal neurons expressing a mutant huntingtin fragment, genetic or pharmacological inhibition of SIRT2 was associated with significant reduction of polyglutamine inclusions. The reduction of inclusion formation, which is believed to be dependent on microtubule transport, may represent a benign biomarker. Another major finding from this study is that this protective effect is intimately related to transcriptional regulation of genes controlling metabolism, including sterol and fatty acid biosynthesis, carbohydrate metabolism, and purine metabolism (Luthi-Carter et al., 2010). More specifically, SIRT2 inhibition reduced sterol levels via decreased nuclear trafficking of sterol regulatory element-binding protein 2 (SREBP-2) and resulted in lower cholesterol levels. Notably, in the same model, manipulation of sterol biosynthesis at the transcriptional level mimicked SIRT2 inhibition, demonstrating that the metabolic effects are sufficient to diminish mutant huntingtin toxicity. Genetic manipulation of SREBP-2 expression levels and/or subcellular localization had no effect on the aggregation state of mutant huntingtin fragments. In follow-up studies, pharmacological inhibition of SIRT2 in wild type primary neurons resulted in SREBP-2 cytoplasmic retention, transcriptional downregulation of cholesterol biosynthetic genes, and reduction of neuronal cholesterol (Taylor et al., 2011). The experiments were extended to Neuro-2a (N2a) neuroblastoma cells and to hippocampal slice cultures from wild type mice, where SIRT2 inhibition-dependent reduction of cholesterol levels was observed as well (Taylor et al., 2011). These results illuminate an emerging novel role of SIRT2 in regulation of neuronal metabolism, and specifically of cholesterol biosynthesis. Accordingly, in a HD fly model, decreased levels of SIRT2 promoted viability of photoreceptor neurons (Pallos et al., 2008).
The fact that the same small molecule is protective in PD and HD, two devastating disorders, is highly promising, suggesting it might also have a beneficial impact on other neurodegenerative diseases.
Despite one decade of research, the precise function of SIRT2 in age-related disorders remains unclear. Therefore, elucidating the cellular function of SIRT2 and the mechanisms underlying its protective and/or pathogenic effects in those disorders is essential for the development of efficient therapies for preventing and treating age-related maladies. The identification of novel SIRT2 substrates will be crucial in pursuing such endeavor. Although a lack of direct evidence supporting the hypothesis that SIRT2 can regulate lifespan in mice exists, it deacetylates a variety of proteins, which play diverse roles in fundamental cellular processes related to the healthy state of the organisms. In this list we have FOXO transcription factors, α-tubulin, PEPCK1, and NF-κB. The effect of SIRT2 seems to vary in a tissue-specific and disease-specific manner, indicating we are still lacking a complete understanding of its mode of action.
Although persuasive, when considering the usage of pharmacological inhibitors/activators of sirtuins as candidates for protection against aging-related maladies, one has to be aware of the apparent opposite effects of SIRT1 and SIRT2 as well as the possible cross talk between other family members. We might not be able to use general inhibitors/activators since they might evoke opposite response from different family members. The development of specific inhibitors/activators for the mammalian sirtuins will enclose the potential to be beneficial to neurodegenerative diseases, cancer, diabetes, and cardiovascular diseases.
Currently unanswered questions in this fast growing field are: (i) How is SIRT2 transcription, stability, and enzymatic activity regulated by pathological stress conditions? (ii) How does SIRT2 communicate with other sirtuins to modulate the pathophysiology of diseases such as cancer, metabolic syndrome, PD, and HD? (iii) Can SIRT2 regulate lifespan in mice? (iiii) Is SIRT2 together with SIRT1 a mediator of CR?
Regardless of the current lack of answers to these questions, SIRT2 biology grew substantially in the last decade. It will be exciting to see which new pieces to the puzzle will undoubtedly be added in the years to come. This will yield tremendous profits for both basic and clinically applied age-related research. In addition, understanding the molecular mechanisms underlying the protective role of all sirtuins in different organs could bring us closer to the development of novel drug targets, which could be used to design new and more successful therapies for these diseases and even postpone the normal aging process.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are grateful to Hugo Vicente Miranda for graphical support in the execution of Figure 1. Tiago Fleming Outeiro was supported by the Michael J. Fox Foundation, by a Marie Curie International Reintegration Grant (Neurofold) from the European Commission and an EMBO Installation Grant. Rita Machado de Oliveira is supported by FCT (SFRH/BPD/41416/2007). Aleksey G. Kazantsev is supported by NIH 1U01NS066912-01A.
Alim, M. A., Ma, Q. L., Takeda, K., Aizawa, T., Matsubara, M., Nakamura, M., Asada, A., Saito, T., Kaji, H., Yoshii, M., Hisanaga, S., and Ueda, K. (2004). Demonstration of a role for alpha-synuclein as a functional microtubule-associated protein. J. Alzheimers Dis. 6, 435–442; discussion 443–439.
Bae, N. S., Swanson, M. J., Vassilev, A., and Howard, B. H. (2004). Human histone deacetylase SIRT2 interacts with the homeobox transcription factor HOXA10. J. Biochem. 135, 695–700.
Bates, G. (2003). Huntingtin aggregation and toxicity in Huntington’s disease. Lancet 361, 1642–1644.
Beirowski, B., Gustin, J., Armour, S. M., Yamamoto, H., Viader, A., North, B. J., Michan, S., Baloh, R. H., Golden, J. P., Schmidt, R. E., Sinclair, D. A., Auwerx, J., and Milbrandt, J. (2011). Sir-two-homolog 2 (Sirt2) modulates peripheral myelination through polarity protein Par-3/atypical protein kinase C (aPKC) signaling. Proc. Natl. Acad. Sci. U.S.A. 108, E952–E961.
Black, J. C., Mosley, A., Kitada, T., Washburn, M., and Carey, M. (2008). The SIRT2 deacetylase regulates autoacetylation of p300. Mol. Cell 32, 449–455.
Blander, G., and Guarente, L. (2004). The Sir2 family of protein deacetylases. Annu. Rev. Biochem. 73, 417–435.
Bosch-Presegue, L., and Vaquero, A. (2011). The dual role of sirtuins in cancer. Genes Cancer 2, 648–662.
Burnett, C., Valentini, S., Cabreiro, F., Goss, M., Somogyvari, M., Piper, M. D., Hoddinott, M., Sutphin, G. L., Leko, V., Mcelwee, J. J., Vazquez-Manrique, R. P., Orfila, A. M., Ackerman, D., Au, C., Vinti, G., Riesen, M., Howard, K., Neri, C., Bedalov, A., Kaeberlein, M., Soti, C., Partridge, L., and Gems, D. (2011). Absence of effects of Sir2 overexpression on lifespan in C. elegans and Drosophila. Nature 477, 482–485.
Chen, D., and Guarente, L. (2007). SIR2: a potential target for calorie restriction mimetics. Trends Mol. Med. 13, 64–71.
Chen, D., Steele, A. D., Lindquist, S., and Guarente, L. (2005). Increase in activity during calorie restriction requires Sirt1. Science 310, 1641.
Cohen, H. Y., Miller, C., Bitterman, K. J., Wall, N. R., Hekking, B., Kessler, B., Howitz, K. T., Gorospe, M., De Cabo, R., and Sinclair, D. A. (2004). Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science 305, 390–392.
Das, C., Lucia, M. S., Hansen, K. C., and Tyler, J. K. (2009). CBP/p300-mediated acetylation of histone H3 on lysine 56. Nature 459, 113–117.
Di Giovanni, S., Knights, C. D., Rao, M., Yakovlev, A., Beers, J., Catania, J., Avantaggiati, M. L., and Faden, A. I. (2006). The tumor suppressor protein p53 is required for neurite outgrowth and axon regeneration. EMBO J. 25, 4084–4096.
Dryden, S. C., Nahhas, F. A., Nowak, J. E., Goustin, A. S., and Tainsky, M. A. (2003). Role for human SIRT2 NAD-dependent deacetylase activity in control of mitotic exit in the cell cycle. Mol. Cell. Biol. 23, 3173–3185.
Ford, E., Voit, R., Liszt, G., Magin, C., Grummt, I., and Guarente, L. (2006). Mammalian Sir2 homolog SIRT7 is an activator of RNA polymerase I transcription. Genes Dev. 20, 1075–1080.
Ford, E. S., Giles, W. H., and Dietz, W. H. (2002). Prevalence of the metabolic syndrome among US adults: findings from the third National Health and Nutrition Examination Survey. JAMA 287, 356–359.
Forman, M. S., Lee, V. M., and Trojanowski, J. Q. (2005). Nosology of Parkinson’s disease: looking for the way out of a quagmire. Neuron 47, 479–482.
Frye, R. A. (2000). Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem. Biophys. Res. Commun. 273, 793–798.
Gelb, D. J., Oliver, E., and Gilman, S. (1999). Diagnostic criteria for Parkinson disease. Arch. Neurol. 56, 33–39.
Haigis, M. C., and Guarente, L. P. (2006). Mammalian sirtuins – emerging roles in physiology, aging, and calorie restriction. Genes Dev. 20, 2913–2921.
Haigis, M. C., and Sinclair, D. A. (2010). Mammalian sirtuins: biological insights and disease relevance. Annu. Rev. Pathol. 5, 253–295.
Han, Y., Jin, Y. H., Kim, Y. J., Kang, B. Y., Choi, H. J., Kim, D. W., Yeo, C. Y., and Lee, K. Y. (2008). Acetylation of Sirt2 by p300 attenuates its deacetylase activity. Biochem. Biophys. Res. Commun. 375, 576–580.
Harting, K., and Knoll, B. (2010). SIRT2-mediated protein deacetylation: An emerging key regulator in brain physiology and pathology. Eur. J. Cell Biol. 89, 262–269.
Hiratsuka, M., Inoue, T., Toda, T., Kimura, N., Shirayoshi, Y., Kamitani, H., Watanabe, T., Ohama, E., Tahimic, C. G., Kurimasa, A., and Oshimura, M. (2003). Proteomics-based identification of differentially expressed genes in human gliomas: down-regulation of SIRT2 gene. Biochem. Biophys. Res. Commun. 309, 558–566.
Imai, S., Armstrong, C. M., Kaeberlein, M., and Guarente, L. (2000). Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403, 795–800.
Inoue, T., Hiratsuka, M., Osaki, M., and Oshimura, M. (2007). The molecular biology of mammalian SIRT proteins: SIRT2 in cell cycle regulation. Cell Cycle 6, 1011–1018.
Iseki, E., Marui, W., Sawada, H., Ueda, K., and Kosaka, K. (2000). Accumulation of human alpha-synuclein in different cytoskeletons in Lewy bodies in brains of dementia with Lewy bodies. Neurosci. Lett. 290, 41–44.
Jiang, W., Wang, S., Xiao, M., Lin, Y., Zhou, L., Lei, Q., Xiong, Y., Guan, K. L., and Zhao, S. (2011). Acetylation regulates gluconeogenesis by promoting PEPCK1 degradation via recruiting the UBR5 ubiquitin ligase. Mol. Cell 43, 33–44.
Jin, Y. H., Kim, Y. J., Kim, D. W., Baek, K. H., Kang, B. Y., Yeo, C. Y., and Lee, K. Y. (2008). Sirt2 interacts with 14-3-3 beta/gamma and down-regulates the activity of p53. Biochem. Biophys. Res. Commun. 368, 690–695.
Jing, E., Gesta, S., and Kahn, C. R. (2007). SIRT2 regulates adipocyte differentiation through FoxO1 acetylation/deacetylation. Cell Metab. 6, 105–114.
Kaeberlein, M., Mcvey, M., and Guarente, L. (1999). The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 13, 2570–2580.
Kim, H. S., Vassilopoulos, A., Wang, R. H., Lahusen, T., Xiao, Z., Xu, X., Li, C., Veenstra, T. D., Li, B., Yu, H., Ji, J., Wang, X. W., Park, S. H., Cha, Y. I., Gius, D., and Deng, C. X. (2011). SIRT2 maintains genome integrity and suppresses tumorigenesis through regulating APC/C activity. Cancer Cell 20, 487–499.
Kim, M. J., Ahn, K., Park, S. H., Kang, H. J., Jang, B. G., Oh, S. J., Oh, S. M., Jeong, Y. J., Heo, J. I., Suh, J. G., Lim, S. S., Ko, Y. J., Huh, S. O., Kim, S. C., Park, J. B., Kim, J., Kim, J. I., Jo, S. A., and Lee, J. Y. (2009). SIRT1 regulates tyrosine hydroxylase expression and differentiation of neuroblastoma cells via FOXO3a. FEBS Lett. 583, 1183–1188.
Kraft, C., Peter, M., and Hofmann, K. (2010). Selective autophagy: ubiquitin-mediated recognition and beyond. Nat. Cell Biol. 12, 836–841.
Lamming, D. W., Latorre-Esteves, M., Medvedik, O., Wong, S. N., Tsang, F. A., Wang, C., Lin, S. J., and Sinclair, D. A. (2005). HST2 mediates SIR2-independent life-span extension by calorie restriction. Science 309, 1861–1864.
Landry, J., Sutton, A., Tafrov, S. T., Heller, R. C., Stebbins, J., Pillus, L., and Sternglanz, R. (2000). The silencing protein SIR2 and its homologs are NAD-dependent protein deacetylases. Proc. Natl. Acad. Sci. U.S.A. 97, 5807–5811.
Lee, I. H., Cao, L., Mostoslavsky, R., Lombard, D. B., Liu, J., Bruns, N. E., Tsokos, M., Alt, F. W., and Finkel, T. (2008). A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. U.S.A. 105, 3374–3379.
Lennerz, V., Fatho, M., Gentilini, C., Frye, R. A., Lifke, A., Ferel, D., Wolfel, C., Huber, C., and Wolfel, T. (2005). The response of autologous T cells to a human melanoma is dominated by mutated neoantigens. Proc. Natl. Acad. Sci. U.S.A. 102, 16013–16018.
Li, W., Zhang, B., Tang, J., Cao, Q., Wu, Y., Wu, C., Guo, J., Ling, E. A., and Liang, F. (2007). Sirtuin 2, a mammalian homolog of yeast silent information regulator-2 longevity regulator, is an oligodendroglial protein that decelerates cell differentiation through deacetylating alpha-tubulin. J. Neurosci. 27, 2606–2616.
Li, Y., Matsumori, H., Nakayama, Y., Osaki, M., Kojima, H., Kurimasa, A., Ito, H., Mori, S., Katoh, M., Oshimura, M., and Inoue, T. (2011). SIRT2 down-regulation in HeLa can induce p53 accumulation via p38 MAPK activation-dependent p300 decrease, eventually leading to apoptosis. Genes Cells 16, 34–45.
Lin, S. J., Ford, E., Haigis, M., Liszt, G., and Guarente, L. (2004). Calorie restriction extends yeast life span by lowering the level of NADH. Genes Dev. 18, 12–16.
Liszt, G., Ford, E., Kurtev, M., and Guarente, L. (2005). Mouse Sir2 homolog SIRT6 is a nuclear ADP-ribosyltransferase. J. Biol. Chem. 280, 21313–21320.
Luthi-Carter, R., Taylor, D. M., Pallos, J., Lambert, E., Amore, A., Parker, A., Moffitt, H., Smith, D. L., Runne, H., Gokce, O., Kuhn, A., Xiang, Z., Maxwell, M. M., Reeves, S. A., Bates, G. P., Neri, C., Thompson, L. M., Marsh, J. L., and Kazantsev, A. G. (2010). SIRT2 inhibition achieves neuroprotection by decreasing sterol biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 107, 7927–7932.
Mattson, M. P., and Magnus, T. (2006). Ageing and neuronal vulnerability. Nat. Rev. Neurosci. 7, 278–294.
Maxwell, M. M., Tomkinson, E. M., Nobles, J., Wizeman, J. W., Amore, A. M., Quinti, L., Chopra, V., Hersch, S. M., and Kazantsev, A. G. (2011). The Sirtuin 2 microtubule deacetylase is an abundant neuronal protein that accumulates in the aging CNS. Hum. Mol. Genet. 20, 3986–3996.
Moller, D. E., and Kaufman, K. D. (2005). Metabolic syndrome: a clinical and molecular perspective. Annu. Rev. Med. 56, 45–62.
Nahhas, F., Dryden, S. C., Abrams, J., and Tainsky, M. A. (2007). Mutations in SIRT2 deacetylase which regulate enzymatic activity but not its interaction with HDAC6 and tubulin. Mol. Cell. Biochem. 303, 221–230.
Nakagawa, T., Lomb, D. J., Haigis, M. C., and Guarente, L. (2009). SIRT5 Deacetylates carbamoyl phosphate synthetase 1 and regulates the urea cycle. Cell 137, 560–570.
North, B. J., Marshall, B. L., Borra, M. T., Denu, J. M., and Verdin, E. (2003). The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol. Cell 11, 437–444.
North, B. J., and Verdin, E. (2007a). Interphase nucleo-cytoplasmic shuttling and localization of SIRT2 during mitosis. PLoS ONE 2, e784. doi:10.1371/journal.pone.0000784
North, B. J., and Verdin, E. (2007b). Mitotic regulation of SIRT2 by cyclin-dependent kinase 1-dependent phosphorylation. J. Biol. Chem. 282, 19546–19555.
Outeiro, T. F., Kontopoulos, E., Altmann, S. M., Kufareva, I., Strathearn, K. E., Amore, A. M., Volk, C. B., Maxwell, M. M., Rochet, J. C., Mclean, P. J., Young, A. B., Abagyan, R., Feany, M. B., Hyman, B. T., and Kazantsev, A. G. (2007). Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease. Science 317, 516–519.
Pallos, J., Bodai, L., Lukacsovich, T., Purcell, J. M., Steffan, J. S., Thompson, L. M., and Marsh, J. L. (2008). Inhibition of specific HDACs and sirtuins suppresses pathogenesis in a Drosophila model of Huntington’s disease. Hum. Mol. Genet. 17, 3767–3775.
Pandithage, R., Lilischkis, R., Harting, K., Wolf, A., Jedamzik, B., Luscher-Firzlaff, J., Vervoorts, J., Lasonder, E., Kremmer, E., Knoll, B., and Luscher, B. (2008). The regulation of SIRT2 function by cyclin-dependent kinases affects cell motility. J. Cell Biol. 180, 915–929.
Payton, J. E., Perrin, R. J., Clayton, D. F., and George, J. M. (2001). Protein-protein interactions of alpha-synuclein in brain homogenates and transfected cells. Brain Res. Mol. Brain Res. 95, 138–145.
Perrod, S., Cockell, M. M., Laroche, T., Renauld, H., Ducrest, A. L., Bonnard, C., and Gasser, S. M. (2001). A cytosolic NAD-dependent deacetylase, Hst2p, can modulate nucleolar and telomeric silencing in yeast. EMBO J. 20, 197–209.
Peters, C. J., Rees, J. R., Hardwick, R. H., Hardwick, J. S., Vowler, S. L., Ong, C. A., Zhang, C., Save, V., O’Donovan, M., Rassl, D., Alderson, D., Caldas, C., and Fitzgerald, R. C. (2010). A 4-gene signature predicts survival of patients with resected adenocarcinoma of the esophagus, junction, and gastric cardia. Gastroenterology 139, 1995–2004 e1915.
Rasheed, B. K., Wiltshire, R. N., Bigner, S. H., and Bigner, D. D. (1999). Molecular pathogenesis of malignant gliomas. Curr. Opin. Oncol. 11, 162–167.
Renault, V. M., Rafalski, V. A., Morgan, A. A., Salih, D. A., Brett, J. O., Webb, A. E., Villeda, S. A., Thekkat, P. U., Guillerey, C., Denko, N. C., Palmer, T. D., Butte, A. J., and Brunet, A. (2009). FoxO3 regulates neural stem cell homeostasis. Cell Stem Cell 5, 527–539.
Rogina, B., and Helfand, S. L. (2004). Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc. Natl. Acad. Sci. U.S.A. 101, 15998–16003.
Rothgiesser, K. M., Erener, S., Waibel, S., Luscher, B., and Hottiger, M. O. (2010). SIRT2 regulates NF-kappaB dependent gene expression through deacetylation of p65 Lys310. J. Cell Sci. 123, 4251–4258.
Salminen, A., and Kaarniranta, K. (2009). NF-kappaB signaling in the aging process. J. Clin. Immunol. 29, 397–405.
Shi, T., Wang, F., Stieren, E., and Tong, Q. (2005). SIRT3, a mitochondrial sirtuin deacetylase, regulates mitochondrial function and thermogenesis in brown adipocytes. J. Biol. Chem. 280, 13560–13567.
Sinclair, D. A., and Guarente, L. (1997). Extrachromosomal rDNA circles – a cause of aging in yeast. Cell 91, 1033–1042.
Smith, J. S., Brachmann, C. B., Celic, I., Kenna, M. A., Muhammad, S., Starai, V. J., Avalos, J. L., Escalante-Semerena, J. C., Grubmeyer, C., Wolberger, C., and Boeke, J. D. (2000). A phylogenetically conserved NAD+-dependent protein deacetylase activity in the Sir2 protein family. Proc. Natl. Acad. Sci. U.S.A. 97, 6658–6663.
Southwood, C. M., Peppi, M., Dryden, S., Tainsky, M. A., and Gow, A. (2007). Microtubule deacetylases, SirT2 and HDAC6, in the nervous system. Neurochem. Res. 32, 187–195.
Spillantini, M. G., Schmidt, M. L., Lee, V. M., Trojanowski, J. Q., Jakes, R., and Goedert, M. (1997). Alpha-synuclein in Lewy bodies. Nature 388, 839–840.
Tang, B. L., and Chua, C. E. (2008). SIRT2, tubulin deacetylation, and oligodendroglia differentiation. Cell Motil. Cytoskeleton 65, 179–182.
Tanner, K. G., Landry, J., Sternglanz, R., and Denu, J. M. (2000). Silent information regulator 2 family of NAD- dependent histone/protein deacetylases generates a unique product, 1-O-acetyl-ADP-ribose. Proc. Natl. Acad. Sci. U.S.A. 97, 14178–14182.
Tao, R., Coleman, M. C., Pennington, J. D., Ozden, O., Park, S. H., Jiang, H., Kim, H. S., Flynn, C. R., Hill, S., Hayes Mcdonald, W., Olivier, A. K., Spitz, D. R., and Gius, D. (2010). Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol. Cell 40, 893–904.
Taylor, D. M., Balabadra, U., Xiang, Z., Woodman, B., Meade, S., Amore, A., Maxwell, M. M., Reeves, S., Bates, G. P., Luthi-Carter, R., Lowden, P. A., and Kazantsev, A. G. (2011). A brain-permeable small molecule reduces neuronal cholesterol by inhibiting activity of sirtuin 2 deacetylase. ACS Chem. Biol. 6, 540–546.
Tedeschi, A., and Di Giovanni, S. (2009). The non-apoptotic role of p53 in neuronal biology: enlightening the dark side of the moon. EMBO Rep. 10, 576–583.
Tissenbaum, H. A., and Guarente, L. (2001). Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature 410, 227–230.
Vaquero, A., Scher, M., Lee, D., Erdjument-Bromage, H., Tempst, P., and Reinberg, D. (2004). Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Mol. Cell 16, 93–105.
Vaquero, A., Scher, M. B., Lee, D. H., Sutton, A., Cheng, H. L., Alt, F. W., Serrano, L., Sternglanz, R., and Reinberg, D. (2006). SirT2 is a histone deacetylase with preference for histone H4 Lys 16 during mitosis. Genes Dev. 20, 1256–1261.
Viswanathan, M., and Guarente, L. (2011). Regulation of Caenorhabditis elegans lifespan by sir-2.1 transgenes. Nature 477, E1–E2.
Vogelstein, B., and Kinzler, K. W. (2004). Cancer genes and the pathways they control. Nat. Med. 10, 789–799.
von Deimling, A., Nagel, J., Bender, B., Lenartz, D., Schramm, J., Louis, D. N., and Wiestler, O. D. (1994). Deletion mapping of chromosome 19 in human gliomas. Int. J. Cancer 57, 676–680.
Wang, F., Chan, C. H., Chen, K., Guan, X., Lin, H. K., and Tong, Q. (2012). Deacetylation of FOXO3 by SIRT1 or SIRT2 leads to Skp2-mediated FOXO3 ubiquitination and degradation. Oncogene 31, 1546–1557.
Wang, F., Nguyen, M., Qin, F. X., and Tong, Q. (2007). SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell 6, 505–514.
Wang, F., and Tong, Q. (2009). SIRT2 suppresses adipocyte differentiation by deacetylating FOXO1 and enhancing FOXO1’s repressive interaction with PPARgamma. Mol. Biol. Cell 20, 801–808.
Werner, H. B., Kuhlmann, K., Shen, S., Uecker, M., Schardt, A., Dimova, K., Orfaniotou, F., Dhaunchak, A., Brinkmann, B. G., Mobius, W., Guarente, L., Casaccia-Bonnefil, P., Jahn, O., and Nave, K. A. (2007). Proteolipid protein is required for transport of sirtuin 2 into CNS myelin. J. Neurosci. 27, 7717–7730.
Wong, E., and Cuervo, A. M. (2010). Autophagy gone awry in neurodegenerative diseases. Nat. Neurosci. 13, 805–811.
Yeung, F., Hoberg, J. E., Ramsey, C. S., Keller, M. D., Jones, D. R., Frye, R. A., and Mayo, M. W. (2004). Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 23, 2369–2380.
Zhao, Y., Wang, L., Yang, J., Zhang, P., Ma, K., Zhou, J., Liao, W., and Zhu, W. G. (2010a). Anti-neoplastic activity of the cytosolic FoxO1 results from autophagic cell death. Autophagy 6, 988–990.
Zhao, Y., Yang, J., Liao, W., Liu, X., Zhang, H., Wang, S., Wang, D., Feng, J., Yu, L., and Zhu, W. G. (2010b). Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat. Cell Biol. 12, 665–675.
Keywords: SIRT2, aging, metabolic syndrome, cancer, neurodegenerative disorder
Citation: de Oliveira RM, Sarkander J, Kazantsev AG and Outeiro TF (2012) SIRT2 as a therapeutic target for age-related disorders. Front. Pharmacol. 3:82. doi: 10.3389/fphar.2012.00082
Received: 23 December 2011; Paper pending published: 05 March 2012;
Accepted: 14 April 2012; Published online: 03 May 2012.
Edited by:
Salvatore Cuzzocrea, University of Messina, ItalyReviewed by:
Won Suk Lee, Pusan National University School of Medicine, South KoreaCopyright: © 2012 de Oliveira, Sarkander, Kazantsev and Outeiro. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Aleksey G. Kazantsev, Harvard Medical School and Mass General Institute for Neurodegenerative Disease, Massachusetts General Hospital, 55 Fruit Street, Boston, MA 02114, USA. e-mail:YWthemFudHNldkBwYXJ0bmVycy5vcmc=; Tiago Fleming Outeiro, Cell and Molecular Neuroscience Unit, Instituto de Medicina Molecular, 1649-028 Lisboa, Lisboa, Portugal. e-mail:dG91dGVpcm9AZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.