 Stefanie Kaffai1
Stefanie Kaffai1 Daniela Angelova-Toshkin1Andreas B. Weins2Sonja Ickinger1
Daniela Angelova-Toshkin1Andreas B. Weins2Sonja Ickinger1 Verena Steinke-Lange3Kurt Vollert4
Verena Steinke-Lange3Kurt Vollert4 Michael C. Frühwald1
Michael C. Frühwald1 Michaela Kuhlen1*
Michaela Kuhlen1*
- 1Pediatrics and Adolescent Medicine, Faculty of Medicine, University of Augsburg, Augsburg, Germany
- 2Augsburger Zentrum für Seltene Erkrankungen, University of Augsburg, Augsburg, Germany
- 3MGZ-Medizinisch Genetisches Zentrum, Munich, Germany
- 4Department of Diagnostic and Interventional Radiology, University of Augsburg, Augsburg, Germany
Introduction: Genetic disposition is a major etiologic factor in childhood cancer. More than 100 cancer predisposing syndromes (CPS) are known. Surveillance protocols seek to mitigate morbidity and mortality. To implement recommendations in patient care and to ascertain that the constant gain of knowledge forces its way into practice specific pediatric CPS programs were established.
Patients and methods: We retrospectively analyzed data on children, adolescents, and young adults referred to our pediatric CPS program between October 1, 2021, and March 31, 2023. Follow-up ended on December 31, 2023.
Results: We identified 67 patients (30 male, 36 female, 1 non-binary, median age 9.5 years). Thirty-five patients were referred for CPS surveillance, 32 for features suspicious of a CPS including café-au-lait macules (n = 10), overgrowth (n = 9), other specific symptoms (n = 4), cancer suspicious of a CPS (n = 6), and rare neoplasms (n = 3). CPS was confirmed by clinical criteria in 6 patients and genetic testing in 7 (of 13). In addition, 6 clinically unaffected at-risk relatives were identified carrying a cancer predisposing pathogenic variant. A total of 48 patients were eventually diagnosed with CPS, surveillance recommendations were on record for 45. Of those, 8 patients did not keep their appointments for various reasons. Surveillance revealed neoplasms (n = 2) and metachronous tumors (n = 4) by clinical (n = 2), radiological examination (n = 2), and endoscopy (n = 2). Psychosocial counselling was utilized by 16 (of 45; 35.6%) families.
Conclusions: The diverse pediatric CPSs pose several challenges necessitating interdisciplinary care in specified CPS programs. To ultimately improve outcome including psychosocial well-being joint clinical and research efforts are necessary.
1 Introduction
Genetic disposition is a major etiologic factor in childhood cancer (1–6). Over recent years, there has been increasing awareness and recognition of childhood cancer predisposition syndromes (CPSs) (7). Considerable research efforts have led to substantial progress in the identification of CPSs in children affected by cancer (8–11). Tailored interventions implemented by rational surveillance protocols seek to mitigate morbidity and mortality through early detection and less toxic therapies (12). Most recommendations are based on history and physical examination combined with imaging and biochemical/metabolic studies. In some instances, whole body magnetic resonance imaging is indicated, e.g., in the case of Li-Fraumeni syndrome (LFS) or rhabdoid tumor predisposition syndrome (13–15).
Incidental findings resulting from those studies may lead to additional and potentially rather invasive diagnostic tests (16, 17). Both the anticipation of a particular test and the waiting time to results elicit anxiety (also referred to as “scanxiety”) (18, 19). The phenomenon of scanxiety, however, remains understudied. In addition, specialized centers such as CPS centers, pediatric oncology clinics, and institutes of human genetics are severely under-resourced in psychology support.
Individuals with CPSs carry a significantly increased risk of developing one or more cancers in a metachronous fashion. In 2016, the American Association of Cancer Research sponsored a pediatric cancer predisposition workshop to develop international consensus recommendations for cancer surveillance of children and adolescents for the 50 most common CPSs (12, 20). The multi-disciplinary working group decided that surveillance should be recommended when there is a ≥ 5% risk of developing cancer during the first 20 years of life and when effective screening modalities are available (12). Surveillance recommendations focused on the type(s) of cancer to which the individual is most likely predisposed and the time of greatest risk. For conditions with a cancer risk between 1%–5% surveillance may be indicated on an individual basis.
As more than 100 childhood CPSs are currently known, an abundance of CPS is on record for which no surveillance protocol exists. Many of those CPSs are (very) rare complex diseases with severe coexisting conditions. Information regarding cancer predisposition is not always in place and used to guide management (11).
We previously reported a variety of rare diseases included in the spectrum of CPSs in a tertiary-care children's hospital (21). Given the clinical relevance of a CPS in a child (e.g., surveillance, prevention, treatment, psychologic support, counselling, and identification of relatives at risk), awareness of cancer risk needs to be enhanced (22).
In order to improve tumor surveillance and, thus, care of children and adolescents with CPSs we established a dedicated pediatric CPS program. We here report our initial experiences asking which patients were referred, by whom, and for what reason. We further analyzed CPS diagnoses, surveillance recommendations, adherence to guidelines, and (psychosocial) challenges.
2 Patients and methods
We included all children, adolescents, and young adults referred to our pediatric CPS program at the Swabian Children's Cancer Centre with a proven or suspected CPS between October 1, 2021, and March 31, 2023. Follow-up for this study was completed on 31 December 2023. No exclusion criterion was defined. The study was performed in accordance with the Declaration of Helsinki and Good Clinical Practice and was approved by the ethics committee of the Ludwig Maximilian University of Munich (IRB number 23-0463), Germany.
Patients, parents, and physicians, respectively, were informed about the CPS program by flyers, information sessions, and personal contact. Information in layman's terms was provided on the hospital's homepage. A podcast and a television report about CPS including the program were promoted by the Bavarian Cancer Research Center (BZKF). Via the Augsburg Center for Rare Diseases (AZeSe), self-help groups were informed about the program.
Surveillance included clinical examination and anthropometric measurements in all patients complemented by neurological examination in patients at brain tumor risk. Abdominal ultrasound was performed trimonthly in patients at risk of Wilms tumor, hepatoblastoma, adrenocortical carcinoma, and pheochromocytoma/paraganglioma, neck ultrasound in patients at risk of thyroid carcinoma or paraganglioma. Whole body magnetic resonance imaging (MRI) was performed annually in patients at risk of sarcoma and once in patients with Neurofibromatosis type 1 (NF1) in case the patient did not need anesthesia, brain MRI in patients at risk of brain tumors, and abdominal MRI in patients at risk of pheochromocytoma/paraganglioma. Surveillance included x-ray of the lungs in patients with DICER1 syndrome, and biochemical testing (metanephrines and methoxytyramine) in patients at risk of pheochromocytoma/paraganglioma. No routine measurement of alpha fetoprotein was conducted. Patients with predisposition to leukemia or myelodysplastic syndrome had a complete blood count with manual differential trimonthly and bone marrow evaluation as appropriate. Upper gastroduodenal endoscopy and/or colonoscopy were performed in patients with gastrointestinal cancer syndromes. Biannual ophthalmologic examination was recommended in patients with NF1. Investigations depended on the patient´s age, if applicable the youngest age at cancer onset in affected family members, the underlying pathogenic variant (e.g., brain MRI in Gorlin-Goltz syndrome), and previous findings. The intervals and diagnostics varied age- and CPS-dependent. We reviewed medical reports to assess demographic data, presenting features, patient characteristics, family history, circumstances of referral, suspected diagnosis, diagnostics, recommendations, psychosocial support, and follow-up. Data were retrospectively categorized for referring (sub)specialities, referral reasons including leading presenting features, and circumstances of CPS diagnosis.
3 Results
A total of 67 children and adolescents with suspected or proven CPS were identified with a mean age of 8.4 years [standard deviation, 6.1 years] at initial presentation. Gender ratio showed a small female predominance [36 (53.7%) female, 30 (44.8%) male, 1 (1.5%) non-binary]. As of December 31, 2023, one patient with Noonan syndrome had died of respiratory failure as part of viral infection. Details on demographic data and presenting features at initial presentation are given in Table 1.
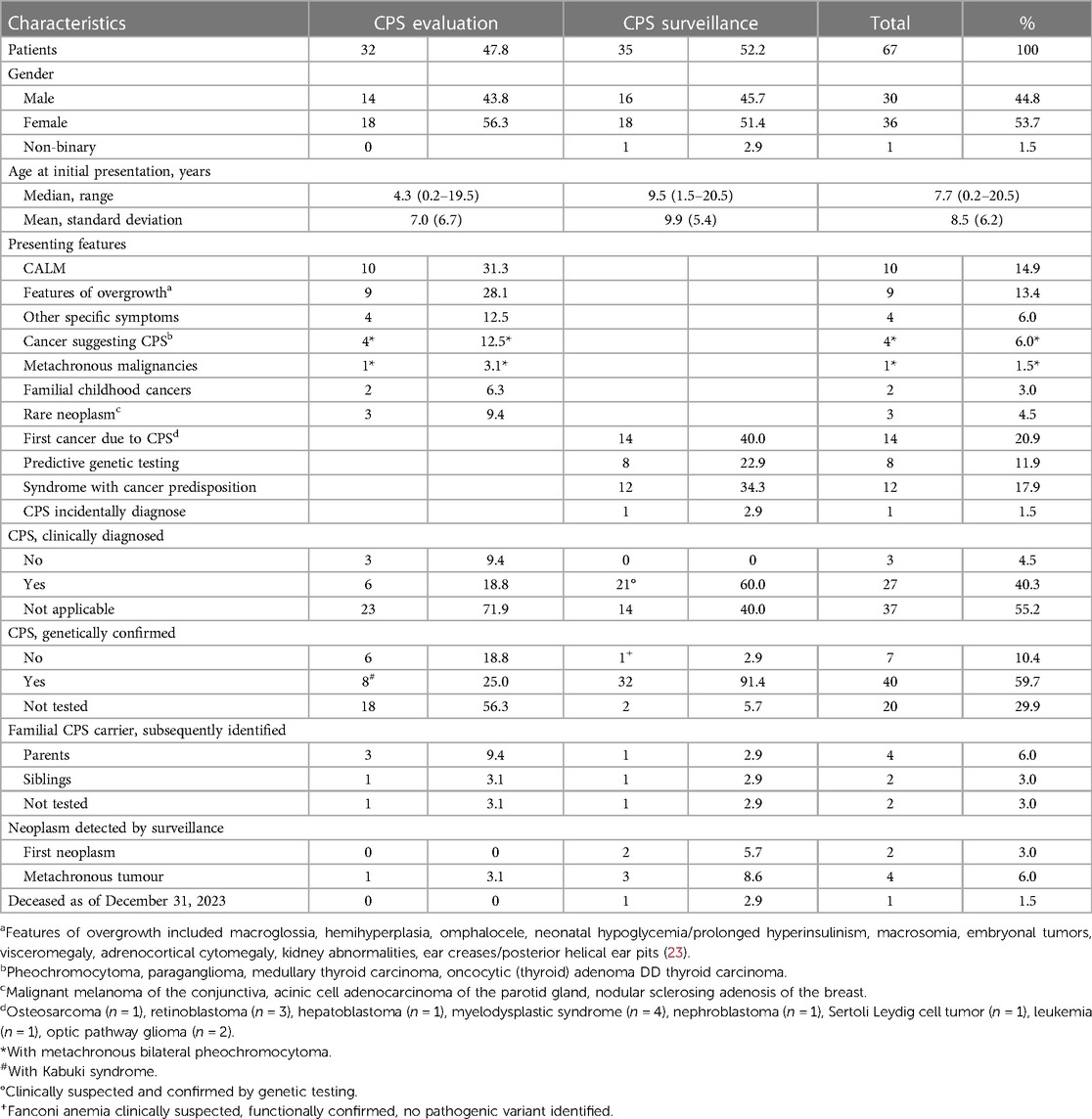
Table 1. Demographic details and presenting features on 67 children and adolescents referred to the specified paediatric CPS program.
Of 67 patients, 21 (31.3%) patients were referred by pediatric oncologists, 9 (13.4%) patients by licensed pediatricians, and 8 (11.9%) patients presented on their individual initiative. Details on referring (sub)specialities are depicted in Figure 1.
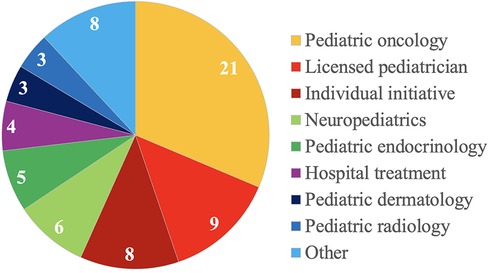
Figure 1. Details on referring specialties of 67 children and adolescents with suspected and proven cancer predisposition syndromes.
In total, 35 (52.2%) patients presented for CPS surveillance, 32 (47.8%) patients for evaluation of an underlying CPS (Figure 2). Of those patients referred for CPS evaluation, 10 (31.3%) patients presented with café-au-lait macules (CALM), 9 (28.1%) patients with suspicion of an overgrowth syndrome, 4 (12.5%) patients with other specific symptoms/congenital anomalies, 4 (12.5%) patients with a childhood cancer highly correlated with specific genetic syndromes and/or molecular analysis suggesting CPS including 1 (3.1%) patient with two metachronous malignancies, 2 (6.2%) siblings with cancer <18 years of age, and 3 (9. 4%) patients with rare neoplasms.
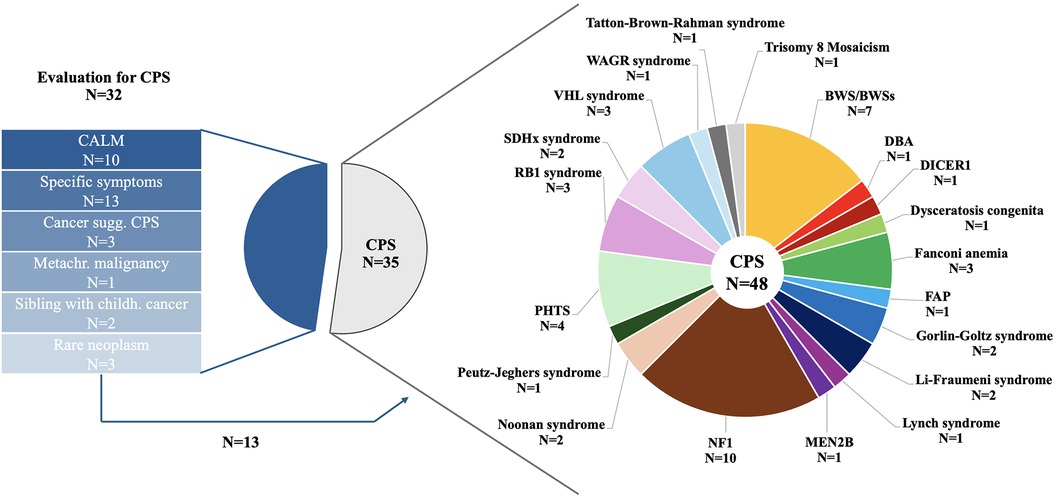
Figure 2. Reasons for referral in 67 children and adolescents presenting to the CPS program and details on CPS diagnosis in 48 patients.
Genetic testing was initiated in 5 of 10 patients with CALM, 3 of 13 with other specific symptoms, 4 of 6 patients with high suspicion of an underlying CPS, both siblings, and none of the 3 patients with rare neoplasms. Genetic testing confirmed CPS diagnosis in 7 (of 13; 53.8%) of those patients and Kabuki syndrome in one patient.
Of 10 patients with CALM, the diagnostic criteria ≥6 CALM >5 mm in greatest diameter in prepubertal individuals and >15 mm in greatest diameter in postpubertal individuals was not fulfilled in 4 patients. One further patient presented with clinical diagnosis of segmental NF1. A total of 6 patients with suspicion of an overgrowth syndrome met diagnostic criteria, 3 patients did not show signs of an overgrowth syndrome.
Of 35 patients referred for CPS surveillance, 14 (40.0%) patients were diagnosed after first cancer onset, 8 (22.9%) patients had undergone predictive genetic testing because one parent carried a pathogenic variant (PV) in a cancer predisposing gene (CPG; PTCH1 n = 2, VHL n = 2, SDHC n = 1, PTEN n = 3) with risk of cancer onset during childhood, 12 (34.3%) patients were referred for an underlying syndrome with increased cancer risk, and 1 (2.9%) patient was incidentally diagnosed with trisomy 8 mosaicism.
In total, 6 other so far unaffected relatives carrying the CPG PV were subsequently identified in LFS (parent, adult sibling), Lynch syndrome (parent), DICER1 syndrome (parent), and PTEN hamartoma tumor syndrome (PHTS; parent, sibling). One parent was subsequently diagnosed with cancer. In two other patients (LFS, PHTS), parents refused genetic testing of themselves. These data highlight the importance of genetic counselling and testing.
Investigating these 48 patients with CPS in more detail, surveillance protocols were available for 45 (93.8%) including 2 with Noonan syndrome, for which surveillance is currently not recommended. Surveillance protocols were not in place for patients with pediatric Lynch syndrome, Tatton-Brown-Rahman syndrome (TBRS), and hematopoietic trisomy 8 mosaicism. The latter patient presented with thrombocytopenia and increased mean corpuscular volume and was therefore included in the CPS program. The parents of one patient with Noonan syndrome and TBRS explicitly wished to be included in the CPS program.
A total of 45 patients were eventually included into our dedicated CPS program. One patient with LFS did not present for follow-up due to parental refusal, 8 patients including 5 adolescents and young adults only sporadically kept their appointments.
Psychosocial counselling was offered to all patients included in the CPS program; 16 (35.6%) families made use of this support for psychological (n = 2), social (n = 9), and psychosocial (n = 5) reasons. Support was provided to parents (n = 12) and families (n = 5) at a median of 2.5 times (range, 1–20).
During the study period, surveillance revealed one first neoplasia by clinical suspicion (highly suspicious vision in clinical examination through fun and games in a toddler with NF1, subsequently confirmed as opticus glioma by MR imaging) and a further one by radiological examination (suspicion of thyroid carcinoma by neck ultrasound; PHTS). Three metachronous tumors were identified by radiological examination (n = 1; follicular-patterned thyroid tumor in DICER1 syndrome) and endoscopy (n = 2; colon cancer in Familial Adenomatous Polyposis, carcinoma in situ in Peutz-Jeghers syndrome). In addition, in one patient with PHTS, a mass in the foot was detected by clinical examination and subsequently confirmed as arteriovenous malformation by radiological examination and pathological evaluation.
4 Discussion
We report our experiences of a newly established program specifically dedicated towards children, adolescents, and their families affected by cancer predisposition at a tertiary care children's hospital in Germany over a 1.5-years-period. Of 67 children and adolescents, 48 (71.6%) patients were diagnosed with a CPS, whereas the diagnosis of a CPS was not confirmed in 19 (28.4%) patients.
As might be expected, most patients were referred by pediatric oncologists (21, 24). However, 13.4% of patients were referred by licensed pediatricians, 11.9% presented on the parents’ initiative. It remains difficult to calculate numbers of children and adolescents affected by one of the various CPS and, thus, to estimate “real numbers” of children and adolescents living with a CPS in our catchment area (24). To raise awareness for children and adolescents with CPS, we provided information on our CPS program at various levels (21). And indeed, most licensed pediatricians and in-house pediatric subspecialists referred patients for that reason. It must be assumed, that there is still a significant number of patients who do not present to our CPS program yet.
Evaluation for an underlying CPS was mostly initiated for CALM and suspicion of overgrowth. Diagnostic criteria of NF1 were established by the National Institute of Health (NIH) in 1987 and revised in 2021 by Legius et al. (25). The NIH criteria relied on clinical features mainly not being present in early infancy although surveillance should be initiated at birth and diagnosis of NF1, respectively. In addition, opticus glioma manifest at a median age of 5 years whereas most diagnostic criteria only later in life (26). We previously demonstrated earlier diagnosis of NF1 by applying the revised diagnostic criteria, in particular by performing genetic testing in infants not fulfilling diagnostic criteria hitherto (27). We thus initiated genetic testing in 5 infants fulfilling the criterion `CALM´ as defined previously (25) or presenting with otherwise suspicious CALM, e.g., constitutional mismatch repair deficiency (28), and arranged re-evaluation in some young patients presenting with CALM not yet meeting the diagnostic criterion of NF1.
Overgrowth syndromes are associated with cancer onset in infancy (29–32). Thus, starting surveillance as early as possible is of crucial importance (29, 33). Recommendations for surveillance in the several overgrowth syndromes vary substantially including advice for genetic testing (33–35). We decided upon inclusion in our CPS program on clinical criteria and performed genetic testing on an individual basis after careful discussion with both caregivers.
Of 48 patients with CPS, this was only diagnosed after first cancer onset in 40% of patients including three patients with otherwise typical signs and symptoms (Multiple Endocrine Neoplasia type 2B, PHTS, Fanconi anemia) (36–38). In addition, in both PHTS families other family members presented with symptoms of PHTS before (37). This once more highlights that more awareness to and knowledge about the various CPS is still needed among pediatricians and other experts (21).
Of 8 patients diagnosed by predictive testing, 2 patients each with von Hippel-Lindau (VHL) syndrome and Gorlin-Goltz syndrome had previously been cared for by non-CPS specialists. The siblings with Gorlin-Goltz syndrome due to a PV in PTCH1 underwent sequential cranial MR imaging in anesthesia though this is recommended for children with PVs in SUFU only (39, 40). In the siblings with VHL syndrome, biochemical testing had comprised catecholamines instead of metanephrines and 3-methoxytyramine (41–43). This impressively illustrates the need for specialized CPS care.
By referring at-risk relatives for genetic testing, we additionally identified 6 so far unaffected family members. One parent subsequently felt ill with cancer underlining the importance of secondary genetic testing of at-risk relatives (17, 44).
Management strategies encompass regular clinical examination, biochemical testing, radiological examination including MR imaging, and endoscopy for early detection of neoplasms (14, 15, 33, 41, 45–50). This requires both extensive human and instrument-based resources. However, lack of insurance coverage limits its availability. By clinical and radiological assessment, we identified 6 patients with new neoplastic manifestations during the study period. This highlights the potential of regular surveillance (51, 52).
We identified 8 patients not regularly keeping their appointments for various reasons. To what extent the phenomenon of “scanxiety” may be accountable is beyond the scope of this study. However, coping strategies and specialized psychosocial support are essential to individuals carrying a cancer predisposing PV (53–55). On the one hand undergoing tailored surveillance for an underlying CPS can lead to increased feelings of control and security (56, 57). On the other hand, it may prompt uncertainties while waiting for the test results and cause worries to the reminder of cancer risk as well as the practical aspects of screening (57).
Beyond that, genetic testing itself represents a major life event while the implications of testing might not be fully understood at the time of decision-making (58, 59). In addition, individuals may experience distress or clinically significant anxiety, especially those who have lost a close relative to the CPS (60, 61).
Survey instruments for psychosocial aspects of pediatric CPS are not available. In addition, psychosocial support within the frame of dedicated pediatric CPS programs is not covered by health insurance funds in Germany to date. Considering the number of individuals in our study who made use of psychosocial counselling, did not keep their surveillance appointments, or refused genetic testing, there are several psychosocial shortcomings which urgently need to be addressed in the future.
5 Conclusions
Pediatric CPSs include several challenges and opportunities necessitating interdisciplinary care in dedicated CPS programs. The multifaceted features of CPSs manifesting in childhood and adolescence merit increased awareness to facilitate identifying patients and at-risk relatives as early as possible. To ultimately improve outcome and psychosocial well-being of affected families joint clinical and research efforts—accompanied by education programs for patients, relatives, and physicians—are necessary.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Ludwig Maximilian 51 University of Munich (IRB 23-0463), Germany. The studies were conducted in accordance with the local legislation and institutional requirements. The ethics committee/institutional review board waived the requirement of written informed consent for participation from the participants or the participants’ legal guardians/next of kin because as a retrospective study according to Bavarian Hospital Law written informed consent was not necessary.
Author contributions
SK: Formal Analysis, Visualization, Writing – original draft. DA-T: Investigation, Writing – review & editing. AW: Investigation, Writing – review & editing. SI: Investigation, Writing – review & editing. VS-L: Investigation, Writing – review & editing. KV: Investigation, Writing – review & editing. MF: Investigation, Resources, Writing – review & editing. MK: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article.
The research of MK on pediatric cancer predisposition is supported by Deutsche Krebshilfe (70115888), Deutsche Forschungsgemeinschaft (KU3764/3-1), and intramurale Forschungsförderung, University of Augsburg. MF's research on rhabdoid tumor disposition is supported by Deutsche Krebshilfe 70113981.
Acknowledgments
Parts of this work were done within the context of the scientific traineeship of the University of Augsburg and as part of the doctoral thesis of SK. This work was partly done within the Zentrum für Kinder mit Tumordispositionssyndromen (ZeKiTDS) and the Augsburger Zentrum für Seltene Erkrankungen, University of Augsburg (AZeSe). VS-L is a member of the European Reference Network on Genetic Tumor Risk Syndromes (ERN GENTURIS).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Zhang J, Walsh MF, Wu G, Edmonson MN, Gruber TA, Easton J, et al. Germline mutations in predisposition genes in pediatric cancer. N Engl J Med. (2015) 373:2336–46. doi: 10.1056/NEJMoa1508054
2. Wagener R, Taeubner J, Walter C, Yasin L, Alzoubi D, Bartenhagen C, et al. Comprehensive germline-genomic and clinical profiling in 160 unselected children and adolescents with cancer. Eur J Hum Genet. (2021) 29:1301–11. doi: 10.1038/s41431-021-00878-x
3. Grobner SN, Worst BC, Weischenfeldt J, Buchhalter I, Kleinheinz K, Rudneva VA, et al. The landscape of genomic alterations across childhood cancers. Nature. (2018) 555:321–7. doi: 10.1038/nature25480
4. Diets IJ, Waanders E, Ligtenberg MJ, van Bladel DAG, Kamping EJ, Hoogerbrugge PM, et al. High yield of pathogenic germline mutations causative or likely causative of the cancer phenotype in selected children with cancer. Clin Cancer Res. (2018) 24:1594–603. doi: 10.1158/1078-0432.CCR-17-1725
5. Akhavanfard S, Padmanabhan R, Yehia L, Cheng F, Eng C. Comprehensive germline genomic profiles of children, adolescents and young adults with solid tumors. Nat Commun. (2020) 11:2206. doi: 10.1038/s41467-020-16067-1
6. Byrjalsen A, Hansen TVO, Stoltze UK, Mehrjouy MM, Barnkob NM, Hjalgrim LL, et al. Nationwide germline whole genome sequencing of 198 consecutive pediatric cancer patients reveals a high incidence of cancer prone syndromes. PLoS Genet. (2020) 16:e1009231. doi: 10.1371/journal.pgen.1009231
7. McGee RB, Oak N, Harrison L, Xu K, Nuccio R, Blake AK, et al. Pathogenic variants in adult-onset cancer predisposition genes in pediatric cancer: prevalence and impact on tumor molecular features and clinical management. Clin Cancer Res. (2023) 29(7):1243–51. doi: 10.1158/1078-0432.CCR-22-2482
8. Jongmans MC, Loeffen JL, Waanders E, Hoogerbrugge PM, Ligtenberg MJ, Kuiper RP, et al. Recognition of genetic predisposition in pediatric cancer patients: an easy-to-use selection tool. Eur J Med Genet. (2016) 59:116–25. doi: 10.1016/j.ejmg.2016.01.008
9. Kuhlen M, Wieczorek D, Siebert R, Fruhwald MC. How I approach hereditary cancer predisposition in a child with cancer. Pediatr Blood Cancer. (2019) 66:e27916. doi: 10.1002/pbc.27916
10. Ripperger T, Bielack SS, Borkhardt A, Brecht IB, Burkhardt B, Calaminus G, et al. Childhood cancer predisposition syndromes-a concise review and recommendations by the cancer predisposition working group of the society for pediatric oncology and hematology. Am J Med Genet A. (2017) 173:1017–37. doi: 10.1002/ajmg.a.38142
11. Goudie C, Coltin H, Witkowski L, Mourad S, Malkin D, Foulkes WD. The McGill interactive pediatric OncoGenetic guidelines: an approach to identifying pediatric oncology patients most likely to benefit from a genetic evaluation. Pediatr Blood Cancer. (2017) 64:e26441. doi: 10.1002/pbc.26441
12. Brodeur GM, Nichols KE, Plon SE, Schiffman JD, Malkin D. Pediatric cancer predisposition and surveillance: an overview, and a tribute to Alfred G. Knudson Jr. Clin Cancer Res. (2017) 23:e1–5. doi: 10.1158/1078-0432.CCR-17-0702
13. Fruhwald MC, Nemes K, Boztug H, Cornips MCA, Evans DG, Farah R, et al. Current recommendations for clinical surveillance and genetic testing in rhabdoid tumor predisposition: a report from the SIOPE host genome working group. Fam Cancer. (2021) 20:305–16. doi: 10.1007/s10689-021-00229-1
14. Kratz CP, Achatz MI, Brugieres L, Frebourg T, Garber JE, Greer MC, et al. Cancer screening recommendations for individuals with Li-Fraumeni syndrome. Clin Cancer Res. (2017) 23:e38–45. doi: 10.1158/1078-0432.CCR-17-0408
15. Greer MC, Voss SD, States LJ. Pediatric cancer predisposition imaging: focus on whole-body MRI. Clin Cancer Res. (2017) 23:e6–e13. doi: 10.1158/1078-0432.CCR-17-0515
16. Kuhlen M, Borkhardt A. Trio sequencing in pediatric cancer and clinical implications. EMBO Mol Med. (2018) 10:e8641. doi: 10.15252/emmm.201708641
17. Druker H, Zelley K, McGee RB, Scollon SR, Kohlmann WK, Schneider KA, et al. Genetic counselor recommendations for cancer predisposition evaluation and surveillance in the pediatric oncology patient. Clin Cancer Res. (2017) 23:e91–7. doi: 10.1158/1078-0432.CCR-17-0834
18. Derry-Vick HM, Heathcote LC, Glesby N, Stribling J, Luebke M, Epstein AS, et al. Scanxiety among adults with cancer: a scoping review to guide research and interventions. Cancers (Basel). (2023) 15(5):1381. doi: 10.3390/cancers15051381
19. Mindrup R. Imprints of “scanxiety”. AMA J Ethics. (2022) 24:E685–693. doi: 10.1001/amajethics.2022.685
20. Malkin D, Nichols KE, Schiffman JD, Plon SE, Brodeur GM. The future of surveillance in the context of cancer predisposition: through the murky looking glass. Clin Cancer Res. (2017) 23:e133–7. doi: 10.1158/1078-0432.CCR-17-2026
21. Huber S, Schimmel M, Dunstheimer D, Nemes K, Richter M, Streble J, et al. The need for tumor surveillance of children and adolescents with cancer predisposition syndromes: a retrospective cohort study in a tertiary-care children’s hospital. Eur J Pediatr. (2022) 181:1585–96. doi: 10.1007/s00431-021-04347-x
22. Chen C, Qin N, Wang M, Dong Q, Tithi SS, Hui Y, et al. Cancer germline predisposing variants and late mortality from subsequent malignant neoplasms among long-term childhood cancer survivors: a report from the St Jude lifetime cohort and the childhood cancer survivor study. Lancet Oncol. (2023) 24:1147–56. doi: 10.1016/S1470-2045(23)00403-5
23. Shuman C, Kalish JM, Weksberg R. Beckwith-Wiedemann syndrome. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews®. Seattle, WA: University of Washington, Seattle (1993–2024).
24. Dutzmann CM, Palmaers NE, Muntnich LJ, Struwe FJ, Penkert J, Sanger B, et al. Research on rare diseases in Germany—the cancer predisposition syndrome registry. J Health Monit. (2023) 8:17–23. doi: 10.25646/11828
25. Legius E, Messiaen L, Wolkenstein P, Pancza P, Avery RA, Berman Y, et al. Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome: an international consensus recommendation. Genet Med. (2021) 23:1506–13. doi: 10.1038/s41436-021-01170-5
26. Hernaiz Driever P, von Hornstein S, Pietsch T, Kortmann R, Warmuth-Metz M, Emser A, et al. Natural history and management of low-grade glioma in NF-1 children. J Neurooncol. (2010) 100:199–207. doi: 10.1007/s11060-010-0159-z
27. Angelova-Toshkina D, Holzapfel J, Huber S, Schimmel M, Wieczorek D, Gnekow AK, et al. Neurofibromatosis type 1: a comparison of the 1997 NIH and the 2021 revised diagnostic criteria in 75 children and adolescents. Genet Med. (2022) 24:1978–85. doi: 10.1016/j.gim.2022.05.013
28. Suerink M, Ripperger T, Messiaen L, Menko FH, Bourdeaut F, Colas C, et al. Constitutional mismatch repair deficiency as a differential diagnosis of neurofibromatosis type 1: consensus guidelines for testing a child without malignancy. J Med Genet. (2019) 56:53–62. doi: 10.1136/jmedgenet-2018-105664
29. Connolly GK, Harris RD, Shumate C, Rednam SP, Canfield MA, Plon SE, et al. Pediatric cancer incidence among individuals with overgrowth syndromes and overgrowth features: a population-based assessment in seven million children. Cancer. (2024) 130:467–75. doi: 10.1002/cncr.35041
30. Hol JA, Kuiper RP, van Dijk F, Waanders E, van Peer SE, Koudijs MJ, et al. Prevalence of (epi)genetic predisposing factors in a 5-year unselected national Wilms tumor cohort: a comprehensive clinical and genomic characterization. J Clin Oncol. (2022) 40:1892–902. doi: 10.1200/JCO.21.02510
31. Duffy KA, Getz KD, Hathaway ER, Byrne ME, MacFarland SP, Kalish JM. Characteristics associated with tumor development in individuals diagnosed with beckwith-wiedemann spectrum: novel tumor-(epi)genotype-phenotype associations in the BWSp population. Genes (Basel). (2021) 12(11):1839. doi: 10.3390/genes12111839
32. Coktu S, Spix C, Kaiser M, Beygo J, Kleinle S, Bachmann N, et al. Cancer incidence and spectrum among children with genetically confirmed Beckwith-Wiedemann spectrum in Germany: a retrospective cohort study. Br J Cancer. (2020) 123:619–23. doi: 10.1038/s41416-020-0911-x
33. Kalish JM, Doros L, Helman LJ, Hennekam RC, Kuiper RP, Maas SM, et al. Surveillance recommendations for children with overgrowth syndromes and predisposition to Wilms tumors and hepatoblastoma. Clin Cancer Res. (2017) 23:e115–22. doi: 10.1158/1078-0432.CCR-17-0710
34. Klein SD, Nisbet AF, Hathaway ER, Kalish JM. Simpson-Golabi-Behmel syndrome type 1. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews®. Seattle, WA: University of Washington, Seattle (1993–2024).
35. Brioude F, Toutain A, Giabicani E, Cottereau E, Cormier-Daire V, Netchine I. Overgrowth syndromes—clinical and molecular aspects and tumour risk. Nat Rev Endocrinol. (2019) 15:299–311. doi: 10.1038/s41574-019-0180-z
36. Redlich A, Lessel L, Petrou A, Mier P, Vorwerk P. Multiple endocrine neoplasia type 2B: frequency of physical stigmata-results of the GPOH-MET registry. Pediatr Blood Cancer. (2020) 67:e28056. doi: 10.1002/pbc.28056
37. Plamper M, Gohlke B, Woelfle J. PTEN Hamartoma tumor syndrome in childhood and adolescence-a comprehensive review and presentation of the German pediatric guideline. Mol Cell Pediatr. (2022) 9:3. doi: 10.1186/s40348-022-00135-1
38. Giampietro PF, Adler-Brecher B, Verlander PC, Pavlakis SG, Davis JG, Auerbach AD. The need for more accurate and timely diagnosis in Fanconi anemia: a report from the international Fanconi Anemia registry. Pediatrics. (1993) 91:1116–20. doi: 10.1542/peds.91.6.1116
39. Foulkes WD, Kamihara J, Evans DGR, Brugieres L, Bourdeaut F, Molenaar JJ, et al. Cancer surveillance in Gorlin syndrome and rhabdoid tumor predisposition syndrome. Clin Cancer Res. (2017) 23:e62–7. doi: 10.1158/1078-0432.CCR-17-0595
40. Waszak SM, Northcott PA, Buchhalter I, Robinson GW, Sutter C, Groebner S, et al. Spectrum and prevalence of genetic predisposition in medulloblastoma: a retrospective genetic study and prospective validation in a clinical trial cohort. Lancet Oncol. (2018) 19:785–98. doi: 10.1016/S1470-2045(18)30242-0
41. Rednam SP, Erez A, Druker H, Janeway KA, Kamihara J, Kohlmann WK, et al. Von Hippel-Lindau and hereditary pheochromocytoma/paraganglioma syndromes: clinical features, genetics, and surveillance recommendations in childhood. Clin Cancer Res. (2017) 23:e68–75. doi: 10.1158/1078-0432.CCR-17-0547
42. Eisenhofer G, Deutschbein T, Constantinescu G, Langton K, Pamporaki C, Calsina B, et al. Plasma metanephrines and prospective prediction of tumor location, size and mutation type in patients with pheochromocytoma and paraganglioma. Clin Chem Lab Med. (2020) 59:353–63. doi: 10.1515/cclm-2020-0904
43. Rao JU, Engelke UF, Sweep FC, Pacak K, Kusters B, Goudswaard AG, et al. Genotype-specific differences in the tumor metabolite profile of pheochromocytoma and paraganglioma using untargeted and targeted metabolomics. J Clin Endocrinol Metab. (2015) 100:E214–22. doi: 10.1210/jc.2014-2138
44. Brozou T, Taeubner J, Velleuer E, Dugas M, Wieczorek D, Borkhardt A, et al. Genetic predisposition in children with cancer—affected families’ acceptance of trio-WES. Eur J Pediatr. (2018) 177:53–60. doi: 10.1007/s00431-017-2997-6
45. Achatz MI, Porter CC, Brugieres L, Druker H, Frebourg T, Foulkes WD, et al. Cancer screening recommendations and clinical management of inherited gastrointestinal cancer syndromes in childhood. Clin Cancer Res. (2017) 23:e107–14. doi: 10.1158/1078-0432.CCR-17-0790
46. Kamihara J, Bourdeaut F, Foulkes WD, Molenaar JJ, Mosse YP, Nakagawara A, et al. Retinoblastoma and neuroblastoma predisposition and surveillance. Clin Cancer Res. (2017) 23:e98–e106. doi: 10.1158/1078-0432.CCR-17-0652
47. Porter CC, Druley TE, Erez A, Kuiper RP, Onel K, Schiffman JD, et al. Recommendations for surveillance for children with leukemia-predisposing conditions. Clin Cancer Res. (2017) 23:e14–22. doi: 10.1158/1078-0432.CCR-17-0428
48. Tabori U, Hansford JR, Achatz MI, Kratz CP, Plon SE, Frebourg T, et al. Clinical management and tumor surveillance recommendations of inherited mismatch repair deficiency in childhood. Clin Cancer Res. (2017) 23:e32–7. doi: 10.1158/1078-0432.CCR-17-0574
49. Villani A, Greer MC, Kalish JM, Nakagawara A, Nathanson KL, Pajtler KW, et al. Recommendations for cancer surveillance in individuals with RASopathies and other rare genetic conditions with increased cancer risk. Clin Cancer Res. (2017) 23:e83–90. doi: 10.1158/1078-0432.CCR-17-0631
50. Wasserman JD, Tomlinson GE, Druker H, Kamihara J, Kohlmann WK, Kratz CP, et al. Multiple endocrine neoplasia and hyperparathyroid-jaw tumor syndromes: clinical features, genetics, and surveillance recommendations in childhood. Clin Cancer Res. (2017) 23:e123–32. doi: 10.1158/1078-0432.CCR-17-0548
51. Kratz CP, Villani A, Nichols KE, Schiffman J, Malkin D. Cancer surveillance for individuals with Li-Fraumeni syndrome. Eur J Hum Genet. (2020) 28:1481–2. doi: 10.1038/s41431-020-00709-5
52. Ghorbanoghli Z, van Kouwen M, Versluys B, Bonnet D, Devalck C, Tinat J, et al. High yield of surveillance in patients diagnosed with constitutional mismatch repair deficiency. J Med Genet. (2023) 60:679–84. doi: 10.1136/jmg-2022-108829
53. Wilharm C, Pralong A, Weiss M, Hallek M, Voltz R, Tuchscherer A, et al. Facing life-threat during youth: a qualitative study on challenges, coping, and needs among adolescents and young adults with cancer. Support Care Cancer. (2024) 32:179. doi: 10.1007/s00520-024-08370-0
54. Rowland E, Metcalfe A. Communicating inherited genetic risk between parent and child: a meta-thematic synthesis. Int J Nurs Stud. (2013) 50:870–80. doi: 10.1016/j.ijnurstu.2012.09.002
55. Kiermeier S, Schott S, Nees J, Dutzmann C, Struwe F, Kratz CP, et al. Health-related quality of life and fear of progression in individuals with Li-Fraumeni syndrome. J Genet Couns. (2024). doi: 10.1002/jgc4.1859
56. Lammens CR, Bleiker EM, Aaronson NK, Wagner A, Sijmons RH, Ausems MG, et al. Regular surveillance for Li-Fraumeni syndrome: advice, adherence and perceived benefits. Fam Cancer. (2010) 9:647–54. doi: 10.1007/s10689-010-9368-z
57. van Engelen K, Barrera M, Wasserman JD, Armel SR, Chitayat D, Druker H, et al. Tumor surveillance for children and adolescents with cancer predisposition syndromes: the psychosocial impact reported by adolescents and caregivers. Pediatr Blood Cancer. (2021) 68:e29021. doi: 10.1002/pbc.29021
58. Weber E, Shuman C, Wasserman JD, Barrera M, Patenaude AF, Fung K, et al. A change in perspective": exploring the experiences of adolescents with hereditary tumor predisposition. Pediatr Blood Cancer. (2019) 66:e27445. doi: 10.1002/pbc.27445
59. Duncan RE, Gillam L, Savulescu J, Williamson R, Rogers JG, Delatycki MB. The challenge of developmentally appropriate care: predictive genetic testing in young people for familial adenomatous polyposis. Fam Cancer. (2010) 9:27–35. doi: 10.1007/s10689-009-9294-0
60. Lammens CR, Aaronson NK, Wagner A, Sijmons RH, Ausems MG, Vriends AH, et al. Genetic testing in Li-Fraumeni syndrome: uptake and psychosocial consequences. J Clin Oncol. (2010) 28:3008–14. doi: 10.1200/JCO.2009.27.2112
Keywords: predisposition, children, surveillance, preventive testing, cancer, hereditary
Citation: Kaffai S, Angelova-Toshkin D, Weins AB, Ickinger S, Steinke-Lange V, Vollert K, Frühwald MC and Kuhlen M (2024) Cancer predisposing syndromes in childhood and adolescence pose several challenges necessitating interdisciplinary care in dedicated programs. Front. Pediatr. 12:1410061. doi: 10.3389/fped.2024.1410061
Received: 31 March 2024; Accepted: 20 May 2024;
Published: 3 June 2024.
Edited by:
Giovana Tardin Torrezan, A.C.Camargo Cancer Center, BrazilReviewed by:
Angela Mastronuzzi, Bambino Gesù Children’s Hospital (IRCCS), ItalyLisa States, Children’s Hospital of Philadelphia, United States
© 2024 Kaffai, Angelova-Toshkin, Weins, Ickinger, Steinke-Lange, Vollert, Frühwald and Kuhlen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michaela Kuhlen, bWljaGFlbGEua3VobGVuQHVrLWF1Z3NidXJnLmRl