
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr., 25 January 2024
Sec. Genetics of Common and Rare Diseases
Volume 12 - 2024 | https://doi.org/10.3389/fped.2024.1339664
 Zuzana Kolkova1
Zuzana Kolkova1 Peter Durdik2,3*
Peter Durdik2,3* Veronika Holubekova1
Veronika Holubekova1 Anna Durdikova2,3Milos Jesenak2,3,4,5Peter Banovcin2,3
Anna Durdikova2,3Milos Jesenak2,3,4,5Peter Banovcin2,3
Background: The mutations in the RPGR (retinitis pigmentosa GTPase regulator) gene are the most common cause of X-linked retinitis pigmentosa (XLRP), a rare genetic disorder affecting the photoreceptor cells in the retina. Several reported cases identified this gene as a genetic link between retinitis pigmentosa (RP) and primary ciliary dyskinesia (PCD), characterised by impaired ciliary function predominantly in the respiratory tract. Since different mutations in the same gene can result in various clinical manifestations, it is important to describe a correlation between the gene variant and the observed phenotype.
Methods: Two young brothers from a non-consanguineous Slovak family with diagnosed retinal dystrophy and recurrent respiratory infections were examined. Suspected PCD was diagnosed based on a PICADAR questionnaire, nasal nitric oxide analysis, transmission electron microscopy, high-speed video microscopy analysis, and genetic testing.
Results: We identified a novel frameshift RPGR mutation NM_001034853: c.309_310insA, p.Glu104Argfs*12, resulting in a complex X-linked phenotype combining PCD and RP. In our patients, this mutation was associated with normal ultrastructure of respiratory cilia, reduced ciliary epithelium, more aciliary respiratory epithelium, shorter cilia, and uncoordinated beating with a frequency at a lower limit of normal beating, explaining the clinical manifestation of PCD in our patients.
Conclusion: The identified novel pathogenic mutation in the RPGR gene expands the spectrum of genetic variants associated with the X-linked PCD phenotype overlapping with RP, highlighting the diversity of mutations contributing to the disorder. The described genotype–phenotype correlation can be useful in clinical practice to recognise a broader spectrum of PCD phenotypes as well as for future research focused on the genetic basis of PCD, gene interactions, the pathways implicated in PCD pathogenesis, and the role of RPGR protein for the proper functioning of cilia in various tissues throughout the body.
Primary ciliary dyskinesia (PCD) is a rare, clinically and genetically heterogeneous multisystem disorder characterised by recurrent respiratory infections, chronic inflammation, bronchiectasis, sinusitis, and impaired fertility. The cause of the respiratory conditions is ineffective mucociliary clearance resulting from defects in the structure, assembly, and function of motile cilia as a result of pathogenic mutations in more than 50 PCD relevant genes described up to now (1). Since the structure of motile and nodal cilia is similar, gene mutations negatively affect the motility and function of nodal cilia during embryogenesis, leading to laterality defects associated with PCD (2). PCD is typically inherited in an autosomal recessive manner, but rare cases of X-linked inheritance have also been described, especially in a syndromic phenotype linked to retinitis pigmentosa (3).
Retinitis pigmentosa (RP) is a hereditary disease characterised by progressive retinal degeneration caused by cell death of photoreceptors. Patients suffer from slowly progressing night blindness, loss of peripheral vision, and even loss of central vision. RP can be inherited in an autosomal dominant, recessive, X-linked, and even mitochondrial pattern (4). More than 70% of X-linked RP (XLRP) and about 10%–15% of all RP cases are caused by mutations in the RPGR gene that encodes the retinitis pigmentosa GTPase regulator located within the connecting cilium between the inner and outer segments of the human rod and cone cells (5, 6). The function of the RPGR protein is not completely clear, but it is proposed that, together with its interacting proteins, it plays an important role in the regulation of protein transport across the connecting cilium, and mutations causing its dysfunction can lead to photoreceptor cell death (7, 8). In addition to the retina, RPGR isoforms resulting from alternative transcription and post-translational modifications were also detected in other tissues, such as the lung, kidney, and brain (9–11). Based on the role of the RPGR protein in intraflagellar transport, it can be assumed that mutations negatively affect the assembly and viability of motile and immotile cilia. Several cases of XLRP associated with recurrent respiratory infections and impaired hearing have been observed (3, 12–18).
Here, we describe a case of two brothers with complex X-linked clinical phenotypes combining PCD with RP, two rare genetic disorders linked by a novel hemizygous frameshift mutation in the RPGR gene. This case report contributes to the suggestion that RPGR protein plays an essential role in ciliary function in the retina and other tissues. Understanding the relationship between RPGR mutations and PCD is important for improving diagnosis, treatment, and genetic counselling for families affected by these conditions.
We examined two brothers, a 13-year-old (P1) and a 10-year-old (P2), from a non-consanguineous Slovak family in the national PCD centre. They were recruited to identify suspicious PCD due to retinal dystrophy, recurrent mostly upper respiratory infections, almost recurrent rhinosinusitis, and bronchiectasis of unknown origin on computed tomography (CT) examination in the case of the older boy. The routine clinical examinations excluded the usual causes of recurrent respiratory infections, including developmental congenital defects, allergic diseases, primary immunodeficiency, gastroesophageal reflux disease, and otorhinolaryngological causes (polyps, adenoid vegetation). Cystic fibrosis was excluded by negative newborn screening provided as standard in our country (the determination of immunoreactive trypsinogen in a dry drop of blood) and by demonstration of normal sweat tests. The PCD diagnostic algorithm was based on the ERS (European Respiratory Society) guidelines (19).
Both boys were term-birth babies with no neonatal respiratory distress and no neonatal rhinitis. There were no congenital heart defects, hydrocephalus, or laterality defects. A history of multi-trigger persistent wheezing, recurrent respiratory tract infections, persistent nasal congestion, and chronic rhinosinusitis was presented mostly from 1 year of life and worsened after joining the collective in both boys. The initial retinal degenerative symptoms started in the older boy (P1) at the age of 5 years with decreased night vision following the worsening of the mid-peripheral visual field. The retinal degeneration with visual impairment, RP, was diagnosed at the age of 8 years by a combination of visual field tests, visual acuity tests, and fluorescein angiography. Approximately 1 year later, the bronchiectasis localised in the right middle lobe was confirmed by a high-resolution CT examination. At the age of 10 years, the older boy was sent to the national PCD centre. Neurological examination, hearing, and lung functions were normal. In the younger brother (P2), deterioration of night vision occurred at the age of 6 years and was confirmed in the same way as in the older brother at 7 years. He was sent to the national PCD centre 1 year later.
Patients were interviewed and scored for probable PCD using the PICADAR questionnaire (20). The PICADAR level for both patients was three points (two points for term-baby and one point for chronic rhinosinusitis), which is not significant for PCD patients.
For nasal nitric oxide (nNO), an electrochemical analyser NIOX VERO (Circassia AB, Uppsala, Sweden) was used. The measurement was performed under standard conditions in the laboratory. The sampling rate was 0.3 nL/min, and the ambient nitric oxide (NO) was 6 ppb. The results were also confirmed by the breath-hold manoeuvre, and a difference of less than 10% was considered normal. The examination was graded C depending on the ERS technical standard (21). The nNO was 249 ppb (74.7 nL/min) on the left nostril and 261 ppb (78.3 nL/min) on the right nostril in the older patient (P1) and 234 ppb (70.2 nL/min) on the left nostril and 252 ppb (75.6 nL/min) on the right nostril in the younger patient (P2). The measured values were at the limit of significance for PCD.
After 3 weeks of no infection or no relapse of chronic symptoms, ciliary cells were obtained by brushing the inferior turbinate using a cytology brush (CytobrushCYTST Changzhou Shunfeng Plastic Co., LM, China). The samples of ciliated epithelium were suspended in RPMI 1640 Medium warmed up to 35°C. The ciliary beat was measured using a digital high-speed imaging workstation, designed by a team of researchers from Jessenius Medical Faculty in Martin and the University of Žilina (both in Slovakia), for the high-speed video microscopy analysis (HSVMA). A detailed description of this method (Utility Model Number 6811) (22) has been published in several papers (23–25). The warmed-up samples were evaluated under an inverted phase-contrast microscope (Zeiss, Oberkochen, Germany) at × 400 magnification, and ciliary movement was recorded with a digital high-speed video (Basler A504KC; Basler AG, Germany) with a frame rate of 500 Hz. Each microscope slide was examined within the first 10 min after its preparation, and more than 10 sequences with the best ciliary kinematics were recorded. The video sequences were played back frame by frame using the software application Ciliary Analysis (National Instruments LabVIEW development environment). The cilia beat frequency (CBF) was determined by calculating the mean of all recorded cilia beat cycles, and the cilia beating pattern (CBP) was evaluated on slow-motion playbacks (up to 10 times slower) and determined by two independent expert operators. The range of mean ciliary beat frequency was 5.5–7.5 Hz in both patients, which represented the lower limit of the normal ciliary frequency. There was a higher percentage of aciliary respiratory epithelium, and most of the cilia were shorter (Supplementary Video S1). The ciliary beat pattern analysis revealed a strong increase in ciliary bundles beating in an uncoordinated manner (Supplementary Video S2).
Respiratory epithelial cells from bronchoscopy or brushing from the middle nasal turbinate were collected and fixed in 3% glutaraldehyde. The transmission electron microscopy (TEM) analysis was performed by certified laboratory CYTOPATHOS, the centre for ciliary ultrastructure analysis. Our patients did not reveal ciliary ultrastructure abnormalities, with no changes of class 1 (Hallmark diagnostic defects) and class 2 defects for the ultrastructural diagnosis (Figure 1A). Reduced ciliary epithelium, more aciliary respiratory epithelium, and shorter cilia were described (Figure 1B).
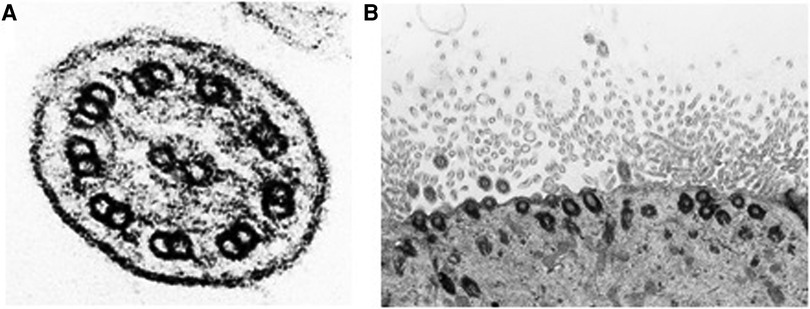
Figure 1. (A) Cross section of respiratory cilia. No structural abnormalities were observed by TEM; normal 9 + 2 configuration of microtubules and the presence of outer and inner arms are shown. (B) Reduced ciliary epithelium and shorter cilia are described.
DNA was isolated from peripheral venous blood samples of both patients and their parents. In the older boy (P1), genetic analysis of coding and splicing-relevant regions of 35 genes associated with PCD was performed by the targeted next generation sequencing (NGS) method using hybridisation solution Nextera Flex Pre-enrichment and TruSight One expanded panel (Illumina, USA) on NextSeq 550 (Illumina, USA). Illumina bcl2fastq v2.20 and SEQNEXT v5.2.0 (JSI medical systems) software were used for bioinformatical analysis with reference genome Homo sapiens GRCh37/hg19 and the RefSeq database. NGS analysis detected one nucleotide insertion in exon 4 of the RPGR gene in the hemizygous state, NM_001034853: c.309_310insA, p.Glu104Argfs*12. Sanger sequencing was used to verify the detected variant in the patient and his younger brother, as well as for segregation analysis in the family. The sequences of primers designed to intron regions were as follows: forward 5′-GTCCTGGACTACTGTTCATTTT-3′, reverse 5′-AAGCCACGTTACTGGAATGAG-3′. Sequencing analysis was performed on the ABI3500 Genetic Analyzer (Applied Biosystems, USA). The pathogenicity and functional effect of the identified variant were assessed by the in silico predictive software MutationTaster (26). The detected variant was confirmed by Sanger sequencing in the older patient (P1) as well as in his younger brother (P2) (Figures 2A, B). The segregation analysis confirmed the maternal origin of the mutation (Figure 2C). As expected, the father has a wild-type genotype (Figure 2D). The identified RPGR mutation NM_001034853: c.309_310insA, p.Glu104Argfs*12 located at the end of exon 4 was not found in databases ExAC, 1000G, or ClinVar and to the best of our knowledge, no report with this variant has been published so far. The consequence of the insertion of nucleotide A within the coding sequence is a shift in the reading frame and the creation of a premature stop codon. Based on the prediction in the MutationTaster program, premature termination of translation leads to nonsense-mediated decay, and this variant is predicted to be disease-causing. According to the ACMG (American College of Medical Genetics and Genomics) guidelines, this variant is classified as pathogenic (PVS1 and PM2 category) (27).
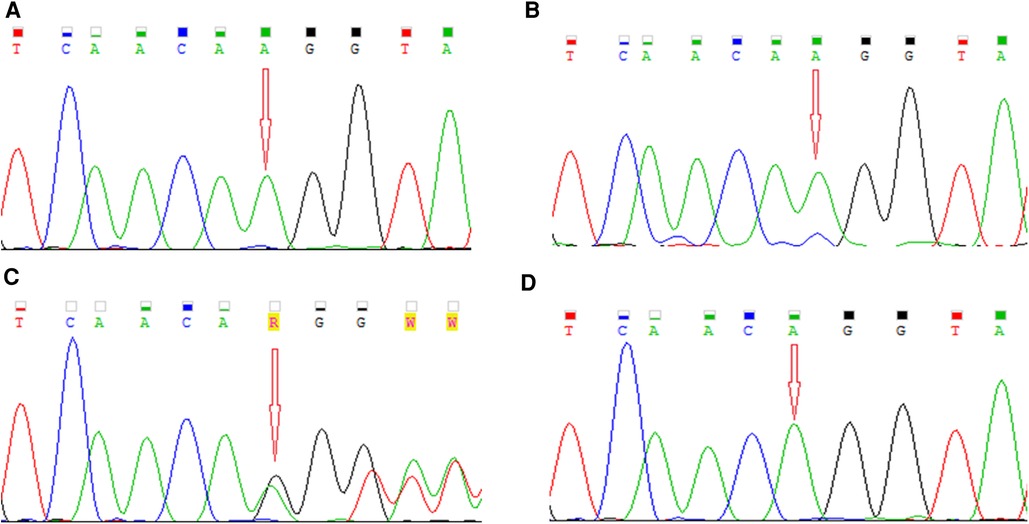
Figure 2. Electropherograms showing detected mutation NM_001034853: c.309_310insA, p.Glu104Argfs*12 in the RPGR gene in (A, B) a hemizygous state in our patients, (C) in a heterozygous state in their mother, and (D) the wild-type allele in their father.
Due to X-linked inheritance, we also analysed other members of the boys' family for the presence of typical respiratory symptoms or visual disturbances typical for RP. On the basis of medical data, a slight visual impairment, myopia, was detected in the boys' mother and grandmother without respiratory symptoms. Subsequently, we searched the complete Slovak PCD database with a focus on the connection between RP and PCD and identified five more boys who had an identical mutation in the RPGR gene (c.309_310insA) and similar clinical features. Based on the family tree provided by the family, distant kinship relationships between patients were clarified. The six-generation pedigree chart is shown in Figure 3. In the diagram, the two patients described above are marked as VII.1 (P1) and VII.2 (P2). The family members VI.2, VI.3, VI.4, VI.5, and VI.7 were also examined in the Slovak PCD centre, owing to the presence of RP and PCD symptoms, by the methods described above. The complete clinical features and results of the structural and functional analysis of the respiratory cilia of all affected boys are summarised in Table 1. From the available medical records, it is known that affected men in the family (III.4, IV.3, IV.4, IV.8, IV.9, IV.10, IV.13, IV.10) suffered from night blindness, but the presence of PCD signs is unknown. In female carriers, varying degrees of myopia were present.
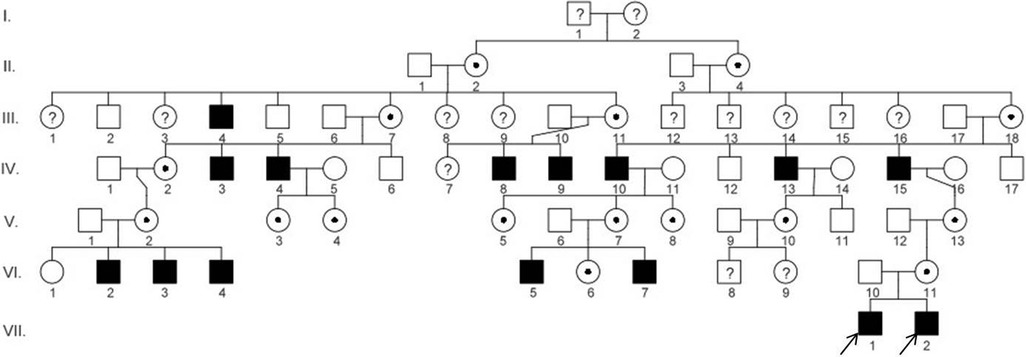
Figure 3. Pedigree diagram of the family showing disease segregation. The arrows indicate the patients (P1 and P2) described in this case report. The pedigree chart was created in the program QuickPed (28).
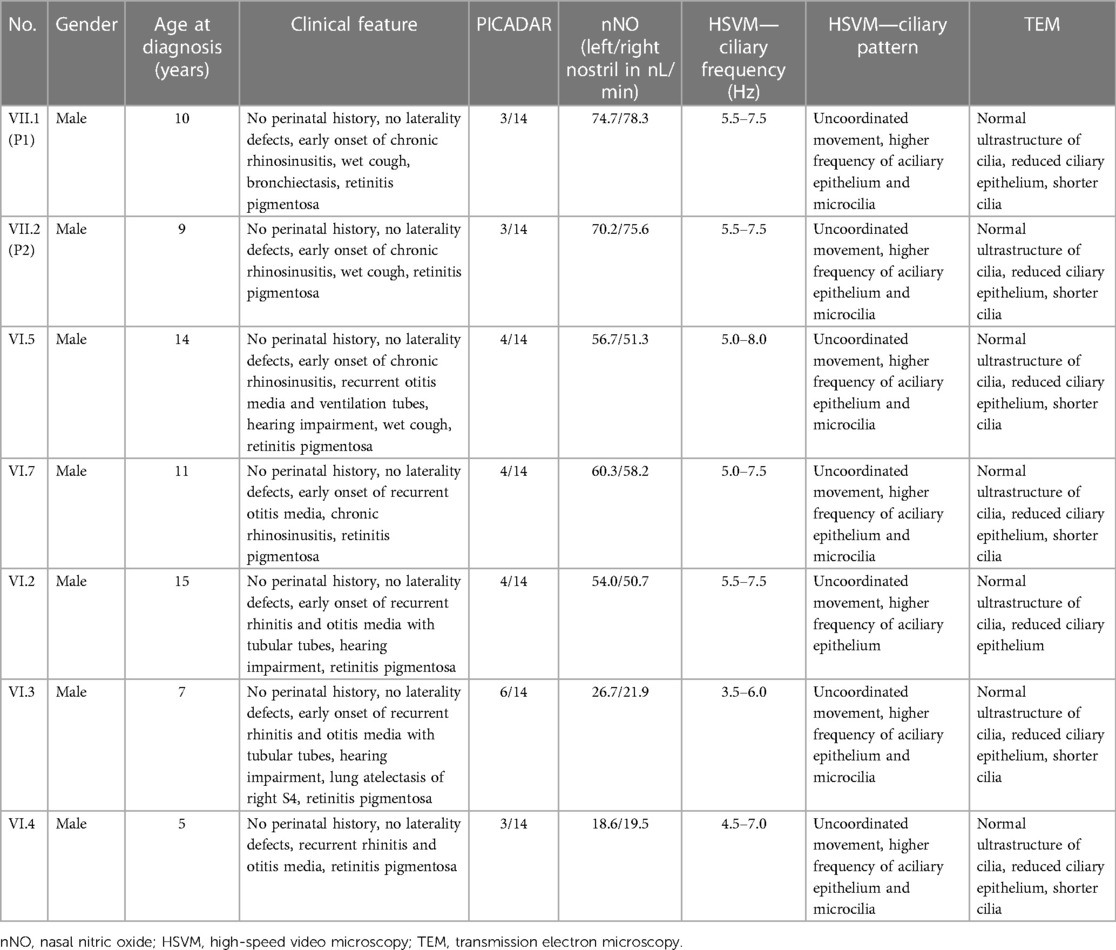
Table 1. Characteristics and results of diagnostic test for PCD in all examined patients.
Both RP and PCD are rare genetic diseases belonging to a very heterogeneous group of inherited defects in ciliary function called ciliopathies. RP, a progressive degenerative disorder of the retina, is caused by the loss of viability and cell death of photoreceptors that are modified primary cilia with sensory function. Conversely, PCD is a multisystem disorder of motile cilia characterised by impaired mucociliary clearance, leading to recurrent respiratory infections. Since motile cilia are also present outside the airways, patients often suffer from laterality abnormalities and subfertility. Since primary and motile cilia share structural features as well as many proteins necessary for their proper function, it is not surprising that mutations in some genes influence the function of both types of cilia, leading to syndromic manifestation with overlapping features of motile and non-motile ciliopathies. This is also the case of the RPGR gene associated with X-linked RP, but several cases of RPGR mutations leading to RP with respiratory symptoms have been published (3, 12, 14–17, 29, 30). This complex interplay between RPGR mutations, RP, and PCD underscores the importance of understanding the underlying molecular mechanisms and potential therapeutic approaches.
PCD is typically inherited in an autosomal recessive manner, but rare cases with autosomal dominant (31) or X-linked inheritance have also been described (3, 32, 33). We report the cases of two brothers with X-linked PCD associated with RP as a result of a novel frameshift mutation in the RPGR gene. Both patients were diagnosed with RP and were monitored because of the early onset of rhinosinusitis, recurrent respiratory infections, and bronchiectasis in the older brother. None of the boys suffered from neonatal respiratory distress, congenital heart defects, or laterality defects. For suspected PCD, they were examined for nNO measurement, ciliary ultrastructure, CBF, and PICADAR score. Lastly, genetic testing revealed mutation c.309_310insA (p.Glu104Argfs*12) located in exon 4 of the RPGR gene, which has not been found in databases or reported in the literature so far. The insertion of nucleotide A into the coding sequence results in a frameshift and premature termination of translation and subsequently presumably leads to nonsense-mediated decay. The character of this mutation was determined to be deleterious and disease-causing, which was also predicted by in silico analysis. Based on these diagnostic tests, we were able to confirm PCD diagnosis with X-linked inheritance.
The RPGR gene located on the X chromosome encodes more than 20 various isoforms resulting from alternative splicing and post-translational modifications that are tissue-specific (9, 10, 34). This explains the different levels of these isoforms in different tissues. There are two major widely expressed isoforms, RPGR1−19 and RPGRORF, that share exons 1–14 but differ in the C-terminus. In humans, the RPGRORF isoform is expressed predominantly in the outer segment of photoreceptors in the retina and harbours the majority of mutations leading to XLRP (35). The constitutive RPGR1−19 isoform is encoded by 19 exons and contains an isoprenylation signal in the C-terminus for its membrane trafficking and ciliary localisation (36–38). This isoform was detected, among others, in motile cilia of the respiratory tract, where it is localised in the transitional zone at the base of the cilium (10). The presence of defective RPGR protein in the respiratory cilia explains respiratory symptoms in RP patients with an RPGR mutation. The function of RPGR protein is not completely understood, but it is proposed that together with its interacting partners, it plays an important role in intraflagellar transport, ensuring protein trafficking in cilia, thereby maintaining their viability and proper function (39–41). Therefore, it can be assumed that mutations in the RPGR gene disrupt ciliary transport and signalling pathways, contributing to the development of both conditions. Nevertheless, it should be noted that not all RPGR mutations result in the PCD phenotype, and the clinical manifestation may vary between individuals.
All examined boys in the family had similar clinical manifestations of PCD: repeated rhinosinusitis and otitis, which started in early childhood in some of them. Eye symptoms such as decreased night vision and the worsening of the mid-peripheral visual field appeared at a young school age. In all patients, we observed normal ultrastructure of respiratory cilia, reduced or even aciliary epithelium, and shorter cilia. The HSVMA showed beat frequency at the lower limit of the normal range but an uncoordinated pattern of movement. McCray et al. reported normal ultrastructure of respiratory cilia but abnormal ciliary orientation associated with defects in the CBP in patients with XLRP without PCD symptoms (42). No abnormalities in ultrastructure were observed in PCD patients with RPGR mutations either, but in the case of the c.154G>A mutation, impaired ciliary orientation and uncoordinated beating of cilia were revealed (12). Normal ciliary beating and orientation are determined during ciliogenesis by the correct anchoring and orientation of the basal body in the plasma membrane, which is controlled through planar cell polarity (PCP) signalling (43, 44). It is suggested that RPGR protein is involved in cilia orientation by interacting with the components of the PCP pathway. Patnaik et al. demonstrated the role of RPGR and its interacting proteins (RPGRIP1 and RPGRIP2l) in actin cytoskeleton arrangement by regulating the stability of components in the PCP pathway (45). This can be one of the mechanisms contributing to the ciliopathies caused by RPGR mutations because the organisation of the actin cytoskeleton is important for proper ciliogenesis and ciliary orientation of the primary retinal cilia (40) as well as motile cilia (46).
The segregation analysis showed that the mother of our patients is a heterozygous carrier of the RPGR mutation consistent with X-linked inheritance. She suffers from myopia but has no respiratory symptoms, and PCD tests were negative, without abnormalities of HSVMA. The manifestation of retinal dystrophy was also recorded in the maternal grandmother as well as in other heterozygous carriers in the family. In the literature, a wide spectrum of visual dysfunction and abnormalities in the function and structure of photoreceptors were observed in female carriers, which is probably caused by mosaicism of inactivated X chromosomes (47, 48). Theoretically, epigenetic silencing of the X chromosome in the early stages of embryonic development is random, and the ratio of inactivated X chromosomes of maternal and paternal origin is 50:50. However, pathogenic mutations in X-linked genes can cause skewing of X-inactivation, leading to disproportionality in the distribution of X chromosomes with mutant alleles (49). The presence of RP symptoms, but not respiratory symptoms in the heterozygous mother, can be explained by two hypotheses. The first one assumes skewing of X chromosome inactivation in the respiratory cells of females as the result of preferential selection of wild-type gene alleles in respiratory cells. Another hypothesis proposes that a small amount of normal RPGR protein is sufficient for the function of respiratory cilia (3).
This case report contributes to the assumption that RPGR mutations disrupt the function of cilia in multiple tissues, including the retina and respiratory tract, highlighting the essential role of RPGR protein in ciliary biology. Understanding the underlying molecular mechanisms can help in the development of targeted therapies and also improve therapeutic approaches and genetic counselling. Moreover, the identification of RPGR mutations in patients with RP and PCD and described genotype–phenotype correlations have important implications for clinical practice to recognise a broader spectrum of PCD phenotypes as well as for future research focused on the genetic basis of PCD, gene interactions, the pathways implicated in PCD pathogenesis, and the role of RPGR protein for the proper functioning of cilia in various tissues throughout the body.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
The studies involving humans were approved by Ethics Committee of the University Hospital Martin. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
ZK: Conceptualization, Investigation, Methodology, Validation, Writing—original draft. PD: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Project administration, Writing—original draft. VH: Methodology, Writing—review and editing. AD: Investigation, Methodology, Writing—review and editing. MJ: Investigation, Writing—review and editing. PB: Writing—review and editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article.
This publication has been produced with the support of the Integrated Infrastructure Operational Program for the project Systemic Public Research Infrastructure—Biobank for Cancer and Rare Diseases, ITMS: 313011AFG5, co-financed by the European Regional Development Fund.
We wish to thank Assoc. Prof. Zora Lasabova and the Department of Molecular Biology and Genomics JFM CU for technical support and Oľga Nyitrayova for electron microscopy.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2024.1339664/full#supplementary-material
1. Mani R, Gomes M, González AR, Hogg C, Morris-Rosendahl D, Maitre B, et al. Development and first results of the BEAT-PCD international primary ciliary dyskinesia gene variant database: CiliaVar. Eur Respir J. (2021) 58. doi: 10.1183/13993003.congress-2021.PA3458
2. Shapiro AJ, Davis SD, Ferkol T, Dell SD, Rosenfeld M, Olivier KN, et al. Laterality defects other than situs inversus totalis in primary ciliary dyskinesia: insights into situs ambiguus and heterotaxy. Chest. (2014) 146:1176–86. doi: 10.1378/chest.13-1704
3. Moore A, Escudier E, Roger G, Tamalet A, Pelosse B, Marlin S, et al. RPGR is mutated in patients with a complex X linked phenotype combining primary ciliary dyskinesia and retinitis pigmentosa. J Med Genet. (2006) 43:326–33. doi: 10.1136/jmg.2005.034868
4. Daiger SP, Bowne SJ, Sullivan LS. Perspective on genes and mutations causing retinitis pigmentosa. Arch Ophthalmol. (2007) 125:151–8. doi: 10.1001/archopht.125.2.151
5. Stone EM, Andorf JL, Whitmore SS, DeLuca AP, Giacalone JC, Streb LM, et al. Clinically focused molecular investigation of 1000 consecutive families with inherited retinal disease. Ophthalmology. (2017) 124:1314–31. doi: 10.1016/j.ophtha.2017.04.008
6. Zhang Q, Giacalone JC, Searby C, Stone EM, Tucker BA, Sheffield VC. Disruption of RPGR protein interaction network is the common feature of RPGR missense variations that cause XLRP. Proc Natl Acad Sci U S A. (2019) 116:1353–60. doi: 10.1073/pnas.1817639116
7. Megaw RD, Soares DC, Wright AF. RPGR: its role in photoreceptor physiology, human disease, and future therapies. Exp Eye Res. (2015) 138:32–41. doi: 10.1016/j.exer.2015.06.007
8. Hosch J, Lorenz B, Stieger K. RPGR: role in the photoreceptor cilium, human retinal disease, and gene therapy. Ophthalmic Genet. (2011) 32:1–11. doi: 10.3109/13816810.2010.535889
9. He S, Parapuram SK, Hurd TW, Behnam B, Margolis B, Swaroop A, et al. Retinitis pigmentosa GTPase regulator (RPGR) protein isoforms in mammalian retina: insights into X-linked retinitis pigmentosa and associated ciliopathies. Vision Res. (2008) 48:366–76. doi: 10.1016/j.visres.2007.08.005
10. Hong D-H, Pawlyk B, Sokolov M, Strissel KJ, Yang J, Tulloch B, et al. RPGR isoforms in photoreceptor connecting cilia and the transitional zone of motile cilia. Invest Ophthalmol Visual Sci. (2003) 44:2413–21. doi: 10.1167/iovs.02-1206
11. Schmid F, Glaus E, Cremers FPM, Kloeckener-Gruissem B, Berger W, Neidhardt J. Mutation- and tissue-specific alterations of RPGR transcripts. Invest Ophthalmol Visual Sci. (2010) 51:1628–35. doi: 10.1167/iovs.09-4031
12. Bukowy-Bieryłło Z, Ziętkiewicz E, Loges NT, Wittmer M, Geremek M, Olbrich H, et al. RPGR mutations might cause reduced orientation of respiratory cilia. Pediatr Pulmonol. (2013) 48:352–63. doi: 10.1002/ppul.22632
13. Dry KL, Manson FD, Lennon A, Bergen AA, Van Dorp DB, Wright AF. Identification of a 5’ splice site mutation in the RPGR gene in a family with X-linked retinitis pigmentosa (RP3). Hum Mutat. (1999) 13:141–5. doi: 10.1002/(SICI)1098-1004(1999)13:2%3C141::AID-HUMU6%3E3.0.CO;2-Q
14. Iannaccone A, Breuer DK, Wang XF, Kuo SF, Normando EM, Filippova E, et al. Clinical and immunohistochemical evidence for an X linked retinitis pigmentosa syndrome with recurrent infections and hearing loss in association with an RPGR mutation. J Med Genet. (2003) 40:e118. doi: 10.1136/jmg.40.11.e118
15. van Dorp DB, Wright AF, Carothers AD, Bleeker-Wagemakers EM. A family with RP3 type of X-linked retinitis pigmentosa: an association with ciliary abnormalities. Hum Genet. (1992) 88:331–4. doi: 10.1007/BF00197269
16. Zito I, Downes SM, Patel RJ, Cheetham ME, Ebenezer ND, Jenkins SA, et al. RPGR mutation associated with retinitis pigmentosa, impaired hearing, and sinorespiratory infections. J Med Genet. (2003) 40:609–15. doi: 10.1136/jmg.40.8.609
17. Sengillo JD, Fridman G, Cho GY, Buchovecky C, Tsang SH. Novel mutation in retinitis pigmentosa GTPase regulator gene causes primary ciliary dyskinesia and retinitis pigmentosa. Ophthalmic Surg Lasers Imaging Retina. (2018) 49:548–52. doi: 10.3928/23258160-20180628-14
18. Krawczyński MR, Witt M. PCD and RP: X-linked inheritance of both disorders? Pediatr Pulmonol. (2004) 38:88–9. doi: 10.1002/ppul.30001
19. Lucas JS, Barbato A, Collins SA, Goutaki M, Behan L, Caudri D, et al. European respiratory society guidelines for the diagnosis of primary ciliary dyskinesia. Eur Respir J. (2017) 49. doi: 10.1183/13993003.01090-2016
20. Behan L, Dimitrov BD, Kuehni CE, Hogg C, Carroll M, Evans HJ, et al. PICADAR: a diagnostic predictive tool for primary ciliary dyskinesia. Eur Respir J. (2016) 47:1103–12. doi: 10.1183/13993003.01551-2015
21. Beydon N, Kouis P, Marthin JK, Latzin P, Colas M, Davis SD, et al. Nasal nitric oxide measurement in children for the diagnosis of primary ciliary dyskinesia: European Respiratory Society technical standard. Eur Respir J. (2023) 61:2202031. doi: 10.1183/13993003.02031-2022
22. Koniar D, inventor; Jessenius Faculty of Medicine of Comenius University, University of Zilina, assignee. Automatic lighting system of inverted microscope for high-speed cinematography. Slovak Republic. Utility Model No 6811. (2014).
23. Hargaš L, Koniar D, Štofan S. Advanced methodology for frequency description of biomechanical systems. Procedia Eng. (2012) 48:205–12. doi: 10.1016/j.proeng.2012.09.506
24. Koniar D, Hargaš L, Stofan S, Hrianka M. High speed video system for tissue measurement based on PWM regulated dimming and virtual instrumentation. Elektronika Ir Elektrotechnika. (2015) 106:169–72. doi: 10.5755/J01.EEE.106.10.9154
25. Loncova Z, Hargas L, Koniar D, Simonova A, Kozacek B. Objects detection and recognition in biomedical microscopic images for the purpose of non-invasive and more precise diagnostic. 2017 Progress in Electromagnetics Research Symposium—Spring (PIERS) (2017). p. 3159–65. doi: 10.1109/PIERS.2017.8262301
26. MutationTaster. Available at: https://www.mutationtaster.org/ (Accessed November 14, 2022).
27. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
28. Vigeland MD. QuickPed: an online tool for drawing pedigrees and analysing relatedness. BMC Bioinformatics. (2022) 23:220. doi: 10.1186/s12859-022-04759-y
29. Krawczyński MR, Dmeńska H, Witt M. Apparent X-linked primary ciliary dyskinesia associated with retinitis pigmentosa and a hearing loss. J Appl Genet. (2004) 45:107–10. doi: 10.1002/ppul.30001
30. De Brauwer PJ, Blaise P, Hermans G, Boniver V, Bartsch P, Rakic JM. Retinitis pigmentosa and bronchiectasis: a case report on a rare association suggestive of a common underlying primary ciliary dyskinesia (PCD). Bull Soc Belge Ophtalmol. (2010) 314:9–14.
31. Wallmeier J, Frank D, Shoemark A, Nöthe-Menchen T, Cindric S, Olbrich H, et al. De Novo mutations in FOXJ1 result in a motile ciliopathy with hydrocephalus and randomization of left/right body asymmetry. Am J Hum Genet. (2019) 105:1030–9. doi: 10.1016/j.ajhg.2019.09.022
32. Paff T, Loges NT, Aprea I, Wu K, Bakey Z, Haarman EG, et al. Mutations in PIH1D3 cause X-linked primary ciliary dyskinesia with outer and inner dynein arm defects. Am J Hum Genet. (2017) 100:160–8. doi: 10.1016/j.ajhg.2016.11.019
33. Aprea I, Raidt J, Höben IM, Loges NT, Nöthe-Menchen T, Pennekamp P, et al. Defects in the cytoplasmic assembly of axonemal dynein arms cause morphological abnormalities and dysmotility in sperm cells leading to male infertility. PLoS Genet. (2021) 17:e1009306. doi: 10.1371/journal.pgen.1009306
34. Kirschner R, Rosenberg T, Schultz-Heienbrok R, Lenzner S, Feil S, Roepman R, et al. RPGR transcription studies in mouse and human tissues reveal a retina-specific isoform that is disrupted in a patient with X-linked retinitis pigmentosa. Hum Mol Genet. (1999) 8:1571–8. doi: 10.1093/hmg/8.8.1571
35. Vervoort R, Lennon A, Bird AC, Tulloch B, Axton R, Miano MG, et al. Mutational hot spot within a new RPGR exon in X-linked retinitis pigmentosa. Nat Genet. (2000) 25:462–6. doi: 10.1038/78182
36. Yan D, Swain PK, Breuer D, Tucker RM, Wu W, Fujita R, et al. Biochemical characterization and subcellular localization of the mouse retinitis pigmentosa GTPase regulator (mRpgr). J Biol Chem. (1998) 273:19656–63. doi: 10.1074/jbc.273.31.19656
37. Rao KN, Zhang W, Li L, Anand M, Khanna H. Prenylated retinal ciliopathy protein RPGR interacts with PDE6δ and regulates ciliary localization of Joubert syndrome-associated protein INPP5E. Hum Mol Genet. (2016) 25:4533–45. doi: 10.1093/hmg/ddw281
38. Lee J-J, Seo S. PDE6D Binds to the C-terminus of RPGR in a prenylation-dependent manner. EMBO Rep. (2015) 16:1581–2. doi: 10.15252/embr.201541220
39. Khanna H. More than meets the eye: current understanding of RPGR function. In: Ash JD, Anderson RE, LaVail MM, Bowes Rickman C, Hollyfield JG, Grimm C, editors. Retinal Degenerative Diseases. Advances in Experimental Medicine and Biology. Cham: Springer International Publishing (2018). p. 521–38. doi: 10.1007/978-3-319-75402-4_64
40. Gakovic M, Shu X, Kasioulis I, Carpanini S, Moraga I, Wright AF. The role of RPGR in cilia formation and actin stability. Hum Mol Genet. (2011) 20:4840–50. doi: 10.1093/hmg/ddr423
41. Patnaik SR, Raghupathy RK, Zhang X, Mansfield D, Shu X. The role of RPGR and its interacting proteins in ciliopathies. J Ophthalmol. (2015) 2015:e414781. doi: 10.1155/2015/414781
42. McCray G, Griffin P, Martinello P, de Iongh R, Ruddle J, Robinson P. Altered airway ciliary orientation in patients with X-linked retinitis pigmentosa. Thorax. (2019) 74:914–6. doi: 10.1136/thoraxjnl-2018-212584
43. Apodaca G. Role of polarity proteins in the generation and organization of apical surface protrusions. Cold Spring Harb Perspect Biol. (2018) 10:a027813. doi: 10.1101/cshperspect.a027813
44. Marshall WF. Chapter 1 basal bodies: platforms for building cilia. In: Yoder BY, editor. Current Topics in Developmental Biology. Ciliary Function in Mammalian Development. Academic Press (2008). p. 1–22 doi: 10.1016/S0070-2153(08)00801-6
45. Patnaik SR, Zhang X, Biswas L, Akhtar S, Zhou X, Kusuluri DK, et al. RPGR protein complex regulates proteasome activity and mediates store-operated calcium entry. Oncotarget. (2018) 9:23183–97. doi: 10.18632/oncotarget.25259
46. Werner ME, Hwang P, Huisman F, Taborek P, Yu CC, Mitchell BJ. Actin and microtubules drive differential aspects of planar cell polarity in multiciliated cells. J Cell Biol. (2011) 195:19–26. doi: 10.1083/jcb.201106110
47. Talib M, van Schooneveld MJ, Van Cauwenbergh C, Wijnholds J, ten Brink JB, Florijn RJ, et al. The spectrum of structural and functional abnormalities in female carriers of pathogenic variants in the RPGR gene. Invest Ophthalmol Visual Sci. (2018) 59:4123–33. doi: 10.1167/iovs.17-23453
48. Nanda A, Salvetti AP, Clouston P, Downes SM, MacLaren RE. Exploring the variable phenotypes of RPGR carrier females in assessing their potential for retinal gene therapy. Genes (Basel). (2018) 9:643. doi: 10.3390/genes9120643
Keywords: RPGR gene, mutation, retinitis pigmentosa, primary ciliary dyskinesia, X-linked inheritance
Citation: Kolkova Z, Durdik P, Holubekova V, Durdikova A, Jesenak M and Banovcin P (2024) Identification of a novel RPGR mutation associated with retinitis pigmentosa and primary ciliary dyskinesia in a Slovak family: a case report. Front. Pediatr. 12:1339664. doi: 10.3389/fped.2024.1339664
Received: 16 November 2023; Accepted: 10 January 2024;
Published: 25 January 2024.
Edited by:
Ammar Husami, Cincinnati Children’s Hospital Medical Center, United StatesReviewed by:
Emilia Stellacci, National Institute of Health (ISS), Italy© 2024 Kolkova, Durdik, Holubekova, Durdikova, Jesenak and Banovcin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter Durdik cGV0ZXIuZHVyZGlrQGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.