
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
SYSTEMATIC REVIEW article
Front. Pediatr. , 17 January 2024
Sec. Genetics of Common and Rare Diseases
Volume 12 - 2024 | https://doi.org/10.3389/fped.2024.1315229
 Vivian van Wijngaarden1*
Vivian van Wijngaarden1* Hester de Wilde2
Hester de Wilde2 Dieuwke Mink van der Molen3Jildo Petter3
Dieuwke Mink van der Molen3Jildo Petter3 Inge Stegeman4,5Ellen Gerrits6,7
Inge Stegeman4,5Ellen Gerrits6,7 Adriana L. Smit4,6
Adriana L. Smit4,6 Marie-José van den Boogaard1
Marie-José van den Boogaard1
Introduction: Developmental language disorder (DLD) is a common childhood condition negatively influencing communication and psychosocial development. An increasing number of pathogenic variants or chromosomal anomalies possibly related to DLD have been identified. To provide a base for accurate clinical genetic diagnostic work-up for DLD patients, understanding the specific genetic background is crucial. This study aims to give a systematic literature overview of pathogenic variants or chromosomal anomalies causative for DLD in children.
Methods: We conducted a systematic search in PubMed and Embase on available literature related to the genetic background of diagnosed DLD in children. Included papers were critically appraised before data extraction. An additional search in OMIM was performed to see if the described DLD genes are associated with a broader clinical spectrum.
Results: The search resulted in 15,842 papers. After assessing eligibility, 47 studies remained, of which 25 studies related to sex chromosome aneuploidies and 15 papers concerned other chromosomal anomalies (SCAs) and/or Copy Number Variants (CNVs), including del15q13.1–13.3 and del16p11.2. The remaining 7 studies displayed a variety of gene variants. 45 (candidate) genes related to language development, including FOXP2, GRIN2A, ERC1, and ATP2C2. After an additional search in the OMIM database, 22 of these genes were associated with a genetic disorder with a broader clinical spectrum, including intellectual disability, epilepsy, and/or autism.
Conclusion: Our study illustrates that DLD can be related to SCAs and specific CNV's. The reported (candidate) genes (n = 45) in the latter category reflect the genetic heterogeneity and support DLD without any comorbidities and syndromic language disorder have an overlapping genetic etiology.
A delayed or deviant language development is one of the most common disabilities in childhood (1). However, there has been a lack of agreement in literature and between practitioners about the exact criteria and terminology related to children's language problems.
The complex nature of language contributes to the challenges of identifying and classifying language impairments. Language involves understanding and using words and sentences to convey thoughts and information and should not be conflated with speech, which pertains to the production of vocal sounds.
In the context of scientific literature, there are challenges in precisely defining these concepts. Not only are different terms used to refer to language problems (e.g., specific language impairment, language delay, developmental verbal dyspraxia), at times terms referring to speech problems like “speech delay” are used to refer to problems in language. This lack of agreement about criteria and terminology for children's language problems hinders research and practice (2).
In their Delphi consensus study CATALISE of 2017, Bishop et al. (3, 4) aimed to reach international and multidisciplinary consensus in terminology regarding language impairment in children. Although the discussion about terminology and classification of language disorders continues to date, Bishop and colleagues propose the use of the term “Developmental Language Disorder” (DLD) to refer to cases of language disorder with no known differentiating conditions. These differentiating conditions include autism, epilepsy and intellectual disability. In the case of a language disorder co-occurring with one of these differentiating conditions, they recommend the use of the term “Language disorder (LD) associated with X”, where X is the differentiating condition. In their study, it was agreed that the presence of biological or environmental risk factors did not preclude a diagnosis of DLD, that DLD can co-occur with other neurodevelopmental disorders not related to language development and that DLD does not require a mismatch between language and nonverbal intelligence.
Although the debate on the terminology surrounding language issues remains unresolved, we adopt the terminology suggested by Bishop et al. for the sake of clarity. This means that in this article, we will use a strict definition of the term Developmental Language Disorder (DLD) for a language disorder with no known differentiating conditions.
Much is still unknown about the precise mechanisms behind language disorders. It is suggested that there may be multiple risk factors, which can reinforce each other, like chronic otitis media (5), socio-economic status (6), and oral-motor difficulties (7). As stated above, it is well known that language problems can co-occur with autism spectrum disorder (8), behavioral disorders (9) and certain epilepsy disorders (10). Furthermore, family and twin studies have indicated that DLD have a strong genetic component (11).
In the last decade, there has been an enormous progress in knowledge about the clinical features and genetic background of DLD, and conditions associated with DLD (12, 13). Furthermore, an increasing number of (candidate) genes possibly involved in the underlying mechanisms of DLD are reported. In particular, advances in molecular technologies are shedding new light on the genetic architecture underlying language-related disorders (14). Linkage and genome wide association studies (GWAS) resulted in several interesting genetic loci that seem to be associated with DLD and contribute to the risk of developing DLD with a multifactorial basis (15).
However, language disorders can also be Mendelian (or monogenic) in nature, which implies that the disorder involves a single genetic locus or gene. Mendelian disorders are caused by rare genetic variants with a large effect size. Several Mendelian causes for DLD have been identified over the years (13).
At this moment, it is not yet clear to what extent DLD is the result of monogenic causes and what part can be explained by multifactorial causes.
Mountford et al. (16) provided an overview of the genetic landscape of language disorders associated with a medical condition. However, in children with DLD, without any possibly explanatory comorbidities, a comprehensive overview of genes specifically related to DLD is missing. Such an overview could help in the diagnostic work-up of these patients. An early genetic diagnosis can be important for early treatment and better outcomes. Besides this, having an overview of genetic causes of DLD would be a good starting point for elaborating on the role of DLD within clinical genetic diagnostics, and it would provide a basis for further research regarding the combination of DLD and comorbidities like intellectual disability, epilepsy, and/or autism spectrum disorder. Gaining a deeper understanding of the genetic origins of DLD would offer valuable theoretical insights into the connection between genes, brain functionality, cognition, and particularly the language faculty.
With this review, we aim to give an overview of the literature in children on pathogenic variations or chromosomal anomalies related to DLD.
This review is reported according to Preferred Reporting Items for Systematic Review and Meta-Analysis PRISMA-statement (17).
Which specific pathogenic variants and chromosomal anomalies are reported in the literature to be related to developmental language disorder in children?
In the present review study, we used the term “developmental language disorder” or “DLD” as an umbrella term for the different fields in which language problems can occur: phonology, (morpho)syntax, semantics and pragmatics. We used a strict definition of “developmental language disorder” or “DLD”, to refer to language problems in the absence of other neurodevelopmental disorders or other confounding issues.
Inclusion criteria included original research describing children with developmental language disorder associated with a specific genetic variation written in English or Dutch for which a full text was available. Inclusion was based on the existence of problems in language development in described cases, resulting from language tests or because this was stated by the authors, in the absence of any known confounding variables.
Reviews, linkage studies, genetic association studies, articles based on animal models, articles in which insufficient details were given regarding the genetic variant, and articles in which the subjects were adults or had hearing problems, clefts, dyslexia, anatomical issues that could affect speech (e.g., velopharyngeal insufficiency (VPI), or when the speech language problems solely consisted of motor speech problems, and not language problems, were excluded. Furthermore, articles in which the language problems were not isolated but patients presented with comorbidities like intellectual disability (IQ <70), epilepsy/seizures, autism spectrum disorder (ASD) and/or ataxia were excluded.
We searched the PubMed- and Embase-databases for studies on the genetics of DLD. All articles published until April 1, 2023 were included. An overview of the search queries used, both for PubMed and Embase, can be found in Appendix 1.
The articles resulting from the search query in Embase and PubMed were deduplicated by two reviewers (VW and HW) and screened independently by four reviewers (VW, HW, DM and JP) using title and abstract on relevance to the topic of interest. Articles not related to the topic were excluded. Reviewer 1 (VW) is a linguist/speech, language, and hearing researcher; reviewer 2 (HW) is a linguist/speech language pathologist; reviewer 3 (DM) and 4 (JP) are medical students. The full text of the screened articles was assessed independently by two reviewers (VW and DM). Only articles that met the inclusion and exclusion criteria were included for qualitative analysis. Conflicts were resolved by discussion and in the case of disagreement, a third researcher weighed in.
The articles included in the review were critically appraised using the relevant Critical Appraisal Tools (CAT) of the Joanna Briggs Institute for the relevant study design. The papers regarding sex chromosome aneuploidies were assessed independently by reviewer VW and reviewer JP. The papers regarding other pathogenic variants were assessed independently by reviewer VW and reviewer HW.
As overall consideration for possible bias were considered a clear description and demographic information of the patients and appropriate identification and measurement of the condition and valid statistical analysis. Furthermore, in cases series, clear criteria for inclusion and complete and consecutive inclusion were considered. In case control studies, it was considered if both the group of patients in which the condition was present and the group in which the condition was absent were otherwise comparable. In prevalence studies, it was considered if the sampling was appropriate.
Since intervention procedures, treatment and exposure parameters are not relevant for the topic of interest of this review, we have not scored these checklist items in the study designs case reports (checklist items 4–6 regarding intervention and treatment), case control studies (checklist items 4, 5 and 9 regarding exposure) and case series (checklist item 8).
Relevant data including the study characteristics (first author's name, year of publication), study design, subject information (number of subjects, age), genetic background (type of mutation, genetic tests), linguistic background (language test, language profile) were extracted from the studies included in the qualitative review. Data extraction was done by VW and HW for double checking.
In the synthesis, reported genetic backgrounds of the subjects described in the included studies were described. Subsequently, an descriptive analysis was performed for interpretation of the found mutations. The found genes were checked in OMIM, to see if they were known OMIM morbid genes related to a genetic disorder. An additional search was performed in PubMed to reveal any possible relation between described genetic variants and intellectual disability, epilepsy, autism, and/or dyslexia. Outcomes of genetic findings on different subgroups were described, including clinical spectrum of reported genes and the relation with linguistic background of reported children.
The search query in Embase and PubMed resulted in 15,842 studies after deduplication. After screening on title and abstract, a total of 14,725 were excluded and 1,117 were reviewed on full text. This screening on full text resulted in 47 studies included for critical appraisal (see Figure 1).
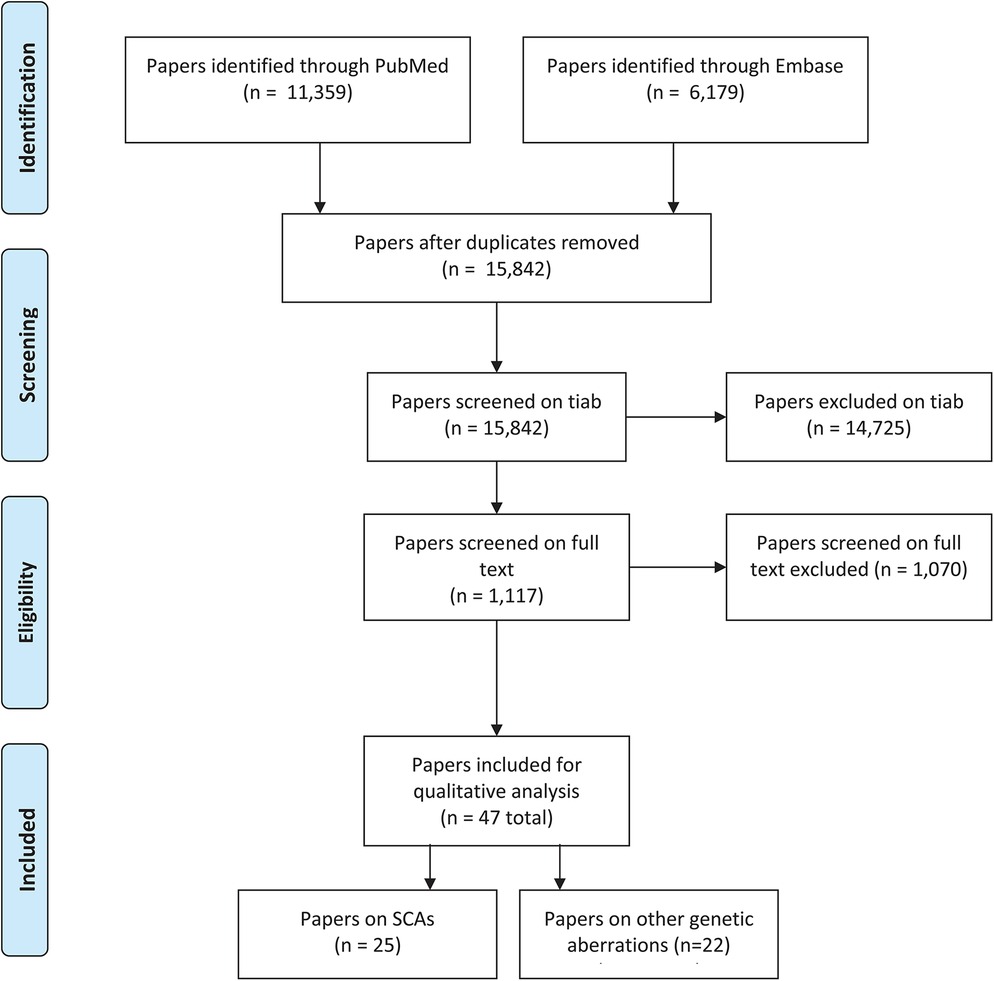
Figure 1. PRISMA-flowchart screening.
The vast majority of papers (11,039) were excluded because of the exclusion criteria linkage studies, genetic association studies, articles in a language other than English or Dutch, articles based on animal models, articles in which insufficient details were given regarding the genetic mutation, articles in which the subjects had ataxia, motor speech problems, hearing problems, clefts, dyslexia, anatomical issues that could affect speech (e.g., velopharyngeal insufficiency (VPI) or adult subjects or the article completely lacked relevance to the topic of interest. Relatively many papers (n = 3,686) were excluded because of the combination of DLD with intellectual disability (IQ <70) (n = 2,727), epilepsy/seizures (n = 1,237), and/or autism spectrum disorder (n = 888). (Please note that papers can be excluded because of multiple exclusion criteria, e.g., both ID and epilepsy, see Table 1).

Table 1. Papers excluded based on exclusion criteria ID, epilepsy, and/or autism.
A total of 47 papers were finally included describing children with developmental language disorders in relation to a specific pathogenic variants or chromosomal anomalies, without any other possibly explanatory risk factor. Concerning the study design, 18 papers were case control studies, 14 papers were case reports, 9 papers were case series and 6 papers were prevalence studies. Studies were published between 1973 and 2023.
Of the 47 studies included, 34 studies have met 80%–100% of the JBI-criteria and were classified as low risk of bias, 9 studies met 60%–80% of the JBI-criteria and were classified as moderate risk of bias, and 4 studies met less than 60% of the JBI-criteria and were therefore classified as high risk of bias.
There were 14 case reports: 1 paper with a high risk of bias, 1 with a moderate risk of bias and 12 with a low risk of bias. The moderate and high risk of bias was mainly due to insufficient description of the condition and the fact that diagnostic tests/assessments were not clearly described.
9 papers were case series: 1 paper had a high risk of bias, 5 a moderate risk of bias and 3 a low risk of bias. The high and moderate risk of bias was due to the fact that it was not clear if inclusion of participants was complete and consecutive and because of unclear reporting of the demographic and/or clinical information of the participants.
Of the 18 case control studies, 1 had a high risk of bias, 3 a moderate risk of bias and 14 a low risk of bias. The high and moderate risk of bias was mainly because it was unclear if the groups were appropriately matched and because confounding factors were not mentioned or dealt with appropriately.
Finally, there were 6 prevalence studies. 1 of these papers had a high risk of bias, the other 5 papers had a low risk of bias. In the study with the high risk of bias, it was unclear if the sampling was done appropriately.
The results of the critical appraisal of each individual study are depicted in Tables 2A–D.
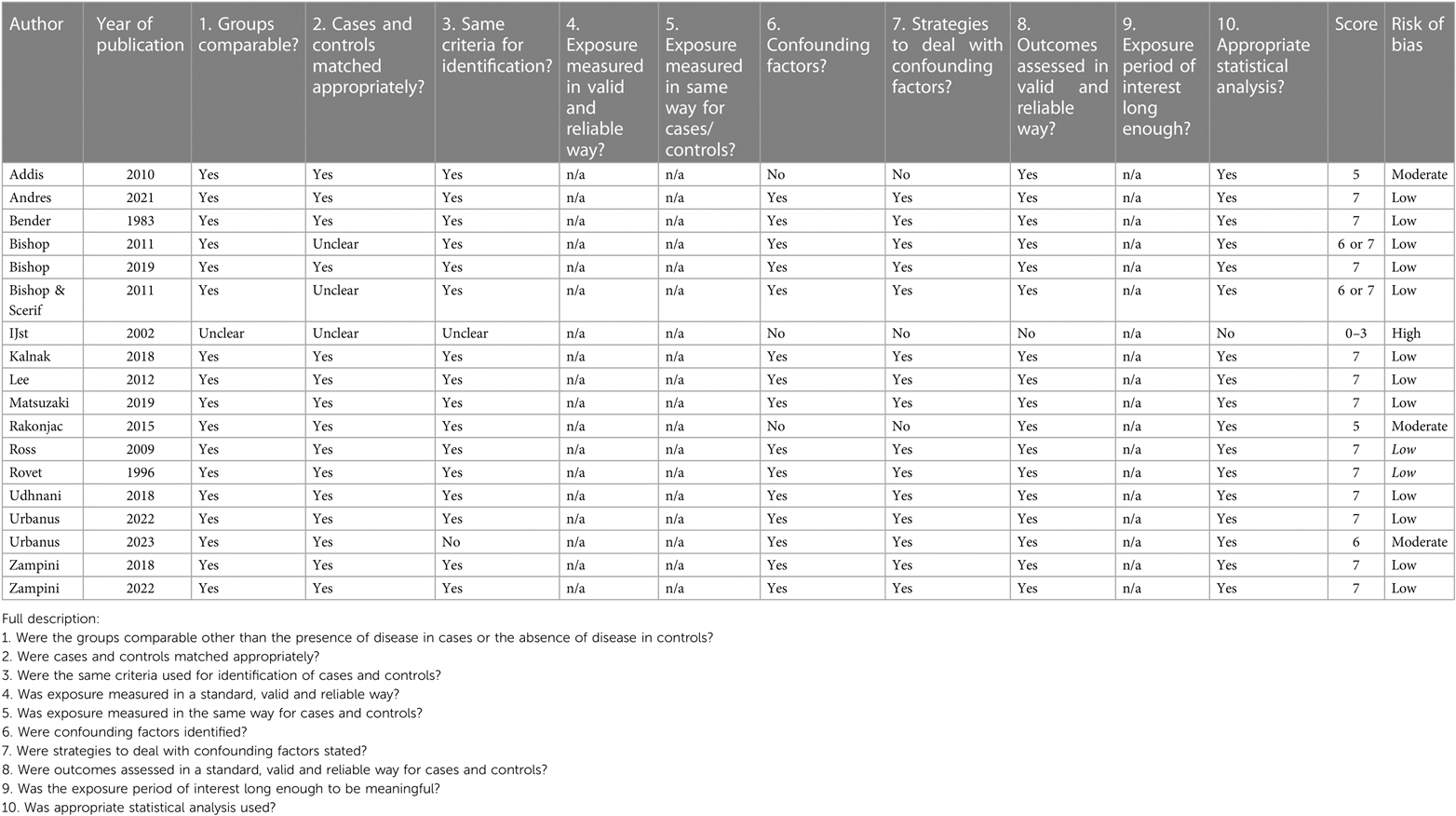
Table 2A. Critical appraisal of papers: CAT case control.
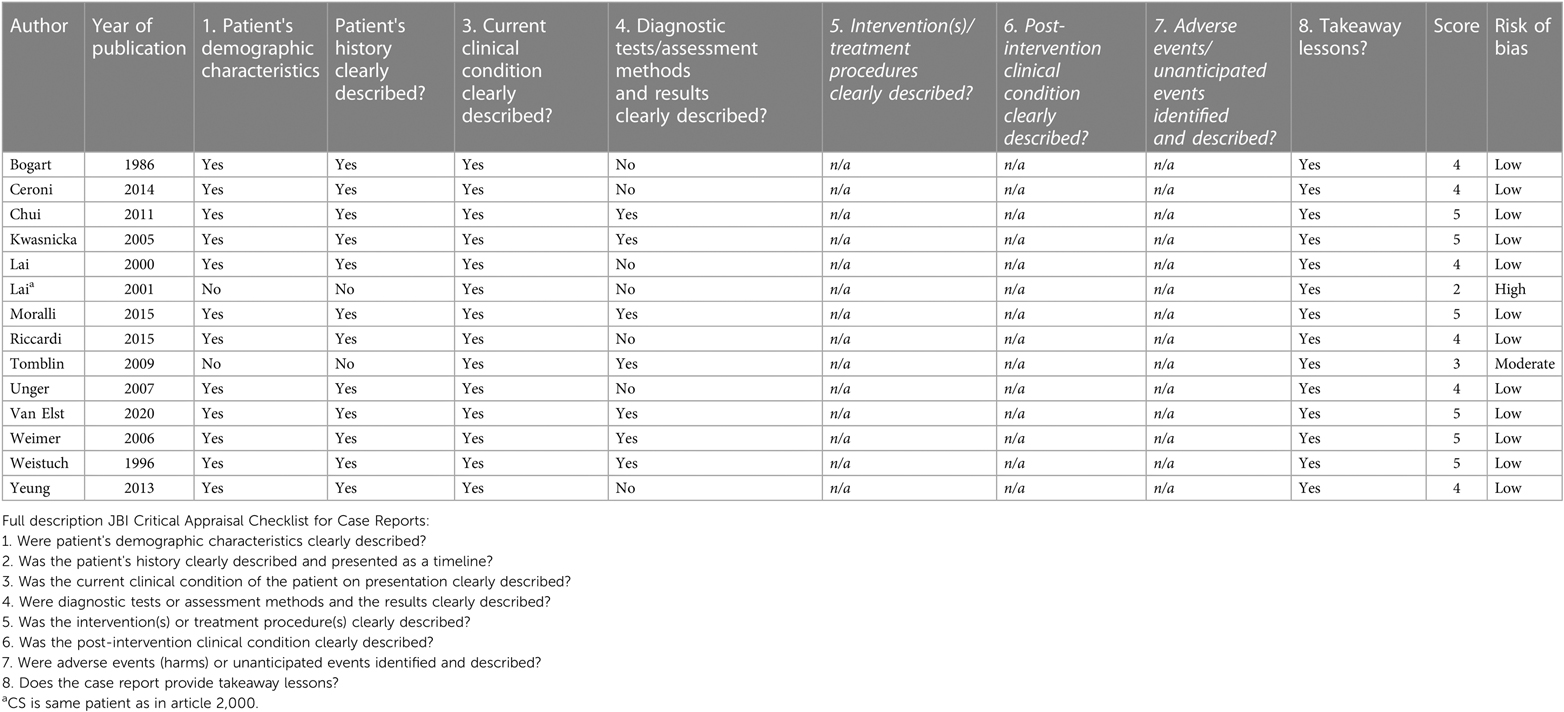
Table 2B. Critical appraisal of papers: CAT case reports.
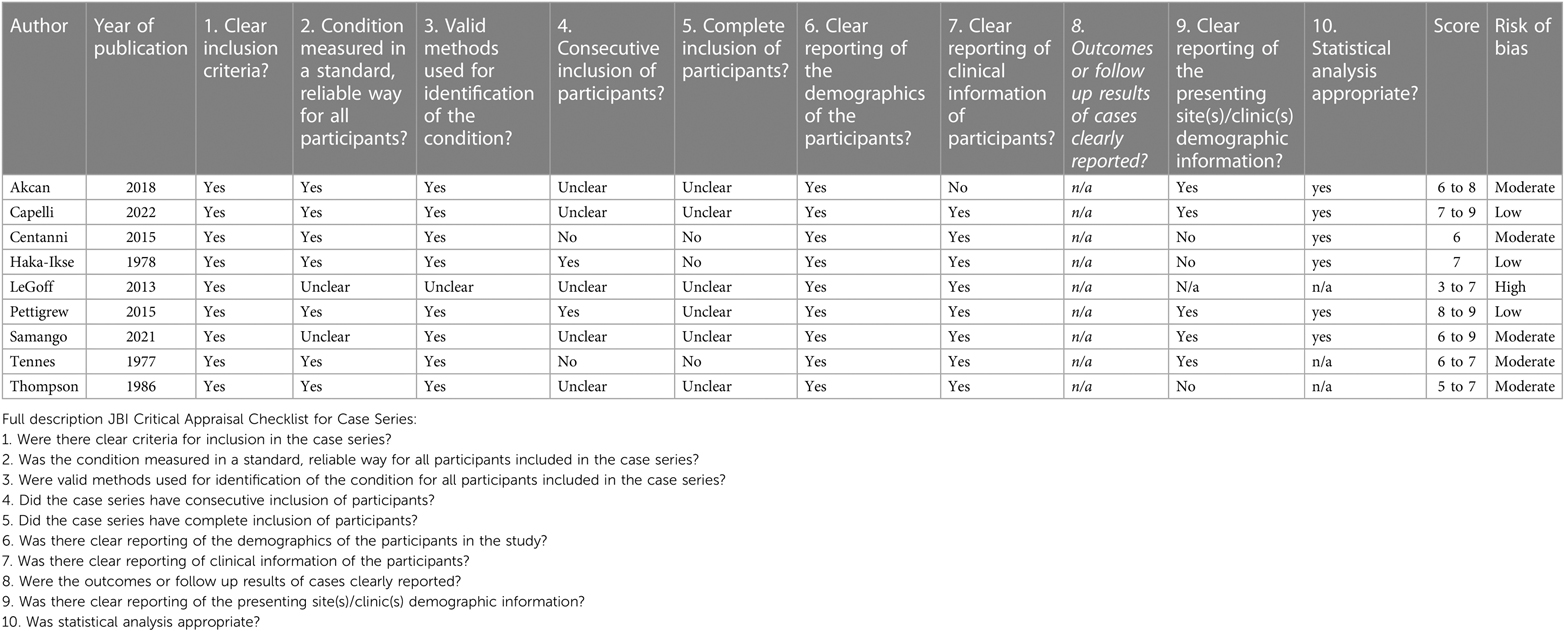
Table 2C. Critical appraisal of papers: CAT case series.
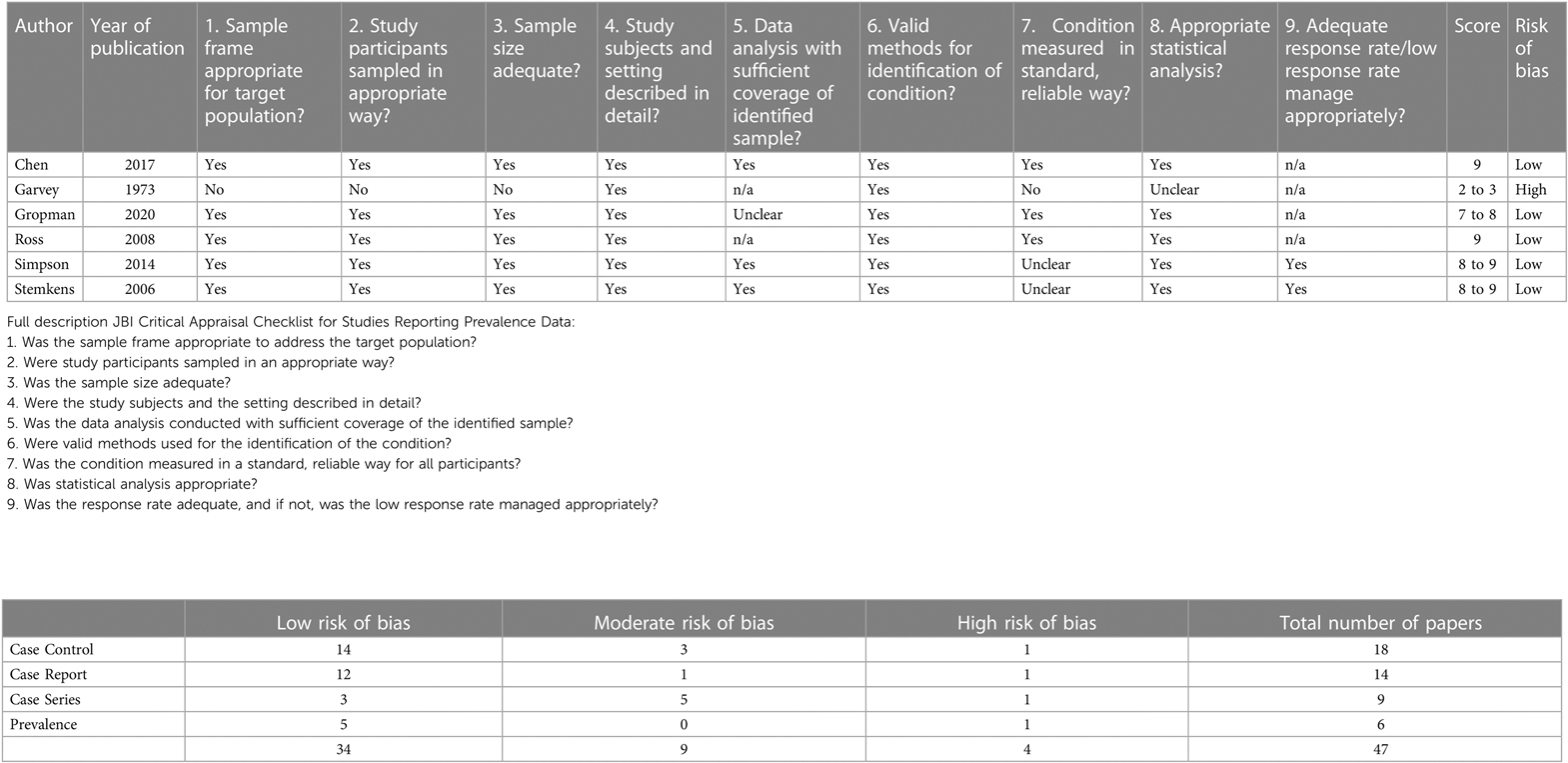
Table 2D. Critical appraisal of papers: prevalence.
Of the 47 included studies, 25 papers reported on sex chromosome aneuploidies (SCAs). The remaining 22 studies discuss a variety of structural chromosomal anomalies (7 studies), Copy Number Variants (CNVs) (7 studies) and gene variants (7 studies) (Supplementary Tables 3A,B).
Data of study characteristics, study design, subject information (number of subjects, age), genetic background (type of mutation, genetic tests), and linguistic background (language test, language profile) of cases out of included papers can be found in Supplementary Table S5.
In the following section, we will elaborate on different subgroups of genetic findings, clinical spectrum of reported genes and the relation with linguistic background as reported in included studies.
25 out of 47 included studies reported on cases with different sex chromosomes aneuplodies (see Table 3A): (18–42).
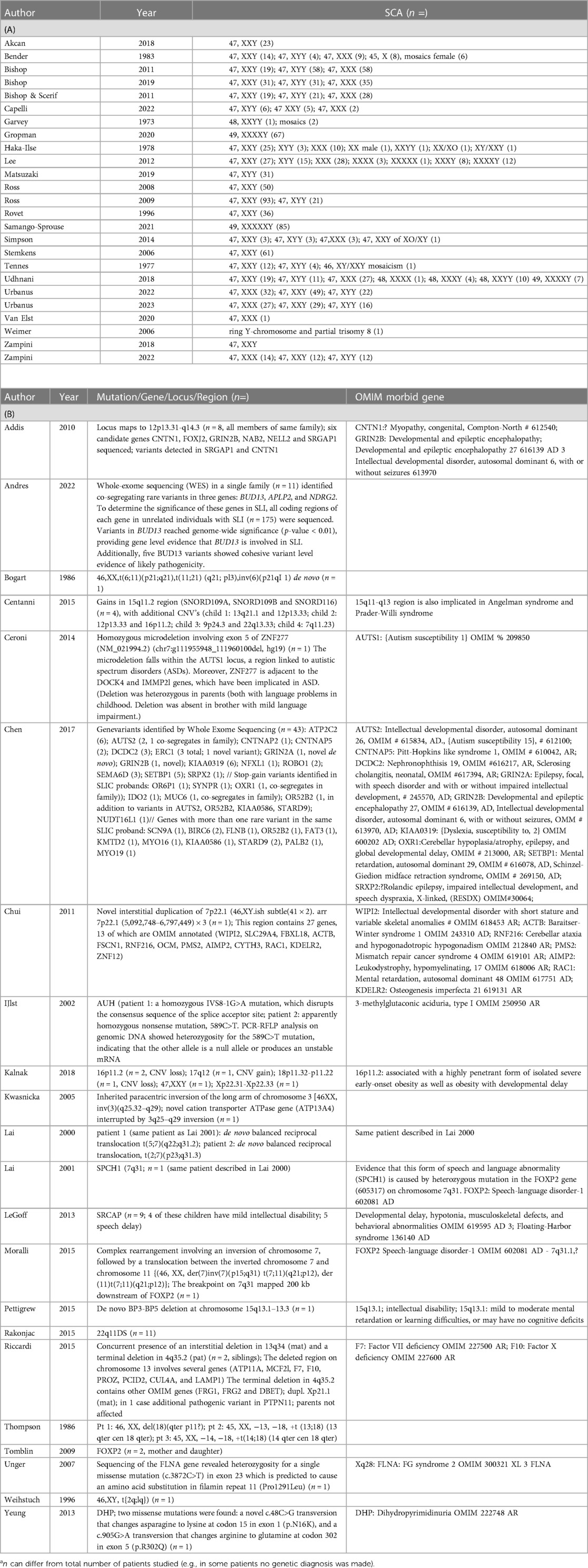
Table 3. Found SCAs and other chromosomal anomalies or pathogenic variants.
Reported SCAs related to DLD were 45,X (Turner-syndrome); 47,XXY (Klinefelter-syndrome), 47,XYY; 47,XXX; 48,XXYY; 48,XXXX; 48,XXXY; 49,XXXXX;49,XXXXY; XX/XO, XY/XXY mosaicism, and a ring Y-chromosome.
Seven studies (3 case reports, 2 case series and 1 cohort study) reported on an identified chromosomal anomaly encompassing a chromosomal deletion (43), chromosomal translocations (n = 5) (44–46), chromosomal inversions (n = 2) (47, 48), and a complex chromosomal rearrangement (49), for details see Table 3B.
Thompson and colleagues (43) discussed three children with a chromosome 18p deletion. All children presented with a speech and (both expressive and receptive) language delay with a striking delayed articulation. There was a marked difference between verbal and non-verbal abilities. The non-verbal IQ in one case was 90, compared to 60 and 68 in the two other cases. The clinical features were consistent with the 18p- syndrome, including short stature and facial dysmorphisms.
Bogart and colleagues (47) presented a girl with a prenatally detected de novo complex chromosomal rearrangement karyotype: 46,XX,t(6;11) (p21;q21),t(11;21) (q21;p13),inv(6) (p21q11). Her “speech development” was delayed. At 2.5 years of age, she spoke her first words. At 2 ¾ years, she did not make complete sentences. Growth was normal until 30 months of age. Eventually, growth was below the 5th centile. No further follow-up was presented.
Weistuch and colleagues (44) reported on a five-year-old boy with a balanced chromosomal translocation (46, XY, t(2q;1q) and a “specific expressive language impairment with verbal apraxia”. Other developmental milestones were normal. No specific physical features were mentioned.
Lai and colleagues (45) discussed two cases with different chromosomal translocations, but with a similar chromosomal breakpoint (the SPCH1-region on 7q31) (45). This locus corresponded with the locus co-segregation in the affected family members in the large three-generation pedigree of the well-known K.E.-family, (K.E.), in which a severe speech and language disorder is transmitted as an autosomal dominant monogenic trait. In a second included publication of the same author, Lai and colleagues identified FOXP2 as the causative gene for the language disorder in the KE-family and in one of the above included patients, unrelated to the KE-family (50).
Moralli and colleagues (49) reported on a young female, presenting with a severe speech and language disorder with a non-verbal IQ above the mean for her age, with a novo complex chromosomal rearrangement with breakpoint on 7q31, mapping 200 kb downstream of the gene FOXP2. Although no splice site or non-synonymous coding variants could be found in the FOXP2 coding sequence, expression of some of the FOXP2 targets (EFNB2, INHBB, NTN4, ROBO2, and SLC14A1) were altered, suggesting a possible role of FOXP2 and/or its downstream targets in the etiology of the speech and language disorder in this girl.
Tomblin and colleagues (46) discussed a mother and daughter with DLD due to a chromosomal translocation disrupting the gene FOXP2. The language phenotype of mother and daughter was more or less comparable with KE-family, both also demonstrated significant impairments of speech.
Kwasnicka and colleagues (48) described a girl with an expressive and receptive language delay and a paracentric inversion of the long arm of chromosome 3 [46XX, inv(3)(q25.32–q29), leading to the identification of the gene ATP13A4 (transporter ATPase gene) as a possible cause of language delay.
Seven studies reported on a CNV detected by array CGH or SNP array in cases with isolated DLD (51–57), for details see Table 3.
In one of these studies, by Centanni et al. (53), the chromosome region 15q11.2 was found to be a susceptibility region for “specific language impairment” based on genome wide analysis for CNVs in eight children aged 4–17 years with DLD without any comorbid conditions.
Ceroni and colleagues (52) presented the identification of a homozygous microdeletion of exon 5 in the gene ZNF277, which falls within the AUTS1 locus, in a girl with “specific language impairment”. They performed a genome-wide CNV screen in 512 individuals from families with SLI, who all formed part of the SLI Consortium. Screening of an additional 321 SLI families showed an increased allelic frequency of ZNF277 microdeletions. However, due to the rarity of the microdeletion this difference did not reach significance.
Chui and colleagues (51) described a boy with a “deficit … best characterized as significant isolated speech delay” with a dup7p22.1.
Kalnak et al. (57) analyzed rare and de novo CNVs in 58 children with severe DLD and their siblings. Clinically significant CNVs or chromosomal anomalies were found in 4 of these children, of which 2 carried 16p11.2-deletions. One sibling who also carried a deletion at this locus did not have any neurodevelopmental problems reported. The other two clinically significant variants that were found in affected probands were a 17q12 duplication and 47, XXY respectively.
Pettigrew and colleagues (54) identified a deletion of 15q13.1–13.3, as a possible cause of isolated DLD, in a girl with clinical concerns regarding speech and language development. She spoke her first words at 21 months (>1 SD of the cohort mean age at first word) and presented at 3 years and 7 months with difficulty producing speech-sounds coupled with problems of expressive and receptive language development. Her IQ was above 70. The deletion in this girl was de novo and no family history of language impairment or dyslexia was reported. Both parents and her sibling scored above average on cognitive tests.
Rakonjac et al. (55) compared speech and language abilities of children with a 22q11.2 microdeletion to children with a phenotype resembling 22q11.2DS but without the microdeletion. Their results revealed that children from the group with the microdeletion had a lower level of speech and language abilities compared to the group without the microdeletion.
Riccardi and colleagues (56) reported on a patient and a sibling with DLD, both with the same constellation of familial CNVs (dupXp22.11; del4q35.2 and del13q34). These CNVs were respectively inherited from an unaffected mother and father. However, one of the affected siblings had an additional pathogenic variant in PTPN11, associated with Noonan syndrome.
In total, seven studies discussed a variety of candidate genes and gene-variants (50, 58–63), for details see Table 3.
Addis and colleagues (59) presented a three-generation family with language impairment with “auditory processing difficulties” as a core deficit. They compared the results of this family with the results of a control group. All affected family members reported a language development delay. In all tested affected family members, auditory discrimination deficits for tone duration were detected by psychoacoustic tests. Non-verbal IQ was normal. Linkage analysis mapped the disorder to the chromosomal region 12p13.31-q14.3 with a maximum LOD score of 2.2. The haplotype at this locus fully co-segregated following an autosomal dominant pattern. Further sequencing of six relevant candidate genes in this region (CNTN1, FOXJ2, GRIN2B, NELL2, NAB2, SRGAP1) revealed no causative pathogenic gene variants. To exclude possible underlying causative CNVs, genome wide CNV analysis was performed, yielding no novel inherited copy number variants.
Andres and colleagues (63) performed whole-exome sequencing (WES) in a single family (n = 11). They identified co-segregating rare variants in three genes: BUD13, APLP2, and NDRG2. To determine the significance of these genes in SLI, the authors Sanger sequenced all coding regions of each gene in unrelated individuals with SLI (n = 175). Variants in BUD13 reached genome-wide significance (p-value < 0.01) upon comparison with similar variants in the 1,000 Genomes Project, providing gene level evidence that BUD13 is involved in SLI. Additionally, five BUD13 variants showed cohesive variant level evidence of likely pathogenicity.
Chen and colleagues (61) reported on the results of broad genetic analysis in 43 unrelated probands with severe “specific language impairment”. These probands were selected from the SLI consortium cohort, recruited from five centers across the UK. The group of probands had a mean verbal IQ of 84.2 (−1.1 SD), compared to a mean non-verbal IQ of 98.7 (−0.1 SD). Expressive language was more affected than receptive language. The mean scores were 65.9 (−2.3 SD below expected for chronological age) and 73.8 (−1.7 SD) for expressive and receptive language respectively. Genome wide exome sequencing was performed, followed by Sanger validations and segregation analyses. In several cases, analysis of a pre-defined set of 19 known candidates implicated in language related syndromes, could identify probable pathogenic gene variants in the genes ERC1, GRIN2A and SRPX2. Potential pathogenic variants, identified in the genes SEMA6D, AUT2 and ROBO1, co-segregated with the language disorder in affected relatives of the respective probands. Furthermore, novel variants were identified in GRIN2B and CNTNAP2. Six rare SNVs were identified in the genes ATP2C2, AUTS2, CNTNAP5, ROBO1 and SRPX2. Analysis beyond the known candidate genes revealed 7 rare or novel stop-gain variants in the genes OR6P1, NUDT16l, SYNPR, OXR1, IDO2, MUC6 and OR52B2. Two of these variants, in the genes OXR1 and MUC6, showed co-segregation with the disorder in affected family members. Compound heterozygous variants were identified in 11 genes. Four cases carried more than one rare gene variants probably reflecting “multiple hits”. One proband showed a rare coding variant in AUTS2, with in addition a stop-gain in OR52B2, and a rare variant the genes OR52B2, KIAA0586 (OMIM*610178), and STARD9.
Unger and colleagues (58) reported on a boy, 18 months of age, with a mild delay in language acquisition and a normal psychomotor development and a possible causative variant in the gene FLNA.
Two studies reported on cases with DLD with gene variants causative for a metabolic disorder. Yeung and colleagues (60) presented a 2-year-old boy diagnosed with Dihydropyrimidinase deficiency (OMIM # 222748); compound heterozygous for pathogenic variants in the gene DPYS. The boy could not produce a single recognizable word, although his motor development was appropriate for his age. Neurological examination was normal. He had no dysmorphic features.
IJlst and colleagues (62) reported on two patients with “retardation in speech development” diagnosed with 3-methylglutaconic aciduria type I (OMIM # 250950). In one of them a motor development delay became evident in retrospect. In both cases, homozygosity for a pathogenic variant in the gene AUH confirmed the diagnosis.
The 45 genes found within the retrieved studies were evaluated within the OMIM database. By this it was revealed that 22 of these 45 DLD (candidate) genes were identified as a cause for a genetic disorder (see Supplementary Table S4).
Two of these genes, FOXP2 and GRIN2A, are OMIM morbid genes strongly related to speech and language development. ATP2C2 is identified as a susceptibility locus (OMIM #606711). To date, FOXP2 is well-known as monogenic cause for the autosomal dominant disorder Speech-language disorder 1 (SPCH1; OMIM # 602081).
Twenty-two of the 45 genes are a known cause for intellectual disability, 17 for epilepsy or seizures, 18 for autism spectrum disorder, and 8 for dyslexia.
Four out of 45 described genes do not seem to be directly related to speech and language development; for more details see Supplementary Table S4.
With this systematic review of the literature, we aimed to gain more insight into the genes found in children with a language disorder without differentiating conditions (e.g., intellectual disability, epilepsy, autism and anatomical issues like clefts).
Our search resulted in 47 studies that met the inclusion criteria and that subsequently were critically appraised. A total of 34 studies had a low risk of bias, 9 studies a moderate risk of bias and 4 studies a high risk of bias. The included studies displayed a large spectrum of genetic causes of DLD.
Interestingly, more than half of the papers (25/47) concerned findings of SCA, including 45, XO (Turner syndrome) and 47, XXY (Klinefelter syndrome). The large number of papers on SCA can be explained by the high frequency of SCA in the general population and seems in line with the increasing evidence SCA is often associated with mild clinical features (64).
Fifteen out of the 47 papers reported on chromosomal anomalies (8/47) and CNV's (7/47) as a possible cause of DLD. Chromosomal rearrangements at 7q31, eventually, led to identification of the DLD gene FOXP2. Furthermore, a chromosomal inversion and an intragenic CNV revealed ATP13A4 and ZNF277, respectively, as candidate genes for DLD.
The reported CNV's display both deletions and duplications on different chromosomes. The CNV 15q13.1-q13.3 deletion reported by Pettigrew and colleagues (54) and the 16p11.2 deletion reported by Kalnak and colleagues (57), respectively, was also, recently, mentioned by Plug and colleagues (12) as causative CNV's in their assessment of the genetic work-up in 127 patients diagnosed with DLD.
Interestingly, the region 15q11.2 was previously reported by Centanni and colleagues (53) as a susceptibility locus, thus not as a causative CNV for isolated DLD. They describe four cases with a 15q11.2 duplication who all had one or more additional CNV's (13q21.1 dup and 12p13.33 del; 10q21.1del and 16p11.2del; 9p24.3 and 22q13.33 and 7q11.23 dup, respectively). Moreover, these CNV's are often associated with a broader clinical spectrum including apraxia, autism, epilepsy and/or intellectual disability (65, 66).
In addition, the 17q12 duplication and 22q11.2 deletion reported in relation to LD by Kalnak and colleagues (57) and Rakonjac and colleagues (55), respectively, are associated with a broad and variable clinical spectrum with a mild to severe development delay [Mitchel et al. (67)]. In our review, studies on 22q11.2DS were generally excluded because of these and other risk factors.
The role of the reported combination of familial CNVs (dupXp22.11; del4q35.2 and del13q34) in the cause of DLD (56) can be argued, because one of the sibs of the proband was described to be diagnosed with Noonan syndrome. On the other hand, one might hypothesize these CNV's contribute to a multifactorial etiologic model.
From the 21 selected papers not related to SCAs, 45 (candidate) genes for DLD, including FOXP2, ATP2C2, ERC1, GRIN2A and KMT2D (Tables 3B, Supplementary Table S4).
Most of these genes (31 out of 45) were reported and discussed by Chen and colleagues (61).
Of these, FOXP2 is acknowledged as a monogenic cause for specific Speech-Language disorder-1 (OMIM# 602081) and ATP2C2 as a susceptibility locus (OMIM % 606711) contributing in the etiology of DLD. On the other hand, ERC1 was supported by the authors as an interesting candidate for DLD, by the identification of a novel probably pathogenic variant in a child with DLD and its affected mother. However, since the father also reported a history of DLD and both sibs needed special education, although they were not carrying the ERC1 variant, the exact inheritance pattern still needs to be unraveled. KMTD2 was also identified as candidate gene. The authors hypothesized specific missense variants might lead to DLD, because the described cases did not show features of Kabuki syndrome (OMIM#147920), caused by loss of function in KMTD2 gene.
The genes SEMA6D, AUT2 and ROBO1, OXR1 and MUC6 were reported as interesting candidate genes because potential pathogenic variants co-segregated with the language disorder in affected relatives of the respective probands (61). The genes FAT3, KMTD2, SCN9A and PALB2 were considered as interesting candidate genes because compound heterozygous potential deleterious variants, inherited from the opposite parent, were identified in these genes (61). Most of the reported genes, when checked in OMIM, were also related to intellectual disability, epilepsy and/or autism. This might be related to the study design. Chen and colleagues first focused on a pre-defined set of known candidates from the literature, and identified potentially pathogenic variants in genes already implicated in diverse language-related syndromes, including ERC1, GRIN2A, and SRPX2. They performed whole-exome sequencing in 43 unrelated probands affected by severe specific language impairment, followed by independent validations with Sanger sequencing, focusing on these candidate genes.
Andres and colleagues (63) found gene level evidence that BUD13 is involved in SLI.
Further studies are needed to support the exact contribution of the reported candidate genes in the etiology of isolated DLD nd to gain more insight in to what extent these genes can be defined as a monogenic cause or a contributing factor in a multifactorial etiology of DLD. The identification of more than one rare gene variant probably representing “multiple hits” in four cases, support a multifactorial inheritance in some cases of DLD (61).
Notably, 22 out of the 45 reported (candidate) genes are known to be related to a broader phenotype than DLD, including intellectual disability, epilepsy, and/or autism (e.g., GRIN2A, CNTNAP2, CNTNAP5, AUTS2). Even FOXP2, often considered the “language gene” and in OMIM recognized as specific Speech-Language disorder-1 (OMIM; # 602081), has been linked to other neurodevelopmental disorders, like intellectual disability and autism (68, 69).
Although most of the study designs used in the included papers were case-related, and symptoms in individual cases might emerge at a later stage, the results in this review suggest that the etiology of DLD may be heterogeneous, and that DLD can actually be part of a broader phenotype.
Interestingly, this systematic review revealed two papers reporting cases with apparently DLD and pathogenic gene variants in the genes DPYS and AUH, which are known to be responsible for the metabolic disorders Dihydropyrimidinase deficiency (OMIM # 222748) and 3-methylglutaconic aciduria type I (OMIM # 250950), respectively (60, 62). These cases might demonstrate that both metabolic disorders can result in a mild to severe phenotype. However, the absence of significant clinical features in the reported cases, besides DLD, might be related to their young age.
In conclusion, this systematic review supports the theory that DLD is a heterogeneous disorder and that different genetic underlying mechanisms, including SCA, CNV's, and a variety of gene variants, can play a role in the etiology of DLD. Common underlying etiologic mechanisms seem to be involved in a broader phenotypic spectrum which includes DLD. If this is the case, DLD could be the first manifestation of a broader underlying etiology, and this would mean that it could constitute a starting point for further genetic diagnostics to unravel the full spectrum of the disorder.
As a result of the increasing awareness of a possible genetic cause for DLD and the technological advancements made in genetic analysis, DNA analysis is increasingly being implemented in the diagnostic work-up in patients with DLD seen in clinical practice.
An early genetic diagnosis can be important for early treatment and better outcomes. For example, DLD might be a first clinical presentation of epileptic activity without clinically apparent seizures (70), which needs treatment to improve outcomes. Also, other genetic disorders, like SCAs (e.g., 47, XXY, Klinefelter syndrome and 45, XO; Turner syndrome) and microdeletions (e.g., 22q11.2 deletion), require a specific management and adequate follow-up. For example, early hormonal therapy was found to be associated with a positive effect on, amongst others, expressive and receptive language in boys with Klinefelter syndrome (71).
When genetic testing in children with DLD is considered, we recommend to start with SNP array analysis based on the frequently reported chromosomal disorders as the cause of DLD. Nowadays, SNP array, identifying sex chromosomal disorders and microdeletions/-deletions, is broadly implemented in clinical care. Next generation sequencing (NGS) can detect underlying pathogenic gene variants. It has been demonstrated that NGS, analyzing a large number of genes simultaneously, proves to be highly efficient and cost-effective (72, 73). By implementation of novel analysis tools, CNVs can also be detected by NGS.
Especially in patients with DLD with a significant discrepancy in IQ with the parents, with a high IQ in the parents, broader genetic testing by NGS should be considered. In these cases, a de novo CNV or de novo pathogenic variant in a gene associated with intellectual disability might be present.
Although we thoroughly evaluated the literature on developmental language disorder to create unique knowledge on genetic causes of DLD, we realize this study has some important limitations. An important issue is the fact that different definitions of DLD are used in different studies. Furthermore, as the main topic of the study in most cases was not language development, it is not always clear how language was measured and how isolated the language problems really were. Based on the outcomes and descriptions of cases in the included studies, it was not possible to reach a good overview of possible differences in language phenotype between the different underlying genetic causes, which could be used in a more personalized genetic diagnostic work-up in clinical practice.
In addition, since techniques for clinical genetic diagnostics have evolved rapidly during the last years, aberrations that can be detected by relatively “old” techniques like karyotyping (e.g., SCAs) may be overrepresented in the search query, simply as a consequence of a greater number of studies performed using this technique because of the larger time frame. Aneuploidies can be detected since the 1950s by general karyotyping (74) and array CGH detecting CNV's is developed in 1992. The earliest study included in this review was the study of Garvey, from 1973 (25).
Finally, only articles with full texts in Dutch and English were included, as these are the languages all reviewers have a good command of, which means that we might have missed an important article in any other language.
With these limitations in mind, our review clearly exemplifies that DLD that appears to be isolated can have an underlying genetic etiology and that genetic testing should be considered in the diagnostic work-up in patients with DLD without any apparent comorbidities. More knowledge on possibly different developmental language profiles associated with different genetic genotypes could be useful in adequate and efficient genetic counseling.
It would be helpful if in studies regarding a genetic disorder which is associated with DLD, a consistent terminology is used to describe the language profile of the patient and more details on the parents are given. A more elaborate description of the language profile associated with specific genotypes in combination with the relevant gene function could also lead to a better understanding of the neurobiological causes of DLD.
Larger and more controlled studies like cohort studies directed at specific genetic variants and their relation to a deviant language development in children are recommended.
In this systematic review, we found 47 studies reporting on genetic findings found in children with isolated DLD. Twenty-five were related to numerical sex chromosome aberrations. The other 22 papers concerned a highly diversified group of CNV's and (candidate) genes. A possible explanation for the low yield would be that if a genetic background for DLD exists, the phenotype is broader and also includes (potential partially explanatory) issues like intellectual disability, epilepsy, and/or autism. Besides this, at the current stage, genetic diagnostic testing is not part of standard diagnostic work-up in children with DLD. Based on the findings of our review, we hypothesize that in most cases DLD has a multifactorial inheritance and not a monogenic etiology. DLD and LD associated with comorbidities could have an overlapping genetic etiology, in which case DLD could be the first manifestation of a broader underlying etiology.
When genetic testing in children with DLD is considered, we recommend to start with SNP array analysis based on the frequency of chromosomal disorders as the cause of DLD found in this study.
Especially in patients with DLD with a significant discrepancy in IQ with the parents, with a high IQ in the parents, broader genetic testing by NGS should be considered.
More research is needed regarding the questions what role clinical genetic research could play in diagnosing DLD, the relation between genetic cause and DLD profile or treatment outcome, and the preferences or perspectives of patients/parents on genetic testing for DLD.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
VW: Conceptualization, Methodology, Writing – original draft, Writing – review & editing, Data curation. HW: Writing – review & editing. DM: Writing – review & editing, Data curation. JP: Writing – review & editing, Data curation. IS: Conceptualization, Methodology, Writing – review & editing. EG: Conceptualization, Writing – review & editing. AS: Conceptualization, Methodology, Writing – original draft, Writing – review & editing. M-JB: Conceptualization, Methodology, Writing – original draft, Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2024.1315229/full#supplementary-material
1. Tomblin JB, Records NL, Buckwalter P, Zhang X, Smith E, O'Brien M. Prevalence of specific language impairment in kindergarten children. J Speech Lang Hear Res. (1997) 40(6):1245–60. doi: 10.1044/jslhr.4006.1245
2. Bishop DVM. Why is it so hard to reach agreement on terminology? The case of developmental language disorder (DLD). Int J Lang Commun Disord. (2017) 52(6):671–80. doi: 10.1111/1460-6984.12335
3. Bishop DA-O, Snowling MJ, Thompson PA, Greenhalgh T. CATALISE: a multinational and multidisciplinary delphi consensus study. Identifying language impairments in children. PLoS One. (2016) 11(7):e0158753. doi: 10.1371/journal.pone.0158753. eCollection 2016
4. Bishop DVM, Snowling MJ, Thompson PA, Greenhalgh T. Phase 2 of CATALISE: a multinational and multidisciplinary delphi consensus study of problems with language development: terminology. J Child Psychol Psychiatry. (2017) 58(10):1068–80. doi: 10.1111/jcpp.12721
5. Callahan CW Jr., Lazoritz S. Otitis media and language development. Am Fam Physician. (1988) 37(5):186–90.3284303
6. McNally S, McCrory C, Quigley J, Murray A. Decomposing the social gradient in children’s vocabulary skills at 3 years of age: a mediation analysis using data from a large representative cohort study. Infant Behav Dev. (2019) 57:101326. doi: 10.1016/j.infbeh.2019.04.008
7. Narayanan S, Vijayan K, Vastare Guruprasad M, Prabhu PP, Barman A. Oral and verbal praxis in impaired language learners. Percept Mot Skills. (2022) 129(1):33–46. doi: 10.1177/00315125211056421
8. Williams D, Botting N, Boucher J. Language in autism and specific language impairment: where are the links? Psychol Bull. (2008) 134(6):944–63. doi: 10.1037/a0013743
9. Nudel R, Appadurai V, Buil A, Nordentoft M, Werge T. Pleiotropy between language impairment and broader behavioral disorders-an investigation of both common and rare genetic variants. J Neurodev Disord. (2021) 13(1):54. doi: 10.1186/s11689-021-09403-z
10. Caplan R, Siddarth P, Vona P, Stahl L, Bailey C, Gurbani S, et al. Language in pediatric epilepsy. Epilepsia. (2009) 50(11):2397–407. doi: 10.1111/j.1528-1167.2009.02199.x
11. Bishop DV, North T, Donlan C. Genetic basis of specific language impairment: evidence from a twin study. Dev Med Child Neurol. (1995) 37(1):56–71. doi: 10.1111/j.1469-8749.1995.tb11932.x
12. Plug MB, van Wijngaarden V, de Wilde H, van Binsbergen E, Stegeman I, van den Boogaard MH, et al. Clinical characteristics and genetic etiology of children with developmental language disorder. Front Pediatr. (2021) 9:651995. doi: 10.3389/fped.2021.651995
13. Barnett CP, van Bon BW. Monogenic and chromosomal causes of isolated speech and language impairment. J Med Genet. (2015) 52(11):719–29. doi: 10.1136/jmedgenet-2015-103161
14. Deriziotis P, Fisher SE. Speech and language: translating the genome. Trends Genet. (2017) 33(9):642–56. doi: 10.1016/j.tig.2017.07.002
15. Reader RH, Covill LE, Nudel R, Newbury DF. Genome-wide studies of specific language impairment. Curr Behav Neurosci Rep. (2014) 1(4):242–50. doi: 10.1007/s40473-014-0024-z
16. Mountford HS, Braden R, Newbury DF, Morgan AT. The genetic and molecular basis of developmental language disorder: a review. Children (Basel). (2022) 9(5):586. doi: 10.3390/children9050586
17. Moher D, Liberati A, Tetzlaff J, Altman DG. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. Br Med J. (2009) 339:b2535. doi: 10.1136/bmj.b2535
18. Akcan N, Poyrazoglu S, Bas F, Bundak R, Darendeliler F. Klinefelter syndrome in childhood: variability in clinical and molecular findings. J Clin Res Pediatr Endocrinol. (2018) 10(2):100–7. doi: 10.4274/jcrpe.5121
19. Bender B, Fry E, Pennington B, Puck M, Salbenblatt J, Robinson A. Speech and language development in 41 children with sex chromosome anomalies. Pediatrics. (1983) 71(2):262–7. doi: 10.1542/peds.71.2.262
20. Bishop DVM, Brookman-Byrne A, Gratton N, Gray E, Holt G, Morgan L, et al. Language phenotypes in children with sex chromosome trisomies. Wellcome Open Res. (2019) 3:143. doi: 10.12688/wellcomeopenres.14904.2
21. Bishop DV, Scerif G. Klinefelter syndrome as a window on the aetiology of language and communication impairments in children: the neuroligin-neurexin hypothesis. Acta Paediatr. (2011) 100(6):903–7. doi: 10.1111/j.1651-2227.2011.02150.x
22. Bishop DV, Jacobs PA, Lachlan K, Wellesley D, Barnicoat A, Boyd PA, et al. Autism, language and communication in children with sex chromosome trisomies. Arch Dis Child. (2011) 96(10):954–9. doi: 10.1136/adc.2009.179747
23. Capelli E, Silibello G, Ajmone PF, Altamore E, Lalatta F, Vizziello PG, et al. Language development in sex chromosome trisomies: developmental profiles at 2 and 4 years of age, and predictive measures. Dev Neurorehabil. (2022) 25(5):337–48. doi: 10.1080/17518423.2021.2020925
24. van Elst PC, Otter M, Wijnen F, Junge C. Evaluating the scope of language impairments in a patient with triple X syndrome: a brief report. Dev Neurorehabil. (2020) 23(6):402–6. doi: 10.1080/17518423.2020.1764652
25. Garvey M, Mutton DE. Sex chromosome aberrations and speech development. Arch Dis Child. (1973) 48(12):937–41. doi: 10.1136/adc.48.12.937
26. Gropman AL, Porter GF, Lasutschinkow PC, Sadeghin T, Tipton ES, Powell S, et al. Neurocognitive development and capabilities in boys with 49,XXXXY syndrome. Am J Med Genet A. (2021) 185(12):3541–6. doi: 10.1002/ajmg.a.61736
27. Haka-Ikse K, Stewart DA, Cripps MH. Early development of children with sex chromosome aberrations. Pediatrics. (1978) 62(5):761–6. doi: 10.1542/peds.62.5.761
28. Lee NR, Wallace GL, Adeyemi EI, Lopez KC, Blumenthal JD, Clasen LS, et al. Dosage effects of X and Y chromosomes on language and social functioning in children with supernumerary sex chromosome aneuploidies: implications for idiopathic language impairment and autism spectrum disorders. J Child Psychol Psychiatry. (2012) 53(10):1072–81. doi: 10.1111/j.1469-7610.2012.02573.x
29. Matsuzaki J, Bloy L, Blaskey L, Miller J, Kuschner ES, Ku M, et al. Abnormal auditory mismatch fields in children and adolescents with 47,XYY syndrome. Dev Neurosci. (2019) 41(1):123–31. doi: 10.1159/000500799
30. Ross JL, Roeltgen DP, Stefanatos G, Benecke R, Zeger MP, Kushner H, et al. Cognitive and motor development during childhood in boys with Klinefelter syndrome. Am J Med Genet . (2008) 146(6):708–19. doi: 10.1002/ajmg.a.32232
31. Ross JL, Zeger MP, Kushner H, Zinn AR, Roeltgen DP. An extra X or Y chromosome: contrasting the cognitive and motor phenotypes in childhood in boys with 47,XYY syndrome or 47,XXY Klinefelter syndrome. Dev Disabil Res Rev. (2009) 15(4):309–17. doi: 10.1002/ddrr.85
32. Rovet J, Netley C, Keenan M, Bailey J, Stewart D. The psychoeducational profile of boys with Klinefelter syndrome. J Learn Disabil. (1996) 29(2):180–96. doi: 10.1177/002221949602900208
33. Simpson NH, Addis L, Brandler WM, Slonims V, Clark A, Watson J, et al. Increased prevalence of sex chromosome aneuploidies in specific language impairment and dyslexia. Dev Med Child Neurol. (2014) 56(4):346–53. doi: 10.1111/dmcn.12294
34. Stemkens D, Roza T, Verrij L, Swaab H, van Werkhoven MK, Alizadeh BZ, et al. Is there an influence of X-chromosomal imprinting on the phenotype in Klinefelter syndrome? A clinical and molecular genetic study of 61 cases. Clin Genet. (2006) 70(1):43–8. doi: 10.1111/j.1399-0004.2006.00635.x
35. Tennes K, Puck M, Orfanakis D, Robinson A. The early childhood development of 17 boys with sex chromosome anomalies: a prospective study. Pediatrics. (1977) 59(4):574–83. doi: 10.1542/peds.59.4.574
36. Udhnani M, Maiman M, Blumenthal JD, Clasen LS, Wallace GL, Giedd JN, et al. Phonemic and semantic verbal fluency in sex chromosome aneuploidy: contrasting the effects of supernumerary X versus Y chromosomes on performance. J Int Neuropsychol Soc. (2018) 24(9):917–27. doi: 10.1017/S1355617718000723
37. Urbanus E, Swaab H, Tartaglia N, Boada R, van Rijn S. [Formula: see text]. A cross-sectional study of early language abilities in children with sex chromosome trisomy (XXY, XXX, XYY) aged 1-6 years. Child Neuropsychol. (2022) 28(2):171–96. doi: 10.1080/09297049.2021.1960959
38. Weimer J, Metzke-Heidemann S, Plendl H, Caliebe A, Grunewald R, Ounap K, et al. Characterization of two supernumerary marker chromosomes in a patient with signs of Klinefelter syndrome, mild facial anomalies, and severe speech delay. Am J Med Genet A. (2006) 140(5):488–95. doi: 10.1002/ajmg.a.31104
39. Zampini L, Draghi L, Silibello G, Dall'Ara F, Rigamonti C, Suttora C, et al. Vocal and gestural productions of 24-month-old children with sex chromosome trisomies. Int J Lang Commun Disord. (2018) 53(1):171–81. doi: 10.1111/1460-6984.12334
40. Zampini L, Lorini A, Silibello G, Zanchi P, Dall'Ara F, Ajmone PF, et al. Language development in the second year of life: the case of children with sex chromosome trisomies diagnosed before birth. Int J Environ Res Public Health. (2022) 19(3):1831. doi: 10.3390/ijerph19031831
41. Samango-Sprouse CA, Lasutschinkow PC, McLeod M, Porter GF, Powell S, St. Laurent J, et al. Speech and language development in children with 49,XXXXY syndrome. Am J Med Genet A. (2021) 185(12):3567–75. doi: 10.1002/ajmg.a.61767
42. Urbanus E, Swaab H, Tartaglia N, Stumpel C, van Rijn S. Structural and pragmatic language in young children with sex chromosome trisomy (XXX, XXY, XYY): predictive value for neurobehavioral problems one year later. Clin Neuropsychol. (2023) 37(3):650–75. doi: 10.1080/13854046.2022.2067078
43. Thompson RW, Peters JE, Smith SD. Intellectual, behavioral, and linguistic characteristics of three children with 18p- syndrome. J Dev Behav Pediatr. (1986) 7(1):1–7. doi: 10.1097/00004703-198602000-00001
44. Weistuch L, Schiff-Myers N. Chromosomal translocation in a child with SLI and apraxia. J Speech Hear Res. (1996) 39(3):668–71. doi: 10.1044/jshr.3903.668
45. Lai CS, Fisher SE, Hurst JA, Levy ER, Hodgson S, Fox M, et al. The SPCH1 region on human 7q31: genomic characterization of the critical interval and localization of translocations associated with speech and language disorder. Am J Hum Genet. (2000) 67(2):357–68. doi: 10.1086/303011
46. Tomblin JB, O'Brien M, Shriberg LD, Williams C, Murray J, Patil S, et al. Language features in a mother and daughter of a chromosome 7;13 translocation involving FOXP2. J Speech Lang Hear Res. (2009) 52(5):1157–74. doi: 10.1044/1092-4388(2009/07-0162)
47. Bogart MH, Bradshaw CL, Jones OW, Schanberger JE. Prenatal diagnosis and follow up of a child with a complex chromosome rearrangement. J Med Genet. (1986) 23(2):180–3. doi: 10.1136/jmg.23.2.180
48. Kwasnicka-Crawford D, Carson AR, Roberts W, Summers AM, Rehnstrom K, Jarvela I, et al. Characterization of a novel cation transporter ATPase gene (ATP13A4) interrupted by 3q25-q29 inversion in an individual with language delay. Genomics. (2005) 86(2):182–94. doi: 10.1016/j.ygeno.2005.04.002
49. Moralli D, Nudel R, Chan MT, Green CM, Volpi EV, Benitez-Burraco A, et al. Language impairment in a case of a complex chromosomal rearrangement with a breakpoint downstream of FOXP2. Mol Cytogenet. (2015) 8:1, eCollection 2015. doi: 10.1186/s13039-015-0148-1
50. Lai CS, Fisher SE, Hurst JA, Vargha-Khadem F, Monaco AP. A forkhead-domain gene is mutated in a severe speech and language disorder. Nature. (2001) 413(6855):519–23. doi: 10.1038/35097076
51. Chui JV, Weisfeld-Adams J, Tepperberg J, Mehta L. Clinical and molecular characterization of chromosome 7p22.1 microduplication detected by array CGH. Am J Med Genet A. (2011) 155(10):2508–11. doi: 10.1002/ajmg.a.34180
52. Ceroni F, Simpson NH, Francks C, Baird G, Conti-Ramsden G, Clark A, et al. Homozygous microdeletion of exon 5 in ZNF277 in a girl with specific language impairment. Eur J Hum Genet. (2014) 22(10):1165–71. doi: 10.1038/ejhg.2014.4
53. Centanni TM, Green JR, Iuzzini-Seigel J, Bartlett CW, Hogan TP. Evidence for the multiple hits genetic theory for inherited language impairment: a case study. Front Genet. (2015) 6:272. doi: 10.3389/fgene.2015.00272
54. Pettigrew KA, Reeves E, Leavett R, Hayiou-Thomas M, Sharma A, Simpson NH, et al. Copy number variation screen identifies a rare De Novo deletion at chromosome 15q13.1–13.3 in a child with language impairment. PLoS One. (2015) 10(8):e0134997. doi: 10.1371/journal.pone.0134997
55. Rakonjac M, Cuturilo G, Stevanovic M, Jelicic L, Subotic M, Jovanovic I, et al. Differences in speech and language abilities between children with 22q11.2 deletion syndrome and children with phenotypic features of 22q11.2 deletion syndrome but without microdeletion. Res Dev Disabil. (2016) 55:322–9. doi: 10.1016/j.ridd.2016.05.006
56. Riccardi F, Rivolta GF, Uliana V, Grati FR, La Starza R, Marcato L, et al. Cryptic 13q34 and 4q35.2 deletions in an Italian family. Cytogenet Genome Res. (2015) 147(1):24–30. doi: 10.1159/000442068
57. Kalnak N, Stamouli S, Peyrard-Janvid M, Rabkina I, Becker M, Klingberg T, et al. Enrichment of rare copy number variation in children with developmental language disorder. Clin Genet. (2018) 94(3–4):313–20. doi: 10.1111/cge.13389
58. Unger S, Mainberger A, Spitz C, Bahr A, Zeschnigk C, Zabel B, et al. Filamin A mutation is one cause of FG syndrome. Am J Med Genet A. (2007) 143(16):1876–9. doi: 10.1002/ajmg.a.31751
59. Addis L, Friederici AD, Kotz SA, Sabisch B, Barry J, Richter N, et al. A locus for an auditory processing deficit and language impairment in an extended pedigree maps to 12p13.31-q14.3. Genes Brain Behav. (2010) 9(6):545–61. doi: 10.1111/j.1601-183X.2010.00583.x
60. Yeung CW, Yau MM, Ma CK, Siu TS, Tam S, Lam CW. Diagnosis of dihydropyrimidinase deficiency in a Chinese boy with dihydropyrimidinuria. Hong Kong Med J. (2013) 19(3):272–5. doi: 10.12809/hkmj133598
61. Chen XS, Reader RH, Hoischen A, Veltman JA, Simpson NH, Francks C, et al. Next-generation DNA sequencing identifies novel gene variants and pathways involved in specific language impairment. Sci Rep. (2017) 7:46105. doi: 10.1038/srep46105
62. Ijlst L, Loupatty FJ, Ruiter JP, Duran M, Lehnert W, Wanders RJ. 3-methylglutaconic aciduria type I is caused by mutations in AUH. Am J Hum Genet. (2002) 71(6):1463–6. doi: 10.1086/344712
63. Andres EM, Earnest KK, Zhong C, Rice ML, Raza MH. Family-based whole-exome analysis of specific language impairment (SLI) identifies rare variants in BUD13, a component of the retention and splicing (RES) Complex. Brain Sci. (2021) 12(1):47. doi: 10.3390/brainsci12010047
64. Berglund A, Stochholm K, Gravholt CH. The epidemiology of sex chromosome abnormalities. Am J Med Genet C Semin Med Genet. (2020) 184(2):202–15. doi: 10.1002/ajmg.c.31805
65. Mervis CB, Morris CA, Klein-Tasman BP, Velleman SL, Osborne LR. 7q11.23 duplication syndrome. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, et al., editors. GeneReviews(®) [Internet]. Seattle, WA: University of Washington, Seattle (1993–2024). PMID: 26610320.
66. Rein B, Yan Z. 16p11.2 copy number variations and neurodevelopmental disorders. Trends Neurosci. (2020) 43(11):886–901. doi: 10.1016/j.tins.2020.09.001
67. McDonald-McGinn DM, Hain HS, Emanuel BS, Zackai EH. 22q11.2 Deletion Syndrome. BTI—GeneReviews(®).
68. Reuter MS, Riess A, Moog U, Briggs TA, Chandler KE, Rauch A, et al. FOXP2 variants in 14 individuals with developmental speech and language disorders broaden the mutational and clinical spectrum. J Med Genet. (2017) 54(1):64–72. doi: 10.1136/jmedgenet-2016-104094
69. Morgan A, Fisher SE, Scheffer I, Hildebrand M. FOXP2-related speech and language disorder. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, et al., editors. GeneReviews(®). [Internet]. Seattle, WA: University of Washington, Seattle (1993–2024). PMID: 27336128.
70. Meulemans J, Goeleven A, Zink I, Loyez L, Lagae L, Debruyne F. Underlying neurological dysfunction in children with language, speech or learning difficulties and a verbal IQ–performance IQ discrepancy. B-ENT. (2012) 8(2):71–6. PMID: 22896925.22896925
71. Samango-Sprouse CA, Tran SL, Lasutschinkow PC, Sadeghin T, Powell S, Mitchell FL, et al. Neurodevelopmental outcome of prenatally diagnosed boys with 47,XXY (Klinefelter syndrome) and the potential influence of early hormonal therapy. Am J Med Genet A. (2020) 182(8):1881–9. doi: 10.1002/ajmg.a.61561
72. Mellone S, Puricelli C, Vurchio D, Ronzani S, Favini S, Maruzzi A, et al. The usefulness of a targeted next generation sequencing gene panel in providing molecular diagnosis to patients with a broad Spectrum of neurodevelopmental disorders. Front Genet. (2022) 13:875182. doi: 10.3389/fgene.2022.875182
73. Ontario Health (Quality). Genome-wide sequencing for unexplained developmental disabilities or multiple congenital anomalies: a health technology Assessment. Ont Health Technol Assess Ser. (2020) 20(11):1–178. PMID: 32194879.
74. Durmaz AA, Karaca E, Demkow U, Toruner G, Schoumans J, Cogulu O. Evolution of genetic techniques: past, present, and beyond. Biomed Res Int. (2015) 2015:461524. doi: 10.1155/2015/461524
Search queries used are:
PubMed
(((((genetics[MeSH Terms]) AND syndrom*[Title/Abstract])) OR (((((((gene[Title/Abstract]) OR genes[Title/Abstract]) OR genetic*[Title/Abstract]) OR chromosom*[Title/Abstract]) OR mutation*[Title/Abstract]) OR human genetics[MeSH Terms])))) AND (((((((((((delay*[Title/Abstract]) OR impair*[Title/Abstract]) OR disorder*[Title/Abstract]) OR abilit*[Title/Abstract]) OR problem*[Title/Abstract]) OR development*[Title/Abstract])) AND language[Title/Abstract])) OR ((((((((delay*[Title/Abstract]) OR impair*[Title/Abstract]) OR disorder*[Title/Abstract]) OR abilit*[Title/Abstract]) OR problem*[Title/Abstract]) OR development*[Title/Abstract])) AND speech[Title/Abstract])) OR language disorder[MeSH Terms])
Embase
#1: Language
("Language Disorders"[Mesh] OR language impairment[Title/Abstract] OR communication disorder[Title/Abstract] OR semantic disorder[Title/Abstract] OR syntactic disorder[Title/Abstract] OR speech disorder[Title/Abstract] OR sound disorder[Title/Abstract] OR phonological disorder[Title/Abstract] OR speech impairment[Title/Abstract] OR speech delay[Title/Abstract] OR language delay[Title/Abstract] OR pragmatic disorder[Title/Abstract] OR language problem[Title/Abstract] OR language difficulties[Title/Abstract] OR language difficulty[Title/Abstract] OR language retardation[Title/Abstract] OR communication disorder[Title/Abstract] OR communication delay[Title/Abstract] OR language disorder[Title/Abstract] OR language learning[Title/Abstract] OR language development[Title/Abstract] OR language abilities[Title/Abstract] OR language ability[Title/Abstract])
AND
#2: Genetics
(genes[Title/Abstract] OR genetic[Title/Abstract] OR chromosome[Title/Abstract] OR chromosomes[Title/Abstract] OR chromosomal[Title/Abstract] OR syndrome[Title/Abstract] OR syndromes[Title/Abstract] or syndromal[Title/Abstract] OR mutation[Title/Abstract] OR heredit*[Title/Abstract] OR inherit*[Title/Abstract] OR "Genetic Diseases, Inborn"[Mesh] OR "Genes"[Mesh] OR "Genetic Loci"[Mesh] OR "Inheritance Patterns"[Mesh])
AND
#3: Children
("Infant"[Mesh:noexp] OR "Child"[Mesh] OR infant[Title/Abstract] OR infants[Title/Abstract] OR toddler[Title/Abstract] OR child[Title/Abstract] OR children[Title/Abstract] OR boy[Title/Abstract] OR girl[Title/Abstract])
Keywords: children, developmental language disorder (DLD), diagnostic, genes, genetic etiology
Citation: van Wijngaarden V, de Wilde H, Mink van der Molen D, Petter J, Stegeman I, Gerrits E, Smit AL and van den Boogaard M-J (2024) Genetic outcomes in children with developmental language disorder: a systematic review. Front. Pediatr. 12:1315229. doi: 10.3389/fped.2024.1315229
Received: 10 October 2023; Accepted: 2 January 2024;
Published: 17 January 2024.
Edited by:
Ammar Husami, Cincinnati Children's Hospital Medical Center, United StatesReviewed by:
Dejan Budimirovic, Johns Hopkins University, United States© 2024 van Wijngaarden, de Wilde, Mink van der Molen, Petter, Stegeman, Gerrits, Smit and van den Boogaard. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vivian van Wijngaarden di52YW53aWpuZ2FhcmRlbkB1bWN1dHJlY2h0Lm5s
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.