
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr. , 03 April 2024
Sec. Genetics of Common and Rare Diseases
Volume 12 - 2024 | https://doi.org/10.3389/fped.2024.1360867
 Takuya Hiraide1
Takuya Hiraide1 Taiju Hayashi1,2Yusuke Ito1Rei Urushibata1Hiroshi Uchida1Ryoichi Kitagata1Hidetoshi Ishigaki1
Taiju Hayashi1,2Yusuke Ito1Rei Urushibata1Hiroshi Uchida1Ryoichi Kitagata1Hidetoshi Ishigaki1 Tsutomu Ogata1,2,3
Tsutomu Ogata1,2,3 Hirotomo Saitsu2
Hirotomo Saitsu2 Tokiko Fukuda1,4*
Tokiko Fukuda1,4*
Background: Galloway-Mowat syndrome (GAMOS) is a rare genetic disease characterized by early-onset nephrotic syndrome and microcephaly with central nervous system abnormalities. Pathogenic variants in genes encoding kinase, endopeptidase, and other proteins of small size (KEOPS) complex subunits cause GAMOS. The subunit TPRKB (TP53RK binding protein) has been reported in only two patients with GAMOS with homozygous missense variants.
Clinical report: Herein, we described a three-year-old male with GAMOS. He exhibited developmental delay, developmental regression, microcephaly, distinctive facial features, skeletal abnormalities, and epilepsy. Brain magnetic resonance imaging revealed progressive brain atrophy, delayed myelination, T2-hypointense signals in the thalamus, and multiple intracranial abnormal signals on diffusion-weighted imaging. He presented with relapsing nephrotic proteinuria exacerbated by upper respiratory tract infections and progressive renal function decline. Exome sequencing identified compound heterozygous missense and frameshift variants in TPRKB: c.224dup, p.(Ser76IlefsTer3) and c.247C>T, p.(Leu83Phe).
Conclusions: Our study supports that pathogenic TPRKB variants cause KEOPS complex-related GAMOS.
Galloway-Mowat syndrome (GAMOS; MIM#251300) is a rare genetic disease characterized by early-onset nephrotic syndrome (NS), microcephaly, and brain anomalies that result in severely delayed psychomotor development (1). The renal prognosis of GAMOS is poor, and renal replacement therapy or renal transplantation is necessary for survival (2). GAMOS is clinically and genetically heterogeneous. Other clinical features include dysmorphic facial features, skeletal anomalies, and esophageal hiatal hernias. Several genes have been reported to be associated with GAMOS, including WDR73, LAGE3, OSGEP, TP53RK, TPRKB, WDR4, NUP107, NUP133, GON7, YRDC, and PRDM15 (2–8).
TPRKB (TP53RK binding protein) (MIM*608680) encodes a subunit of the highly conserved kinase, endopeptidase, and other proteins of small size (KEOPS) complex. The human KEOPS complex comprises OSGEP, TP53RK, TPRKB, LAGE3, and GON7 (9). It regulates the universal chemical modification of tRNA, N6-threonylcarbamoyl adenosine (t6A), which is essential for normal cell growth and accurate translation by ribosomes (10). The KEOPS complex is involved in human podocyte migration through impaired cell proliferation, increased apoptosis, genomic instability, and defects in actin regulation (4). Biallelic pathogenic variants in genes of the KEOPS complex are responsible for GAMOS in many patients (3, 4). Renal biopsies of patients with KEOPS complex-related GAMOS reveals mainly focal segmental glomerulosclerosis or diffuse mesangial sclerosis, with partial podocyte foot process effacement on electron microscopy (4). In contrast, GAMOS 5 (MIM#617731) caused by TPRKB has only been reported in two patients with homozygous missense variants (4). Herein, we report the case of a patient with GAMOS with compound heterozygous missense and frameshift variants in TPRKB.
After 40 weeks of gestation without asphyxia, a Japanese boy was born to nonconsanguineous, healthy parents as their first child. His birth weight was 3,450 g [+1.04 standard deviation (SD)], his length was 51.0 cm (+0.90 SD), and his occipitofrontal circumference was 35.0 cm (+1.25 SD). Distinctive facial features such as widely spaced eyes and pointed chin, pectus excavatum, tapered fingers, esotropia, and muscle hypotonia, were noted (Figures 1A–D). He could hold his head up at five months of age and sit with support at eight months of age. At nine months of age, he babbled but could not maintain a sitting position, even with support. At one year and one month of age, antiepileptic drugs were initiated because of the onset of epileptic spasms and focal to bilateral tonic-clonic seizures. His development regressed, losing interest in toys at one year and three months of age and becoming unable to hold his head up at one year and eight months of age. Tube feeding was initiated because of dysphagia and weight loss at two years and one month of age, followed by gastrostomy at three years of age.
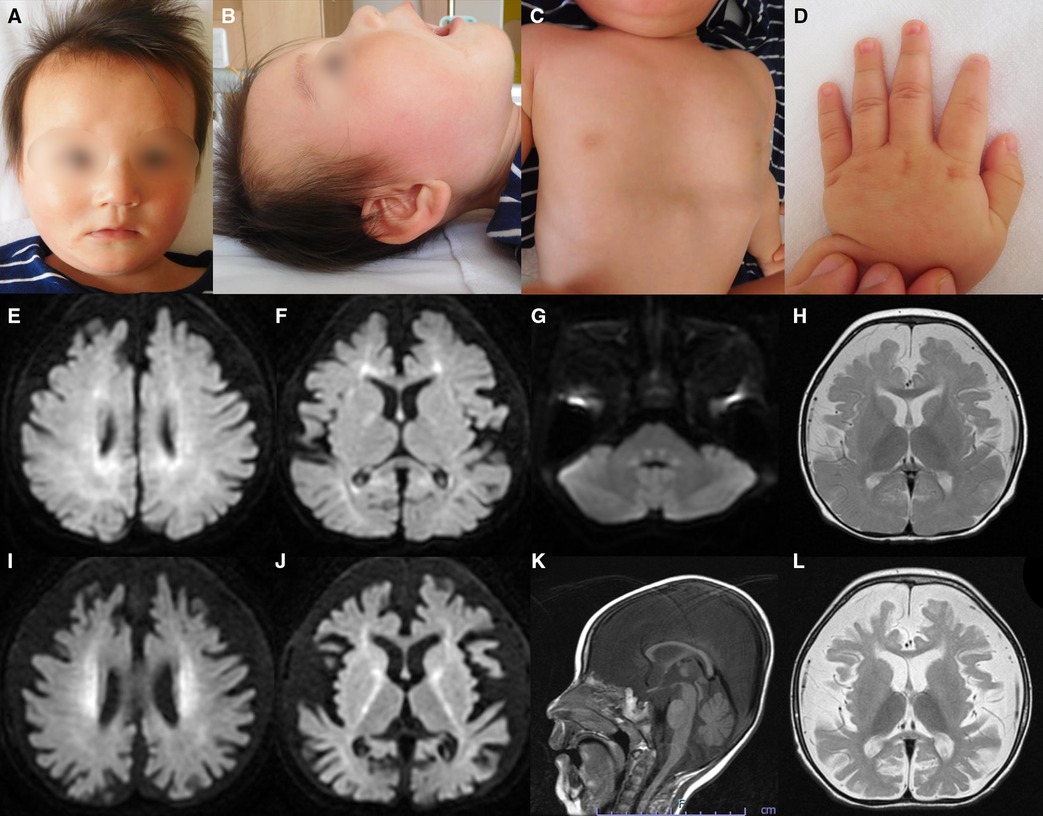
Figure 1. Representative clinical findings. (A–D) Photographs of the patient at ten months of age. Widely spaced eyes, epicanthus, left esotropia, medial sparse eyebrow, pointed chin, and thin upper lip vermilion are observed (A, B). Pectus excavatum (C) and tapered fingers (D) are recognized. (E–L) Brain magnetic resonance imaging (MRI) at ten months of age (E–H) and one year and eight months of age (I–L). Axial diffusion-weighted imaging (DWI) demonstrated elevated signal in the bilateral periventricular white matter, deep and subcortical white matter of the bilateral posterior lobe, posterior limb of the internal capsule, and dorsal part of the pons (E–G). An axial T2-weighted image shows brain atrophy with frontal white matter dominance and diffuse hypointense signal of the thalamus (H) DWI hyperintensities of the bilateral corona radiata and internal capsule are more distinct (I, J). A sagittal T1-weighted image shows atrophy of cerebellar vermis and thinning of the corpus callosum (K). An axial T2-weighted image reveals progressive brain atrophy and no hypointensity in the subcortical white matter. Hypointensity in the anterior limb of the internal capsule is obscured (L).
He was diagnosed with proteinuria at six months of age. The spot urine protein/creatinine ratio (UPCR) was 3.33 g/g at one year and one month of age. At 2 years and eight months of age, he was diagnosed with NS due to increased proteinuria (UPCR 17.23 g/g) and hypoalbuminemia (Alb 2.9 g/dl) induced by an acute upper respiratory tract infection. As the infection improved, the proteinuria decreased (UPCR 0.97 g/g). He initiated treatment with valsartan. Subsequently, NS relapsed during respiratory infections, with serum albumin ranging from 2.2 to 3.9 g/dl and UPCR ranging from 0.73 to 17.23 g/g. Serum creatine was low (0.15 mg/dl), while the cystatin C value, which was 0.46 mg/L at 2 years of age, increased to 0.66 mg/L at 3 years of age, suggesting a progressive decline in renal function. Renal biopsy was not performed because of respiratory depression during anesthesia.
Metabolic screenings of blood and cerebrospinal fluid, including amino acids, lactic acid, and pyruvic acid, were unremarkable. Interictal electroencephalogram at one year and eight months of age revealed multifocal spikes with slow background activity. Bilateral visual evoked potentials were absent. Electrocardiography, auditory brainstem response, nerve conduction studies, electroretinography, and echocardiography findings were normal. Radiography revealed no bone abnormalities other than scoliosis and pectus excavatum. Abdominal computed tomography scans of the kidneys, bladder, liver, and spleen revealed no abnormalities. Brain magnetic resonance imaging (MRI) showed a diffuse T2-hypointense signal in the thalamus and numerous characteristic aberrant DWI signals in the white matter, internal capsule, and dorsal part of the pons (Figures 1E–J, L). At one year and eight months of age, there was no T2-hypointensity in the subcortical white matter, suggesting hypomyelination (Figure 1L). The T2-hypointensity in the anterior limb of the internal capsule observed at 10 months of age was indistinct at one year and eight months of age, suggesting demyelination (Figures 1H, L). Progressive atrophy of the cerebral hemispheres and cerebellum was observed (Figures 1K, L).
The last physical examination at three years of age showed a body weight of 12.0 kg (−1.1 SD), height of 97.0 cm (+1.1 SD), and head circumference of 42.5 cm (−4.4 SD). He had a social smile, but his visual tracking was poor. He had constant stridor. Hypotonia of the upper limbs and trunk and spasticity of the lower limbs were noted. His deep tendon reflexes were hyperactive with lower-extremity dominance, and the Babinski reflex and ankle clonus were observed. These findings were suggestive of GAMOS. Progression of neuronal symptoms and repeated worsening of urinary proteinuria due to infectious disease are shown along the timeline in Figure 2.
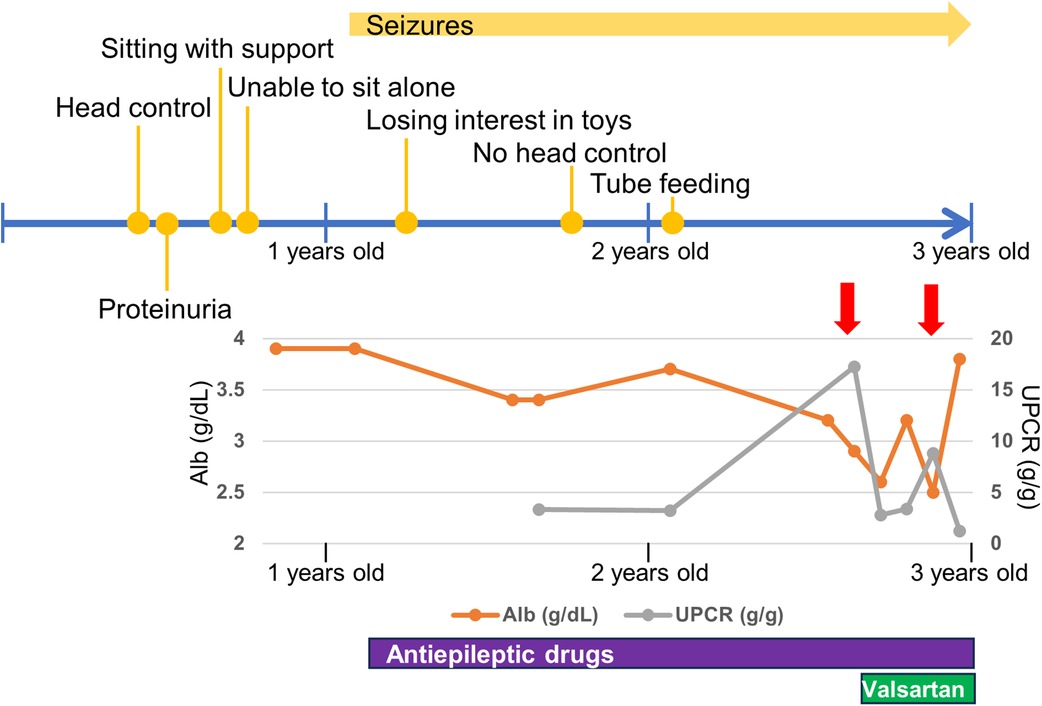
Figure 2. Timeline of neurological and renal symptoms. Red allows indicate respiratory infections. Alb, serum albumin; UPCR, spot urine protein/creatinine ratio.
This study was approved by the Institutional Review Board Committee of Hamamatsu University School of Medicine (16-076) and was performed after obtaining written informed consent. Consent to publish identifiable images was obtained from the patient's parents.
Genomic DNA was extracted from the blood leukocytes of this family. The patient's DNA for exome sequencing was captured using an xGen Exome Research Panel v2 kit (IDT, Coralville, IA, USA) and sequenced on a NextSeq500 (Illumina, San Diego, CA, USA) with 75-bp paired-end reads. Data processing, variant calling, annotation, and filtering were performed as previously described (11). We identified compound heterozygous variants of TPRKB (NM_016058.5), c.224dup, p.(Ser76IlefsTer3) and c.247C > T, p.(Leu83Phe) (Figure 3A). The allele frequencies of the c.224dup and c.247C>T variants were 0.0055% (6/108,604 alleles) and 0.0064% (7/108,604 alleles), respectively, in the ToMMo 54KJPN Allele Frequency Panel (v20230626) (https://jmorp.megabank.tohoku.ac.jp/). The allele frequencies in an East Asian population in the gnomAD v4.0.0 (http://gnomad.broadinstitute.org/) were 0.005039% (2/39,694 alleles) and 0.002520% (1/39,684 alleles), respectively. The c.247C>T, p.(Leu83Phe) variant was predicted to be deleterious using in silico pathogenicity prediction tools (Supplementary Table S1), and the Leu83 residue is highly evolutionarily conserved (Figure 3B). The Leu83Phe substitution, like the previously reported TPRKB variants Leu136Pro (α6) and Tyr149Cys (α7), is located at the α helix (α3), a deeply buried position that may affect the structural integrity of the protein (4, 12) (Figure 2B; Supplementary Figure S1). No other likely pathogenic variants were identified among the candidate variants (Supplementary Tables S1, S2). No candidate pathogenic copy number variants were detected using the eXome-hidden Markov model or jNord methods (13, 14). According to the American College of Medical Genetics and Genomics guideline 2015, the c.224dup variant was classified as likely pathogenic and the c.247C>T variant uncertain significance (Supplementary Tables S1, S2). However, since our case had characteristic GAMOS symptoms such as nephrotic proteinuria and central nervous system abnormalities, we concluded that the TPRKB variants were likely responsible in this case.
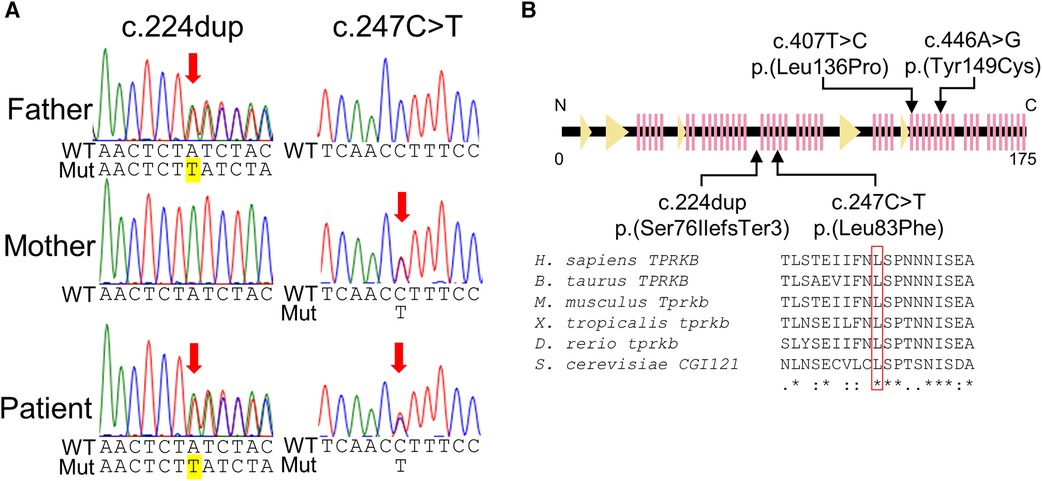
Figure 3. TPRKB variants in the patients with Galloway-Mowat syndrome. (A) Compound heterozygous variants in TPRKB. Sanger sequencing shows a paternal T duplication (yellow) on the left and a maternal missense variant on the right. (B) Schematic presentation of the TPRKB protein and location of altered residues. Previously reported TPRKB variants are depicted above, and the variants identified in our case are shown below. The predicted secondary structure modified from the PDB (code 6WQX; https://www.rcsb.org/) is indicated for β-strand and α-helix conformation as triangle and zigzag lines, respectively. Multiple amino acid sequences of TPRKB were aligned using the ClustalW tool (http://www.genome.jp/tools/clustalw).
In this study, we describe the case of a patient with novel compound heterozygous TPRKB variants. Two individuals with GAMOS with TPRKB variants have been previously described, and the clinical manifestations of the three cases of TPRKB variants, including our patient, are summarized in Table 1 (4). Microcephaly, global developmental delay, spasticity, and distinctive facial features were common to all three cases. Most patients with GAMOS associated with the KEOPS complex had microcephaly, developmental delay, and distinctive facial features, whereas spasticity was not commonly reported (9/33) (4). This may be because patients with GAMOS with TPRKB variants were relatively older children in whom spasticity was evident due to progression of brain atrophy. In our case, proteinuria was diagnosed earlier than in other patients with TPRKB variants, possibly because of the loss-of-function variant. The age of onset of proteinuria in patients with TPRKB-related GAMOS was later than that in patients with KEOPS complex-related GAMOS other than TPRKB-related GAMOS (median, three months) (4). Furthermore, the median age of death for GAMOS cases with variants of KEOPS complex-encoding genes other than TPRKB was reported to be six months of age. In contrast, one individual with TPRKB variant died at 6.8 years of age, and two cases survived at three and six years. TPRKB-related GAMOS may be milder than GAMOS caused by other KEOPS complex-related genes.
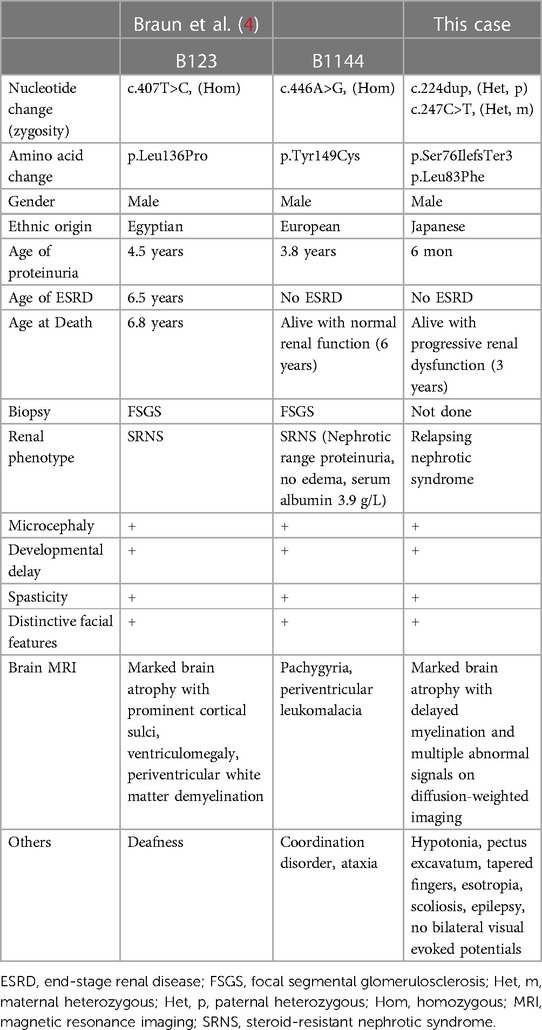
Table 1. Clinical features of TPRKB-associated Galloway-Mowat syndrome.
The most frequently observed brain MRI anomalies in the GAMOS associated with the KEOPS complex include cortical and cerebellar atrophy, gyration abnormalities, and myelination defects (4). Abnormal DWI signals, as in our case, have been reported in patients with GAMOS with pathogenic variants of TP53RK and OSGEP, which belong to the KEOPS complex (15, 16). MRI of another case of GAMOS attributed to TP53RK showed diffuse T2-hypointense signals in the thalamus (17). In animal studies, knockout of tprkb in zebrafish larvae recapitulated microcephaly, and Tprkb knockout mouse embryos showed a significant reduction in brain size, cortex length, cortex-midbrain midline length, and cortex width (4). Proteomic profiling of fibroblasts from patients with GAMOS caused by OSGEP variants revealed upregulation of the P2X7 receptor signaling complex pathway (15). In multiple sclerosis, the sustained activation of P2X7 receptors induces oligodendrocyte death, demyelination, neuroinflammatory processes, and neurodegeneration (18). The activation of P2X7 receptors may be involved in the brain phenotype of KEOPS complex-related GAMOS.
Our patient had NS with repeated relapses concurrently with respiratory infections. Increased edema in patients with congenital NS is especially common during infection (19). Relapsing NS with mild persistent proteinuria and relapses of the nephrotic range associated with upper respiratory tract infections have been described in familial cases of the NPHS1 variants encoding nephrin, an essential component of the interpodocyte-spanning slit diaphragm (20). The molecular mechanism of NS relapse caused by genetic defects remains to be elucidated and requires further investigation.
The patients with KEOPS complex-related GAMOS have severe renal and neurological symptoms and die in early childhood (4). Kidney complications are the direct cause of most GAMOS deaths, and the patients with GAMOS should be screened regularly for proteinuria and renal function and evaluated closely (21). The genetic diagnosis of GAMOS is useful for family planning, and prenatal and preimplantation testing is available after genetic counseling.
In conclusion, we identified a patient with GAMOS and compound heterozygous TPRKB variants who had nephrotic proteinuria, developmental delay, developmental regression, microcephaly, distinctive facial features, skeletal abnormalities, epilepsy, progressive brain atrophy, delayed myelination, T2-hypointense signals in the thalamus, and multiple intracranial abnormal signals. This is the first case of a patient with GAMOS associated with a frameshift variant in TPRKB. Further accumulation of cases is necessary to establish the common clinical features in patients with pathogenic TPRKB variants.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
The studies involving humans were approved by Institutional Review Board Committee of Hamamatsu University School of Medicine (16-076). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
THi: Data curation, Formal Analysis, Funding acquisition, Investigation, Resources, Visualization, Writing – original draft, Writing – review & editing. THa: Data curation, Investigation, Resources, Visualization, Writing – original draft, Writing – review & editing. YI: Resources, Writing – original draft, Writing – review & editing. RU: Data curation, Resources, Writing – original draft, Writing – review & editing. HU: Data curation, Investigation, Resources, Writing – original draft, Writing – review & editing. RK: Data curation, Investigation, Writing – original draft, Writing – review & editing. HI: Data curation, Resources, Writing – original draft, Writing – review & editing. TO: Conceptualization, Funding acquisition, Project administration, Supervision, Validation, Writing – original draft, Writing – review & editing. HS: Conceptualization, Formal Analysis, Funding acquisition, Methodology, Project administration, Software, Supervision, Validation, Writing – original draft, Writing – review & editing. TF: Conceptualization, Data curation, Funding acquisition, Methodology, Project administration, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article.
This research is supported by a donation from Hamamatsu City to the Endowed chair of Hamamatsu Child Health and Development. This work was supported by the Japan Agency for Medical Research and Development (AMED) (JP23ek0109549 to TO, JP23ek0109674 and JP23ek0109637 to HS), Grants-in-Aid for Scientific Research (B) (JP20H03641 and JP23H02875 to HS), Grant-in-Aid for Early-Career Scientists (23K14944 to TH), Grant-in-Aid for Research Activity Start-up (22K20852 to TH) from the Japan Society for the Promotion of Science (JSPS) KAKENHI, Japan Intractable Diseases (Nanbyo) Research Foundation (2020A02), and the Takeda Science Foundation.
We would like to thank the patients for participating in this work. We thank Editage (www.editage.jp) for English language editing.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2024.1360867/full#supplementary-material
1. Galloway WH, Mowat AP. Congenital microcephaly with hiatus hernia and nephrotic syndrome in two sibs. J Med Genet. (1968) 5(4):319–21. doi: 10.1136/jmg.5.4.319
2. Braun DA, Shril S, Sinha A, Schneider R, Tan W, Ashraf S, et al. Mutations in Wdr4 as a new cause of Galloway-Mowat syndrome. Am J Med Genet A. (2018) 176(11):2460–5. doi: 10.1002/ajmg.a.40489
3. Arrondel C, Missoury S, Snoek R, Patat J, Menara G, Collinet B, et al. Defects in T(6)a trna modification due to Gon7 and yrdc mutations lead to Galloway-Mowat syndrome. Nat Commun. (2019) 10(1):3967. doi: 10.1038/s41467-019-11951-x
4. Braun DA, Rao J, Mollet G, Schapiro D, Daugeron MC, Tan W, et al. Mutations in keops-complex genes cause nephrotic syndrome with primary microcephaly. Nat Genet. (2017) 49(10):1529–38. doi: 10.1038/ng.3933
5. Colin E, Huynh Cong E, Mollet G, Guichet A, Gribouval O, Arrondel C, et al. Loss-of-function mutations in Wdr73 are responsible for microcephaly and steroid-resistant nephrotic syndrome: Galloway-Mowat syndrome. Am J Hum Genet. (2014) 95(6):637–48. doi: 10.1016/j.ajhg.2014.10.011
6. Fujita A, Tsukaguchi H, Koshimizu E, Nakazato H, Itoh K, Kuraoka S, et al. Homozygous splicing mutation in Nup133 causes Galloway-Mowat syndrome. Ann Neurol. (2018) 84(6):814–28. doi: 10.1002/ana.25370
7. Mann N, Mzoughi S, Schneider R, Kühl SJ, Schanze D, Klämbt V, et al. Mutations in Prdm15 are a novel cause of Galloway-Mowat syndrome. J Am Soc Nephrol. (2021) 32(3):580–96. doi: 10.1681/asn.2020040490
8. Rosti RO, Sotak BN, Bielas SL, Bhat G, Silhavy JL, Aslanger AD, et al. Homozygous mutation in Nup107 leads to microcephaly with steroid-resistant nephrotic condition similar to Galloway-Mowat syndrome. J Med Genet. (2017) 54(6):399–403. doi: 10.1136/jmedgenet-2016-104237
9. Wan LC, Maisonneuve P, Szilard RK, Lambert JP, Ng TF, Manczyk N, et al. Proteomic analysis of the human keops complex identifies C14orf142 as a core subunit homologous to yeast Gon7. Nucleic Acids Res. (2017) 45(2):805–17. doi: 10.1093/nar/gkw1181
10. Perrochia L, Guetta D, Hecker A, Forterre P, Basta T. Functional assignment of keops/ekc complex subunits in the biosynthesis of the universal T6a trna modification. Nucleic Acids Res. (2013) 41(20):9484–99. doi: 10.1093/nar/gkt720
11. Komatsu K, Fukumura S, Minagawa K, Nakashima M, Saitsu H. A new case of concurrent existence of Prrt2-associated paroxysmal movement disorders with C.649dup variant and 16p11.2 microdeletion syndrome. Brain Dev. (2022) 44(7):474–9. doi: 10.1016/j.braindev.2022.03.008
12. Li J, Ma X, Banerjee S, Chen H, Ma W, Bode AM, et al. Crystal structure of the human prpk-tprkb complex. Commun Biol. (2021) 4(1):167. doi: 10.1038/s42003-021-01683-4
13. Fromer M, Moran JL, Chambert K, Banks E, Bergen SE, Ruderfer DM, et al. Discovery and statistical genotyping of copy-number variation from whole-exome sequencing depth. Am J Hum Genet. (2012) 91(4):597–607. doi: 10.1016/j.ajhg.2012.08.005
14. Uchiyama Y, Yamaguchi D, Iwama K, Miyatake S, Hamanaka K, Tsuchida N, et al. Efficient detection of copy-number variations using exome data: batch- and sex-based analyses. Hum Mutat. (2021) 42(1):50–65. doi: 10.1002/humu.24129
15. Alghamdi M A, Benabdelkamel H, Masood A, Saheb Sharif-Askari N, Hachim MY, Alsheikh H, et al. Genomic, proteomic, and phenotypic spectrum of novel O-sialoglycoprotein endopeptidase variant in four affected individuals with Galloway-Mowat syndrome. Front Genet. (2022) 13:806190. doi: 10.3389/fgene.2022.806190
16. Chen J, Ye GB, Huang JR, Peng M, Gu WY, Xiong P, et al. Novel Tp53rk variants cause varied clinical features of Galloway-Mowat syndrome without nephrotic syndrome in three unrelated Chinese patients. Front Mol Neurosci. (2023) 16:1116949. doi: 10.3389/fnmol.2023.1116949
17. Treimer E, Kalayci T, Schumann S, Suer I, Greco S, Schanze D, et al. Functional characterization of a novel Tp53rk mutation identified in a family with Galloway-Mowat syndrome. Hum Mutat. (2022) 43(12):1866–71. doi: 10.1002/humu.24472
18. Andrejew R, Oliveira-Giacomelli Á, Ribeiro DE, Glaser T, Arnaud-Sampaio VF, Lameu C, et al. The P2x7 receptor: central hub of brain diseases. Front Mol Neurosci. (2020) 13:124. doi: 10.3389/fnmol.2020.00124
19. Hölttä T, Jalanko H. Congenital nephrotic syndrome: is early aggressive treatment needed? Yes. Pediatr Nephrol. (2020) 35(10):1985–90. doi: 10.1007/s00467-020-04578-4
20. Kitamura A, Tsukaguchi H, Hiramoto R, Shono A, Doi T, Kagami S, et al. A familial childhood-onset relapsing nephrotic syndrome. Kidney Int. (2007) 71(9):946–51. doi: 10.1038/sj.ki.5002110
Keywords: TPRKB, Galloway-Mowat syndrome, exome sequencing, KEOPS complex, nephrotic proteinuria
Citation: Hiraide T, Hayashi T, Ito Y, Urushibata R, Uchida H, Kitagata R, Ishigaki H, Ogata T, Saitsu H and Fukuda T (2024) Case Report: Novel compound heterozygous TPRKB variants cause Galloway-Mowat syndrome. Front. Pediatr. 12:1360867. doi: 10.3389/fped.2024.1360867
Received: 24 December 2023; Accepted: 20 March 2024;
Published: 3 April 2024.
Edited by:
Pranoot Tanpaiboon, Quest Diagnostics, United StatesReviewed by:
Daniela Anne Braun, University Hospital Münster, Germany© 2024 Hiraide, Hayashi, Ito, Urushibata, Uchida, Kitagata, Ishigaki, Ogata, Saitsu and Fukuda. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tokiko Fukuda dG9raS1ma2RAaGFtYS1tZWQuYWMuanA=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.