
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pediatr., 06 December 2024
Sec. Pediatric Hematology and Hematological Malignancies
Volume 12 - 2024 | https://doi.org/10.3389/fped.2024.1505060
 Estefânia Rodrigues Biojone1*
Estefânia Rodrigues Biojone1* Bruna Cândido Guido2
Bruna Cândido Guido2 Larissa Lemos Mendanha Cavalcante2Agenor de Castro Moreira dos Santos Júnior2Robéria Mendonça de Pontes2Felipe Magalhães Furtado1,2,3José Carlos Córdoba1,2Isis Maria Quezado Magalhães1,2
Larissa Lemos Mendanha Cavalcante2Agenor de Castro Moreira dos Santos Júnior2Robéria Mendonça de Pontes2Felipe Magalhães Furtado1,2,3José Carlos Córdoba1,2Isis Maria Quezado Magalhães1,2 Diêgo Madureira de Oliveira4
Diêgo Madureira de Oliveira4 Ricardo Camargo2
Ricardo Camargo2
Introduction: There is consistent evidence that FLT3 may be a driver gene in B-ALL and that selected cases may benefit from the use of FLT3 inhibitors. Our study was conducted to evaluate the frequency and types of FLT3 mutations in pediatric patients with B-ALL, the relative expression of this gene, and their influence on clinical evolution.
Methods: We evaluated 156 children with B-ALL treated between July 2018 and September 2023. Screening for FLT3 mutations was performed using RFLP and fragment analysis, while FLT3 expression was assessed by qPCR.
Results: FLT3-TKD and/or FLT3-JM-INDEL mutations were found in 8 patients (5.1%). We did not identify any ITD-type mutations. None of the patients with identified FLT3 mutations presented recurrent rearrangements in B-ALL or alterations in the IKZF1, PAX5, or ERG genes, suggesting that FLT3 mutation may serve as the driving mechanism for leukemia in these cases. Two (2/8) patients with FLT3 mutations experienced disease relapse. Although we did not observe FLT3 overexpression among patients with FLT3 mutations, FLT3 expression levels were higher in these patients compared to WT patients. Four FLT3-WT patients presented FLT3 overexpression, defined as RQ > 10. FLT3 mutations or overexpression were not associated with relapses or survival rates.
Discussion: Our findings do not support the inclusion of FLT3 as a routine marker in the risk stratification of B-ALL patients; nevertheless, FLT3 alterations may be relevant for guiding personalized treatment approaches in specific clinical contexts.
B-cell Acute Lymphoblastic Leukemia (B-ALL) is the most common cancer in the pediatric population, accounting for approximately 25% of malignant neoplasms in patients up to 18 years of age (1, 2). Despite significant improvements in survival rates, relapsed or refractory disease remains a frequent cause of death among B-ALL patients (3). Currently, the assessment of leukemic cells through molecular biology techniques and genetic evaluation, including next-generation sequencing (NGS), enables the identification of over thirty ALL subtypes characterized by specific gene expression profiles or biological markers (4). This characterization not only enhances diagnostic accuracy but also provides opportunities for treatment optimization, either by adjusting chemotherapy intensity or by introducing targeted therapies (5).
The FLT3 (FMS-like tyrosine kinase 3) gene, located on chromosome 13q12, encodes a type III receptor tyrosine kinase predominantly expressed in the bone marrow, particularly in hematopoietic precursor cells. The protein consists of four distinct regions: an extracellular domain, a transmembrane region, a juxtamembrane region, and an intracellular portion containing a tyrosine kinase domain. Upon binding to the FLT3 ligand (FL), the receptor is activated through dimerization and autophosphorylation, initiating a cascade of signaling pathways, including PI3K/AKT, RAS/MAPK, and STAT5 (6, 7). This signaling promotes cell proliferation and inhibits apoptosis. The FLT3 gene plays a critical role in the survival, proliferation, and differentiation of hematopoietic cells across both myeloid and lymphoid lineages (6, 8–11).
Mutations in FLT3, occurring either in the juxtamembrane domain or within the tyrosine kinase domain, lead to constitutive activation of FLT3 and are associated with leukemogenesis (12–20). There are four main types of activating mutations in the FLT3 gene: internal tandem duplications in the juxtamembrane domain (FLT3-ITD), in-frame insertions or deletions in the juxtamembrane domain (FLT3-JM-INDEL), point mutations in the juxtamembrane domain (FLT3 JM-PM), and mutations in the tyrosine kinase domain (FLT3-TKD) (21).
FLT3-activating mutations are commonly found in Acute Myeloid Leukemias (AML), accounting for approximately one-third of adult AML cases and 10%–15% of pediatric AML cases (19). Among AML patients, FLT3-ITD is a recurrent driver mutation (present in about 25% of all AML cases) and is associated with higher relapse rates and reduced overall survival (12–26). In contrast, TKD domain mutations are likely secondary events with uncertain prognostic impact (22, 23). FLT3 inhibitor drugs are approved for AML patients with FLT3 mutations, and their use has been associated with improved overall survival and event-free survival rates (26–30).
In B-ALL, FLT3 mutations are less common, reported in 0.2%–12.5% of cases when evaluated by conventional techniques (RFLP and fragment analysis) (31–36) and in up to 25% when investigated using NGS (20). The prognostic impact of these alterations remains poorly defined (20, 30–36). ITD-type mutations are rare in this context, and recent studies have described in-frame indels in the juxtamembrane domain as the most common type of FLT3 genetic variant in patients with B-ALL (21).
FLT3 overexpression, in addition to its activating mutations, has been documented in B-ALL, particularly in specific subtypes such as r-KMT2A, r-ZNF384, and high hyperdiploid ALL (20, 37–41). The mechanisms contributing to elevated FLT3 expression in certain B-ALL subtypes are not yet fully understood. Epigenetic modifications, such as enhancer hijacking due to deletions at 13q12.2, have been linked to higher expression levels in hyperdiploid patients and in cases of relapse (42). Although the surface expression of the FLT3 receptor does not correlate with FLT3 transcript levels, total cellular FLT3 protein levels generally reflect transcript levels. Overexpressed wild-type FLT3 proteins have been observed to undergo tyrosine phosphorylation (43). Some studies have associated elevated FLT3 expression with poorer outcomes in B-ALL (20, 36–40, 44). Encouragingly, the autophosphorylation of wild-type FLT3 induced by its overexpression was shown to be inhibited by a potent FLT3 kinase inhibitor, with sensitivity comparable to that observed in mutant forms (43, 45). Additionally, evidence suggests therapeutic responsiveness to FLT3 inhibitors in certain subsets of relapsed B-ALL patients exhibiting FLT3 overexpression (46–48). Nevertheless, few studies have investigated therapeutic strategies involving FLT3 inhibitors specifically for B-ALL (41, 49).
Given the current uncertainty regarding the prognostic value of FLT3 alterations in pediatric B-ALL and considering recent reports of the therapeutic efficacy of FLT3 inhibitors in patients with relapsed B-cell ALL (46–48), it is essential to deepen our understanding in this field of research. The objective of this study was to identify FLT3 alterations in patients with B-ALL and to correlate these alterations with their clinical course to clarify whether FLT3 is a molecular marker of clinical relevance in children with B-ALL.
This is a retrospective, descriptive clinical study involving pediatric patients diagnosed with B-ALL at a public pediatric oncology referral hospital in Brasília, Brazil. A convenience sampling was performed, including patients admitted between July 2018 and September 2023. Data collected comprised clinical characteristics (age, gender, white blood cell count at diagnosis, and the presence of Central Nervous System—CNS—infiltration), biological characterization (cytogenetic alterations, recurrent rearrangements, and mutations in IKZF1, PAX5, and ERG), and treatment response measured by Minimal Residual Disease (MRD). Outcomes were categorized as remission, relapse or dead in remission.
Patients aged 1–18 years with a primary diagnosis of B-ALL and no prior treatment were included. Between July 2018 and September 2022, treatment was based on the BFM ALLIC 2009 protocol, locally adapted for B-ALL management at our institution. Since September 2022, the GBTLI 2021 protocol—a Brazilian multicenter research protocol in which our institution participates—has been used. As a result, patients in this study followed two different protocols. The backbone strategy for both protocols is similar, though there are differences in the criteria used for risk classification. The GBTLI 2021 protocol reduces the intensity of induction therapy for patients classified as low and intermediate risk (Supplementary Frames S1, S2, and S3).
Bone marrow samples were collected via aspiration to confirm ALL diagnosis. Immediately after collection, smears were prepared for morphological evaluation. Bone marrow aspirate samples (or peripheral blood in cases of high white blood cell counts and patient severity) were sent to the Translational Research Laboratory for immunophenotyping by flow cytometry, cytogenetics (cell culture), and molecular biology analysis.
MRD was evaluated by flow cytometry according to Euroflow Consortium guidelines (50). Cellular events were acquired using the FACS Canto II cytometer (BD), with data analysis performed using FACS Diva (BD) and Infinicyt (Cytognos, version 2.0) software. MRD values were measured at the mid-point and conclusion of the induction phase. For patients treated with the adapted ALLIC BFM 2009 protocol, MRD was assessed on days 15, 33, and 78. For patients following the GBTLI 2021 protocol, MRD quantification was conducted on days 19, 26 (for the low-risk subgroup), and 49.
Mononuclear cells were isolated using a Ficoll gradient (GE Healthcare Life Sciences), washed in 1X PBS, and aliquoted into two aliquots. One tube was used for total RNA extraction via the Trizol® method (Invitrogen), and the other for DNA extraction. DNA was extracted using the Wizard Genomic DNA Purification Kit (Promega) following manufacturer instructions. Samples were quantified via spectrophotometry and stored at −20°C (for DNA) and −80°C (for RNA) until assays were performed.
Screening for genetic variants in the FLT3 tyrosine kinase domain (D835) was conducted on all patients with available samples (n = 155/156) via restriction fragment length polymorphism (RFLP) analysis (51). The PCR reaction included 1x PCR buffer, 200 nM each dNTP, 1.5 mM MgCl₂, 0.75 U Platinum™ Taq DNA Polymerase (Thermo Fisher Scientific), and 0.2 mM of each primer (FWD-CCGCCAGGAACGTGCTTG, REV-CAGCCTCACATTGCCCC). PCR conditions were as follows: 95°C for 3 min, 35 cycles of 95°C for 30 s, 56°C for 30 s, and 72°C for 1 min, followed by a final extension at 72°C for 5 min. PCR products were digested using the restriction enzyme EcoRV (NEB), and samples showing alterations by RFLP were subjected to Sanger sequencing.
Detection of genetic variants in the juxtamembrane domain was performed by fragment analysis (51) on the ABI3500 Genetic Analyzer (Applied Biosystems). PCR was performed under the same conditions described above, but with 27 cycles, using FWD-6-FAM-GCAATTTAGGTATGAAAGCCAGC and REV-CTTTCAGCATTTTGACGGCAACC primers. Fragment size was estimated using the GeneScan™ 500 LIZ size standard (Thermo Fisher Scientific).
For the classification of somatic variant pathogenicity in cancer (oncogenicity), we applied the Standard Operating Procedure developed in accordance with recommendations from the Clinical Genome Resource (ClinGen), the Cancer Genomics Consortium (CGC), and the Variant Interpretation for Cancer Consortium (VICC) (52).
FLT3 expression was analyzed in patients admitted between July 2018 and December 2022 with available samples (n = 112/130) using real-time RT-PCR (RT-qPCR) on bone marrow samples collected at diagnosis. FLT3 expression was also evaluated in in 10 samples collected at relapse. One microgram of RNA was used for cDNA synthesis, followed by PCR with 200 nM primers and 2x PCR MasterMix containing SybrGreen®. Relative quantification was performed using the 2-ΔΔCq method, with HPRT1 and B2M as reference genes. The calibrator was the median ΔCq from all cases in the study, excluding relapses. Singleplex reactions were conducted on a QuantStudio 5 Real-Time PCR System (Thermo Fisher Scientific) with primers for FLT3 (FWD-AGGGACAGTGTACGAAGCTG; REV-GTCGTGCTTAAAGACCCAGAG), HPRT1 (FWD-TGACACTGGCAAAACAATGCA; REV-GGTCCTTTTCACCAGCAAGCT), and B2M (FWD-TGCTGTCTCCATGTTTGATGTATCT; REV-TCTCTGCTCCCCACCTCTAAGT). Amplification conditions were: 2 min at 50°C, 10 min at 95°C for the holding stage, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. Melt curve analysis was performed to assess amplicon specificity.
Data were tested for normal distribution, by the D'Agostino and Pearson normality test and analyses of skewness and kurtosis, when applicable. Data were expressed as average ± SD (qPCR), mean ± SEM or median and ranges according to the distribution. Statistical analysis was performed using GraphPad Prism version 5.00 for Windows (GraphPad Software, San Diego California USA). The statistical approach adopted for each analysis is described in the figure legends. Non-parametric tests were used for data with non-normal distribution and probability values of p < 0.05 were accepted as indication of statistically significant difference.
The study was approved by the local research ethics committee (protocol code 44796221.9.0000.0144. July 04, 2021). Informed consent was obtained from guardians, and assent forms were signed by patients over 5 years old.
A total of 208 patients diagnosed with B-ALL were treated at our institution from July 2018–September 2023, with 156 meeting the inclusion criteria for this study (Figure 1). Patient characterization included age, sex, CNS status, treatment protocol applied, initial and post-induction risk classification, MRD values at mid-induction and post-induction, disease progression and current clinical status, cytogenetic alterations, recurrent rearrangements, presence of iAMP21, IKZF1 deletions (whether associated with the IKZF1 Plus subtype or not), PAX5 alterations, P2RY8::CRLF2 rearrangement, FLT3 mutations, and FLT3 expression levels.

Figure 1. FLT3 mutations and expression assessment in 156 children with B ALL diagnosis (RQ: relative quantification).
The patients' ages ranged from 1–17 years, with a peak incidence between ages 2 and 5, and a median age of 4 years and 5 months. Thirty-four patients (21.8%) were over 10 years old at admission, and the sex ratio was 1.05 (80 males to 76 females). Regarding white blood cell counts at admission, 133 patients (85.2%) presented counts below 50,000 leukocytes/mm³, while 33 patients had counts above 50,000/mm³, including 12 with counts exceeding 100,000/mm³.
Most children (122) received treatment following the ALLIC BFM 2009 protocol. In terms of risk classification at the end of induction, 25 patients (20.4%) were classified as low risk, 74 (60.6%) as intermediate risk, and 23 (18.8%) as high risk. Among those treated under the GBTLI 2021 protocol, risk classifications were as follows: 16 (47%) intermediate risk, 12 (33%) high risk, 4 low risk, and 2 very high risk (Supplementary Table S1). Differences in risk classification criteria and treatment intensity between the two protocols led to a higher frequency of “high-risk” patients under the GBTLI protocol; however, this difference does not imply greater disease aggressiveness in this group.
The 5-year overall survival (OS) and event-free survival (EFS) rates for this cohort were 87.5% and 78%, respectively, based on a minimum 5-year follow-up for 24 patients from the time of diagnosis. The clinical and laboratory data of the patients are presented in Figure 2.
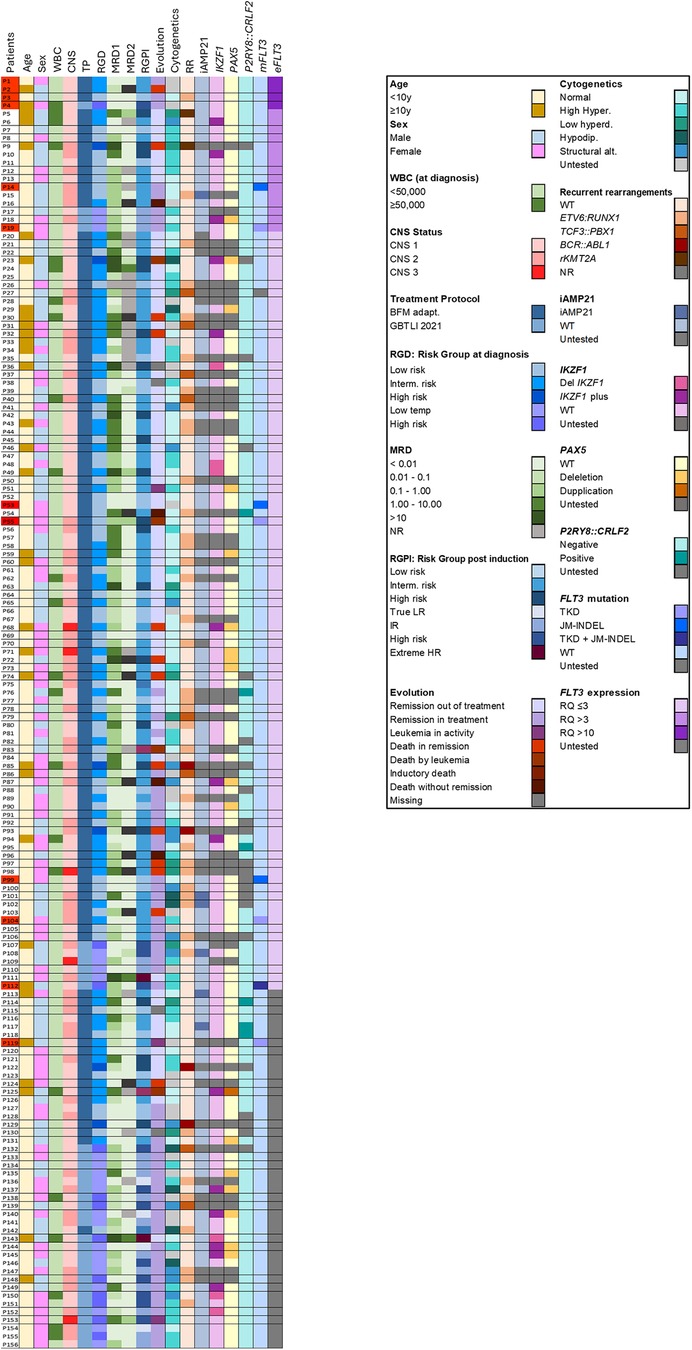
Figure 2. Clinical and molecular profile of the 156 patients included in the study. Interm. Risk: intermediary risk. Low temp: low risk temporary (GBTLI protocol). True LR: true low risk (GBTLI protocol). IR: intermediary risk. Extreme HR: extreme high risk. High hyperd: high hyplerdiploidy; Low hyperdip: Low hyperdiploidy. Hipodip: hypodiploidy. Strutural alt: structural alteration. mFLT3: FLT3 mutation. eFLT3: FLT3 expression. In red, in the left column, patients with FLT3 overexpression (RQ > 10) or mutation are highlighted.
FLT3 mutation screening was conducted in 155 patients (Figure 1), revealing nine mutations in eight patients (5.1%). One type of mutation in the tyrosine kinase domain (p.Ile836del) was identified in two patients. Four patients (2.58%) presented with FLT3-TKD mutations, three patients (1.93%) had in-frame insertions and deletions in the juxtamembrane domain (FLT3-JM-INDEL), and one patient (0.64%) had mutations in both the tyrosine kinase domain (FLT3-TKD) and the juxtamembrane domain (FLT3-JM-INDEL) (Figure 3). No FLT3-ITD variants were found in our cohort.
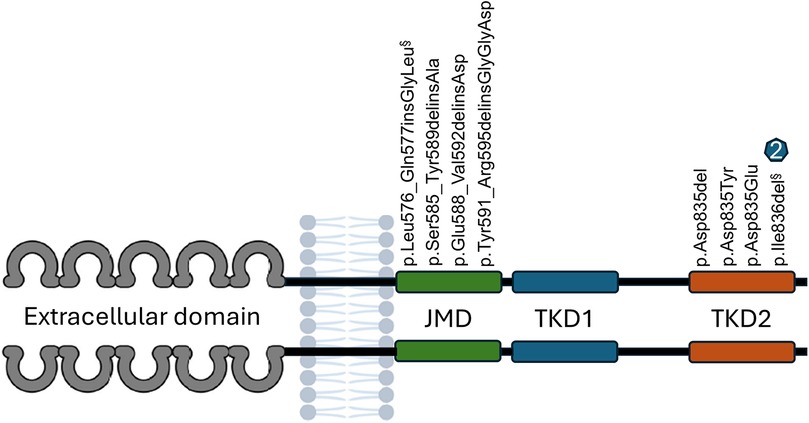
Figure 3. Description of eight distinct FLT3 Gene Mutations in eight (8) patients with B-ALL (nine occurrences). FLT3-JM-INDEL mutations were identified in four patients. Five patients had mutations in the tyrosine kinase domain (FLT3-TKD). One patient exhibited two mutations: one in the juxtamembrane domain (§) and another in the tyrosine kinase domain (§). Two patients presented the p.Ile836del mutation, with one patient having this mutation exclusively and the other having it in conjunction with a FLT3-JM-INDEL (§).
The four mutations identified in the tyrosine kinase domain had been previously documented. Among these, three mutations have been associated with gain of function and oncogenic potential, while one mutation is likely to exhibit oncogenic potential due to a probable gain of function. None of the four mutations identified in the juxtamembrane region had been reported prior to this study. These alterations demonstrated moderate oncogenic potential attributed to a possible gain of function (Table 1).
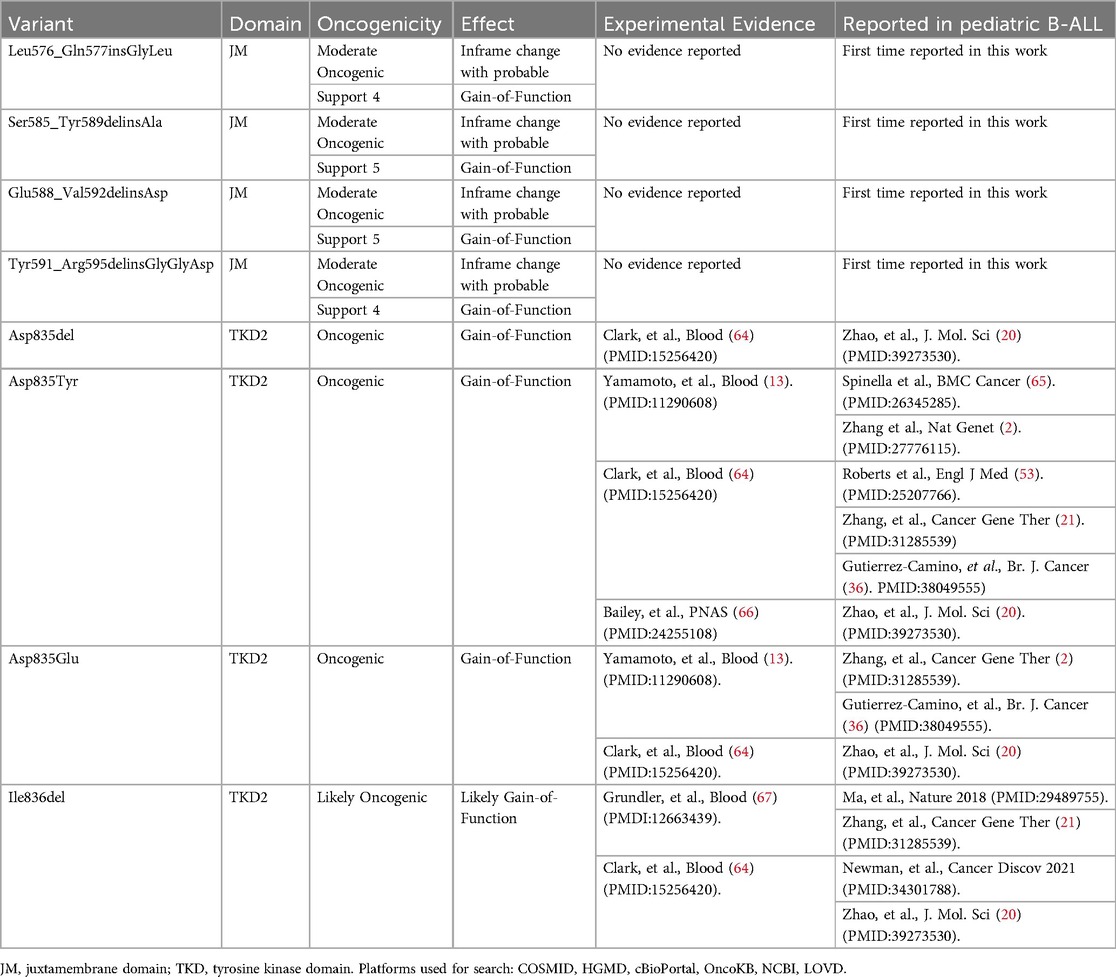
Table 1. Description of FLT3 mutations in patients with B-ALL.
No association was found between FLT3 mutations and age, gender, white blood cell (WBC) count at diagnosis, or minimal residual disease (MRD) values measured at the mid-point and at the end of induction therapy. Table 2 presents the clinical features and detailed descriptions of the FLT3 mutations.
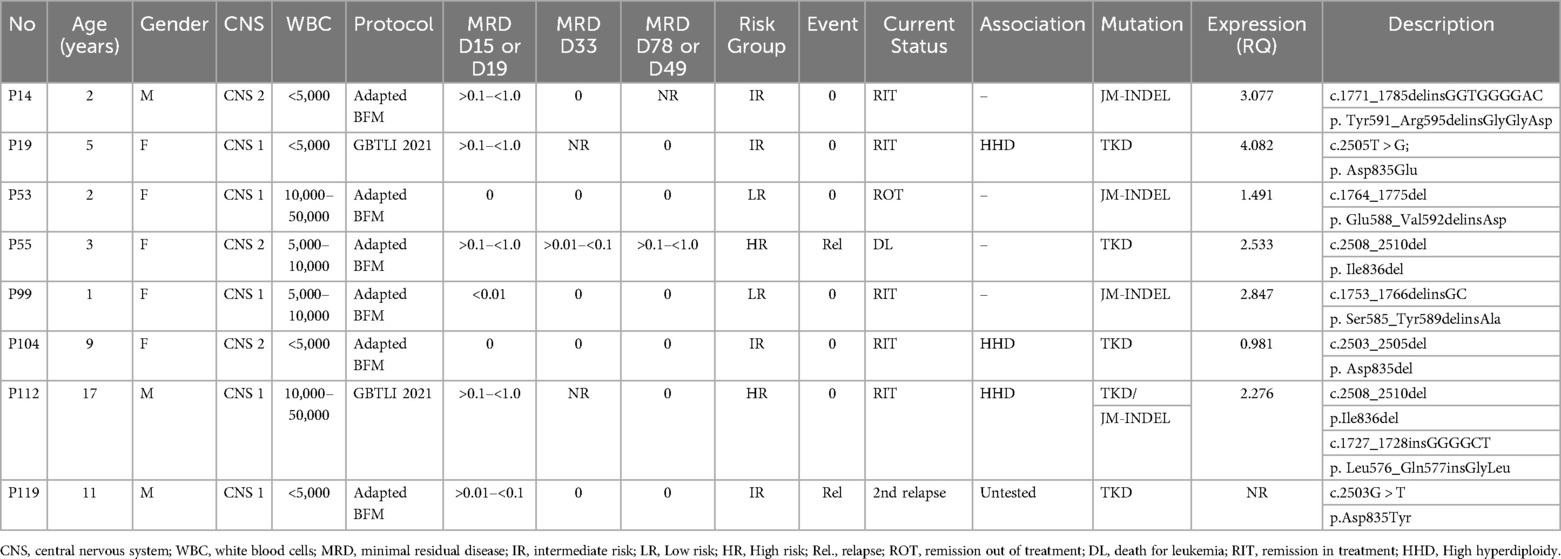
Table 2. Characteristics of patients with FLT3 mutations.
FLT3 mutations were associated with high hyperdiploidy in 3 out of 8 patients (37.5%) (patients P19, P104, P112,). All three cases had TKD-type mutations, and one patient presented with a JM-INDEL associated with a TKD mutation (P112). No FLT3 mutations were identified in any patients with ETV6::RUNX1 (n = 38), BCR::ABL1 (n = 4), TCF3::PBX1 (n = 9), or r-KMT2A (n = 2). Additionally, none of the patients with P2RY8::CRLF2 (n = 7), IKZF1 deletions (n = 21), including IKZF1plus (n = 11), PAX5 alterations (n = 21), or ERG deletions (n = 9) presented with FLT3 mutations (Table 2, Figure 2).
Among patients with FLT3 mutations, a relapse rate of 25% (2/8) was observed, with both cases occurring in patients with the FLT3-TKD mutation and no associated high hyperdiploidy. Although this difference was not statistically significant compared to FLT3 wild-type (WT) patients (p = 0.08), it is noteworthy given the rarity of the mutation. The relapse rate among FLT3 WT patients was 5.4% (8/147) (Figure 4).
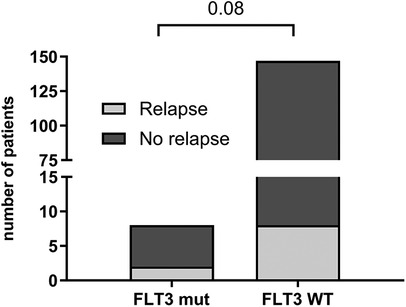
Figure 4. Relapse occurrence in patients with FLT3 gene mutation (n = 8) and wild type—WT (n = 147). Statistically analyzed by Fisher's exact test.
Follow-up of the two relapsed patients with FLT3-TKD mutations revealed that one experienced a very early relapse, failed to achieve remission, and died three months after the relapse diagnosis (FLT3 mutation screening was not conducted on the relapse sample). The other patient achieved remission but experienced a second relapse with an orbital lesion. Treatment included chemotherapy and orbital radiotherapy (20 Gy). The mutation in FLT3 identified in the initial diagnosis sample was also detected in the bone marrow sample collected at the time of relapse. This patient is currently alive, with a follow-up period of 55 months.
There was no difference in survival rates when comparing patients with FLT3 mutations to those with FLT3 WT (Figure 5).

Figure 5. Survival curves for B-ALL pediatric patients according to status of FLT3 mutation (A) (global survival) and (B) (event free survival). Statistically analyzed by Log-rank (Mantel-Cox) test.
FLT3 expression was evaluated in RNA samples of 112 patients at diagnosis. Additionally, FLT3 expression was assessed in 10 samples collected at relapse.
The distribution of relative FLT3 expression in the samples showed a non-Gaussian pattern, with a strong rightward skew (Figure 6A), indicating high expression levels without correspondingly low levels, as would be expected for independent variables with a normal distribution. This pattern is typical of genes with biological significance in carcinogenesis, where elevated expression levels confer an advantage, and lower levels are incompatible with disease maintenance (54, 55).
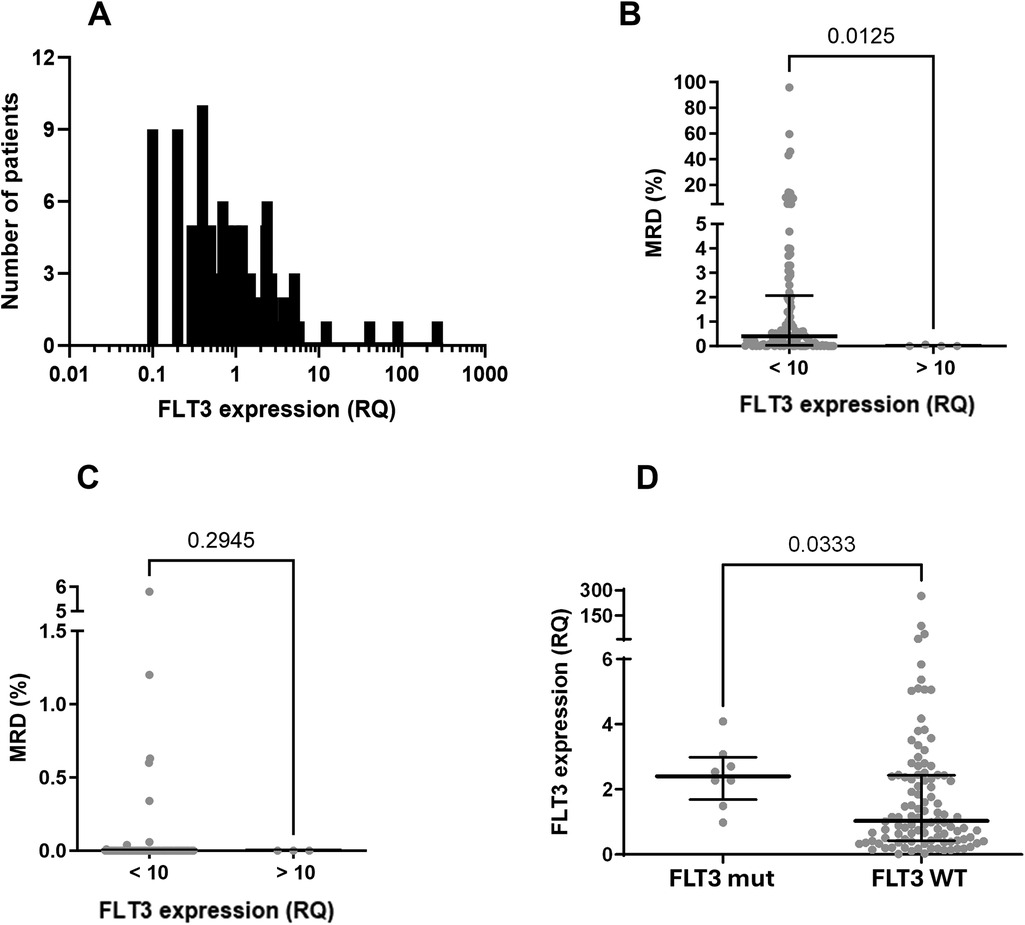
Figure 6. FLT3 expression analysis in samples of children with B-cell ALL. (A) Frequence distribution of FLT3 expression (relative quantification—RQ) in the studied population (attention to the log scale). Data did not pass in Shapiro-Wilk normality test and showed skewness of +8.69. (B) Evaluation of MRD at mid-induction (D15 for patients treated with the adapted BFM ALLIC 2009 protocol and D19 for patients treated with the Brazilian GBTLI 2021 protocol) among patients overexpressing FLT3 (>10) and all the others (<10). Statistical analyses were performed with Mann Whitney test. (C) Evaluation of MRD at the end of induction (D78 for patients treated with the BFL ALLIC 2009 and D49 for the group treated with the GBTLI protocol) among patients overexpressing FLT3 (>10) and all the others (<10). Statistical analyses were performed with Mann Whitney test. (D) FLT3 expression between patients with (FLT3 mut) and without (FLT3 WT) mutations. Statistical analyses were performed with Mann Whitney test.
In 93 patients, the expression ratio (RQ) ranged from 0.017–3. Fifteen patients had expression ratios between 3 and 6.13, and in four patients, the expression ratio exceeded 10, which we classified as “hyperexpression.” These elevated RQ values were observed exclusively in four wild-type (WT) FLT3 patients. We were unable to identify factors associated with FLT3 hyperexpression in these four patients with RQ > 10 (see Table 3), but we observed that these individuals demonstrated a rapid response to treatment, as indicated by MRD values assessed during mid-induction (D15 or D19, depending on the protocol) (Figure 6B). Conversely, there was no statistically significant difference in MRD values at the end of induction between patients with hyperexpression and those without (Figure 6C).
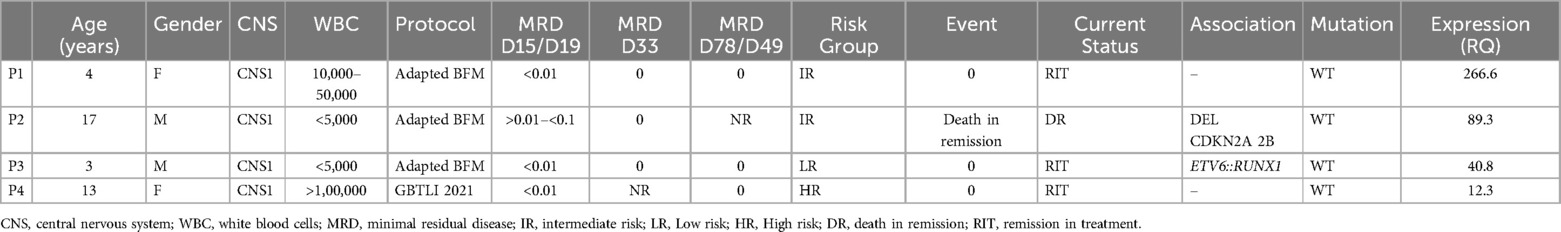
Table 3. Characteristics of patients with FLT3 overexpression.
Although none of the patients with FLT3 mutations exhibited hyperexpression of the gene (RQ > 10), FLT3 expression values were higher among patients with FLT3 mutations compared to wild-type patients (median 2.53 vs. 1.03, p = 0.03) (Figure 6D).
No significant differences in FLT3 expression values were observed among patients across different risk groups (data not shown).
A noteworthy finding is that FLT3 expression levels were significantly lower in relapse cases. Patients who relapsed had relative expression levels (at initial diagnosis) comparable to the group median (Figure 7A). However, these values decreased markedly after relapse (Figure 7B). This difference is even more pronounced when comparing paired expression values at diagnosis and at relapse for each patient (Figure 7C). Despite this, FLT3 expression levels had no significant impact on survival rates (EFS and OS) (Figure 8, Supplementary Table S2).
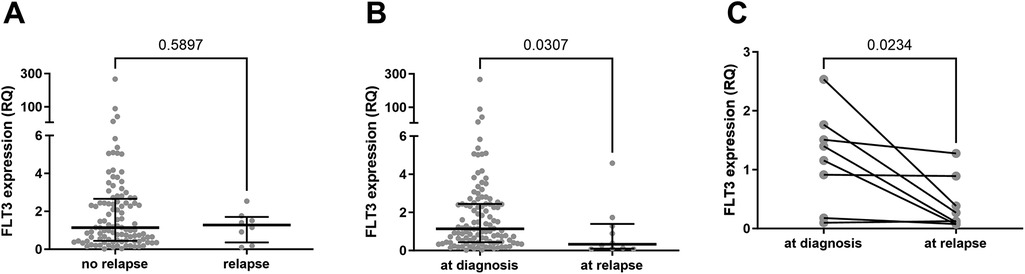
Figure 7. FLT3 expression levels analysis in relapsed patients vs. patients in remission. (A) Comparison between FLT3 expression values of samples from 103 patients initially diagnosed with B-cell ALL who did not experience relapse until last data assessment and FLT3 expression values in BM samples collected at the initial diagnosis from 8 patients who relapsed. Statistically analyzed by Mann Whitney test. (B) Comparison between FLT3 expression values of samples from 112 patients initially diagnosed with B-cell ALL and FLT3 expression values of BM samples collected at the time of relapse from 10 patients. Statistically analyzed by Mann Whitney test. (C) FLT3 expression levels of 8 samples from patients at the initial diagnosis and at the time of relapse. The patient with the highest RQ value (2.5) at diagnosis is the only one in the group with FLT3 mutation. Statistically analyzed by Wilcoxon matched-pairs signed rank test.
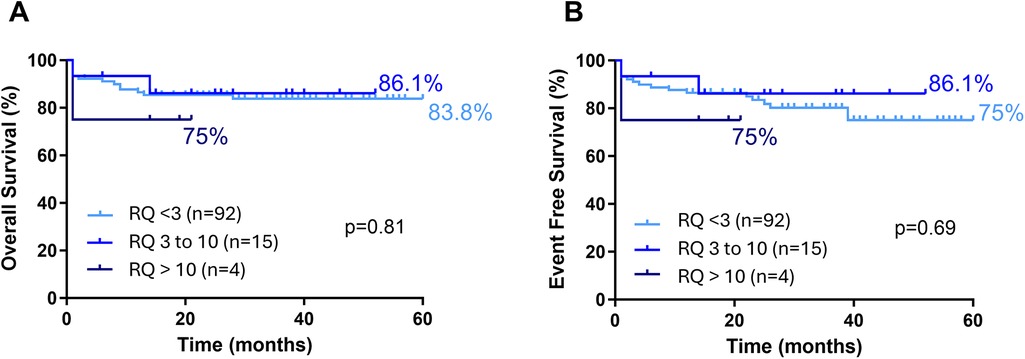
Figure 8. Survival curves for B ALL pediatric patients according to FLT3 expression status. (A) (Overal Survival) and (B) (Event free survival). Statistically analyzed by Log-rank (Mantel-Cox) test.
Limited studies have investigated the implications of the FLT3 gene in B-cell Acute Lymphoblastic Leukemia (B-ALL), likely due to the infrequent occurrence of FLT3 alterations. Nonetheless, in recent years, FLT3 has emerged as a significant marker for enhancing the biological characterization of patients with ALL, particularly within specific subtypes such as Ph-like and r-KMT2A ALL (31, 32, 44, 53, 56). Furthermore, several researchers have emphasized the need for additional studies to elucidate the role of FLT3 in B-ALL patients (20, 31, 33). Although this study is based on data from a single institution, it complements and builds upon previous research. Moreover, it is notable for evaluating a population with a distinct genetic composition, characterized by a mixture of various ethnic backgrounds (indigenous, African, Caucasian).
In our study, we found FLT3 mutations in 5.1% of the samples (8/155), with 3.2% (5/155) of mutations occurring in the TKD domain (in one case, associated with JM-INDEL). Unlike acute myeloid leukemia (AML), the variants found in the juxtamembrane domain were indels rather than internal tandem duplications (ITDs).
These findings are consistent with those reported in previous studies regarding both the frequency of mutations and the different spectrum of FLT3 mutations in B-cell acute lymphoblastic leukemia (B-ALL) compared to AML (20, 21). Taketani et al. (2004) found FLT3-TKD mutations in 6 (5.4%) of 112 children with ALL older than 1 year and in 8 (16.0%) of 50 infants with ALL, but no FLT3-ITD mutations were detected (32). Zhang et al. (2020) reported a predominance of JM-INDELs in B-ALLs (21). In Saudi Arabia, 4.7% of children with B-ALL had FLT3 mutations (2.4% ITD) (33), and in Brazil, Barbosa et al. found FLT3 mutations in 6.7% of 134 B-ALL patients (34). Additionally, this frequency was reported to be 5.5% among Canadian children and adolescents with ALL, including 1.1% FLT3-ITD and 4.3% FLT3-TKD point mutations, with an observed association between FLT3 mutations and hyperdiploidy (36).
We recognize that broader horizontal coverage and the ability to simultaneously detect multiple genetic alterations [single nucleotide variants [SNVs], copy number variations [CNVs], fusions, and indels] using highly sensitive techniques like next-generation sequencing (NGS) allow for the identification of FLT3 mutations at higher frequencies than restriction fragment length polymorphism (RFLP) and fragment analysis (20, 44). While NGS provides comprehensive genomic insights, it is time-consuming and may produce false negatives due to amplification issues or software limitations in detecting FLT3 internal tandem duplications (FLT3-ITD). In contrast, PCR fragment analysis and PCR-RFLP targeting FLT3-TKD (D835/I836) mutations are robust, cost-effective methods that deliver faster results, making them a viable and efficient alternative to NGS (57, 58).
In a recently published study, Zhao et al. described a higher frequency of FLT3 mutations in ALL patients when evaluated by NGS (6.3% ITD and 18.8% TKD) and identified new non-canonical genetic variants, such as point mutations outside the TKD and insertion/deletion variants causing in-frame amino acid alterations. In the same study, a higher proportion of patients with negative minimal residual disease (MRD) at mid-induction (D19) was observed among patients with FLT3 mutations compared to wild-type patients (20). In our sample, we did not identify differences in MRD levels between patients with or without FLT3 mutations.
Regarding initial presentation, unlike what is well established in patients with AML, where FLT3 mutations are associated with elevated leukocyte counts at diagnosis (22, 23, 59), none of the patients with FLT3 mutations in our study presented with hyperleukocytosis. Furthermore, no patients with FLT3 mutations had central nervous system (CNS) involvement at diagnosis.
In our sample, FLT3 mutations were not detected in patients with recurrent genetic alterations commonly associated with B-ALL, including ETV6::RUNX1, TCF3::PBX1, BCR::ABL1, r-KMT2A, P2RY8::CRLF2 rearrangements, PAX5 alterations, or deletions involving IKZF1. This finding supports the hypothesis that, contrary to the prevailing concept of kinase alterations being secondary (31, 60), FLT3 mutations may function as leukemogenic drivers in a small subset of B-ALL (5, 61). Further studies utilizing bone marrow samples collected during treatment monitoring or at relapse could clarify the role of FLT3 mutations in disease progression. Preliminary studies suggest that FLT3 mutations negatively impact prognosis in infants with r-KMT2A B-ALL (31, 49). However, in patients with hyperdiploidy, the presence of FLT3 mutations did not affect the natural course of the disease in this subgroup, which generally has a favorable outcome (31–33). In our study, among the five patients with TKD mutations, three who also had hyperdiploidy achieved remission, while the other two without hyperdiploidy experienced relapse. In light of these results and based on evidence of an association between hyperdiploidy and higher levels of FLT3 expression, as well as previous studies linking elevated FLT3 expression with certain subtypes of B-ALL (r-KMT2A, Ph-like), we decided to complement our research by evaluating FLT3 expression levels.
Higher FLT3 expression levels are described and considered recurrent alterations in acute leukemias. The expression levels of this gene are higher in cells from patients with acute leukemias compared to normal bone marrow samples and other types of neoplasms (37–43). In the context of B-ALL, subgroups of patients with r-KMT2A, high hyperdiploidy, and r-ZNF384 subtypes exhibit higher FLT3 expression levels (5, 36, 37, 40, 42). In our study, which was limited to patients older than one year, only two patients had r-KMT2A, both with expression values above the 75th percentile of our cohort.
Although the association between hyperdiploidy and higher levels of FLT3 expression has been documented in some studies (44, 61, 62), we did not find differences in FLT3 expression in this group. Similarly, there was no correlation between FLT3 expression and indicators of aggressiveness in B-ALL, such as white blood cell count at diagnosis or CNS involvement. This lack of association may be specific to our study population; however, expanding the sample size is necessary to confirm these findings.
Additionally, we were unable to identify any common characteristics among the four patients with extremely high expression levels (RQ > 10). Yang and colleagues demonstrated that epigenetic alterations with enhancer hijacking secondary to the deletion of the PAN3 gene (13q12.2) explain elevated FLT3 expression in B-ALL patients, particularly among those with hyperdiploidy or those who experienced relapse. Although none of the four patients with RQ > 10 had associated hyperdiploidy or experienced disease relapse, this could be a possible mechanism to explore in these cases (42).
The implications of FLT3 expression levels on prognosis remain uncertain and controversial. Among patients with r-KMT2A B-ALL, high FLT3 expression levels have been associated with poorer outcomes (38, 40, 49). However, in 2017, Fedders and colleagues found an opposite association (63). In our study, we were unable to identify any influence on survival rates in patients with B-ALL and FLT3 overexpression detected in bone marrow samples at diagnosis. The majority of authors also did not find an association between FLT3 expression levels at diagnosis and survival or relapse rates (20, 36). On the other hand, Garza Veloz reported that high FLT3 expression levels at the end of induction were associated with higher relapse and mortality rates (40).
We identified evidence that FLT3 may be a marker influencing the biological behavior of B-ALL, reinforcing the need for more in-depth studies to better elucidate FLT3's relationship with mechanisms involved in the genesis, survival, or resistance of leukemic cells to chemotherapeutic effects: 1. the frequency distribution of relative FLT3 expression (Figure 6); 2. the low MRD values observed during mid-induction in patients with high FLT3 expression (Figure 6); and 3. significantly lower FLT3 expression levels in samples from relapsed patients (Figure 7).
Given these results, the detailed biological effects and prognostic impact of FLT3 expression levels should be further investigated, particularly in the context of the potential use of FLT3 inhibitors. Additionally, it is important to not only focus on transcript expression levels but also evaluate the true functional impact of FLT3 by assessing receptor saturation or activation.
The small sample size, particularly given the low frequency of FLT3 mutations, along with the limited follow-up time for the patients, constrains the interpretation of our findings and underscores the necessity for continued research in this area. We acknowledge the inherent limitations of Restriction Fragment Length Polymorphism (RFLP) and fragment analysis in detecting low-frequency FLT3 mutations, especially when compared to the enhanced sensitivity of Next-Generation Sequencing (NGS).
Nevertheless, considering the potential for direct and short-term benefits for selected patients, we believe it is crucial to disseminate our findings, even with the limited follow-up period. In light of these limitations, we assert that our findings contribute meaningfully to the understanding of FLT3 mutation prevalence and advocate for further research and investment in comprehensive molecular diagnostics in similar contexts.
Preclinical studies and case reports provide evidence supporting the use of FLT3 inhibitors in specific subgroups of B-ALL patients (3, 45, 46, 56). However, although the use of FLT3 inhibitors has been suggested as a therapeutic option for B-ALL patients for nearly two decades, studies exploring this approach remain scarce (46, 56).
We propose that in selected cases, alongside MRD assessment, evaluating FLT3 status (mutation or expression) may serve as an additional tool in guiding treatment strategies. For instance, in patients with persistent MRD following induction or in relapse cases with FLT3 mutations or increased expression, FLT3 inhibitors could be considered as salvage treatment. Another option is to use FLT3 inhibitors in combination with conventional chemotherapy to reduce the required doses in patients with a favorable prognosis. FLT3 inhibitors may also be beneficial for patients with FLT3 mutation or overexpression where high-dose chemotherapy is contraindicated, such as in cases of infection or toxicity.
In conclusion, although we did not find consistent data regarding the impact of FLT3 mutations or elevated FLT3 expression levels on patient response (measured by MRD values after induction) or relapse rates, our findings suggest that FLT3 alterations — whether genetic or expression-related —exert a biological influence on the behavior of leukemic cells and may complement traditional tools used to enhance B-ALL characterization. Moreover, these alterations may help guide therapeutic strategies for selected B-ALL patients by utilizing FLT3 inhibitors.
Currently, there is no evidence supporting the inclusion of FLT3 inhibitors as a first-line therapeutic approach for B-ALL patients with FLT3 alterations. However, we believe that in cases of persistent MRD positivity following induction therapy or in instances of relapse, the use of FLT3 inhibitors should be considered as salvage therapy for patients with FLT3 mutations or elevated FLT3 expression. An additional, somewhat more ambitious possibility worth exploring is the combination of FLT3 inhibitors with conventional treatment to reduce chemotherapy doses.
This hypothesis requires validation in larger cohorts and through studies utilizing samples collected at different stages of leukemia treatment. Despite these limitations, our study highlights a relatively unexplored aspect of B-ALL with promising translational potential, paving the way for more personalized treatment approaches.
The authors acknowledge that the data presented in this study must be deposited and made publicly available in an acceptable repository, prior to publication. Frontiers cannot accept a manuscript that does not adhere to our open data policies.
The studies involving humans were approved by Local research ethics committee of Children's Hospital of Brasilia. Protocol code 44796221.9.0000.0144 (July 04, 2021). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants' legal guardians/next of kin.
EB: Writing – review & editing, Writing – original draft, Investigation, Formal Analysis, Data curation, Conceptualization. BG: Writing – original draft, Writing – review & editing, Methodology, Investigation, Formal Analysis, Data curation. LC: Writing – original draft, Writing – review & editing, Visualization, Investigation, Formal Analysis, Data curation. AS: Writing – original draft, Writing – review & editing, Visualization, Methodology, Formal Analysis. RP: Writing – original draft, Writing – review & editing, Visualization, Formal Analysis. FF: Writing – original draft, Writing – review & editing, Visualization, Investigation, Formal Analysis. JC: Writing – original draft, Writing – review & editing, Supervision, Investigation, Funding acquisition. IM: Writing – original draft, Writing – review & editing, Supervision, Investigation. DO: Writing – review & editing, Writing – original draft, Methodology, Formal Analysis, Conceptualization. RC: Writing – original draft, Writing – review & editing, Supervision, Resources, Project administration, Methodology, Funding acquisition, Formal Analysis.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was funded by FUNDAÇÃO DE APOIO À PESQUISA DO DISTRITO FEDERAL (FAPDF) and FUNDAÇÃO DE ENSINO E PESQUISA EM CIÊNCIAS DA SAÚDE (FEPECS).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Gen AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2024.1505060/full#supplementary-material
1. Inaba H, Pui CH. Advances in the diagnosis and treatment of pediatric acute lymphoblastic leukemia. J Clin Med. (2021) 10(9):1926. doi: 10.3390/jcm10091926
2. Zhang L, Habeebu SSM, Li W. Chapter 10. Prognostic and predictive biomarkers in precursor B-cell acute lymphoblastic leukemia. In: Li W, editor. Leukemia. Brisbane, AU: Exon Publications (2022). p. 165–93.
3. Mullighan CG. How advanced are we in targeting novel subtypes of ALL? Best Pract Res Clin Haematol. (2019) 32(4):101095. doi: 10.1016/j.beha.2019.101095
4. Iacobucci I, Kimura S, Mullighan CG. Biologic and therapeutic implications of genomic alterations in acute lymphoblastic leukemia. J Clin Med. (2021) 10(17):3792. doi: 10.3390/jcm10173792
5. Pieters R, Mullighan CG, Hunger SP. Advancing diagnostics and therapy to reach universal cure in childhood ALL. J Clin Oncol. (2023) 41(36):5579–91. doi: 10.1200/JCO.23.01286
6. Rosnet O, Schiff C, Pébusque MJ, Marchetto S, Tonnelle C, Toiron Y, et al. Human FLT3/FLK2 gene: cDNA cloning and expression in hematopoietic cells. Blood. (1993) 82(4):1110–9. doi: 10.1182/blood.V82.4.1110.1110
7. Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. (2010) 141(7):1117–34. doi: 10.1016/j.cell.2010.06.011
8. Matthews W, Jordan CT, Wiegand GW, Pardoll D, Lemischka IR. A receptor tyrosine kinase specific to hematopoietic stem and progenitor cell-enriched populations. Cell. (1991) 65(7):1143–52. doi: 10.1016/0092-8674(91)90010-v
9. Lyman SD, James L, Vanden Bos T, de Vries P, Brasel K, Gliniak B, et al. Molecular cloning of a ligand for the flt3/flk-2 tyrosine kinase receptor: a proliferative factor for primitive hematopoietic cells. Cell. (1993) 75(6):1157–67. doi: 10.1016/0092-8674(93)90325-K
10. Lyman SD, James L, Johnson L, Brasel K, de Vries P, Escobar SS, et al. Cloning of the human homologue of the murine FLT3 ligand: a growth factor for early hematopoietic progenitor cells. Blood. (1994) 83(10):2795–801. doi: 10.1182/blood.V83.10.2795.2795
11. Lyman SD. Biology of FLT3 ligand and receptor. Int J Hematol. (1995) 62(2):63–73. doi: 10.1016/0925-5710(95)00389-a
12. Stirewalt DL, Radich JP. The role of FLT3 in haematopoietic malignancies. Nat Rev Cancer. (2003) 3(9):650–65. doi: 10.1038/nrc1169
13. Yamamoto Y, Kiyoi H, Nakano Y, Suzuki R, Kodera Y, Miyawaki S, et al. Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood. (2001) 97(8):2434–9. doi: 10.1182/blood.v97.8.2434
14. Vu HA, Xinh PT, Masuda M, Motoji T, Toyoda A, Sakaki Y, et al. FLT3 is fused to ETV6 in a myeloproliferative disorder with hypereosinophilia and a t(12;13)(p13;q12) translocation. Leukemia. (2006) 20(8):1414–21. doi: 10.1038/sj.leu.2404266
15. Spitzer B, Dela Cruz FS, Ibanez Sanchez GD, Zhang Y, Xiao W, Benayed R, et al. ETV6-FLT3-positive myeloid/lymphoid neoplasm with eosinophilia presenting in an infant: an entity distinct from JMML. Blood Adv. (2021) 5(7):1899–902. doi: 10.1182/bloodadvances.2020003699
16. Reilly JT. Class III receptor tyrosine kinases: role in leukaemogenesis. Br J Haematol. (2002) 116(4):744–57. doi: 10.1046/j.0007-1048.2001.03294.x
17. Huang K, Yang M, Pan Z, Heidel FH, Scherr M, Eder M, et al. Leukemogenic potency of the novel FLT3-N676K mutant. Ann Hematol. (2016) 95(5):783–91. doi: 10.1007/s00277-016-2616-z
18. Ge SS, Qiu QC, Dai HP, Shen XD, Wu TM, Du JH, et al. Mutation spectrum of FLT3 and significance of non-canonical FLT3 mutations in haematological malignancy. Br J Haematol. (2023) 202(3):539–49. doi: 10.1111/bjh.18877
19. Annesley CE, Brown P. The biology and targeting of FLT3 in pediatric leukemia. Front Oncol. (2014) 4:263. doi: 10.3389/fonc.2014.00263
20. Zhao L, Chen H, Lan F, Hao J, Zhang W, Li Y, et al. Distinct FLT3 pathways gene expression profiles in pediatric de novo acute lymphoblastic and myeloid leukemia with FLT3 mutations: implications for targeted therapy. Int J Mol Sci. (2024) 25(17):9581. doi: 10.3390/ijms25179581
21. Zhang Y, Zhang Y, Wang F, Wang M, Liu H, Chen X, et al. The mutational spectrum of FLT3 gene in acute lymphoblastic leukemia is different from acute myeloid leukemia. Cancer Gene Ther. (2020) 27(1-2):81–8. doi: 10.1038/s41417-019-0120-z
22. Kennedy VE, Smith CC. FLT3 mutations in acute myeloid leukemia: key concepts and emerging controversies. Front Oncol. (2020) 10:612880. doi: 10.3389/fonc.2020.612880
23. Knight TE, Edwards H, Meshinchi S, Taub JW, Ge Y. “FLipping” the story: FLT3-mutated acute myeloid leukemia and the evolving role of FLT3 inhibitors. Cancers. (2022) 14(14):3398. doi: 10.3390/cancers14143398
24. Fedorov K, Maiti A, Konopleva M. Targeting FLT3 mutation in acute myeloid leukemia: current strategies and future directions. Cancers. (2023) 15(8):2312. doi: 10.3390/cancers15082312
25. Kottaridis PD, Gale RE, Frew ME, Harrison G, Langabeer SE, Belton AA, et al. The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: analysis of 854 patients from the United Kingdom medical research council AML 10 and 12 trials. Blood. (2001) 98(6):1752–9. doi: 10.1182/blood.v98.6.1752
26. Ge SS, Liu SB, Xue SL. Developments and challenges of FLT3 inhibitors in acute myeloid leukemia. Front Oncol. (2022) 12:996438. doi: 10.3389/fonc.2022.996438
27. Pollard JA, Alonzo TA, Gerbing R, Brown P, Fox E, Choi J, et al. Sorafenib in combination with standard chemotherapy for children with high allelic ratio FLT3/ITD+ acute myeloid leukemia: a report from the children’s oncology group protocol AAML1031. J Clin Oncol. (2022) 40(18):2023–35. doi: 10.1200/JCO.21.01612
28. Negotei C, Colita A, Mitu I, Lupu AR, Lapadat ME, Popovici CE, et al. A review of FLT3 kinase inhibitors in AML. J Clin Med. (2023) 12(20):6429. doi: 10.3390/jcm12206429
29. Kantarjian H, Borthakur G, Daver N, DiNardo CD, Issa G, Jabbour E, et al. Current status and research directions in acute myeloid leukemia. Blood Cancer J. (2024) 14(1):163. doi: 10.1038/s41408-024-01143-2
30. Stone RM, Mandrekar SJ, Sanford BL, Laumann K, Geyer S, Bloomfield CD, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med. (2017) 377(5):454–64. doi: 10.1056/NEJMoa1614359
31. Armstrong SA, Mabon ME, Silverman LB, Li A, Gribben JG, Fox EA, et al. FLT3 mutations in childhood acute lymphoblastic leukemia. Blood. (2004) 103(9):3544–6. doi: 10.1182/blood-2003-07-2441
32. Taketani T, Taki T, Sugita K, Furuichi Y, Ishii E, Hanada R, et al. FLT3 mutations in the activation loop of tyrosine kinase domain are frequently found in infant ALL with MLL rearrangements and pediatric ALL with hyperdiploidy. Blood. (2003) 103(3):1085–8. doi: 10.1182/blood-2003-02-0418
33. Elyamany G, Awad M, Alsuhaibani O, Fadalla K, Al Sharif O, Al Shahrani M, et al. FLT3 internal tandem duplication and D835 mutations in patients with acute lymphoblastic leukemia and its clinical significance. Mediterr J Hematol Infect Dis. (2014) 6(1):e2014038. doi: 10.4084/MJHID.2014.038
34. Barbosa TC, Andrade FG, Lopes BA, de Andrade CF, Mansur MB, Emerenciano M, et al. Impact of mutations in FLT3, PTPN11 and RAS genes on the overall survival of pediatric B cell precursor acute lymphoblastic leukemia in Brazil. Leuk Lymphoma. (2014) 55(7):1501–9. doi: 10.3109/10428194.2013.847934
35. Alkhayat N, Elborai Y, Al Sharif O, Al Shahrani M, Alsuhaibani O, Awad M, et al. Cytogenetic profile and FLT3 gene mutations of childhood acute lymphoblastic leukemia. Clin Med Insights Oncol. (2017) 11:1179554917721710. doi: 10.1177/1179554917721710
36. Gutierrez-Camino A, Richer C, Ouimet M, Fuchs C, Langlois S, Khater F, et al. Characterisation of FLT3 alterations in childhood acute lymphoblastic leukaemia. Br J Cancer. (2024) 130(2):317–26. doi: 10.1038/s41416-023-02511-8
37. Stam RW, Schneider P, de Lorenzo P, Valsecchi MG, den Boer ML, Pieters R. Prognostic significance of high-level FLT3 expression in MLL-rearranged infant acute lymphoblastic leukemia. Blood. (2007) 110(7):2774–5. doi: 10.1182/blood-2007-05-091934
38. Chillón MC, Gómez-Casares MT, López-Jorge CE, Rodriguez-Medina C, Molines A, Sarasquete ME, et al. Prognostic significance of FLT3 mutational status and expression levels in MLL-AF4+ and MLL-germline acute lymphoblastic leukemia. Leukemia. (2012) 26(11):2360–6. doi: 10.1038/leu.2012.161
39. Kang H, Wilson CS, Harvey RC, Chen IM, Murphy MH, Atlas SR, et al. Gene expression profiles predictive of outcome and age in infant acute lymphoblastic leukemia: a children’s oncology group study. Blood. (2012) 119(8):1872–81. doi: 10.1182/blood-2011-10-382861
40. Garza-Veloz I, Martinez-Fierro ML, Jaime-Perez JC, Carrillo-Sanchez K, Ramos-Del Hoyo MG, Lugo-Trampe A, et al. Identification of differentially expressed genes associated with prognosis of B acute lymphoblastic leukemia. Dis Markers. (2015) 2015:828145. doi: 10.1155/2015/828145
41. Poubel CP, Mansur MB, Boroni M, Emerenciano M. FLT3 Overexpression in acute leukaemias: new insights into the search for molecular mechanisms. Biochim Biophys Acta Rev Cancer. (2019) 1872(1):80–8. doi: 10.1016/j.bbcan.2019.06.001
42. Yang M, Safavi S, Woodward EL, Duployez N, Olsson-Arvidsson L, Ungerbäck J, et al. 13q12.2 deletions in acute lymphoblastic leukemia lead to upregulation of FLT3 through enhancer hijacking. Blood. (2020) 136(8):946–56. doi: 10.1182/blood.2019004684
43. Ozeki K, Kiyoi H, Hirose Y, Iwai M, Ninomiya M, Kodera Y, et al. Biologic and clinical significance of the FLT3 transcript level in acute myeloid leukemia. Blood. (2004) 103(5):1901–8. doi: 10.1182/blood-2003-06-1845
44. Brady SW, Roberts KG, Gu Z, Shi L, Pounds S, Pei D, et al. The genomic landscape of pediatric acute lymphoblastic leukemia. Nat Genet. (2022) 54(9):1376–89. doi: 10.1038/s41588-022-01159-z
45. Armstrong SA, Kung AL, Mabon ME, Silverman LB, Stam RW, Den Boer ML, et al. Inhibition of FLT3 in MLL. Validation of a therapeutic target identified by gene expression based classification. Cancer Cell. (2003) 3(2):173–83. doi: 10.1016/s1535-6108(03)00003-5
46. Rinella SP, Bell HC, Hess NJ, Hoang NM, Nguyen TT, Turicek DP, et al. Combination fedratinib and venetoclax has activity against human B-ALL with high FLT3 expression. bioRxiv [Preprint]. 2023.06.07.544058 (2023). doi: 10.1101/2023.06.07.544058
47. Hu M, Li W, Li P, Tan J, Wang Y. A case report of secondary B-cell acute lymphoblastic leukemia treated with a combination of FLT3 inhibitor and decitabine. Front Oncol. (2024) 14:1329279. doi: 10.3389/fonc.2024.1329279
48. Madero-Marroquin R, DuVall AS, Saygin C, Wang P, Gurbuxani S, Larson RA, et al. Durable responses in acute lymphoblastic leukaemia with the use of FLT3 and IDH inhibitors. Br J Haematol. (2024) 204(4):1238–42. doi: 10.1111/bjh.19250
49. Brown PA, Kairalla JA, Hilden JM, Dreyer ZE, Carroll AJ, Heerema NA, et al. FLT3 Inhibitor lestaurtinib plus chemotherapy for newly diagnosed KMT2A-rearranged infant acute lymphoblastic leukemia: children’s oncology group trial AALL0631. Leukemia. (2021) 35(5):1279–90. doi: 10.1038/s41375-021-01177-6 Erratum in: Leukemia. 2021 May;35(5):1527. doi: 10.1038/s41375-021-01245-x.33623141
50. Theunissen P, Mejstrikova E, Sedek L, van der Sluijs-Gelling AJ, Gaipa G, Bartels M, et al. Euroflow consortium. standardized flow cytometry for highly sensitive MRD measurements in B-cell acute lymphoblastic leukemia. Blood. (2017) 129(3):347–57. doi: 10.1182/blood-2016-07-726307
51. Murphy KM, Levis M, Hafez MJ, Geiger T, Cooper LC, Smith BD, et al. Detection of FLT3 internal tandem duplication and D835 mutations by a multiplex polymerase chain reaction and capillary electrophoresis assay. J Mol Diagn. (2003) 5(2):96–102. doi: 10.1016/S1525-1578(10)60458-8
52. Horak P, Griffith M, Danos AM, Pitel BA, Madhavan S, Liu X, et al. Standards for the classification of pathogenicity of somatic variants in cancer (oncogenicity): joint recommendations of clinical genome resource (ClinGen), cancer genomics consortium (CGC), and variant interpretation for cancer consortium (VICC). Genet Med. (2022) 24(5):986–98. doi: 10.1016/j.gim.2022.01.001 Erratum in: Genet Med. 2022 Sep;24(9):1991. doi: 10.1016/j.gim.2022.07.001.35101336
53. Roberts KG, Li Y, Payne-Turner D, Harvey RC, Yang YL, Pei D, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. New Engl J Med. (2014) 371:1005–15. doi: 10.1056/NEJMoa1403088
54. Feinberg AP, Levchenko A. Epigenetics as a mediator of plasticity in cancer. Science. (2023) 379(6632):eaaw3835. doi: 10.1126/science.aaw3835
55. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
56. Brown P, Levis M, Shurtleff S, Campana D, Downing J, Small D. FLT3 Inhibition selectively kills childhood acute lymphoblastic leukemia cells with high levels of FLT3 expression. Blood. (2005) 105(2):812–20. doi: 10.1182/blood-2004-06-2498
57. Sakaguchi M, Nakajima N, Yamaguchi H, Najima Y, Shono K, Marumo A, et al. The sensitivity of the FLT3-ITD detection method is an important consideration when diagnosing acute myeloid leukemia. Leuk Res Rep. (2020) 13:100198. doi: 10.1016/j.lrr.2020.100198
58. Bergeron J, Capo-Chichi JM, Tsui H, Mahe E, Berardi P, Minden MD, et al. The clinical utility of FLT3 mutation testing in acute leukemia: a Canadian consensus. Curr Oncol. (2023) 30(12):10410–36. doi: 10.3390/curroncol30120759
59. Kiyoi H, Kawashima N, Ishikawa Y. FLT3 mutations in acute myeloid leukemia: therapeutic paradigm beyond inhibitor development. Cancer Sci. (2020) 111(2):312–22. doi: 10.1111/cas.14274
60. Malard F, Mohty M. Acute lymphoblastic leukaemia. Lancet. (2020) 395(10230):1146–62. doi: 10.1016/S0140-6736(19)33018-1
61. Yeoh EJ, Ross ME, Shurtleff SA, Williams WK, Patel D, Mahfouz R, et al. Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell. (2002) 1(2):133–43. doi: 10.1016/s1535-6108(02)00032-6
62. Yang M, Vesterlund M, Siavelis I, Moura-Castro LH, Castor A, Fioretos T, et al. Proteogenomics and Hi-C reveal transcriptional dysregulation in high hyperdiploid childhood acute lymphoblastic leukemia. Nat Commun. (2019) 10(1):1519. doi: 10.1038/s41467-019-09469-3
63. Fedders H, Alsadeq A, Schmah J, Vogiatzi F, Zimmermann M, Moricke A, et al. The role of constitutive activation of FMS-related tyrosine kinase-3 and NRas/KRas mutational status in infants with KMT2A-rearranged acute lymphoblastic leukemia. Haematologica. (2017) 102:e438–e42. doi: 10.3324/haematol.2017.169870
64. Clark JJ, Cools J, Curley DP, Yu JC, Lokker NA, Giese NA, et al. Variable sensitivity of FLT3 activation loop mutations to the small molecule tyrosine kinase inhibitor MLN518. Blood. (2004) 104(9):2867–72. doi: 10.1182/blood-2003-12-4446
65. Spinella JF, Cassart P, Garnier N, Rousseau P, Drullion C, Richer C, et al. A novel somatic mutation in ACD induces telomere lengthening and apoptosis resistance in leukemia cells. BMC Cancer. (2015) 15:621. doi: 10.1186/s12885-015-1639-5
66. Bailey E, Li L, Duffield AS, Ma HS, Huso DL, Small D. FLT3/D835Y mutation knock-in mice display less aggressive disease compared with FLT3/internal tandem duplication (ITD) mice. Proc Natl Acad Sci U S A. (2013) 110(52):21113–8. doi: 10.1073/pnas.1310559110
Keywords: precursor B-cell lymphoblastic leukemia-lymphoma, precision medicine, molecular biology, tumor biomarkers, child health
Citation: Biojone ER, Guido BC, Cavalcante LLM, Santos Júnior AdCMd, Pontes RMd, Furtado FM, Córdoba JC, Magalhães IMQ, de Oliveira DM and Camargo R (2024) Prevalence of FLT3 gene mutation and its expression in Brazilian pediatric B-ALL patients: clinical implications. Front. Pediatr. 12:1505060. doi: 10.3389/fped.2024.1505060
Received: 2 October 2024; Accepted: 19 November 2024;
Published: 6 December 2024.
Edited by:
Tomasz Szczepanski, Medical University of Silesia, PolandReviewed by:
Katarzyna Pawińska-Wąsikowska, University Children's Hospital in Krakow, PolandCopyright: © 2024 Biojone, Guido, Cavalcante, Santos Junior, Pontes, Furtado, Córdoba, Magalhães, de Oliveira and Camargo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Estefânia Rodrigues Biojone, ZXN0ZWZhbmlhYmlvam9uZUBnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.