
- Respiratory Disease Unit, Catholic University, Rome, Italy
Pulmonary hypertension (PH) is a life-threatening condition characterized by elevated pulmonary arterial pressure. It is clinically classified into five groups: patients in the first group are considered to have pulmonary arterial hypertension (PAH) whereas patients of the other groups have PH that is due to cardiopulmonary or other systemic diseases. The management of patients with PH has advanced rapidly over the last decade and the introduction of specific treatments especially for PAH has lead to an improved outcome. However, despite the progress in the treatment, the functional limitation and the survival of these patients remain unsatisfactory and there is no cure for PAH. Therefore the search for an “ideal” therapy still goes on. At present, two levels of treatment can be identified: primary and specific therapy. Primary therapy is directed at the underlying cause of the PH. It also includes a supportive therapy consisting in oxygen supplementation, diuretics, and anticoagulation which should be considered in all patients with PH. Specific therapy is directed at the PH itself and includes treatment with vasodilatators such as calcium channel blockers and with vasodilatator and pathogenetic drugs such as prostanoids, endothelin receptor antagonists and phosphodiesterase type-5 inhibitors. These drugs act in several pathogenetic mechanisms of the PH and are specific for PAH although they might be used also in the other groups of PH. Finally, atrial septostomy and lung transplantation are reserved for patients refractory to medical therapy. Different therapeutic approaches can be considered in the management of patients with PH. Therapy can be established on the basis of both the clinical classification and the functional class. It is also possible to adopt a goal-oriented therapy in which the timing of treatment escalation is determined by inadequate response to known prognostic indicators.
Definition and Classification
Pulmonary hypertension (PH) is characterized by elevated pulmonary arterial pressure and secondary right ventricular (RV) failure. It is a life-threatening condition with a poor prognosis if untreated. According to the most recent guidelines published jointly by the European Society of Cardiology (ESC) and the European Respiratory Society (ERS) PH is an hemodynamic and pathophysiological state defined as an increase in mean pulmonary arterial pressure (PAPm) ≥25 mmHg at rest as assessed by right heart catheterization (RHC; Galiè et al., 2009a). PH can be found in multiple clinical conditions and for this reason it is clinically classified into five groups, as shown in Table 1 (Simonneau et al., 2009). Patients in the first group are considered to have pulmonary arterial hypertension (PAH), whereas patients in the remaining four groups are considered to have PH.

Table 1. Clinical classification of pulmonary hypertension.
On the basis of hemodynamic parameters, PH is classified into two groups. Pre-capillary PH is characterized by PAPm ≥25 mmHg, pulmonary wedge pressure (PWP) ≤15 mmHg, cardiac output (CO) normal or reduced. It is typical of the group 1, i.e., PAH; group 3, i.e., PH due to lung diseases and/or hypoxemia; group 4, i.e., chronic thromboembolic PH (CTEPH); group 5, i.e., PH with unclear and/or multifactorial mechanisms. Post-capillary PH is characterized by PAPm ≥25 mmHg, PWP >15 mmHg, CO normal or reduced. It is typical of group 2, i.e., PH due to left heart disease.
This review focuses on the main therapeutic options to date available for PH of the adult and on emerging and potential therapies under development.
Treatment
The management of patients with PH has advanced rapidly over the last decade and the introduction of specific treatments especially for PAH has lead to an improved outcome for patients with this disease. A recent meta-analysis of 23 randomized controlled trial showed a 43% reduction in mortality and a 61% decrease in the rate of hospitalizations in patients with PAH treated with specific therapy in comparison with placebo (Galiè et al., 2009b). However, some patients still have a very poor prognosis and a rapid deterioration of their condition. For these reasons, early identification and treatment of PH are crucial because advanced disease may be less responsive to therapy.
Treatment begins with a baseline assessment of disease severity which is essential because the response to therapy will be measured as changes from baseline. Functional impairment and hemodynamic derangement are the key determinants of disease severity: the former is determined by measuring the exercise capacity by the 6-min walking test and assessing the patient’s World Health Organization (WHO) functional class as reported in Table 2; the latter is based both on echocardiography that is useful to estimate pulmonary artery systolic pressure and RV function and RHC that is necessary to confirm that PH exists. However, RHC is often deferred until specific therapy is indicated because it is an invasive procedure.

Table 2. Functional classification of pulmonary hypertension according to World Health Organization (WHO).
The goal of the treatment recommended by the latest ESC/ERS guidelines is to obtain and maintain for the patient a “stable and satisfactory” condition which is associated with a better prognosis (Galiè et al., 2009a). This condition is characterized by absence of clinical signs of RV failure, stable WHO functional class I or II, a 6-min walking distance >500 m depending on the individual patient, a peak oxygen consumption >15 mL/min/kg, normal or near-normal brain natriuretic peptide (BNP) and N-terminal fragment of pro-BNP (NT-proBNP) plasma levels, no pericardial effusion, right atrial pressure (RAP) <8 mmHg, and a cardiac index (CI) ≥2.5 L/min/m2 (Table 3).

Table 3. Parameters with established importance for assessing disease severity, stability, and prognosis.
Two levels of treatment can be identified: primary and specific therapy. Primary therapy is directed at the underlying cause of the PH. It also includes a supportive therapy which should be considered in all patients with PH. Specific therapy is directed at the PH itself and includes treatment with vasodilatators such as calcium channel blockers (CCBs) and with vasodilatator and pathogenetic drugs such as prostanoids, endothelin receptor antagonists and phosphodiesterase type-5 inhibitors. These drugs act in several pathogenetic mechanisms of the PH and are specific for PAH although they might be used also in the other groups of PH. A schematic diagram of the main mechanisms which trigger PAH and represent targets for pharmacological treatments is shown in Figure 1.
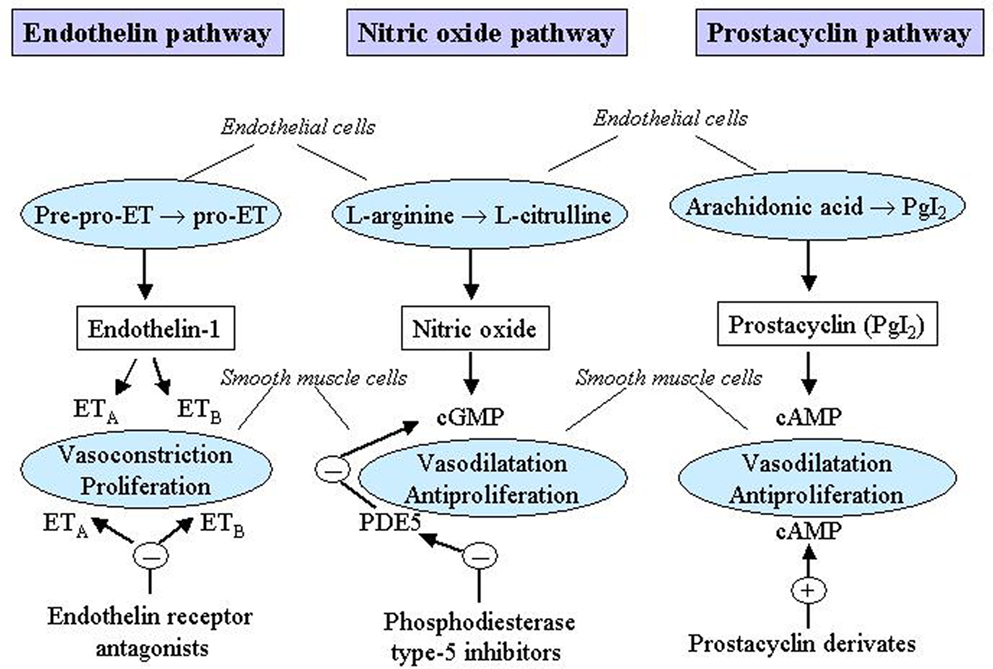
Figure 1. Mechanisms which trigger pulmonary arterial hypertension (PAH) as targets for pharmacological treatments. cAMP, cyclic adenosine monophosphate; cGMP, cyclic guanosine monophosphate; ET, endothelin; ETA, endothelin receptor A; ETB, endothelin receptor B; PDE5, phosphodiesterase type-5; PgI2, prostaglandin I2. Reproduced with permission from Humbert et al. (2004a) and Boutet et al. (2008).
Primary Therapy
It is warranted in patients with groups 2 and 3 who have PH secondary to left heart disease and lung diseases, respectively, and in patients with group 5 who have PH secondary to hematologic, systemic or metabolic disorders. In patients with group 4 who have PH due to thromboembolic occlusion of the proximal or distal pulmonary vasculature, anticoagulation is primary medical therapy, whereas surgical thromboendarterectomy is primary surgical therapy for selected patients with thromboembolic obstruction of the proximal pulmonary arteries.
Supportive therapy which should be considered in all patients with PH includes oxygen supplementation, diuretics, anticoagulation and exercise.
Oxygen
Oxygen supplementation is of fundamental importance in all patients with arterial hypoxemia. The indication to the oxygen therapy is generally set at a value of arterial pressure of oxygen (PaO2) less than 60 mmHg. Continuous oxygen administration represents the cornerstone of the therapy in patients with group 3 PH but it should also be considered for all patients with PH plus hypoxemia at rest, during exercise and/or at night. Indeed, oxygen is the only treatment with proven mortality benefit in some patients with group 3 PH as shown by two large trials which studied patients with chronic obstructive pulmonary disease (COPD), the most common cause of group 3 PH (NOTT, 1980; MRC, 1981). On the contrary, there are no randomized data to suggest that long-term oxygen therapy is beneficial in patients with PAH, although oxygen administration has been demonstrated to reduce the pulmonary vascular resistance in these patients (Galiè et al., 2009a). Oxygen is generally administered via nasal prongs or Venturi masks at a flow rate or concentration as to maintain saturation above 90%.
Diuretics
Diuretics are used to treat fluid retention due to PH and to reduce hepatic congestion and peripheral edema. Moreover, diuresis can prevent a distended right ventricle from impending left ventricular filling. Diuretics are especially useful in patients with groups 2 and 3 PH. Although there are no randomized controlled trials of diuretics in PAH, clinical experience shows symptomatic benefits also in patients with group 1 PH when a decompensated right heart failure occurs. However, they should be administered with caution to avoid decreased CO (due to decreased right and/or left ventricular preload), arrhythmias induced by hypokalemia, and metabolic alkalosis. The most commonly used loop diuretic is furosemide, usually at a starting dose of 20–40 mg daily.
Anticoagulation
Patients with PH are at increased risk for intrapulmonary thrombosis and thromboembolism, due to sluggish pulmonary blood flow, dilated right heart chambers, venous stasis, and a sedentary lifestyle. These risk factors and the consideration that even a small thrombus can produce hemodynamic deterioration in a patient with a compromised pulmonary vascular bed that is unable to dilate or recruit unused vasculature, represent the rationale for oral anticoagulation in PH. It is generally accepted that anticoagulation is indicated in patients with idiopathic PAH, hereditary PAH and drug-induced PAH (Barst et al., 2009; Galiè et al., 2009a; McLaughlin et al., 2009), or group 4 PH. However, most of the evidence comes from observational studies of patients with idiopathic PAH (Kawut et al., 2005; Barst et al., 2009). In a systematic review of seven observational studies that evaluated the effect of warfarin in patients with group 1 PH, five studies found a mortality benefit (Johnson et al., 2006). In patients with other groups of PH the potential benefits of anticoagulation should be weighed against the risk of bleeding.
The anticoagulant of choice is warfarin, with a therapeutic goal of an International Normalized Ratio (INR) of approximately two.
Exercise
Exercise training appears to be beneficial for patients with PH (Mereles et al., 2006; de Man et al., 2009). A randomized controlled trial has demonstrated an improvement in exercise capacity in patients with PAH who took part in a training program (Mereles et al., 2006). Exercise training improved the WHO functional class and peak oxygen consumption although it didn’t improve hemodynamic abnormalities (Mereles et al., 2006). Moreover, there is growing evidence supporting loss of peripheral muscle mass in patients with advanced PAH, and this may be corrected by a defined rehabilitation program (Galiè et al., 2009a). Thus, skeletal muscle training may play a role in the treatment of patients with PH.
Specific Therapy
It is directed at the PH itself and is considered for patients who have evidence of persistent PH and a WHO functional class II, III, or IV despite adequate primary therapy (Badesch et al., 2007; Barst et al., 2009). Specific therapy is often needed for patients with group 1 PH, because there are not effective primary therapies for these patients.
Patients with PH who are selected for specific therapy should undergo an invasive hemodynamic assessment, i.e., RHC, prior to the initiation of treatment. It is recommended that patients with group 1 PH also undergo a vasoreactivity test with intravenous adenosine, intravenous epoprostenol, or inhaled nitric oxide (NO, Barst et al., 2009). Patients with a positive vasoreactivity test can be given a trial of oral CCB therapy. In contrast, patients with a negative vasoreactivity test require therapy with a prostanoid, endothelin receptor antagonist, or phosphodiesterase type-5 inhibitor. Combination specific therapy may be appropriate in refractory cases, although data are limited. Some patients are refractory to all medical interventions. In such cases, lung transplantation or creation of a right to left shunt by atrial septostomy may be considered (Keogh et al., 2009). The main pharmacological characteristics of the specific drugs are reported in Table 4.
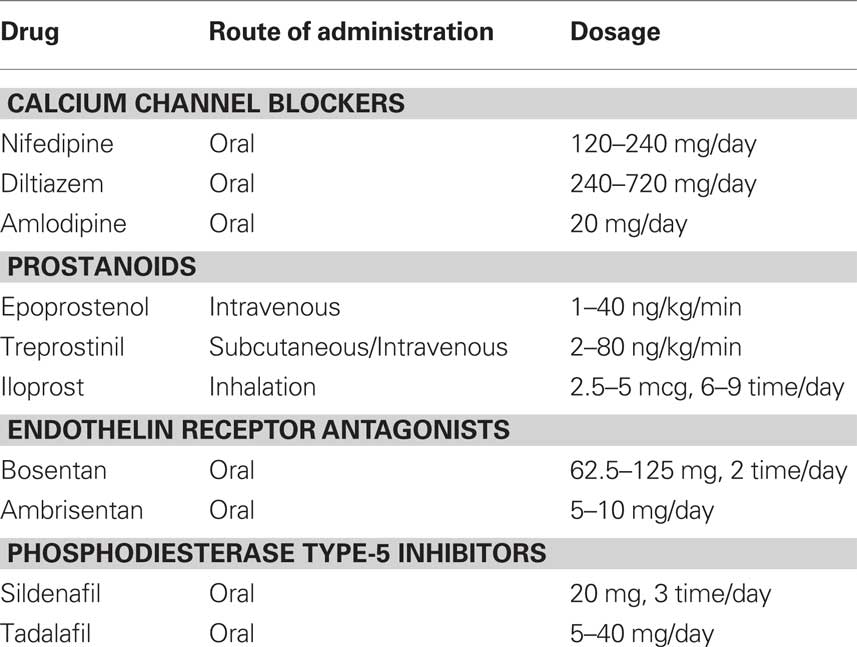
Table 4. Main pharmacological characteristics of the drugs used in the specific therapy.
Calcium Channel Blockers
The rationale to use vasodilatators such as CCBs is that vasoconstriction and smooth muscle cell hypertrophy contribute to the pathogenesis of PH. However, it has been recognized that these drugs are of benefit only in a small number of patients with PAH who demonstrate a positive response to the vasoreactivity test. These patients can achieve prolonged survival, sustained functional improvement and hemodynamic improvement by taking nifedipine or diltiazem (Rich et al., 1992; Humbert et al., 2004a; Sitbon et al., 2005). The daily doses of these drugs that have shown efficacy in PAH are relatively high, 120–240 mg for nifedipine, 240–720 mg for diltiazem. CCB therapy can be initiated with either long-acting nifedipine (30 mg/day) or diltiazem (120 mg/day), then increased to the maximal tolerated dose. Sustained release preparations of both nifedipine and diltiazem are recommended because they minimize the adverse effects of therapy, especially systemic hypotension. Amlodipine, a long-acting dihydropyridine, is a useful alternative at a dosage up to 20 mg/day. Patients who respond to CCB therapy should be reassessed after 3–6 months of treatment.
Prostanoids
Prostanoids are prostacyclin analogs. Prostacyclin is produced by the endothelial cells, induces vasodilatation and is a powerful platelet aggregation inhibitor. Its synthesis is reduced in patients with PAH (Galiè et al., 2003). Prostanoid formulations used to treat PH include epoprostenol, treprostinil, and iloprost. All have the limitation of a short half-life and a heterogeneous response to therapy.
Epoprostenol used intravenous improves hemodynamic parameters and functional capacity in patients with PAH (Rubin et al., 1990; Barst et al., 1996; Shapiro et al., 1997; Higenbottam et al., 1998) and is the only treatment shown to improve survival in idiopathic PAH in a randomized trial (Barst et al., 1996). The main limitation of its use is that it has a short half-life (3–5 min) and is stable for only 8 h. Therefore it needs to be administered continuously by an infusion pump and a permanently implanted central venous catheter. It is usually initiated at doses of 2–4 ng/kg/min and increased by 1–2 ng/kg/min every 1 to 2 days as tolerated. The optimal dose varies between 20 and 40 ng/kg/min. Side effects include both diarrhea, arthralgias, flushing, headache and effects related to the delivery system such as thrombosis, local site infection, sepsis. For these side effects and for its administration disadvantages, epoprostenol perfusion cannot be considered an ideal treatment and this drug is generally used as first-line agent only in patients with severe disease (i.e., WHO functional class IV).
Treprostinil is a epoprostenol analog with a longer half-life (58–80 min) which can be given intravenously or subcutaneously although subcutaneous administration, that is accomplished by a microinfusion pump and a small subcutaneous catheter, frequently induces local side effects such as severe pain. It is available also for aerosol administration. Inhaled treprostinil has recently been approved for patients with group 1 PH who are WHO functional class III. Several randomized controlled trial have been shown that treprostinil improves hemodynamic parameters, symptoms, and exercise capacity (Barst et al., 2006a; Tapson et al., 2006; Benza et al., 2008). Treatment with subcutaneous treprostinil is initiated at a dose of 1–2 ng/kg/min with doses increasing at a rate limited by side effects (flushing, headache, local pain). The optimal dose varies between 20 and 80 ng/kg/min. The use of intravenous treprostinil is approved by the US Food and Drug Administration (FDA) in WHO functional class II, III, and IV PAH patients in whom subcutaneous infusion in not tolerated.
Iloprost is a prostacyclin analog available for aerosol administration. Inhaled iloprost has theoretical advantages in targeting the lung vasculature and not require intravenous administration. The main disadvantages is the need for frequent administration (six to nine time per day) due to its short half-life. Iloprost has been evaluated in a randomized controlled trial in which patients with group 1 and 4 PH who were in WHO functional class III and IV, inhaled iloprost (median 30 mcg daily) or placebo for 12 weeks (Olschewski et al., 2002). The primary endpoint (improvement in WHO functional class and in exercise capacity at the 6-min walking test) was greater in the iloprost group compared to the placebo group. Overall, inhaled iloprost is well tolerated with flushing and cough being the most frequent side effects. Inhaled iloprost is approved by the FDA for PAH patients in WHO functional class III and IV.
Beraprost is the first orally active prostacyclin analog. A randomized controlled trial showed that this compound improved the exercise capacity in patients with PAH. However this improvement persisted only up to 3–6 months and there were no hemodynamic benefits (Galiè et al., 2002).
Endothelin Receptor Antagonists
Endothelin-1 is a potent vasoconstrictor and smooth muscle mitogen. High concentrations of endothelin-1 have been recorded in the lungs of patients with PAH (Channick et al., 2004). Two distinct receptors with different characteristics are present in the pulmonary vascular smooth muscle cells and, in part, in the endothelial cells: endothelin-A and endothelin-B receptors. Despite potential differences in receptor activity, the efficacy in PAH of the dual endothelin-A and B receptor antagonist drugs and of selective endothelin receptor antagonist compounds appears to be comparable.
Bosentan is an oral active dual endothelin-A and B receptor antagonist. It has been evaluated in several randomized controlled trials that have shown improvement in exercise capacity, hemodynamic variables and time to clinical worsening in patients with PAH (Rubin et al., 2002; Galiè et al., 2006, 2008a). The major advantage of bosentan is its oral administration. It appears to be effective in group 1 PH patients both in WHO functional class III or IV and in WHO functional class II. The initial dose is 62.5 mg twice daily and uptitrated to 125 mg twice daily after 4 weeks. Increases in hepatic aminotransferases occurred in about 10% of the patients but were found to be dose dependent and reversible after discontinuation of the drug.
Sitaxentan is a selective orally active endothelin-A receptor antagonist which has been assessed in two randomized controlled trials on PAH patients with WHO functional class II and III (Barst et al., 2004, 2006b). The studies demonstrated the efficacy of sitaxentan in improving exercise capacity and hemodynamics. The commonly used dose is 100 mg once daily. However, sitaxentan has been very recently withdrawn from the market worldwide after two cases of fatal liver injury and all ongoing clinical trials have also been discontinued.
Ambrisentan is another selective orally active endothelin-A receptor antagonist. It has shown to improve exercise tolerance, WHO functional class, hemodynamics and quality of life in patients with PAH with a durability of the effects for at least 1 year (Galiè et al., 2008b; Oudiz et al., 2009). Ambrisentan has been approved for the treatment of patients in WHO functional class II and III at a dose of 5 mg once daily which can be increased to 10 mg once daily. As reported above, the main adverse effect of the endothelin receptor antagonists is hepatotoxicity, which appears to be more severe at higher doses. According to one observational study ambrisentan may be a safe and effective alternative for patients who previously discontinued bosentan or sitaxsentan due to abnormal liver function test results (McGoon et al., 2009), suggesting the hepatotoxicity may differ among the endothelin receptor antagonists. Liver function tests should be closely monitored during therapy and endothelin receptor antagonists should be avoided in patients with moderate or severe hepatic dysfunction.
Phosphodiesterase Type-5 Inhibitors
The pulmonary vasculature contains substantial amounts of phosphodiesterase type-5, an enzyme that specifically contributes to cyclic guanosine monophosphate (cGMP) breakdown, thus decreasing smooth muscle cell capacity for vasodilatation. Inhibition of the cGMP degrading enzyme results in vasodilatation through the increase of NO which is a potent vasodilatator.
Sildenafil is an orally active inhibitor of phosphodiesterase type-5 that improves pulmonary hemodynamics, symptoms, and exercise capacity in patients with group 1 PH (Galiè et al., 2005; Pepke-Zaba et al., 2008). However its effect on mortality has not been adequately evaluated. Indeed in a randomized controlled trial patients treated with sildenafil demonstrated a significant improvement in hemodynamics and 6-min walking distance in comparison with patients treated with placebo (Galiè et al., 2005). This beneficial effects persisted during 1 year of follow-up but mortality was not reported (Galiè et al., 2005). The drug is approved for patients with WHO functional class II and III at a dose of 20 mg three times daily. However the durability of effect up to 1 year has been demonstrated only with the dose of 80 mg three times daily. The side effects of sildenafil are mainly related to vasodilatation (flushing, headache). It should be used with caution in patients with arterial hypotension.
Tadalafil is a once-daily dispensed phosphodiesterase type-5 inhibitor currently approved for the treatment of erectile dysfunction which appears to improve outcomes in patients with group 1 PH. A randomized clinical trial has shown favorable results on exercise capacity, hemodynamics, and time to clinical worsening in patients treated with the largest dose of tadalafil (40 mg daily) compared to patients receiving placebo (Galiè et al., 2009c).
Combination Therapy
It has been proposed that combining pharmacologic agents with different mechanisms of action may produce an additive effect or may induce the same effect at lower doses of each agent. Clinical studies have begun to evaluate combination therapy.
Bosentan added to either epoprostenol or treprostinil
Limited experience suggests that bosentan can be used safely and effectively added to epoprostenol or subcutaneous treprostinil therapy (Humbert et al., 2004b; Benza et al., 2008).
Bosentan plus iloprost
The effect of combining bosentan with iloprost is less clear. Early observational studies suggested that the combination was both safe and effective when bosentan was added to preexisting inhaled iloprost therapy (Hoeper et al., 2003). However, a subsequent trial that randomly assigned 40 patients with idiopathic PAH to receive bosentan alone or bosentan plus iloprost for 12 weeks, demonstrated no difference in the 6-min walking distance, the trial’s primary endpoint (Hoeper et al., 2006a).
Treprostinil added to either bosentan or sildenafil
The addition of inhaled treprostinil may improve the exercise capacity and quality of life of patients with persistent symptoms despite bosentan or sildenafil therapy. In a randomized controlled trial, 235 patients with group 1 PH, a WHO functional class III or IV, and a 6-min walking distance of only 200–450 m despite bosentan or sildenafil therapy were randomly assigned to receive either inhaled treprostinil or placebo for 12 weeks (McLaughlin et al., 2010). The treprostinil group had a larger improvement in their 6-min walking distance and quality of life, but there were no differences in the time to clinical worsening, dyspnea, or WHO functional class.
Sildenafil added to epoprostenol
The addition of sildenafil to long-term epoprostenol therapy improves clinical outcomes. A trial randomly assigned 267 patients with group 1 PH who were receiving epoprostenol to have sildenafil or placebo added for 16 weeks (Simonneau et al., 2008). Sildenafil improved hemodynamic parameters, exercise capacity, quality of life, and time to clinical worsening, compared to placebo but there was no difference in dyspnea.
Sildenafil added to bosentan
A prospective cohort study followed 25 patients with group 1 PH who were initially treated with bosentan monotherapy, but developed clinical deterioration and had sildenafil added (Mathai et al., 2007). Clinical improvement occurred after the addition of sildenafil, as measured by symptoms, exercise capacity, and WHO functional class. Improvement was more frequent and of greater magnitude in patients with idiopathic PAH, compared to patients with scleroderma-associated PAH.
Sildenafil added to iloprost
The combination of iloprost plus sildenafil may improve outcomes compared to either agent alone (Ghofrani et al., 2002a, 2003).
There are many open questions regarding combination therapy, including the choice of combination agents, the optimal timing (initial combination in naive patients or sequential combination according to the response to the first drug), when to switch, and when to combine. Combination therapy of established PAH drugs is recommended for patients not responding adequately to monotherapy, but combination therapy should be instituted by expert centers only. Whether the response to monotherapy is sufficient or not can only be decided on an individual basis. This is judged in an individual patient who, despite monotherapy and optimized background treatment, has an inadequate clinical response (Galiè et al., 2009a).
Non-Medical Treatment
Therapeutic options in PH other than medical therapies are mainly represented by surgical treatment which should be considered only in selected patients.
Atrial Septostomy
The creation of a right-to-left shunt by atrial septostomy can decompress the right heart chambers and increase left ventricular preload. The goal is to increase systemic blood flow by by-passing the pulmonary vascular obstruction. The consequent arterial desaturation that follows the procedure is offset by increased CO and augmentation of systemic oxygen delivery (Reichenberger et al., 2003). Atrial septostomy may be considered in individuals with refractory severe PAH and right heart failure, despite aggressive specific therapy and maximal diuretic therapy (Doyle et al., 2004). It may also be considered in patients who have signs of impaired systemic blood flow (such as syncope) due to reduced left heart filling. Stepwise balloon dilatation is the procedure of choice (Keogh et al., 2009). However, procedure-related mortality may be as high as 15–20% (Sandoval et al., 2001; Reichenberger et al., 2003) and it is difficult to predict which patients will benefit and which will deteriorate after this therapy.
Transplantation
The advent of disease-specific therapy for severe PAH has reduced patient referral for lung transplant programs (Keogh et al., 2009). However, the long-term outcomes of medically treated patients remains uncertain and transplantation should remain an important option for those who fail on such therapy. Studies indicate that up to 25% of patients with idiopathic PAH may fail to improve on disease-specific therapy and the prognosis of patients who remain in WHO functional class III or IV is poor (McLaughlin et al., 2002; Sitbon et al., 2002). Thus transplantation is considered by some to be the final effective treatment for selected patients with idiopathic PAH. Bilateral lung or heart–lung transplantation is the procedure of choice (Keogh et al., 2009). The 3 year survival of patients who had a lung or heart–lung transplant for idiopathic PAH is approximately 50% (Doyle et al., 2004; Trulock et al., 2007). The timing of transplantation is critical. Guidelines for when to refer a patient for transplant evaluation are as follows (Orens et al., 2006): WHO functional class III or IV; mean right atrial pressure > 10 mmHg; PAPm > 50 mmHg; CI < 2.5 L/min/m2; failure to improve functionally despite medical therapy; rapidly progressive disease.
Transplant organizations and organ allocation policies are influenced by medical, ethical, geographical, and political factors, and systems vary from country to country. In the United States, since May 2005, the order of patients on the waiting list for lung transplantation has been based on a Lung Allocation Score (LAS) that was developed to address high waiting list mortality and progressively earlier placement of patients on the waiting list. The three main objectives of LAS include: (1) reducing the number of deaths on the lung transplant waiting list; (2) increasing transplant benefit for lung recipients; (3) ensuring the efficient and equitable allocation of lungs to active transplant candidates. The LAS system works by assigning a score ranging from 0 to 100 to all candidates older than age 12. It is a weighted combination of the predicted risk of death during the following year on the waiting list and the predicted likelihood of survival during the first year following transplantation (Davis and Garrity, 2007). Higher scores represent higher urgency and greater potential transplant benefit. In the 2 years following implementation of the LAS system, wait-list times decreased and the mean LAS score of transplant recipients increased, consistent with a greater urgency for transplantation; the total number of patients transplanted also increased (Iribarne et al., 2009).
Thromboendarterectomy
This treatment is specifically indicated for patients with group 4 PH, i.e., patients with CTEPH. CTEPH is one of the most prevalent forms of PH. Nevertheless its overall prevalence is very difficult to determine since not all patients have a history of acute pulmonary embolism, acute embolism may be clinically silent, and finally CTEPH may develop in the absence of previous pulmonary embolism (Hoeper et al., 2006b; Galiè et al., 2009a). In these cases the disease is probably initiated by thrombotic or inflammatory lesions in the pulmonary vasculature and then a process of pulmonary vascular remodeling is started which self-perpetuates the progression of PH (Rubin et al., 2006).
Oral anticoagulation is the most important medical therapy for these patients but pulmonary thromboendarterectomy remains the treatment of choice as it is a potentially curative option. However the surgical approach is possible only in selected patients (Dartevelle et al., 2004). Indeed, the decision to proceed to pulmonary thromboendarterectomy in patients suffering from CTEPH is based upon both objective and subjective factors, which are carefully defined during the preoperative evaluation (Moser et al., 1992; Dunning and McNeil, 1999). The first and most important criterion for potential surgical intervention is the accessibility of the thrombi, as defined by angiography. Present surgical techniques allow removal of chronic thrombi whose proximal location extends to the main, lobar, and segmental arteries. The second criterion involves the presence of a significant hemodynamic or ventilatory impairment as a consequence of the chronic thromboembolic pulmonary vascular obstruction. The third criterion is the absence of co-morbid conditions.
Approximately 4,000 operations of pulmonary thromboendarterectomy have been performed worldwide and the results are excellent with post-operative mortality rates ranging from <5 to 10% (Keogh et al., 2009). A significant trend of improved survival with increasing levels of experience of the surgical team has been shown by Mayer (2010) who reported an early mortality of 10.9% in 210 operations performed from 1994 to 2003 and of 5.2% in 210 operations performed from 2003 to 2009. Other observational studies confirmed that perioperative mortality decreased in the last decades (Jamieson et al., 2003; Condliffe et al., 2008). In one study of 500 consecutive patients who underwent thromboendarterectomy, perioperative mortality was 16% before 1990, 7% between 1990 and 1999, and 4% between 1998 and 2002 (Jamieson et al., 2003). Long-term mortality is low among patients who survive the 3-month post-operative period. In an observational study that followed 137 such patients, only six (4%) died during the 4-year follow-up period (Corsico et al., 2008). A similar study demonstrated 1- and 3-year mortality rates of 1 and 6%, respectively, among patients who survived the post-operative period (Condliffe et al., 2008).
Treatment Algorithms
Different therapeutic approaches can be considered in the management of patients with PH. Therapy can be established on the basis of both the clinical classification and the functional class. It is also possible to adopt a goal-oriented therapy in which the timing of treatment escalation is determined by inadequate response to known prognostic indicators.
Therapy According to Clinical Classification
There are special considerations and different therapeutic indications for each group of PH, as shown in Table 5.
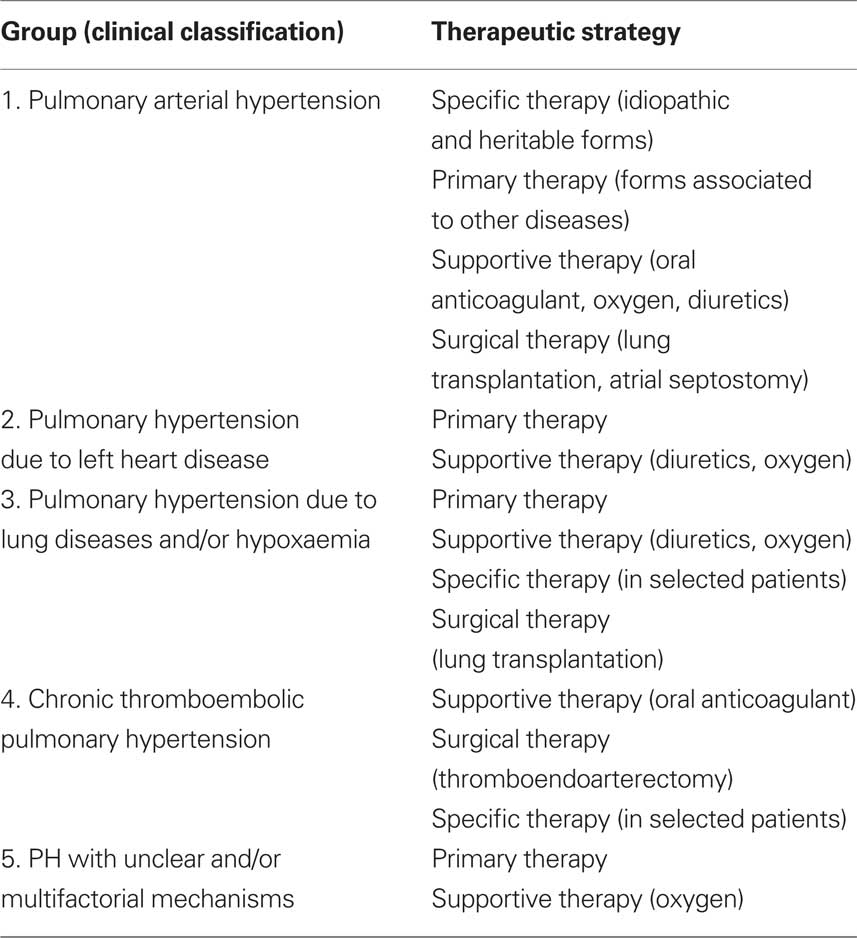
Table 5. Therapy according to the clinical classification.
Group 1
Treatment in patients of this group is based essentially on specific therapy because there are no effective primary therapies especially for patients with idiopathic PAH. Primary therapy is possible only in PAH associated with other diseases. Supportive therapy with oxygen to correct hypoxemia and diuretics to treat fluid retention, may be used, as needed. It is generally accepted that anticoagulation is indicated in patients with idiopathic PAH (Barst et al., 2009; Galiè et al., 2009a; McLaughlin et al., 2009).
Group 2
Primary therapy is the main treatment in patients of this group and consists in the treatment of the underlying heart disease. Specific therapy should be avoided in most of these patients because it may be harmful, as demonstrated in a randomized trial that showed the detrimental impact of epoprostenol on mortality of patients with PH and severe left ventricular dysfunction (McLaughlin et al., 2009). A possible explanation of this effect includes the inability of a compromised left heart to handle the increased flow across a newly dilated pulmonary vascular bed (Badesch et al., 2009).
Group 3
Primary therapy represents the main treatment also for patients of this group. Indeed, any treatment able to delay the natural progression of pulmonary disease and to reduce the loss of pulmonary function seems to have a favorable impact on PH. Moreover, the correction of hypoxemia with supplemental oxygen plays an important role in these patients. Continuous oxygen therapy improves survival in patients with COPD and PaO2 below 55 mmHg, despite seemingly mild effects on pulmonary hemodynamics (Ashutosh and Dunsky, 1987). Specific therapy is not approved by the US FDA for patients with group 3 PH and several guideline panels recommend against its use in this population, except in the context of a clinical trial (Galiè et al., 2009a; Hoeper et al., 2009). Despite this, it is occasionally considered for patients who remain WHO functional class III or IV despite correction of hypoxemia and optimization of therapy of the underlying disease, especially if the severity of the PH is out of proportion to the severity of the parenchymal lung disease. Specific therapy for patients with group 3 PH should administered cautiously due to its potential to worsen ventilation-perfusion mismatch and increase hypoxemia (Ghofrani et al., 2002b; Blanco et al., 2010).
Group 4
Anticoagulation is primary medical therapy for patients with group 4 PH and its value is based on the clinical evidence that anticoagulation prevents recurrent pulmonary embolism. Nevertheless, data suggesting that anticoagulation is beneficial in patients with CTEPH are lacking. Surgical thromboendarterectomy is primary surgical therapy for selected patients (Keogh et al., 2009). Specific therapy can be considered for patients with group 4 PH who remain WHO functional class III or IV even after anticoagulation or thromboendarterectomy. Pharmacologic therapy can also act as a bridge to surgical intervention or can be used in patients with inoperable disease and those with persistent or recurrent PH after pulmonary thromboendarterectomy (Piazza and Goldhaber, 2011). Epoprostenol, and sildenafil have been tested in patients with CTEPH with some beneficial hemodynamic effects (Nagaya et al., 2003; Suntharalingam et al., 2008). The efficacy of bosentan has been investigated in a randomized controlled trial (Jais et al., 2008). Bosentan significantly improved the pulmonary vascular resistance and CI, compared to placebo. However, exercise capacity (measured by a 6-min walking test) did not improve.
Group 5
Primary therapy, directed at the underlying cause, represents the main treatment in patients of this group. Small studies have addressed the role of specific therapy for patients with PH related to sarcoidosis. One series of eight patients described a favorable response to intravenous epoprostenol in most patients (Fisher et al., 2006).
Therapy According to Functional Class
A therapeutic approach based on severity of the disease is actually recommended in patients with PAH as shown in Figure 2 (Galiè et al., 2009a).
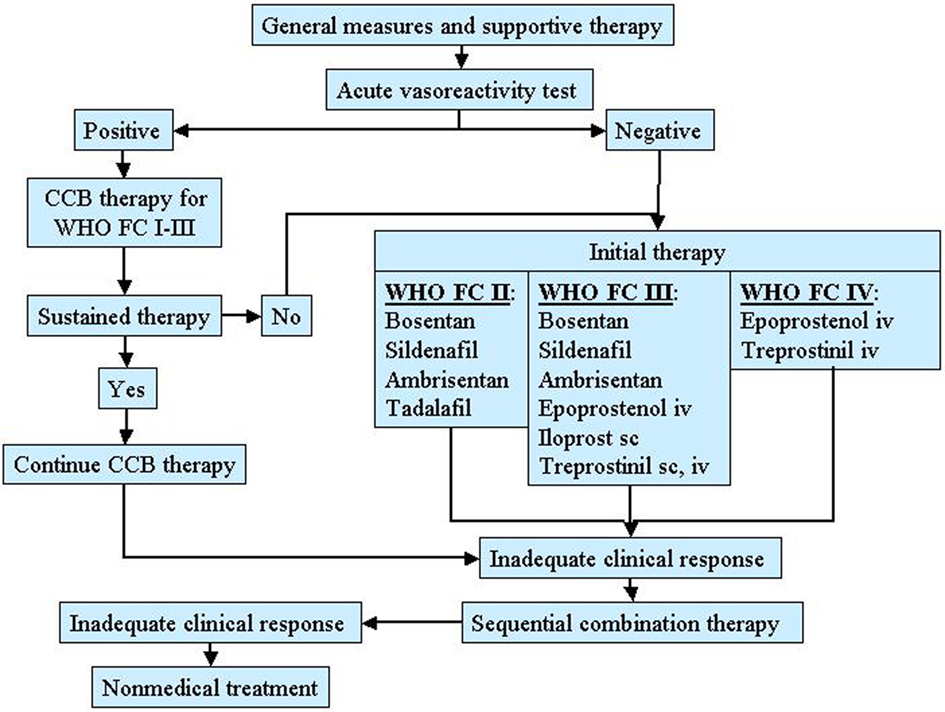
Figure 2. Therapeutic approach for pulmonary arterial hypertension (PAH) based on the WHO functional class. CCB, calcium channel blocker.
Functional class I
Considering the mild functional limitation, there is not a clear indication to treatment in these patients except for primary therapy if there is an associated disease causing PAH. In patients with idiopathic PAH and a positive response to the vasoreactivity test, CCBs may be effective. Supportive therapy is rarely indicated.
Functional class II, III, IV
These patients should to be evaluated for specific therapy. In addition, the need for oxygen, diuretic, and anticoagulant therapy should be assessed. All patients selected for specific therapy should undergo RHC and vasoreactivity test. For patients who have a positive vasoreactivity test, a trial of CCB therapy is recommended. Patients who respond to such therapy should be reassessed after 3–6 months of treatment. For patients who have a negative vasoreactivity test or fail CCB therapy, specific therapy with prostanoids, endothelin receptor antagonists, and phosphodiesterase 5 inhibitors is indicated. The preferred agent is related to the functional severity of the disease and the choice should also be done according to the physician experience, drug’s availability, and patient preference. For patients who are WHO functional class II, acceptable first-line agents include oral bosentan, oral sildenafil, oral ambrisentan. For patients who are WHO functional class III, acceptable first-line agents include oral bosentan, oral sildenafil, oral ambrisentan, inhaled iloprost, intravenous or subcutaneous treprostinil, intravenous epoprostenol. For patients who are WHO functional class IV intravenous epoprostenol has been suggested. An inadequate clinical response to a single agent represents an indication for a combination therapy. Finally, atrial septostomy and lung transplantation are reserved for patients refractory to medical therapy.
Goal-Oriented Therapy
This approach concerns patients with PAH in whom the objective of treatment is to reduce functional impairment and prolong life (Sitbon and Galiè, 2010). Goal-oriented therapy uses known prognostic indicators as treatment targets thus facilitating early intervention and therapeutic escalation before patients deteriorate. A crucial role in this strategy is played by the identification of parameters that correlate with the risk of deterioration and mortality. Several data indicate that WHO functional class is the most appropriate prognostic marker, with 6-min walking distance and some hemodynamic parameters representing alternatives (Sitbon and Galiè, 2010). The goal-oriented treatment algorithm proposed by Hoeper et al. (2005) identifies 6-min walking distance, peak oxygen consumption, and peak systolic blood pressure as prognostic indicators and set values for each of them that act as goals (Figure 3). According to this algorithm, patients were started on first-line bosentan monotherapy: if patients met the three treatment goals during follow-up, monotherapy was continued but, if a goal was not met, additional therapy was added. The authors reported that after 3 years of follow-up survival improved by using this goal-oriented therapy (80 versus 63% of historical controls; Hoeper et al., 2005).
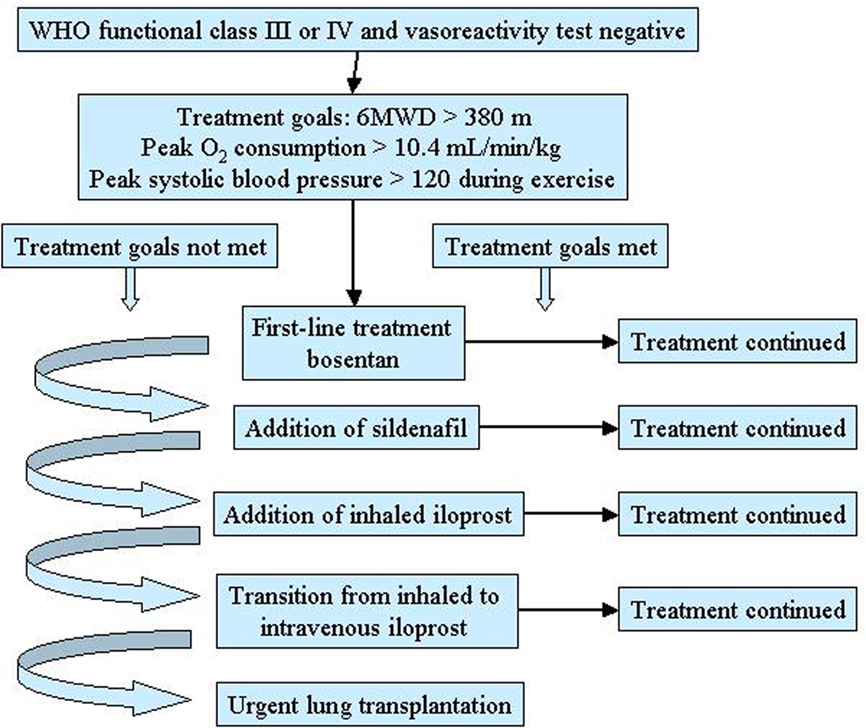
Figure 3. Goal-oriented treatment algorithm for patients with pulmonary arterial hypertension (PAH). 6MWD, 6-min walking distance. Reproduced with permission from Hoeper et al. (2005).
New Classes of Drugs in Development
Despite the progress in the treatment of PH, the functional limitation and the survival of these patients remain unsatisfactory and there is no cure for PAH. Therefore the search for an “ideal” treatment still goes on, triggered by research aiming to identify new molecular pathways which are important in both initiating and aggravating the disease process.
The following is a brief presentation of new classes of drugs tested in experimental models and already subject to ongoing clinical trials.
Rho-Kinase Inhibitors: Fasudil
Rho-kinase plays an important role in PAH: it enhances the contraction of the vascular smooth muscle cells through inhibition of myosin phosphatase and activation of its downstream effectors. On the contrary, an over expression of the endothelial NO synthase (eNOS) and a lower migration of inflammatory cells have been shown when Rho-kinase is inhibited (Fukumoto et al., 2007).
A recent clinical study showed a Rho-kinase activation in patients with PAH (Do et al., 2009). Rho-kinase activity in circulating neutrophils was significantly increased in PAH patients compared with controls. Analogously, the expression and activity of Rho-kinase in isolated lung tissues were significantly increased in PAH patients compared with controls. Finally, significant correlations were noted between Rho-kinase activity and the severity and duration of PAH (Do et al., 2009).
In animal models, the treatment with Rho-kinase inhibitors seems to improve the endothelium dysfunction enhancing eNOS expression, suppressing hypercontraction and proliferation of the vascular smooth muscle cells and reducing the migration of inflammatory cells (Fukumoto et al., 2007).
A more recent study compared fasudil, a potent and selective Rho-kinase inhibitor, to bosentan and sildenafil in a rat model of monocrotaline (MCT) induced PAH (Mouchaers et al., 2010). Both fasudil in monotheraphy and fasudil in association with sildenafil or bosentan preserved heart rate, stroke volume and RV contractility, and reduced pulmonary vascular resistance and RV dilatation. Fasudil significantly lowered PAPm by reducing pulmonary vascular remodeling and RV hypertrophy. Combining bosentan or sildenafil with fasudil had no synergistic effects (Mouchaers et al., 2010). These results suggest that Rho-kinase inhibitors might be a novel therapeutic agent in PAH. A schematic representation of the mechanisms of pulmonary dilatation in response to some specific drugs and Rho-kinase inhibitors is illustrated in Figure 4.
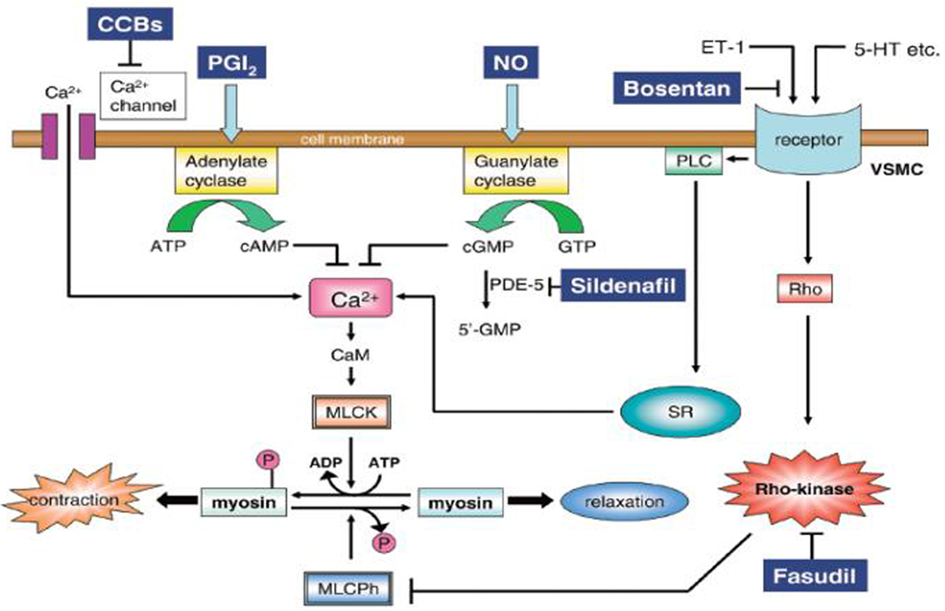
Figure 4. Mechanism of pulmonary dilatation in response to some specific drugs and Rho-kinase inhibitors. 5-HT, serotonin; CaM, calmodulin; CCBs, calcium channel blockers; ET-1, endothelin-1; MLCK, myosin light chain kinase; MLCPh, myosin light chain phosphatase; NO, nitric oxide; PDE-5, phosphodiesterase-5; VSMC, vascular smooth muscle cell; PGI2, prostacyclin; PLC, phospholipase C; SR, sarcoplasmic reticulum. Reproduced with permission from Fukumoto et al. (2007).
Stimulator and Activator of Soluble Guanylate Cyclase: Riociguat
As well known, NO synthetized by eNOS is a potent vasodilator and is considered to play an important role in regulating pulmonary vascular tone. The downstream effector of NO is soluble guanylate cyclase (sGC), which synthetizes the secondary messenger cGMP. Increased levels of cGMP result in acute vasodilatation and reduction in platelet aggregation. Its chronic effects are prevention of the pulmonary vascular remodeling as well as reduction in the RV afterload.
An animal model study conducted on mice with PAH induced by chronic hypoxia and rats with PAH induced by MCT, showed that both the sGC stimulator “Bay 41-2272” and sGC activator “Bay 58-2667” could reverse PH and pulmonary vascular remodeling (Figure 5; Dumitrascu et al., 2006).
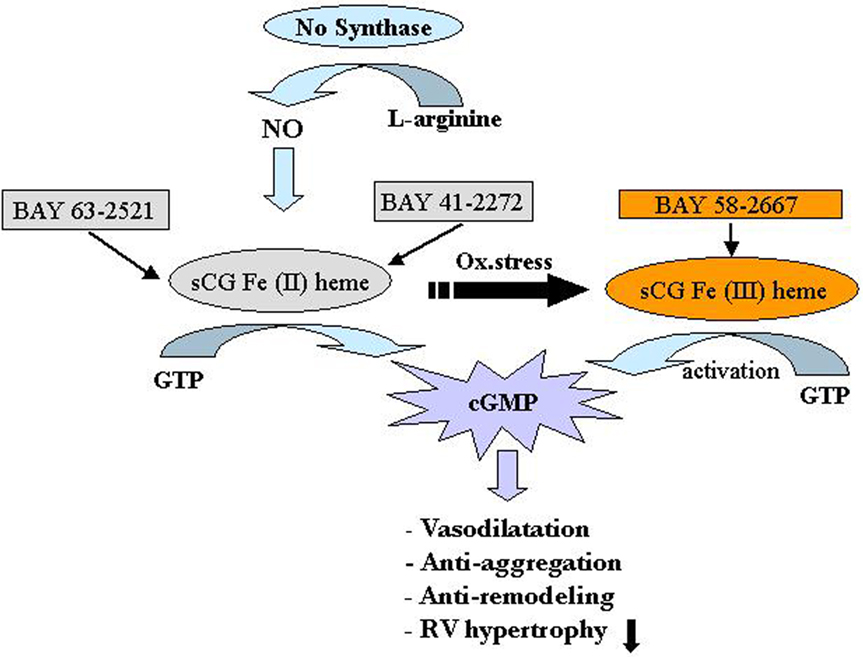
Figure 5. Soluble guanylate cyclase (sGC) as a target in pulmonary arterial hypertension (PAH). The sGC stimulator Bay41-2272 and the activator Bay58-2667 increase cGMP production, thereby regulating smooth muscle function. Bay41-2272 is an NO-dependent stimulator acting preferentially on the physiological form of sGC containing the iron II heme [Fe(II) heme] (left). In contrast, Bay58-2667 is a NO-independent activator preferably addressing the oxidized (and therefore NO-insensitive) iron III heme form [Fe(III) heme] of sGC (right). Increased levels of cGMP then result acutely in vasodilatation and antiaggregation and result chronically in antiremodeling of the vascular wall as well as unloading of the right ventricle. cGMP, cyclic guanosine monophosphate; GTP, guanosine triphosphate; NO, nitric oxide; Ox. stress, oxidative stress; RV, right ventricle. Reproduced with permission from Dumitrascu et al. (2006).
Riociguat is an oral stimulator of the NO receptor sCG which has demonstrated vasodilatory and antiremodeling properties in preclinical studies (Ghofrani and Grimminger, 2009).
A recent phase II trial is the first report that evaluated safety, tolerability and efficacy of riociguat on patients with moderate to severe PH of different etiology (PAH, CTEPH, PH due to interstizial lung disease; Grimminger et al., 2009). Riociguat had a favorable safety profile at single dose ≤ 2.5 mg. It was well tolerated and significantly improved pulmonary hemodynamic parameters measured by RHC in a dose dependent manner and to a greater extent than inhaled NO. Although riociguat had also systemic effects and showed no pulmonary selectivity, mean systolic pressure remained > 110 mmHg.
Further investigations are necessary but riociguat can be considered an emerging option in PH therapy, included in patients with CTEPH (Kim, 2010).
Serotonin Antagonists
In the 1960s, serotonin was associated with PAH caused by certain diet pills (anorexigenic drugs aminorex and dexfenfluramine), but, more recently, it has been the subject of a renewed interest in the field of PAH (MacLean and Dempsie, 2010). Serotonin can be synthetized in the pulmonary endothelium with the rate-limiting step being the activity of the tryptophan hydroxylase1 (Tph1). Then it can pass into the underlying pulmonary smooth muscle cells through the serotonin transporter (SERT) to initiate proliferation and/or it can activate serotonin receptors on pulmonary smooth muscle cells to evoke proliferation and/or contraction. Serotonin may also mediate pulmonary fibroblast proliferation via the SERT and/or serotonin receptors. This is the basis of the so called “serotonin hypothesis” of PAH (MacLean, 2007). The activity of serotonin receptors, SERT and Tph1 can all be elevated in clinical and experimental PAH and each offers a potentially unique therapeutic target. Both serotonin receptor antagonists and serotonin-transporter inhibitors are currently under consideration in order to verify the relevance of this new therapeutic strategy specifically directed against vascular proliferation.
Phosphodiesterase-1 Inhibitors
PH is associated with increased vascular resistance due to sustained contraction and enhanced proliferation of pulmonary vascular smooth muscle cells. The abnormal tone and remodeling in the pulmonary vasculature may relate, at least in part, to decreased cyclic nucleotide levels. Cyclic nucleotide phosphodiesterases (PDEs) catalyze the hydrolysis of both cyclic adenosine monophosphate (cAMP) and cGMP. Several studies analyzed the hypothesis that pulmonary vascular smooth muscle cells isolated from patients with either idiopathic PAH or secondary PH increased expression and activity of PDE isoforms so reducing the responsiveness to agents that raise cellular cAMP. It has been demonstrated that the expression of several families of PDE was enhanced in pulmonary vascular smooth muscle cells of patients with PH compared with controls (Murray et al., 2007). Consistent with this enhanced expression of PDEs, agonist-stimulated cAMP levels were significantly reduced in pulmonary vascular smooth muscle cells of both idiopathic PAH and secondary PH unless a PDE inhibitor was present. The use of specific PDE inhibitors revealed that an increase in PDE1 and PDE3 activity largely accounted for reduced agonist-induced cAMP levels and increased proliferation in pulmonary vascular smooth muscle cells. Treatment with PDE1C-targeted small interference RNA enhanced cAMP accumulation and inhibited cellular proliferation to a greater extent in pulmonary vascular smooth muscle cells of patients with PH than of controls. The results imply that an increase in PDE isoforms, in particular PDE1C, contributes to decreased cAMP and increased proliferation of pulmonary vascular smooth muscle cells in patients with PH. Therefore, PDE1 isoforms may provide novel targets for the treatment of both idiopathic PAH and secondary PH.
Vasoactive Intestinal Peptide (VIP)
A further new concept for treatment of PH is based on VIP, a neuropeptide primarily functioning as a neurotransmitter that acts as a potent systemic and pulmonary vasodilator. Rationale for the use of VIP is the finding of a deficiency of this peptide in serum and lung tissue of patients with idiopathic PAH with an upregulation of the corresponding receptor sites (Petkov et al., 2003). Consequently, the use of VIP results in a substantial improvement of hemodynamic parameters without significant side effects. It has been shown that VIP decreased PAPm in eight patients, increased CO and mixed venous oxygen saturation (Petkov et al., 2003). These data provide enough proof for further investigation of VIP as a novel therapy in idiopathic PAH.
Conclusion
Current available drugs improve exercise capacity and functional class in patients with PH. Randomized trials have also shown improvement in hemodynamic parameters and time to clinical worsening. Although the impact on mortality has been less well established, advances in therapeutic strategies have allowed a better prognosis especially for patients with PAH. It is likely that combination therapy will be the cornerstone of PAH treatment in the future and some novel agents will become part of the therapeutic armamentarium to improve the quality of life, slow the progression of disease and improve survival. Future directions are oriented to better elucidate the pathogenesis of PAH which remains incompletely understood. Indeed, pathophysiology and genetics of this disease can represent a link to new treatments. Although it is clear that platelets, fibroblasts, and circulating cells are involved in the progression of PAH, changes in pulmonary arterial smooth muscle cells and in pulmonary arterial endothelial cells resulting from multiple genetic and acquired defects are probably the major cause for the onset of the disease (Yuan and Rubin, 2005). Accordingly, efforts directed at developing effective therapeutic strategies should target genes, molecular mechanisms, and pathogenetic pathways involved in the disease.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ashutosh, K., and Dunsky, M. (1987). Noninvasive tests for responsiveness of pulmonary hypertension to oxygen. Prediction of survival in patients with chronic obstructive lung disease and cor pulmonale. Chest 92, 393–399.
Badesch, D. B., Abman, S. H., Simonneau, G., Rubin, L. J., and McLaughlin, V. V. (2007). Medical therapy for pulmonary arterial hypertension: updated ACCP evidence-based clinical practice guidelines. Chest 131, 1917–1928.
Badesch, D. B., Champion, H. C., Sanchez, M. A., Hoeper, M. M., Loyd, J. E., Manes, A., McGoon, M., Naeije, R., Olschewski, H., Oudiz, R. J., and Torbicki, A. (2009). Diagnosis and assessment of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 54(Suppl. 1), S55–S66.
Barst, R. J., Galiè, N., Naeije, R., Simonneau, G., Jeffs, R., Arneson, C., and Rubin, L. J. (2006a). Long-term outcome in pulmonary arterial hypertension patients treated with subcutaneous treprostinil. Eur. Respir. J. 28, 1195–1203.
Barst, R. J., Langleben, D., Badesch, D., Frost, A., Lawrence, E. C., Shapiro, S., Naeije, R., and Galiè, N. (2006b). Treatment of pulmonary arterial hypertension with the selective endothelin-A receptor antagonist sitaxsentan. J. Am. Coll. Cardiol. 47, 2049–2056.
Barst, R. J., Gibbs, J. S., Ghofrani, H. A., Hoeper, M. M., McLaughlin, V. V., Rubin, L. J., Sitbon, O., Tapson, V. F., and Galiè, N. (2009). Updated evidence-based treatment algorithm in pulmonary arterial hypertension. J. Am. Coll. Cardiol. 54(Suppl. 1), S78–S84.
Barst, R. J., Langleben, D., Frost, A., Horn, E. M., Oudiz, R., Shapiro, S., McLaughlin, V., Hill, N., Tapson, V. F., Robbins, I. M., Zwicke, D., Duncan, B., Dixon, R. A., and Frumkin, L. R. (2004). Sitaxsentan therapy for pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 169, 441–447.
Barst, R. J., Rubin, L. J., Long, W. A., McGoon, M. D., Rich, S., Badesch, D. B., Groves, B. M., Tapson, V. F., Bourge, R. C., and Brundage, B. H. (1996). A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. The Primary Pulmonary Hypertension Study Group. N. Engl. J. Med. 334, 296–302.
Benza, R. L., Rayburn, B. K., Tallaj, J. A., Pamboukian, S. V., and Bourge, R. C. (2008). Treprostinil-based therapy in the treatment of moderate-to-severe pulmonary arterial hypertension: long-term efficacy and combination with bosentan. Chest 134, 139–145.
Blanco, I., Gimeno, E., Munoz, P. A., Pizarro, S., Gistau, C., Rodriguez-Roisin, R., Roca, J., and Barbera, J. A. (2010). Hemodynamic and gas exchange effects of sildenafil in patients with chronic obstructive pulmonary disease and pulmonary hypertension. Am. J. Respir. Crit. Care Med. 181, 270–278.
Boutet, K., Montani, D., Jais, X., Yaici, A., Sitbon, O., Simonneau, G., and Humbert, M. (2008). Therapeutic advances in pulmonary arterial hypertension. Ther. Adv. Respir. Dis. 2, 249–265.
Channick, R. N., Sitbon, O., Barst, R. J., Manes, A., and Rubin, L. J. (2004). Endothelin receptor antagonists in pulmonary arterial hypertension. J. Am. Coll. Cardiol. 43(Suppl. S12), 62S–67S.
Condliffe, R., Kiely, D. G., Gibbs, J. S., Corris, P. A., Peacock, A. J., Jenkins, D. P., Hodgkins, D., Goldsmith, K., Hughes, R. J., Sheares, K., Tsui, S. S., Armstrong, I. J., Torpy, C., Crackett, R., Carlin, C. M., Das, C., Coghlan, J. G., and Pepke-Zaba, J. (2008). Improved outcomes in medically and surgically treated chronic thromboembolic pulmonary hypertension. Am. J. Respir. Crit. Care Med. 177, 1122–1127.
Corsico, A. G., D’Armini, A. M., Cerveri, I., Klersy, C., Ansaldo, E., Niniano, R., Gatto, E., Monterosso, C., Morsolini, M., Nicolardi, S., Tramontin, C., Pozzi, E., and Vigano, M. (2008). Long-term outcome after pulmonary endarterectomy. Am. J. Respir. Crit. Care Med. 178, 419–424.
Dartevelle, P., Fadel, E., Mussot, S., Chapelier, A., Herve, P., de Perrot, M., Cerrina Ladurie, F. L., Lehouerou, D., Humbert, M., Sitbon, O., and Simonneau, G. (2004). Chronic thromboembolic pulmonary hypertension. Eur. Respir. J. 23, 637–648.
Davis, S. Q., and Garrity, E. R. Jr. (2007). Organ allocation in lung transplant. Chest 132, 1646–1651.
de Man, F. S., Handoko, M. L., Groepenhoff, H., van ′t Hul, A. J., Abbink, J., Koppers, R. J., Grotjohan, H. P., Twisk, J. W., Bogaard, H. J., Boonstra, A., Postmus, P. E., Westerhof, N., van der Laarse, W. J., and Vonk-Noordegraaf, A. (2009). Effects of exercise training in patients with idiopathic pulmonary arterial hypertension. Eur. Respir. J. 34, 669–675.
Do, E. Z., Fukumoto, Y., Takaki, A., Tawara, S., Ohashi, J., Nakano, M., Tada, T., Saji, K., Sugimura, K., Fujita, H., Hoshikawa, Y., Nawata, J., Kondo, T., and Shimokawa, H. (2009). Evidence for Rho-kinase activation in patients with pulmonary arterial hypertension. Circ. J. 73, 1731–1739.
Doyle, R. L., McCrory, D., Channick, R. N., Simonneau, G., and Conte, J. (2004). Surgical treatments/interventions for pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines. Chest 126(Suppl. 1), 63S–71S.
Dumitrascu, R., Weissmann, N., Ghofrani, H. A., Dony, E., Beuerlein, K., Schmidt, H., Stasch, J. P., Gnoth, M. J., Seeger, W., Grimminger, F., and Schermuly, R. T. (2006). Activation of soluble guanylate cyclase reverses experimental pulmonary hypertension and vascular remodeling. Circulation 113, 286–295.
Dunning, J., and McNeil, K. (1999). Pulmonary thromboendarterectomy for chronic thromboembolic pulmonary hypertension. Thorax 54, 755–756.
Fisher, K. A., Serlin, D. M., Wilson, K. C., Walter, R. E., Berman, J. S., and Farber, H. W. (2006). Sarcoidosis-associated pulmonary hypertension: outcome with long-term epoprostenol treatment. Chest 130, 1481–1488.
Fukumoto, Y., Tawara, S., and Shimokawa, H. (2007). Recent progress in the treatment of pulmonary arterial hypertension: expectation for Rho-kinase inhibitors. Tohoku J. Exp. Med. 211, 309–320.
Galiè, N., Beghetti, M., Gatzoulis, M. A., Granton, J., Berger, R. M. F., Lauer, A., Chiossi, E., Landzberg, M. for the Bosentan Randomized Trial of Endothelin Antagonist Therapy. (2006). Bosentan therapy in patients with Eisenmenger syndrome: a multicenter, double-blind, randomized, placebo-controlled study. Circulation 114, 48–54.
Galiè, N., Ghofrani, H. A., Torbicki, A., Barst, R. J., Rubin, L. J., Badesch, D., Fleming, T., Parpia, T., Burgess, G., Branzi, A., Grimminger, F., Kurzyna, M., and Simonneau, G. (2005). Sildenafil citrate therapy for pulmonary arterial hypertension. N. Engl. J. Med. 353, 2148–2157.
Galiè, N., Humbert, M., Vachiery, J. L., Vizza, C. D., Kneussl, M., Manes, A., Sitbon, O., Torbicki, A., Delcroix, M., Naeije, R., Hoeper, M., Chaouat, A., Morand, S., Besse, B., and Simonneau, G. (2002). Effects of beraprost sodium, an oral prostacyclin analogue, in patients with pulmonary arterial hypertension: a randomised, double-blind placebo-controlled trial. J. Am. Coll. Cardiol. 39, 1496–1502.
Galiè, N., Manes, A., and Branzi, A. (2003). Prostanoids for pulmonary arterial hypertension. Am. J. Respir. Med. 2, 123–137.
Galiè, N., Rubin, L. J., Hoeper, M., Jansa, P., Al-Hiti, H., Meyer, G. M. B., Chiossi, E., Kusic-Pajic, A., and Simonneau, G. (2008a). Treatment of patients with mildly symptomatic pulmonary arterial hypertension with bosentan (EARLY study): a double-blind, randomised controlled trial. Lancet 371, 2093–2100.
Galiè, N., Olschewski, H., Oudiz, R. J., Torres, F., Frost, A., Ghofrani, H. A., Badesch, D. B., McGoon, M. D., McLaughlin, V. V., Roecker, E. B., Gerber, M. J., Dufton, C., Wiens, B. L., and Rubin, L. J. (2008b). Ambrisentan for the treatment of pulmonary arterial hypertension: results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation 117, 3010–3019.
Galiè, N. the Task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS). (2009a). Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur. Heart J. 30, 2493–2537.
Galiè, N., Manes, A., Negro, L., Palazzini, M., Bacchi-Reggiani, M. L., and Branzi, A. (2009b). A meta-analysis of randomised controlled trial in pulmonary arterial hypertension. Eur. Heart J. 30, 394–403.
Galiè, N., Brundage, B., Ghofrani, A., Oudiz, R., Simonneau, G., Safdar, Z., Shapiro, R. S., White, J., Chan, M., Beardsworth, A., Frumkin, L. R., and Barst, R. (2009c). Tadalafil therapy for pulmonary arterial hypertension. Circulation 119, 2894–2903.
Ghofrani, H. A., and Grimminger, F. (2009). Soluble guanylate cyclase stimulation: an emerging option in pulmonary hypertension therapy. Eur. Respir. Rev. 18, 35–41.
Ghofrani, H. A., Rose, F., Schermuly, R. T., Olschewski, H., Wiedemann, R., Kreckel, A., Weissmann, N., Ghofrani, S., Enke, B., Seeger, W., and Grimminger, F. (2003). Oral sildenafil as long-term adjunct therapy to inhaled iloprost in severe pulmonary arterial hypertension. J. Am. Coll. Cardiol. 42, 158–164.
Ghofrani, H. A., Wiedemann, R., Rose, F., Olschewski, H., Schermuly, R. T., Weissmann, N., Seeger, W., and Grimminger, F. (2002a). Combination therapy with oral sildenafil and inhaled iloprost for severe pulmonary hypertension. Ann. Intern. Med. 136, 515–522.
Ghofrani, H. A., Wiedemann, R., Rose, F., Schermuly, R. T., Olschewski, H., Weissmann, N., Gunther, A., Walmrath, D., Seeger, W., and Grimminger, F. (2002b). Sildenafil for treatment of lung fibrosis and pulmonary hypertension: a randomised controlled trial. Lancet 360, 895–900.
Grimminger, F., Weimann, G., Frey, R., Voswinckel, R., Thamm, M., Bölkow, D., Weissmann, N., Mück, W., Unger, S., Wensing, G., Schermuly, R. T., and Ghofrani, H. A. (2009). First acute haemodynamic study of soluble guanylate cyclase stimulator riociguat in pulmonary hypertension. Eur. Respir. J. 33, 785–792.
Higenbottam, T., Butt, A. Y., McMahon, A., Westerbeck, R., and Sharples, L. (1998). Long-term intravenous prostaglandin (epoprostenol or iloprost) for treatment of severe pulmonary hypertension. Heart 80, 151–155.
Hoeper, M. M., Barbera, J. A., Channick, R. N., Hassoun, P. M., Lang, I. M., Manes, A., Martinez, F. J., Naeije, R., Olschewski, H., Pepke-Zaba, J., Redfield, M. M., Robbins, I. M., Souza, R., Torbicki, A., and McGoon, M. (2009). Diagnosis, assessment, and treatment of non-pulmonary arterial hypertension pulmonary hypertension. J. Am. Coll. Cardiol. 54(Suppl. 1), S85–S96.
Hoeper, M. M., Leuchte, H., Halank, M., Wilkens, H., Meyer, F. J., Seyfarth, H. J., Wensel, R., Ripken, F., Bremer, H., Kluge, S., Hoeffken, G., and Behr, J. (2006a). Combining inhaled iloprost with bosentan in patients with idiopathic pulmonary arterial hypertension. Eur. Respir. J. 28, 691–694.
Hoeper, M. M., Mayer, E., Simonneau, G., and Rubin, L. J. (2006b). Chronic thromboembolic pulmonary hypertension. Circulation 113, 2011–2020.
Hoeper, M. M., Markevych, I., Spiekerkoetter, E., Welte, T., and Niedermeyer, J. (2005). Goal-oriented treatment and combination therapy for pulmonary arterial hypertension. Eur. Respir. J. 26, 858–863.
Hoeper, M. M., Taha, N., Bekjarova, A., Gatzke, R., and Spiekerkoetter, E. (2003). Bosentan treatment in patients with primary pulmonary hypertension receiving nonparenteral prostanoids. Eur. Respir. J. 22, 330–334.
Humbert, M., Sitbon, O., and Simonneau, G. (2004a). Treatment of pulmonary arterial hypertension. N. Engl. J. Med. 351, 1425–1436.
Humbert, M., Barst, R. J., Robbins, I. M., Channick, R. N., Galiè, N., Boonstra, A., Rubin, L. J., Horn, E. M., Manes, A., and Simonneau, G. (2004b). Combination of bosentan with epoprostenol in pulmonary arterial hypertension: BREATHE-2. Eur. Respir. J. 24, 353–359.
Iribarne, A., Russo, M. J., Davies, R. R., Hong, K. N., Gelijns, A. C., Bacchetta, M. D., D’Ovidio, F., Arcasoy, S., and Sonett, J. R. (2009). Despite decreased wait-list times for lung transplantation, lung allocation scores continue to increase. Chest 135, 923–928.
Jais, X., D’Armini, A. M., Jansa, P., Torbicki, A., Delcroix, M., Ghofrani, H. A., Hoeper, M. M., Lang, I. M., Mayer, E., Pepke-Zaba, J., Perchenet, L., Morganti, A., Simonneau, G., and Rubin, L. J. (2008). Bosentan for treatment of inoperable chronic thromboembolic pulmonary hypertension: BENEFiT (Bosentan Effects in iNopErable Forms of chronIc Thromboembolic pulmonary hypertension), a randomized, placebo-controlled trial. J. Am. Coll. Cardiol. 52, 2127–2134.
Jamieson, S. W., Kapelanski, D. P., Sakakibara, N., Manecke, G. R., Thistlethwaite, P. A., Kerr, K. M., Channick, R. N., Fedullo, P. F., and Auger, W. R. (2003). Pulmonary endarterectomy: experience and lessons learned in 1,500 cases. Ann. Thorac. Surg. 76, 1457–1462.
Johnson, S. R., Mehta, S., and Granton, J. T. (2006). Anticoagulation in pulmonary arterial hypertension: a qualitative systematic review. Eur. Respir. J. 28, 999–1004.
Kawut, S. M., Horn, E. M., Berekashvili, K. K., Garofano, R. P., Goldsmith, R. L., Widlitz, A. C., Rosenzweig, E. B., Kerstein, D., and Barst, R. J. (2005). New predictors of outcome in idiopathic pulmonary arterial hypertension. Am. J. Cardiol. 95, 199–203.
Keogh, A. M., Mayer, E., Benza, R. L., Corris, P., Dartevelle, P. G., Frost, A. E., Kim, N. H., Lang, I. M., Pepke-Zaba, J., and Sandoval, J. (2009). Interventional and surgical modalities of treatment in pulmonary hypertension. J. Am. Coll. Cardiol. 54(Suppl. 1), S67–S77.
Kim, N. H. (2010). Riociguat: an upcoming therapy in chronic thromboembolic pulmonary hypertension? Eur. Respir. Rev. 19, 68–71.
MacLean, M. R. (2007). Pulmonary hypertension and the serotonin hypothesis: where are we now? Int. J. Clin. Pract. Suppl. 156, 27–31.
MacLean, M. R., and Dempsie, Y. (2010). The serotonin hypothesis of pulmonary hypertension revisited. Adv. Exp. Med. Biol. 661, 309–322.
Mathai, S. C., Girgis, R. E., Fisher, M. R., Champion, H. C., Housten-Harris, T., Zaiman, A., and Hassoun, P. M. (2007). Addition of sildenafil to bosentan monotherapy in pulmonary arterial hypertension. Eur. Respir. J. 29, 469–475.
Mayer, E. (2010). Surgical and post-operative treatment of chronic thromboembolic pulmonary hypertension. Eur. Respir. Rev. 19, 64–67.
McGoon, M. D., Frost, A. E., Oudiz, R. J., Badesch, D. B., Galiè, N., Olschewski, H., McLaughlin, V. V., Gerber, M. J., Dufton, C., Despain, D. J., and Rubin, L. J. (2009). Ambrisentan therapy in patients with pulmonary arterial hypertension who discontinued bosentan or sitaxsentan due to liver function test abnormalities. Chest 135, 122–129.
McLaughlin, V. V., Archer, S. L., Badesch, DB, Barst, R. J., Farber, H. W., Lindner, J. R., Mathier, M. A., McGoon, M. D., Park, M. H., Rosenson, R. S., Rubin, L. J., Tapson, V. F., and Varga, J. (2009). ACCF/AHA 2009 expert consensus document on pulmonary hypertension a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association developed in collaboration with the American College of Chest Physicians; American Thoracic Society, Inc; and the Pulmonary Hypertension Association. J. Am. Coll. Cardiol. 53, 1573–1619.
McLaughlin, V. V., Benza, R. L., Rubin, L. J., Channick, R. N., Voswinckel, R., Tapson, V. F., Robbins, I. M., Olschewski, H., Rubenfire, M., and Seeger, W. (2010). Addition of inhaled treprostinil to oral therapy for pulmonary arterial hypertension: a randomized controlled clinical trial. J. Am. Coll. Cardiol. 55, 1915–1922.
McLaughlin, V. V., Shillington, A., and Rich, S. (2002). Survival in primary pulmonary hypertension: the impact of epoprostenol therapy. Circulation 106, 1477–1482.
Mereles, D., Ehlken, N., Kreuscher, S., Ghofrani, S., Hoeper, M. M., Halank, M., Meyer, F. J., Karger, G., Buss, J., Juenger, J., Holzapfel, N., Opitz, C., Winkler, J., Herth, F. F., Wilkens, H., Katus, H. A., Olschewski, H., and Grunig, E. (2006). Exercise and respiratory training improve exercise capacity and quality of life in patients with severe chronic pulmonary hypertension. Circulation 114, 1482–1489.
Moser, K. M., Auger, W. R., Fedullo, P. F., and Jamieson, S. W. (1992). Chronic thromboembolic pulmonary hypertension: clinical picture and surgical treatment. Eur. Respir. J. 5, 334–342.
Mouchaers, K. T., Schalij, I., de Boer, M. A., Postmus, P. E., van Hinsbergh, V. W., van Nieuw Amerongen, G. P., Vonk Noordegraaf, A., and van der Laarse, W. J. (2010). Fasudil reduces monocrotaline-induced pulmonary arterial hypertension: comparison with bosentan and sildenafil. Eur. Respir. J. 36, 800–807.
MRC (Medical Research Council) Working Party. (1981). Long term domiciliary oxygen therapy in chronic hypoxic cor pulmonale complicating chronic bronchitis and emphysema. Lancet 1, 681–686.
Murray, F., Patel, H. H., Suda, R. Y., Zhang, S., Thistlethwaite, P. A., Yuan, J. X., and Insel, P. A. (2007). Expression and activity of cAMP phosphodiesterase isoforms in pulmonary artery smooth muscle cells from patients with pulmonary hypertension: role for PDE1. Am. J. Physiol. Lung Cell. Mol. Physiol. 292, L294–L303.
Nagaya, N., Sasaki, N., Ando, M., Ogino, H., Sakamaki, F., Kyotani, S., and Nakanishi, N. (2003). Prostacyclin therapy before pulmonary thromboendarterectomy in patients with chronic thromboembolic pulmonary hypertension. Chest 123, 338–343.
NOTT (Nocturnal Oxygen Therapy Trial) Group. (1980). Continuous or nocturnal oxygen therapy in hypoxemic chronic obstructive lung disease: a clinical trial. Ann. Intern. Med. 93, 391–398.
Olschewski, H., Simonneau, G., Galiè, N., Higenbottam, T., Naeije, R., Rubin, L. J., Nikkho, S., Sitbon, O., Speich, R., Hoeper, M., Behr, J., Winkler, J., Seeger, W. for the AIR Study Group. (2002). Inhaled iloprost in severe pulmonary hypertension. N. Engl. J. Med. 347, 322–329.
Orens, J. B., Estenne, M., Arcasoy, S., Conte, J. V., Corris, P., Egan, J. J., Egan, T., Keshavjee, S., Knoop, C., Kotloff, R., Martinez, F. J., Nathan, S., Palmer, S., Patterson, A., Singer, L., Snell, G., Studer, S., Vachiery, J. L., and Glanville, A. R. (2006). International guidelines for the selection of lung transplant candidates: 2006 update. J. Heart Lung Transplant. 25, 745–755.
Oudiz, R. J., Galiè, N., Olschewski, H., Torres, F., Frost, A., Ghofrani, H. A., Badesch, D. B., McGoon, M. D., McLaughlin, V. V., Roecker, E. B., Harrison, B. C., Despain, D., Dufton, C., and Rubin, L. J. (2009). Long-term ambrisentan therapy for the treatment of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 54, 1971–1981.
Pepke-Zaba, J., Gilbert, C., Collings, L., and Brown, M. C. (2008). Sildenafil improves health-related quality of life in patients with pulmonary arterial hypertension. Chest 133, 183–189.
Petkov, V., Mosgoeller, W., Ziesche, R., Raderer, M., Stiebellehner, L., Vonbank, K., Funk, G. C., Hamilton, G., Novotny, C., Burian, B., and Block, L. H. (2003). Vasoactive intestinal peptide as a new drug for treatment of primary pulmonary hypertension. J. Clin. Invest. 111, 1339–1346.
Piazza, G., and Goldhaber, S. Z. (2011). Chronic thromboembolic pulmonary hypertension. N. Engl. J. Med. 364, 351–360.
Reichenberger, F., Pepke-Zaba, J., McNeil, K., Parameshwar, J., and Shapiro, L. M. (2003). Atrial septostomy in the treatment of severe pulmonary arterial hypertension. Thorax 58, 797–800.
Rich, S., Kaufmann, E., and Levy, P. S. (1992). The effect of high doses of calcium-channel blockers on survival in primary pulmonary hypertension. N. Engl. J. Med. 327, 76–81.
Rubin, L. J., Badesch, D. B., Barst, R. J., Galiè, N., Black, C. M., Keogh, A., Pulido, T., Frost, A., Roux, S., Leconte, I., Landzberg, M., and Simonneau, G. (2002). Bosentan therapy for pulmonary arterial hypertension. N. Engl. J. Med. 346, 896–903.
Rubin, L. J., Hoeper, M. M., Klepetko, W., Galiè, N., Lang, I. M., and Simonneau, G. (2006). Current and future management of chronic thromboembolic pulmonary hypertension: from diagnosis to treatment responses. Proc. Am. Thorac. Soc. 3, 601–607.
Rubin, L. J., Mendoza, J., Hood, M., McGoon, M., Barst, R., Williams, W. B., Diehl, J. H., Crow, J., and Long, W. (1990). Treatment of primary pulmonary hypertension with continuous intravenous prostacyclin (epoprostenol). Results of a randomized trial. Ann. Intern. Med. 112, 485–491.
Sandoval, J., Rothman, A., and Pulido, T. (2001). Atrial septostomy for pulmonary hypertension. Clin. Chest Med. 22, 547–560.
Shapiro, S. M., Oudiz, R. J., Cao, T., Romano, M. A., Beckmann, X. J., Georgiou, D., Mandayam, S., Ginzton, L. E., and Brundage, B. H. (1997). Primary pulmonary hypertension: improved long-term effects and survival with continuous intravenous epoprostenol infusion. J. Am. Coll. Cardiol. 30, 343–349.
Simonneau, G., Robbins, I. M., Beghetti, M., Channick, R. N., Delcroix, M., Denton, C. P., Elliott, C. G., Gaine, S. P., Gladwin, M. T., Jing, Z. C., Krowka, M. J., Langleben, D., Nakanishi, N., and Souza, R. (2009). Updated clinical classification of pulmonary hypertension. J. Am. Coll. Cardiol. 54(Suppl. 1), S43–S54.
Simonneau, G., Rubin, L. J., Galiè, N., Barst, R. J., Fleming, T. R., Frost, A. E., Engel, P. J., Kramer, M. R., Burgess, G., Collings, L., Cossons, N., Sitbon, O., and Badesch, D. B. (2008). Addition of sildenafil to long-term intravenous epoprostenol therapy in patients with pulmonary arterial hypertension: a randomized trial. Ann. Intern. Med. 149, 521–530.
Sitbon, O., and Galiè, N. (2010). Treat-to-target strategies in pulmonary arterial hypertension: the importance of using multiple goals. Eur. Respir. Rev. 19, 272–278.
Sitbon, O., Humbert, M., Jais, X., Ioos, V., Hamid, A. M., Provencher, S., Garcia, G., Parent, F., Herve, P., and Simonneau, G. (2005). Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation 111, 3105–3111.
Sitbon, O., Humbert, M., Nunes, H., Parent, F., Garcia, G., Herve, P., Rainisio, M., and Simonneau, G. (2002). Long-term intravenous epoprostenol infusion in primary pulmonary hypertension: prognostic factors and survival. J. Am. Coll. Cardiol. 40, 780–788.
Suntharalingam, J., Treacy, C. M., Doughty, N. J., Goldsmith, K., Soon, E., Toshner, M. R., Sheares, K. K., Hughes, R., Morrell, N. W., and Pepke-Zaba, J. (2008). Long-term use of sildenafil in inoperable chronic thromboembolic pulmonary hypertension. Chest 134, 229–236.
Tapson, V. F., Gomberg-Maitland, M., McLaughlin, V. V., Benza, R. L., Widlitz, A. C., Krichman, A., and Barst, R. J. (2006). Safety and efficacy of IV treprostinil for pulmonary arterial hypertension: a prospective, multicenter, open-label, 12-week trial. Chest 129, 683–688.
Trulock, E. P., Christie, J. D., Edwards, L. B., Boucek, M. M., Aurora, P., Taylor, D. O., Dobbels, F., Rahmel, A. O., Keck, B. M., and Hertz, M. I. (2007). Registry of the International Society for Heart and Lung Transplantation: twenty-fourth official adult lung and heart-lung transplantation report-2007. J. Heart Lung Transplant. 26, 782–795.
Keywords: pulmonary hypertension, specific therapy, combination therapy, prostanoids, endothelin receptor antagonists, phosphodiesterase type-5 inhibitors
Citation: Fuso L Baldi F and Perna AD (2011) Therapeutic strategies in pulmonary hypertension. Front. Pharmacol. 2:21. doi: 10.3389/fphar.2011.00021
Received: 21 February 2011;
Paper pending published: 14 March 2011;
Accepted: 30 March 2011;
Published online: 20 April 2011.
Edited by:
Stelios Loukides, University of Athens Medical School, GreeceCopyright: © 2011 Fuso, Baldi and Di Perna. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Leonello Fuso, Servizio di Fisiopatologia Respiratoria, Università Cattolica S. Cuore, Largo A. Gemelli 8, 00168 Roma, Italy. e-mail: leofuso@rm.unicatt.it