
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr. , 16 August 2024
Sec. Pediatric Immunology
Volume 12 - 2024 | https://doi.org/10.3389/fped.2024.1434076
This article is part of the Research Topic Meaningful Cases of Primary Immunodeficiencies, volume IV View all 11 articles
 Geoffrey Hall1
Geoffrey Hall1 Janet G. Markle2
Janet G. Markle2 James Maiarana2
James Maiarana2 Paul L. Martin3
Paul L. Martin3 Jennifer A. Rothman4
Jennifer A. Rothman4 John W. Sleasman1Howard Lederman5
John W. Sleasman1Howard Lederman5 Antoine E. Azar6
Antoine E. Azar6 Robert A. Brodsky7
Robert A. Brodsky7 Talal Mousallem1*
Talal Mousallem1*
A 20-year-old male patient with a history of celiac disease came to medical attention after developing profound fatigue and pancytopenia. Evaluation demonstrated pan-hypogammaglobulinemia. There was no history of significant clinical infections. Bone marrow biopsy confirmed hypocellular marrow consistent with aplastic anemia. Oncologic and hematologic evaluations were unremarkable for iron deficiency, paroxysmal nocturnal hemoglobinuria, myelodysplastic syndromes, T-cell clonality, and leukemia. A next generation genetic sequencing immunodeficiency panel revealed a heterozygous variant of uncertain significance in CTLA4 c.385T >A, p.Cys129Ser (C129S). Cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) is an inhibitory receptor important in maintaining immunologic homeostasis. To determine the functional significance of the C129S variant, additional testing was pursued to assess for diminished protein expression, as described in other pathogenic CTLA4 variants. The results demonstrated severely impaired CTLA-4 expression and CD80 transendocytosis, consistent with other variants causing CTLA-4 haploinsufficiency. He was initially treated with IVIG and cyclosporine, and became transfusion independent for few months, but relapsed. Treatment with CTLA-4-Ig fusion protein (abatacept) was considered, however the patient opted for definitive therapy through reduced-intensity haploidentical hematopoietic stem cell transplant, which was curative.
Inborn errors of immunity (IEIs) encompass a broad and heterogenous group of genetic disorders which disrupt immunologic homeostasis (1). Traditionally, these conditions have been characterized by which arm(s) of the immune system (e.g., innate, humoral, phagocytic, complement, etc.) are impacted resulting in increased susceptibility to infectious agents. However, coinciding autoimmune/autoinflammatory disease states are increasingly being recognized as a primary or secondary feature among IEIs (2). In particular, immune dysregulation is a primary feature of pathologic variants involving genes responsible for maintaining immunologic homeostasis.
Cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) is an inhibitory receptor present on T-cells and serves a fundamental role in regulation of immune responses. The process of T-cell (CD4+) activation requires both primary and costimulatory signals via interaction with antigen presenting cells (APCs). One such costimulatory signal involves CD28 (present on T-cells) and CD80/86 (present on APCs). Once activated, T-cells upregulate expression of CTLA-4 on their surface which binds to CD80/86 resulting in transendocytosis of the receptors thereby limiting further activation (Figure 1) (3). This coinhibitory mechanism prevents unregulated T-cell activation. Additional immune regulation occurs via T-cell subpopulations such as T-regulatory-cells (Tregs). These cells constitutively express CTLA-4 providing an additional checkpoint in immune activation (4). In this way, CTLA-4 acts to maintain immunologic homeostasis, with disruptions caused by CTLA4 variants leading to altered protein expression and clinical disease states.
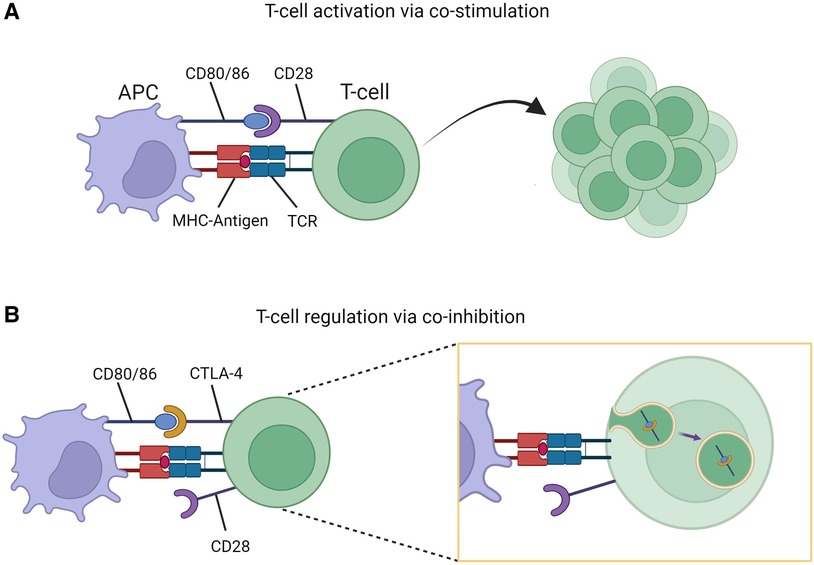
Figure 1. T-cell stimulation and regulation. (A) Co-stimulatory signaling through CD80/86-CD28 interaction resulting in T-cell activation and proliferation. (B) Co-inhibition through CD80/86-CTLA-4 interaction resulting T-cell regulation due to transendocytosis of CD80/86-CTLA-4 complex. Figure created with Biorender.com.
To our knowledge, no cases of complete CTLA-4 deficiency have been reported in humans. Targeted genetic deletion leading to complete CTLA-4 loss of function in mice leads to fatal multiorgan lymphocytic infiltration primarily due to expansion of unregulated CD4+ T-cells (5–8). Complete CTLA-4 deficiency may be incompatible with life in humans. However, pathogenic heterozygous variants in CTLA4 can lead to disease phenotypes with variable clinical penetrance and expressivity. Phenotypic features reported in patients with CTLA-4 haploinsufficiency include: autoimmune cytopenias, hypogammaglobulinemia, lymphadenopathy, and organ dysfunction (enteropathy, splenomegaly, etc.) from lymphocytic infiltration (9). Additionally, polymorphisms in CTLA4 have been associated with risk for type 1 diabetes mellitus, Graves' disease, multiple sclerosis, and malignancies (10–13).
In this report, we present a novel CTLA4 variant manifesting as aplastic anemia and provide functional testing that confirms this novel variant is deleterious to CTLA-4 expression resulting in severely reduced transendocytosis.
A 20-year-old male of self-reported White race (ancestry unavailable) presented to medical care for 1 week of persistent and profound incapacitating fatigue. Additional symptoms included palpitations, lightheadedness and exertional dyspnea with ambulation, which all resulted in an inability to participate in collegiate athletics. His medical history was pertinent for celiac disease (confirmed via endoscopic biopsy with symptom resolution after implementing a gluten-free diet), pityriasis alba, and idiopathic wet macular degeneration status-post successful treatment with aflibercept. He did not have a history of other autoimmune disease, immune deficiency or severe/atypical infections. Paternal history was positive for rheumatologic/autoimmune disease including psoriasis, arthritis, and hypothyroidism. Maternal history was unremarkable. There is no history of consanguinity. The remaining family history was pertinent only for thyroid disease of unclear specificity in maternal and paternal grandparents.
Upon presentation, he was anemic (hemoglobin 8.4 mg/dl) and thrombocytopenic (platelet count 18,000/ml). The subsequent day, his white blood cell counts decreased from 4,400 cells/ml to 2,600 cells/ml, consistent with pancytopenia. Iron studies were consistent with mild iron overload and serum copper levels were slightly elevated. He had no prior history of blood transfusions and genetic testing for hemochromatosis demonstrated a heterozygous variant in HFE (c.187C >G, p.His63Asp) (H63D). The H63D carrier-status rarely results in clinically significant iron overload (14, 15). The mild elevation in serum copper based on internal lab reference ranges were not thought to be clinically significant and levels up to 158.9 μg/dl are considered normal (16). Additional evaluation including folate, B12, lactate dehydrogenase, and haptoglobin were unremarkable (Table 1). Bone marrow biopsy demonstrated hypocellularity with near absence of erythroid precursors and megakaryocytes, consistent with aplastic anemia. Further hematologic testing was unremarkable including: bone marrow chromosomal analysis, fluorescence in situ hybridization for BCR/ABL1 and chromosomal abnormalities (monosomy 5 and 7, trisomy 8, and 20q deletion), myelodysplastic syndrome mutation sequencing, telomere length studies, leukemia flow cytometry immunophenotyping, and T-cell clonality. PNH flow cytometry revealed a loss of GPI-anchored proteins on 0.04% and 0.6% of granulocytes and monocytes, respectively, suggesting that the aplastic anemia may be immune-mediated (18).
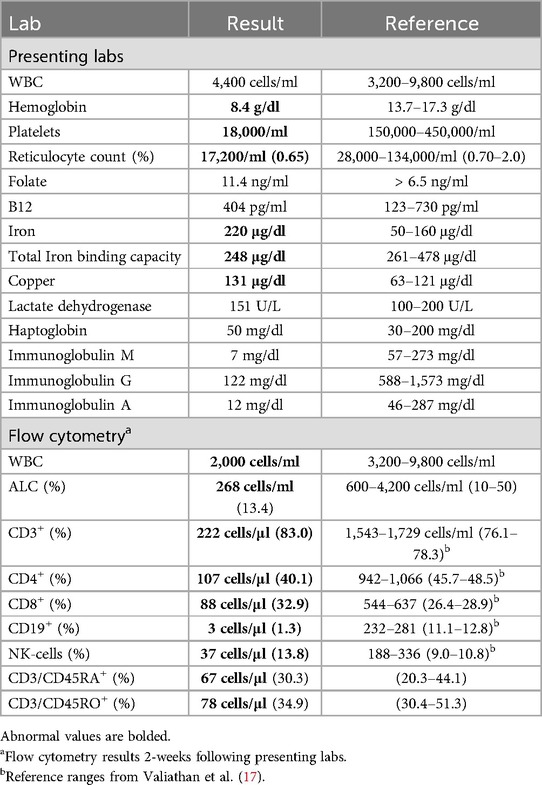
Table 1. Diagnostic evaluation summary.
Blood count differential demonstrated profound lymphopenia with an absolute lymphocyte count of 268 cells/ml. Lymphocyte enumeration through flow cytometry revealed a T-cell (CD3+) count of 222 cells/μl (83%), CD4+ count of 107 cells/μl (40.1%), CD8+ count of 88 cells/μl (32.9%), naïve T-cell (CD45RA+/CD4+/CD62l+) count of 15 cells/μl (14.4%), B-cell (CD19+) count of 3 cells/μl (1.3%), and NK-cell (CD16/56+) count of 17 cells/μl (13.8%). T-cell proliferation to phytohemagglutinin (PHA) and tetanus was normal. Immunoglobulin evaluation demonstrated diffuse hypogammaglobulinemia with: IgG (122 mg/dl), IgM (7 mg/dl) and IgA (12 mg/dl). Of note, immunoglobulin levels collected 16-months prior demonstrated a similar pattern with an IgG of 176 mg/dl, an IgM <25 mg/dl, and IgA of 15.7 mg/dl. Antibody titers to tetanus and diphtheria toxoid were protective, and pneumococcal-23 antibody titers were protective to greater than 75% of serotypes tested.
A primary immunodeficiency next generation sequencing panel was sent and revealed a novel heterozygous variant of uncertain significance in CTLA4 (c.385T >A, p.Cys129Ser) (C129S). Paternal testing revealed the same variant. Maternal and sibling testing was negative. Given the patient's clinical phenotype, known risk for immune dysregulation with CTLA4 variants, and without other identifiable etiologies for the aplastic anemia, a research-based functional assay was pursued.
A CTLA-4 functional assay was performed as previously described (19–21). pCMV6-CTLA4-MycDDK plasmid was obtained from Origene (#RC213631). Construct carrying C129S mutant allele was generated from the wild type (WT) plasmid by site-directed mutagenesis (QuikChange II XL; Agilent Technologies, #200523) according to manufacturer's instructions and validated by Sanger Sequencing. WT or C129S mutant CTLA4 plasmids were transfected into CHO cells (ATCC # CCL-61) using Lipofectamine 2000 (ThermoFisher Scientific, #11668027), per manufacturer's protocol.
For transendocytosis experiments, transfected CHO cells were co-cultured 1:1 with CellTraceTM Violet (ThermoFisher Scientific #C34557) labelled CHO-CD80GFP cells (A gift of Bodo Grimbacher and David Sansom) for 16 h. CTLA-4 expression and transendocytosis of CD80GFP were measured by flow cytometry on the MACSQuant Analyzer 16 (Miltenyi Biotec). A known loss of function pathogenic variant in CTLA4, R51X, was used as a positive control and untransfected CHO cells were used as a negative control. Flow cytometry comparing CTLA-4 expression and CD80 uptake through transendocytosis were run in triplicate.
Results demonstrated that the C129S variant drastically impaired expression of CTLA-4 with a resulting decrease in transendocytosis as compared to wild type. These findings are similar to those seen with the causal allele R51X (Figure 2). Thus, the patient's C129S variant exhibited results consistent with CTLA-4 haploinsufficiency. Moreover, analysis of peripheral blood mononuclear cells (PBMCs) from the patient and his father confirmed reduced CTLA-4 protein levels (Figures 2D,E).
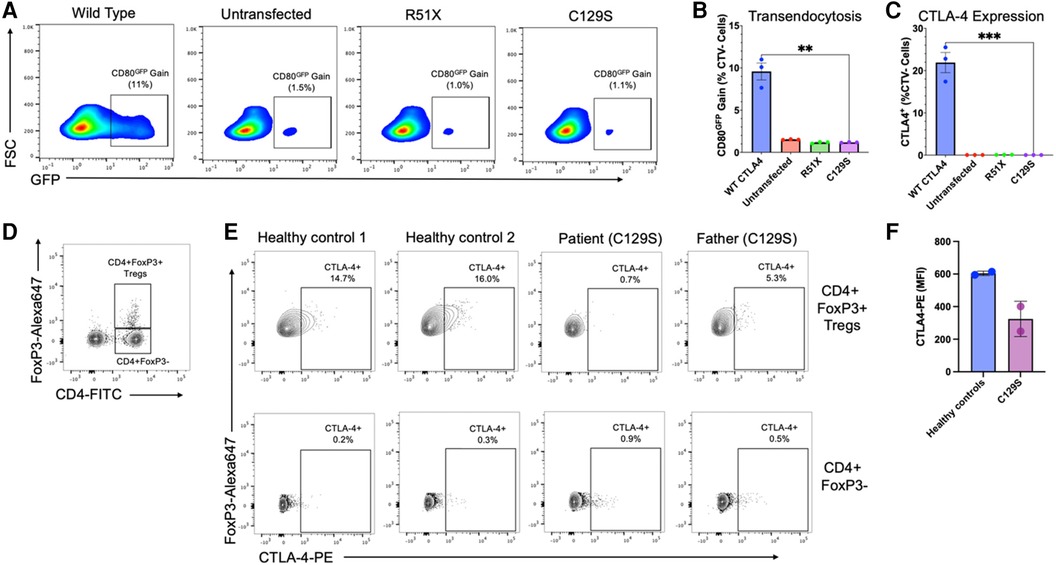
Figure 2. The C129S missense mutation dramatically impairs CTLA-4 expression resulting in decreased transendocytosis. (A) Representative example of CD80GFP transendocytosis assay results. These plots show cells previously gated on forward and side scatter profile, and negative for Cell Trace Violet. GFP signal represents percent transendocytosis of CD80. (B) Summary results from n = 3 independent experiments, performed as in A. (C) Percent CTLA-4+ CHO cells after transfection with WT or mutant CTLA-4 plasmids (n = 3),**p < 0.01, unpaired t-tests. (D) Flow cytometry analysis of peripheral blood mononuclear cells (PBMCs) to identify CD4 + FoxP3 + regulatory T-cells (T¬¬regs) and CD4 + CD3- cells. (E) Analysis of CTLA-4 protein expression by Tregs (upper row) and CD4 + FoxP3 + cells (lower row), gated as shown in (D). (F) Mean fluorescence intensity for CTLA-4 on Tregs from two healthy donors, and the patient and his father with the C129S variant.
Based on the results from the transendocytosis assay, and the patient's clinical history, the C129S variant was determined to be pathogenic, suggesting that the patient's phenotype was likely due to CTLA-4 haploinsufficiency. Initial therapy consisted of high-dose IVIG (1 g/kg × 2 doses), cyclosporine (maximum dose of 275 mg twice daily), and systemic corticosteroids (maximum dose of 30 mg daily). He responded well to these therapies with notable improvement in red blood cell production seen on bone marrow biopsy. Cyclosporine was then decreased to 175 mg twice weekly with a target trough level of 150–300 ng/ml, and systemic corticosteroids were discontinued. However, 6 months into therapy, he developed renal impairment and was transitioned to 5 mg daily of sirolimus with a trough goal of 5–15 ng/ml, but it was poorly tolerated. In the subsequent 2 months, his aplastic anemia relapsed. Off-label use of abatacept was considered given clinical reports showing positive responses in patients with CTLA-4 haploinsufficiency (22–24). However, given disease severity and the patient's preference, definitive therapy in the form of a haploidentical (sibling, variant negative) hematopoietic stem cell transplantation (HSCT) was pursued (25). Use of haploidentical donor marrow was favored to a matched-unrelated donor for the following reasons: earlier time to transplant, improved total stem cell dose which is critical in non-malignant disease such as aplastic anemia, reduced risk for graft-vs.-host disease due to lower T-cell concentration as compared to peripheral stem cell harvest, and comparable outcomes with use of post-transplant cyclophosphamide. The non-myeloablative conditioning regimen included standard anti-thymocyte globulin (0.5 mg/kg/day on day −9 and 2 mg/kg/day on day −8 and −7), fludarabine (30 mg/m2/day on days −6 to −2), cyclophosphamide (14.5 mg/kg/day on days −6 and −5), and total body radiation (400 cGy) on day −1 as previously described (25). Post-transplant course was uncomplicated and graft-vs.-host disease prophylaxis included cyclophosphamide, tacrolimus and mycophenolate mofetil as previously described (25). He achieved >95% CD3+ donor chimerism one-month post-transplant with subsequent increase to 100% at around 3-months post-transplant. His most recent chimerism study (1-year post HSCT) continues to show 100% donor chimerism in both peripheral blood and CD3+ compartments. He acquired a primary Epstein-Barr infection at around 4-months post-transplantation with mild intermittent clinical symptoms of rash and pharyngitis. He now has resolution of clinical symptoms without intervention, and his most recent EBV DNA is below the threshold of assay detection. Now, 24-months post-transplant, infectious prophylaxes have been discontinued. Due to previous adverse reactions to immunoglobulin replacement, it was not administered post-transplantation. Endogenous immunoglobulin levels normalized by 14-months post-transplant with an IgG of 823 mg/dl, IgM of 69 mg/dl, and IgA of 214 mg/dl. Platelet counts remain appropriate (>150,000/ml) and hemoglobin has been stable (12.5–15.4 g/dl). He remains transfusion independent. Tetanus and diphtheria toxoid titers remain protective. Inactivated vaccinations have been well-tolerated, with plans to administer live vaccines in the future. Growth has been appropriate with a body mass index of 19.7 kg/m2. He has resumed normal activities.
CTLA-4 haploinsufficiency is an autosomal dominant condition characterized by reduced CTLA-4 expression and/or function due to variants in CTLA4 (26). Due to variability in expressivity and penetrance, the condition can be under recognized and underdiagnosed. This was well demonstrated in the case of our patient, where both he and his father shared a common variant in CTLA4 and exhibited dissimilar phenotypes. While the patient developed severe aplastic anemia, his father has psoriasis, arthritis and hypothyroidism, likely due to CTLA-4 haploinsufficiency as well. In addition, the family history of thyroid dysfunction suggests this variant may be present across generations. Other inborn errors of immunity can also present with similar clinical features to CTLA-4 haploinsufficiency. An example is lipopolysaccharide-responsive beige-like anchor, or LRBA, deficiency. LRBA is a protein responsible for recycling cellular components, including CTLA-4, thereby preventing lysosomal degradation (27). Thus, deficiency of LRBA can lead to reduced CTLA-4 (28).
Therapeutic options for management of immune dysregulatory conditions can be challenging for the practicing immunologist. Immunosuppression to manage autoimmune and autoinflammatory symptoms must be balanced with heightened risk of infection and further marrow suppression. Targeted therapeutic options are limited in management of immune dysregulation, but sometimes can be tailored when the underlying mechanistic pathways are identified. Abatacept is a promising option for patients with CTLA-4 haploinsufficiency. Abatacept is a fusion protein consisting of CTLA-4 fused to the Fc region of human IgG (29). As such, it (at least partially) compensates for the insufficient endogenous expression and/or function of CTLA-4 and binds to CD80/CD86, thereby regulating T-cell stimulation. While abatacept has shown beneficial results in clinical reports, the absence of clinical trials renders it an off-label agent. Moreover, as a replacement therapy, it necessitates life-long treatment in managing CTLA-4 haploinsufficiency, with no available data regarding the long-term clinical implications. In this case, the patient presented with severe clinical disease. Severe or treatment refractory disease including cytopenias and aplastic anemia should prompt an early search for a potential stem cell donor. While abatacept could have been used, the only definitive cure is HSCT. Overall, his post-transplantation outcome has been excellent.
This report outlines a novel CTLA4 variant with functional confirmation of a pathogenic aberration resulting in CTLA-4 haploinsufficiency. Furthermore, it highlights the diverse phenotypic spectrum increasingly recognized for inborn errors of immunity, especially those involved in immune regulation. Clinical immunologists need to maintain a high index of suspicion when evaluating patients presenting with autoimmunity, autoinflammation, and lymphoproliferation. Genetic sequencing should especially be considered in patients presenting with severe, non-malignant hematologic disease. Establishing a multidisciplinary collaboration is critical to early recognition, management, and occasionally curative intervention for these patients.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
The studies involving humans were approved by Duke University Institutional Review Board. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article. The legal representatives for all participants provided written consent for enrollment into protocol, Genetic and Functional Analysis of Primary Immune Deficiencies (Pro00066839), which was approved by the institutional review board of Duke University.
GH: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Project administration, Writing – original draft, Writing – review & editing, Software. JG-M: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Software, Writing – original draft, Writing – review & editing, Resources, Supervision, Validation. JM: Formal Analysis, Writing – review & editing, Data curation, Investigation, Methodology, Software, Validation, Writing – original draft. PM: Investigation, Writing – review & editing. JR: Investigation, Writing – review & editing. JS: Formal Analysis, Funding acquisition, Resources, Writing – review & editing. HL: Writing – review & editing. AA: Writing – review & editing. RB: Writing – review & editing. TM: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article.
The disclosed funders were not involved in the study design, collection, analysis, interpretation of data, the writing of this article, or the decision to submit it for publication. Funding received from the Duke Jeffrey Modell Diagnostic and Research Center and the Division of Intramural Research, National Institute of Allergy and Infectious Disease. Dr. G. Hall declares he received funding from an NIH T32 training grant (T32AI007062). Dr. J. Maiarana declares he received funding from the NIH R38 Stimulating Access to Research in Residency program. Dr. J. Markle declares she received funding from the Vanderbilt University Medical Center’s Digestive Disease Research Center supported by NIH grant P30DK058404, an American Cancer Society Institutional Research Grant to the Vanderbilt Ingram Cancer Center, and the Indiana University-Ohio State University Maternal and Pediatric Precision in Therapeutics Data, Model, Knowledge, and Research Coordination Center (IU-OSU MPRINT DMKRCC), in part by Grant Number P30HD106451 from the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) Obstetrics and Pediatric Pharmacology and Therapeutics Branch (OPPTB). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The authors would like to acknowledge Dr. Bodo Grimbacher (U. Freiburg) for sharing CHO-CD80GFP cells, which were originally developed by Dr. David Sansom (UCL).
The authors disclose the following commercial and/or financial relationships which were not related to study design, collection, analysis, interpretation of data, the writing of this article, or the decision to submit it for publication.
Dr. Mousallem received funding from Chiesi for a project entitled: A Single Arm, Open-Label, Multicenter, Registry Study of Revcovi Treatment in ADA-SCID Patients Requiring Enzyme Replacement Therapy. Dr. Mousallem is the Duke Site PI for PIDTC (NIAID-University of California, San Francisco U54 AI082973 Puck (PI) 09/2019-08/2024)- Prospective Study of SCID Infants who receive Hematopoietic Cell Therapy. Dr. Sleasman receives grant funding from Sumitomo Pharma America, Inc. Dr. G. Hall received funding from Thermo Fisher Scientific, Inc for the 2023 Prestige Award.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Notarangelo LD, Bacchetta R, Casanova JL, Su HC. Human inborn errors of immunity: an expanding universe. Sci Immunol. (2020) 5(49):eabb1662. doi: 10.1126/sciimmunol.abb1662
2. Chan AY, Torgerson TR. Primary immune regulatory disorders: a growing universe of immune dysregulation. Curr Opin Allergy Clin Immunol. (2020) 20(6):582–90. doi: 10.1097/ACI.0000000000000689
3. Abbas AK, Lichtman AH, Pillai S. Cellular and Molecular Immunology. 10th ed Philadelphia, PA: Elsevier (2022).
4. Van Coillie S, Wiernicki B, Xu J. Molecular and Cellular Functions of CTLA-4. Singapore: Springer (2020). p. 7–32.
5. Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. (1995) 3(5):541–7. doi: 10.1016/1074-7613(95)90125-6
6. Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, et al. Lymphoproliferative disorders with early lethality in mice deficient in ctla-4. Science. (1995) 270(5238):985–8. doi: 10.1126/science.270.5238.985
7. Chambers CA, Cado D, Truong T, Allison JP. Thymocyte development is normal in CTLA-4-deficient mice. Proc Natl Acad Sci USA. (1997) 94(17):9296–301. doi: 10.1073/pnas.94.17.9296
8. Chambers CA, Sullivan TJ, Allison JP. Lymphoproliferation in CTLA-4-deficient mice is mediated by costimulation-dependent activation of CD4+ T cells. Immunity. (1997) 7(6):885–95. doi: 10.1016/S1074-7613(00)80406-9
9. Kuehn HS, Ouyang W, Lo B, Deenick EK, Niemela JE, Avery DT, et al. Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science. (2014) 345(6204):1623–7. doi: 10.1126/science.1255904
10. Kavvoura FK, Ioannidis JPA. CTLA-4 gene polymorphisms and susceptibility to type 1 diabetes mellitus: a HuGE review and meta-analysis. Am J Epidemiol. (2005) 162(1):3–16. doi: 10.1093/aje/kwi165
11. Ligers A, Xu C, Saarinen S, Hillert J, Olerup O. The CTLA-4 gene is associated with multiple sclerosis. J Neuroimmunol. (1999) 97(1–2):182–90. doi: 10.1016/S0165-5728(99)00072-7
12. Pawlak-Adamska E, Frydecka I, Bolanowski M, Tomkiewicz A, Jonkisz A, Karabon L, et al. CD28/CTLA-4/ICOS haplotypes confers susceptibility to Graves’ disease and modulates clinical phenotype of disease. Endocrine. (2017) 55(1):186–99. doi: 10.1007/s12020-016-1096-1
13. Egg D, Schwab C, Gabrysch A, Arkwright PD, Cheesman E, Giulino-Roth L, et al. Increased risk for malignancies in 131 affected CTLA4 mutation carriers. Front Immunol. (2018) 9:2012. doi: 10.3389/fimmu.2018.02012
14. Neghina AM, Anghel A. Hemochromatosis genotypes and risk of iron overload–a meta-analysis. Ann Epidemiol. (2011) 21(1):1–14. doi: 10.1016/j.annepidem.2010.05.013
15. Gochee PA, Powell LW, Cullen DJ, Du Sart D, Rossi E, Olynyk JK. A population-based study of the biochemical and clinical expression of the H63D hemochromatosis mutation. Gastroenterology. (2002) 122(3):646–51. doi: 10.1016/S0016-5085(02)80116-0
16. Otten JJ, Hellwig JP, Meyers LD. DRI, Dietary Reference Intakes: The Essential Guide to Nutrient Requirements. Washington, DC: National Academies Press (2006). xiii, 543 p.
17. Valiathan R, Deeb K, Diamante M, Ashman M, Sachdeva N, Asthana D. Reference ranges of lymphocyte subsets in healthy adults and adolescents with special mention of T cell maturation subsets in adults of south Florida. Immunobiology. (2014) 219(7):487–96. doi: 10.1016/j.imbio.2014.02.010
18. DeZern AE, Symons HJ, Resar LS, Borowitz MJ, Armanios MY, Brodsky RA. Detection of paroxysmal nocturnal hemoglobinuria clones to exclude inherited bone marrow failure syndromes. Eur J Haematol. (2014) 92(6):467–70. doi: 10.1111/ejh.12299
19. Hou TZ, Qureshi OS, Wang CJ, Baker J, Young SP, Walker LS, et al. A transendocytosis model of CTLA-4 function predicts its suppressive behavior on regulatory T cells. J Immunol. (2015) 194(5):2148–59. doi: 10.4049/jimmunol.1401876
20. Maiarana J, Moncada-Velez M, Malbran E, Torre MG, Elonen C, Malbran A, et al. Deep immunophenotyping shows altered immune cell subsets in CTLA-4 haploinsufficiency. Pediatr Allergy Immunol. (2023) 34(7):e13994. doi: 10.1111/pai.13994
21. Duke S, Maiarana J, Yousefi P, Burks E, Gerrie S, Setiadi A, et al. Expanding the molecular and phenotypic spectrum of CTLA-4 insufficiency. Pediatr Allergy Immunol. (2024) 35(2):e14077. doi: 10.1111/pai.14077
22. Uzel G, Karanovic D, Su H, Rump A, Agharahimi A, Holland SM, et al. Management of cytopenias in CTLA4 haploinsufficiency using abatacept and sirolimus. Blood. (2018) 132:2409. doi: 10.1182/blood-2018-99-120185
23. Lee S, Moon JS, Lee C-R, Kim H-E, Baek S-M, Hwang S, et al. Abatacept alleviates severe autoimmune symptoms in a patient carrying a de novo variant in CTLA-4. J Allergy Clin Immunol. (2016) 137(1):327–30. doi: 10.1016/j.jaci.2015.08.036
24. van Leeuwen EM, Cuadrado E, Gerrits AM, Witteveen E, de Bree GJ. Treatment of intracerebral lesions with abatacept in a CTLA4-haploinsufficient patient. J Clin Immunol. (2018) 38(4):464–7. doi: 10.1007/s10875-018-0511-1
25. DeZern AE, Zahurak M, Symons HJ, Cooke KR, Huff CA, Jain T, et al. Alternative donor BMT with posttransplant cyclophosphamide as initial therapy for acquired severe aplastic anemia. Blood. (2023) 141(25):3031–8. doi: 10.1182/blood.2023020435
26. Schubert D, Bode C, Kenefeck R, Hou TZ, Wing JB, Kennedy A, et al. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat Med. (2014) 20(12):1410–6. doi: 10.1038/nm.3746
27. Tesch VK, Abolhassani H, Shadur B, Zobel J, Mareika Y, Sharapova S, et al. Long-term outcome of LRBA deficiency in 76 patients after various treatment modalities as evaluated by the immune deficiency and dysregulation activity (IDDA) score. J Allergy Clin Immunol. (2020) 145(5):1452–63. doi: 10.1016/j.jaci.2019.12.896
28. Lo B, Zhang K, Lu W, Zheng L, Zhang Q, Kanellopoulou C, et al. AUTOIMMUNE DISEASE. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science. (2015) 349(6246):436–40. doi: 10.1126/science.aaa1663
Keywords: Aplastic anemia, inborn error of immunity (IEI), novel variant, CTLA-4, haploinsufficiency, hematopoietic stem cell transplantation (HSCT)
Citation: Hall G, Markle JG, Maiarana J, Martin PL, Rothman JA, Sleasman JW, Lederman H, Azar AE, Brodsky RA and Mousallem T (2024) Case Report: Aplastic anemia related to a novel CTLA4 variant. Front. Pediatr. 12:1434076. doi: 10.3389/fped.2024.1434076
Received: 17 May 2024; Accepted: 24 July 2024;
Published: 16 August 2024.
Edited by:
Jordan Orange, Columbia University, United StatesReviewed by:
Jeffrey J. Bednarski, Washington University in St. Louis, United StatesCopyright: © 2024 Hall, Markle, Maiarana, Martin, Rothman, Sleasman, Lederman, Azar, Brodsky and Mousallem. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Talal Mousallem, dGFsYWwubW91c2FsbGVtQGR1a2UuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.