 Jian Zha
Jian Zha Yong Chen1,†
Yong Chen1,† Jianmin Zhong
Jianmin Zhong- 1Department of Neurology, Jiangxi Provincial Children’s Hospital, Nanchang, China
- 2Department of Radiology, Jiangxi Provincial Children’s Hospital, Nanchang, China
METTL23 belongs to a family of protein lysine methyltransferases that methylate non-histone proteins. Recently, the METTL23 gene has been reported to be related to an intellectual developmental disorder, autosomal recessive 44. Patients present with developmental delay, intellectual disability (ID), and variable dysmorphic features. Here, we report on a Chinese girl who presented with global developmental delay, abnormal brain structure, and multiple facial deformities, including a short/upturned nose with a sunken bridge, thin lips, and flat occiput. Whole-exome sequencing identified a novel variant (NM_001080510.5: c.322+1del) on the METTL23 gene. This variant was not collected on public human variants databases such as gnomAD, predicted to influence the splicing as a classical splicing variant, and classified as Pathogenic according to the American College of Medical Genetics and Genomics (ACMG) guidelines. Since patients with METTL23-related ID are rare, we summarize and compare the clinical phenotype of reported patients with METTL23 variants. Our report further expands the METTL23 variants and provides new evidence for clinical diagnosis of METTL23-related ID.
Introduction
Intellectual disability (ID) is a frequently observed phenotype in clinical genetics, with an estimated prevalence of 1%–3% worldwide (1). It occurs independently or as a component within a more intricate neurological or systemic syndrome. It is believed that genetic factors contribute significantly to a substantial portion of ID patients (2). Transcription plays a pivotal role in the consolidation of memory and is essential for the establishment of long-term synaptic plasticity and memory retention. Recently, more and more transcriptional regulations were identified as ID genes (3).
METTL23 encodes a transcription factor regulator, first identified as the pathogenic gene of ID (OMIM: 615942) from a large Yemen consanguineous family (4). After its initial identification, several variations of the gene were reported (5–8). The main clinical phenotypes include developmental delay in motor and/or language, seizures, specific facial features, abnormal brain MRI, and other behavioral problems, such as autism and attention-deficit hyperactivity disorder (ADHD).
Here, we identify a novel METTL23 gene variant (NM_001080510.5: c.322+1del) in our patient. She presented with developmental delay in motor and language. Her white matter myelination might be delayed compared to children of the same age, and the splenium of the corpus callosum developed thinly. Our report summarizes and compares the clinical phenotypes of patients with METTL23 gene variants and expands the spectrum of METTL23 gene variants related to ID.
Case report
Our patient is a girl from a consanguineous family aged 1 year 10 months who was born at full-term by normal delivery without an abnormal birth history or other family history of disease. She was able to lift her head at 4 months, roll over at 5 months, sit unassisted at 10 months, and presented developmental delay aged 1 year 3 months. When she was admitted to our hospital, she could not crawl or stand, her hands were not flexible in grasping objects, and she could only make “da, ma, ba” sounds.
She has specific facial features, with a short, upturned nose with a sunken bridge, thin lips, and a flat occiput (Figures 1A,B). The electroencephalography results showed that the background activity was slow, but no seizures were present. Brain MRI results suggested that white matter myelination might be delayed when compared with children of the same age, and the splenium of the corpus callosum developed thinly (Figures 1C,D). She has been undergoing rehabilitation training locally and has not returned to the clinic. During telephone follow-up, the child's development gradually improved. Currently, she can imitate adults’ speech, has learned to say goodbye, and learned body language, such as clapping and shaking her head. She is currently crawling steadily and can stand with support. We also incorporated previously reported METTL23 gene variant cases in our analysis. Additional phenotypic and genetic findings in individuals are summarized in Table 1.
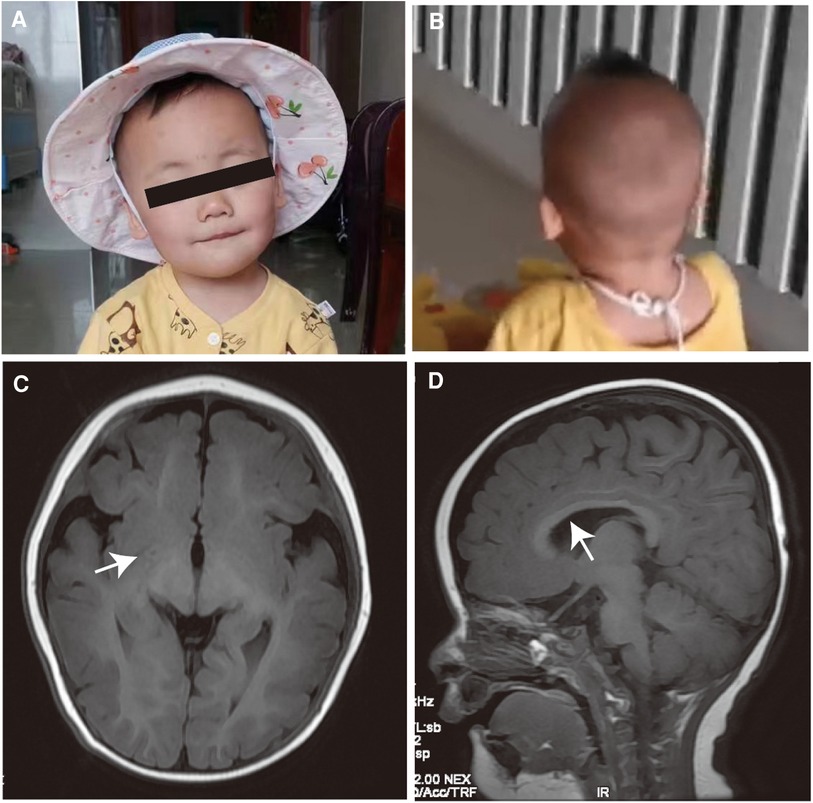
Figure 1 Clinical features. (A,B) Facial features of the affected individual. She has a short, upturned nose with a sunken bridge, thin lips, and flat occiput. (C,D) The brain MRI results in an infant aged 1 year 3 months. The white arrows show delayed white matter myelination and the thinly developed splenium of the corpus callosum.
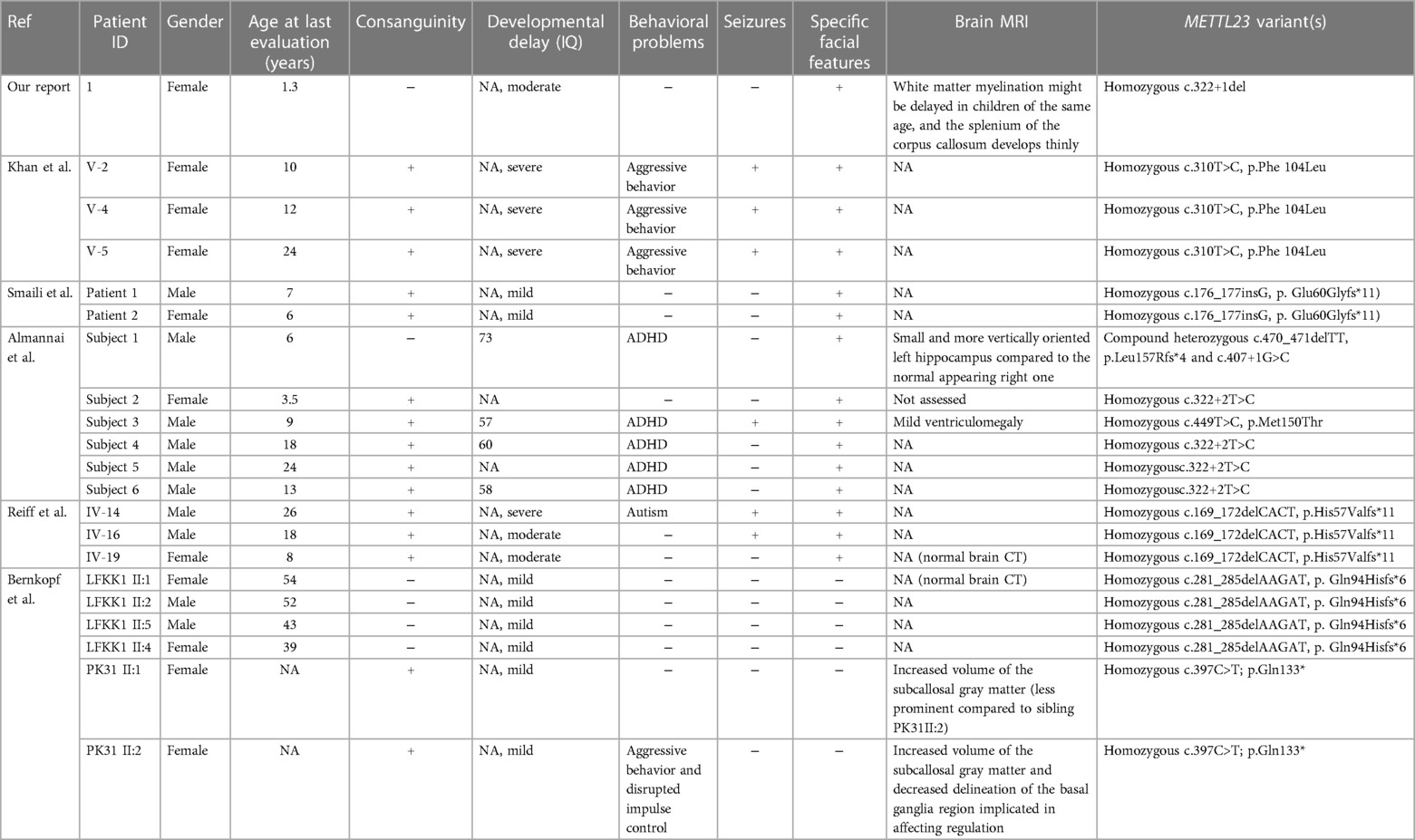
Table 1 Summary of the clinical features of patients with METTL23 variants.
Genetic testing
Whole-exome sequencing was performed to further clarify the cause for our patient. A homozygous splicing variant of the METTL23 gene (NM_001080510.5: c.322+1del) was identified by further genetic testing. The METTL23 variant found in our patient was inherited from her father (Figure 2A), which was confirmed using Sanger sequencing (Figure 2B). The classical splicing variant c.322+1del has a Splice AI score >0.5 (Donor Loss score = 0.83). The METTL23 variants were rare, and the variant in our patient was not included in gnomAD, ExAC, ClinVar, or other databases. Therefore, c.322+1del in our patient was classified as pathogenic according to the American College of Medical Genetics and Genomics guidelines (ACMG) (9) (PVS1+PM3_Supporting+PM2_Supporting).
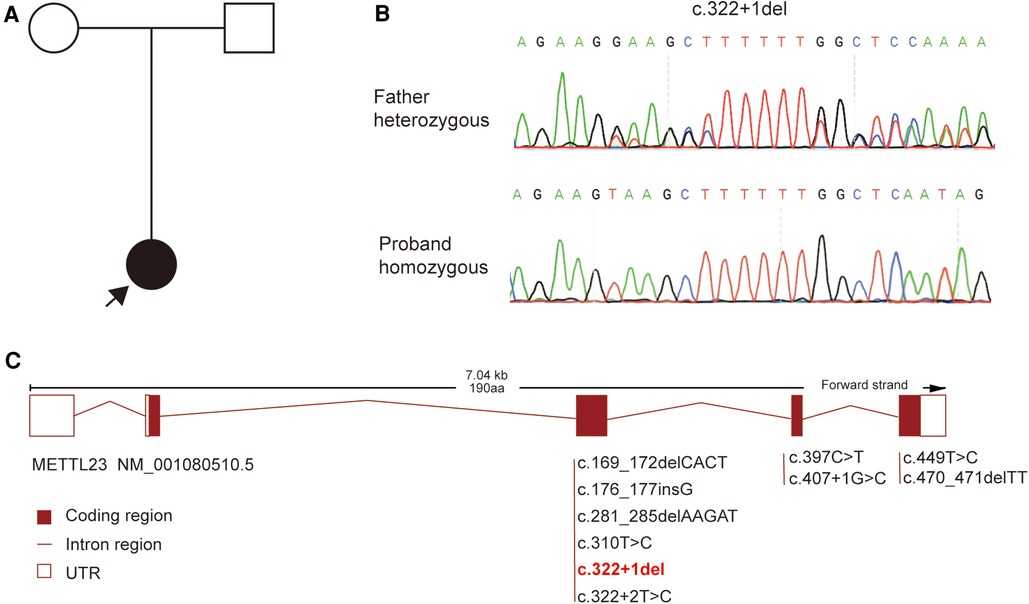
Figure 2 The genetic finding for our patient. (A) The pedigree for our patient. (B) Sanger sequencing verified the variant we found. (C) The spectrum of METTL23 gene variants has been found.
Discussion
The METTL23 gene is deduced based on the METTL22 sequence (10) and as a member of the non-histone methylating protein lysine methyltransferase family. It encodes a 190 (isoform 1) or 123 (isoform 2) amino acid protein, but the function of this gene is largely unknown. It was first reported as a pathogenic gene to ID in 2014, and it has a positive regulatory effect on GABP (GA-binding protein transcription factor) function through its interaction with the subunit GABPA (4). Genes regulated by GABP, such as THPO and ATP5B, may play roles in cognitive and neurodevelopment (4). THPO is thought to be involved in neuroprotection, apoptosis, development, and nerve cell differentiation (11), while ATP5B is downregulated in the thalamus of autistic patients (12). To further elucidate the catalytic performance of METTL23, Bernkopf et al. modeled the three-dimensional structure of human METTL23 and clarified the methyltransferase function of METTL23 (5).
With further exploration of the function of METTL23 protein and the discovery of clinical cases, METTL23 gene variants have been associated with an intellectual developmental disorder, autosomal recessive 44 (OMIM: 615942). Currently, there are 10 reported mutations in the METTL23 gene, including our report. These mutations consist of two missense variants, five nonsense variants, and three splice site mutations. The mutation spectrum of the METTL23 gene is primarily dominated by homozygous mutations, with only one instance of compound heterozygous mutations. The mutation in our patient is c.322+1del, which affects the classical splicing site. The METTL23-related disease is reported to be caused by loss of function (LOF) (5).
The main clinical features related to METTL23 variants are developmental delay in motor and/or language and specific facial features. Patients with METTL23 variants exhibit different levels of ID. Reiff et al. (4) reported their patient's ID was moderate to severe, but in the study by Bernkopf et al. (5), it was mild. The patient in our report showed mild ID. Her development gradually improved through rehabilitation training. We did not find an association between the severity of ID and the type of variant, and it may require additional functional verification to understand. Symptoms may include behavioral problems, such as ADHD, aggressive behavior, and seizures (6, 7). Neither behavioral problems nor seizures have occurred in our patient. She presented with developmental delay and specific facial features with a short, upturned nose with a sunken bridge, thin lips, and a flat occiput. This facial deformity exhibits a high level of consistency in previous reports. It confirms that METTL23 may be a distinct clinical entity associating ID with specific facial dysmorphia (7). In addition, our patient had abnormal MRI results, which showed white matter myelination delay and thin splenium of the corpus callosum. This MRI abnormality was not reported previously, nor were small and more vertically oriented left hippocampus, mild ventriculomegaly (6), and increased volume of subcallosal gray matter (5). This indicates that the brain structural abnormalities caused by METTL23 gene variants may be diverse.
In conclusion, we have summarized the clinical characteristics of all previously reported cases. Patients with developmental delay, varying degrees of intellectual disability, behavioral abnormalities, and epilepsy should undergo genetic testing. This will help clarify the cause of their condition and provide early clinical diagnosis and treatment. Our study contributes a new case of ID with METTL23 variants, making this the first reported case in China. At present, the specific mechanism of METTL23 mutation and the occurrence of ID are unknown, and more cases and further experiments are needed to understand the specific pathogenesis.
Data availability statement
The variant site presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found here: https://www.ncbi.nlm.nih.gov/clinvar/variation/2571608/?oq=SCV003936901m=NM_015166.4(MLC1):c.838_843delinsATTTTA%20(p.Ser280_Phe281delinsIleLeu), SCV004041809.
Ethics statement
The studies involving humans were approved by the Human Ethics Committees of Jiangxi Provincial Children's Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
JZha: Writing – original draft. YC: Writing – original draft. FC: Writing – review & editing, Data curation. YY: Writing – review & editing, Data curation. RW: Writing – review & editing, Writing – original draft. JZho: Writing – review & editing, Writing – original draft.
Funding
The authors declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
We would like to thank CIPHER GENE for their support of the whole-exome sequencing and the patient's family for participating in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Maulik PK, Mascarenhas MN, Mathers CD, Dua T, Saxena S. Prevalence of intellectual disability: a meta-analysis of population-based studies. Res Dev Disabil. (2011) 32(2):419–36. doi: 10.1016/j.ridd.2010.12.018
2. Ellison JW, Rosenfeld JA, Shaffer LG. Genetic basis of intellectual disability. Annu Rev Med. (2013) 64:441–50. doi: 10.1146/annurev-med-042711-140053
3. Vissers LE, Gilissen C, Veltman JA. Genetic studies in intellectual disability and related disorders. Nat Rev Genet. (2016) 17(1):9–18. doi: 10.1038/nrg3999
4. Reiff RE, Ali BR, Baron B, Yu TW, Ben-Salem S, Coulter ME, et al. METTL23, a transcriptional partner of GABPA, is essential for human cognition. Hum Mol Genet. (2014) 23(13):3456–66. doi: 10.1093/hmg/ddu054
5. Bernkopf M, Webersinke G, Tongsook C, Koyani CN, Rafiq MA, Ayaz M, et al. Disruption of the methyltransferase-like 23 gene METTL23 causes mild autosomal recessive intellectual disability. Hum Mol Genet. (2014) 23(15):4015–23. doi: 10.1093/hmg/ddu115
6. Almannai M, Obaid O, Faqeih E, Alasmari A, Samman MM, Pinz H, et al. Further delineation of METTL23-associated intellectual disability. Am J Med Genet A. (2020) 182(4):785–91. doi: 10.1002/ajmg.a.61503
7. Khan A, Miao Z, Umair M, Ullah A, Alshabeeb MA, Bilal M, et al. Two cases of recessive intellectual disability caused by NDST1 and METTL23 variants. Genes (Basel). (2020) 11(9):1021. doi: 10.3390/genes11091021
8. Smaili W, Elalaoui SC, Zrhidri A, Raymond L, Egéa G, Taoudi M, et al. Exome sequencing revealed a novel homozygous METTL23 gene mutation leading to familial mild intellectual disability with dysmorphic features. Eur J Med Genet. (2020) 63(7):103951. doi: 10.1016/j.ejmg.2020.103951
9. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17(5):405–24. doi: 10.1038/gim.2015.30
10. Cloutier P, Lavallée-Adam M, Faubert D, Blanchette M, Coulombe B. A newly uncovered group of distantly related lysine methyltransferases preferentially interact with molecular chaperones to regulate their activity. PLoS Genet. (2013) 9(1):e1003210. doi: 10.1371/journal.pgen.1003210
11. Diederich K, Schäbitz WR, Minnerup J. Seeing old friends from a different angle: novel properties of hematopoietic growth factors in the healthy and diseased brain. Hippocampus. (2012) 22(5):1051–7. doi: 10.1002/hipo.20904
Keywords: METTL23, intellectual disability, developmental delay, magnetic resonance imaging, gene variant
Citation: Zha J, Chen Y, Cao F, Yu Y, Wang R and Zhong J (2024) Identification of a novel METTL23 gene variant in a patient with an intellectual development disorder: a literature review and case report. Front. Pediatr. 12:1328063. doi: 10.3389/fped.2024.1328063
Received: 26 October 2023; Accepted: 12 June 2024;
Published: 4 July 2024.
Edited by:
Santasree Banerjee, Jilin University, ChinaReviewed by:
Zhanqi Hu, Shenzhen Children's Hospital, ChinaJuan Pociecha, Garrahan Hospital, Argentina
Jing Gan, Sichuan University, China
© 2024 Zha, Chen, Cao, Yu, Wang and Zhong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ruiyan Wang, d3J5XzE5OTlfMjAwNEAxNjMuY29t; Jianmin Zhong, emhvbmdqbUAxNjMuY29t
†These authors have contributed equally to this work