 Myrsini Chamakioti
Myrsini Chamakioti Luc P. Brion
Luc P. Brion Pranav Viswanathan
Pranav Viswanathan Cheryl S. Lair4
Cheryl S. Lair4 Dimitrios Angelis
Dimitrios Angelis- 1Health and Precision Medicine, Choremion Laboratory, Aghia Sophia Children’s Hospital, University Research Institute of Maternal and Child, Athens, Greece
- 2Department of Pediatrics, Division of Neonatology, University of Texas Southwestern Medical Center, Dallas, TX, United States
- 3Medical School, University of Texas Southwestern Medical Center, Dallas, TX, United States
- 4Neonatal Nutrition, Parkland Health, Dallas, TX, United States
Zinc (Zn) is one of the most prevalent and essential micronutrients, found in 10% of all human proteins and involved in numerous cellular enzymatic pathways. Zn is important in the neonatal brain, due to its involvement in neurotransmission, synaptic plasticity, and neural signaling. It acts as a neuronal modulator and is highly concentrated in certain brain regions, such as the hippocampus, and the retina. Low Zn intake is frequent in several countries and in populations with high poverty index. Preterm infants are at risk for Zn deficiency for prenatal (missing fetal Zn) and postnatal reasons (less intestinal absorption and insufficient intake in maternal milk to match fetal accretion). The amount of Zn needed for preterm infants is not known and remains the subject of controversy. Recent nutritional recommendations favored an increase in daily Zn supplementation. Systematic reviews of randomized trials have shown that Zn supplementation in preterm infants increases weight gain and may decrease mortality. In this review we will summarize the role of Zn in brain functions and outcomes in preterm newborns, gaps in knowledge and areas of future research.
Introduction
Zinc (Zn) is intrinsically related to human brain development and function from fetal life to adulthood and is one of the most prevalent micronutrients, involved in numerous cellular enzymatic pathways. In recent years, there is an increasing interest in neonatal research community regarding the correct Zn dose and timing of initiation, Zn level interpretation, effectiveness, and its overall role in improving neonatal outcomes. Zn is considered safe in a wide range of doses, but the actual amount of Zn intake needed to optimize basic cellular functions, growth and neurodevelopmental outcomes, and neonatal morbidities and mortality is not well studied. Recent nutritional recommendations favored an increase in daily Zn supplementation (1–3), supporting the importance of this micronutrient in the well-being of the newborn.
This review is structured in three parts. In part A we describe the roles of Zn in key physiologic and pathophysiologic pathways, enzymes and mechanisms of action related to brain functions as well as data on fetal accretion, neonatal absorption, and homeostasis of Zn. In part B we describe the role of Zn in areas of the brain, neuronal populations and conditions that could be associated with or result from Zn deficiency. In Part C we describe the possible neurotoxic effects of Zn, clinical neurodevelopmental outcomes after Zn supplementation and dosing schedules and rationale in preterm neonates. Although the emphasis is given on preterm newborns, pathophysiologic data are usually derived from animals of other in vitro studies and are reported separately.
Methods
For the literature search, we performed a comprehensive search pertaining to Zn and the nervous system, with emphasis if available on the developing brain. The search involved Pubmed and OVID Medline with the inclusion of a broad variety of terms: “Zn or nervous system,” “brain,” “neurodevelopment,” in combination with the individual search terms for each section of this review such as: “glucose and Zn,” “autoregulation,” “carbonic anhydrase,” “nitric oxide synthase,” etc. Preclinical data are reported separately when available. A summary including controversies and gaps in knowledge is presented in the last part of each paragraph when available, in italic font.
Part A
A1 fetal accretion of Zn and neonatal absorption
The placenta and the fetal liver play a pivotal role in fetal Zn accretion, transfer, and utilization. The proximal bowel is responsible for postnatal accretion. The overall goal is to maintain homeostasis, and steady Zn tissue provision according to the needs of the developing fetus and newborn. The transfer of Zn at the cellular level and among tissues occurs via specific Zn-irk like receptors- ZIP, Solute Carrier family 39A (SLC39A) and receptors Zn transporters (ZnT) (SLC30A), the role of which will be expanded later in this review (Figure 1).
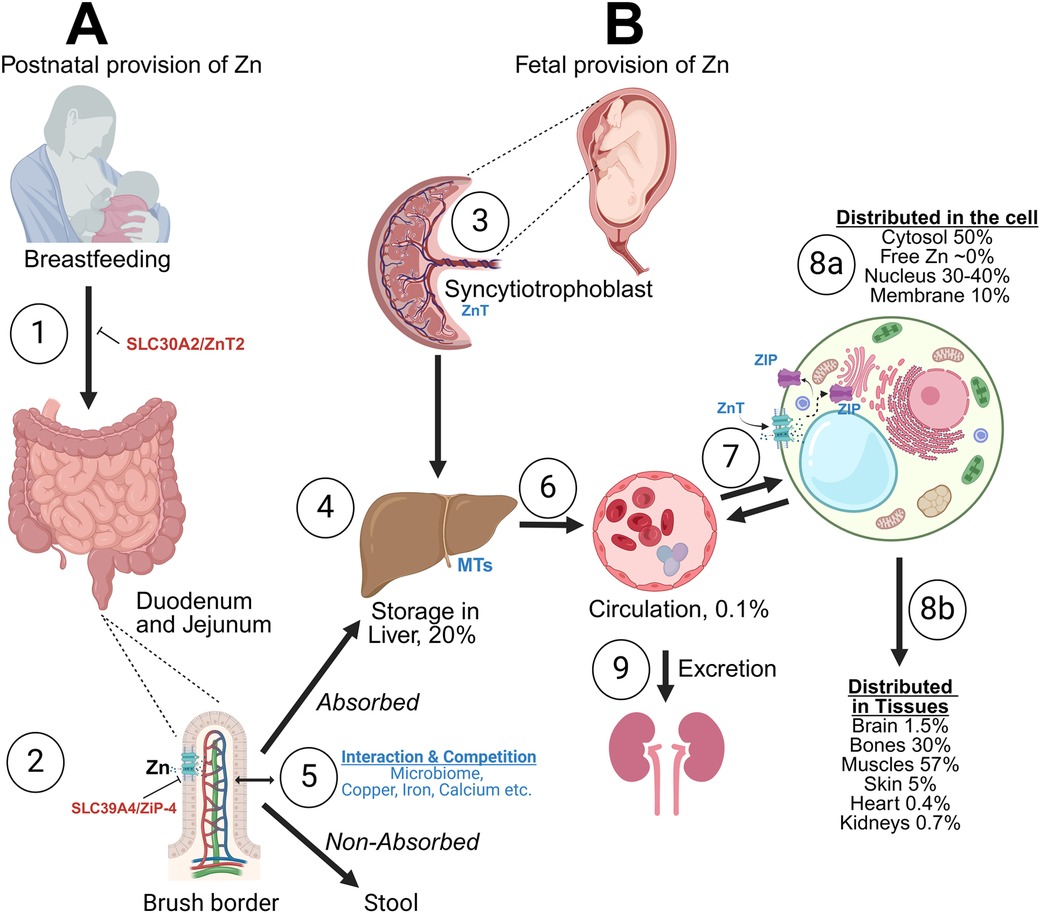
Figure 1. Brief description of perinatal zinc (Zn) transport and storage. (A) Zinc (Zn) is provided enterally via human milk or formula and/or intravenously via parenteral nutrition. The absorption occurs with Zn-irk like receptors (ZIP), with competition with other minerals (Cu, Mg, Ca, Fe). Zn transporter 2 mutation can lead to no Zn in human milk (1), while ZIP subtype 4 mutation can prevent Zn absorption in the gut (2). Both conditions can result in severe Zn deficiency. After interactions with the microbiome Zn is absorbed mainly in duodenum and jejunum. About 30% of Zn is absorbed (5). The liver remains the main storage organ, important for Zn homeostasis up to 2 months of life (4). (B) In the fetus Zn is transferred to the fetal liver via the placenta. Placenta (syncytiotrophoblast) is important for active transport of Zn against gradients from mother to fetus (3), while the fetal liver is the major storage area throughout the fetal life (4). In circulation (6), Zn is transferred in the red blood cell (RBC) (especially as part of the carbonic anhydrase—CA) and as Zn in serum (mostly bound to proteins, such as albumin, 6). 99% of Zn is intracellular. Interaction at the cellular level and preservation of homeostasis occurs via the receptors Zn transporters (ZnT) (decrease cytoplasmic levels) or ZIP (increase cytoplasmic levels) (7). Intracellularly, Zn is stored in metallothioneins (MT), key proteins for Zn homeostasis. Excess Zn is excreted via the kidneys (9). Intracellular distribution of Zn (8a) and tissue distribution (8b) are shown. Fetal accretion of Zn occurs mainly during the last trimester and hence extreme preterm newborns are at very high risk of Zn deficiency, despite having high serum Zn levels at birth. Created in BioRender.com. Angelis, D. (2024) Agreement number RH27AYOZX5.
Transplacental transport
The syncytiotrophoblast is responsible for Zn uptake from the maternal circulation and the subsequent release into the fetal circulation. The placenta has abilities to adjust the absorption of Zn not only during the progression of gestation (high uptake in premature vs. term vesicles), which supports the needs of the growing fetus, but also in conditions of low maternal Zn consumption or deficiency, which may limit fetal Zn deficiency (4). Zn accretion in mg/day increases progressively from 24 to 36 weeks' gestation but accretion in mg/kg/day (factored for fetal weight) decreases from 24 to 30 weeks and then remains constant (5). Both the Zn importers (ZIP, SLC39) and Zn exporters (ZnT, SLC30) are expressed in the placental syncytiotrophoblast, but the processes that facilitate Zn absorption and especially the mechanisms that control the adaptation in absorption of Zn are not well studied in humans (6).
Early studies in artificially perfused human placental lobules, found that tissue Zn concentration was 10 times higher than the concentrations of perfused (plasma) Zn and suggested that transfer of Zn in the syncytiotrophoblast is active, while transfer towards fetal circulation is passive, via simple diffusion (7). The active transport of Zn against gradients from maternal to fetal circulation is supported by several studies that reported higher Zn level in umbilical vein (UV) when compared to maternal blood (8–11). One mechanism that was recently proposed and can explain how Zn is transferred from the maternal circulation to the placenta against gradients is endocytosis via micro-vesicles. These exhibit saturable characteristics, have a biphasic response (initially rapid and later slow phase of accretion) and depend on potassium gradients (voltage gate properties) (7, 12).
Interestingly, total fetal Zn (bound and free) in term newborns, as measured in UV via atomic spectrometry, was found to be higher when compared to maternal or amniotic Zn, but free Zn was not significantly different (13), and Zn variations were attributed to differences in binding of Zn in plasma proteins. Pregnant women with high serum Zn levels >10.7 μmol/L (0.7 μg/ml) were found to have a higher percentage of Zn bound to alpha 2-macroglobulin compared to women with lower Zn levels, while they also had more bound Zn in albumin in their cord blood (70%) when compared to serum (56%) (4). The differences of protein binding of Zn and their potential effects on placenta transfer need further investigation. Mixed models of Zn transfer (passive and active) have also been suggested in experiments with human perfused placentas, where transport fractions of Zn averaged 0.21% of maternal loading concentration (14).
The factors that influence the transfer of Zn via the placenta are also not well understood. The serum fetal Zn level does not appear to significantly affect the transplacental transfer from the mother to the fetus when in situ perfused placentas from guinea pigs are utilized (15, 16). UV concentration was higher than maternal plasma levels, while the maternal to fetal transfer of Zn was directly related to maternal plasma Zn concentrations as well as blood flow in the uterine and umbilical vessels (16).
Zn placenta absorption when mother is Zn deficient
Zn deficiency worldwide is common and is encountered more frequently in lower income countries. In the US, Zn deficiency is less common but lower Zn intake could be encountered in mothers with low poverty index, and in women of Mexican origin (17).
Myers et al. described that nutritional content of Zn are expected to deteriorate with rising atmospheric carbon dioxide levels (CO2). More than 2 billion people live in countries that receive at least 70% of iron (Fe) and Zn from C3 crops (in which photosynthesis starts with 3 carbons, e.g., soybean, rice). Most temperate, non-legume C3 crops are unable to extract sufficient nitrogen (N) from soil at high ambient CO2 to maintain tissue carbon (C): N ratio. Rising ambient CO2 is likely to yield C3 crops with less proteins, Fe and Zn content (18).
Zn deficiency is also encountered in high income societies, as recently noted in Japan, where ∼33% of all women and ∼20% of those in reproductive age had Zn levels <0.6 μg/ml (19). In a Japanese study that included mothers with high baseline Zn deficiency, the ratio of UV to maternal plasma Zn was found to be ∼2:1 and there was no difference in normally grown neonates vs. those with intrauterine growth restriction (IUGR), while umbilical arterial (UA) to UV Zn was <1 in normally grown neonates and ∼1 in IUGR (20). There is evidence in both animal and human studies that placenta can adjust Zn absorption when maternal Zn deficiency is present. For example in Zn deprived pregnant mice, oral provision of radioactive Zn in the last part of pregnancy, resulted in higher total fetal body Zn retention, than those with had Zn rich diets (21). In the previously mentioned study, pregnant women with serum Zn >0.7 μg/ml had higher UV Zn when compared with those with lower levels (4). In a randomized controlled trial (RCT), in Gambian pregnant women who received diets poor in Zn, women in the control group had significantly higher mRNA concentrations of placental Zn transporters, when compared to those who were randomized to Zn supplementation (22). In one study in mice IUGR occurred in association with maternal Zn deficiency despite regulation of placenta Zn transporters at mRNA and protein levels, suggesting that regulation of placenta transporters may be insufficient to prevent fetal Zn deficiency (23).
Role of liver in Zn storage and usage
The fetal liver can retain large quantities of Zn during fetal life (Figure 1B), accounting for approximately 25% of total body Zn content, in contrast to placenta which operates only as a transient storage area. In rodents, placenta retention of radioactive Zn is limited. Immediately after Zn provision, Zn very quickly distributes from the placenta (high at 2 h, minimal at 24 h) to various tissues especially the fetal liver (quadrupled at 24 h after injection) (24). In humans, liver Zn concentration peaks is about 200–1,020 μg/g at 22–30 weeks' gestation and decreases to 140–380 μg/g at term in countries without endemic Zn deficiency (5, 25). In contrast, fetal liver Zn in Brazil, where Zn deficiency is endemic, is 30–304 μg/g at 26–34 weeks gestation and 13–268 μg/g at 40–41 weeks (26). The role of the fetal/neonatal liver as a major storage area for Zn is maintained for up to the first few months postnatal, assuring Zn homeostasis, despite poor Zn provision through enteral nutrition (27).
Postnatal Zn homeostasis
After birth, to meet nutritional needs, the newborn must receive Zn either via mother's own milk (MoM), donor human milk (DHM) or formula and/or via parenteral nutrition including trace elements (PN). In Figure 1, we depict the routes of Zn transfer and storage. Zn is transferred into human milk via the receptor Zn transporter 2 (ZnT2) and absorbed in the duodenum and jejunum via ZIP subtype 4. ZnT2 mutation results in transient Zn deficiency in exclusively breastfed infants, while ZIP4 mutation results in a lifelong disorder, acrodermatitis enteropathica (28). In circulation, Zn is transferred into the red blood cell (RBC) [especially as part of the carbonic anhydrase (CA) system] and as Zn in serum, bound to albumin and other proteins (29). It is likely that Zn is transported across cell membranes via its receptors as free Zn, after release from its ligands, although the exact mechanisms are unknown (6).
Absorption of Zn occurs in the brush border of intestinal mucosa especially in the duodenum and jejunum (30). Differentiation of the enterocyte border and effective length of the jejunum increases with GA (31). For the above reasons, bioavailability of enteral Zn in preterm newborns is only 10%−30%; therefore, enteral Zn needs in preterm infants are estimated as minimum of 4–5 mg/kg/day to match fetal accretion (32–35). Zn content in MoM after term delivery decreases postpartum from a content of 8–12 mg/L in colostrum to 0.7 to 1.6 mg/L by 1 month (36, 37). Zn content in MoM after preterm delivery ranges between 3 and 10 mg/ml in 2 studies (38, 39). The Zn content in DHM (2.14 ± 0.73 mg/L in 11 DHM samples) (40)—which is recommended for preterm infants when the amount of MoM is insufficient- is lower than in preterm MoM, but similar to mature term MoM. Fortification with human milk fortifiers and preterm formula provides about 2 mg/kg Zn per day, thus less than what is needed to match in utero accretion (41).
The total intracellular concentration of Zn is high, reaching 200 μM (42), while the free intracellular Zn is in the femtomolar range showing the tight sequestration of intracellular Zn (43). The interaction of Zn at the cellular level and preservation of strict cellular homeostasis occurs via the aforementioned specialized transporters, specifically, ZIP, SLC39A (which increase cytoplasmic Zn levels) and ZnT (SLC30A) (which decrease cytoplasmic Zn levels) (44, 45). Hormones, cytokines, and the availability of Zn dynamically control the subcellular localization and expression of Zn transporters. The highest tissue concentration, beyond the liver, is in bones and pancreas (200 μg/g), compared with most other organs (1–23 µg/g) (29, 44). Areas of the brain with high Zn content include the olfactory apparatus, the cerebrum and hippocampi, while cerebellum has the lowest (46). About 99% of the total body Zn is intracellular. Inside the cell, Zn is distributed between the membrane (10%), the nucleus (30%–40%), and the cytoplasm (50%) (29) (Figure 1).
Metallothioneins
Most Zn is stored transiently in specific proteins (metallothioneins, MTs). MTs bind 20% of intracellular Zn as their cysteine sulfur groups create two Zn-sulfur complexes, which can bind up to 7 Zn ions per protein (47, 48). The MT family of proteins includes four isoforms, designated as MT I–IV, with I and II being ubiquitous in all tissues and III being present in central nervous system (CNS), while IV is less well described. The hepatocyte has high content of MTs I and II. Zn can be released from liver MTs upon net deficit, contributing to its homeostasis (25, 49). MTs concentration peaks between 14 and 23 weeks of GA (50) and subsequently decreases with gestation, but the total Zn tissue content is maintained (27). In mammals, MT I expression is downregulated by the increasing maternal estrogen (51). In rodents, placental MTs do not seem to play a significant physiolologic role in binding and transferring of Zn (24). However, induction of placenta MTs after toxic metal exposure (e.g., cadmium) or very high Zn concentrations shows that MTs might play secondary protective roles as will be described later in this chapter (52).
In summary: The placental transfer of Zn from maternal blood to fetal blood and subsequently to the fetal tissues likely occurs against gradient at the syncytiotrophoblast. The fetal liver is a key organ for storage of Zn and maintains this role in the first few months of life, until adequate Zn intake is established. Specific brain areas have very high Zn content. Preterm infants are at risk for Zn deficiency for prenatal (missing Zn accretion during the second and third trimesters) and postnatal reasons (less absorption in the proximal intestine and insufficient Zn even in fortified MoM and DHM to match fetal accretion).
A2 how do we assess Zn status in preterm newborns?
Measurement of the level of serum Zn, despite representing a subfraction (∼0.1%) of the total Zn pool, is considered the gold standard for monitoring its deficiency. The accretion of Zn occurs mainly during the second and third trimesters and hence extreme preterm newborns are at high risk of Zn deficiency, despite having high serum Zn levels at birth (53). The American Society for Parenteral and Enteral Nutrition (ASPEN) recommends a normal range for Zn concentration of 0.74–1.46 μg/ml (54). Normal serum Zn levels decrease with postnatal age (32, 55–57).
Several investigators including our group have shown that levels <0.7–0.74 μg/ml (deficiency) are associated with abnormal outcomes [poor growth, retinopathy of prematurity (ROP) etc.] (58, 59). The upper limit of normal (>1.46 μg/ml) has not been validated in large studies. UA and UV Zn levels have been utilized to estimate fetal Zn content and as explained earlier UV Zn levels are higher than UA levels, do not always correlate with maternal Zn levels and are affected by conditions associated with the birthing process (60). In a systematic review, cord Zn levels were found to be lower in pregnancies with various pathological outcomes [small for GA (SGA), IUGR, preeclampsia, smoking, etc.] and to correlate with maternal Zn levels (R = 0.4365) (61).
Zn concentration in hair could be valuable since it correlates with maternal Zn stores (62); however, obtaining enough sample size for Zn measurement may be a challenge in very preterm newborns. Zn supplementation in preterm newborns increases serum levels (total and skeletal subfraction) of alkaline phosphatase (ALP) (63, 64), a Zn-dependent enzyme. Bone alkaline phosphatase derives mainly from osteoblasts and plays a critical role in bone mineralization (65, 66). Zn deficiency can be suspected in infants with low serum concentration of ALP in the absence of rickets (67). Interestingly, Zn increased the activity and the half-life of ALP in vitro (68), without interfering with transcription of ALP protein. These findings could provide a link between Zn, ALP, growth and bone mineralization. It would be compelling to use ALP as an indirect, surrogate method to monitor Zn status since ALP levels are low in association with Zn deficiency and increase with Zn supplementation. However, this method is unlikely to be effective in preterm infants, since ALP may decrease with Zn deficiency and increase with cholestasis, vitamin D deficiency or fractures (63, 64). This may explain why studies in preterm infants have not shown a correlation between serum Zn levels and ALP (66, 69, 70). In addition, Zn deficiency can be encountered more frequently when DHM is provided to preterm neonates (40).
In summary: Measurement of serum Zn level (with normative values: 0.74–1.46 μg/ml) remains the most common and gold standard method for evaluation of Zn, despite its limitations. Serum Zn levels are affected by physiologic factors (GA, postmenstrual age, PMA, birthing process, growth etc.), but also from pathologic factors (inflammation, infection etc.). Umbilical serum Zn level should be used with caution (differences between UA, UV, not well standardized levels, influenced by birthing process etc.). Tissue Zn is ideal but difficult to obtain in neonates.
A3 mechanisms, enzymatic pathways and biochemical effects of Zn
Zn is an important micronutrient that binds up to 10% of the proteins in the body and operates as a cofactor in a variety of enzymes (45). Zn interferes with or is structural part of enzymes and signaling pathways that as a net effect facilitate euglycemia, cellular cross talk, growth, brain functions and development. In these subsequent sections we summarize the evidence of specific enzymatic and biochemical pathways that could explain some of the benefits of Zn supplementation and some of the potential harms. The vast part of this evidence arises from preclinical animal studies or cell cultures.
Nitric oxide (NO) synthase (NOS) pathway
NO pathway dysregulation is important cause of endothelial dysfunction and vascular disease (71, 72). Zn is a part of the structure of all three NOS isoforms (inducible, iNOS, endothelial, eNOS and neuronal, nNOS) (73, 74) and a crucial regulator of the production and activity of these enzymes. The inducible NOS is related to neuroinflammation. Zn and NO operate in a feedback loop. NO produced by iNOS nitrosates Zn-containing intracellular proteins, such as MT. It increases the levels of labile Zn by freeing bound Zn (75). The increased Zn in turn, inhibits the iNOS, reducing NO production and protecting the cells from NO-induced endothelial damage (76).
The mechanism by which Zn inhibits iNOS is by limiting the nuclear factor (NF)-κB transactivation activity. This has been shown by a decrease in the activity of NF-κB-driven luciferase reporter and the NF-κB target genes expression, such as interleukin (IL)-1β and cyclooxygenase (COX) 2. Another mechanism of Zn-mediated inhibition of iNOS is the inhibition of the cytokine-induced activation of the iNOS promoter. Apart from these, Zn facilitates the activity of MTs, which covalently bind NO to form S-nitrothiols, thereby scavenging the cytotoxic NO (77). Zn is also essential for the formation and function of the eNOS in the dimeric—active—form (78). Zn deficiency in the fetal period might cause decreased expression of eNOS in rats (79).
In summary: By inhibiting the NF-κΒ activity, Zn limits iNOS expression, acting as a cytoprotective element against NO-induced inflammation. In addition, Zn deficiency decreases expression of eNOS, a key enzyme for brain autoregulation. The role of Zn as part of the nNOS is not well understood.
Carbonic anhydrase (CA)
CA is the first discovered Zn-containing metalloenzyme. It is one of the most catalytically efficient enzymes. Its prominent role is catalyzing the carbon dioxide (CO2) conversion into bicarbonate (HCO3−) and water. CA is abundant across all kingdoms of life (80, 81). CA type II (CAII), the predominant isoform expressed in RBCs, mediates the transport of CO2, playing a crucial role in respiration. CA (including several isoforms) is expressed in several tissues including the placenta, kidney, liver, brain, gut and bone, mediates CO2/HCO3− equilibrium and blood and tissue acid-base balance (82).
In the brain, CA is expressed in the choroid plexus, oligodendrocytes, myelin, glial cells, and several specialized cells (83). CA has a role in production of cerebrospinal fluid (CSF), in regulation of cerebral blood flow (CBF) (84) and in brain electric activity (85).
CA comprises an active site with a Zn-binding site, an entrance conduit, and various hydrophobic and hydrophilic parts (86). Zn has a vital role in CA catalytic function. More specifically, Zn-bound hydroxide creates Zn-bound bicarbonate by reacting with the carbonyl carbon of CO2. As a next step, the Zn-bound bicarbonate is displaced with water. The Zn-bound water releases H+, which is transported to the external buffer to regenerate the Zn-bound hydroxide (87). Alternative transition metal ions, like Ni2+ and Mn2+, can replace Zn. However, this drastically decreases CA catalytic activity, rendering it sometimes completely inactive (88).
Because of the high turnover rate of CA activity, a significant change in its concentration needs to occur before any clinical effect is observed. Severe Zn deficiency in animals decreases the amount of CA protein and CA activity in RBCs and has been associated with tachypnea and even gasping in pullets and rats (89). Zn deficiency decreases CA activity in submandibular gland, tongue epithelium as well as trigeminal and chorda tympani response to carbonated water in rats (90–92). In adults, Zn deficiency decreases RBC CA activity, maximum exercise capacity and taste (93, 94). Zn supplementation improves taste in adults with CA VI deficiency (95).
In summary: Zn is a key component of CA. Severe Zn deficiency can result in decreased CA activity and overt symptomatology in animal studies and in limited human studies, and include fatigue, decreased muscle strength and cardiopulmonary effects. Zn deficiency can also affect the neuronal CA but these effects remain to be investigated.
Transcription factors
Zn-finger proteins (ZFPs) are nuclear transcription factors. Like other transcription factor families, they regulate gene expression and affect cell proliferation, differentiation, and cell death (96). ZFPs are DNA-binding domains arranged in various formations. They can read various DNA sequences and use their kinase-binding domains to participate in protein-protein interactions and signaling pathways. In addition, ZFPs play a role in further specialized processes, such as chromatin remodeling, cytoskeleton organization, epithelial development, mRNA trafficking, and cell adhesion (97).
While ZFPs differ in structure, it is widely accepted that in a ZFP, a specific combination of cysteines and histidine amino acid residues chelates a Zn ion, creating a complex that consolidates the domain's 3D structure. The identity and spacing of the Zn-binding amino acids determine the type and specificity of each ZFP (98).
In archetypal DNA binding ZFPs, such as transcription factor IIIA (TFIIIA) and GATA-binding factor 1 (GATA-1, which binds the DNA sequence “GATA”), we see that a beta-hairpin and an alpha-helix are folded around a Zn ion and that it is the alpha-helix that binds with the major groove of DNA. In this way, Zn facilitates the three-dimensional (3D) conformation of these transcription factors. The 3D structures of the ZFP in TFIIIA and GATA-1 are crucial for their DNA-binding and regulatory functions. In both cases, the spatial configuration of these ZFPs allows for high specificity in DNA recognition, influencing the regulation of gene expression. Specifically, the alpha-helix of the ZFP forms hydrogen bonds with three DNA bases of the guanine-rich strand of the DNA major groove. This way, the transcription factor can loop around the oligonucleotide sequence in one turn (99).
In summary: Zn homeostasis is important for the function of transcription factors (such as TFIIIA and GATA-1), facilitating their 3D conformation and their highly selective binding in the DNA grooves, assuring appropriate and tightly regulated gene expression. In this way, Zn can control important processes of gene expression and the following downstream effects.
A4 immunity
Zn plays a critical role in the immune system. Acute dysregulation of Zn levels hinders the proliferation, maturation, and activation of cells in innate and adaptive immune responses, while chronically disrupted Zn homeostasis increases the risk of inflammation and disease (100, 101). Inflammation (fetal or neonatal) can affect fetal brain development as it relates with abnormal white matter growth and development of periventricular leukomalacia (PVL) (102, 103).
Innate immunity
Zn controls various aspects of the innate immune response. In vitro extremely high Zn levels (500 μM) induce polymorphonuclear leukocytes (PMN) chemotaxis while very low Zn levels decrease PMN chemotaxis (104). Cell cultures with human cells show variable effects of Zn on cytokines depending on its concentration and cell status (105, 106). Zn overall increases interferon (IFN)-γ, IL-10, IL-1β and tumor necrosis factor (TNF)-α in lipopolysaccharides (LPS)-stimulated cells, while Zn down-regulated levels of IL-1β and TNF-α in peripheral blood mononuclear cells (PBMC) when stimulated with superantigens (107).
Zn, through proteins like the early endosome antigen 1 (EEA1), affects phagocytosis. During sickness and stress serum Zn rapidly declines due to rapid redistribution into the cells, which can be used for protein synthesis as antioxidant and as anti-microbial. Cytokines, such as IL-6 and TNF-α appear to contribute to this physiologic phenomenon (108, 109). Under conditions of Zn deficiency, LPS stimulated mononuclear cells produced higher levels of IL-1β (110, 111). Similar results were shown for TNF-α (111).
Zn excess promotes phagocytes' activity, whereas Zn deficiency has the opposite effect. As a next step after phagocytosis, Zn plays a role in the neutralization of pathogens; abnormal Zn levels, either low or high, inhibit the nicotinamide adenine dinucleotide phosphate oxidases (NADPH). NADPH is crucial for destroying pathogens after phagocytosis, as it controls the production of superoxide anions (112). Regarding monocytes, Zn facilitates their adhesion to the endothelium of the vessels and participates in the production of the pro-inflammatory IL-1β, IL-6, and TNF-α. Zn promotes the expression of ZFPs with anti-inflammatory properties, such as A20, which hinders the activity of NF-κΒ and NF-κΒ target genes, such as IL-1β and TNF, and thus prevents TNF-induced programmed cell death (113, 114). Lastly, Zn is a key element in dendritic cells' maturation process. Downregulation of ZIP-6 decreases the intracellular Zn levels, which subsequently affects the maturation process of dendritic cells and the further activation of the adaptive immune system.
Adaptive immunity
T cell progenitors mature in the thymus, and Zn deficiency causes thymic atrophy and T cell lymphopenia. During maturation, pre-T cells are the most susceptible to Zn deficiency, which can lead to a loss of 50% of them in mice (115, 116). Zn is crucial for the adaptive immune response and, most of all, for the development and function of the T-cells. Zn is a co-factor for the activity of the hormone thymulin, which regulates T-cell differentiation and function (47, 117). Moreover, in activated T-cells, Zn is required for signal transmission during IL-2-induced proliferation (118). Zn affects the TH1/TH2 balance. In the case of Zn deficiency, TH1 cytokines such as IFN-γ, IL-2, and TNF- α are reduced. However, the production of TH2 interleukins, like IL-4, IL-6, and IL-10, does not change. This results in an imbalance between TH1 and TH2 interleukin levels, which Zn supplementation restores (119, 120). Lastly, in cases of Zn deficiency, the levels of glucocorticoid hormones are elevated. The anti-apoptotic protein Bcl-2 is also reduced. This combination promotes pre-T cell apoptosis (115, 121).
Although B cells are much less affected by Zn levels than T cells, Zn is necessary for the survival of premature B cells and antibody production and, thus, crucial for the B-cell antigen-specific immune response (47, 122).
In summary: Modulation of immunity under conditions of Zn deficiency could impact neuronal functions via the direct impact on neuroinflammation. Unfortunately, clinical evidence is lacking so far.
A5 metallothioneins (MTs) and the brain
MTs can act as Zn acceptors and donors inside the cell, exchanging metal ions with proteins. MTs control the intracellular Zn levels with their sulfur cysteine groups, which release Zn after undergoing oxidation (123).
MTs provide age-dependent protection against neuronal toxicity with higher protective effect with advancing age as shown in rodents (124). Oxidizing conditions promote the release of Zn while reducing conditions restore Zn binding to MTs (125). By upregulating the expression of the metal regulatory transcription factor 1 (MTF-1), Zn increases the synthesis of MTs (126). MTs, in turn, act as metal scavengers and prevent the Fenton reaction and reactive oxygen species (ROS) production by binding active redox metals (123, 127). However, the age-induced increase in ROS finally compromises the Zn-binding capacity of MTs (128). Similarly, Zn deficiency increases ROS and inhibits MT activity, resulting in compromised mitochondrial function and cytochrome c oxidase activity (129).
MTs can store and release Zn depending on cellular needs contributing to its homeostasis. The stored Zn is important for the rapid growth of the brain that occurs during the latter part of gestation. The localization of MTs in the brain was first described by Suzuki et al. (130), which identified that the onset of the system starts at about 21–22 weeks GA. MTs I and II also play a role also in the distribution of Zn in this phase as they are found in specific glial populations, located in the periventricular zones. These cells migrate towards the cortex starting at 21 weeks of GA till 35 weeks GA, with possible completion and maturation of the system up to 10th postnatal month (130, 131). The role of MTs in these cases, with clear co-localization with glial proteins might relate with processes that involve these populations such as myelin production and neuronal migration.
In Summary: MTs play a role not only as ROS scavengers and prevention of toxicity in the fetal brain but also maintain homeostasis of Zn and help match the energy and Zn demand in the developing brain and migrating glial cells.
A6 insulin and glucose metabolism
Zn binds to insulin and contributes to its biosynthesis, crystallization, and post translational maturation (132, 133). Zn is transferred inside the insulin secretory granules of β-cells via the ZnT8 transporter. Lack of ZnT8 activity prevents insulin crystallization and secretion and may be associated with type 1 and 2 diabetes mellitus (DM) (134, 135). As mentioned in the previous section, Zn decreases the expression of pro-inflammatory cytokines of IL-1β, TNF-α, and IL-6. In Zn deficiency, the long-term activity of these cytokines results in apoptosis of β-pancreatic cells and insulin resistance.
Apart from insulin control, Zn also participates in glucose metabolism. By activating GLUT4 in cell plasma membranes, it facilitates the uptake of glucose by insulin-dependent tissues (136, 137). In addition, Zn can act as an insulin-mimetic, inhibiting forkhead box transcription factors (FOXO) and regulating necessary gluconeogenic enzymes (138). Lastly, Zn inhibits glucagon secretion by inhibiting voltage-gated channels in pancreatic α-cells (139–141).
The role of Zn concentration on hyper- or hypoglycemia is not well established in preterm newborns. Limited evidence from our group showed that after increasing dose of Zn in PN to recommended dose in 23–28-week GA neonates the number of hyperglycemic episodes decreased, without a change in hypoglycemic events (142, 143).
Finally, association of hyperglycemia and ROP (144, 145) could be explained by abnormal modulation of HIF-1 and VGEF, two key factors for the development of ROP which also dysregulate after hyperglycemic conditions (146–148).
In summary: Zn affects glucose metabolism through insulin maturation, GLUT4 activation and glucagon inhibition. Zn sufficiency could improve glucose metabolism, decrease hyperglycemia, and improve downstream abnormal signals related to ROP. These effects need further exploration.
A7 superoxide dismutase
Superoxide dismutases (SODs) are crucial antioxidant enzymes that protect cells from ROS that arise in oxygen-rich environments from mitochondria, peroxisomes, and cytoplasm. SODs catalyze the conversion of superoxide to oxygen and hydrogen peroxide. Hydrogen peroxide is then eliminated by other antioxidant enzymes, such as catalase and glutathione peroxidases (149).
The catalytic properties of SODs depend on their metalation and disulfide bonding during posttranslational modifications (150). Zn and Cu are the catalytic metal ions for the human Cu-Zn-SOD (SOD1). The binding of the SOD1 homodimer to Zn offers structure stability, whereas Cu is responsible for enzymatic functionality (151). A stable connection with the His63 residue keeps Zn and Cu together and ensures the functionality of SOD1 even at extreme pH levels.
If SOD1 does not function normally, high ROS levels will cause oxidative damage, such as protein carbonylation, DNA breakage, and membrane lipid peroxidation (152). In adults, these can lead to different diseases, from cancer and amyotrophic lateral sclerosis to Parkinson's disease (153–155). Oxidative stress can also act as a stimulus for SOD1, so that it acts as a transcription factor. When hydrogen peroxide levels are high, SOD1 is phosphorylated via the cascade of Mec1, DNA Damage Response (DDR) kinase. The phosphorylated SOD1 then translocates to the nucleus, where, by binding to gene promoters, it controls the transcription of genes related to oxidative stress resistance (156).
SOD1 mutations can affect RNA metabolism. Mutant SOD1 can bind to the mRNAs of Neurofilament Light Chain (NFL) and Vascular Endothelial Growth Factor (VEGF) and form ribonucleoproteins complexes that then aggregate (157, 158). One of these RNA-binding proteins is HuR, which is protective against stress in motor neurons. Its interaction with the mutant SOD1 impairs its neuroprotective function and, along with the downregulation of VEGF, contributes to cytotoxicity and the appearance of neurodegenerative diseases like amyotrophic lateral sclerosis (159, 160).
In summary: Zn is the catalytic metal ion for SOD, a key antioxidant enzyme. If SOD is not adequate, high ROS levels can emerge resulting in oxidative stress, DNA breakage, lipid peroxidation and neuronal apoptosis.
Part B Zn and the neonatal brain
Zn is abundant in various brain parts, especially the hippocampus, cerebral cortex, amygdala, olfactory bulb and retina (161). It plays crucial roles in synaptic transmission, neuronal signaling, and brain development in these regions. Zn deficiency or excess can result in abnormalities in neurodevelopment, behavior, CNS formation, and a variety of neurological diseases.
B1 Zn in synaptic transmission and plasticity
The hippocampus, which contains a high concentration of Zn, acts as the center of learning and memory and houses neuronal machinery responsible for stress responses. Accordingly, hippocampal Zn appears to be directly associated with learning, memory, and behavior (162, 163). Zn interacts with ligand-gated ion channels post-synaptically and modulates ion transport. Glutamatergic Zn-enriched neurons (ZENs) contain high densities of the excitatory glutamate receptors N-methyl-D-aspartate (NMDA) and alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), which have several modulatory Zn-binding sites (163). ZENs are found in the hippocampus, the olfactory bulb, and the dorsal cochlear nucleus (162) (Figure 2).
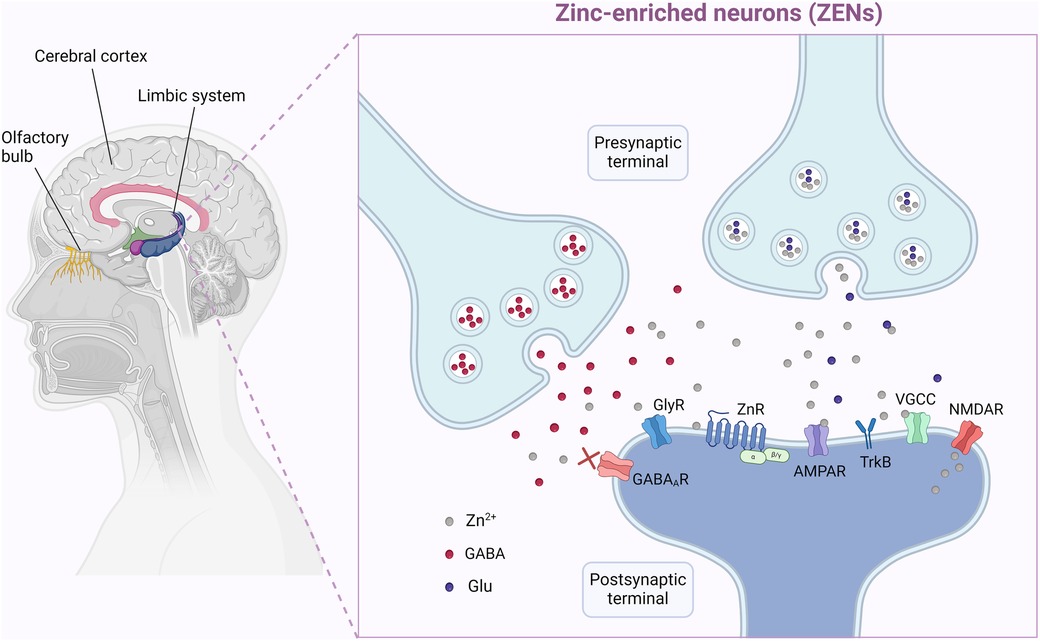
Figure 2. Zinc (Zn) exists in very high concentration is specific tissues and plays a key role in synaptogenesis, plasticity, neuronal repair and cellular migration and development. Here on the left we depict some of these such as the olfactory apparatus, the limbic system and cerebrum. These actions are mediated with specific Zn-enriched neurons (ZEN). ZENs contain high densities of the excitatory glutamate receptors, the N-methyl-D-aspartate (NMDA) and the α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA), which have several modulatory Zn-binding sites. The NMDA and AMPA glutamate receptors regulate various calcium-, sodium-, and potassium-dependent intracellular pathways and play critical roles in brain function and development. In certain brain regions their function is greatly affected by the extracellular Zn levels. Created in BioRender. Chamakioti, M and Asimakopoulos, T. Abbreviations: (2024). AMPAR, α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate receptor; GABA (R), gamma-aminobutyric acid (receptor); Glu, glutamate; GlyR, glycine receptor; NMDAR, N-methyl-D-aspartate receptor; TrkR, tropomyosin receptor kinase receptor; VGCC, voltage-gated calcium channel; ZEN, zinc-enriched neuron; Zn, zinc; ZnR, zinc receptor. Created in BioRender.com. Asimakopoulos, T and Chamakioti, M (2024) Agreement Number MB26TVCEGU.
The NMDA and AMPA glutamate receptors regulate various calcium-, sodium-, and potassium-dependent intracellular pathways. In certain brain regions, namely the neocortex, the amygdala, and the hippocampus, their function is greatly affected by the extracellular Zn concentrations (164). Zn has a dual effect and can bind differently to the glutamate receptors, attenuating or amplifying their signals, depending on their levels. Because NMDA receptors are pivotal in triggering long-term changes in synaptic efficacy, it is evident that Zn participates as a co-transmitter in cognition and plasticity of the synapses (128). Apart from glutamate receptors, Zn also interacts with gamma (γ)-aminobutyric acid (GABA) receptors (165), glycine receptors and metabotrophic Zn receptors (166–170).
Preclinical data show that gestational Zn deficiency in rodents caused significant alternation in the production of and expression of NMDA subunits, brain derived neurotrophic factor (BDNF) and nerve growth factor (NGF) which caused memory and learning impairments later in life (171).
In summary: Zn affects synaptogenesis, neuronal plasticity and synaptic efficacy and in these ways plays a role in memory and behavior.
B2 Zn in brain development
Neurogenesis, the CNS development process, occurs during gestational and neonatal periods. During this period, the radial and tangential migration and the division of neural stem cells (NSCs) create the neural tube, the neural crest, and the notochord. More specifically, the invagination of the neural plate creates the neural tube, and this process is called neurulation (172). When the neural tube closes, the neuroepithelial cells of the ventricular zone become radial glial progenitor cells (RGCs), which later turn into glial cells and neurons (173).
B3 Zn finger proteins (ZFPs) and neurogenesis
Zn, in the form of ZFPs, plays a crucial role in brain morphogenesis. ZFPs are involved in transcriptional regulation, signal transduction, actin targeting, and cell migration (96, 174). During neurogenesis, ZFPs direct the fate of embryonic stem cells (175). Zac1 protein participates in neuronal development in the cerebellum, Zn finger and BTB domain-containing protein 20 (Zbtb20) in the hippocampus, and fasciculation and elongation zeta protein (Fez) f1 and Fezf2 in the brain's olfactory region (176). ZFPs, e.g., Glioma-associated oncogene family Zn finger 3 (Gli3), regulate the cell cycle of RGCs, affecting their differentiation into neural cells. Interaction with the Sonic hedgehog protein (SHH) can alter the length of the G1 phase in the cell cycle of RGCs (153, 177). Mutations in Gli3 result in a shortened cell cycle, impaired cortical lamination, and cortical neuron formation and are responsible for various morphological CNS abnormalities, such as Greig cephalopolysyndactyly syndrome (GCPS), Pallister-Hall syndrome (PHS) and polydactyly and syndactyly (178).
Other characteristic examples are Zeb Zn fingers, Zeb1, and Zeb2. Zeb1 controls the development of the neocortex by acting as a transcriptional repressor that regulates the division mode of RGCs, their proliferation, migration, and differentiation. Zeb1 also affects the development of electrophysiological properties in developing neurons (179). Zeb2 participates in the differentiation of RGCs to Bergmann glial cells and astrocytes (180). Lastly, the Zic-type poly-ZNFs (Zic1, Zic2, Zic3, Zic4, and Zic5) take part in the closing of neural plates, the formation of the neural crests, the proliferation and the differentiation of NPCs in the medial forebrain, and cerebellar morphogenesis (181–183). Mutations in the Zic-type poly-ZNFs can hinder the physiological division of the brain into two hemispheres. They can also cause the Dandy-Walker malformation, which is associated with specific brain abnormalities such as cerebellar vermis hypoplasia and delayed motor and cognitive development (184).
B4 the role of Zn in the different stages of neurogenesis
In short, neurogenesis can be divided into three main stages: NSC proliferation and migration, neuronal differentiation, and cell survival (185–188).
Zn in stem cell proliferation, glucocorticoid modulation and neuronal survival
Zn affects genes associated with the cell cycle, such as transcription factor AP-1, nuclear factor κB (NF-κB), and nuclear factor of activated T cells (NFAT) responsible for cell proliferation, differentiation, and apoptosis. This way, Zn controls the proliferation of neuronal precursor cells (189, 190).
Zn influences the levels of glucocorticoid hormones in the brain, and Zn deficiency increases their levels, resulting in numerous harmful effects on CNS development. Glucocorticoids induce extracellular accumulation of glutamate in hippocampal neurons, triggering excitotoxicity and cell death. Chronically increased glucocorticoid levels also damage the hippocampus via metabolic alterations that result in ischemia, hypoxia, and hypoglycemia and limited proliferation of NSC in the hippocampal dentate gyrus of primates (191–194). Finally, glucocorticoids interact with the Hedgehog signaling (HHS) pathway, as both low and high glucocorticoid levels result in the inhibition of HHS. This inhibition occurs via promotion of Notch/Hes-signaling and downstream inhibition of transforming growth factor (TGF) β- suppressor of Mothers against Decapentaplegic (SMAD)2/3-signaling respectively (195).
The p53 protein is a Zn-binding cell regulator and tumor suppressor. In Zn deficiency, p53 is translocated to the nucleus, resulting in cell cycle arrest (190). The extracellular signal-regulated kinase 1/2 (ERK1/2) pathway is down-regulated, hindering NSC proliferation. Zn deficiency during gestation affects neurogenesis by limiting the proliferation and differentiation of neurons in the fetal rat brain cortex and disrupting the cortical excitatory/inhibitory balance. This disrupts the formation of the physiological cortical structure, resulting in cognitive impairment (189, 196).
In summary: Zn directly affects the survival of primitive neurons by interfering with key intracellular processes and pathways such as transcription factors, glucocorticoids and the p53 system.
Neuronal differentiation
During early development, Zn plays an essential role in neuronal differentiation. It promotes the proliferation and differentiation of the pluripotent Adipose-derived mesenchymal stem cells (ADMSCs) into neurons. In the differentiated stem cells, Zn triggers neurite outgrowth, inactivates RhoA (a key guanosine triphosphate, GTPase), downregulates the ERK1/2 pathway, and promotes the expression of migration/neuronal genes such as the microtubule-associated proteins (MAP)2 and nestin (197). Lastly, it delays the radial migration of neurons during the cerebral cortex formation by elevating the glucocorticoid levels (198).
As expected, low Zn levels hinder neuronal differentiation during early development. Zn deficiency inhibits the dendritic differentiation of various cells, such as Purkinje and stellate cells, in the cerebellar cortex of 21-day-old rats and hinders the differentiation of human-induced pluripotent stem cells by modifying the Zn transporter gene expression (199–201).
TGFβ receptors (TGFβR) are thought to play a role in Zn regulation of neuronal differentiation. Zn deficiency impairs TGFβR subtype 2 induction after retinoic acid in human cell lines (202). TGFβR deficiency relates with neurodegenerative disorders, β amyloid peptide production and Alzheimer's disease (203). Zn deficiency reduces the expression of ZnT in young/multipotent neurons, increases cholinergic signaling among others and could change their developmental potential (202). Changes in cholinergic function of neurons has been shown in autism spectrum disorders (ASD) (204) and the role of Zn in these diseases processes has been previously reviewed (205).
In summary: These findings suggest that Zn is intrinsically involved in key pathways that affect neuronal differentiation such as TGFβ and cholinergic systems and its deficiency causes disruption of these pathways. Behavioral disorders such as ASD might be linked with these pathways.
Neuronal precursor survival
Zn also plays a role in the survival of neuronal precursor cells, as it regulates the expression of both pro-survival pathways, like the ERK, Ak strain transforming (Akt), and nuclear factor (NF)-kB pathways, and pro-apoptotic cascades, such as the Jun N-terminal kinase (JNK) and p53 pathways. In this context, Zn deficiency is responsible for altered neuronal differentiation, limited cell survival, and impaired synapse function (199). Low Zn levels are associated with activation of Caspase-3 and downregulation of the ERK pathway, reduced Ki67-positive cells, increased TUNEL-labeled cells in the subgranular zone (SGZ), arrest of the cell cycle in the G0 G1 phase, promotion of apoptosis in neurons, and modification of cell signals that control the pro-survival and pro-apoptotic gene expression, such as NF-κΒ and p53 (189).
Zn and gyration; cerebrum and cerebellar effects
The hippocampus and cerebellum are among the brain regions affected by Zn deficiency. Specifically, low Zn levels act in the cerebellar granular layer of rats and reduce the density of neurons, which also have shorter and less branched dendrites (200).
Zn deprivation significantly inhibits the development of the cerebellar cortex and delays the withdrawal of the external cell layer, the acquisition of granule cells, and the differentiation of Purkinje cells (200). It also impairs the dendritic growth of basket and stellate cells, interneurons of the cerebellar molecular layer that form and differentiate during the initial postnatal period (206, 207). The effects of Zn deficiency on the dendritic differentiation of these cells were studied in 21-day-old rats. It was shown that the total dendritic length of neurons and the dendritic field area of Zn-deficient animals were 43% and 30% smaller in the lower half of the molecular layer, respectively. This could be possibly explained by the delayed onset of dendritic differentiation and the slow rate of dendritic growth (200).
In summary: In preclinical studies, the hippocampus and cerebellum are affected when Zn deficiency is present. Specifically, the granule cells, and the differentiation of Purkinje cells are very sensitive to lack of Zn. These result in delayed dendritic development and cerebellar cortex abnormalities.
B5 Zn and white matter development
Zn deficiency can affect multiple cell types in the brain.
Astrocytes
Zn deficiency during the early developmental period in rats downregulates the STAT3 signaling pathway, inhibiting astrogliogenesis and producing a low number of astrocyte cells in the early postnatal cortex (208). When these rats reach adulthood, the astrocyte number does not increase; it instead remains the same, representing an excellent example of the long-term consequences of Zn deficiency in the early stages of life (209). The homeostasis of Zn in astrocytes is maintained by a previously described Zn transporter system, with one subtype (Zn T1) to act as a protective mechanism against extreme accumulation of intracellular Zn (210).
Myelin production, oligodendrocytes and Zn
Zn is also critical for the development of oligodendrocytes (161, 211–214). Zfp488 is a ZFP specific for oligodendrocyte cells that co-regulates the gene expression during differentiation (215). Zn levels in developing oligodendrocytes remain high during differentiation and fall significantly after maturation is achieved, indicating a role of Zn in the differentiation of this cell type and a possible restorative potential of Zn for the failure of pre-oligodendrocyte maturation in premature infants with white matter injury (216, 217). Gestational deprivation of Zn has been shown to affect various myelin components in rodents and primates (218, 219).
In a severe but rare form of Zn deficiency in humans, as occurs in acrodermatitis enteropathica, several case reports have described cerebral atrophy, irritability, apathy, and psychomotor delay in the affected individuals (220, 221). On some occasions the findings are reversible upon Zn supplementation.
In summary: These effects show that Zn deficiency, especially if it occurs early in fetal development, can disrupt astrocytes, oligodendrocytes and myelin production, with potential lingering effects towards adulthood.
B6 Zn and retina
Although Zn exists in many ocular tissues, such as the choroid, ciliary body, and iris, the highest Zn concentrations in the eye are found in the retina-choroid complex (464–472 μg/g Zn of dry weight) (222). Zn is an element necessary for many ocular metalloenzymes, both from a structural and functional aspect. Deficient Zn status can hinder ocular development, especially during the early prenatal stages (222). In the eye, vitamin A is converted to its active form, retinal, by a Zn metalloenzyme, alcohol dehydrogenase (ADH). Retinal is needed for the synthesis of rhodopsin, the photopigment found in the retinal rods responsible for night vision. Therefore, depressed ADH activity in cases of Zn deficiency would result in night blindness (222). In pregnant women with nyctalopia due to combined vitamin A and Zn deficiency responded to vitamin A and Zn supplementation but not to vitamin A or Zn alone (223). In addition, Zn has an important protective role against glutamate-induced toxicity in the retina. The retina is abundant in glutamatergic neurons. Zn is released along with glutamate in the retina, just like in the brain. By binding on the NMDA glutamate receptors of retinal ganglion cells and blocking excitation, Zn prevents the toxic effects of glutamate on the retina (224).
In preterm infants, the main concern is the possible association of Zn deficiency with the development of ROP. The pathophysiology of ROP evolves in critical hyperoxic and hypoxic phases with key enzymatic pathways such as VEGF, erythropoietin (EPO) and IGF-1 among others to play important roles in each phase (225–227). The role of Zn in the development of ROP is controversial and not well studied but could affect the development of ROP by several mechanisms: (1) as a potent antioxidant; (2) as modulator of glutamate receptors and amelioration of related toxicity as noted above, especially since glutamate can induce VEGF, a key molecule for development of ROP (228); (3) via modulation of transcription factors. Zn has antioxidant properties, as it is part of certain protective antioxidant enzymes, such as the previously mentioned enzyme SOD1, which has a protective effect against oxygen-induced retinopathy in mice (229). However, preterm neonates often have deficient Zn levels and subsequent reduced SOD activity. Moreover, Li et al. recently identified through a genome wide association study a novel lead single nucleotide polymorphism (SNP) that fell in an intronic region within the GLi3 gene. GLi3 is critical for the differentiation of the retinal pigment epithelium (RPE) and rod photoreceptor layer, functioning as both a transcriptional activator and repressor of canonical SHH signaling (230, 231). It also controls both the innate and adaptive immune response (232, 233), and, as aberrant inflammation is involved in the pathophysiology of ROP (234, 235), there is a possible association between GLi3 and ROP. The role of Zn related to EPO is quite complex, and to our knowledge indirect, and will be discussed briefly in the next paragraph. Clinically, exogenous human EPO initiated shortly after birth, although could result in angiogenesis, was not found to increase the risk of severe ROP (Preterm Erythropoietin Neuroprotection Trial, PENUT) (236).
Several clinical observational studies in premature newborns attempted to investigate the role of Zn with ROP, with so far mixed results. In one study, preterm newborns with ROP had lower maternal Zn levels, cord levels and serum levels at 40 weeks PMA when compared to those who did not develop ROP (237). In a retrospective study that involved infants 28–37 weeks GA, investigators measured one serum Zn level at less than 24 h after birth and in those newborns with levels <0.7 μg/ml, a higher proportion developed ROP when compared to those with higher Zn level (42% vs. 24%, P = 0.02) (59). However, in a systematic review of RCTs, enteral Zn supplementation compared to no supplementation or placebo had no effect on ROP (238, 239). Notably, several of the included studies also involved infants >32 weeks GA, with low baseline risk of ROP. The efficacy of maternal Zn supplementation for the prevention of ROP remains to be investigated.
In summary: Preclinical data link Zn deficiency with the development of ROP and other developmental eye disorders. Specifically, Zn could improve ROP via its antioxidant properties, modulation of glutamate pathway and excitotoxicity and via changes of downstream transcription factors. Nevertheless, robust clinical data to support this mechanistic link, is lacking so far.
B7 Zn and Vitamin D
Vitamin D is a lipid soluble vitamin with a steroid structure. Its structural integrity and functions are tightly regulated by Zn. Cholecalciferol is hydroxylated in the liver into 25-hydroxycholecalciferol, which is further hydroxylated in the kidney into 1,25-dihydroxycholecalciferol (active form). The latter interacts with vitamin D receptors (VDR). Once vitamin D interacts with its receptor, VDR dimerizes with the retinoid X receptor (RXR), to upregulate several downstream genes, including the calcium stimulated ATPase and alkaline phosphatase, causing increased intestinal absorption of calcium. VDR uses Zn to regulate the actions of vitamin D-dependent genes. In vitro, in the absence of Zn, VDR conformation cannot occur, and vitamin D function fails (240, 241). Zn supplementation increases Vit D levels and there a positive correlation between vitamin D levels and Zn levels (242, 243). Low Zn levels were found to predict vitamin D deficiency in adolescent women (244). On the other hand, vitamin D regulates Zn homeostasis by affecting the expression of Zn transporters (245). The role of vitamin D in normal brain function and development has been reviewed and demonstrated in several studies (246–248).
In summary: The intrinsic role of vitamin D in the regulation of Zn and vice versa should be taken into consideration when interpreting studies with Zn deficiency.
B8 Zn and factors that regulate hemopoiesis—possible roles in neuronal development
Zn participates in hemopoiesis in the form of ZFPs. Different ZFPs act on different cell lineages and maturation stages. ZFPs like GATA, Ikaros, FOG-1, Snail, MOZ, Gfi, and Zfp521 regulate the survival and differentiation of hematopoietic cells at the stem cell level until they commit to the lymphoid or myeloid lineage (249). Lymphopoiesis is regulated mainly by the ZFPs of the Ikaros family while myelopoiesis is controlled by a more complicated and diverse expression of ZFPs and has not yet been thoroughly studied. Among the 6 members of GATA-binding transcription factors of ZFPs, GATA-1 is important for erythroid cell line development (249). Zn is essential for lymphopoiesis and myelopoiesis, and it is also very important for erythropoiesis, mainly through the previously mentioned transcription factor GATA-1, the apoptotic proteins such as caspase 3, and the hypoxia-inducible factor (HIF) (250).
In the stage of proerythroblasts, GATA-1 release is triggered by the binding of erythropoietin to the erythropoietin receptor. Then GATA interacts with its co-factor, friend of GATA protein 1 (FOG-1), and together as a complex, they control gene expression (251). The formation of the complex requires the presence of Zn (252, 253). In addition, Zn, along with carnitine, inhibits the cleavage of caspase 3, a key component of apoptosis that is expressed when stimulation by erythropoietin is interrupted (254, 255). In this way, Zn prevents RBC apoptosis and promotes RBC survival, whereas Zn depletion results in RBC death (256, 257).
Erythropoiesis is initiated in hypoxic states when the HIF triggers the expression of the EPO gene. When oxygen levels are low, the HIF-1 subunit becomes stabilized and assembled. In contrast, in physiologic oxygen levels, HIF-1 is degraded. An HIF-1-specific prolyl-hydroxylase (PHD) hydroxylases proline-564 and/or −402 residues of HIF-1, signaling its ubiquitination and degradation by the proteasome. PHD2, also known as EGLN1, the main enzyme in control of hydroxylating HIF-1, has a conserved catalytic domain similar to that of other prolyl-4-hydroxylases. However, it also has a distinctive N-terminal MYND-type Zn finger domain (258) that has been anticipated to have either a positive or a negative regulatory function. Varying roles of this domain have been indicated, and according to the most recent results from Sinnema et al., the Zn finger ordinarily has a positive regulatory effect on the catalytic activity of PHD2. Specifically, a human PHD2 Zn finger mutation results in a loss of PHD2 function and is associated with congenital erythrocytosis (259).
HIF not only has functional roles but also takes part in the morphogenesis and development of the CNS. As shown by preclinical experiments, brain angiogenesis depends on the HIF signaling pathway since new vessel development is affected by oxygen regulation in neurons (260). In addition, HIF has a significant role in the neurogenesis of the autonomic/sympathetic nervous system (ANS), as the loss of HIF-1α hindered the survival and proliferation of pre- and post-ganglionic neurons. Specifically, lack of HIF-1α in the cardiac outflow tract, right ventricle and atrium, pharyngeal mesoderm, peripheral neurons, and hindlimbs caused hypoplasia of the sympathetic ganglion chain and diminished chromaffin cell number in the adrenal medulla (261). Astrocytes interact with HIF-1a in the opposite way; the deletion of HIF-1α prevents a hypoxia-induced cell death (262). Interestingly, the deletion of HIF-1α in neurons facilitates their hypoxia-induced cell death (262). Finally, HIF can induce hypomyelination by inhibiting the differentiation of oligodendrocyte precursor cells (263, 264).
In summary: The role of Zn in factors that affect hemopoiesis and lymphopoiesis is well supported by preclinical studies. HIF-1 is a regulator of erythropoiesis under conditions of hypoxia, but also a key regulator of brain morphogenesis, astrocyte function, myelination and vascular development of the CNS and ANS. Specific Zn containing enzymes (Zn-fingers) affect HIF-1 function, but whether Zn deficiency has significant effects on those pathways remain to be investigated.
Part C clinical aspects of Zn provision
C1 can Zn induce neuronal toxicity?
Zn reaches physiologically high but transient concentrations in synaptic cleft. The synaptic clefts are enclosed low volume cylindrical compartments which represent only 1% of the extracellular fluid (ECF) of the brain (265). In these areas, Zn concentration is tightly regulated by receptors and local MTs (type III) (266) and is estimated to be 1–100 μM (267).
Zn is released and possibly reaches toxic levels during pathological events such as cerebral ischemia or seizures. Nolte et al. assessed the effect of extracellular Zn on cultured astrocytes in vitro and found that Zn at concentration of 200–250 μM induced cell death within 1.5–2 h of exposure (210). The efflux of Zn after noxious stimuli in a confined ECF space may alter the pH of the microenvironment, leading to cellular damage, with subfield CA3 hippocampal neurons being very sensitive to early destruction (268–270). Induction of convulsions with kainic acid (KA) in rats can induce an increase in serum and ECF Zn but overall depletion of tissue Zn (271), while Zn appeared to accelerate brain infarction after focal ischemia in rats (272).
During global brain ischemia, Zn might become toxic and result in selective neuronal loss independently of other mechanisms of brain damage such as excitotoxicity. In rats, after brain ischemia, Zn accumulated in the hippocampal hilus and CA1, as well as in the cerebral cortex, thalamus, striatum, and amygdala, while Zn chelation showed reduced ischemic neuronal degeneration (273).
High neuronal synaptic Zn concentrations (up to 300 μM) are present transiently in specific areas in rodent CNS (such as the hippocampal mossy fibers) after neuronal hyperexcitation with kainic acid (KA), a process that is calcium mediated and might have a role in physiologic neuronal excitation (267, 274). Lower Zn concentrations (3–30 μM) in the synaptic cleft of cultured rat retinal neurons, was sufficient to protect from the toxicity of glutamate or NMDA (224).
Zn neurotoxicity might be associated with the proto-oncogene c-Src (abbreviation for cellular sarcoma), a non-receptor ubiquitous cellular tyrosine kinase. Src kinase is an essential regulator of cellular physiological processes ranging from differentiation, mitogenic signaling to motility and neuroinflammation (275–277). The brain expresses 200-fold higher levels of this protein than most other cells (276). Zn is released along with glutamate in the CNS and inhibits NMDA receptor activation. Following this blockade, use of high (but sublethal) concentrations of Zn in rodent cortex can cause a Src kinase-mediated up-regulation of NMDA receptor activity and subsequent cytotoxicity (278).
Inflammation could also contribute to Zn toxicity, since high extracellular Zn concentrations may be pro-inflammatory in primary mononuclear cells. In a study of human PBMCs where high concentrations of Zn (>100 μM) were achieved, all types of cytokines were increased, and pro-apoptotic genes were induced (279).
The above studies show that Zn has the potential to induce neuronal toxicity especially after pathologic conditions (seizures, stroke and ischemia), either directly or indirectly via excitotoxicity or/and inflammation. High Zn extracellular concentrations can be physiologic, with trophic and receptor modulatory properties in specific neurons and not related to toxicity. Of note Zn concentrations of 100 μM (μmoles L) correspond to 6.53 μg/ml—although these levels are reported in the ECF or synaptic cleft of CNS and cannot be compared directly with serum levels, they are much higher than the upper normative serum Zn level that was previously discussed (1.43 μg/ml) and 100 times more than Zn in CSF.
Indirect Zn toxicity
While Zn may interact with many elements, attention has been drawn to its relationship with Fe, Cu, calcium absorption, vitamin D and vitamin A. Even though Zn supplementation is considered relatively safe, enteral administration has the potential to negatively influence Cu and Fe absorption in the GI tract (69, 280, 281). By competing for the same receptors, Zn reduces Cu uptake (2, 282–285). Although neonatal data are lacking, in adults a dose of Zn: Fe of 5:1, significantly decreased Fe absorption (286, 287). Limited data in rats show that Zn could interfere with Vitamin A, by decreasing its liver mobilization and release, whereas similar studies in neonates are lacking (288). Therefore, Zn supplementation over and above the recommended daily intake requires careful monitoring and evaluation for patients dependent on a balanced micronutrient intake. Although definition of “prolonged” Zn provision is not well defined, any newborn that has received >2 weeks of Zn provision could be at risk for Cu deficiency unless Cu is supplemented at a 10:1 Zn: Cu weight ratio and at alternate times of administration. A recent systematic review of RCTs in children 6 months-6 years demonstrated a small decrease in Cu concentration in Zn-supplemented patients when compared to no Zn. This result of unclear clinical relevance was based on studies that provided prolonged Zn supplementation with no supplemented Cu (289).
In summary: Direct neuronal Zn toxicity is unlikely to occur under physiologic conditions with routine Zn supplementation but could be encountered under conditions of cerebral ischemia, stroke, or seizures. In these circumstances, Zn supplementation needs to be held. Indirect Zn toxicity, secondary to its effect on Cu and Fe absorption needs to be carefully monitored especially in an era of increasing provision of Zn. Adequately powered studies that will investigate the potential toxicity of Zn provision in premature neonates, given the trends towards higher dosing supplementation, are urgently needed.
C2 alcohol fetal exposure, fetal alcohol syndrome (FAS) and Zn
Alcohol consumption may interfere with Zn tissue utilization and can have significant effects on the fetus (290, 291). Alcohol consumption can result in deficiency of several micronutrients including Zn. Whether Zn deficiency can contribute to the development of fetal alcohol syndrome (FAS) - a devastating disease with serious neurodevelopmental effects- is controversial (292). Zn is part of ADH, which detoxifies alcohol and generates aldehydes and ketones as metabolites. Alcohol in high concentrations can deactivate this enzyme (293). In addition, alcohol can induce increased urinary Zn excretion and low plasma levels in pregnant women (294), while alcohol abstinence reverses zincuria in adults (295). Affected infants from FAS could also present with low plasma and high urinary Zn excretion (296). In alcohol-exposed primates, when adequate Zn (3.5 mg/day) was given, maternal and neonatal brain Zn concentrations were similar to non-exposed controls, indicating that placental Zn transfer was not significantly affected (297). In rodents, whether alcohol affects Zn placental transfer is controversial (298, 299) and Zn supplementation might not be able to restore Zn transplacental movement in alcohol exposed mothers (300). In addition, ethanol-exposed rats had similar Zn placental transfer when compared to non-ethanol fed controls (301).
In summary: Alcohol consumption could be associated with multinutrient and/or isolated Zn deficiency. Although some of the manifestations of FAS may be related to co-existent or alcohol induced Zn deficiency, further research is needed to identify its significance in brain outcomes in the affected neonates.
C3 smoking, Zn and the role of Cadmium (Cd)
The effects of smoking during pregnancy on fetal Zn are largely mediated by the Cd content of cigarettes. Cd and Zn are closely related in many metabolic pathways and often antagonize one another in their usage. Cd strongly induces MTs, which bind both Cd and Zn and can lower systemic Zn levels (302, 303). Smoking during pregnancy increases Cd levels in maternal blood and in placenta (302, 303). However, a large quantity of Cd is sequestered in the maternal liver and kidneys, with only a fraction accumulating in the placenta and the fetus (303). Mouse models have shown an increase in MT in the placenta after MT elevations in the liver have taken place, which tends to sequester Zn in the process, decreasing delivery of Zn to the fetus (52).
Additionally, cord blood analyses have shown that in mothers that do not smoke, elevations in placental Zn are associated with elevations in cord Zn, whereas, in mothers who smoke, elevations in placental Zn are not accompanied by elevations in cord Zn (302). Another notable feature of Cd's effect on maternal-fetal Zn delivery is through Zn transporters. In mouse models, Cd downregulates Zn transporters, which possibly could decrease Zn fetal uptake (303, 304). Mouse models have further associated maternal Cd exposure with a decreased BDNF and Zn level in the brain (304), indicating that smoking during pregnancy may impact fetal brain development. Supplementation of Zn appears to restore BDNF levels in fetuses (304).
In summary: Smoking affects Zn levels in the fetus primarily via Cd-mediated upregulation of MTs, and downregulation of Zn transporters, which together, act to sequester Zn in placental tissue and can harm fetal brain development as a downstream effect.
C4 clinical data in premature neonates, infants and children—neurodevelopmental outcomes and Zn
The neurodevelopmental effects of Zn in preterm newborns can be related to (1) its deficiency (low Zn level) (2) to its adequate supplementation or (3) to indirect effects on growth. Systematic reviews have shown that Zn supplementation increases growth and decreases mortality in preterm infants (238, 239). These findings are summarized in a Cochrane review (238) including one study with high-dose Zn intake [(305), ∼10 mg/day] and 4 studies with low (currently recommended) dose were included (63, 64, 306, 307). Other studies with high-dose Zn intake [(308, 309, 310) were not included]. The included studies were relatively small but had good methodological quality. The meta-analysis showed that enteral Zn supplementation may decrease all-cause mortality [Relative Risk (RR) 0.55, 95% Confidence Intervals (CI) 0.31–0.97; 3 studies, 345 infants; low-certainty evidence based on high-dose Zn study by Terrin et al. (305)], while had little or no effect on common morbidities such as bronchopulmonary dysplasia, ROP, bacterial sepsis, or NEC (low certainty of evidence). The authors concluded that Zn supplementation probably improves weight gain [standardized mean difference (SMD) 0.46, 95% CI 0.28–0.64; 5 studies, 481 infants; moderate-certainty evidence]; and may slightly improve linear growth (SMD 0.75, 95% CI 0.36–1.14, 3 studies, 289 infants; low-certainty evidence), but had minimal effect on fronto-occipital head circumference (FOC) (SMD 0.21, 95% CI −0.02 to 0.44, 3 studies, 289 infants; moderate-certainty evidence). No data was available on long-term neurodevelopmental outcomes at 18–24 months of age. In a systematic review that focused on anthropometrics and neurodevelopment only, Alshaikh et al. (239), included 8 RCTs studies (63, 64, 305–308, 311, 312) two of which utilized high dose (∼10 mg/day) Zn (305, 308). 7 out of 8 RCTs (742 infants), reported growth data at 3–6 months corrected age and 2 reported neurodevelopmental outcomes at 6–12 months, and will be further discussed later in this chapter (306, 312). Zn supplementation was associated with increased weight z-score (SMD 0.50; 95% CI 0.23–0.76), length z-score (SMD 1.12; 95% CI 0.63–1.61) and motor developmental score (SMD 9.54; 95% CI 6.6–12.4). There was no effect of Zn supplementation on FOC.
Most of the studies reported in both systematic reviews included older premature and low birth infants and as result, outcomes that occur more frequently in very low birth weight (VLBW) and extremely low birth weight (ELBW) infants and very preterm (28–32 weeks or extremely preterm (<28 weeks) infants (such as ROP, NEC etc.) were not assessed adequately.
Our group showed that In Zn-deficient ELBW preterm newborns, low dose Zn provision for at least 2 weeks improved the Z-score of FOC, but not length and weight (58). In another study in Japan, routine enteral Zn supplementation at 3 mg/kg/day, starting at 2 weeks of life to discharge without any PN Zn intake, showed no effect on growth parameters (313). In contrast, higher and incremental enteral doses of Zn (5–10 mg/day) in VLBW infants, improved linear growth in a study from India (314).
Differences in Zn dosing, duration of treatment and initiation and differences in baseline maternal Zn deficiency could all contribute to the variation in the above outcomes. Overall, though, Zn provision in premature infants has been shown to improve growth quite consistently (238, 239).
Isolated or balanced postnatal growth patterns in various parameters [FOC, length, weight, growth velocity and/or body mass index (BMI)] are all associated, and possibly independently, with improved neurodevelopmental outcomes in preterm infants (315–318). From those growth parameters, isolated linear growth in VLBW infants is associated with improved language scores in the Bayley-III scores and less chance for future developmental deficits (319, 320). Improved anti-inflammatory milieu and glucose-regulatory hormones (321–323) are important factors for adequate growth in preterm newborns. Zn provision could relate via its well-described anti-inflammatory and glucose regulatory properties towards this effect (148).
In summary: Several studies have shown consistently improved weight gain and linear growth in preterm newborns even after low or moderate (as currently recommended) Zn supplementation dosing schedules. Since normal growth can be independently associated with improved neurodevelopment, and Zn improves growth, it needs to be further investigated if and to what extent Zn supplementation can contribute or mediate a potential independent positive effect towards neurodevelopment.
So far, the direct link between Zn provision and neurodevelopment in newborns is not well established, and most data are derived from older infants and children. The overall effect of Zn supplementation in infants or children on neurodevelopmental outcomes is not convincing, possibly since brain development and growth is less dependent on Zn later in life.
Clinical data in infants and children: In infants, 1–12 months of age, Zn intake improves length and weight (324, 325) while higher Zn dose (10 mg/day vs. 5 mg/day) had a higher impact on those growth parameters (325). The effect of Zn supplementation—especially when administered alone- was investigated in a systematic review of 96 studies in 219,584 children (age: 6 months to 6 years) and showed benefits in Zn status, diarrhea- related morbidity, linear growth and possibly a small positive impact on all-cause mortality (289). Regarding neurodevelopment, Zn supplementation was found to improve minimally the executive function and motor development (326) or have no global effect (327), while supplementing mothers during pregnancy was not found to improve long term developmental outcomes (324, 325). On the other hand, Zn supplementation in infants who reside in areas with high baseline Zn deficiency, improved cognitive and sensorimotor developmental outcomes (328). A systematic review that included children 0–5 years (25 studies and 11,559 patients), showed no significant efficacy of Zn with and without Fe co-supplementation on child mental and motor development up to 9 years old age (329). Finally, a systematic review in children with attention-deficit/hyperactivity disorder (ADHD) has shown that Zn supplementation may improve total ADHD scores (330).
In summary: These studies in older infants and children show that the effects of Zn supplementation on neurodevelopmental outcomes are minimal and possibly more pronounced in individuals with baseline Zn deficiency.
In newborns, only a few RCTs have reported limited neurodevelopmental outcomes (Table 1). Of them, three reported neurodevelopmental outcomes at 6–12 months prematurity-corrected age and one at ≤3 months.
Table 1. Neonatal randomized controlled studies (RCT) that assessed neurodevelopment with zinc provision.
Ragab et al. (312) in a study completed in Egypt, used the parents-completed Ages and Stages Questionnaires (ASQ) at 4 and 6 months of life and showed improvement in communication, gross and fine motor skills, problem solving and social interaction in infants who received Zn supplementation. Friel et al. (306), conducted a study in the US in 1993 and reported a significant increase in motor developmental score using Griffiths mental development scale using Amiel-Tison neurologic assessment at 3, 6, 9 and 12 months of life (331). Mathur et al. (64), conducted a study in India and found that Zn provision improved alertness and attention pattern at 40 weeks post menstrual age and decreased hyper-excitability at 3 months corrected GA. As mentioned earlier, two of these studies (64, 306) were evaluated in a systematic review by Alshaikh et al. who showed an improvement in motor developmental scores, but not in global development (239).
In summary: The above-mentioned studies overall show benefits in early neurodevelopmental outcomes after Zn provision, although the time frame of observation is up to 12 months after birth, which does not meet current standard for follow-up to 22–28 months corrected age or school age.
Whether Zn levels could be associated with poor developmental outcomes is also poorly investigated. Terrin et al. (37) showed that in preterm newborns 23–34 weeks GA, there was a significantly positive correlation between total composite motor score (with Bayley III scale) and serum Zn levels at 28 days of life (DOL) (R = 0.467, P < 0.05) (33758400). The same authors found that serum Zn levels at 28 DOL were dependent on energy (β −0.650; P < 0.001) and protein (β −0.669; P < 0.001) intake received through PN in the first week of life.
In summary: Currently there is limited evidence to suggest low Zn levels (early, or later in life) relate to poor neurodevelopment. The interaction of Zn levels, energy consumption and neurodevelopment need further research.
C5 optimal dosing in premature neonates
Recent nutritional recommendations favored an increase in daily Zn supplementation (enteral Zn 2–3 mg/kg/day, parenteral Zn 500 μg/kg/day) (1–3). The enteral recommendation might not match physiologic data of transplacental fetal Zn accretion and actual needs for Zn might not be met, especially given the limited amount of Zn absorbed in the premature gut (32, 37). In addition, current recommendations, do not suggest different dosing schedules in cases of maternal Zn deficiency as commonly encountered in mothers with high poverty index and in women living in or recently immigrated from countries with endemic Zn deficiency (17). High Zn dosing (10 mg/dose) has been implemented by some investigators (305, 308, 332), but no neurodevelopmental outcomes have been provided so far. These studies have not reported major toxicity related to Zn and some have reported improved early outcomes, e.g., mortality, sepsis, necrotizing enterocolitis, feeding tolerance and/or growth. In Table 2 we summarize possible gaps of knowledge and future research directions.
Table 2. Gaps of knowledge and future research directions in Zn and neonates.
In summary: We believe that well organized RCTs that include early Zn provision, at different - possibly higher - dosing schedules and include adequate neurodevelopmental follow up, need to be undertaken as soon as possible, in low-, middle- and high-income countries.
Conclusions
In conclusion, Zn as a micronutrient, has a variety of physiologic roles that affect brain function and autoregulation but also in neuronal growth, migration and survival. Preterm newborns lose the period of highest fetal Zn accretion—the last two trimesters—and as result represent a population that is at very high risk for Zn deficiency. Additional risk factors, such as maternal Zn deficiency can exacerbate neonatal Zn deficiency, while feeding practices—such as exclusive unfortified DHM - can also result Zn deficiency.
Despite the emerging role of Zn as a key nutritional supplement, there are only a few, well organized published studies that investigate its role in short term neonatal morbidities and long-term neurodevelopmental outcomes. We suggest that multicenter RCTs be undertaken to investigate the above issues.
Author contributions
MC: Writing – original draft, Writing – review & editing. LB: Conceptualization, Writing – original draft, Writing – review & editing. PV: Writing – original draft, Writing – review & editing. CL: Supervision, Writing – original draft, Writing – review & editing. DA: Conceptualization, Funding acquisition, Investigation, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received from the Flora Miller Parill Award” of the University of Texas, Southwestern Medical Center for the research, authorship, and/or publication of this article.
Acknowledgments
We would like to acknowledge the Crystal Charity Ball and the NeuroNICU program at the University of Texas Southwestern Medical Center for supporting this project. We also thank Mr. Thalis Asimakopoulos for contributing to the creation of Figure 2.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Embleton ND, Jennifer Moltu S, Lapillonne A, van den Akker CHP, Carnielli V, Fusch C, et al. Enteral nutrition in preterm infants (2022): a position paper from the ESPGHAN committee on nutrition and invited experts. J Pediatr Gastroenterol Nutr. (2023) 76(2):248–68. doi: 10.1097/MPG.0000000000003642
2. Bhatia J, Griffin I, Anderson D, Kler N, Domellof M. Selected macro/micronutrient needs of the routine preterm infant. J Pediatr. (2013) 162(3 Suppl):S48–55. doi: 10.1016/j.jpeds.2012.11.053
3. Klein CJ. Nutrient requirements for preterm infant formulas. J Nutr. (2002) 132(6 Suppl 1):1395S–577. doi: 10.1093/jn/132.6.1395
4. Zapata CL, Melo MR, Donangelo CM. Maternal, placental and cord zinc components in healthy women with different levels of serum zinc. Biol Neonate. (1997) 72(2):84–93. doi: 10.1159/000244470
5. Shaw JC. Trace elements in the fetus and young infant. I. Zinc. Am J Dis Child. (1979) 133(12):1260–8. doi: 10.1001/archpedi.1979.02130120052011
6. Ford D. Intestinal and placental zinc transport pathways. Proc Nutr Soc. (2004) 63(1):21–9. doi: 10.1079/PNS2003320
7. Page KR, Abramovich DR, Aggett PJ, Todd A, Dacke CG. The transfer of zinc across the term dually perfused human placental lobule. Q J Exp Physiol. (1988) 73(4):585–93. doi: 10.1113/expphysiol.1988.sp003178
8. Jariwala M, Suvarna S, Kiran Kumar G, Amin A, Udas AC. Study of the concentration of trace elements fe, zn, cu, se and their correlation in maternal serum, cord serum and colostrums. Indian J Clin Biochem. (2014) 29(2):181–8. doi: 10.1007/s12291-013-0338-8
9. Khoushabi F, Shadan MR, Miri A, Sharifi-Rad J. Determination of maternal serum zinc, iron, calcium and magnesium during pregnancy in pregnant women and umbilical cord blood and their association with outcome of pregnancy. Mater Sociomed. (2016) 28(2):104–7. doi: 10.5455/msm.2016.28.104-107
10. Iqbal AS, Shahidullah M, Islam MN, Akhter S, Banu S. Serum zinc and copper levels in the maternal blood and cord blood of neonates. Indian J Pediatr. (2001) 68(6):523–6. doi: 10.1007/BF02723246
11. Yamashita K, Ohno H, Doi R, Mure K, Ishikawa M, Shimizu T, et al. Distribution of zinc and copper in maternal and cord blood at delivery. Biol Neonate. (1985) 48(6):362–5. doi: 10.1159/000242195
12. Aslam N, McArdle HJ. Mechanism of zinc uptake by microvilli isolated from human term placenta. J Cell Physiol. (1992) 151(3):533–8. doi: 10.1002/jcp.1041510312
13. Henkin RI, Marshall JR, Meret S. Maternal-fetal metabolism of copper and zinc at term. Am J Obstet Gynecol. (1971) 110(1):131–4. doi: 10.1016/0002-9378(71)90234-1
14. Nandakumaran M, Dashti HM, Al-Saleh E, Al-Zaid NS. Transport kinetics of zinc, copper, selenium, and iron in perfused human placental lobule in vitro. Mol Cell Biochem. (2003) 252(1–2):91–6. doi: 10.1023/A:1025565720489
15. Paterson PG, Sarkar B, Zlotkin SH. The effect of zinc levels in fetal circulation on zinc clearance across the in situ perfused Guinea pig placenta. Can J Physiol Pharmacol. (1990) 68(11):1401–6. doi: 10.1139/y90-213
16. Simmer K, Dwight JS, Brown IM, Thompson RP, Young M. Placental handling of zinc in the Guinea pig. Biol Neonate. (1985) 48(2):114–21. doi: 10.1159/000242162
17. Mejia-Rodriguez F, Shamah-Levy T, Villalpando S, Garcia-Guerra A, Mendez-Gomez Humaran I. Iron, zinc, copper and magnesium deficiencies in Mexican adults from the national health and nutrition survey 2006. Salud Publica Mex. (2013) 55(3):275–84. doi: 10.21149/spm.v55i3.7210
18. Myers SS, Wessells KR, Kloog I, Zanobetti A, Schwartz J. Effect of increased concentrations of atmospheric carbon dioxide on the global threat of zinc deficiency: a modelling study. Lancet Glob Health. (2015) 3(10):e639–45. doi: 10.1016/S2214-109X(15)00093-5
19. Yokokawa H, Morita Y, Hamada I, Ohta Y, Fukui N, Makino N, et al. Demographic and clinical characteristics of patients with zinc deficiency: analysis of a nationwide Japanese medical claims database. Sci Rep. (2024) 14(1):2791. doi: 10.1038/s41598-024-53202-0
20. Osada H, Watanabe Y, Nishimura Y, Yukawa M, Seki K, Sekiya S. Profile of trace element concentrations in the feto-placental unit in relation to fetal growth. Acta Obstet Gynecol Scand. (2002) 81(10):931–7. doi: 10.1034/j.1600-0412.2002.811006.x
21. Matsusaka N, Sakamoto H, Sato I, Shinagawa K, Kobayashi H, Nishimura Y. Whole-body retention and fetal uptake of 65Zn in pregnant mice fed a zn-deficient diet. J Radiat Res. (1995) 36(3):196–202. doi: 10.1269/jrr.36.196
22. Jobarteh ML, McArdle HJ, Holtrop G, Sise EA, Prentice AM, Moore SE. mRNA levels of placental iron and zinc transporter genes are upregulated in Gambian women with low iron and zinc status. J Nutr. (2017) 147(7):1401–9. doi: 10.3945/jn.116.244780
23. Helston RM, Phillips SR, McKay JA, Jackson KA, Mathers JC, Ford D. Zinc transporters in the mouse placenta show a coordinated regulatory response to changes in dietary zinc intake. Placenta. (2007) 28(5–6):437–44. doi: 10.1016/j.placenta.2006.07.002
24. Mas A, Sarkar B. The metabolism of metals in rat placenta. Biol Trace Elem Res. (1988) 18:191–9. doi: 10.1007/BF02917503
25. Klein D, Scholz P, Drasch GA, Muller-Hocker J, Summer KH. Metallothionein, copper and zinc in fetal and neonatal human liver: changes during development. Toxicol Lett. (1991) 56(1–2):61–7. doi: 10.1016/0378-4274(91)90090-S
26. Dorea JG, Brito M, Araujo MO. Concentration of copper and zinc in liver of fetuses and infants. J Am Coll Nutr. (1987) 6(6):491–5. doi: 10.1080/07315724.1987.10720208
27. Zlotkin SH, Cherian MG. Hepatic metallothionein as a source of zinc and cysteine during the first year of life. Pediatr Res. (1988) 24(3):326–9. doi: 10.1203/00006450-198809000-00010
28. Brion LP, Heyne R, Lair CS. Role of zinc in neonatal growth and brain growth: review and scoping review. Pediatr Res. (2021) 89(7):1627–40. doi: 10.1038/s41390-020-01181-z
29. Vallee BL, Falchuk KH. The biochemical basis of zinc physiology. Physiol Rev. (1993) 73(1):79–118. doi: 10.1152/physrev.1993.73.1.79
30. Lee HH, Prasad AS, Brewer GJ, Owyang C. Zinc absorption in human small intestine. Am J Physiol. (1989) 256(1 Pt 1):G87–91. doi: 10.1152/ajpgi.1989.256.1.G87
31. Carlson SJ, Chang MI, Nandivada P, Cowan E, Puder M. Neonatal intestinal physiology and failure. Semin Pediatr Surg. (2013) 22(4):190–4. doi: 10.1053/j.sempedsurg.2013.10.007
32. Terrin G, Berni Canani R, Di Chiara M, Pietravalle A, Aleandri V, Conte F, et al. Zinc in early life: a key element in the Fetus and preterm neonate. Nutrients. (2015) 7(12):10427–46. doi: 10.3390/nu7125542
33. Friel JK, Andrews WL, Simmons BS, Miller LV, Longerich HP. Zinc absorption in premature infants: comparison of two isotopic methods. Am J Clin Nutr. (1996) 63(3):342–7. doi: 10.1093/ajcn/63.3.342
34. Higashi A, Ikeda T, Iribe K, Matsuda I. Zinc balance in premature infants given the minimal dietary zinc requirement. J Pediatr. (1988) 112(2):262–6. doi: 10.1016/S0022-3476(88)80067-2
35. Altigani M, Murphy JF, Gray OP. Plasma zinc concentration and catch up growth in preterm infants. Acta Paediatr Scand Suppl. (1989) 357:20–33. doi: 10.1111/j.1651-2227.1989.tb11271.x
36. Walravens PA, Chakar A, Mokni R, Denise J, Lemonnier D. Zinc supplements in breastfed infants. Lancet. (1992) 340(8821):683–5. doi: 10.1016/0140-6736(92)92229-9
37. Terrin G, Boscarino G, Di Chiara M, Iacobelli S, Faccioli F, Greco C, et al. Nutritional intake influences zinc levels in preterm newborns: an observational study. Nutrients. (2020) 12(2):1–9. doi: 10.3390/nu12020529
38. Gates A, Marin T, Leo G, Stansfield BK. Review of preterm human-milk nutrient composition. Nutr Clin Pract. (2021) 36(6):1163–72. doi: 10.1002/ncp.10570
39. Kim SY, Park JH, Kim EA, Lee-Kim YC. Longitudinal study on trace mineral compositions (selenium, zinc, copper, manganese) in Korean human preterm milk. J Korean Med Sci. (2012) 27(5):532–6. doi: 10.3346/jkms.2012.27.5.532
40. Sanchez-Rosado M, Lair CS, Edwards A, Jacob T, Heyne R, Brown LS, et al. Growth after implementing a donor breast milk program in neonates <33 weeks gestational age or birthweight <1,500 grams: retrospective cohort study. J Perinatol. (2023) 43(5):608–15. doi: 10.1038/s41372-023-01627-2
41. Abrams SA. Zinc for preterm infants: who needs it and how much is needed? Am J Clin Nutr. (2013) 98(6):1373–4. doi: 10.3945/ajcn.113.076489
42. Palmiter RD, Findley SD. Cloning and functional characterization of a mammalian zinc transporter that confers resistance to zinc. EMBO J. (1995) 14(4):639–49. doi: 10.1002/j.1460-2075.1995.tb07042.x
43. Outten CE, O’Halloran TV. Femtomolar sensitivity of metalloregulatory proteins controlling zinc homeostasis. Science. (2001) 292(5526):2488–92. doi: 10.1126/science.1060331
44. Rink L, Haase H. Zinc homeostasis and immunity. Trends Immunol. (2007) 28(1):1–4. doi: 10.1016/j.it.2006.11.005
45. Tamura Y. The role of zinc homeostasis in the prevention of diabetes mellitus and cardiovascular diseases. J Atheroscler Thromb. (2021) 28(11):1109–22. doi: 10.5551/jat.RV17057
46. Ebadi M, Itoh M, Bifano J, Wendt K, Earle A. The role of Zn2+ in pyridoxal phosphate mediated regulation of glutamic acid decarboxylase in brain. Int J Biochem. (1981) 13(10):1107–12. doi: 10.1016/0020-711X(81)90174-9
47. Stefanidou M, Maravelias C, Dona A, Spiliopoulou C. Zinc: a multipurpose trace element. Arch Toxicol. (2006) 80(1):1–9. doi: 10.1007/s00204-005-0009-5
48. Hamer DH. Metallothionein. Annu Rev Biochem. (1986) 55:913–51. doi: 10.1146/annurev.bi.55.070186.004405
49. Brown KH. Effect of infections on plasma zinc concentration and implications for zinc status assessment in low-income countries. Am J Clin Nutr. (1998) 68(2 Suppl):425S–9. doi: 10.1093/ajcn/68.2.425S
50. Clough SR, Mitra RS, Kulkarni AP. Qualitative and quantitative aspects of human fetal liver metallothioneins. Biol Neonate. (1986) 49(5):241–54. doi: 10.1159/000242538
51. Rosenthal MD, Albrecht ED, Pepe GJ. Estrogen modulates developmentally regulated gene expression in the fetal baboon liver. Endocrine. (2004) 23(2–3):219–28. doi: 10.1385/ENDO:23:2-3:219
52. Wade JV, Agrawal PR, Poisner AM. Induction of metallothionein in a human trophoblast cell line by cadmium and zinc. Life Sci. (1986) 39(15):1361–6. doi: 10.1016/0024-3205(86)90334-6
53. Levenson CW, Morris D. Zinc and neurogenesis: making new neurons from development to adulthood. Adv Nutr. (2011) 2(2):96–100. doi: 10.3945/an.110.000174
54. Finch CW. Review of trace mineral requirements for preterm infants: what are the current recommendations for clinical practice? Nutr Clin Pract. (2015) 30(1):44–58. doi: 10.1177/0884533614563353
55. Gibson RS, DeWolfe MS. Changes in serum zinc concentrations of some Canadian full term and low birthweight infants from birth to six months. Acta Paediatr Scand. (1981) 70(4):497–500. doi: 10.1111/j.1651-2227.1981.tb05729.x
56. Tyrala EE, Manser JI, Brodsky NL, Tran N. Serum zinc concentrations in growing premature infants. Acta Paediatr Scand. (1983) 72(5):695–8. doi: 10.1111/j.1651-2227.1983.tb09795.x
57. McMaster D, Lappin TR, Halliday HL, Patterson CC. Serum copper and zinc levels in the preterm infant. A longitudinal study of the first year of life. Biol Neonate. (1983) 44(2):108–13. doi: 10.1159/000241703
58. Brion LP, Heyne R, Steven Brown L, Lair CS, Edwards A, Burchfield PJ, et al. Zinc deficiency limiting head growth to discharge in extremely low gestational age infants with insufficient linear growth: a cohort study. J Perinatol. (2020) 40(11):1694–704. doi: 10.1038/s41372-020-00778-w
59. Mishra S, Shrivastava N, Agrawal A, Shrivastava J. Serum zinc levels in preterm newborns and its relation with retinopathy of prematurity. J Neonatol. (2023) 37(4):365–70. doi: 10.1177/09732179231173774
60. Katz O, Paz-Tal O, Lazer T, Aricha-Tamir B, Mazor M, Wiznitzer A, et al. Severe pre-eclampsia is associated with abnormal trace elements concentrations in maternal and fetal blood. J Matern Fetal Neonatal Med. (2012) 25(7):1127–30. doi: 10.3109/14767058.2011.624221
61. Akdas S, Yazihan N. Cord blood zinc status effects on pregnancy outcomes and its relation with maternal serum zinc levels: a systematic review and meta-analysis. World J Pediatr. (2020) 16(4):366–76. doi: 10.1007/s12519-019-00305-8
62. Friel JK, Gibson RS, Balassa R, Watts JL. A comparison of the zinc, copper and manganese status of very low birth weight pre-term and full-term infants during the first twelve months. Acta Paediatr Scand. (1984) 73(5):596–601. doi: 10.1111/j.1651-2227.1984.tb09981.x
63. Diaz-Gomez NM, Domenech E, Barroso F, Castells S, Cortabarria C, Jimenez A. The effect of zinc supplementation on linear growth, body composition, and growth factors in preterm infants. Pediatrics. (2003) 111(5 Pt 1):1002–9. doi: 10.1542/peds.111.5.1002
64. Mathur NB, Agarwal DK. Zinc supplementation in preterm neonates and neurological development, a randomized controlled trial. Indian Pediatr. (2015) 52(11):951–5. doi: 10.1007/s13312-015-0751-6
65. Vimalraj S. Alkaline phosphatase: structure, expression and its function in bone mineralization. Gene. (2020) 754:144855. doi: 10.1016/j.gene.2020.14485
66. Koo WW, Succop P, Hambidge KM. Serum alkaline phosphatase and serum zinc concentrations in preterm infants with rickets and fractures. Am J Dis Child. (1989) 143(11):1342–5. doi: 10.1001/archpedi.1989.02150230100032
67. Heinen F, Matern D, Pringsheim W, Leititis JU, Brandis M. Zinc deficiency in an exclusively breast-fed preterm infant. Eur J Pediatr. (1995) 154(1):71–5. doi: 10.1007/BF01972977
68. Hall SL, Dimai HP, Farley JR. Effects of zinc on human skeletal alkaline phosphatase activity in vitro. Calcif Tissue Int. (1999) 64(2):163–72. doi: 10.1007/s002239900597
69. Obladen M, Loui A, Kampmann W, Renz H. Zinc deficiency in rapidly growing preterm infants. Acta Paediatr. (1998) 87(6):685–91. doi: 10.1111/j.1651-2227.1998.tb01531.x
70. Thorp JW, Boeckx RL, Robbins S, Horn S, Fletcher AB. A prospective study of infant zinc nutrition during intensive care. Am J Clin Nutr. (1981) 34(6):1056–60. doi: 10.1093/ajcn/34.6.1056
71. Franco C, Canzoniero LMT. Zinc homeostasis and redox alterations in obesity. Front Endocrinol (Lausanne). (2023) 14:1273177. doi: 10.3389/fendo.2023.1273177
72. Angelis D, Savani R, Chalak L. Nitric oxide and the brain. Part 2: effects following neonatal brain injury-friend or foe? Pediatr Res. (2021) 89(4):746–52. doi: 10.1038/s41390-020-1021-4
73. Hemmens B, Goessler W, Schmidt K, Mayer B. Role of bound zinc in dimer stabilization but not enzyme activity of neuronal nitric-oxide synthase. J Biol Chem. (2000) 275(46):35786–91. doi: 10.1074/jbc.M005976200
74. Li H, Raman CS, Glaser CB, Blasko E, Young TA, Parkinson JF, et al. Crystal structures of zinc-free and -bound heme domain of human inducible nitric-oxide synthase. Implications for dimer stability and comparison with endothelial nitric-oxide synthase. J Biol Chem. (1999) 274(30):21276–84. doi: 10.1074/jbc.274.30.21276
75. Aravindakumar CT, Ceulemans J, De Ley M. Nitric oxide induces Zn2+ release from metallothionein by destroying zinc-sulphur clusters without concomitant formation of S-nitrosothiol. Biochem J. (1999) 344(Pt 1):253–8. doi: 10.1042/0264-6021:3440253
76. Cortese-Krott MM, Kulakov L, Oplander C, Kolb-Bachofen V, Kroncke KD, Suschek CV. Zinc regulates iNOS-derived nitric oxide formation in endothelial cells. Redox Biol. (2014) 2:945–54. doi: 10.1016/j.redox.2014.06.011
77. Spahl DU, Berendji-Grun D, Suschek CV, Kolb-Bachofen V, Kroncke KD. Regulation of zinc homeostasis by inducible NO synthase-derived NO: nuclear metallothionein translocation and intranuclear Zn2+ release. Proc Natl Acad Sci U S A. (2003) 100(24):13952–7. doi: 10.1073/pnas.2335190100
78. Chreifi G, Li H, McInnes CR, Gibson CL, Suckling CJ, Poulos TL. Communication between the zinc and tetrahydrobiopterin binding sites in nitric oxide synthase. Biochemistry. (2014) 53(25):4216–23. doi: 10.1021/bi5003986
79. Mendes Garrido Abregu F, Gobetto MN, Castanon A, Lucero D, Caniffi C, Elesgaray R, et al. Fetal and postnatal zinc restriction: sex differences in metabolic alterations in adult rats. Nutrition. (2019) 65:18–26. doi: 10.1016/j.nut.2019.01.022
80. Davenport HW. The early days of research on carbonic anhydrase. Ann N Y Acad Sci. (1984) 429:4–9. doi: 10.1111/j.1749-6632.1984.tb12310.x
81. Boone CD, Habibzadegan A, Gill S, McKenna R. Carbonic anhydrases and their biotechnological applications. Biomolecules. (2013) 3(3):553–62. doi: 10.3390/biom3030553
82. Kim JK, Lee C, Lim SW, Adhikari A, Andring JT, McKenna R, et al. Elucidating the role of metal ions in carbonic anhydrase catalysis. Nat Commun. (2020) 11(1):4557. doi: 10.1038/s41467-020-18425-5
83. Cammer WB, Brion LP. Carbonic anhydrase in the nervous system. EXS. (2000) 90:475–89. doi: 10.1007/978-3-0348-8446-4_24
84. Stella C, Hachlouf A, Calabro L, Cavalli I, Schuind S, Gouvea Bogossian E, et al. The effects of acetazolamide on cerebral hemodynamics in adult patients with an acute brain injury: a systematic review. Brain Sci. (2023) 13(12). doi: 10.3390/brainsci13121678
85. Ozsoy HZ. Anticonvulsant effects of carbonic anhydrase inhibitors: the enigmatic link between carbonic anhydrases and electrical activity of the brain. Neurochem Res. (2021) 46(11):2783–99. doi: 10.1007/s11064-021-03390-2
86. Liljas A, Kannan KK, Bergsten PC, Waara I, Fridborg K, Strandberg B, et al. Crystal structure of human carbonic anhydrase C. Nat New Biol. (1972) 235(57):131–7. doi: 10.1038/newbio235131a0
87. McCall KA, Huang C, Fierke CA. Function and mechanism of zinc metalloenzymes. J Nutr. (2000) 130(5S Suppl):1437S–46. doi: 10.1093/jn/130.5.1437S
88. Krishnamurthy VM, Kaufman GK, Urbach AR, Gitlin I, Gudiksen KL, Weibel DB, et al. Carbonic anhydrase as a model for biophysical and physical-organic studies of proteins and protein-ligand binding. Chem Rev. (2008) 108(3):946–1051. doi: 10.1021/cr050262p
89. Huber AM, Gershoff SN. Effects of dietary zinc on zinc enzymes in the rat. J Nutr. (1973) 103(8):1175–81. doi: 10.1093/jn/103.8.1175
90. Chaudhry IM, Gandor DW, Gerson SJ. Reduction of carbonic anhydrase activity in the submandibular salivary glands of zinc-deficient rats. Arch Oral Biol. (1981) 26(5):399–402. doi: 10.1016/0003-9969(81)90036-4
91. Gandor DW, Fanslow DJ, Meyer J. Effects of zinc deficiency on developmental changes in alkaline phosphatase and carbonic anhydrase activities in the submandibular gland of the rat. Arch Oral Biol. (1983) 28(7):609–15. doi: 10.1016/0003-9969(83)90009-2
92. Goto T, Komai M, Bryant BP, Furukawa Y. Reduction in carbonic anhydrase activity in the tongue epithelium and submandibular gland in zinc-deficient rats. Int J Vitam Nutr Res. (2000) 70(3):110–8. doi: 10.1024/0300-9831.70.3.110
93. Lukaski HC. Low dietary zinc decreases erythrocyte carbonic anhydrase activities and impairs cardiorespiratory function in men during exercise. Am J Clin Nutr. (2005) 81(5):1045–51. doi: 10.1093/ajcn/81.5.1045
94. Ohno H, Yamashita K, Doi R, Yamamura K, Kondo T, Taniguchi N. Exercise-induced changes in blood zinc and related proteins in humans. J Appl Physiol (1985). (1985) 58(5):1453–8. doi: 10.1152/jappl.1985.58.5.1453
95. Henkin RI, Martin BM, Agarwal RP. Efficacy of exogenous oral zinc in treatment of patients with carbonic anhydrase VI deficiency. Am J Med Sci. (1999) 318(6):392–405. doi: 10.1016/S0002-9629(15)40664-0
96. Laity JH, Lee BM, Wright PE. Zinc finger proteins: new insights into structural and functional diversity. Curr Opin Struct Biol. (2001) 11(1):39–46. doi: 10.1016/S0959-440X(00)00167-6
97. Malgieri G, Palmieri M, Russo L, Fattorusso R, Pedone PV, Isernia C. The prokaryotic zinc-finger: structure, function and comparison with the eukaryotic counterpart. FEBS J. (2015) 282(23):4480–96. doi: 10.1111/febs.13503
98. Baglivo I, Russo L, Esposito S, Malgieri G, Renda M, Salluzzo A, et al. The structural role of the zinc ion can be dispensable in prokaryotic zinc-finger domains. Proc Natl Acad Sci U S A. (2009) 106(17):6933–8. doi: 10.1073/pnas.0810003106
99. Mackay JP, Crossley M. Zinc fingers are sticking together. Trends Biochem Sci. (1998) 23(1):1–4. doi: 10.1016/S0968-0004(97)01168-7
100. Maares M, Haase H. Zinc and immunity: an essential interrelation. Arch Biochem Biophys. (2016) 611:58–65. doi: 10.1016/j.abb.2016.03.022
101. Bonaventura P, Benedetti G, Albarede F, Miossec P. Zinc and its role in immunity and inflammation. Autoimmun Rev. (2015) 14(4):277–85. doi: 10.1016/j.autrev.2014.11.008
102. Rezaie P, Dean A. Periventricular leukomalacia, inflammation and white matter lesions within the developing nervous system. Neuropathology. (2002) 22(3):106–32. doi: 10.1046/j.1440-1789.2002.00438.x
103. Volpe JJ. Neurobiology of periventricular leukomalacia in the premature infant. Pediatr Res. (2001) 50(5):553–62. doi: 10.1203/00006450-200111000-00003
104. Zhao H, Alam A, San CY, Eguchi S, Chen Q, Lian Q, et al. Molecular mechanisms of brain-derived neurotrophic factor in neuro-protection: recent developments. Brain Res. (2017) 1665:1–21. doi: 10.1016/j.brainres.2017.03.029
105. Metz CH, Schroder AK, Overbeck S, Kahmann L, Plumakers B, Rink L. T-helper type 1 cytokine release is enhanced by in vitro zinc supplementation due to increased natural killer cells. Nutrition. (2007) 23(2):157–63. doi: 10.1016/j.nut.2006.10.007
106. Poleganov MA, Pfeilschifter J, Muhl H. Expanding extracellular zinc beyond levels reflecting the albumin-bound plasma zinc pool potentiates the capability of IL-1beta. IL: 18, and IL-12 to Act as IFN-gamma-inducing factors on PBMC. J Interferon Cytokine Res. (2007) 27(12):997–1001. doi: 10.1089/jir.2007.0037
107. Driessen C, Hirv K, Wellinghausen N, Kirchner H, Rink L. Influence of serum on zinc, toxic shock syndrome toxin-1, and lipopolysaccharide-induced production of IFN-gamma and IL-1 beta by human mononuclear cells. J Leukoc Biol. (1995) 57(6):904–8. doi: 10.1002/jlb.57.6.904
108. Gaetke LM, McClain CJ, Talwalkar RT, Shedlofsky SI. Effects of endotoxin on zinc metabolism in human volunteers. Am J Physiol. (1997) 272(6 Pt 1):E952–6. doi: 10.1152/ajpendo.1997.272.6.E952
109. Young B, Ott L, Kasarskis E, Rapp R, Moles K, Dempsey RJ, et al. Zinc supplementation is associated with improved neurologic recovery rate and visceral protein levels of patients with severe closed head injury. J Neurotrauma. (1996) 13(1):25–34. doi: 10.1089/neu.1996.13.25
110. Prasad AS, Beck FW, Grabowski SM, Kaplan J, Mathog RH. Zinc deficiency: changes in cytokine production and T-cell subpopulations in patients with head and neck cancer and in noncancer subjects. Proc Assoc Am Physicians. (1997) 109(1):68–77.9010918
111. Beck FW, Li Y, Bao B, Prasad AS, Sarkar FH. Evidence for reprogramming global gene expression during zinc deficiency in the HUT-78 cell line. Nutrition. (2006) 22(10):1045–56. doi: 10.1016/j.nut.2006.08.001
112. Szewczyk B, Poleszak E, Wlaz P, Wrobel A, Blicharska E, Cichy A, et al. The involvement of serotonergic system in the antidepressant effect of zinc in the forced swim test. Prog Neuropsychopharmacol Biol Psychiatry. (2009) 33(2):323–9. doi: 10.1016/j.pnpbp.2008.12.011
113. Prasad AS. Discovery of human zinc deficiency: its impact on human health and disease. Adv Nutr. (2013) 4(2):176–90. doi: 10.3945/an.112.003210
114. Jarosz M, Olbert M, Wyszogrodzka G, Mlyniec K, Librowski T. Antioxidant and anti-inflammatory effects of zinc. Zinc-dependent NF-kappaB signaling. Inflammopharmacology. (2017) 25(1):11–24. doi: 10.1007/s10787-017-0309-4
115. Fraker PJ, King LE. Reprogramming of the immune system during zinc deficiency. Annu Rev Nutr. (2004) 24:277–98. doi: 10.1146/annurev.nutr.24.012003.132454
116. King LE, Frentzel JW, Mann JJ, Fraker PJ. Chronic zinc deficiency in mice disrupted T cell lymphopoiesis and erythropoiesis while B cell lymphopoiesis and myelopoiesis were maintained. J Am Coll Nutr. (2005) 24(6):494–502. doi: 10.1080/07315724.2005.10719495
117. Prasad AS, Meftah S, Abdallah J, Kaplan J, Brewer GJ, Bach JF, et al. Serum thymulin in human zinc deficiency. J Clin Invest. (1988) 82(4):1202–10. doi: 10.1172/JCI113717
118. Kaltenberg J, Plum LM, Ober-Blobaum JL, Honscheid A, Rink L, Haase H. Zinc signals promote IL-2-dependent proliferation of T cells. Eur J Immunol. (2010) 40(5):1496–503. doi: 10.1002/eji.200939574
119. Prasad AS. Effects of zinc deficiency on Th1 and Th2 cytokine shifts. J Infect Dis. (2000) 182(Suppl 1):S62–8. doi: 10.1086/315916
120. Beck FW, Prasad AS, Kaplan J, Fitzgerald JT, Brewer GJ. Changes in cytokine production and T cell subpopulations in experimentally induced zinc-deficient humans. Am J Physiol. (1997) 272(6 Pt 1):E1002–7. doi: 10.1152/ajpendo.1997.272.6.E1002
121. Haase H, Rink L. Zinc signals and immune function. Biofactors. (2014) 40(1):27–40. doi: 10.1002/biof.1114
122. DePasquale-Jardieu P, Fraker PJ. Interference in the development of a secondary immune response in mice by zinc deprivation: persistence of effects. J Nutr. (1984) 114(10):1762–9. doi: 10.1093/jn/114.10.1762
123. Choi S, Liu X, Pan Z. Zinc deficiency and cellular oxidative stress: prognostic implications in cardiovascular diseases. Acta Pharmacol Sin. (2018) 39(7):1120–32. doi: 10.1038/aps.2018.25
124. Natale JE, Knight JB, Cheng Y, Rome JE, Gallo V. Metallothionein I and II mitigate age-dependent secondary brain injury. J Neurosci Res. (2004) 78(3):303–14. doi: 10.1002/jnr.20265
125. Krezel A, Maret W. Different redox states of metallothionein/thionein in biological tissue. Biochem J. (2007) 402(3):551–8. doi: 10.1042/BJ20061044
126. Chasapis CT, Ntoupa PA, Spiliopoulou CA, Stefanidou ME. Recent aspects of the effects of zinc on human health. Arch Toxicol. (2020) 94(5):1443–60. doi: 10.1007/s00204-020-02702-9
127. Olechnowicz J, Tinkov A, Skalny A, Suliburska J. Zinc status is associated with inflammation, oxidative stress, lipid, and glucose metabolism. J Physiol Sci. (2018) 68(1):19–31. doi: 10.1007/s12576-017-0571-7
128. Sensi SL, Paoletti P, Bush AI, Sekler I. Zinc in the physiology and pathology of the CNS. Nat Rev Neurosci. (2009) 10(11):780–91. doi: 10.1038/nrn2734
129. Kang M, Zhao L, Ren M, Deng M, Li C. Reduced metallothionein expression induced by zinc deficiency results in apoptosis in hepatic stellate cell line LX-2. Int J Clin Exp Med. (2015) 8(11):20603–9.26884979
130. Suzuki K, Nakajima K, Otaki N, Kimura M. Metallothionein in developing human brain. Biol Signals. (1994) 3(4):188–92. doi: 10.1159/000109544
131. Cozzi B, Giacomello M, Zambenedetti P, Bolognin S, Rossipal E, Peruffo A, et al. Ontogenesis and migration of metallothionein I/II-containing glial cells in the human telencephalon during the second trimester. Brain Res. (2010) 1327:16–23. doi: 10.1016/j.brainres.2010.02.073
132. Chabosseau P, Rutter GA. Zinc and diabetes. Arch Biochem Biophys. (2016) 611:79–85. doi: 10.1016/j.abb.2016.05.022
133. Hardy AB, Prentice KJ, Froese S, Liu Y, Andrews GK, Wheeler MB. Zip4 mediated zinc influx stimulates insulin secretion in pancreatic beta cells. PLoS One. (2015) 10(3):e0119136. doi: 10.1371/journal.pone.0119136
134. Wijesekara N, Dai FF, Hardy AB, Giglou PR, Bhattacharjee A, Koshkin V, et al. Beta cell-specific Znt8 deletion in mice causes marked defects in insulin processing, crystallisation and secretion. Diabetologia. (2010) 53(8):1656–68. doi: 10.1007/s00125-010-1733-9
135. Shan Z, Bao W, Zhang Y, Rong Y, Wang X, Jin Y, et al. Interactions between zinc transporter-8 gene (SLC30A8) and plasma zinc concentrations for impaired glucose regulation and type 2 diabetes. Diabetes. (2014) 63(5):1796–803. doi: 10.2337/db13-0606
136. Wu Y, Lu H, Yang H, Li C, Sang Q, Liu X, et al. Zinc stimulates glucose consumption by modulating the insulin signaling pathway in L6 myotubes: essential roles of Akt-GLUT4, GSK3β and mTOR-S6K1. J Nutr Biochem. (2016) 34:126–35. doi: 10.1016/j.jnutbio.2016.05.008
137. Fukunaka A, Fujitani Y. Role of zinc homeostasis in the pathogenesis of diabetes and obesity. Int J Mol Sci. (2018) 19(2):126–35. doi: 10.3390/ijms19020476
138. Ruz M, Carrasco F, Rojas P, Basfi-Fer K, Hernandez MC, Perez A. Nutritional effects of zinc on metabolic syndrome and type 2 diabetes: mechanisms and main findings in human studies. Biol Trace Elem Res. (2019) 188(1):177–88. doi: 10.1007/s12011-018-1611-8
139. Ramracheya R, Ward C, Shigeto M, Walker JN, Amisten S, Zhang Q, et al. Membrane potential-dependent inactivation of voltage-gated ion channels in alpha-cells inhibits glucagon secretion from human islets. Diabetes. (2010) 59(9):2198–208. doi: 10.2337/db09-1505
140. Ilouz R, Kaidanovich O, Gurwitz D, Eldar-Finkelman H. Inhibition of glycogen synthase kinase-3beta by bivalent zinc ions: insight into the insulin-mimetic action of zinc. Biochem Biophys Res Commun. (2002) 295(1):102–6. doi: 10.1016/S0006-291X(02)00636-8
141. Cameron AR, Anil S, Sutherland E, Harthill J, Rena G. Zinc-dependent effects of small molecules on the insulin-sensitive transcription factor FOXO1a and gluconeogenic genes. Metallomics. (2010) 2(3):195–203. doi: 10.1039/B914984H
142. Brion LP, Rosenfeld CR, Heyne R, Brown SL, Lair CS, Burchfield PJ, et al. Adjustable feedings plus accurate serial length measurements decrease discharge weight-length disproportion in very preterm infants: quality improvement project. J Perinatol. (2019) 39(8):1131–9. doi: 10.1038/s41372-019-0424-8
143. Brion LP, Rosenfeld CR, Heyne R, Brown SL, Lair CS, Burchfield PJ, et al. Correction to: adjustable feedings plus accurate serial length measurements decrease discharge weight-length disproportion in very preterm infants: quality improvement project. J Perinatol. (2019) 39(12):1694. doi: 10.1038/s41372-019-0521-8
144. Au SC, Tang SM, Rong SS, Chen LJ, Yam JC. Association between hyperglycemia and retinopathy of prematurity: a systemic review and meta-analysis. Sci Rep. (2015) 5:9091. doi: 10.1038/srep09091
145. Lee JH, Hornik CP, Testoni D, Laughon MM, Cotten CM, Maldonado RS, et al. Insulin, Hyperglycemia, And severe retinopathy of prematurity in extremely low-birth-weight infants. Am J Perinatol. (2016) 33(4):393–400. doi: 10.1055/s-0035-1565999
146. Conejo R, Lorenzo M. Insulin signaling leading to proliferation, survival, and membrane ruffling in C2C12 myoblasts. J Cell Physiol. (2001) 187(1):96–108. doi: 10.1002/1097-4652(2001)9999:9999%3C::AID-JCP1058%3E3.0.CO;2-V
147. Tacchini L, Dansi P, Matteucci E, Desiderio MA. Hepatocyte growth factor signalling stimulates hypoxia inducible factor-1 (HIF-1) activity in HepG2 hepatoma cells. Carcinogenesis. (2001) 22(9):1363–71. doi: 10.1093/carcin/22.9.1363
148. Angelis D, Jaleel MA, Brion LP. Hyperglycemia and prematurity: a narrative review. Pediatr Res. (2023) 94(3):892–903. doi: 10.1038/s41390-023-02628-9
149. Fridovich I. Superoxide radical and superoxide dismutases. Annu Rev Biochem. (1995) 64:97–112. doi: 10.1146/annurev.bi.64.070195.000525
150. Arnesano F, Banci L, Bertini I, Martinelli M, Furukawa Y, O’Halloran TV. The unusually stable quaternary structure of human Cu,Zn-superoxide dismutase 1 is controlled by both metal occupancy and disulfide status. J Biol Chem. (2004) 279(46):47998–8003. doi: 10.1074/jbc.M406021200
151. Wright GSA, Antonyuk SV, Hasnain SS. The biophysics of superoxide dismutase-1 and amyotrophic lateral sclerosis. Q Rev Biophys. (2019) 52:e12. doi: 10.1017/S003358351900012X
152. Eleutherio ECA, Silva Magalhaes RS, de Araujo Brasil A, Monteiro Neto JR, de Holanda Paranhos L. SOD1, more than just an antioxidant. Arch Biochem Biophys. (2021) 697:108701. doi: 10.1016/j.abb.2020.108701
153. Theil T. Gli3 is required for the specification and differentiation of preplate neurons. Dev Biol. (2005) 286(2):559–71. doi: 10.1016/j.ydbio.2005.08.033
154. Papa L, Manfredi G, Germain D. SOD1, an unexpected novel target for cancer therapy. Genes Cancer. (2014) 5(1–2):15–21. doi: 10.18632/genesandcancer.4
155. Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, et al. Mutations in cu/zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. (1993) 362(6415):59–62. doi: 10.1038/364362c0
156. Tsang CK, Liu Y, Thomas J, Zhang Y, Zheng XF. Superoxide dismutase 1 acts as a nuclear transcription factor to regulate oxidative stress resistance. Nat Commun. (2014) 5:3446. doi: 10.1038/ncomms4446
157. Butti Z, Patten SA. RNA dysregulation in amyotrophic lateral sclerosis. Front Genet. (2018) 9:712. doi: 10.3389/fgene.2018.00712
158. Strong MJ, Volkening K, Hammond R, Yang W, Strong W, Leystra-Lantz C, et al. TDP43 is a human low molecular weight neurofilament (hNFL) mRNA-binding protein. Mol Cell Neurosci. (2007) 35(2):320–7. doi: 10.1016/j.mcn.2007.03.007
159. Lu L, Zheng L, Viera L, Suswam E, Li Y, Li X, et al. Mutant cu/zn-superoxide dismutase associated with amyotrophic lateral sclerosis destabilizes vascular endothelial growth factor mRNA and downregulates its expression. J Neurosci. (2007) 27(30):7929–38. doi: 10.1523/JNEUROSCI.1877-07.2007
160. Lu L, Wang S, Zheng L, Li X, Suswam EA, Zhang X, et al. Amyotrophic lateral sclerosis-linked mutant SOD1 sequesters hu antigen R (HuR) and TIA-1-related protein (TIAR): implications for impaired post-transcriptional regulation of vascular endothelial growth factor. J Biol Chem. (2009) 284(49):33989–98. doi: 10.1074/jbc.M109.067918
161. Frederickson CJ, Koh JY, Bush AI. The neurobiology of zinc in health and disease. Nat Rev Neurosci. (2005) 6(6):449–62. doi: 10.1038/nrn1671
162. Barr CA, Burdette SC. The zinc paradigm for metalloneurochemistry. Essays Biochem. (2017) 61(2):225–35. doi: 10.1042/EBC20160073
163. Mathie A, Sutton GL, Clarke CE, Veale EL. Zinc and copper: pharmacological probes and endogenous modulators of neuronal excitability. Pharmacol Ther. (2006) 111(3):567–83. doi: 10.1016/j.pharmthera.2005.11.004
164. Pochwat B, Nowak G, Szewczyk B. Relationship between zinc (zn (2+)) and glutamate receptors in the processes underlying neurodegeneration. Neural Plast. (2015) 2015:591563. doi: 10.1155/2015/591563
165. Carver CM, Chuang SH, Reddy DS. Zinc selectively blocks neurosteroid-sensitive extrasynaptic deltaGABAA receptors in the hippocampus. J Neurosci. (2016) 36(31):8070–7. doi: 10.1523/JNEUROSCI.3393-15.2016
166. Hwang JJ, Park MH, Choi SY, Koh JY. Activation of the trk signaling pathway by extracellular zinc. Role of metalloproteinases. J Biol Chem. (2005) 280(12):11995–2001. doi: 10.1074/jbc.M403172200
167. Trombley PQ, Blakemore LJ, Hill BJ. Zinc modulation of glycine receptors. Neuroscience. (2011) 186:32–8. doi: 10.1016/j.neuroscience.2011.04.021
168. Chorin E, Vinograd O, Fleidervish I, Gilad D, Herrmann S, Sekler I, et al. Upregulation of KCC2 activity by zinc-mediated neurotransmission via the mZnR/GPR39 receptor. J Neurosci. (2011) 31(36):12916–26. doi: 10.1523/JNEUROSCI.2205-11.2011
169. Saadi RA, He K, Hartnett KA, Kandler K, Hershfinkel M, Aizenman E. SNARE-dependent upregulation of potassium chloride co-transporter 2 activity after metabotropic zinc receptor activation in rat cortical neurons in vitro. Neuroscience. (2012) 210:38–46. doi: 10.1016/j.neuroscience.2012.03.001
170. Besser L, Chorin E, Sekler I, Silverman WF, Atkin S, Russell JT, et al. Synaptically released zinc triggers metabotropic signaling via a zinc-sensing receptor in the hippocampus. J Neurosci. (2009) 29(9):2890–901. doi: 10.1523/JNEUROSCI.5093-08.2009
171. Chowanadisai W, Kelleher SL, Lonnerdal B. Maternal zinc deficiency reduces NMDA receptor expression in neonatal rat brain, which persists into early adulthood. J Neurochem. (2005) 94(2):510–9. doi: 10.1111/j.1471-4159.2005.03246.x
172. Bergstrom T, Forsberg-Nilsson K. Neural stem cells: brain building blocks and beyond. Ups J Med Sci. (2012) 117(2):132–42. doi: 10.3109/03009734.2012.665096
173. Gotz M, Huttner WB. The cell biology of neurogenesis. Nat Rev Mol Cell Biol. (2005) 6(10):777–88. doi: 10.1038/nrm1739
174. Cassandri M, Smirnov A, Novelli F, Pitolli C, Agostini M, Malewicz M, et al. Zinc-finger proteins in health and disease. Cell Death Discov. (2017) 3:17071. doi: 10.1038/cddiscovery.2017.71
175. Fidalgo M, Shekar PC, Ang YS, Fujiwara Y, Orkin SH, Wang J. Zfp281 functions as a transcriptional repressor for pluripotency of mouse embryonic stem cells. Stem Cells. (2011) 29(11):1705–16. doi: 10.1002/stem.736
176. Chung SH, Marzban H, Aldinger K, Dixit R, Millen K, Schuurmans C, et al. Zac1 plays a key role in the development of specific neuronal subsets in the mouse cerebellum. Neural Dev. (2011) 6:25. doi: 10.1186/1749-8104-6-25
177. Hasenpusch-Theil K, West S, Kelman A, Kozic Z, Horrocks S, McMahon AP, et al. Gli3 controls the onset of cortical neurogenesis by regulating the radial glial cell cycle through Cdk6 expression. Development. (2018) 145(17):1–12. doi: 10.1242/dev.163147
178. Biesecker LG. What you can learn from one gene: gLI3. J Med Genet. (2006) 43(6):465–9. doi: 10.1136/jmg.2004.029181
179. Yan L, Li Y, Shi Z, Lu X, Ma J, Hu B, et al. The zinc finger E-box-binding homeobox 1 (Zeb1) promotes the conversion of mouse fibroblasts into functional neurons. J Biol Chem. (2017) 292(31):12959–70. doi: 10.1074/jbc.M116.771493
180. He L, Yu K, Lu F, Wang J, Wu LN, Zhao C, et al. Transcriptional regulator ZEB2 is essential for Bergmann Glia development. J Neurosci. (2018) 38(6):1575–87. doi: 10.1523/JNEUROSCI.2674-17.2018
181. Inoue T, Ogawa M, Mikoshiba K, Aruga J. Zinc deficiency in the cortical marginal zone and meninges results in cortical lamination defects resembling those in type II lissencephaly. J Neurosci. (2008) 28(18):4712–25. doi: 10.1523/JNEUROSCI.5735-07.2008
182. Aruga J. The role of zinc genes in neural development. Mol Cell Neurosci. (2004) 26(2):205–21. doi: 10.1016/j.mcn.2004.01.004
183. Merzdorf CS. Emerging roles for zic genes in early development. Dev Dyn. (2007) 236(4):922–40. doi: 10.1002/dvdy.21098
184. Grinberg I, Northrup H, Ardinger H, Prasad C, Dobyns WB, Millen KJ. Heterozygous deletion of the linked genes ZIC1 and ZIC4 is involved in dandy-walker malformation. Nat Genet. (2004) 36(10):1053–5. doi: 10.1038/ng1420
185. Taupin P. Neurogenesis in the adult central nervous system. C R Biol. (2006) 329(7):465–75. doi: 10.1016/j.crvi.2006.04.001
186. Xu C, Zhang Y, Zheng H, Loh HH, Law PY. Morphine modulates mouse hippocampal progenitor cell lineages by upregulating miR-181a level. Stem Cells. (2014) 32(11):2961–72. doi: 10.1002/stem.1774
187. Taverna E, Gotz M, Huttner WB. The cell biology of neurogenesis: toward an understanding of the development and evolution of the neocortex. Annu Rev Cell Dev Biol. (2014) 30:465–502. doi: 10.1146/annurev-cellbio-101011-155801
188. Augusto-Oliveira M, Arrifano GPF, Malva JO, Crespo-Lopez ME. Adult hippocampal neurogenesis in different taxonomic groups: possible functional similarities and striking controversies. Cells. (2019) 8(2):1–25. doi: 10.3390/cells8020125
189. Adamo AM, Liu X, Mathieu P, Nuttall JR, Supasai S, Oteiza PI. Early developmental marginal zinc deficiency affects neurogenesis decreasing neuronal number and altering neuronal specification in the adult rat brain. Front Cell Neurosci. (2019) 13:62. doi: 10.3389/fncel.2019.00062
190. Corniola RS, Tassabehji NM, Hare J, Sharma G, Levenson CW. Zinc deficiency impairs neuronal precursor cell proliferation and induces apoptosis via p53-mediated mechanisms. Brain Res. (2008) 1237:52–61. doi: 10.1016/j.brainres.2008.08.040
191. Conrad CD, McLaughlin KJ, Harman JS, Foltz C, Wieczorek L, Lightner E, et al. Chronic glucocorticoids increase hippocampal vulnerability to neurotoxicity under conditions that produce CA3 dendritic retraction but fail to impair spatial recognition memory. J Neurosci. (2007) 27(31):8278–85. doi: 10.1523/JNEUROSCI.2121-07.2007
192. Tamano H, Kan F, Oku N, Takeda A. Ameliorative effect of Yokukansan on social isolation-induced aggressive behavior of zinc-deficient young mice. Brain Res Bull. (2010) 83(6):351–5. doi: 10.1016/j.brainresbull.2010.08.013
193. Elliott EM, Sapolsky RM. Corticosterone impairs hippocampal neuronal calcium regulation–possible mediating mechanisms. Brain Res. (1993) 602(1):84–90. doi: 10.1016/0006-8993(93)90245-I
194. Tauber SC, Schlumbohm C, Schilg L, Fuchs E, Nau R, Gerber J. Intrauterine exposure to dexamethasone impairs proliferation but not neuronal differentiation in the dentate gyrus of newborn common marmoset monkeys. Brain Pathol. (2006) 16(3):209–17. doi: 10.1111/j.1750-3639.2006.00021.x
195. Anacker C, Cattaneo A, Luoni A, Musaelyan K, Zunszain PA, Milanesi E, et al. Glucocorticoid-related molecular signaling pathways regulating hippocampal neurogenesis. Neuropsychopharmacology. (2013) 38(5):872–83. doi: 10.1038/npp.2012.253
196. Adamo AM, Zago MP, Mackenzie GG, Aimo L, Keen CL, Keenan A, et al. The role of zinc in the modulation of neuronal proliferation and apoptosis. Neurotox Res. (2010) 17(1):1–14. doi: 10.1007/s12640-009-9067-4
197. Moon MY, Kim HJ, Choi BY, Sohn M, Chung TN, Suh SW. Zinc promotes adipose-derived mesenchymal stem cell proliferation and differentiation towards a neuronal fate. Stem Cells Int. (2018) 2018:5736535. doi: 10.1155/2018/5736535
198. Fukumoto K, Morita T, Mayanagi T, Tanokashira D, Yoshida T, Sakai A, et al. Detrimental effects of glucocorticoids on neuronal migration during brain development. Mol Psychiatry. (2009) 14(12):1119–31. doi: 10.1038/mp.2009.60
199. Pfaender S, Fohr K, Lutz AK, Putz S, Achberger K, Linta L, et al. Cellular zinc homeostasis contributes to neuronal differentiation in human induced pluripotent stem cells. Neural Plast. (2016) 2016:3760702. doi: 10.1155/2016/3760702
200. Dvergsten CL, Fosmire GJ, Ollerich DA, Sandstead HH. Alterations in the postnatal development of the cerebellar cortex due to zinc deficiency. II. Impaired maturation of Purkinje cells. Brain Res. (1984) 318(1):11–20. doi: 10.1016/0165-3806(84)90057-9
201. Dvergsten CL, Johnson LA, Sandstead HH. Alterations in the postnatal development of the cerebellar cortex due to zinc deficiency. III. Impaired dendritic differentiation of basket and stellate cells. Brain Res. (1984) 318(1):21–6. doi: 10.1016/0165-3806(84)90058-0
202. Gower-Winter SD, Corniola RS, Morgan TJ Jr, Levenson CW. Zinc deficiency regulates hippocampal gene expression and impairs neuronal differentiation. Nutr Neurosci. (2013) 16(4):174–82. doi: 10.1179/1476830512Y.0000000043
203. Tesseur I, Zou K, Esposito L, Bard F, Berber E, Can JV, et al. Deficiency in neuronal TGF-beta signaling promotes neurodegeneration and Alzheimer’s pathology. J Clin Invest. (2006) 116(11):3060–9. doi: 10.1172/JCI27341
204. Lippiello PM. Nicotinic cholinergic antagonists: a novel approach for the treatment of autism. Med Hypotheses. (2006) 66(5):985–90. doi: 10.1016/j.mehy.2005.11.015
205. Vela G, Stark P, Socha M, Sauer AK, Hagmeyer S, Grabrucker AM. Zinc in gut-brain interaction in autism and neurological disorders. Neural Plast. (2015) 2015:972791. doi: 10.1155/2015/972791
206. Altman J. Autoradiographic and histological studies of postnatal neurogenesis. 3. Dating the time of production and onset of differentiation of cerebellar microneurons in rats. J Comp Neurol. (1969) 136(3):269–93. doi: 10.1002/cne.901360303
207. Altman J. Postnatal development of the cerebellar cortex in the rat. I. The external germinal layer and the transitional molecular layer. J Comp Neurol. (1972) 145(3):353–97. doi: 10.1002/cne.901450305
208. Supasai S, Adamo AM, Mathieu P, Marino RC, Hellmers AC, Cremonini E, et al. Gestational zinc deficiency impairs brain astrogliogenesis in rats through multistep alterations of the JAK/STAT3 signaling pathway. Redox Biol. (2021) 44:102017. doi: 10.1016/j.redox.2021.102017
209. Willekens J, Runnels LW. Impact of zinc transport mechanisms on embryonic and brain development. Nutrients. (2022) 14(12):1–48. doi: 10.3390/nu14122526
210. Nolte C, Gore A, Sekler I, Kresse W, Hershfinkel M, Hoffmann A, et al. ZnT-1 expression in astroglial cells protects against zinc toxicity and slows the accumulation of intracellular zinc. Glia. (2004) 48(2):145–55. doi: 10.1002/glia.20065
211. Maret W. Zinc in cellular regulation: the nature and significance of “zinc signals”. Int J Mol Sci. (2017) 18(11):2285. doi: 10.3390/ijms18112285
212. Fukada T, Yamasaki S, Nishida K, Murakami M, Hirano T. Zinc homeostasis and signaling in health and diseases: zinc signaling. J Biol Inorg Chem. (2011) 16(7):1123–34. doi: 10.1007/s00775-011-0797-4
213. Li Y, Maret W. Transient fluctuations of intracellular zinc ions in cell proliferation. Exp Cell Res. (2009) 315(14):2463–70. doi: 10.1016/j.yexcr.2009.05.016
214. Kuspert M, Wegner M. SomethiNG 2 talk about-transcriptional regulation in embryonic and adult oligodendrocyte precursors. Brain Res. (2016) 1638(Pt B):167–82. doi: 10.1016/j.brainres.2015.07.024
215. Wang SZ, Dulin J, Wu H, Hurlock E, Lee SE, Jansson K, et al. An oligodendrocyte-specific zinc-finger transcription regulator cooperates with Olig2 to promote oligodendrocyte differentiation. Development. (2006) 133(17):3389–98. doi: 10.1242/dev.02522
216. Volpe JJ. Iron and zinc: nutrients with potential for neurorestoration in premature infants with cerebral white matter injury. J Neonatal Perinatal Med. (2019) 12(4):365–8. doi: 10.3233/NPM-190369
217. Bourassa D, Elitt CM, McCallum AM, Sumalekshmy S, McRae RL, Morgan MT, et al. Chromis-1, a ratiometric fluorescent probe optimized for two-photon microscopy reveals dynamic changes in labile zn(II) in differentiating oligodendrocytes. ACS Sens. (2018) 3(2):458–67. doi: 10.1021/acssensors.7b00887
218. Liu H, Oteiza PI, Gershwin ME, Golub MS, Keen CL. Effects of maternal marginal zinc deficiency on myelin protein profiles in the suckling rat and infant rhesus monkey. Biol Trace Elem Res. (1992) 34(1):55–66. doi: 10.1007/BF02783898
219. Prohaska JR, Luecke RW, Jasinski R. Effect of zinc deficiency from day 18 of gestation and-or during lactation on the development of some rat brain enzymes. J Nutr. (1974) 104(11):1525–31. doi: 10.1093/jn/104.11.1525
220. Kharfi M, El Fekih N, Aounallah-Skhiri H, Schmitt S, Fazaa B, Kury S, et al. Acrodermatitis enteropathica: a review of 29 Tunisian cases. Int J Dermatol. (2010) 49(9):1038–44. doi: 10.1111/j.1365-4632.2010.04566.x
221. Ohlsson A. Acrodermatitis enteropathica reversibility of cerebral atrophy with zinc therapy. Acta Paediatr Scand. (1981) 70(2):269–73. doi: 10.1111/j.1651-2227.1981.tb05556.x
222. Karcioglu ZA. Zinc in the eye. Surv Ophthalmol. (1982) 27(2):114–22. doi: 10.1016/0039-6257(82)90195-3
223. Christian P, Khatry SK, Yamini S, Stallings R, LeClerq SC, Shrestha SR, et al. Zinc supplementation might potentiate the effect of vitamin A in restoring night vision in pregnant Nepalese women. Am J Clin Nutr. (2001) 73(6):1045–51. doi: 10.1093/ajcn/73.6.1045
224. Kikuchi M, Kashii S, Honda Y, Ujihara H, Sasa M, Tamura Y, et al. Protective action of zinc against glutamate neurotoxicity in cultured retinal neurons. Invest Ophthalmol Vis Sci. (1995) 36(10):2048–53.7657543
225. Pierce EA, Foley ED, Smith LE. Regulation of vascular endothelial growth factor by oxygen in a model of retinopathy of prematurity. Arch Ophthalmol. (1996) 114(10):1219–28. doi: 10.1001/archopht.1996.01100140419009
226. Chen J, Connor KM, Aderman CM, Willett KL, Aspegren OP, Smith LE. Suppression of retinal neovascularization by erythropoietin siRNA in a mouse model of proliferative retinopathy. Invest Ophthalmol Vis Sci. (2009) 50(3):1329–35. doi: 10.1167/iovs.08-2521
227. Hellstrom A, Perruzzi C, Ju M, Engstrom E, Hard AL, Liu JL, et al. Low IGF-I suppresses VEGF-survival signaling in retinal endothelial cells: direct correlation with clinical retinopathy of prematurity. Proc Natl Acad Sci U S A. (2001) 98(10):5804–8. doi: 10.1073/pnas.101113998
228. Upreti S, Nag TC, Ghosh MP. Trolox aids coenzyme Q(10) in neuroprotection against NMDA induced damage via upregulation of VEGF in rat model of glutamate excitotoxicity. Exp Eye Res. (2024) 238:109740. doi: 10.1016/j.exer.2023.109740
229. Spierer A, Rabinowitz R, Pri-Chen S, Rosner M. An increase in superoxide dismutase ameliorates oxygen-induced retinopathy in transgenic mice. Eye (Lond). (2005) 19(1):86–91. doi: 10.1038/sj.eye.6701424
230. Sun J, Yoon J, Lee M, Lee HK, Hwang YS, Daar IO. Zic5 stabilizes Gli3 via a non-transcriptional mechanism during retinal development. Cell Rep. (2022) 38(5):110312. doi: 10.1016/j.celrep.2022.110312
231. Reinhardt R, Centanin L, Tavhelidse T, Inoue D, Wittbrodt B, Concordet JP, et al. Sox2, tlx, Gli3, and Her9 converge on Rx2 to define retinal stem cells in vivo. EMBO J. (2015) 34(11):1572–88. doi: 10.15252/embj.201490706
232. Matissek SJ, Elsawa SF. GLI3: a mediator of genetic diseases, development and cancer. Cell Commun Signal. (2020) 18(1):54. doi: 10.1186/s12964-020-00540-x
233. Hager-Theodorides AL, Dessens JT, Outram SV, Crompton T. The transcription factor Gli3 regulates differentiation of fetal CD4- CD8- double-negative thymocytes. Blood. (2005) 106(4):1296–304. doi: 10.1182/blood-2005-03-0998
234. Holm M, Morken TS, Fichorova RN, VanderVeen DK, Allred EN, Dammann O, et al. Systemic inflammation-associated proteins and retinopathy of prematurity in infants born before the 28th week of gestation. Invest Ophthalmol Vis Sci. (2017) 58(14):6419–28. doi: 10.1167/iovs.17-21931
235. Al-Kharashi AS. Role of oxidative stress, inflammation, hypoxia and angiogenesis in the development of diabetic retinopathy. Saudi J Ophthalmol. (2018) 32(4):318–23. doi: 10.1016/j.sjopt.2018.05.002
236. Juul SE, Comstock BA, Wadhawan R, Mayock DE, Courtney SE, Robinson T, et al. A randomized trial of erythropoietin for neuroprotection in preterm infants. N Engl J Med. (2020) 382(3):233–43. doi: 10.1056/NEJMoa1907423
237. Agrawal G, Dutta S, Prasad R, Dogra MR. Fetal oxidative stress, micronutrient deficiency and risk of retinopathy of prematurity: a nested case-control study. Eur J Pediatr. (2021) 180(5):1487–96. doi: 10.1007/s00431-020-03896-x
238. Staub E, Evers K, Askie LM. Enteral zinc supplementation for prevention of morbidity and mortality in preterm neonates. Cochrane Database Syst Rev. (2021) 3(3):CD012797. doi: 10.1002/14651858.CD012797.pub2
239. Alshaikh B, Abo Zeed M, Yusuf K, Guin M, Fenton T. Effect of enteral zinc supplementation on growth and neurodevelopment of preterm infants: a systematic review and meta-analysis. J Perinatol. (2022) 42(4):430–9. doi: 10.1038/s41372-021-01094-7
240. Craig TA, Benson LM, Naylor S, Kumar R. Modulation effects of zinc on the formation of vitamin D receptor and retinoid X receptor alpha-DNA transcription complexes: analysis by microelectrospray mass spectrometry. Rapid Commun Mass Spectrom. (2001) 15(12):1011–6. doi: 10.1002/rcm.332
241. Leon O, Roth M. Zinc fingers: DNA binding and protein-protein interactions. Biol Res. (2000) 33(1):21–30. doi: 10.4067/S0716-97602000000100009
242. Vazquez-Lorente H, Molina-Lopez J, Herrera-Quintana L, Gamarra-Morales Y, Lopez-Gonzalez B, Planells E. Effectiveness of eight-week zinc supplementation on vitamin D(3) status and leptin levels in a population of postmenopausal women: a double-blind randomized trial. J Trace Elem Med Biol. (2021) 65:126730. doi: 10.1016/j.jtemb.2021.126730
243. Shams B, Afshari E, Tajadini M, Keikha M, Qorbani M, Heshmat R, et al. The relationship of serum vitamin D and zinc in a nationally representative sample of Iranian children and adolescents: the CASPIAN-III study. Med J Islam Repub Iran. (2016) 30:430.28210595
244. Gonoodi K, Tayefi M, Saberi-Karimian M, Amirabadi Zadeh A, Darroudi S, Farahmand SK, et al. An assessment of the risk factors for vitamin D deficiency using a decision tree model. Diabetes Metab Syndr. (2019) 13(3):1773–7. doi: 10.1016/j.dsx.2019.03.020
245. da Silva TC, Hiller C, Gai Z, Kullak-Ublick GA. Vitamin D3 transactivates the zinc and manganese transporter SLC30A10 via the vitamin D receptor. J Steroid Biochem Mol Biol. (2016) 163:77–87. doi: 10.1016/j.jsbmb.2016.04.006
246. Montero-Odasso M, Zou G, Speechley M, Almeida QJ, Liu-Ambrose T, Middleton LE, et al. Effects of exercise alone or combined with cognitive training and vitamin D supplementation to improve cognition in adults with mild cognitive impairment: a randomized clinical trial. JAMA Netw Open. (2023) 6(7):e2324465. doi: 10.1001/jamanetworkopen.2023.24465
247. Cui P, Lu W, Wang J, Wang F, Zhang X, Hou X, et al. Microglia/macrophages require vitamin D signaling to restrain neuroinflammation and brain injury in a murine ischemic stroke model. J Neuroinflammation. (2023) 20(1):63. doi: 10.1186/s12974-023-02705-0
248. Zhang H, Wang S, Tuo L, Zhai Q, Cui J, Chen D, et al. Relationship between maternal vitamin D levels and adverse outcomes. Nutrients. (2022) 14(20):4230. doi: 10.3390/nu14204230
249. da Silva Lima F, da Silva Goncalves CE, Fock RA. A review of the role of zinc finger proteins on hematopoiesis. J Trace Elem Med Biol. (2023) 80:127290. doi: 10.1016/j.jtemb.2023.127290
250. Takahashi A. Role of zinc and copper in erythropoiesis in patients on hemodialysis. J Ren Nutr. (2022) 32(6):650–7. doi: 10.1053/j.jrn.2022.02.007
251. Chlon TM, Crispino JD. Combinatorial regulation of tissue specification by GATA and FOG factors. Development. (2012) 139(21):3905–16. doi: 10.1242/dev.080440
252. Bresnick EH, Martowicz ML, Pal S, Johnson KD. Developmental control via GATA factor interplay at chromatin domains. J Cell Physiol. (2005) 205(1):1–9. doi: 10.1002/jcp.20393
253. Fox AH, Liew C, Holmes M, Kowalski K, Mackay J, Crossley M. Transcriptional cofactors of the FOG family interact with GATA proteins by means of multiple zinc fingers. EMBO J. (1999) 18(10):2812–22. doi: 10.1093/emboj/18.10.2812
254. Qi SN, Zhang ZF, Wang ZY, Yoshida A, Ueda T. L-carnitine inhibits apoptotic DNA fragmentation induced by a new spin-labeled derivative of podophyllotoxin via caspase-3 in Raji cells. Oncol Rep. (2006) 15(1):119–22. doi: 10.3892/or.15.1.119
255. Asadi M, Taghizadeh S, Kaviani E, Vakili O, Taheri-Anganeh M, Tahamtan M, et al. Caspase-3: structure, function, and biotechnological aspects. Biotechnol Appl Biochem. (2022) 69(4):1633–45. doi: 10.1002/bab.2233
256. Hyun HJ, Sohn JH, Ha DW, Ahn YH, Koh JY, Yoon YH. Depletion of intracellular zinc and copper with TPEN results in apoptosis of cultured human retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. (2001) 42(2):460–5.11157883
257. Perry DK, Smyth MJ, Stennicke HR, Salvesen GS, Duriez P, Poirier GG, et al. Zinc is a potent inhibitor of the apoptotic protease, caspase-3. A novel target for zinc in the inhibition of apoptosis. J Biol Chem. (1997) 272(30):18530–3. doi: 10.1074/jbc.272.30.18530
258. Song D, Li LS, Heaton-Johnson KJ, Arsenault PR, Master SR, Lee FS. Prolyl hydroxylase domain protein 2 (PHD2) binds a pro-Xaa-leu-glu motif, linking it to the heat shock protein 90 pathway. J Biol Chem. (2013) 288(14):9662–74. doi: 10.1074/jbc.M112.440552
259. Sinnema M, Song D, Guan W, Janssen JWH, van Wijk R, Navalsky BE, et al. Loss-of-function zinc finger mutation in the EGLN1 gene associated with erythrocytosis. Blood. (2018) 132(13):1455–8. doi: 10.1182/blood-2018-06-854711
260. Nasyrov E, Nolan KA, Wenger RH, Marti HH, Kunze R. The neuronal oxygen-sensing pathway controls postnatal vascularization of the murine brain. FASEB J. (2019) 33(11):12812–24. doi: 10.1096/fj.201901385RR
261. Bohuslavova R, Cerychova R, Papousek F, Olejnickova V, Bartos M, Gorlach A, et al. HIF-1alpha is required for development of the sympathetic nervous system. Proc Natl Acad Sci U S A. (2019) 116(27):13414–23. doi: 10.1073/pnas.1903510116
262. Vangeison G, Carr D, Federoff HJ, Rempe DA. The good, the bad, and the cell type-specific roles of hypoxia inducible factor-1 alpha in neurons and astrocytes. J Neurosci. (2008) 28(8):1988–93. doi: 10.1523/JNEUROSCI.5323-07.2008
263. Yuen TJ, Silbereis JC, Griveau A, Chang SM, Daneman R, Fancy SPJ, et al. Oligodendrocyte-encoded HIF function couples postnatal myelination and white matter angiogenesis. Cell. (2014) 158(2):383–96. doi: 10.1016/j.cell.2014.04.052
264. Leu T, Schutzhold V, Fandrey J, Ferenz KB. When the brain yearns for oxygen. Neurosignals. (2019) 27(1):50–61. doi: 10.33594/000000199
265. Kawahara M, Kato-Negishi M, Tanaka KI. Dietary trace elements and the pathogenesis of neurodegenerative diseases. Nutrients. (2023) 15(9):2067. doi: 10.3390/nu15092067
266. Koh JY, Lee SJ. Metallothionein-3 as a multifunctional player in the control of cellular processes and diseases. Mol Brain. (2020) 13(1):116. doi: 10.1186/s13041-020-00654-w
267. Vogt K, Mellor J, Tong G, Nicoll R. The actions of synaptically released zinc at hippocampal mossy fiber synapses. Neuron. (2000) 26(1):187–96. doi: 10.1016/S0896-6273(00)81149-6
268. Pei Y, Zhao D, Huang J, Cao L. Zinc-induced seizures: a new experimental model of epilepsy. Epilepsia. (1983) 24(2):169–76. doi: 10.1111/j.1528-1157.1983.tb04876.x
269. Nadler JV, Perry BW, Cotman CW. Intraventricular kainic acid preferentially destroys hippocampal pyramidal cells. Nature. (1978) 271(5646):676–7. doi: 10.1038/271676a0
270. Aniksztejn L, Charton G, Ben-Ari Y. Selective release of endogenous zinc from the hippocampal mossy fibers in situ. Brain Res. (1987) 404(1–2):58–64. doi: 10.1016/0006-8993(87)91355-2
271. Ben-Ari Y, Tremblay E, Ottersen OP, Naquet R. Evidence suggesting secondary epileptogenic lesion after kainic acid: pre treatment with diazepam reduces distant but not local brain damage. Brain Res. (1979) 165(2):362–5. doi: 10.1016/0006-8993(79)90571-7
272. Lee JM, Zipfel GJ, Park KH, He YY, Hsu CY, Choi DW. Zinc translocation accelerates infarction after mild transient focal ischemia. Neuroscience. (2002) 115(3):871–8. doi: 10.1016/S0306-4522(02)00513-4
273. Koh JY, Suh SW, Gwag BJ, He YY, Hsu CY, Choi DW. The role of zinc in selective neuronal death after transient global cerebral ischemia. Science. (1996) 272(5264):1013–6. doi: 10.1126/science.272.5264.1013
274. Assaf SY, Chung SH. Release of endogenous Zn2+ from brain tissue during activity. Nature. (1984) 308(5961):734–6. doi: 10.1038/308734a0
275. Rous P. A sarcoma of the fowl transmissible by an agent separable from the tumor cells. J Exp Med. (1911) 13(4):397–411. doi: 10.1084/jem.13.4.397
276. Brown MT, Cooper JA. Regulation, substrates and functions of src. Biochim Biophys Acta. (1996) 1287(2–3):121–49. doi: 10.1016/0304-419x(96)00003-0
277. Angelis D, Fontanez-Nieves TD, Delivoria-Papadopoulos M. The role of SRC kinase in the caspase-1 pathway after hypoxia in the brain of newborn piglets. Neurochem Res. (2014) 39(11):2118–26. doi: 10.1007/s11064-014-1404-1
278. Manzerra P, Behrens MM, Canzoniero LM, Wang XQ, Heidinger V, Ichinose T, et al. Zinc induces a src family kinase-mediated up-regulation of NMDA receptor activity and excitotoxicity. Proc Natl Acad Sci U S A. (2001) 98(20):11055–61. doi: 10.1073/pnas.191353598
279. Chang KL, Hung TC, Hsieh BS, Chen YH, Chen TF, Cheng HL. Zinc at pharmacologic concentrations affects cytokine expression and induces apoptosis of human peripheral blood mononuclear cells. Nutrition. (2006) 22(5):465–74. doi: 10.1016/j.nut.2005.11.009
280. Livingstone C. Zinc: physiology, deficiency, and parenteral nutrition. Nutr Clin Pract. (2015) 30(3):371–82. doi: 10.1177/0884533615570376
281. Sugiura T, Goto K, Ito K, Ueta A, Fujimoto S, Togari H. Chronic zinc toxicity in an infant who received zinc therapy for atopic dermatitis. Acta Paediatr. (2005) 94(9):1333–5. doi: 10.1111/j.1651-2227.2005.tb02097.x
282. Donangelo CM, Woodhouse LR, King SM, Viteri FE, King JC. Supplemental zinc lowers measures of iron status in young women with low iron reserves. J Nutr. (2002) 132(7):1860–4. doi: 10.1093/jn/132.7.1860
283. de Brito NJ, Rocha ED, de Araujo Silva A, Costa JB, Franca MC, das gracas almeida M, et al. Oral zinc supplementation decreases the serum iron concentration in healthy schoolchildren: a pilot study. Nutrients. (2014) 6(9):3460–73. doi: 10.3390/nu6093460
284. Griffin IJ, Domellof M, Bhatia J, Anderson DM, Kler N. Zinc and copper requirements in preterm infants: an examination of the current literature. Early Hum Dev. (2013) 89(Suppl 2):S29–34. doi: 10.1016/j.earlhumdev.2013.08.001
285. Agostoni C, Buonocore G, Carnielli VP, De Curtis M, Darmaun D, Decsi T, et al. Enteral nutrient supply for preterm infants: commentary from the European society of paediatric gastroenterology, hepatology and nutrition committee on nutrition. J Pediatr Gastroenterol Nutr. (2010) 50(1):85–91. doi: 10.1097/MPG.0b013e3181adaee0
286. Rossander-Hulten L, Brune M, Sandstrom B, Lonnerdal B, Hallberg L. Competitive inhibition of iron absorption by manganese and zinc in humans. Am J Clin Nutr. (1991) 54(1):152–6. doi: 10.1093/ajcn/54.1.152
287. Olivares M, Pizarro F, Ruz M. Zinc inhibits nonheme iron bioavailability in humans. Biol Trace Elem Res. (2007) 117(1–3):7–14. doi: 10.1007/BF02698079
288. Smith JC Jr, Brown ED, McDaniel EG, Chan W. Alterations in vitamin A metabolism during zinc deficiency and food and growth restriction. J Nutr. (1976) 106(4):569–74. doi: 10.1093/jn/106.4.569
289. Imdad A, Rogner J, Sherwani RN, Sidhu J, Regan A, Haykal MR, et al. Zinc supplementation for preventing mortality, morbidity, and growth failure in children aged 6 months to 12 years. Cochrane Database Syst Rev. (2023) 3(3):CD009384. doi: 10.1002/14651858.CD009384.pub3
290. Masters DG, Keen CL, Lonnerdal B, Hurley LS. Zinc deficiency teratogenicity: the protective role of maternal tissue catabolism. J Nutr. (1983) 113(4):905–12. doi: 10.1093/jn/113.4.905
291. Miller SI, Del Villano BC, Flynn A, Krumhansl M. Interaction of alcohol and zinc in fetal dysmorphogenesis. Pharmacol Biochem Behav. (1983) 18(Suppl 1):311–5. doi: 10.1016/0091-3057(83)90192-2
292. Keppen LD, Pysher T, Rennert OM. Zinc deficiency acts as a co-teratogen with alcohol in fetal alcohol syndrome. Pediatr Res. (1985) 19(9):944–7. doi: 10.1203/00006450-198509000-00016
293. Vallee BL, Wacker WE, Bartholomay AF, Hoch FL. Zinc metabolism in hepatic dysfunction. II. Correlation of metabolic patterns with biochemical findings. N Engl J Med. (1957) 257(22):1055–65. doi: 10.1056/NEJM195711282572201
294. Flynn A, Miller SI, Martier SS, Golden NL, Sokol RJ, Del Villano BC. Zinc status of pregnant alcoholic women: a determinant of fetal outcome. Lancet. (1981) 1(8220 Pt 1):572–51. doi: 10.1016/s0140-6736(81)92029-8
295. Sullivan JF, Lankford HG. Urinary excretion of zinc in alcoholism and postalcoholic cirrhosis. Am J Clin Nutr. (1962) 10:153–7. doi: 10.1093/ajcn/10.2.153
296. Assadi FK, Ziai M. Zinc status of infants with fetal alcohol syndrome. Pediatr Res. (1986) 20(6):551–4. doi: 10.1203/00006450-198606000-00014
297. Fisher SE, Alcock NW, Amirian J, Altshuler HL. Neonatal and maternal hair zinc levels in a nonhuman primate model of the fetal alcohol syndrome. Alcohol Clin Exp Res. (1988) 12(3):417–21. doi: 10.1111/j.1530-0277.1988.tb00219.x
298. Henderson GI, Hoyumpa AM Jr, McClain C, Schenker S. The effects of chronic and acute alcohol administration on fetal development in the rat. Alcohol Clin Exp Res. (1979) 3(2):99–106. doi: 10.1111/j.1530-0277.1979.tb05281.x
299. Ghishan FK, Patwardhan R, Greene HL. Fetal alcohol syndrome: inhibition of placental zinc transport as a potential mechanism for fetal growth retardation in the rat. J Lab Clin Med. (1982) 100(1):45–52.7086268
300. Ghishan FK, Greene HL. Fetal alcohol syndrome: failure of zinc supplementation to reverse the effect of ethanol on placental transport of zinc. Pediatr Res. (1983) 17(7):529–31. doi: 10.1203/00006450-198307000-00002
301. Zidenberg-Cherr S, Rosenbaum J, Keen CL. Influence of ethanol consumption on maternal-fetal transfer of zinc in pregnant rats on day 14 of pregnancy. J Nutr. (1988) 118(7):865–70. doi: 10.1093/jn/118.7.865
302. Kuhnert BR, Kuhnert PM, Lazebnik N, Erhard P. The effect of maternal smoking on the relationship between maternal and fetal zinc status and infant birth weight. J Am Coll Nutr. (1988) 7(4):309–16. doi: 10.1080/07315724.1988.10720248
303. Wang H, Wang Y, Bo QL, Ji YL, Liu L, Hu YF, et al. Maternal cadmium exposure reduces placental zinc transport and induces fetal growth restriction in mice. Reprod Toxicol. (2016) 63:174–82. doi: 10.1016/j.reprotox.2016.06.010
304. Mimouna SB, Boughammoura S, Chemek M, Haouas Z, Banni M, Messaoudi I. Disruption of the zinc metabolism in rat foetal brain after prenatal exposure to cadmium. Chem Biol Interact. (2018) 286:88–95. doi: 10.1016/j.cbi.2018.03.005
305. Terrin G, Berni Canani R, Passariello A, Messina F, Conti MG, Caoci S, et al. Zinc supplementation reduces morbidity and mortality in very-low-birth-weight preterm neonates: a hospital-based randomized, placebo-controlled trial in an industrialized country. Am J Clin Nutr. (2013) 98(6):1468–74. doi: 10.3945/ajcn.112.054478
306. Friel JK, Andrews WL, Matthew JD, Long DR, Cornel AM, Cox M, et al. Zinc supplementation in very-low-birth-weight infants. J Pediatr Gastroenterol Nutr. (1993) 17(1):97–104. doi: 10.1097/00005176-199307000-00015
307. Islam MN, Chowdhury MA, Siddika M, Qurishi SB, Bhuiyan MK, Hoque MM, et al. Effect of oral zinc supplementation on the growth of preterm infants. Indian Pediatr. (2010) 47(10):845–9. doi: 10.1007/s13312-010-0145-8
308. Kumar TVR, Ramji S. Effect of zinc supplementation on growth in very low birth weight infants. J Trop Pediatr. (2012) 58(1):50–4. doi: 10.1093/tropej/fmr036
309. Sahin S, Sari FN, Bidev D, Bozkurt O, Dizdar EA, Oguz SS. Zinc supplementation in very low birth weight infants: a randomized controlled trial. Am J Perinatol. (2024) 41(S 01):3107–e14. doi: 10.1055/s-0043-1776762
310. Kaban R, Azis H, Prawitasari T, Kautsar A, Lusyati S, Insani N. Zinc supplementation in preterm infants and growth indicators in a developing country. Paediatr Indones. (2023) 63(6):443–9. doi: 10.14238/pi63.6.2023.443-9
311. Aminisani N, Barak M, Shamshirgaran SM. Effect of zinc supplementation on growth of low birth weight infants aged 1-6 mo in Ardabil, Iran. Indian J Pediatr. (2011) 78(10):1239–43. doi: 10.1007/s12098-011-0541-7
312. Hegran H, Kassem S, Ragab S. The effect of zinc supplementation on growth and development in preterm neonates. Menoufia Med J. (2014) 27(3):524–8. doi: 10.4103/1110-2098.145500
313. Ogasawara K, Hayato G, Honda Y, Maeda H. Effect of enteral zinc supplementation on the anthropometric measurements of preterm infants at discharge from the neonatal intensive care unit and evaluation of copper deficiency. Nutrients. (2024) 16(11):1612. doi: 10.3390/nu16111612
314. Taneja S, Bhandari N, Rongsen-Chandola T, Mahalanabis D, Fontaine O, Bhan MK. Effect of zinc supplementation on morbidity and growth in hospital-born, low-birth-weight infants. Am J Clin Nutr. (2009) 90(2):385–91. doi: 10.3945/ajcn.2009.27707
315. Belfort MB, Rifas-Shiman SL, Sullivan T, Collins CT, McPhee AJ, Ryan P, et al. Infant growth before and after term: effects on neurodevelopment in preterm infants. Pediatrics. (2011) 128(4):e899–906. doi: 10.1542/peds.2011-0282
316. Ehrenkranz RA, Dusick AM, Vohr BR, Wright LL, Wrage LA, Poole WK. Growth in the neonatal intensive care unit influences neurodevelopmental and growth outcomes of extremely low birth weight infants. Pediatrics. (2006) 117(4):1253–61. doi: 10.1542/peds.2005-1368
317. Kan E, Roberts G, Anderson PJ, Doyle LW, Victorian infant collaborative study G. The association of growth impairment with neurodevelopmental outcome at eight years of age in very preterm children. Early Hum Dev. (2008) 84(6):409–16. doi: 10.1016/j.earlhumdev.2007.11.002
318. Latal-Hajnal B, von Siebenthal K, Kovari H, Bucher HU, Largo RH. Postnatal growth in VLBW infants: significant association with neurodevelopmental outcome. J Pediatr. (2003) 143(2):163–70. doi: 10.1067/S0022-3476(03)00243-9
319. Ramel SE, Demerath EW, Gray HL, Younge N, Boys C, Georgieff MK. The relationship of poor linear growth velocity with neonatal illness and two-year neurodevelopment in preterm infants. Neonatology. (2012) 102(1):19–24. doi: 10.1159/000336127
320. Belfort MB, Gillman MW, Buka SL, Casey PH, McCormick MC. Preterm infant linear growth and adiposity gain: trade-offs for later weight status and intelligence quotient. J Pediatr. (2013) 163(6):1564–9.e2. doi: 10.1016/j.jpeds.2013.06.032
321. Ahmad I, Zaldivar F, Iwanaga K, Koeppel R, Grochow D, Nemet D, et al. Inflammatory and growth mediators in growing preterm infants. J Pediatr Endocrinol Metab. (2007) 20(3):387–96. doi: 10.1515/JPEM.2007.20.3.387
322. Holgersen K, Rasmussen MB, Zamir I, Aunsholt L, Zachariassen G, Sangild PT. Glucose-regulatory hormones and growth in very preterm infants fed fortified human milk. Pediatr Res. (2024) 96(3):713–22. doi: 10.1038/s41390-024-03166-8
323. Eliakim A, Nemet D, Ahmad I, Zaldivar F, Koppel R, Grochow D, et al. Growth factors, inflammatory cytokines and postnatal bone strength in preterm infants. J Pediatr Endocrinol Metab. (2009) 22(8):733–40. doi: 10.1515/JPEM.2009.22.8.733
324. Liu E, Pimpin L, Shulkin M, Kranz S, Duggan CP, Mozaffarian D, et al. Effect of zinc supplementation on growth outcomes in children under 5 years of age. Nutrients. (2018) 10(3):377. doi: 10.3390/nu10030377
325. Nissensohn M, Sanchez-Villegas A, Fuentes Lugo D, Henriquez Sanchez P, Doreste Alonso J, Pena Quintana L, et al. Effect of zinc intake on growth in infants: a meta-analysis. Crit Rev Food Sci Nutr. (2016) 56(3):350–63. doi: 10.1080/10408398.2013.802661
326. Warthon-Medina M, Moran VH, Stammers AL, Dillon S, Qualter P, Nissensohn M, et al. Zinc intake, status and indices of cognitive function in adults and children: a systematic review and meta-analysis. Eur J Clin Nutr. (2015) 69(6):649–61. doi: 10.1038/ejcn.2015.60
327. Gogia S, Sachdev HS. Zinc supplementation for mental and motor development in children. Cochrane Database Syst Rev. (2012) 12:CD007991. doi: 10.1002/14651858.CD007991.pub2
328. Colombo J, Zavaleta N, Kannass KN, Lazarte F, Albornoz C, Kapa LL, et al. Zinc supplementation sustained normative neurodevelopment in a randomized, controlled trial of Peruvian infants aged 6–18 months. J Nutr. (2014) 144(8):1298–305. doi: 10.3945/jn.113.189365
329. Sajedi F, Shahshahani S, Ghiasvand H, Mosallanezhad Z, Fatollahierad S. Does zinc with and without iron co-supplementation have effect on motor and mental development of children? A systematic review and meta-analysis. BMC Pediatr. (2020) 20(1):451. doi: 10.1186/s12887-020-02340-1
330. Talebi S, Miraghajani M, Ghavami A, Mohammadi H. The effect of zinc supplementation in children with attention deficit hyperactivity disorder: a systematic review and dose-response meta-analysis of randomized clinical trials. Crit Rev Food Sci Nutr. (2022) 62(32):9093–102. doi: 10.1080/10408398.2021.1940833
331. Noble Y, Boyd R. Neonatal assessments for the preterm infant up to 4 months corrected age: a systematic review. Dev Med Child Neurol. (2012) 54(2):129–39. doi: 10.1111/j.1469-8749.2010.03903.x
Keywords: zinc, brain, neonates, neurodevelopment, mechanisms
Citation: Chamakioti M, Brion LP, Viswanathan P, Lair CS and Angelis D (2024) The role of zinc in the premature brain: functions, outcomes and future research perspectives. Front. Pediatr. 12:1496846. doi: 10.3389/fped.2024.1496846
Received: 15 September 2024; Accepted: 25 November 2024;
Published: 23 December 2024.
Edited by:
Raul Chavez-Valdez, Johns Hopkins Medicine, United StatesReviewed by:
Johana Diaz, University of Maryland, United StatesJennifer Fundora, Johns Hopkins University, United States
Copyright: © 2024 Chamakioti, Brion, Viswanathan, Lair and Angelis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dimitrios Angelis, ZGltaXRyaW9zLmFuZ2VsaXNAdXRzb3V0d2VzdGVybi5lZHU=