 Yuying Zhu1,†
Yuying Zhu1,† Ke Wu
Ke Wu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr. , 10 February 2025
Sec. Genetics of Common and Rare Diseases
Volume 13 - 2025 | https://doi.org/10.3389/fped.2025.1494530
Background: Genomic or exome sequencing is beneficial for identifying more than one pathogenic variation causing blended atypical and/or severe phenotypes. Herein, we are the first to report a 5-year-old boy with the blended phenotypes of infantile hypotonia, severe neurodevelopmental disorder, patent ductus arteriosus, cryptorchidism, obesity, distinctive facial features, and elevated isovaleryl carnitine.
Methods: Trio-based whole-exome sequencing was performed on genomic DNA from peripheral blood samples from the boy and his parents. Functional analysis of the IVD variant in vitro was performed. Mutant IVD gene pcDNA3.1(+)-MUT-3xFlag and control pcDNA3.1(+)-WT-3xFlag mammalian expression vectors were constructed. Both vectors were transformed into HEK293T cells. The assays of relative IVD gene mRNA expression, IVD protein expression, and enzymatic activity were used.
Results: Whole-exome sequencing identified a novel homozygous missense variant in the IVD gene (NM_002225.5) c.1006T>C (p.Cys336Arg) within a region of homozygosity of 15q11.2-q21.3. Our in vitro functional and computer simulation findings revealed that this variant was associated with haploinsufficiency, which resulted in dramatically reducing the formation of IVD protein due to unstable mutant protein and not a lack of mRNA expression.
Conclusion: The boy was diagnosed with the dual genetic disorders of Prader–Willi syndrome and isovaleric acidemia. This case provides a useful reference for genetic counseling for complex and diverse clinical phenotypes. The presence of two or more likely pathogenic or pathogenic variations in an individual with neurodevelopmental phenotypes is not an “exceptional” phenomenon.
Isovaleric acidemia (IVA, OMIM #243500) is an autosomal recessive inborn error of leucine metabolism caused by homozygous or compound heterozygous variants in the isovaleryl-CoA dehydrogenase gene (IVD; OMIM *607036) on chromosome 15q15 (1). Isovaleryl-CoA dehydrogenase (IVD, EC:1.3.8.4) belongs to the acyl-CoA dehydrogenase family, and it is a mitochondrial matrix enzyme that catalyzes the third step in leucine catabolism. A deficiency of IVD results in the accumulation of isovaleric acid, which is a serious hazard to the central nervous system and leads to IVA. Clinical manifestations of untreated IVA include developmental delay, neuropathological implications, characteristic sweaty foot odor, poor feeding, vomiting, lethargy, and seizures during infancy, childhood, or adolescence (2). Prader–Willi syndrome (PWS, OMIM#176270) is caused by the loss of function of paternally expressed genes in 15q11q13, which could be due to a de novo paternal deletion, maternal uniparental disomy (UPD) of the 15q11-q13 region, or the silencing of the paternal expressed imprinting genes (3). Consensus diagnostic clinical criteria for PWS have been developed (4). The main clinical features of PWS are severe hypotonia and feeding difficulties in early infancy, hyperphagia leading to obesity in later infancy or early childhood, intellectual disability, delayed language development, craniofacial anomalies, hypogonadism/hypogenitalism, short stature, and distinctive behavioral problems (5). Although the co-occurrence of multiple (two or more) rare diseases intuitively appears unlikely, it was estimated that approximately 4%–7.5% of patients analyzed by trio-based whole-exome sequencing (trio-WES) had two or more genetic diagnoses (6, 7), with a higher frequency in consanguineous families. Herein, we report a Chinese boy diagnosed with isovaleric acidemia and Prader–Willi syndrome due to a region of homozygosity (ROH), which has not been previously reported. We conducted this study to expand the mutational IVD spectrum. It should be noted that more than one genome variation may contribute to complex and diverse clinical phenotypes.
Before her pregnancy, a Chinese woman sought genetic counseling from the Prenatal Diagnosis Center of Quzhou Maternity and Child Health Care Hospital (Zhejiang, China). She reported that her first child (the proband) was a 5-year-old boy born with IVA (detected through newborn screening with elevated isovaleryl carnitine), patent ductus arteriosus (PDA), infantile hypotonia, and cryptorchidism. This boy was naturally conceived with an uneventful normal pregnancy. At the age of 5 years and 6 months, tandem mass spectrometry (MS) revealed he still had an isovaleryl carnitine level of 16.08 μmol/L (C5, normal range: 0.01–0.40 μmol/L). A physical examination showed that he had no obvious dysmorphic features of the hands and feet but he had distinctive facial features (almond-shaped eyes, thin upper lip with downturned corners of the mouth, flat nasal bridge, and low-set ears). At the age of approximately 2 years, his constant craving for food resulted in rapid weight gain. At the age of 5 years and 6 months, an anthropometric examination revealed growth retardation with a height of 102 cm (3rd percentile), overweight with a weight of 24.6 kg (90th percentile), and a body mass index (BMI) of 23.6 (>P85) (8). The radiographic atlas method for assessing the skeletal age of the hand and wrist indicated delayed bone age with 3/10 ossification center of the left carpal, ossified metacarpophalangeal bones, and unclosed epiphyseal plate. At the age of 5 years and 10 months, the Griffiths Mental Development Scale-China (GDS-C) (9) showed that this boy had a low gross quotient (GQ) score of 36, which suggested that he was moderately delayed in his cognitive and physical development.
Trio-WES was performed on genomic DNA (gDNA) from peripheral blood samples from the boy and his parents. The xGen™ Exome Research Panel v2 (designed by Integrated DNA Technologies) was used for WES. Quality control (QC) of the DNA library was performed using an Agilent 2100 Bioanalyzer System (Agilent, USA). DNA nanoball (DNB) preps of the clinical samples were sequenced on an ultra-high throughput DNBSEQ-T7 platform (MGI, Shenzhen, China) with a paired-end 150 nt strategy following the manufacturer's protocol.
Sequencing data were analyzed according to our in-house (Chigene Translational Medicine Research Center) procedures. Adapters and low-quality reads were removed, and data quantity and quality were statistically analyzed. The trimmed reads were then mapped to the University of California Santa Cruz (UCSC) GRCh37/hg19 reference genome using the Burrows–Wheeler Aligner (BWA) software. Genome Analysis Toolkit (GATK) software was used for single nucleotide polymorphisms (SNPs) and short (<50 bp) insertion/deletion (indel) calling. The Samtools and Picard software packages were used to generate clean Bam data by removing duplicate data. Variants were annotated for analysis using the single nucleotide polymorphism database (dbSNP), gnomAD exomes database, and Chigene in-house minor allele frequency (MAF) database. Tools for pathogenicity prediction including rare exome variant ensemble learner (REVEL) and phaMissense were used for predicting the possible impact of the variants. DynaMut (10) (a web server) is a well-established normal mode approach used for assessing the stability of a mutant protein. Pathogenicity annotation was conducted according to the American College of Medical Genetics and Genomics (ACMG) guidelines, and the Online Mendelian Inheritance in Man (OMIM), Human Gene Mutation Database (HGMD), and ClinVar databases were used as references for the pathogenicity of each variant.
As per the ACMG guidelines for interpreting sequence variants, the variants were classified. The classification considered the position of the variant in the human genome, MAF, the pathogenicity prediction of the variant, disease mechanism, clinical phenotypes, literature evidence, and evolutionary conservation.
Sanger sequencing (BigDye Terminator v3.1 Cycle Sequencing Kit and ABI 3730 Applied Biosystem) was used for further verification of candidate variants. Methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA) (ME028-B1 PWS/AS probe mixture purchased from MRC-Holland) was used for methylation of imprinted regions in the human genome.
The mutant IVD gene pcDNA3.1(+)-MUT-3xFlag and control pcDNA3.1(+)-WT-3xFlag mammalian expression vectors were constructed and purchased from Wuhan Biorun Biosciences Co., Ltd. HEK293T cells were transfected with expression vectors for two tubes. Then, 6 h after the transfection, we gently removed the lipofectamine-containing medium, replaced the medium with Dulbecco's modified eagle medium (DMEM) containing 5% fetal bovine serum (FBS), and incubated the slide at 37℃ in a 5% CO2 incubator until day 2. On day 3, samples were collected and used for a reverse transcription-polymerase chain reaction (RT-PCR) in the IVD mRNA expression, protein expression, and enzymatic activity assays.
Trio-WES identified an ROH in chromosome 15q11.2-q21.3 (GRCh38, chr15:20534520-58432427) (Figure 1) and a novel homozygous IVD gene variant (NM_002225.5) c.1006T>C (p.Cys336Arg) caused by an ROH of 15q11.2-q21.3. The ROH of 15q11.2-q21.3 was further verified by MS-MLPA, which revealed maternal UPD (Figure 2). The Sanger sequencing confirmed that the homozygous variant was derived from the boy's heterozygous carrier mother and the boy's father was normal (Figure 3).
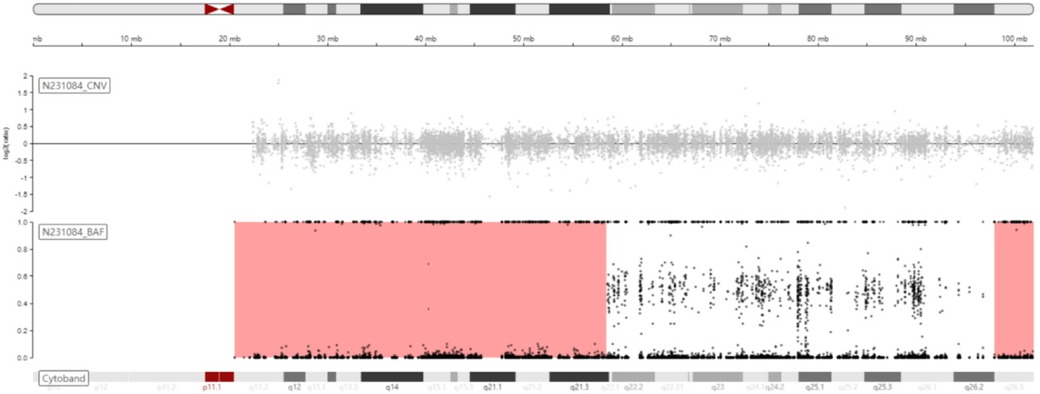
Figure 1. Schematic diagram of B-allele frequency (BAF) on chromosome 15 showing the ROH of 15q11.2-q21.3 (chr15:20534520-58432427, GRCh38).
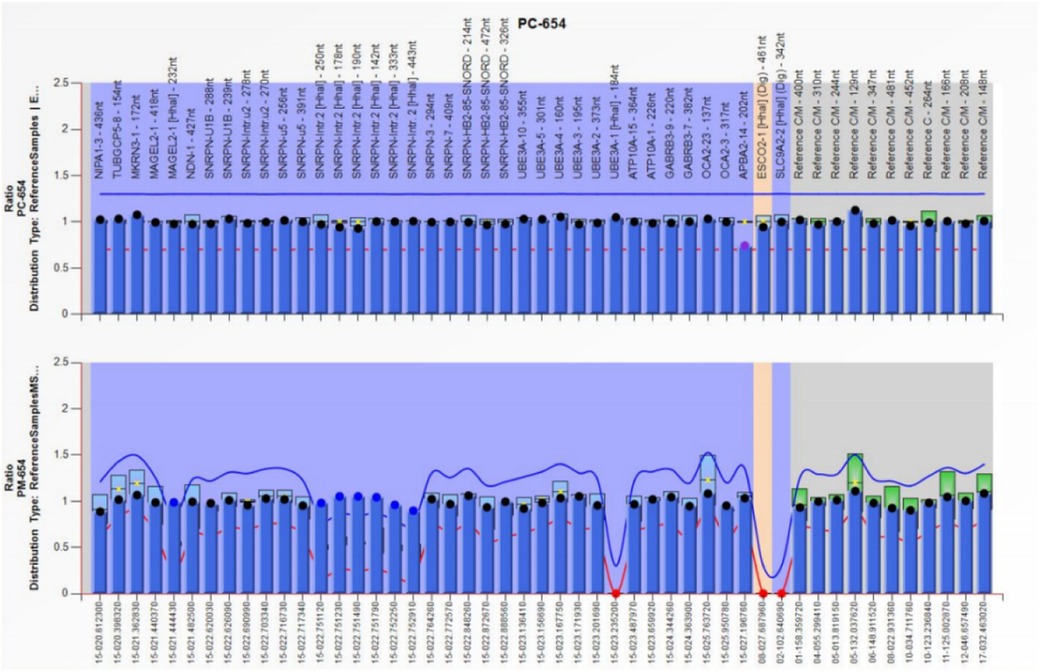
Figure 2. The result of MS-MLPA showing that the copy number of chromosome 15q11.2-q13 was normal but there was abnormal methylation in this region, which suggested this region was maternal UPD.
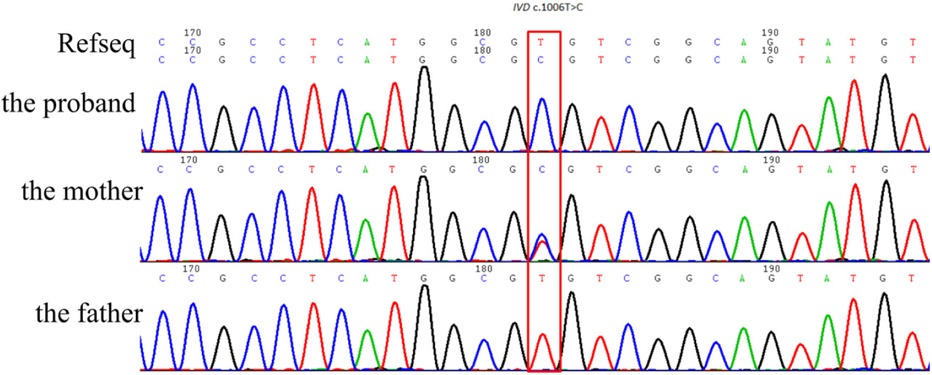
Figure 3. Sanger sequencing confirmed that the homozygous IVD gene variant (NM_002225.5) c.1006T>C (p.Cys336Arg) was derived from the boy's heterozygous carrier mother.
The Chinese boy was diagnosed with isovaleric acidemia, which was highly specific for a disease with a single genetic etiology (PP4_Supporting). The allele frequency of homozygous c.1006T>C has not been registered in population databases (1,000 Genomes Project, gnomAD, and dbSNP) or reported in disease databases (ClinVar, Human Gene Mutation Database, and OMIM) (PM2_Supporting). The in silico predictive algorithm for this variant showed that the missense variant was moderately deleterious (REVEL score 0.83 > 0.64; AlphaMissense score 0.973 > 0.869) (PP3_Moderate). The DynaMut prediction outcome of the IVD stability was ΔΔG: −0.926 kcal/mol (destabilizing) (PDB accession code: 1IVH; length of mature IVD monomer: 394 amino acids; Cys336 residue mapped to Cys307 in the PDB structure). As per the interpretation guidelines of the American College of Medical Genetics and Genomics (11), this novel variant is classified as a “variant of uncertain significance (VUS)” (PP4; PM2; PP3).
To evaluate the effect of this IVD VUS allele, our functional study suggested that this variant (NM_002225.5) c.1006T>C (p.Cys336Arg) did not restrict IVD mRNA expression from the plasmid (Figure 4A), but led to a lack of IVD protein (Figure 4B). The enzymatic activity assay of IVD (human IVD ELISA Kit) suggested that the enzymatic activity was slightly reduced in the mutant HEK293T cell lines and serum extracted from the patient compared to normal controls (Figure 4C).

Figure 4. Relative mRNA expression, protein expression, and enzymatic activity of control, wild-type (WT) IVD, and mutant IVD HEK293T lines. Relative mRNA expression, IVD protein expression, and enzymatic activity assays were all conducted in triplicate. **p < 0.01; ns, no statistical difference. (A) Relative mRNA expression of the IVD gene variant indicated the level of relative mRNA expression in the IVD-WT cell line was significantly lower than in the mutant IVD cell line. (B) Western blot for IVD confirmed it was significantly reduced in the mutant cell line. Abundant IVD protein was observed with expression of the WT-IVD cell line. (C) The enzymatic activity assay of IVD (human IVD ELISA Kit) suggested that the enzymatic activity was not significantly different in the mutant HEK293T cell line and patient's serum compared to normal controls, but it was slightly reduced.
The IVD gene is located in chromosome 15q15.1. The transcript of IVD (NM_002225.5) has 12 exons, a transcript length of 4,350 base pairs, and a translation length of 423 amino acids (12). IVD is a homotetrameric enzyme (172 kDa) with dehydrogenase activity and is composed of four identical mature monomeric peptides (43 kDa). The 43 kDa mature IVD protein is created by posttranslational modification (29 amino acids are removed) of a 45 kDa precursor (in the cytoplasm) initially encoded by the IVD gene and is transferred into the mitochondria through the terminal signal peptide. The IVD protein belongs to the acyl-CoA dehydrogenase family (ACAD) and catalyzes the conversion of isovaleryl-CoA(3-methylbutyryl-CoA) into 3-methylcrotonyl-CoA as the third step in the leucine catabolic pathway (13). Ubiquitous expression of IVD is in the thyroid (RPKM 25.0) and liver (RPKM 18.5) (14). IVD contains three domains of approximately equal size: an N-terminal domain, a middle domain, and a C-terminal domain. The genotype–phenotype correlation in patients with IVA has not yet been clarified (15).
These in vitro and computer simulation findings were consistent with the previous functional analysis (16) of p.Arg337Gln (c.1010G>A) adjacent to p.Cys336Arg (found by this study). The p.Cys336Arg variant may dramatically reduce the formation of IVD protein due to unstable mutant protein and not a lack of mRNA expression. We speculated that this homozygous missense variant was associated with haploinsufficiency and caused a loss of IVD function, which disrupted the normal function of the enzyme. Furthermore, in the functional study and as per the guidelines of ACMG for interpreting the sequence, this variant was upgraded to “likely pathogenic.”
In neurodevelopmental disorders in the Turkish population, Mitani et al. identified that 28.9% of families (51/176) with more than one pathogenic variant were mostly driven by an ROH (17). In this case, the ROH of 15q11.2-q21.3 included the IVD gene variant. The boy presented with two kinds of rare genetic disorders. Thus, it is essential to note that more than one genome variation may contribute to complex and diverse clinical phenotypes.
In conclusion, we are the first to report a boy with dual genetic diagnoses of Prader–Willi syndrome and isovaleric acidemia attributed to a segmental isodisomy. The identification and functional analysis of a novel IVD gene variant in this study expanded the IVD spectrum. Furthermore, the presence of two or more likely pathogenic or pathogenic variations in an individual with neurodevelopmental phenotypes was not an “anomalous” phenomenon.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://databases.lovd.nl/shared/individuals/00446575.
The studies involving humans were approved by the Ethics Committee of Quzhou Maternal and Child Health Care Hospital (Approval KY-2023-11, date 2023.05.08). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
YZ: Writing – original draft. KW: Data curation, Formal Analysis, Writing – original draft, Writing – review & editing. HW: Funding acquisition, Resources, Supervision, Validation, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was supported by grants from two projects of Science and Technology of Quzhou (2023ZD084 and 2023K183).
We are grateful to the patient and their family for their participation in this study and for the help of all the physicians in the course of the medical treatment. We wish to thank the staff of Chigene (Beijing) Translational Medical Research Center Co. Ltd. for assisting with sequencing data analysis.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Vockley J, Parimoo B, Tanaka K. Molecular characterization of four different classes of mutations in the isovaleryl-CoA dehydrogenase gene responsible for isovaleric acidemia. Am J Hum Genet. (1991) 49(1):147–57.2063866
2. Zaki OK, Priya Doss CG, Ali SA, Murad GG, Elashi SA, Ebnou MSA, et al. Genotype-phenotype correlation in patients with isovaleric acidaemia: comparative structural modelling and computational analysis of novel variants. Hum Mol Genet. (2017) 26:3105–15. doi: 10.1093/hmg/ddx195
3. Buiting K, Cassidy SB, Driscoll DJ, Gillessen-Kaesbach G, Kanber D, Tauber M, et al. Clinical utility gene card for: Prader-Willi syndrome. Eur J Hum Genet. (2014) 22:1153. doi: 10.1038/ejhg.2014.66
4. Beygo J, Buiting K, Ramsden SC, Ellis R, Clayton-Smith J, Kanber D. Update of the EMQN/ACGS best practice guidelines for molecular analysis of Prader-Willi and Angelman syndromes. Eur J Hum Genet. (2019) 27(9):1326–40. doi: 10.1038/s41431-019-0435-0
5. Butler MG, Miller JL, Forster JL. Prader-Willi syndrome—clinical genetics, diagnosis and treatment approaches: an update. Curr Pediatr Rev. (2019) 15(4):207–44. doi: 10.2174/1573396315666190716120925
6. Balci TB, Hartley T, Xi Y, Dyment DA, Beaulieu CL, Bernier FP, et al. Debunking Occam’s razor: diagnosing multiple genetic diseases in families by whole-exome sequencing. Clin Genet. (2017) 92(3):281–9. doi: 10.1111/cge.12987
7. Rosina E, Pezzani L, Pezzoli L, Marchetti D, Bellini M, Pilotta A, et al. Atypical, composite, or blended phenotypes: how different molecular mechanisms could associate in double-diagnosed patients. Genes (Basel). (2022) 13(7):1275. doi: 10.3390/genes13071275
8. de Onis M, Onyango AW, Borghi E, Siyam A, Nishida C, Siekmann J. Development of a WHO growth reference for school-aged children and adolescents. Bull World Health Organ. (2007) 85(9):660–7. doi: 10.2471/BLT.07.043497
9. Tso WWY, Wong VCN, Xia X, Faragher B, Li M, Xu X, et al. The Griffiths development scales-Chinese (GDS-C): a cross-cultural comparison of developmental trajectories between Chinese and British children. Child Care Health Dev. (2018) 44(3):378–83. doi: 10.1111/cch.12548
10. Rodrigues CH, Pires DE, Ascher DB. Dynamut: predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res. (2018) 46(W1):W350–5. doi: 10.1093/nar/gky300
11. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
12. Parimoo B, Tanaka K. Structural organization of the human isovaleryl-CoA dehydrogenase gene. Genomics. (1993) 15(3):582–90. doi: 10.1006/geno.1993.1111
13. Mohsen AW, Vockley J. Identification of the active site catalytic residue in human isovaleryl-CoA dehydrogenase. Biochemistry. (1995) 34(32):10146–52. doi: 10.1021/bi00032a007
14. Fagerberg L, Hallström BM, Oksvold P, Kampf C, Djureinovic D, Odeberg J, et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol Cell Proteomics. (2014) 13(2):397–406. doi: 10.1074/mcp.M113.035600
15. Liu X, Liu X, Fan W, Zhang Z, Zhang P, Liu X, et al. Analysis of the genotype-phenotype correlation in isovaleric acidaemia: a case report of long-term follow-up of a Chinese patient and literature review. Front Neurol. (2022) 13:928334. doi: 10.3389/fneur.2022.928334
16. D'Annibale OM, Koppes EA, Alodaib AN, Kochersperger C, Karunanidhi A, Mohsen AW, et al. Characterization of variants of uncertain significance in isovaleryl-CoA dehydrogenase identified through newborn screening: an approach for faster analysis. Mol Genet Metab. (2021) 134(1–2):29–36. doi: 10.1016/j.ymgme.2021.08.012
Keywords: Prader–Willi syndrome, isovaleric acidemia, genetic counseling, case report, IVD gene
Citation: Zhu Y, Wu K and Wen H (2025) Functional analysis of a novel homozygous missense IVD gene variant: a case report with dual genetic diagnoses. Front. Pediatr. 13:1494530. doi: 10.3389/fped.2025.1494530
Received: 11 September 2024; Accepted: 17 January 2025;
Published: 10 February 2025.
Edited by:
Andrea Accogli, McGill University Health Centre, CanadaReviewed by:
Muzammil Ahmad Khan, Gomal University, PakistanCopyright: © 2025 Zhu, Wu and Wen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hanying Wen, NzU0Mjk5MDU4QHFxLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.