 Layla M. Jeries
Layla M. Jeries Tatyana A. Sysoeva
Tatyana A. Sysoeva Lisa Karstens
Lisa Karstens Maryellen S. Kelly
Maryellen S. Kelly- 1Department of Biological Sciences, The University of Alabama in Huntsville, Huntsville, AL, United States
- 2Department of Medical Informatics and Clinical Epidemiology, Oregon Health & Science University, Portland, OR, United States
- 3Department of Obstetrics and Gynecology, Oregon Health & Science University, Portland, OR, United States
- 4Division of Healthcare of Women and Children, School of Nursing, Duke University, Durham, NC, United States
- 5Department of Urology, Duke University Hospital, Durham, NC, United States
The human urinary bladder hosts a complex microbial community of low biomass referred to as the urobiome. While the composition of the urobiome has been investigated in adults for over a decade now, only a few studies have considered the presence and composition of the urobiome in children. It is critical to explore how the urobiome develops throughout the life span and how it changes in the presence of various health conditions. Therefore, we set to review the available data on pediatric urobiome composition and its development with age and disease. In addition, we focused on identifying and reporting specific gaps in our knowledge of the pediatric urobiome that we hope will be addressed by future studies in this swiftly developing field with fast-improving methods and consensus.
1 Introduction
The urinary microbiome, or urobiome, comprises the microbial community of the urinary tract, which includes the kidneys, ureters, bladder, and urethra. Standard urine culture (SUC) long supported the paradigm that urine and the urinary tract were sterile except in the case of infection (1). However, the development of expanded quantitative urine culture (EQUC) and culture-independent 16S rRNA gene sequencing have allowed the detection and characterization of primarily bacteria from urine samples (2, 3). As with other microbial niches of the human body, the urobiome has been suggested to contribute to both urinary health and disease. Elevated or reduced populations of microorganisms that can potentially lead to dysbiosis have been associated with urologic diseases, such as prostate cancer, bladder cancer, chronic prostatitis/chronic pelvic pain syndrome, urge urinary incontinence, overactive bladder, stone disease, and urinary tract infection (UTI) [reviewed in (4)].
The last decade has seen an increase in the study of the urobiome, examining cohorts that include one or both sexes, as well as various infectious states and pre-existing conditions. The urobiome varies greatly between individuals and is expected to be uniquely defined, yet dynamic, within an individual across time, as has been found in a study of premenopausal women (5). Several bacterial taxa can be found in the urobiomes of adult males and females, such as Streptococcus, Staphylococcus, and Corynebacterium (6–9). However, certain bacteria tend to be predominant more frequently within a specific sex, such as lactobacilli, which tend to be predominant bacteria in women (10, 11) but not in men, and Staphylococcus which is sometimes a predominant bacterium in males (12–14), but less often in females. Importantly, the composition of the urobiome can vary by sample collection technique, as it dictates which niche bacteria are collected from. Catheter and suprapubic aspirates sample the bladder directly with little contribution from surrounding areas (2), whereas voided samples typically include bacteria from the genitals. Thus, the former are referred to as bladder microbiome and the latter as urogenital microbiome (15).
Questions remain regarding the development of the urobiome in childhood across age, sex, toilet-training status, and pubertal stage, as well as how urologic and non-urologic diseases might be affecting urobiome composition and abundance. How the urobiome develops during childhood and how this may contribute to disease states or promote health is unknown. This review aims to summarize the current data regarding urobiome composition of different pediatric cohorts. We examine the implications of these results for the health and disease of the urinary tract and discuss how the pediatric cohort influences study design. Finally, we highlight the next steps necessary to further discern the dynamics of the pediatric urobiome.
2 Methods employed in pediatric urobiome studies
Several reviews have summarized important considerations for designing and understanding urobiome studies (15–17). There are numerous variables and factors of interest that can affect urobiome composition and abundance (Figure 1). For example, differences in the urobiome based on sex and age have been observed in adult cohorts, and this question persists in pediatric cohorts, especially in regard to development from birth into childhood, adolescence, and young adulthood. It is also important to consider whether disease, antibiotic exposure, and other environmental/regional factors have influence. There is also the question of whether collection methods and downstream processing of samples can further determine the resolution and accuracy with which these measures are detected. Specific features of pediatric cohorts limit the use of some techniques and require additional considerations. Herein, we highlight those unique features in the context of typical urobiome study and identify current trends in pediatric urobiome study design.

Figure 1 Summary of variables affecting observed urobiome composition.
2.1 Sample collection
Combining the nature of urine being a low biomass sample (15, 16) with the smaller bladder capacity of children compared to adults means sample collection must be carefully considered in pediatric cohorts. The two most common techniques for obtaining urine samples in pediatric and adult urobiome studies are clean-catch midstream urine (CCMSU) and transurethral catheterization (TUC). Suprapubic aspiration is another sampling method in adult urobiome studies and is considered to be the “cleanest” method of sampling (2), but it has not yet been employed in pediatric cohorts due to its high degree of invasiveness and ethical considerations. Across studies presented in this review (Table 1), there is an almost equal share of either CCMSU or TUC collection methods used to sample the urobiome.
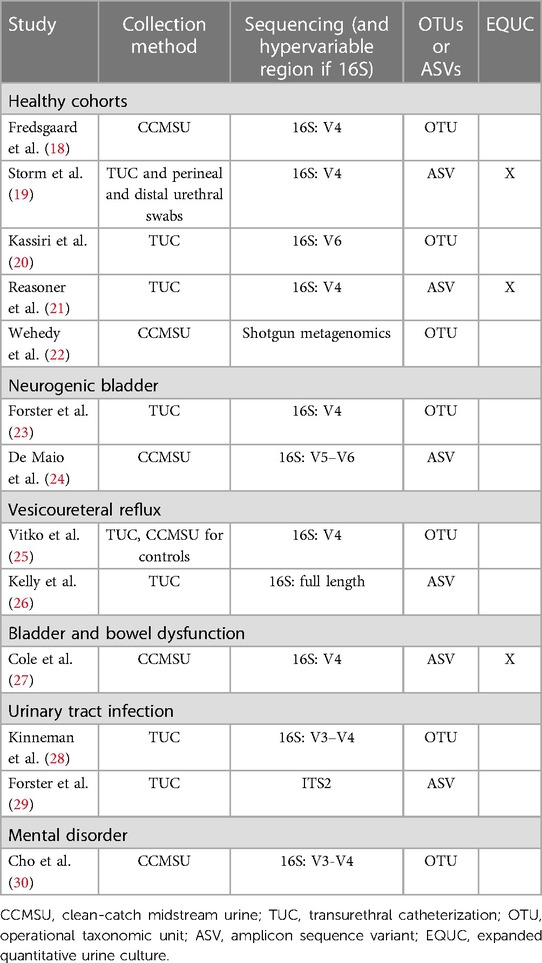
Table 1 Summary of the highlighted methods employed across studies of the pediatric urobiome.
CCMSU is the least invasive method of collecting urobiome samples but it is prone to collecting some microbes from the surrounding urogenital microbiomes, which Storm and colleagues observed to differ from urethral urobiomes (19). Possible skin and adjacent niche contributions, such as from the vagina and anus, can be accounted for via swabbing and culturing or sequencing of the surrounding urogenital regions. Another limiting factor of the CCMSU method is that children must be able to volitionally void. So, this method potentially excludes younger children and infants whose samples could provide greater insight into the acquisition and early-life development of the urobiome. Similarly, younger children also require assistance providing samples and/or can potentially contaminate samples. While, ultimately, CCMSU contains what is more appropriately described as a urogenital microbiome sample than of the urinary tract microbiome (15), it is often the most feasible option in pediatric cohorts.
TUC requires the insertion of a urinary catheter into the bladder. While this allows for sampling of the bladder and opens cohorts to virtually all ages, TUC is much harder to justify ethically for use in healthy children as this carries the risk of infection, inflammation and injury, and can cause undue stress and pain to a child. The studies that have employed this method take advantage of TUC already being ordered for other procedures, such as a voiding cystourethrogram, surgery, or in children who perform catheterization regularly due to urinary retention. While TUC is also prone to possible contribution from surrounding skin during catheter insertion, particularly in studies of uncircumcised males, this is reduced compared to CCMSU.
2.2 Sequencing methods
The available studies thus far have primarily relied on culture-independent 16S rRNA gene amplicon sequencing to identify constituents of pediatric urobiomes (Table 1). This method relies on isolating DNA, amplifying region(s) of the 16S rRNA gene of bacteria and performing high-throughput sequencing, such as on the Illumina sequencing platform. Usually, the amplified regions comprise one to three hypervariable (V) regions out of the nine total V regions identified in the bacterial 16S rRNA gene, with V1–V3 and V4–V6 being the most common ranges. Multiple reads are then processed to produce amplicon sequence variants (ASVs) or operational taxonomic units (OTUs) for downstream analysis. ASVs are generated by employing an error correction model, whereas OTUs are generated by grouping sequences that share identity above a similarity threshold, typically 97% but occasionally 99% (31–33). Benchmarking OTUs vs. ASVs has revealed significant differences in diversity metrics and identified taxa, generally favoring ASVs (34, 35). While OTUs have not been fully abandoned (36, 37), there has been a consistent shift towards the use of ASVs in the most recent pediatric urobiome studies (Table 1).
The use of the 16S rRNA gene for sequencing has several important implications. First, the taxonomic resolution of short 16S amplicons is considerably low. Most analyses distinguish down to the genus level at best, though families or higher can often be the limit. Species resolution is rare but possible for some species (38), and it cannot provide strain level distinction. With the additional factor of potential reclassification of species and genera, mapping reads to recent nomenclature changes further complicates identification (39). Additionally, the 16S rRNA gene is specific to bacteria. Therefore, 16S sequencing overlooks other microbes that might also be present in the urinary tract. Detection and classification of other microbes require applying different detection methods, such as sequencing of the internal transcribed spacer (ITS) sequence for fungi, specific 16S sequencing for archaea, or metagenomic sequencing of the entire microbial biomass including viruses.
Metagenomic sequencing allows for reading of the entire complement of DNA present in a particular niche. This type of sequencing analysis applied to urine is complicated by the low biomass of this microbial community, as well as the high and variable number of host cells and DNA present that complicate the analysis of metagenomic sequencing results. This results in a need to increase the coverage of sequencing, apply complex and expensive methods for microbial DNA enrichment, and involves intricate bioinformatics. Alternative methods that might bring higher taxonomic resolution with a lower cost than true metagenomics include the use of long-read sequencing (PacBio, Nanopore) or synthetic long-read methods that can provide data on longer regions of interest such as the entire 16S rRNA gene (40).
While it might not be feasible to apply the most advanced methods listed above to all samples due to cost or expertise, it is likely possible to run benchmarking studies to compare different methods’ outcomes to obtain an estimate on whether they are comparable and/or biased. This has been done for adult urobiome studies (41, 42). Additionally, a consensus has been released which highlights similar considerations for the study of urobiomes in general (15).
2.3 Expanded quantitative urine culture
Initially SUC was developed to detect major uropathogens for UTI diagnostics. SUC typically involves plating urine samples onto 5% sheep blood agar and MacConkey agar plates and then incubating aerobically at 35°C for 24 h. Therefore, SUC caters mostly to Escherichia coli and other fast-growing, facultative anaerobes with similar growth requirements. EQUC was developed to encompass a greater variety and multiple combinations of incubation times, culture media, and atmospheric environments (3, 17). Therefore, EQUC has enabled the characterization of bacteria otherwise not detected or detected less frequently when using traditional SUC technique (43–45). Unlike culture-independent sequencing methods described above, EQUC allows identification of microbes that are viable while in urine and, at the same time, for isolation and storage of those organisms for further studies. These isolates can be identified to species and strain level using matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) mass spectrometry, 16S rRNA gene sequencing, and whole genome sequencing.
Only three of the identified pediatric urobiome studies use EQUC (Table 1). Storm et al. (19) employed EQUC coupled with MALDI-TOF and culture-dependent 16S sequencing for taxonomic identification. Their culture-independent 16S sequencing alone was able to detect bacteria in 84% of urine samples, and EQUC was able to detect bacteria in 60%. Combined, bacteria were detected in 91% of all urine samples. Swabs of adjacent niches had similar results (>90%). Reasoner et al. (21) modified the EQUC protocol and with its use, identified growth in 65% of urine samples in at least one growth condition. They were also able to detect additional families and species in their samples using MALDI-TOF. Cole et al. (27) similarly employed these methods and were able to verify their 16S-based taxa identification.
This way, the shift to predominance of Lactobacillus and Bifidobacterium in post-pubertal females in the study by Storm et al. (19) was observed by both EQUC and sequencing, and the composition and abundance measured by EQUC by Cole et al. (27) overall agreed with culture-independent 16S sequencing. However, EQUC and culture-independent 16S sequencing do not always reconcile. For example, the families detected most frequently by EQUC by Reasoner et al. (21) were not detected in culture-independent sequencing results at all, and several families detected most frequently by 16S sequencing were not detected in EQUC isolates. 16S sequencing methods are limited by available mapping to known genetic sequences in available databases, and EQUC still is not a fully comprehensive method of culturing all species. At their current level of development, combining EQUC and sequencing can reveal what a single method cannot, but further use and development of these techniques is necessary and can improve analyses of urobiome composition.
3 Summary of current studies
We have identified thirteen published studies that investigate the composition of the pediatric urobiome (Table 1). Herein, we summarize the results for each study and compare these with similar studies, both pediatric and adult. We also provide potential insight into the larger context of the connection between the urobiome and health and disease. The urobiome findings of each study are summarized in Table 2.
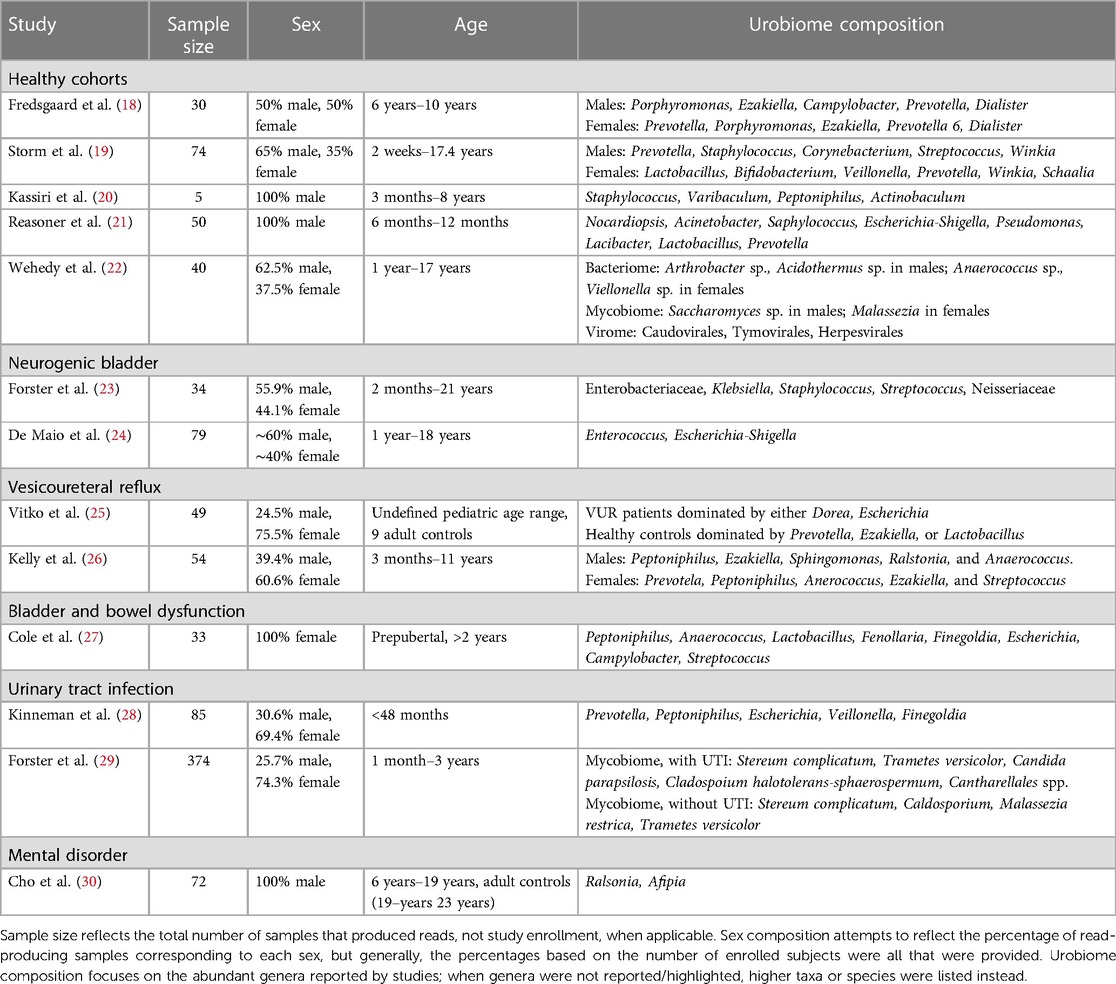
Table 2 Summary of study features and reported urobiome composition based on culture-independent sequencing.
3.1 Healthy prepubertal urobiome
Several studies have examined cohorts of children that were considered “healthy”, meaning that they were asymptomatic for UTI and had urinary continence. Fredsgaard et al. (18) examined a prepubertal cohort of 30 children comprising ages 6 years to 10 years and an equal male-to-female sex ratio. Participants had not been exposed to antibiotics within the 3 months prior to CCMSU sample collection. Diversity was significantly higher in females, but there was no difference in the relative abundance of each genus. Males and females exhibited some similarities in the most abundant genera; however, the relative abundances of these genera were significantly different. Porphyromonas was the most abundant in males, followed by Ezakiella, Campylobacter, Prevotella, and Dialister. Prevotella was the most abundant in females, followed by Porphyromonas, Ezakiella, Prevotella 6, and Dialister.
The cohort examined by Storm et al. (19) consisted of 74 children aged 2 weeks to approximately 17 years undergoing anesthesia for various urologic/specialty procedures. Still, these children were otherwise considered healthy, had no prior urinary tract surgeries, possessed a healthy immune system, and had not been exposed to antibiotics within 3 months prior to sample collection. In addition to TUC urine samples, swabs of the adjacent microbial niches were taken, including the urethra, perineum, vagina for females, and foreskin for males. The most common genera in male urobiomes were Prevotella, Staphylococcus, Corynebacterium, Streptococcus, and Winkia. Neither composition nor diversity appeared to change with age. In prepubertal female urobiomes the most common genera were Lactobacillus, Bifidobacterium, Veillonella, Prevotella, Winkia, and Schaalia. Both composition and diversity did change with age; Lactobacillus and Bifidobacterium became predominant after puberty in the female urobiome and were similarly observed in the urethral and vaginal microbiomes. Males demonstrated reduced diversity in microbiome composition of their urogenital niches compared to females. In both sexes, diversity was observed to be reduced prior to toilet training, but this was especially evident in females.
Two studies have examined exclusively male infants and/or prepubertal males. The earliest, by Kassiri et al. (20), identified evidence of a urobiome in TUC samples of prepubertal males as young as 3 months and as old as 8 years. Staphylococcus and Varibaculum had the highest abundance, followed by Peptoniphilus and Actinobaculum. This cohort included an equal mix of those who did and did not have prior antibiotic exposure, but no statistically significant difference in alpha diversity was observed between these groups. Principal component analysis (PCoA) showed that urobiomes of the two groups clustered separately, which suggested dissimilarities between the composition of the two groups, but the especially small sample size of this study (n = 5) made it impossible to draw definitive conclusions.
The latest of the two studies, by Reasoner et al. (21), also examined a tighter age range of 50 prepubertal males—infants aged 6–12 months—and was the first to examine a cohort that did not contain any individuals possessing any structural or functional urinary tract abnormalities nor had experienced any prior UTI. Nocardiopsis and Acinetobacter were detected in all TUC samples, and Staphylococcus, Escherichia-Shigella, and Pseudomonas were detected in over 90% of samples. Lacibacter, Lactobacillus, and Prevotella were also abundant, though detected variably across samples. Mode of delivery and prior antibiotic exposures did not appear to influence alpha diversity or overall urobiome composition significantly.
Many genera identified in the urobiome of children match those identified in their adult counterparts. For example, Staphylococcus and Corynebacterium have now been observed in the urobiomes of both healthy adult males and healthy male children (6, 7, 20). The question of how the healthy urobiome develops in early life is of notable interest in the study of pediatric urobiomes. Interestingly, the prepubertal cohorts comprising both sexes share many abundant genera, which could suggest that later pubertal changes are an important influence in differentiating the healthy male and female urobiomes. Across studies, Prevotella have been observed frequently in samples, regardless of the sex or age composition of cohorts. Prevotella are Gram-negative, nonmotile anaerobes known to inhabit other niches such as the skin, oral cavity, vagina, and the gastrointestinal tract (46). The studies thus far have not identified strong correlations between urobiome composition or abundances and mode of delivery, unlike the pattern identified in microbiome studies of other anatomical locations (47). In early life, Prevotella, Viellonella, and Clostridium have been transiently detected at the buttock, along with more frequently detected species of Staphylococcus, Streptococcus, and members of the order Enterobacteriales (48). Prevotella and other abundant genera in adjacent niches could contribute to similar observations in the pediatric urobiome, especially in these cohorts that consist of pre-toilet trained children.
3.2 Urologic conditions
Compared to studies of healthy children, there has been more work that has gone into the pediatric urobiome’s potential relationship with urinary tract pathologies, including neurogenic bladder (NB), vesicoureteral reflux (VUR), bladder and bowel dysfunction (BBD), and UTI. These studies have identified potential differences in genera composition and relative abundances compared to those observed in the urobiomes of healthy individuals.
3.2.1 Neurogenic bladder
Several adult studies have identified distinct variations in the urobiomes of individuals with NB compared to those of healthy individuals. Notably, most genera identified at the highest abundance contain many known uropathogens, including Enterococcus, Escherichia, Klebsiella, and Pseudomonas (49–51). Additionally, lactobacilli have been observed to be greatly reduced in adult female neurogenic urobiomes compared to healthy females (51).
Similar genera have been observed in the urobiomes of children with NB, and their overall urobiome composition appears to have varying and overall reduced abundances of identified genera compared to healthy children. Forster et al. (23) aimed to investigate the baseline urobiome of 34 children—aged 2 months to 21 years—with NB and determine compositional differences among those with negative SUC, asymptomatic bacteriuria (ASB), or UTI. The family Enterobacteriaceae (unspecified genera) were the most abundant across all TUC samples, followed by genera Klebsiella, Staphylococcus, Streptococcus, and family Neisseriaceae (unspecified genera). There were too few participants to statistically compare the composition between those who had a Mitrofanoff, had both a Mitrofanoff and an augmented bladder, or neither. There were no statistically significant differences in composition or diversity within samples between participants with negative SUC, ASB, or UTI.
De Maio et al. (24) analyzed CCMSU samples from 79 children aged 1–18 years and found that those with spina bifida (SB) possessed an altered urobiome compared to healthy controls. The phyla Actinobacteria, Bacteroidetes, Firmicutes, and Proteobacteria were detected in both groups; compared to healthy controls, the phylum Proteobacteria was detected in higher abundance, and the phyla Bacteroidetes and Firmicutes were in lower abundance. Genera associated with UTI, such as Enterococcus and Escherichia-Shigella, were also more abundant in the SB samples compared to healthy controls. There were no statistically significant differences in alpha diversity, but beta diversity did differ between the two groups; PCoA also reiterated this difference. Clean intermittent catheterization (CIC) usage also significantly affected abundance of some taxa. The phylum Bacteroidetes, which was reduced in individuals with SB compared to healthy controls, was further reduced in those with SB who performed CIC. Additionally, several genera Faecalibacterium, Lactobacillus, Staphylococcus, and Streptococcus were significantly reduced in those with SB who performed CIC.
Two additional studies have examined the urobiome of children with NB in the context of urobiome intervention and alteration. Because these are intervention studies and not observational of baseline urobiome dynamics, we only briefly highlight their results in the context of NB. Kispal et al. (52) observed a high abundance of Corynebacterium followed by Pseudoxanthomonas, Lactobacillus, Flavobacterium, and Micrococcus post-bladder augmentation with either colon or ileum mucosal tissue across 4 years of observation, regardless of the origin of the mucosal tissues used. Forster et al. (23) administered intravesical instillations of the common probiotic Lactobacillus rhamnosus GG [now reclassified as Lacticaseibacillus rhamnosus (39)] into neurogenic bladders. While initial post-instillation urinary symptoms arose in some children, it was overall well-tolerated, and Lactobacillus was successfully introduced and/or maintained in most. While both studies had small sample sizes, they demonstrate the promise and potential next steps in understanding how the urobiome modulation can be harnessed to treat urinary tract disease.
3.2.2 Vesicoureteral reflux
There have been two studies examining the urobiome in children with VUR. Vitko et al. (25) were the first to report a baseline urobiome profile in VUR patients. They examined a cohort of 49 individuals that consisted of healthy controls and VUR patients, either lacking or accompanied by renal scarring. This cohort was notably a broader, undefined pediatric range that did include 9 adult controls. Regardless of scarring, VUR participants exhibited a predominance of Dorea and/or Escherichia in TUC samples. Healthy controls possessed urobiomes dominated by either Prevotella, Ezakiella, or Lactobacillus. Adjusting for age, sex, and antibiotic usage, composition differed significantly among their participant categories. While some healthy controls did include adult CCMSU samples, the predominant genera of the reported healthy urobiomes matched those detected in abundance in the identified healthy pediatric urobiomes. Meanwhile, the urobiome of children with VUR was dominated by the genera associated with infection, further supporting the idea that the urobiome is tied to urinary tract health and disease.
Kelly et al. (26) examined the urobiome in samples of 54 children aged 3 months to 10.8 years that were referred for a voiding cystourethrogram (VCUG) due to urologic concerns. This study took advantage of TUC samples obtained during the procedure and used synthetic long-read sequencing of the entire 16S rRNA gene for improved taxonomic resolution of the urobiome composition. The majority of subjects were either confirmed to possess VUR or had a history of VUR that resolved, but none utilized intermittent catheterization or had a diagnosis of neurogenic bladder. Male urobiomes were observed to be dominated by Peptoniphilus, Ezakiella, Sphingomonas, Ralstonia, and Anaerococcus. Female urobiomes were dominated by Prevotela, Peptoniphilus, Anerococcus, Ezakiella, and Streptococcus; Anaerococcus and Prevotella were in significantly higher abundance in females compared to males. Females exhibited overall greater diversity than males, but this diversity was relatively consistent across ages in females and increased with age in males.
3.2.3 Bladder and bowel dysfunction
Cole et al. (27) sought to compare the urogenital microbiome of 33 healthy children with that of children with BBD. The study population consisted of 33 prepubertal females over the age of 2 years who had completed toilet training at least 6 months prior to collection, which is especially important in the context of the chosen CCMSU collection method. Both healthy controls and those with BBD had microbiomes consisting of shared core genera: Peptoniphilus, Anaerococcus, Lactobacillus, Fenollaria, and Finegoldia. However, several genera predominantly observed in the BBD group were not observed in the healthy controls, including Escherichia, Campylobacter, and Streptococcus. These genera contain many species found to cause opportunistic infection, although the study exclusion criteria had prevented those with symptomatic infection from enrolling. However, overall alpha and beta diversity did not differ significantly between the healthy and BBD groups, and the authors concluded that the urogenital microbiomes did not differ significantly.
3.2.4 Urinary tract infection
The connection between the urobiome and UTI/recurrent UTI in adults is being elucidated (17, 45, 53–56). Current work has reported distinct urobiome profiles when comparing healthy and infected patients; as similarly noted in our previous discussions, pathogenic genera tend to predominate in UTI, including Klebsiella, Pseudomonas, Enterobacter, and Enterococcus (57, 58). Herein we specifically highlight bacterial composition and abundances; we continue this discussion with the inclusion of fungi in the following section.
Pathogenic genera have also been identified at higher abundances in pediatric urobiome samples that were positive for UTI. Kinneman et al. (28) examined a cohort of 85 children under 2 years of age who were suspected to have a UTI, with most being febrile; they otherwise exhibited no urinary tract pathologies or complications. The study was able to identify the most abundant urobiome members of different taxonomic levels, including phyla, classes, orders, families, and genera. However, the latter is what we choose to highlight exclusively here for the sake of more closely comparing it with other studies. Identified genera overall included Prevotella, Peptoniphilus, Escherichia, Veillonella, and Finegoldia. The nine participants that were indeed diagnosed with UTI all exhibited an increased abundance of Escherichia/Shigella. Alpha diversity was significantly reduced in those with a confirmed UTI or symptom of hematuria. The use of antibiotics within the 2 weeks prior to TUC sample collection significantly affected alpha and beta diversity. Other demographics (including sex) and mode of delivery did not appear to affect either diversity measure. Outside the context of UTI, healthy results align with some of the abundant genera observed in urobiomes of the previously discussed healthy pediatric cohorts, including Prevotella, Veillonella, and Peptoniphilus. The results from the small sample of individuals diagnosed with a UTI aligned with prior works identifying reduced diversity and a predominance of genera typically associated with infection.
Similarly, the previously discussed study by Kelly et al. (26) also examined differences in composition between groups of children who had a history of one, two, or three or more UTIs. Those with three or more UTIs had significantly reduced diversity when compared with those with one UTI. Overall abundances of certain taxa were observed to be significantly decreased in those with 3 or more UTIs, such as Lawsonella, Corynebacterium, and, interestingly, Enterococcus, which have been previously been described to be predominate in UTI. This cohort did include children who had been treated with antibiotics prior to enrollment or were concurrently taking prophylactic antibiotics, which may have contributed to the observed differences in urobiome features, but overall, there was no significant difference between the two groups of exposure.
3.3 Metagenomics and urinary mycobiomes/viromes
Other human microbiome niches have been shown to contain microbes other than bacteria, such as archaea, fungi, protists, and viruses, including bacteriophages (59). There have been several studies that have expanded the analysis of the urobiome of adults to the characterization of present fungi and viruses (57, 60–64). Recently, two studies—Wehedy et al. (22) and Forster et al. (29)—have similarly applied metagenomic and ITS sequencing methods to pediatric cohorts.
Wehedy et al. (22) were the first to use metagenomic sequencing to characterize children's true urinary profile containing the urinary bacteriome, mycobiome, and virome. They examined 40 healthy children, primarily comparing by sex and age (1–5 years, 6–10 years, and 10–18 years). Eight nationalities were included in this study, but no significant differences were observed among them. All participants were considered healthy and had not been exposed to antibiotics within 2 months prior to CCMSU sample collection.
Bacteria were the most abundant domain detected. Differences in beta diversity were significant between males and females. Firmicutes, Actinobacteria, and Proteobacteria dominated detected phyla in both sexes, with Firmicutes were significantly higher in females and Actinobacteria were higher in males. Genera Anaerococcus and Viellonella were significantly higher in females, and genera Arthrobacter and Acidothermus were significantly higher in males. Metagenomic analysis allowed for species-level resolution, revealing higher abundances of Anaerococcus pervotii, Anerococcus vaginalis, and Viellonella parvula in females, and higher abundances of Arthrobacter spp., Arthrobacter aurescens, Thermobifida fusca, and Acidothermus cellulolyticus in males. Across age groups, there were no statistically significant differences in alpha or beta diversity, and there were no significant differences in genera and species composition.
The urinary mycobiomes, or uromycobiomes, of this cohort were dominated by phylum Ascomycota in males and Basidiomycota in females. At the species level, Saccharomyces pastorianus and Saccharomyces cerevisiae were highest in males, and Malassezia lobose was highest in females. Unlike the results observed in bacterial abundances, the two sexes had no significant difference in beta diversity. Age groups also saw no significant diversity differences, except for a significant difference in beta diversity between the 6–10 years and >10 years age groups.
Using sequencing of the ITS region for fungi, Forster et al. (29), also detected uromycobiomes in the largest pediatric cohort examined in a urobiome study to date: 50 prepubertal children (aged 1 month to 3 years) with UTI and 324 without UTI. Both groups shared Stereum complicatum and Trametes versicolor within their top four most abundant fungal species. Candida parapsilosis and Cladosporium halotolerans-sphaerospermum were the other two most detected species in TUC samples of those with UTI, and Malassezia restricta and Cladosporium spp. were the other two most detected in those without UTI. Additionally, Cantharellales spp. were detected exclusively in those with UTI. Younger children and those with UTI were more likely to have detectable mycobiomes, but there was no difference based on sex. Overall, those with UTI had higher total fungal cell counts compared to those without. Interestingly, alpha diversity of fungi was higher in those with UTI than those without, but no difference in beta diversity was observed.
The uromycobiome results of these two studies are especially notable in the context of UTI. Bacteria and fungi can both be etiological agents of infection, and recent decades have seen a rise in UTIs caused by fungal species, such as those of genera Candida (65, 66). This genus was also observed in the adult cohort of Moustafa et al. (57). While the majority of those with UTI in the study by Forster et al. (29) cultured E. coli (92%), Candida parapsilosis differed strongly compared to those without UTI. There have been several reported interactions between E. coli and Candida that suggest their interplay in promoting infection (67–71). Meanwhile, Wehedy et al. (22) did not detect Candida at a similar abundance in their cohort of healthy children. This could further suggest an important link between the composition balance of the uromycobiome and urinary health. More research is needed to confirm this and establish a baseline for the healthy uromycobiome in children.
Wehedy et al. (22) has been the only study, to date, that has examined the urinary virome in children. Caudovirales, Tymovirales, and Herpesvirales were observed to be the most abundant orders, and T4 Virus, Betapartitivirus, Tymovirus, and alpha-partitivirus were the most abundant genera. Like their uromycobiome analyses, there was no significant difference in beta diversity between the two sexes. However, at the species level, Mastadenovirus and the Human Mastadenovirus-A virus were notably identified in males compared to females, and Dill cryptic virus 2 and Chrysochromulina ericina virus were in higher abundance in females compared to males. The greatest differences between age groups were observed in virome composition, with an observed increase in abundance with age of order Caudovirales, genus T4 Virus, and Shigella phage SHFML-11. Compared to the >10 years age group, the 1–5 age group had higher alpha diversity and higher presence of Herpesvirales, Shigella phage SHFML-11 Rosellinia necatrix partitivirus 2, and red clover cryptic virus 2. The urinary virome has been reviewed for its potential connection with human pathology and health (72), and it has been suggested to be an alternative mode of screening in the context of human viruses, such as human papillomaviruses in adults (73). However, no studies have linked the urinary virome to disease in children. Bacteriophages and other viruses have been detected in adult urine samples (64), but the overall connection between viruses to urinary tract health and urobiome health has not been resolved.
3.4 The pediatric urobiome as a biomarker for mental disorders
As previously discussed, the human microbiome has been linked to the health and disease of the host, and extensive work has recently gone into exploring the microbiome as an indicator of shifts between these states (74). reviewed the potential for human microbiomes to be analyzed for biomarkers of a wide variety of diseases and disorders, including those that affect the skin, respiratory system, immune system, and metabolism, as well as in the cases of infection and cancers. Mental disorders such as depression and anxiety have also been included in the research mapping microbiome biomarkers to disease, specifically by linking gut microbiome features to symptoms measured by clinical interviews and other diagnostic criteria, as reviewed by Simpson et al. (75).
Cho et al. (30) has been the only study, to date, to examine the urobiome as a potential biomarker for a mental disorder, specifically for those with attention-deficit hyperactivity disorder (ADHD). 33 males aged 6–15 years diagnosed with ADHD were compared against 39 adult men aged 19–23 years without ADHD. While there were no statistically significant differences in alpha diversity, there were significant differences in abundance and beta diversity between the two groups. Phyla Firmicutes and Actinobacteria were more abundant in CCMSU samples of those with ADHD, while healthy controls exhibited a greater abundance of Proteobacteria. Genera Ralstonia and Afipia were more abundant in those with ADHD, while Corynebacterium and Peptoniphilus were more abundant in healthy controls. Genus Afipia had strong correlations with two subscale t-scores for diagnosing ADHD, Child Behavior Checklist and DMS-oriented ADHD, but not with the ADHD Rating Scale IV scores, which were identified as being the “current gold standard” for diagnosis. Further study is required to evaluate these findings. This study was limited by its lack of age-matched healthy controls. However, the authors identify their conclusions based on the findings by Kassiri et al. (20) and other adult male urobiome studies that suggest certain stability of the healthy male urobiome across ages.
4 Larger gaps to be addressed in future studies
4.1 Genomic and functional studies are needed
The implication of the frequently employed 16S rRNA sequencing is that it only provides taxonomic, compositional information about present microbes. This method cannot provide functional genomic information needed for analysis of traits such as virulence and fitness factors and required for understanding urobiome development. While there are now several compositional studies of the pediatric urobiome, with a couple including EQUC isolation of constituents, only one study proceeded with complete genome sequencing and analyses of identified constituents: Actinotignum schaali and Actinotignum sanguinis in Reasoner et al. (21). In their enhanced culture Actinotignum spp. was obtained most often in the culturable urine samples (28.1%). Notably, their initial 16S rRNA results had also filtered out Actinotignum ASVs, and MALDI-TOF had identified all nine of their culturable isolates as A. schaalii. Further whole genome sequencing of these isolates reclassified five of them as A. sanguinis. All nine isolates possessed several potential factors of urinary tract fitness, including genes for enterobactin transport, heme acquisition, and copper detoxification systems. Additionally, they reported two potential virulence factors: ermX, which confers macrolide resistance, and Esx-1 Type VII secretion/esxA, which is an anti-eukaryotic membrane-permeabilizing toxin found in Mycobacterium tuberculosis pathogen and gram-positive bacteria. This is of special interest because A. schaalii is now being recognized as an opportunistic causative agent of UTI with increasing incidence (21).
To date, no functional studies analyzing behavior or interactions of isolated species from pediatric urobiome were reported in either in vitro or in vivo models, though there have been a few functional studies of the common bacteria found in the adult urobiomes (76–81). However, the genomes of numerous urobiome commensal strains have been established and analyzed bioinformatically, even with establishing new speciation of bacteria specific to the human bladder (8, 82–87).
4.2 Female cohort studies
Due to complications arising from the specifics of the pediatric population, the existing studies either work with both sexes or only male participants; the only exception is the single female-only cohort of Cole et al. (27) examining the urobiome in females with BBD. To date, there are no other female-only studies, and there are no studies at all that examine the healthy urobiome of exclusively females across any age range. While the importance of understanding urobiome development for recurrent UTI studies in female participants is clearly shown (54), no studies have yet addressed this issue carefully in female children.
4.3 Replicated cohorts and methods benchmarking
While we have attempted to identify similarities in the makeup of the pediatric urobiome across studies, it is difficult to draw definitive conclusions, mainly because some studies collect data for either very narrow or very broad age ranges (Table 1) and the analyses of changes observed are done in different cohorts. For example, the broader age range of boys in Storm et al. (19) and reported analyses cannot be truly, directly aligned with the narrow age range of boys in Reasoner et al. (21) study. The physical features of these cohorts regarding the health status of the urinary tract do not always match, which further complicates comparisons. We thus reiterate the importance and need for further, replicated studies of the urobiome in a variety of pediatric cohorts to promote a robust understanding of its development and dynamics. Creation of unified and standardized databases for pediatric and adult urobiome studies will be helpful in comparative analyses going forward (82).
Additionally, it is important to critically evaluate and identify gaps in current benchmarking and optimization studies for pediatric urobiome applications. For example, many of the laboratory methods have been evaluated for optimization on adult human and canine urines (88–90), and several comparisons of detection methods (91), and bioinformatics (38, 41) specifically for the urobiome. It is also essential that investigators in the pediatric urobiome research community employ current recommendations developed by the urobiome (15), kidney microbiome (92), and general microbiome communities (93) and develop consensus on standards and guidelines for the field.
5 Conclusions
The knowledge base of the urobiome is still not as robust as that of more extensively studied human microbiome niches. Nonetheless, the present urobiome research has hit notable milestones in studying the human microbiome, and common patterns in makeup and abundance across cohorts begin to present themselves. While sampling pediatric populations remains a unique challenge, advancements in urobiome sequencing have improved the ability of researchers to discern greater detail regarding urobiome composition and dynamics, especially in regard to the immense diversity of microbial species yet to be explored in greater detail. As the field of urobiome research enters its second decade, a more detailed study of the pediatric urobiome will be vital in understanding the crosstalk between urinary microbiota and a lifetime of urinary health and disease.
Author contributions
LJ: Investigation, Writing – original draft, Writing – review & editing. TS: Resources, Supervision, Writing – original draft, Writing – review & editing. LK: Resources, Writing – review & editing. MK: Conceptualization, Resources, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article.
This work was funded by the National Institutes of Health (NIH) CAIRIBU U24 Interactions Core award U24DK127726 through the Collaboration Awards Program, by Innovative Science Accelerator (ISAC) Award by Augusta University through NIDDK award U24DK128851 to LK, and by the 2021 CNR Pilot Grant from the Duke University School of Nursing to MK. The contents of the article are solely our responsibility and do not represent the official views of the NIH or of any other funding agency.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Zorc JJ, Kiddoo DA, Shaw KN. Diagnosis and management of pediatric urinary tract infections. Clin Microbiol Rev. (2005) 18(2):417–22. doi: 10.1128/cmr.18.2.417-422.2005
2. Wolfe AJ, Toh E, Shibata N, Rong R, Kenton K, FitzGerald M, et al. Evidence of uncultivated Bacteria in the adult female bladder. J Clin Microbiol. (2012) 50(4):1376–83. doi: 10.1128/jcm.05852-11
3. Hilt EE, McKinley K, Pearce MM, Rosenfeld AB, Zilliox MJ, Mueller ER, et al. Urine is not sterile: use of enhanced urine culture techniques to detect resident bacterial flora in the adult female bladder. J Clin Microbiol. (2013) 52(3):871–76. doi: 10.1128/jcm.02876-13
4. Lee KW, Song HY, Kim YH. The microbiome in urological diseases. Investig Clin Urol. (2020) 61(4):338. doi: 10.4111/icu.2020.61.4.338
5. Price TK, Wolff B, Halverson T, Limeira R, Brubaker L, Dong Q, et al. Temporal dynamics of the adult female lower urinary tract microbiota. MBio. (2020) 11:2. doi: 10.1128/mbio.00475-20
6. Dong Q, Nelson DE, Toh E, Diao L, Gao X, Fortenberry JD, et al. The microbial communities in male first catch urine are highly similar to those in paired urethral swab specimens. PLoS One. (2011) 6(5):e19709. doi: 10.1371/journal.pone.0019709
7. Bučević Popović V, Šitum M, Chow C-ET, Chan LS, Roje B, Terzić J. The urinary microbiome associated with bladder cancer. Sci Rep. (2018) 8(1):12157. doi: 10.1038/s41598-018-29054-w
8. Thomas-White K, Forster SC, Kumar N, Van Kuiken M, Putonti C, Stares MD, et al. Culturing of female bladder Bacteria reveals an interconnected urogenital microbiota. Nat Commun. (2018) 9(1):1557. doi: 10.1038/s41467-018-03968-5
9. Lewis DA, Brown R, Williams J, White P, Kim Jacobson S, Marchesi JR, et al. The human urinary microbiome; bacterial DNA in voided urine of asymptomatic adults. Front Cell Infect Microbiol. (2013) 3:41. doi: 10.3389/fcimb.2013.00041
10. Komesu YM, Richter HE, Carper B, Dinwiddie DL, Lukacz ES, Siddiqui NY, et al. The urinary microbiome in women with mixed urinary incontinence compared to similarly aged controls. Int Urogynecol J. (2018) 29(12):1785–95. doi: 10.1007/s00192-018-3683-6
11. Mueller ER, Wolfe AJ, Brubaker L. Female urinary microbiota. Curr Opin Urol. (2017) 27(3):282–86. doi: 10.1097/MOU.0000000000000396
12. Bowie KR, Garzotto M, Orwoll E, Karstens L. BMI and BPH correlate with urinary microbiome diversity and lower urinary tract symptoms in men. BioRxiv. (2023). doi: 10.1101/2023.12.14.571758
13. Bajic P, Van Kuiken ME, Burge BK, Kirshenbaum EJ, Joyce CJ, Wolfe AJ, et al. Male bladder microbiome relates to lower urinary tract symptoms. Eur Urol Focus. (2020) 6(2):376–82. doi: 10.1016/j.euf.2018.08.001
14. Shrestha E, White JR, Yu S-H, Kulac I, Ertunc O, De Marzo AM, et al. Profiling the urinary microbiome in men with positive versus negative biopsies for prostate cancer. J Urol. (2018) 199(1):161–71. doi: 10.1016/j.juro.2017.08.001
15. Brubaker L, Gourdine J-P, Siddiqui NY, Holland A, Halverson T, Limeria R, et al. Forming consensus to advance urobiome research. Msystems. (2021) 6(4):e01371–20. doi: 10.1128/msystems.01371-20
16. Karstens L, Asquith M, Caruso V, Rosenbaum JT, Fair DA, Braun J, et al. Community profiling of the urinary Microbiota: considerations for low-biomass samples. Nat Rev Urol. (2018) 15(12):735–49. doi: 10.1038/s41585-018-0104-z
17. Neugent ML, Hulyalkar NV, Nguyen VH, Zimmern PE, De Nisco NJ. Advances in understanding the human urinary microbiome and its potential role in urinary tract infection. Mbio. (2020) 11(2):e00218–20. doi: 10.1128/mbio.00218-20
18. Fredsgaard L, Thorsteinsson K, Bundgaard-Nielsen C, Ammitzbøll N, Leutscher P, Chai Q, et al. Description of the voided urinary microbiota in asymptomatic prepubertal children—a pilot study. J Pediatr Urol. (2021) 17(4):545.e1–8. doi: 10.1016/j.jpurol.2021.03.019
19. Storm DW, Copp HL, Halverson TM, Du J, Juhr D, Wolfe AJ. A child’s urine is not sterile: a pilot study evaluating the pediatric urinary microbiome. J Pediatr Urol. (2022) 18(3):383–92. doi: 10.1016/j.jpurol.2022.02.025
20. Kassiri B, Shrestha E, Kasprenski M, Antonescu C, Florea LD, Sfanos KS, et al. A prospective study of the urinary and gastrointestinal microbiome in prepubertal males. Urology. (2019) 131:204–10. doi: 10.1016/j.urology.2019.05.031
21. Reasoner SA, Flores V, Van Horn G, Morales G, Peard LM, Abelson B, et al. Survey of the infant male urobiome and genomic analysis of Actinotignum spp. NPJ Biofilms Microbiomes. (2023) 9(91):91. doi: 10.1038/s41522-023-00457-6
22. Wehedy E, Murugesan S, George CR, Shatat IF, Al Khodor S. Characterization of the urinary metagenome and virome in healthy children. Biomedicines. (2022) 10(10):2412–12. doi: 10.3390/biomedicines10102412
23. Forster CS, Panchapakesan K, Stroud C, Banerjee P, Gordish-Dressman H, Hsieh MH. A cross-sectional analysis of the urine microbiome of children with neuropathic bladders. J Pediatr Urol. (2020) 16(5):593.e1–8. doi: 10.1016/j.jpurol.2020.02.005
24. De Maio F, Grotti G, Mariani F, Buonsenso D, Santarelli G, Bianco DM, et al. Profiling the urobiota in a pediatric population with neurogenic bladder secondary to spinal dysraphism. Int J Mol Sci. (2023) 24(9):8261–61. doi: 10.3390/ijms24098261
25. Vitko D, McQuaid JW, Gheinani AH, Hasegawa K, DiMartino S, Davis KH, et al. Urinary tract infections in children with vesicoureteral reflux are accompanied by alterations in urinary microbiota and metabolome profiles. Eur Urol. (2022) 81(2):151–54. doi: 10.1016/j.eururo.2021.08.022
26. Kelly MS, Dahl EM, Jeries L, Sysoeva TA, Karstens L. Characterization of pediatric urinary microbiome at species-level resolution indicates variation due to sex, age, and urologic history. J Pediatr Urol. (2024). doi: 10.1016/j.jpurol.2024.05.016
27. Cole EB, Khemmani M, Liu H, Halverson TM, Noronha MF, Forster CS, et al. Urogenital urobiome of healthy children does not differ from that of children with bladder and bowel dysfunction. J Pediatr Urol. (2023) 19(4):368.e1–8. doi: 10.1016/j.jpurol.2023.04.009
28. Kinneman L, Zhu W, Wong WSW, Clemency N, Provenzano M, Vilboux T, et al. Assessment of the urinary microbiome in children younger than 48 months. Pediatr Infect Dis J. (2020) 39(7):565–70. doi: 10.1097/INF.0000000000002622
29. Forster CS, Liu H, Kurs-Lasky M, Ullmer W, Krumbeck JA, Shaikh N. Uromycobiome in infants and toddlers with and without urinary tract infections. Pediatr Nephrol. (2022) 38(7):2117–23. doi: 10.1007/s00467-022-05844-3
30. Cho YJ, Shin B, Lee S-H, Park S, Kim Y-K, Kim J-J, et al. Altered urine microbiome in male children and adolescents with attention-deficit hyperactivity disorder. Microorganisms. (2023) 11(8):2063–63. doi: 10.3390/microorganisms11082063
31. Callahan B, McMurdie PJ, Holmes SP. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. (2017) 11(12):2639–43. doi: 10.1038/ismej.2017.119
32. Porter TM, Hajibabaei M. Scaling up: a guide to high-throughput genomic approaches for biodiversity analysis. Mol Ecol. (2018) 27(2):313–38. doi: 10.1111/mec.14478
33. Westcott SL, Schloss PD. De novo clustering methods outperform reference-based methods for assigning 16S RRNA gene sequences to operational taxonomic units. PeerJ. (2015) 3:e1487. doi: 10.7717/peerj.1487
34. Chiarello M, McCauley M, Villéger S, Jackson CR. Ranking the biases: the choice of OTUs vs. ASVs in 16S RRNA amplicon data analysis has stronger effects on diversity measures than rarefaction and OTU identity threshold. PLoS One. (2022) 17(2):e0264443. doi: 10.1371/journal.pone.0264443
35. Caruso V, Song X, Asquith M, Karstens L. Performance of microbiome sequence inference methods in environments with varying biomass. MSystems. (2019) 4(1):e00163–18. doi: 10.1128/msystems.00163-18
36. Glassman SI, Martiny JBH. Broadscale ecological patterns are robust to use of exact sequence variants versus operational taxonomic units. Msphere. (2018) 3(4):e00148–18. doi: 10.1128/msphere.00148-18
37. García-López R, Cornejo-Granados F, Lopez-Zavala AA, Cota-Huízar A, Sotelo-Mundo RR, Gómez-Gil B, et al. OTUs and ASVs produce comparable taxonomic and diversity from shrimp microbiota 16S profiles using tailored abundance filters. Genes (Basel). (2021) 12(4):564. doi: 10.3390/genes12040564
38. Hoffman C, Siddiqui NY, Fields I, Thomas Gregory W, Simon HM, Mooney MA, et al. Species-level resolution of female bladder microbiota from 16S RRNA amplicon sequencing. MSystems. (2021) 6(5):e00518–21. doi: 10.1128/msystems.00518-21
39. Zheng J, Wittouck S, Salvetti E, Franz CMAP, Harris HMB, Mattarelli P, et al. A taxonomic note on the genus Lactobacillus: description of 23 novel genera, emended description of the genus Lactobacillus beijerinck 1901, and union of Lactobacillaceae and Leuconostocaceae. Int J Syst Evol Microbiol. (2020) 70(4):2782–858. doi: 10.1099/ijsem.0.004107
40. Tedersoo L, Albertsen M, Anslan S, Callahan B. Perspectives and benefits of high-throughput long-read sequencing in microbial ecology. Appl Environ Microbiol. (2021) 87(17):e00626–21. doi: 10.1128/aem.00626-21
41. Siddiqui NY, Ma L, Brubaker L, Mao J, Hoffman C, Dahl EM, et al. Updating urinary microbiome analyses to enhance biologic interpretation. Front Cell Infect Microbiol. (2022) 12:789439. doi: 10.3389/fcimb.2022.789439
42. Heidrich V, Inoue LT, Asprino PF, Bettoni F, Mariotti ACH, Bastos DA, et al. Choice of 16S ribosomal RNA primers impacts male urinary microbiota profiling. Front Cell Infect Microbiol. (2022) 12:862338. doi: 10.3389/fcimb.2022.862338
43. Thapaliya J, Khadka P, Thapa S, Gongal C. Enhanced quantitative urine culture technique, a slight modification, in detecting under-diagnosed pediatric urinary tract infection. BMC Res Notes. (2020) 13(5):5. doi: 10.1186/s13104-019-4875-y
44. Price TK, Dune T, Hilt EE, Thomas-White KJ, Kliethermes S, Brincat C, et al. The clinical urine culture: enhanced techniques improve detection of clinically relevant microorganisms. J Clin Microbiol. (2016) 54(5):1216–22. doi: 10.1128/jcm.00044-16
45. Hochstedler BR, Burnett L, Price TK, Jung C, Wolfe AJ, Brubaker L. Urinary microbiota of women with recurrent urinary tract infection: collection and culture methods. Int Urogynecol J. (2021) 33(3):563–70. doi: 10.1007/s00192-021-04780-4
46. Tett A, Pasolli E, Masetti G, Ercolini D, Segata N. Prevotella diversity, niches and interactions with the human host. Nat Rev Microbiol. (2021) 19(9):585–99. doi: 10.1038/s41579-021-00559-y
47. Coelho GDP, Arial Ayres LF, Barreto DS, Henriques BD, Cardoso Prado MRM, Dos Passos CM. Acquisition of microbiota according to the type of birth: an integrative review. Rev Lat Am Enfermagem. (2021) 29(3446):e3446. doi: 10.1590/1518.8345.4466.3446
48. Palmer C, Bik EM, DiGiulio DB, Relman DA, Brown PO. Development of the human infant intestinal microbiota. PLoS Biol. (2007) 5(7):e177. doi: 10.1371/journal.pbio.0050177
49. Lane GI, Bassis CM, Greiman SE, Romo PB, Quentin Clemens J, Gupta P, et al. Distinguishing features of the urinary bacterial microbiome in patients with neurogenic lower urinary tract dysfunction. J Urol. (2021) 207(3):627–34. doi: 10.1097/ju.0000000000002274
50. Groah SL, Pérez-Losada M, Caldovic L, Ljungberg IH, Sprague BM, Castro-Nallar E, et al. Redefining healthy urine: a cross-sectional exploratory metagenomic study of people with and without bladder dysfunction. J Urol. (2016) 196(2):579–87. doi: 10.1016/j.juro.2016.01.088
51. Fouts DE, Pieper R, Szpakowski S, Pohl H, Knoblach S, Suh M-J, et al. Integrated next-generation sequencing of 16S RDNA and metaproteomics differentiate the healthy urine microbiome from asymptomatic bacteriuria in neuropathic bladder associated with spinal cord injury. J Transl Med. (2012) 10(1):174. doi: 10.1186/1479-5876-10-174
52. Kispal ZF, Vajda P, Kardos D, Klymiuk I, Moissl-Eichinger C, Castellani C, et al. The local microbiome after pediatric bladder augmentation: intestinal segments and the native urinary bladder host similar mucosal microbiota. J Pediatr Urol. (2019) 15(1):30.e1–7. doi: 10.1016/j.jpurol.2018.07.028
53. Burnett LA, Hochstedler BR, Weldon K, Wolfe AJ, Brubaker L. Recurrent urinary tract infection: association of clinical profiles with urobiome composition in women. Neurourol Urodyn. (2021) 40(6):1479–89. doi: 10.1002/nau.24707
54. Neugent ML, Hulyalkar NV, Kumar A, Xing C, Zimmern PE, Shulaev V, et al. Urinary glycosaminoglycans are associated with recurrent UTI and urobiome ecology in postmenopausal women. ACS Infect Dis. (2023) 9(4):1022–32. doi: 10.1021/acsinfecdis.3c00027
55. Vaughan MH, Mao J, Karstens LA, Ma L, Amundsen CL, Schmader KE, et al. The urinary microbiome in postmenopausal women with recurrent urinary tract infections. J Urol. (2021) 206(5):1222–31. doi: 10.1097/ju.0000000000001940
56. Barnes HC, Wolff B, Abdul-Rahim O, Harrington A, Hilt EE, Price TK, et al. A randomized clinical trial of standard versus expanded cultures to diagnose urinary tract infections in women. J Urol. (2021) 206(5):e1212–21. doi: 10.1097/ju.0000000000001949
57. Moustafa A, Li W, Singh H, Moncera KJ, Torralba MG, Yu Y, et al. Microbial metagenome of urinary tract infection. Sci Rep. (2018) 8:4333. doi: 10.1038/s41598-018-22660-8
58. Hasman H, Saputra D, Sicheritz-Ponten T, Lund O, Svendsen CA, Frimodt-Moller N, et al. Rapid whole-genome sequencing for detection and characterization of microorganisms directly from clinical samples. J Clin Microbiol. (2013) 52(1):139–46. doi: 10.1128/jcm.02452-13
59. Berg G, Rybakova D, Fischer D, Cernava T, Vergès M-CC, Charles T, et al. Microbiome definition re-visited: old concepts and new challenges. Microbiome. (2020) 8(1):103. doi: 10.1186/s40168-020-00875-0
60. Ackerman AL, Underhill DM. The mycobiome of the human urinary tract: potential roles for fungi in urology. Ann Transl Med. (2017) 5(2):31. doi: 10.21037/atm.2016.12.69
61. Ackerman AL, Anger JT, Khalique MU, Ackerman JE, Tang J, Kim J, et al. Optimization of DNA extraction from human urinary samples for mycobiome community profiling. PLoS One. (2019) 14(4):e0210306. doi: 10.1371/journal.pone.0210306
62. Neugent ML, Kumar A, Hulyalkar NV, Lutz KC, Nguyen VH, Fuentes JL, et al. Recurrent urinary tract infection and estrogen shape the taxonomic ecology and function of the postmenopausal urogenital microbiome. Cell Rep Med. (2022) 3(10):100753. doi: 10.1016/j.xcrm.2022.100753
63. Jiang L, Wang H, Luo L, Pang X, Liu T, Sun L, et al. Urogenital microbiota-driven virulence factor genes associated with recurrent urinary tract infection. Front Microbiol. (2024) 15:1344716. doi: 10.3389/fmicb.2024.1344716
64. Garretto A, Thomas-White K, Wolfe AJ, Putonti C. Detecting viral genomes in the female urinary microbiome. J Gen Virol. (2018) 99(8):1141–46. doi: 10.1099/jgv.0.001097
65. Gharanfoli A, Mahmoudi E, Torabizadeh R, Torabizadeh R, Katiraii F, Faraji S. Isolation, characterization, and molecular identification of Candida species from urinary tract infections. Curr Med Mycol. (2019) 5(2):33–6. doi: 10.18502/cmm.5.2.1159
66. Bongomin F, Gago S, Oladele R, Denning D. Global and multi-national prevalence of fungal diseases—estimate precision. J Fungi. (2017) 3(4):57. doi: 10.3390/jof3040057
67. Wang F, Wang Z, Tang J. The interactions of Candida albicans with gut bacteria: a new strategy to prevent and treat invasive intestinal candidiasis. Gut Pathog. (2023) 15(1):30. doi: 10.1186/s13099-023-00559-8
68. Klaerner HG, Uknis ME, Acton RD, Dahlberg PS, Carlone-Jambor C, Dunn DL. Candida albicans and Escherichia coli are synergistic pathogens during experimental microbial peritonitis. J Surg Res. (1997) 70(2):161–65. doi: 10.1006/jsre.1997.5110
69. Bandara HMHN, Yau JYY, Watt RM, Jin LJ, Samaranayake LP. Escherichia coli and its lipopolysaccharide modulate in vitro Candida biofilm formation. J Med Microbiol. (2009) 58(12):1623–31. doi: 10.1099/jmm.0.012989-0
70. Farrokhi Y, Al-Shibli B, Al-Hameedawi DF-J, Neshati Z, Makhdoumi A. Escherichia Coli enhances the virulence factors of Candida albicans, the cause of vulvovaginal candidiasis, in a dual bacterial/fungal biofilm. Res Microbiol. (2021) 172(4-5):103849. doi: 10.1016/j.resmic.2021.103849
71. Díaz-Navarro M, Irigoyen Á, Palomo M, Escribano P, Guinea J, Burillo A, et al. In vitro study to assess modulation of Candida biofilm by Escherichia coli from vaginal strains. Biofilm. (2023) 5:100116–16. doi: 10.1016/j.bioflm.2023.100116
72. Salabura A, Łuniewski A, Kucharska M, Myszak D, Dołęgowska B, Ciechanowski K, et al. Urinary tract virome as an urgent target for metagenomics. Life. (2021) 11(11):1264–64. doi: 10.3390/life11111264
73. Santiago-Rodriguez TM, Ly M, Bonilla N, Pride DT. The human urine virome in association with urinary tract infections. Front Microbiol. (2015) 6:14. doi: 10.3389/fmicb.2015.00014
74. Hajjo R, Sabbah DA, Al Bawab AQ. Unlocking the potential of the human microbiome for identifying disease diagnostic biomarkers. Diagnostics. (2022) 12(7):1742. doi: 10.3390/diagnostics12071742
75. Simpson CA, Diaz-Arteche C, Eliby D, Schwartz OS, Simmons JG, Cowan CSM. The gut microbiota in anxiety and depression—a systematic review. Clin Psychol Rev. (2021) 83:101943. doi: 10.1016/j.cpr.2020.101943
76. Johnson JA, Delaney LF, Ojha V, Rudraraju M, Hintze KR, Siddiqui NY, et al. Commensal urinary lactobacilli inhibit major uropathogens in vitro with heterogeneity at species and strain level. Front Cell Infect Microbiol. (2022) 12:870603. doi: 10.3389/fcimb.2022.870603
77. Szczerbiec D, Słaba M, Torzewska A. Substances secreted by Lactobacillus spp. From the urinary tract microbiota play a protective role against Proteus mirabilis infections and their complications. Int J Mol Sci. (2023) 25(1):103–3. doi: 10.3390/ijms25010103
78. Abdul-Rahim O, Wu Q, Price TK, Pistone G, Diebel K, Bugni TS, et al. Phenyl-lactic acid is an active ingredient in bactericidal supernatants of Lactobacillus crispatus. J Bacteriol. (2021) 203(19):e00360–21. doi: 10.1128/jb.00360-21
79. Song CH, Kim YH, Naskar M, Hayes BW, Abraham MA, Noh JH, et al. Lactobacillus crispatus limits bladder uropathogenic E. coli infection by triggering a host type I interferon response. Proc Natl Acad Sci USA. (2022) 119(33):e2117904119. doi: 10.1073/pnas.2117904119
80. Nguyen VH, Khan F, Shipman BM, Neugent ML, Hulyalkar NV, Cha NY, et al. A semi-quantitative assay to measure glycosaminoglycan degradation by the urinary microbiota. Front Cell Infect Microbiol. (2021) 11:803409. doi: 10.3389/fcimb.2021.803409
81. Hilt EE, Putonti C, Thomas-White K, Lewis AL, Visick KL, Gilbert NM, et al. Aerococcus urinae isolated from women with lower urinary tract symptoms: in vitro aggregation and genome analysis. J Bacteriol. (2020) 202(13):e00170–20. doi: 10.1128/jb.00170-20
82. Du J, Khemmani M, Halverson T, Ene A, Limeira R, Tinawi L, et al. Cataloging the phylogenetic diversity of human bladder bacterial isolates. Genome Biol. (2024) 25(1):75. doi: 10.1186/s13059-024-03216-8
83. Hulyalkar NV, Sharon BM, Shipman BM, Arute AP, Zimmern PE, De Nisco NJ. Complete genome sequences of three Lactobacillus crispatus strains isolated from the urine of postmenopausal women. Microbiol Resour Announc. (2021) 10(48):e01017–21. doi: 10.1128/mra.01017-21
84. Shipman BM, Nguyen A, Sharon BM, Zimmern PE, De Nisco NJ. Complete genome sequences of three Lactobacillus gasseri urine isolates obtained from postmenopausal women. Microbiol Resour Announc. (2022) 11(9):e00481–22. doi: 10.1128/mra.00481-22
85. London LY, Lim CH, Modliszewski JL, Siddiqui NY, Sysoeva TA. Draft genomes of Lactobacillus delbrueckii and Klebsiella pneumoniae coexisting within a female urinary bladder. Microbiol Resour Announc. (2023) 12(10):e00305–23. doi: 10.1128/mra.00305-23
86. Stegman N, Steiling M, Sedano C, Jackson B, Putonti C. Draft genome sequences of five Lactobacillus gasseri strains isolated from voided urine samples. Microbiol Resour Announc. (2024) 13(2):e01119–23. doi: 10.1128/mra.01119-23
87. Johnson JA, Modliszewski JL, Siddiqui NY, Sysoeva TA. Draft genome sequence of a Lactobacillus gasseri strain isolated from the catheterized urine of a healthy postmenopausal woman. Microbiol Resour Announc. (2022) 11(6):e00021–22. doi: 10.1128/mra.00021-22
88. Karstens L, Siddiqui NY, Zaza T, Barstad A, Amundsen CL, Sysoeva TA. Benchmarking DNA isolation kits used in analyses of the urinary microbiome. Sci Rep. (2021) 11(1):6186. doi: 10.1038/s41598-021-85482-1
89. Mrofchak R, Madden C, Evans MV, Hale VL. Evaluating extraction methods to study canine urine microbiota. PLoS One. (2021) 16(7):e0253989. doi: 10.1371/journal.pone.0253989
90. Vendrell JA, Henry S, Cabello-Aguilar S, Heckendorn E, Godreuil S, Solassol J. Determination of the optimal bacterial DNA extraction method to explore the urinary microbiota. Int J Mol Sci. (2022) 23(3):1336. doi: 10.3390/ijms23031336
91. Vaughan MH, Zemtsov GE, Dahl EM, Karstens L, Ma L, Siddiqui NY. Concordance of urinary microbiota detected by 16S ribosomal RNA amplicon sequencing vs expanded quantitative urine culture. Am J Obstet Gynecol. (2022) 227(5):773–75. doi: 10.1016/j.ajog.2022.06.031
92. Kachroo N, Lange D, Penniston KL, Stern JM, Tasian GE, Bajic P, et al. Standardization of microbiome studies for urolithiasis: an international consensus agreement. Nat Rev Urol. (2021) 18(5):303–11. doi: 10.1038/s41585-021-00450-8
Keywords: urinary microbiome, urobiome, pediatric urobiome, pediatric bladder microbiome, urinary mycobiome, urinary virome
Citation: Jeries LM, Sysoeva TA, Karstens L and Kelly MS (2024) Synthesis of current pediatric urinary microbiome research. Front. Pediatr. 12:1396408. doi: 10.3389/fped.2024.1396408
Received: 5 March 2024; Accepted: 29 May 2024;
Published: 18 June 2024.
Edited by:
Walid Farhat, University of Wisconsin-Madison, United StatesReviewed by:
Hannah Zierden, University of Maryland, College Park, United StatesAlan J. Wolfe, Loyola University Chicago, United States
© 2024 Jeries, Sysoeva, Karstens and Kelly. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tatyana A. Sysoeva, dGF0eWFuYS5zeXNvZXZhQHVhaC5lZHU=