 Fan Chen
Fan Chen Yuan Huang
Yuan Huang Zhihua Huang
Zhihua Huang Feng Fang
Feng Fang Sainan Shu
Sainan Shu- Department of Pediatrics, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
Background: This investigation aimed to examine the epidemiological characteristics of children with liver disease hospitalized for the first time between June 2012 and May 2022 in a tertiary hospital.
Methods: The study retrospectively recruited children aged between 29 days and 18 years who had been hospitalized for liver disease. Clinical characteristics were categorized by age and etiology, and time trends were assessed using linear regression analysis.
Results: A total of 4,313 children were recruited, with a median age of 0.7 (0.2–4.5) years, and 54.5% of the cases were in the 0–1 years age group. Infection was the primary cause of liver disease (30.0%), followed by undiagnosed cases (25.8%), biliary obstructive disease (15.9%), inherited metabolic liver disease (13.9%), and non-alcoholic fatty liver disease (NAFLD) (3.2%). Genetic diagnoses were established in 43.9% (478/1,088) of patients. The percentage of NAFLD demonstrated an upward trend from 1.2% in 2012 to 12.6% in 2022 (p = 0.006). In contrast, the percentage of cytomegalovirus hepatitis decreased from 13.3% in 2012 to 3.4% in 2022 (p = 0.002).
Conclusions: Liver disease in infancy makes up the largest group in pediatric liver disease. Infection remains the leading cause of pediatric liver disease. Hospital admissions for NAFLD in children have increased rapidly over the past decade, while cytomegalovirus hepatitis has declined markedly.
Introduction
Pediatric hepatology has branched out into nearby clinical fields, such as metabolic liver diseases and systemic diseases involving the liver (1, 2). Next-generation sequencing (NGS) has become a powerful diagnostic tool in recent years (3), particularly with the application of panel-based NGS and whole-exome sequencing (WES). The growing prevalence of gene sequencing in clinical settings has also altered the range of liver diseases (4, 5). A growing number of novel genetic diseases have been added to the list of pediatric liver disease etiologies. However, pediatric liver disease continues to be a major challenge around the world (6). Certain liver diseases that manifest during childhood serve as antecedents to chronic liver disease, cirrhosis, and hepatocellular carcinoma in adulthood (7). Prompt identification and intervention have the potential to significantly modify the progression of the ailment and enhance the patient's overall well-being (8). Liver disease is not mentioned in the top fifteen causes of death for children, however, approximately two million fatalities per year are attributed to liver disease, which accounts for 4% of global mortality (9). According to 2000 data, approximately 15,000 children in the United States are hospitalized annually due to liver disease (10). To the best of our knowledge, the majority of current research focuses on particular liver disorders, including cholestasis and inherited liver disease, or a particular demographic. Further elaboration is necessary to estimate disease prevalence effectively and prioritize healthcare policies.
Hubei Province is situated in central China and has a resident population of 58.44 million people, making it the tenth-largest province in China by population. To further understand the epidemiological characteristics of pediatric liver disease, to facilitate early diagnosis and treatment, a retrospective study on children admitted to the hospital with liver disease was conducted at Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology between June 2012 and May 2022. Our study aims to analyze the clinical spectrum of a large cohort of hospitalized children with liver disease and investigate the time trend. To the best of our knowledge, this is the first comprehensive elucidation of the disease spectrum of pediatric liver disease using a large sample population in Hubei Province, China.
Materials and methods
Patient selection and study design
Participant information was acquired from medical records. Some of the missing information was collected through follow-up visits to outpatients or via the telephone. The data was extracted by reading the discharge diagnoses, which were categorized according to the 9th/10th revision of the International Classification of Diseases. Subsequently, each medical record was thoroughly reviewed to exclude cases that did not meet the inclusion criteria. Two experienced pediatric hepatologists determined the cause of the disease based on current opinions or guidelines. The participants were eligible if they met the following criteria: (i) they were children admitted to the pediatric ward, pediatric surgical ward, or pediatric intensive care unit (ICU); (ii) they had clinical, biochemical, or imaging evidence of liver disease, which may present as chronic liver disease, an acute hepatic event, or liver damage involved in systemic disorders; iii) they were aged between 29 days and 18 years old. Exclusion criteria included the absence of liver disease-related manifestations such as jaundice, abnormal liver function tests, hepatomegaly/hepatosplenomegaly, or abnormal liver imaging, as well as re-hospitalization for liver disease. Duplicate cases were excluded, meaning that only the medical record from the initial hospitalization for liver disease was considered, even if the patient was hospitalized multiple times (Figure 1).
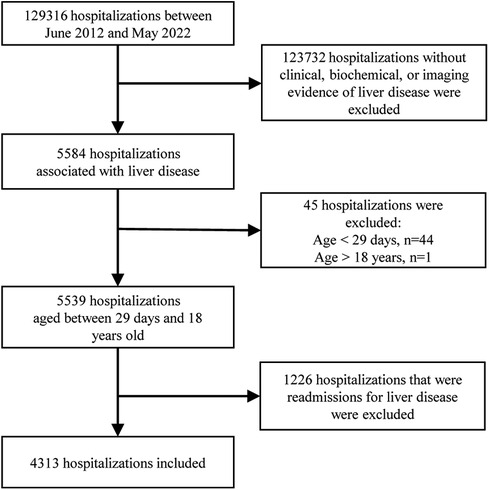
Figure 1. Flowchart of the enrollment of children with liver disease.
The clinical data we collected included demographic information, medical history, physical examination results, laboratory test results, genetic test results, and treatment outcomes. Disease outcome relied on the patient's condition during hospital discharge. Regarding the relevant criteria of the World Health Organization, “malnutrition” in this study refers to a z-score of weight-for-stature less than −2 SD. Cholestasis is defined as the presence of (i) serum conjugated bilirubin >17.1 μmol/L when total bilirubin is <85.5 μmol/L, or (ii) a conjugated component >20% of the total when total bilirubin is >85.5 μmol/L (11, 12). Diagnosis of cirrhosis is based on liver histology or clinical and imaging findings (13). The following criteria are used to define acute liver failure: (i) acute onset of liver illness without signs of chronic liver disease; (ii) biochemical evidence of severe liver injury; and (iii) coagulopathy that is not improved by vitamin K. Prothrombin time (PT) ≥15 s or international normalized ratio (INR) ≥1.5 in the presence of hepatic encephalopathy (HE) or PT ≥ 20 s or INR ≥ 2.0 in the absence of HE (14).
Patients were categorized into five age groups, namely: 0–1 years, 1–3 years, 3–6 years, 6–12 years, and 12–18 years. To determine the etiological distribution, we divided the study population into fourteen sections: infection, biliary obstructive disease, inherited metabolic liver disease (IMLD), non-alcoholic fatty liver disease (NAFLD), autoimmune liver disease, drug-induced liver injury, poison-induced liver injury, parenteral nutrition-associated liver disease, vascular/ischemic liver disease, malignant liver mass, benign liver mass, rheumatologic disease, endocrine disease, and undiagnosed cases.
Laboratory assessment
A laboratory assessment protocol was developed for children with liver disease based on a comprehensive physical examination and accurate clinical history. Examination measurements were conducted to assess a range of parameters including hematology, chemistry, microbiology, immunology, imaging, endoscopy, and histopathology. The selection of tests varied depending on the patient's age, clinical presentation, and the presence or absence of jaundice. Table 1 displays the laboratory methods used in our study.
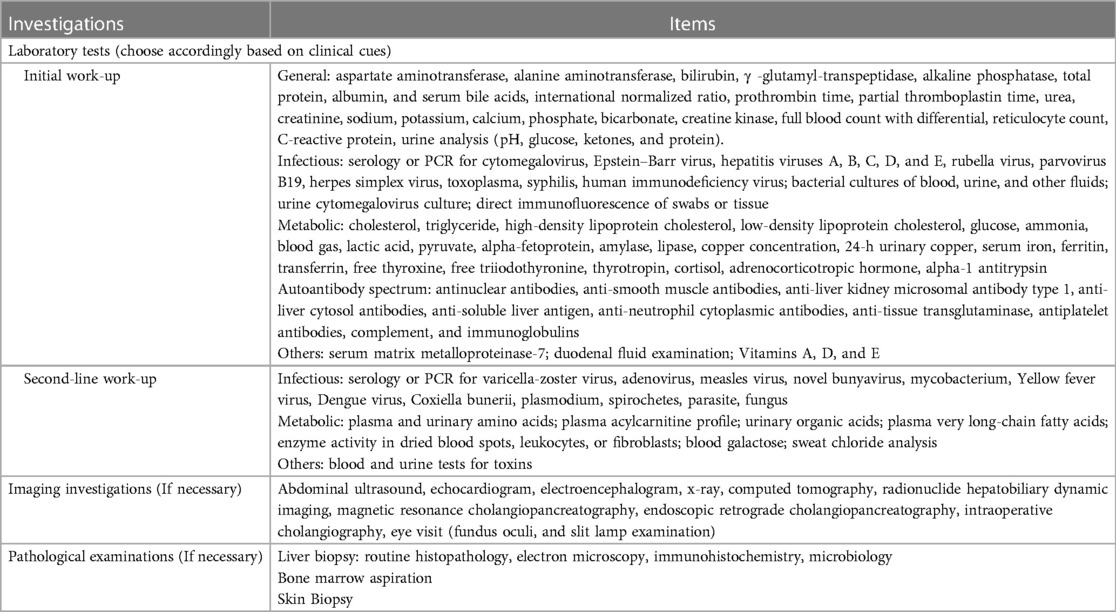
Table 1. Laboratory assessment protocol for children with liver disease.
Genetic sequencing
If clinical, biochemical, or pathology features highly suggest a specific genetic disease, medical practitioners may decide to conduct genetic tests after obtaining the patient or guardian's informed consent. From 2012 to 2014, Sanger sequencing was the dominant method of analysis at our center. However, starting in 2015, there has been a gradual shift towards NGS as the primary test. NGS includes panel-based NGS, clinical exome sequencing (CES), and WES. When selecting Sanger sequencing, we usually performed a single-gene test based on clinical features due to limited resources, particularly for strong phenotypes: SLC25A13 for citrin deficiency; JAG1 and NOTCH2 for Alagille syndrome; ATP8B1, ABCB11, and ABCB4 for phenotypic progressive familial intrahepatic cholestasis; HSD3B7 and AKR1D1 for phenotypic congenital bile acid synthesis defect; ATP7B for Wilson disease; UGT1A1 for unconjugated hyperbilirubinemia. Generally, patients with liver-focused disease phenotypes received panel-based NGS testing, while those with a wider range of disease phenotypes underwent CES or WES testing.
The genomic DNA of the patients and their parents was extracted from peripheral blood samples. The DNA fragments were amplified via polymerase chain reaction (PCR) and then hybridized with capture probes using the Agilent SureSelectXT2 Target Enrichment System (Agilent, Santa Clara, USA). The Illumina platform was used for sequencing in the genetic screening process. The human reference genome (hg19) was used to map the sequencing data, and the pathogenicity of the variations was categorized by the American College of Medical Genetics and Genomics recommendations (15). Sanger sequencing was used to validate all potential pathogenic variants, and quantitative real-time polymerase chain reaction (qPCR) was used to validate copy number variations (CNVs). Studies on cosegregation within the households were conducted. A positive genetic diagnosis was obtained for autosomal-recessive disease through the detection of two allelic genetic variations and for autosomal-dominant disease through the detection of one allelic genetic variation (15).
Statistical analysis
Data processing and analysis were conducted using SPSS 23.0. Continuous variables were expressed as either the mean ± standard deviation or the median and interquartile range, following an assessment of the normality of the distribution. The chi-square or Fisher's exact test was employed to compare counting data. When necessary, the Mann-Whitney U test or an independent t-test was used to assess comparisons in different groups. We calculated the overall number of eligible admissions for each year between 2012 and 2022, categorized by age groups and the specific causes of liver disease. To determine the percentage of admissions by etiology and age group for each year, we conducted comprehensive analyses. We performed time trend analyses using linear regression with each study year as a dependent variable.
This study was reviewed and approved by the Medical Ethics Committee of Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology (TJ-IRB20220607).
Results
Overall distribution characteristics
Medical records of 4,313 patients were used for statistical analysis in this study. The clinical characteristics of this cohort are summarized in Table 2. The median age was 0.7 (0.2–4.5) years (range: 0.1–17.1 years). The largest number of patients in the study (54.5%) were aged 0–1 years. The rest were split between the age groups of 1–3 years (13.2%), 3–6 years (12.6%), 6–12 years (15.6%), and 12–18 years (4.1%). Males made up 58.5% of the participants. 90.8% of patients were referred to our hospital within six months of onset. The chief complaints included jaundice (1,922 cases), abnormal liver function tests (2,018 cases), abnormal liver imaging (218 cases), and organomegaly (155 cases). Upon admission, medical evaluations revealed that 15.5% of the children recruited suffered from malnutrition.
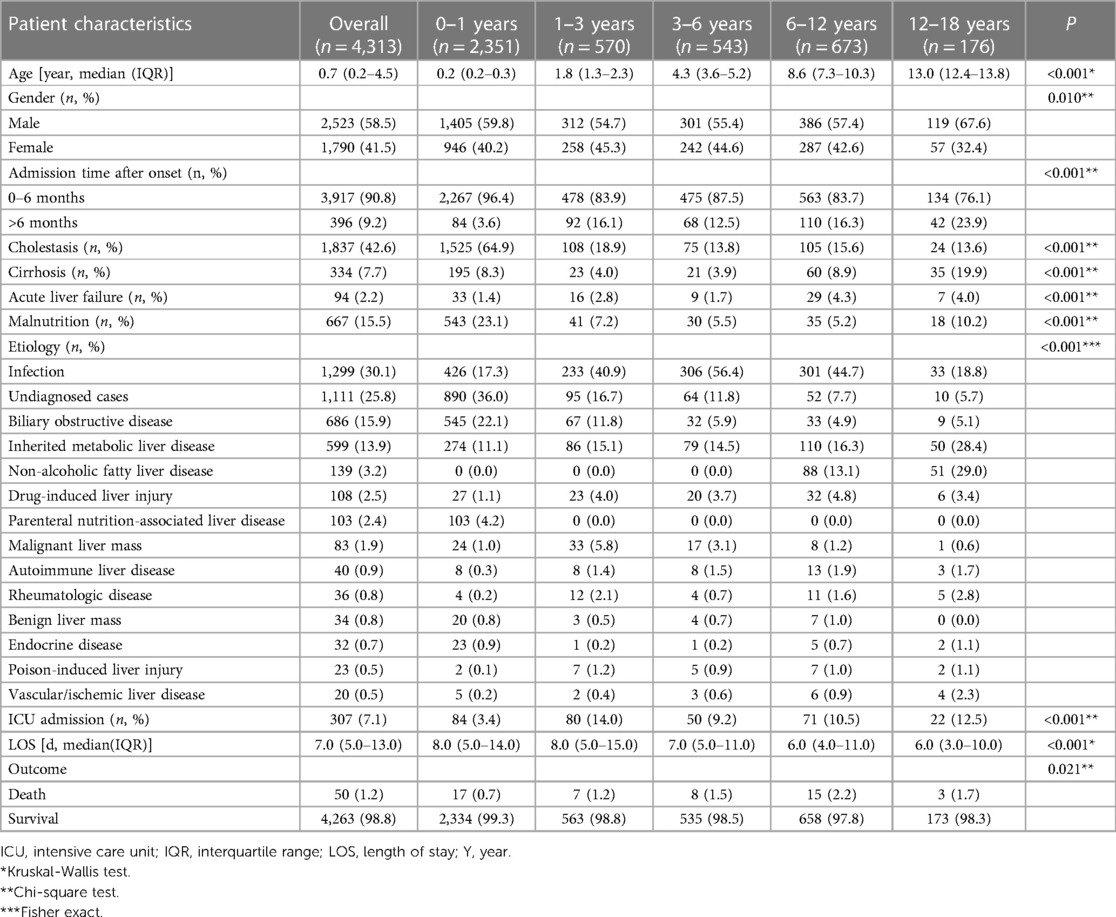
Table 2. Epidemiological characteristics of patients in different age groups.
Furthermore, 42.6% of the participants exhibited cholestasis; pertinently, the 0–1 years age group had the highest incidence at 64.9%, followed by 1–3 years at 18.9%, 6–12 years at 15.6%, 3–6 years at 13.8%, and 12–18 years at 13.6% (p < 0.001). While 7.7% of the study participants presented with cirrhosis, acute liver failure was observed in 2.2% of the cohort. The etiologic composition of cholestasis, cirrhosis, and acute liver failure is shown in Figure 2. Infantile cholestasis accounted for a substantial percentage of pediatric liver disease, including 36.4% (1,568/4,313) of cases in this population, and 85.4% of childhood cholestasis. Figure 3 shows the distribution of causes of infantile cholestasis. Differences in the diagnostic yield among age groups were observed, with the highest rate of diagnosis (94.3%) found in children aged 12–18 years and the lowest rate (62.1%) in the age group of 0–1 years. Among the 4,313 admitted patients, 50 died, corresponding to a mortality rate of 1.2%. Furthermore, 7.1% of the patients needed ICU admission. The median length of stay for this cohort was 7.0 (5.0–13.0) days.
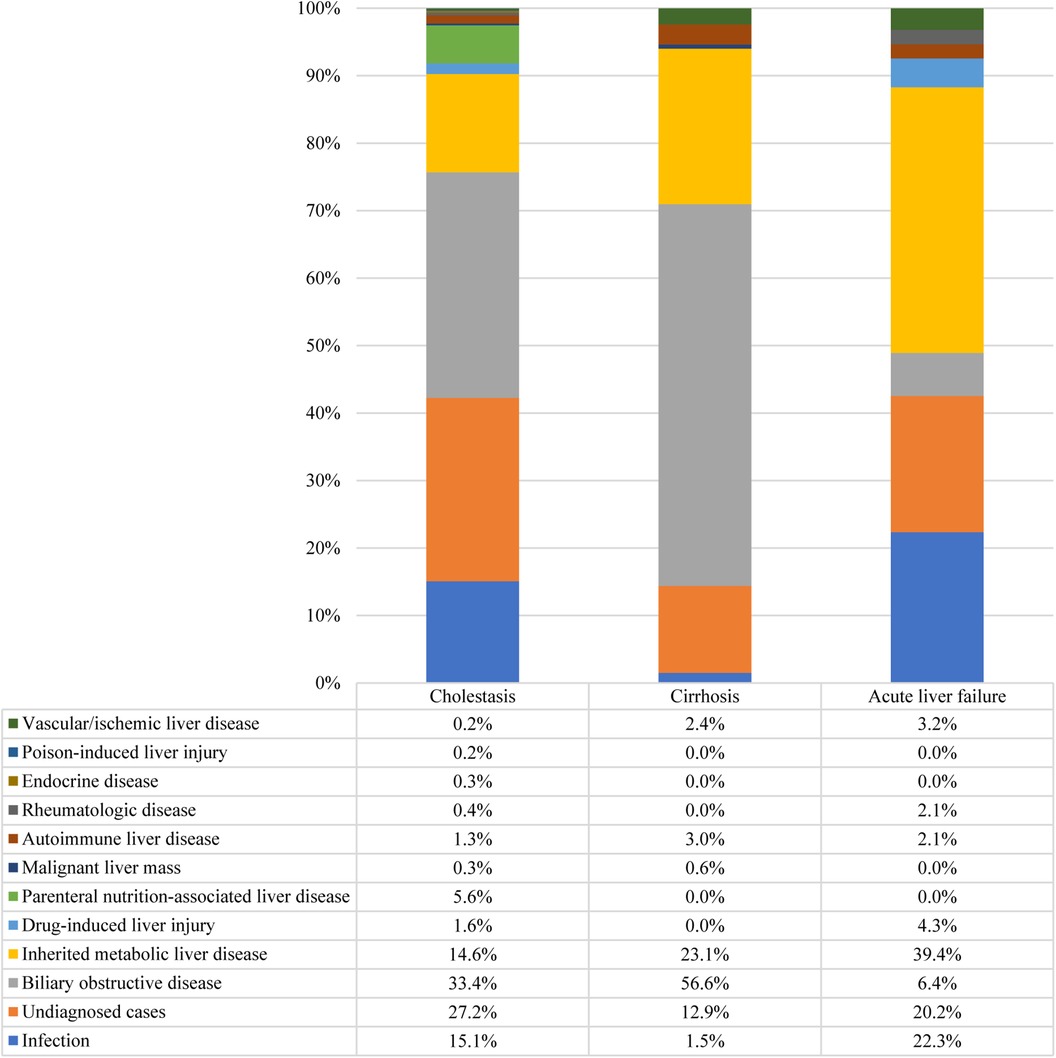
Figure 2. Distribution of causes of cholestasis, cirrhosis, and acute liver failure. There were 1,837 cases of cholestasis, 334 cases of cirrhosis, and 94 cases of acute liver failure.
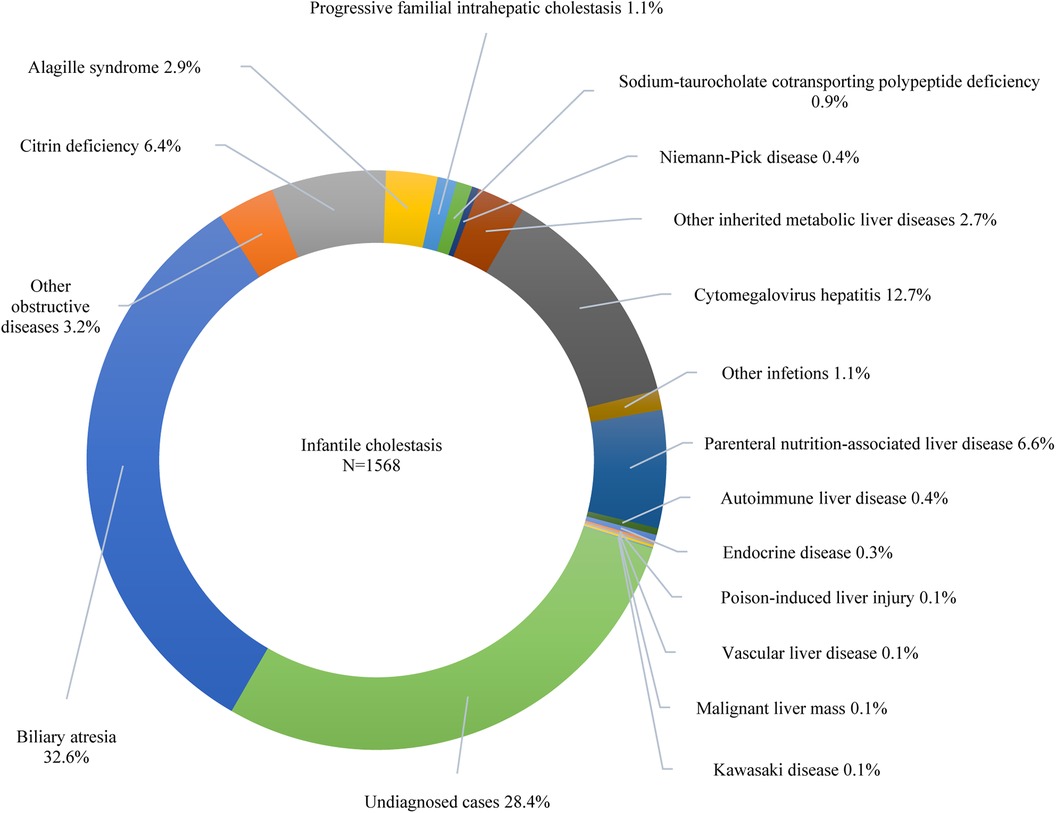
Figure 3. Distribution of causes of infantile cholestasis.
Distributional characteristics by etiology
Infection was found to be the primary causative agent accounting for 30.1% (1,299/4,313) of cases in this cohort. The distribution of pathogens resulted in 1,192 cases of viruses, 70 cases of bacteria, 27 cases of sepsis, seven cases of fungus, and three cases of parasites. Epstein-Barr virus (EBV) hepatitis was the most common cause of infectious liver disease, with 760 reported cases. The median age of affected patients was 4.7 (2.7–7.3) years, with 419 males and 341 females. Cytomegalovirus (CMV) hepatitis (347 cases) ranked second among the causes of infectious diseases, with 204 cases in males and 143 in females. The median age of affected individuals was 0.2 (0.2–0.3) years, with 199 cases presenting as infantile cholestasis, 146 cases with elevated aminotransferases, and two cases with hepatomegaly. The remaining pathogens are detailed in Table 3. Notably, 21 patients presented with acute liver failure. The leading causes were sepsis (11 cases), EBV infection (seven cases), non-A-E virus infection (two cases), and bacterial infection (one case). In addition, cirrhosis was seen in five children with hepatitis B. In this group of patients with infectious liver disease, 18 patients died during hospitalization, resulting in a mortality rate of 1.4%. The causes of death were diverse and included sepsis (eight cases), EBV infection (three cases), and infections caused by non-A-E virus, enterovirus, novel bunyavirus, staphylococcus aureus, klebsiella pneumoniae, acinetobacter baumannii, and candida albicans (one case each).
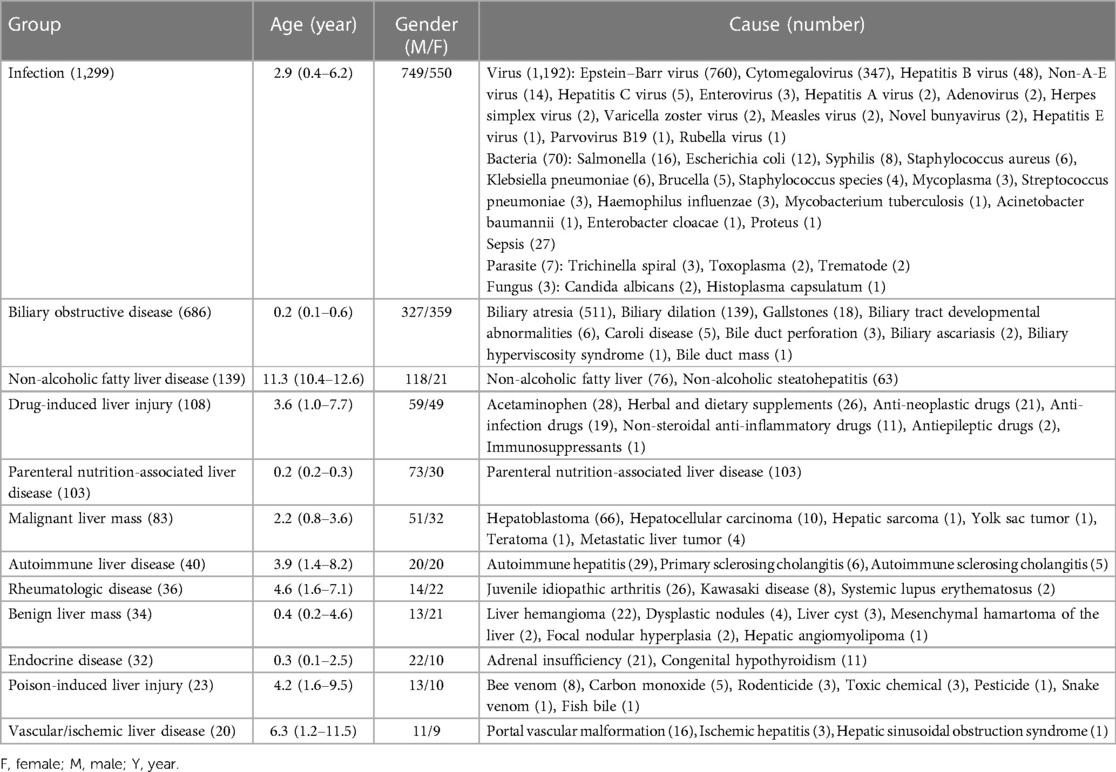
Table 3. Clinical spectrum of non-hereditary liver diseases (n = 2,603).
Among the 1,111 cases without a definitive diagnosis, 890 (80.1%) involved infants. The median age was 0.3 (0.2–0.7) years. There were 683 males and 428 females. Of these, 567 cases exhibited abnormal liver function tests, 499 cases presented with cholestatic jaundice, 42 cases presented with organomegaly, and three cases showed abnormalities found on ultrasound. In addition, 43 cases presented with cryptogenic cirrhosis and 19 with acute liver failure. Genetic testing was performed on 35.3% (392/1,111) of cases and liver biopsy was performed on 17.1% (190/1,111) of cases. Specifically, 125 patients underwent both genetic testing and histological examinations, while 267 underwent only genetic testing, and 65 underwent only histological examinations. During hospitalization, there were 11 mortalities (1.0%), 66 patients showed no improvement (5.9%), and 1,034 patients (93.1%) showed improvement.
Biliary obstructive diseases, mainly consisting of biliary atresia and biliary dilatation, accounted for 15.9% (686/4,313) of the cases. Biliary atresia accounted for 32.6% of infantile cholestasis, with 511 reported cases. The median age of cases with biliary atresia was 0.2 (0.1–0.2) years, with 269 males and 242 females. Biliary dilation ranked second among obstructive causes, with 139 reported cases. The affected individuals had a median age of 1.9 (0.6–3.7) years, with 41 males and 98 females. There were 88 cases of biliary dilatation with cholestasis and 51 with elevated transaminases. The remaining causes are detailed in Table 3. In this group of patients, two children with biliary atresia and four with biliary dilation experienced acute liver failure. Additionally, cirrhosis was observed in 180 children with biliary atresia, six with biliary dilation, two with Caroli disease, and one with biliary tract developmental abnormalities. A total of four fatalities were recorded due to biliary atresia during hospitalization, resulting in a mortality rate of 0.6%.
IMLDs represented 13.9% (599/4,313) of the study population, with sixty diverse forms detected. The median age was 1.5 (0.3–6.6) years. The analysis included 370 males and 229 females. Distribution characteristics according to pathogenesis and age of onset are shown in Tables 4, 5, respectively. In this cohort, 54.4% of cases were initiated during infancy and 80.5% of cases progressed before the age of six years. Wilson disease, citrin deficiency, glycogen storage disease, and Alagille syndrome were the most common IMLDs, accounting for 66.4% of cases. In 154 cases of Wilson disease, the median age was 8.6 (5.4–11.3) years, with a higher proportion of male patients (65.6%). The clinical signs of liver involvement ranged from incidental observations of aberrant liver function to severe hepatic failure. Citrin deficiency was the second most common genetic cause in the cohort with 101 cases. There were 58 males and 43 females, with a median age of 0.2 (0.2–0.3) years, and 99.0% of cases presented with infantile cholestasis. Glycogen storage disease (92 cases) was typically identified through liver enlargement or elevated aminotransferases. The median age was 2.5 (1.5–6.0) years, and there was a male predominance (66.3%). Out of the 51 cases of Alagille syndrome, 29 were male and 22 were female, with a median age of 0.2 (0.2–0.6) years. The majority of patients (88.2%) presented with infantile cholestasis and multisystem involvement. Other infrequent IMLDs presented diverse clinical manifestations. In this group, 37 cases presented with acute liver failure. The cases included 18 cases of Wilson disease, 10 cases of citrin deficiency, three cases of tyrosinemia, two cases of infantile liver failure syndrome, and one case each of ornithine transcarbamylase deficiency, dicarboxylic aminoaciduria, congenital disorder of glycosylation, and Alagille syndrome. In addition, 77 cases of cirrhosis were identified with causes including Wilson disease (55 cases), citrin deficiency (three cases), polycystic kidney disease 4 (three cases), glycogen storage disease (two cases), progressive familial intrahepatic cholestasis (two cases), Niemann-Pick disease (two cases), and cystic fibrosis liver disease (two cases). There was one case each of congenital bile acid synthesis defect, Alstrom syndrome, nephronophthisis 12, Shwachman-Diamond syndrome, immunodeficiency 28 with mycobacteriosis, Adams-Oliver syndrome, familial visceral amyloidosis, and hereditary hemorrhagic telangiectasia syndrome. In this group of patients, one individual died from ornithine transcarbamylase deficiency and four died from Wilson disease, resulting in a mortality rate of 0.8%.
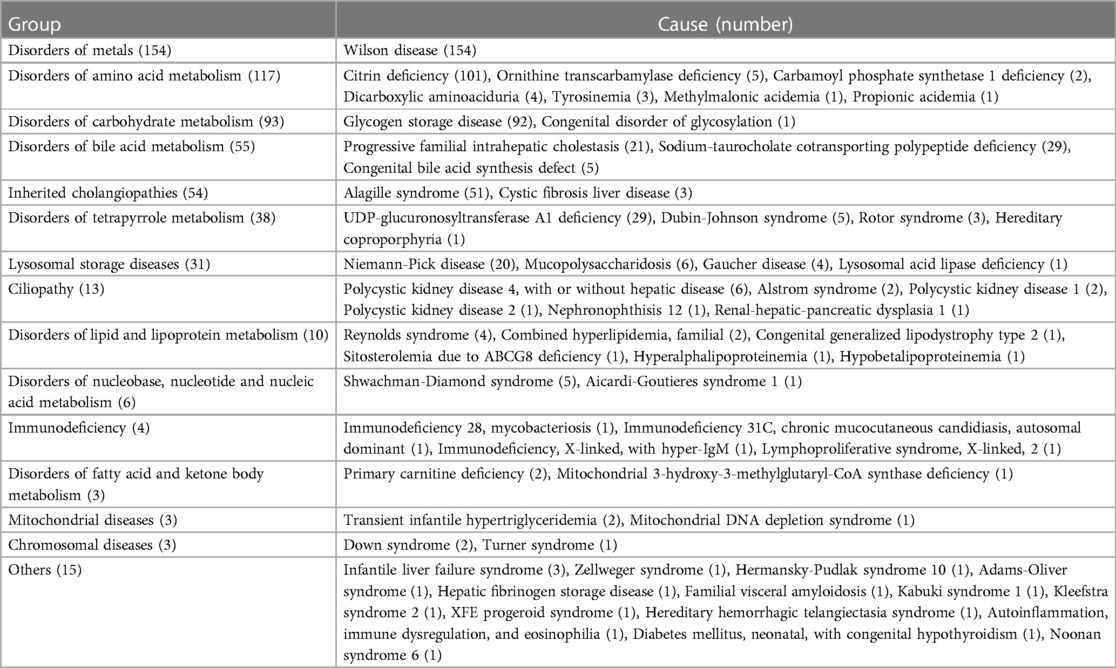
Table 4. Clinical spectrum of inherited metabolic liver diseases (n = 599).

Table 5. Age distribution characteristics of inherited metabolic liver diseases (n = 599).
NAFLD accounted for 3.2% (139/4,313) of the total cases, ranking fifth in the group. The median age was 11.3 (10.4–12.6) years, with a male predominance (84.9%). Assessment of body mass index revealed that 137 individuals were classified as obese and two as overweight. Forty-nine patients exhibited insulin resistance, 24 presented with dyslipidemia, 10 were diagnosed with type 2 diabetes mellitus, and two had hypertension. Non-alcoholic fatty liver was diagnosed in 76 cases and non-alcoholic steatohepatitis in 63 cases.
The remaining etiologies are detailed in Table 3. Drug-induced liver injury, parenteral nutrition-associated liver disease, malignant liver mass, autoimmune liver disease, rheumatologic disease, benign liver mass, endocrine disease, poison-induced liver injury, and vascular/ischemic liver disease were, respectively, seen in 108 (2.5%), 103 (2.4%), 83 (1.9%), 40 (0.9%), 36 (0.8%), 34 (0.8%), 32 (0.7%), 23 (0.5%), 20 (0.5%) cases. Of note, the less common diseases, like drug-induced liver injury and autoimmune liver disease, were difficult to diagnose and required specialized attention. Out of the 108 cases of drug-induced liver disease, 10 were diagnosed more than six months after the onset of the disease, with the longest period being 12 months.
Table 6 presents the distribution of common diseases in different age groups. In our study, biliary atresia and CMV hepatitis were prevalent during infancy, whereas EBV-associated hepatitis was the most frequent in the age group 1–12 years. Moreover, NAFLD and Wilson disease were more common in the age group of 12–18 years.
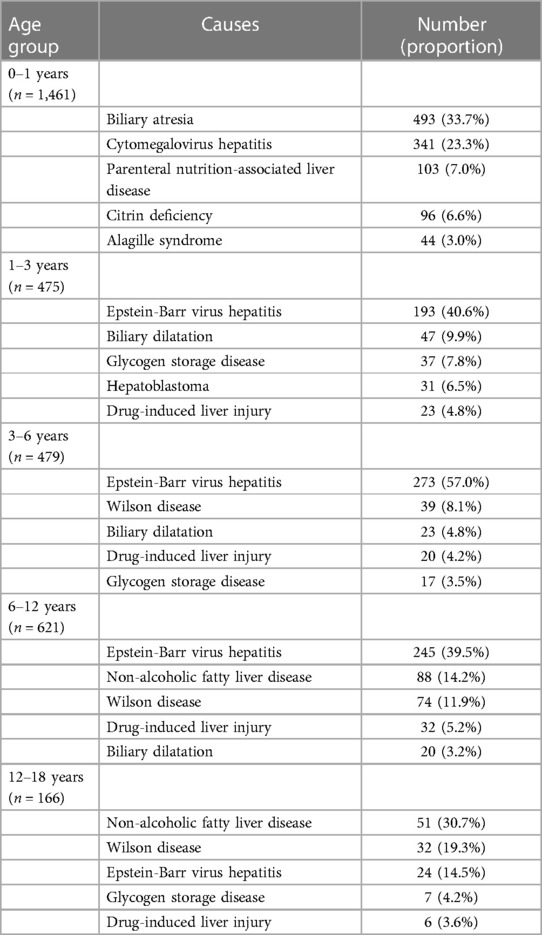
Table 6. Top five causes among cases with a definite diagnosis.
Cases with genetic testing
Genetic results for 1,088 patients were collected from medical records. The temporal distribution of genetic testing is illustrated in Figure 4. Positive genetic diagnoses were established in 478/1,088 (43.9%) of patients, with Sanger sequencing detecting 122/341 cases and NGS identifying 356/747 cases. Out of the 610 cases in which genetic testing was inconclusive, 218 were later diagnosed with other clinical conditions. These included 76 cases of infectious liver diseases, 48 cases of biliary obstructive diseases, 31 cases of IMLDs, 31 cases of parenteral nutrition-associated liver disease, 15 cases of drug-induced liver injury, 13 cases of autoimmune liver disease, two cases of NAFLD, and two cases of portal vascular malformation.
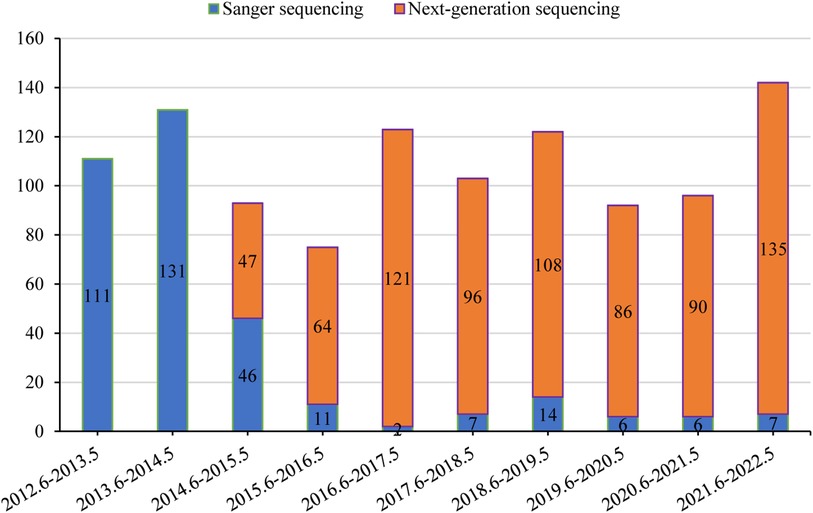
Figure 4. Temporal distribution of genetic testing.
Notably, 31 patients fulfilled the clinical diagnostic criteria but did not receive a conclusive genetic diagnosis. Clinically, 20 patients were diagnosed with citrin deficiency; however, genetic testing detected a heterozygous mutation in the SLC25A13 gene (nine via Sanger sequencing and 11 via panel-based NGS). Genetic testing (two via Sanger sequencing, two via panel-based NGS, and one via WES) identified a heterozygous mutation in the ATP7B gene for five individuals clinically diagnosed with Wilson disease. In three cases of children clinically diagnosed with glycogen storage disease, NGS testing failed to identify the expected pathogenic variants (two via panel-based NGS and one via WES). Additionally, three individuals were clinically diagnosed with Alagille syndrome. No pathogenic variants were identified in the JAG1 or NOTCH2 gene through Sanger sequencing.
Moreover, there were still 392 cases without a definitive diagnosis. Out of these, 269 cases presented with cholestatic jaundice, 106 cases showed abnormal liver function tests, 15 cases presented with organomegaly, and two cases showed abnormalities detected on ultrasound.
Time trends between June 2012 and May 2022
Overall, the annual number of pediatric liver diseases remained stable between 2012 (407 cases) and 2022 (435 cases) (p = 0.145) (Figure 5A). The percentage of infants aged 0–1 with liver diseases significantly fell from 66.6% (n = 271) to 38.6% (n = 168) (p < 0.001). In contrast, there was a rising tendency in the percentage of children aged 3–18. There was a rise in the percentage of children belonging to the age range of 3–6 years, from 5.4% (n = 22) to 16.3% (n = 71) (p = 0.008), in the age range of 6–12 years, from 12.5% (n = 51) to 25.3% (n = 110) (p = 0.001), and in the age category of 12–18 years, from 2.9% (n = 12) to 8.5% (n = 37) (p = 0.002). The percentage of children aged 1–3 years remained relatively constant, declining from 12.5% (n = 51) to 11.3% (n = 49) (p = 0.444) (Figure 5B).
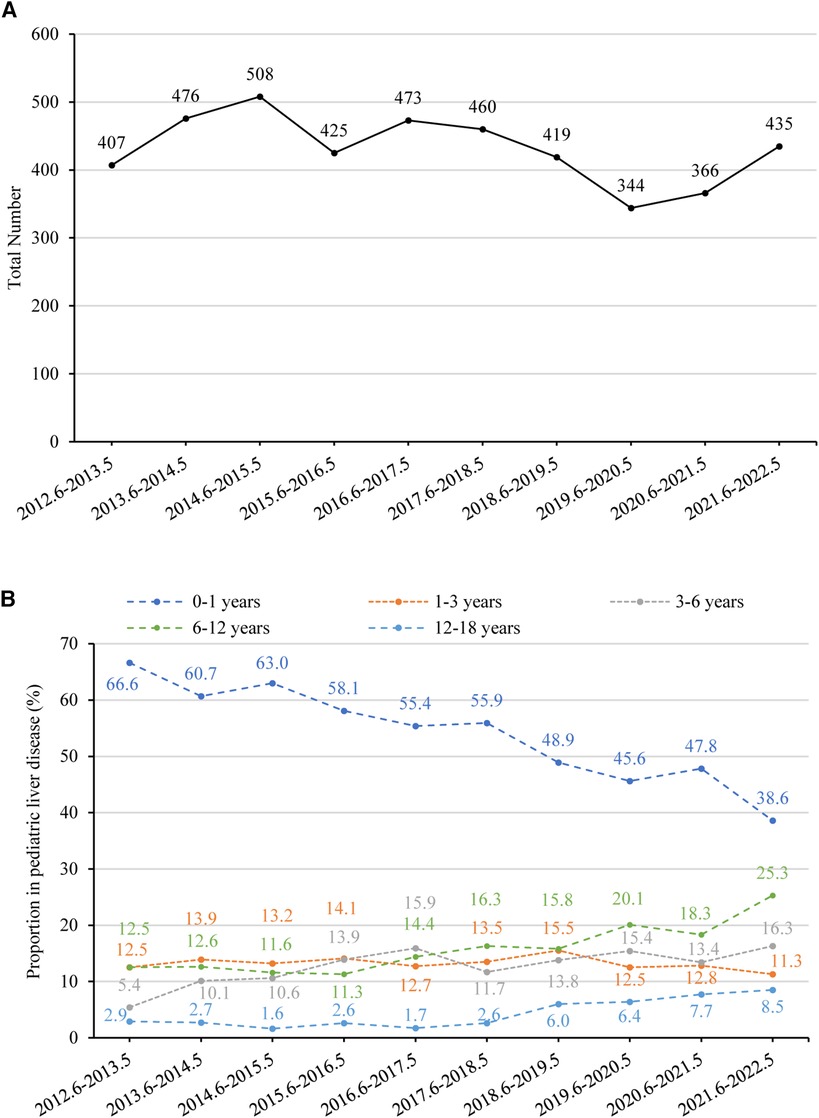
Figure 5. (A) number of pediatric liver disease-related admissions between June 2012 and May 2022. The time trend was assessed using linear regression analysis. The annual number of pediatric liver diseases remained stable (p = 0.145). (B) Percentage of children with liver disease in different age groups between June 2012 and May 2022. The proportion of children aged 0–1 decreased significantly (p < 0.001), while the proportion of those aged 1–3 remained stable (p = 0.444). There was an increasing trend in the proportion of children aged 3–6 (p = 0.008), 6–12 (p = 0.001), and 12–18 (p = 0.002).
Figures 6A,B provide a summary of liver disease with rising and declining trends between 2012 and 2022 (p < 0.05), respectively. The percentage of undiagnosed cases declined from 31.7% (n = 129) to 20.5% (n = 89) (p = 0.031). In contrast, there was a significant upsurge in the percentage of NAFLD, rising from 1.2% (n = 5) to 12.6% (n = 55) (p = 0.006). Additionally, the percentage of rheumatologic disease showed an increasing trend from 0.0% (n = 0) to 3.0% (n = 13) between 2012 and 2022 (p = 0.002). Liver disease that significantly declined between 2012 and 2022 was biliary obstructive disease (from 16.7% to 10.1%, p = 0.003). The overall percentage of infectious liver diseases remained relatively stable, with fluctuations between 31.2% (n = 127) to 27.4% (n = 119) (p = 0.266). However, CMV hepatitis demonstrated a significant decline from 13.3% (n = 54) to 3.4% (n = 15) (p = 0.002). Furthermore, overall IMLDs presented a stable trend, shifting from 11.3% (n = 46) to 15.9% (n = 69) (p = 0.369); however, infrequent IMLDs (excluding Wilson disease, citrin deficiency, glycogen storage disease, and Alagille syndrome) displayed an increased trend from 2.9% (n = 12) to 9.4% (n = 41) (p = 0.020). Diseases related to liver injury with a relatively stable trend between 2012 and 2022 were drug-induced liver injury (from 1.2% to 1.8%, p = 0.170), parenteral nutrition-associated liver disease (from 3.4% to 2.8%, p = 0.596), malignant liver mass (from 0.5% to 2.5%, p = 0.278), autoimmune liver disease (from 1.5% to 1.1%, p = 0.521), benign liver mass (from 0.0% to 0.7%, p = 0.501), endocrine disease (from 0.5% to 0.5%, p = 0.724), poison-induced liver injury (from 0.2% to 0.9%, p = 0.764), and vascular/ ischemic liver disease (from 0.5% to 0.2%, p = 0.462).
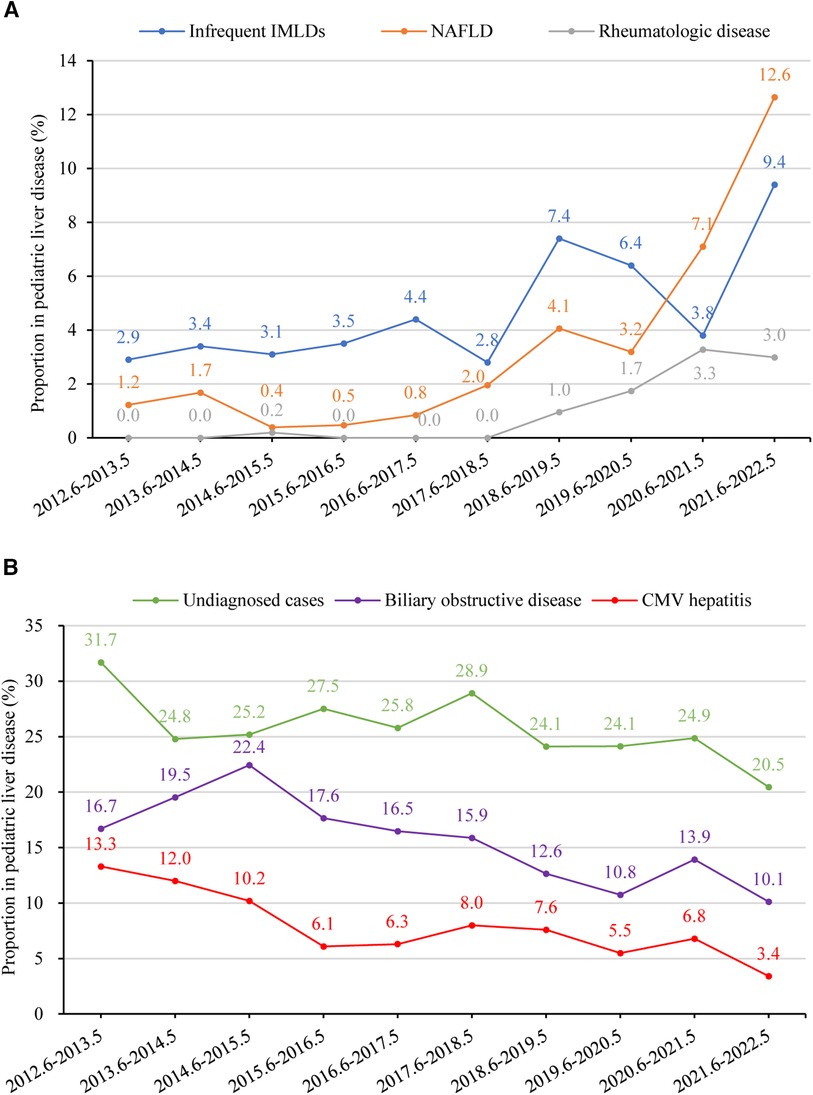
Figure 6. (A) percentage of children with liver disease in different etiology groups from 2012 to 2022 with a rising trend (p < 0.05). Time trends were assessed using linear regression analysis. The proportions of infrequent IMLDs, NAFLD, and rheumatologic disease showed an increasing trend (p-values 0.020, 0.006, and 0.002, respectively). (B) Percentage of children with liver disease in different etiology groups between June 2012 and May 2022 with a declining trend (p < 0.05). The proportions of undiagnosed cases, biliary obstructive disease, and CMV hepatitis showed a declining trend (p-values 0.031, 0.003, and 0.002, respectively). CMV, Cytomegalovirus; IMLD, inherited metabolic liver disease, infrequent IMLDs, defined as IMLDs excluding Wilson disease, citrin deficiency, glycogen storage disease, and Alagille syndrome. NAFLD, non-alcoholic fatty liver disease.
The etiology of liver disease in infancy was analyzed for time trends, given that half of the cases in this cohort occurred during infancy (Table 7). There was a decreasing trend in the percentage of cytomegalovirus hepatitis among liver diseases in infancy (p = 0.023). However, the percentage of undiagnosed cases remained relatively stable (p = 0.147).
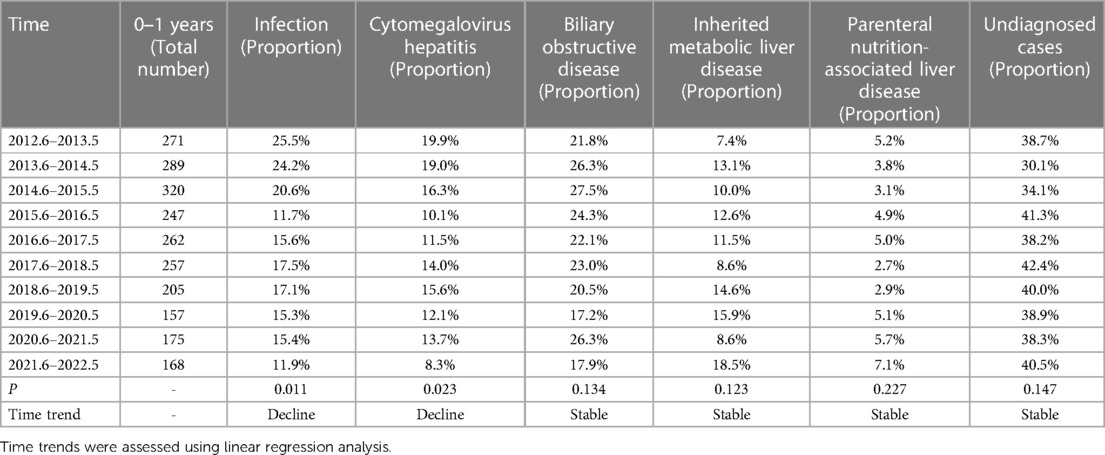
Table 7. Percentage of children aged 0–1 years with liver disease from 2012 to 2022.
Discussion
In this study, we described the epidemiological features in a large cohort of hospitalized children with liver disease in Hubei Province, China. Different age groups exhibit distinct characteristics. While infants typically display cholestatic jaundice, older children may exhibit abnormal liver function tests or organomegaly, in addition to jaundice (7). Liver disease in infancy makes up the largest group in pediatric liver disease. Even if the child does not display severe symptoms, an infant with liver disease ought to be treated as a medical emergency (8). Our study showed that over half of these patients experienced symptoms during infancy. This contradicts a survey conducted in California, USA, which revealed that half of all childhood liver disease occurs in the 16–21 years age group (16). These regional disparities contribute to this phenomenon. In addition, this study shows the lowest etiological diagnosis rate (62.1%) for cases in the 0–1 years age group. Cholestasis is a prevalent and severe condition in childhood (17). The incidence of cholestasis is believed to be 1:2,500 infants in North America (12). In our study population, cholestasis had a prevalence of 64.9% among infants with liver disease. Moreover, due to the young age and invasiveness, examination in infancy is limited. However, several of these maladies may have notable clinical consequences in the long run if they are not diagnosed early and treated promptly (8). Liver disease in infancy should therefore be of great clinical concern.
Infectious hepatitis may arise from a hepatotropic viral infection or as part of a systemic disorder. The symptoms can vary greatly, with a range of presentations including asymptomatic elevation in aminotransferases, cholestasis, acute liver failure, hepatic fibrosis, and cirrhosis (18). Consistent with prior research conducted in another region of China (19), infection remained the most common entity encountered, especially EBV and CMV infections in this study. CMV hepatitis is predominantly seen in young infants, whereas EBV hepatitis is predominantly seen in children over 1 year of age. The prevalence of hepatitis B virus infection in children was extremely low due to the widespread use of vaccinations (20). EBV is a worldwide widespread virus, infecting more than 90% of the world's population by maturity (21). The prevalence of EBV infection in Chinese children is more than 50% before the age of 3 years and more than 90% after the age of 8 years. In comparison, EBV infections occur at an earlier age than in the United States (22). In our cohort, the median age of patients with EBV hepatitis was 4.7 (2.7–7.3) years. Cases of acute liver failure and death were also reported. Adequate attention should also be paid to the most common form of EBV hepatitis in Chinese children.
CMV is a herpes virus with high (40%–100%) seroprevalence rates in adults (23). The incidence of CMV is negatively correlated with a country's level of socioeconomic development. It is more widespread in the Global South and Asia, whereas it is less frequent in Europe and the United States (24). In China, it was previously thought that CMV infection was the main cause of liver disease in infancy. This study also showed that CMV hepatitis was the second most common cause of liver disease in infancy and accounted for 12.7% of infantile cholestasis, behind biliary atresia and undiagnosed cases. However, it is encouraging that our study showed a downward trend in hospitalizations for cytomegalovirus hepatitis over the last decade. Over the past two decades, China has experienced significant demographic changes, including a decline in the birth rate from 14% to 6.8% and accelerated aging (25). In our cohort, the proportion of liver disease in infancy has also decreased. Furthermore, our study revealed a decreasing trend in the proportion of CMV hepatitis in liver disease during infancy. This suggests that demographic changes are not a significant influencing factor. This may be related to the increasing interest in CMV infection and the improved understanding of the etiology of pediatric liver disease. CMV infection is often observed in these cases, but it is not the primary cause of certain liver diseases (26). To our knowledge, no similar time-trend surveys on the proportion of CMV hepatitis among hospitalized children with liver disease in other centers in China have been conducted. Therefore, our findings require further validation through a national multicenter study.
About 20% of pediatric liver transplants are performed for single-gene liver diseases, and almost half of chronic liver diseases that manifest in childhood have a genetic basis (27). Clinical features that are similar and caused by IMLDs may be under-recognized or misdiagnosed due to their non-specific nature (4). This study observed that IMLD presents a range of clinical manifestations. Some cases only or mainly affect the liver, while others exhibit extrahepatic or systemic effects. With the growing popularity and reduced cost of genetic testing, early detection of liver disease is becoming more common (1, 28). In various genetic backgrounds, the spectrum of diseases differs extensively. The prevalent IMLDs in this group were Wilson disease, citrin deficiency, glycogen storage disease, and Alagille syndrome, which aligns with prior reported data (29). The prevalence of common inherited liver diseases varies by age. Citrin deficiency and Alagille syndrome were frequently observed in cases of infantile inherited cholestasis in our cohort. This finding is consistent with the results of a previous study conducted in Shanghai, China (30). Conversely, glycogen storage disease was more prevalent in toddlers and preschoolers with enlarged livers. Wilson disease, however, was more frequently observed in older children. Furthermore, our study showed that an increase in the percentage of infrequent or newly identified IMLDs was noted. For example, Sodium-taurocholate cotransporting polypeptide deficiency, a newly described inborn error of bile acid metabolism, was first reported in 2015 (31). Refractory hypercholanemia, brief cholestatic jaundice in infancy, and unconjugated hyperbilirubinemia in newborns are the primary clinical manifestations of this disease (32, 33). The identification of this condition has strengthened our comprehension of the reasons behind infantile cholestasis.
Biliary atresia is the foremost chronic liver condition afflicting children and responsible for most pediatric liver transplants worldwide. The incidence of it varies from 1 in 5,000 to 1 in 19,000 live births, with a higher prevalence in Asian nations than in European countries (12). In this investigation, biliary atresia emerged as the primary cause of liver diseases among infants, causing 32.6% of infantile cholestasis, consistent with prior research (12). Furthermore, it has been found that biliary atresia is the most frequent cause of liver cirrhosis, which is consistent with previous research (34). However, detecting biliary atresia in its early stages remains challenging, and further investigation is necessary.
NAFLD has become the leading cause of chronic liver disease in high-income nations due to the worldwide outbreak of obesity and overweight (35). While most patients have not exhibited obvious symptoms, pediatric NAFLD demonstrates a potential for decreased survival without liver transplantation when compared to their peers without the condition (36, 37). Anderson et al. reported that worldwide, the prevalence of NAFLD was 7.6% in children within the general population and 34% in obese pediatric populations (38). According to a systematic review, the prevalence of pediatric NAFLD has risen to 7.10%, from 4.42% before 2010 in the last decade in Asia (39). Our research indicates that the proportion of NAFLD cases in hospitalized children with liver disease has increased almost 10 times over the last decade. In addition, there has been a sharp rise in this rate from June 2020 to May 2022, which may be linked to the impact of the COVID-19 pandemic. Notably, a considerable portion of patients remain without diagnosis and treatment due to insufficient diagnostic tools and the absence of effective pharmaceutical remedies (40). Enhancing future care for children with NAFLD necessitates a superior understanding of the disease's natural progression and underlying pathology (41).
The etiological makeup of children admitted to our center with liver disease has undergone significant change in the past decade. Liver disease in patients with systemic disease is increasingly recognized. For example, an increase in the proportion of rheumatologic disease may be related to the severity of the disease and clinical considerations. In addition, there has been a significant decrease in the percentage of undiagnosed cases, from 31.7% to 20.5%. However, the etiological identification of pediatric liver disease remains a challenge, especially in developing countries. In our cohort, out of the 1,111 cases with an unknown diagnosis, only 35.3% underwent genetic testing and only 17.1% underwent liver biopsy, owing to limited resources. Furthermore, as 80.1% of the cases occurred during infancy, the restricted diagnostic options available, combined with vague or non-existent symptoms, make it challenging to determine the cause (4).
The study revealed a diagnostic rate of 43.9% for genetic testing. However, some cases that met clinical diagnostic criteria did not receive a conclusive genetic diagnosis. Furthermore, some patients were suspected of having an underlying hereditary condition, although most had inconclusive genetic test results. This could be a result of flaws in the methodology or causes that have not yet been identified. Clinical follow-up is crucial in pediatric liver disease, and further assessments should be conducted when necessary. In addition, genetic testing provides genotype-phenotype correlation and is particularly useful in cases where there are no clear indications or when the patient's condition is challenging to diagnose. However, it is important for clinicians to avoid excessive reliance on genetic testing as it cannot replace routine biochemical and pathology testing in disease management (42).
There are limitations to this study. Firstly, it was a retrospective study conducted in a single center, and no long-term follow-up data on patients were analyzed. Additionally, cases of patients younger than 29 days were excluded from the analysis in our study due to the unavailability of patient data from the neonatal ward. Therefore, these data can only reflect the real-world circumstances of certain patients admitted to the hospital. Secondly, not all children were diagnosed with the same etiology due to the stepwise approach to etiological diagnosis.
In conclusion, our study provides insight into the disease spectrum of pediatric liver disease in a vast population cohort. The fact that over 50% of childhood liver disease originates in infancy, coupled with the low diagnostic rate of liver disease etiology during this period, should be a clinical concern. The causes of liver disease in children are complex and varied. This research validates that different age groups exhibit different etiological profiles. Identifying the etiologies is critical to commence disease-specific management and facilitate the provision of genetic counseling. The more interesting finding is that infection remains the leading cause of pediatric liver disease. Hospital admissions for NAFLD in children have increased rapidly over the past decade, while cytomegalovirus hepatitis has declined markedly.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Medical Ethics Committee of Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
Author contributions
FC: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Supervision, Validation, Writing – original draft, Writing – review & editing. YH: Validation, Writing – review & editing. ZH: Validation, Writing – review & editing. FF: Validation, Writing – review & editing. HZ: Validation, Writing – review & editing. SS: Conceptualization, Methodology, Resources, Supervision, Validation, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
We express our gratitude to the patients and their families for their participation in this study. Our sincere appreciation goes towards all the healthcare professionals who were involved in providing care to the patients.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Hadzic N, Baumann U, McKiernan P, McLin V, Nobili V. Long-term challenges and perspectives of pre-adolescent liver disease. Lancet Gastroenterol Hepatol. (2017) 2(6):435–45. doi: 10.1016/S2468-1253(16)30160-1
2. Runde J, Azzam RK. Hepatobiliary manifestations in systemic disease. Pediatr Ann. (2018) 47(11):e458–64. doi: 10.3928/19382359-20181023-01
3. Bahassi EM, Stambrook PJ. Next-generation sequencing technologies: breaking the sound barrier of human genetics. Mutagenesis. (2014) 29(5):303–10. doi: 10.1093/mutage/geu031
4. Zou YG, Wang H, Li WW, Dai DL. Challenges in pediatric inherited/metabolic liver disease: focus on the disease spectrum, diagnosis and management of relatively common disorders. World J Gastroenterol. (2023) 29(14):2114–26. doi: 10.3748/wjg.v29.i14.2114
5. Lv T, Jia J. Rare liver diseases are not rare in China. Liver Int. (2022) 42(9):2023–8. doi: 10.1111/liv.15267
6. Taylor-Phillips S, Stinton C, Ferrante DRL, Seedat F, Clarke A, Deeks JJ. Association between use of systematic reviews and national policy recommendations on screening newborn babies for rare diseases: systematic review and meta-analysis. Br Med J. (2018) 361:k1612. doi: 10.1136/bmj.k1612
7. Della CC, Mosca A, Vania A, Alterio A, Alisi A, Nobili V. Pediatric liver diseases: current challenges and future perspectives. Expert Rev Gastroenterol Hepatol. (2016) 10(2):255–65. doi: 10.1586/17474124.2016.1129274
8. Jagadisan B, Dhawan A. Emergencies in paediatric hepatology. J Hepatol. (2022) 76(5):1199–214. doi: 10.1016/j.jhep.2021.12.027
9. Asrani SK, Devarbhavi H, Eaton J, Kamath PS. Burden of liver diseases in the world. J Hepatol. (2019) 70(1):151–71. doi: 10.1016/j.jhep.2018.09.014
10. Arya G, Balistreri WF. Pediatric liver disease in the United States: epidemiology and impact. J Gastroenterol Hepatol. (2002) 17(5):521–5. doi: 10.1046/j.1440-1746.2002.02678.x
11. Moyer V, Freese DK, Whitington PF, Olson AD, Brewer F, Colletti RB, et al. Guideline for the evaluation of cholestatic jaundice in infants: recommendations of the North American society for pediatric gastroenterology, hepatology and nutrition. J Pediatr Gastroenterol Nutr. (2004) 39(2):115–28. doi: 10.1097/00005176-200408000-00001
12. Fawaz R, Baumann U, Ekong U, Fischler B, Hadzic N, Mack CL, et al. Guideline for the evaluation of cholestatic jaundice in infants: joint recommendations of the North American society for pediatric gastroenterology, hepatology, and nutrition and the European society for pediatric gastroenterology, hepatology, and nutrition. J Pediatr Gastroenterol Nutr. (2017) 64(1):154–68. doi: 10.1097/MPG.0000000000001334
13. Liver EAFT. EASL clinical practice guidelines for the management of patients with decompensated cirrhosis. J Hepatol. (2018) 69(2):406–60. doi: 10.1016/j.jhep.2018.03.024
14. Lee WS, McKiernan P, Kelly DA. Etiology, outcome and prognostic indicators of childhood fulminant hepatic failure in the United Kingdom. J Pediatr Gastroenterol Nutr. (2005) 40(5):575–81. doi: 10.1097/01.mpg.0000158524.30294.e2
15. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17(5):405–24. doi: 10.1038/gim.2015.30
16. Martin M, Zou B, Hoang J, Jeong D, Bensen R, Nguyen MH. Racial and socioeconomic disparities in hospitalization of pediatrics with liver disease from 2005 to 2015. Dig Dis Sci. (2021) 66(7):2240–9. doi: 10.1007/s10620-020-06530-w
17. Chen HL, Wu SH, Hsu SH, Liou BY, Chen HL, Chang MH. Jaundice revisited: recent advances in the diagnosis and treatment of inherited cholestatic liver diseases. J Biomed Sci. (2018) 25(1):75. doi: 10.1186/s12929-018-0475-8
18. Talwani R, Gilliam BL, Howell C. Infectious diseases and the liver. Clin Liver Dis. (2011) 15(1):111–30. doi: 10.1016/j.cld.2010.09.002
19. Meng Q, Li N, Yuan L, Gao X. Analysis of common causes of liver damage among children 12 years and younger in Weifang. J Int Med Res. (2021) 49(4):675899707. doi: 10.1177/03000605211006661
20. Zhang M, Wu R, Xu H, Uhanova J, Gish R, Wen X, et al. Changing incidence of reported viral hepatitis in China from 2004 to 2016: an observational study. BMJ Open. (2019) 9(8):e028248. doi: 10.1136/bmjopen-2018-028248
21. Bunchorntavakul C, Reddy KR. Epstein-Barr virus and cytomegalovirus infections of the liver. Gastroenterol Clin North Am. (2020) 49(2):331–46. doi: 10.1016/j.gtc.2020.01.008
22. Xiong G, Zhang B, Huang MY, Zhou H, Chen LZ, Feng QS, et al. Epstein-Barr virus (EBV) infection in Chinese children: a retrospective study of age-specific prevalence. PLoS One. (2014) 9(6):e99857. doi: 10.1371/journal.pone.0099857
23. Da CT, Wu GY. Cytomegalovirus hepatitis in immunocompetent and immunocompromised hosts. J Clin Transl Hepatol. (2021) 9(1):106–15. doi: 10.14218/JCTH.2020.00088
24. Gonzalez HC, Gordon SC. Hepatic manifestations of systemic diseases. Med Clin North Am. (2023) 107(3):465–89. doi: 10.1016/j.mcna.2023.01.008
25. Chen Z, Wang Y, Lan F, Li S, Wang J. An expanded view of infertility: the challenge of the changing profiling of major birth defects in China. Biosci Trends. (2023) 17(4):318–21. doi: 10.5582/bst.2023.01160
26. Neema RY, Sainan S. Re-discussion on the diagnosis and treatment of cytomegalovirus hepatitis in children. Chinese J Hepatol. (2021) 29(1):13–5. doi: 10.3760/cma.j.cn501113-20201226-00680
27. Sze YK, Dhawan A, Taylor RM, Bansal S, Mieli-Vergani G, Rela M, et al. Pediatric liver transplantation for metabolic liver disease: experience at King’s College Hospital. Transplantation. (2009) 87(1):87–93. doi: 10.1097/TP.0b013e31818bc0c4
28. Hadzic N, Verkade HJ. The changing spectrum of neonatal hepatitis. J Pediatr Gastroenterol Nutr. (2016) 63(3):316–9. doi: 10.1097/MPG.0000000000001203
29. Fang Y, Yu J, Lou J, Peng K, Zhao H, Chen J. Clinical and genetic spectra of inherited liver disease in children in China. Front Pediatr. (2021) 9:631620. doi: 10.3389/fped.2021.631620
30. Wang NL, Lu Y, Gong JY, Xie XB, Lin J, Abuduxikuer K, et al. Molecular findings in children with inherited intrahepatic cholestasis. Pediatr Res. (2020) 87(1):112–7. doi: 10.1038/s41390-019-0548-8
31. Vaz FM, Paulusma CC, Huidekoper H, de Ru M, Lim C, Koster J, et al. Sodium taurocholate cotransporting polypeptide (SLC10A1) deficiency: conjugated hypercholanemia without a clear clinical phenotype. Hepatology. (2015) 61(1):260–7. doi: 10.1002/hep.27240
32. Deng M, Mao M, Guo L, Chen FP, Wen WR, Song YZ. Clinical and molecular study of a pediatric patient with sodium taurocholate cotransporting polypeptide deficiency. Exp Ther Med. (2016) 12(5):3294–300. doi: 10.3892/etm.2016.3752
33. Deng LJ, Ouyang WX, Liu R, Deng M, Qiu JW, Yaqub MR, et al. Clinical characterization of NTCP deficiency in paediatric patients: a case-control study based on SLC10A1 genotyping analysis. Liver Int. (2021) 41(11):2720–8. doi: 10.1111/liv.15031
34. Olave MC, Gurung A, Mistry PK, Kakar S, Yeh M, Xu M, et al. Etiology of cirrhosis in the young. Hum Pathol. (2020) 96:96–103. doi: 10.1016/j.humpath.2019.09.015
35. Welsh JA, Karpen S, Vos MB. Increasing prevalence of nonalcoholic fatty liver disease among United States adolescents, 1988–1994 to 2007–2010. J Pediatr. (2013) 162(3):496–500. doi: 10.1016/j.jpeds.2012.08.043
36. Vos MB, Abrams SH, Barlow SE, Caprio S, Daniels SR, Kohli R, et al. NASPGHAN clinical practice guideline for the diagnosis and treatment of nonalcoholic fatty liver disease in children: recommendations from the expert committee on NAFLD (ECON) and the North American Society of Pediatric Gastroenterology, Hepatology and Nutrition (NASPGHAN). J Pediatr Gastroenterol Nutr. (2017) 64(2):319–34. doi: 10.1097/MPG.0000000000001482
37. Feldstein AE, Charatcharoenwitthaya P, Treeprasertsuk S, Benson JT, Enders FB, Angulo P. The natural history of non-alcoholic fatty liver disease in children: a follow-up study for up to 20 years. Gut. (2009) 58(11):1538–44. doi: 10.1136/gut.2008.171280
38. Anderson EL, Howe LD, Jones HE, Higgins JP, Lawlor DA, Fraser A. The prevalence of non-alcoholic fatty liver disease in children and adolescents: a systematic review and meta-analysis. PLoS One. (2015) 10(10):e0140908. doi: 10.1371/journal.pone.0140908
39. Zou ZY, Zeng J, Ren TY, Huang LJ, Wang MY, Shi YW, et al. The burden and sexual dimorphism with nonalcoholic fatty liver disease in Asian children: a systematic review and meta-analysis. Liver Int. (2022) 42(9):1969–80. doi: 10.1111/liv.15080
40. Zhou J, Zhou F, Wang W, Zhang XJ, Ji YX, Zhang P, et al. Epidemiological features of NAFLD from 1999 to 2018 in China. Hepatology. (2020) 71(5):1851–64. doi: 10.1002/hep.31150
41. Goldner D, Lavine JE. Nonalcoholic fatty liver disease in children: unique considerations and challenges. Gastroenterology. (2020) 158(7):1967–83. doi: 10.1053/j.gastro.2020.01.048
Keywords: pediatrics, inpatients, epidemiology, liver disease, non-alcoholic fatty liver disease
Citation: Chen F, Huang Y, Huang Z, Fang F, Zhou H and Shu S (2024) The epidemiological characteristics of liver disease in hospitalized children: a 10-year single-center retrospective study. Front. Pediatr. 12:1344714. doi: 10.3389/fped.2024.1344714
Received: 26 November 2023; Accepted: 26 February 2024;
Published: 6 March 2024.
Edited by:
Runar Almaas, Oslo University Hospital, NorwayReviewed by:
Andrea Pietrobattista, Bambino Gesù Children’s Hospital (IRCCS), ItalyNatalie Lie Berntsen, Oslo University Hospital, Norway
© 2024 Chen, Huang, Huang, Fang, Zhou and Shu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sainan Shu c2h1c2FpbmFuQDE2My5jb20=