
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr., 20 February 2024
Sec. General Pediatrics and Pediatric Emergency Care
Volume 12 - 2024 | https://doi.org/10.3389/fped.2024.1278047
 Ina Kirchberg1*
Ina Kirchberg1* Elke Lainka2
Elke Lainka2 Andrea Gangfuß3Alma Kuechler4Fabian Baertling5Lea D. Schlieben6,7
Andrea Gangfuß3Alma Kuechler4Fabian Baertling5Lea D. Schlieben6,7 Dominic Lenz8
Dominic Lenz8 Eva Tschiedel1
Eva Tschiedel1
Background/purpose: MEGDHEL syndrome is a rare autosomal recessive metabolic disorder, which is characterized by 3-methylglutaconic aciduria with deafness-dystonia, hepatopathy, encephalopathy and Leigh-like syndrome. It is caused by biallelic pathogenic variants in the SERAC1 gene. Due to the unspecific symptoms and the diverse manifestations of the clinical phenotype, the diagnosis is challenging. Infantile MEGDHEL syndrome often has a severe disease course with acute liver failure. Differentiation from other metabolic disorders is difficult and requires a multidisciplinary approach.
Case presentation: A two-day-old small for gestational age neonate was admitted to our pediatric intensive care unit (PICU) due to severe liver failure with distinct hyperammonemia and hypoglycemia without elevation of transaminases or cholestasis. Due to high ammonia level, continuous hemodialysis was established immediately after admission. In addition, protein intake was stopped, and the patient anabolized with intravenous glucose. Temporary stabilization could be achieved after four days. In the further course, severe neurological and cardiocirculatory complications occurred, which ultimately led to the infant's death. In the metabolic diagnostics, a pronounced lactate acidosis and in urine an increased excretion of 3-methylglutaconic acid as well as other metabolites of mitochondrial energy metabolism has been the leading findings besides the hyperammonemia. Post-mortem trio whole genome analysis detected a homozygous pathogenic variant in SERAC1 with evidence of SERAC1 deficiency leading to the diagnosis of infantile MEGDHEL syndrome.
Conclusion: When pediatricians are faced with hepatopathy or even acute liver failure without elevation of transaminases or cholestasis in newborns, SERAC1 deficiency should be considered as a potential differential diagnosis. The initial treatment is based on the recommended management of suspected metabolic disorders. Even while no cure is available yet, patients should be offered proper supportive management through a multidisciplinary team. In addition, genetic confirmation of the diagnosis is important for the families, especially regarding further family planning.
If a newborn presents with hyperammonemia, hypoglycemia and impaired liver synthesis function without elevation of transaminases or cholestasis, the possible presence of MEGDHEL syndrome due to a SERAC1 mutation should be considered.
MEGDHEL (OMIM #614739) syndrome is a rare autosomal recessive metabolic disorder, which is characterized by 3-methylglutaconic aciduria with deafness-dystonia, hepatopathy, encephalopathy and Leigh-like syndrome. First description of 4 patients with MEGDEL (3-methylglutaconic aciduria with deafness-dystonia, encephalopathy and Leigh-like syndrome) was published by Wortmann et al. in (1). In the following years hepatopathy was incorporated into the acronym due to the observation of liver involvement as another clinical feature (2). MEGDHEL syndrome is caused by mutations in the SERAC1 gene (OMIM *614725). SERAC1 deficiency can lead to different clinical phenotypes, varying according to the age of manifestation (3, 4). Most neonates develop problems during their first days of life. Symptoms are typically hypoglycemia, hyperammonemia, elevated serum lactate and sepsis-like picture not linked to infection (5).
We report a case of infantile-onset MEGDHEL syndrome due to a homozygous splice variant in SERAC1 who was admitted to our pediatric intensive care unit (PICU) on the second day of life.
A two-days-old small for gestational age (birth weight 2,580 g (5th percentile) neonate was transferred to our PICU for hemodialysis due to significant hyperammonemia (ammonia level 764 µg/dl). The patient was born as the first child (primigravida) of healthy, non-consanguineous parents. Postnatal cardiorespiratory adaptation was without complications; APGAR was 9/10/10 accordingly. The patient received a 2 mg oral dose vitamin K at birth. No congenital anomalies or dysmorphisms were noted. The maternity hospital is an urban hospital with level II perinatal care. The patient was initially breastfed and presented on the second day of life with recurrent hypothermia, poor sucking, and surpassing weight loss. Biochemical findings were partially compensated lactate acidosis (lactate 7.0 mmol/L, pH 7.14, PaCO2 14.1 mmHg, BE −24.2 mmol/L) and hyperammonemia (589 µg/dl), with transaminases within the reference range.
Due to the distinct hyperammonemia the patient was transferred to a specific metabolic center requiring an ambulance ride of about 45 min. Initial management of hyperammonemia included the stop of protein intake, anabolization with glucose, support of nitrogen detoxification (sodium phenylacetate and sodium benzoate) and supplementation of carglum acid, L-arginine, L-carnitine, vitamin B12 and biotin. Despite intensive therapy, hyperammonemia increased to ammonia level 850 µg/dl and there were signs of acute liver failure (INR 2.1), which was not correctable by supplementation of vitamin K. Metabolic laboratory for investigation of inborn errors of metabolism including acylcarnitine profile analysis in dried blood spots, testing of organic acids in urine and analysis of plasma amino acids was performed and the patient was transferred to our PICU for hemodialysis.
Examination findings: On admission to our PICU, the neonate presented in a severely reduced general condition, marked tachydyspnoea and reduced skin turgor. Other pediatric examination findings were unremarkable. Blood tests revealed evidence of acute liver failure: conjugated bilirubin 0.92 mg/dl (<0.2), total bilirubin 7.0 mg/dl (0.2–1), INR 2.45, aPTT 86.5 s (31–54), thrombin time 26.2 s (16.2–19.1), ammonia level 850 µg/dl (45–109) (Figure 1), while transaminases were still within the reference range [AST 86 U/L (<90), ALT 25 U/L (<60)].
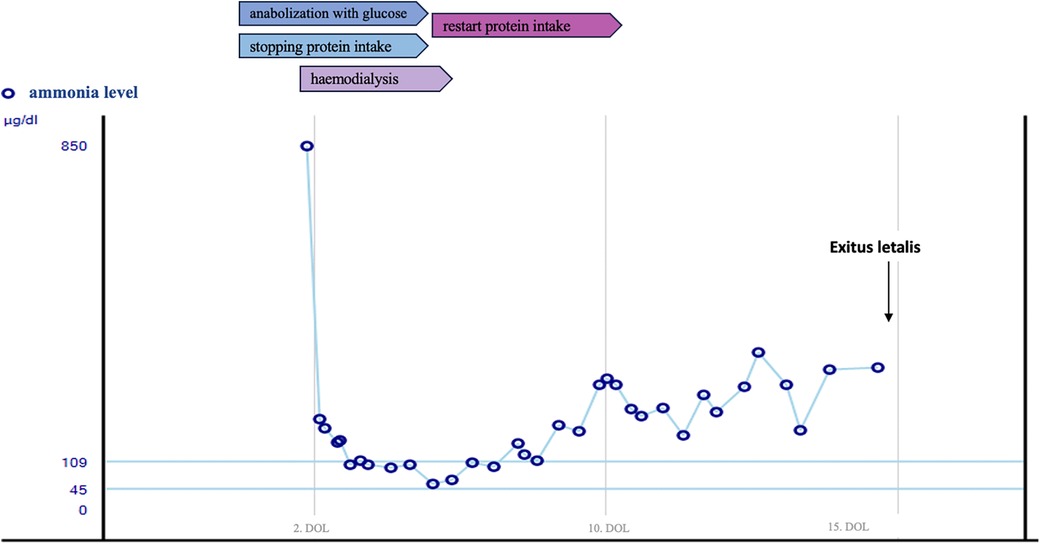
Figure 1. Course of serum ammonia level.
After admission to our PICU intubation and emergency hemodialysis was performed via a Shaldon catheter. Within 7 h after admission the ammonia level reached normal ranges. Repeated supplementation with coagulation factors and fresh frozen plasma were necessary. During hemodialysis, the patient was catecholamine-dependent, therefore requiring noradrenaline max. 0.7 µg/kg/min iv and dobutamine max. 20 µg/kg/min iv. The initial abdominal ultrasound was suspicious for portal vein thrombosis. Etiologically, we assume that this is a complication of umbilical vein/arterial catheter. Partial recanalization was achieved after three days of heparin administration.
Protein fasting and anabolization by increased glucose intake (15 g/kg/d) and hydration were continued. In addition, the patient received L-arginine (2 mmol/kg/d iv), nitrogen scavengers (sodium benzoate (250 mg/kg/d iv), and carglumic acid (77 mg/kg/d PO) because a urea cycle disorder was suspected.
Biochemical results were neither suggestive for urea cycle disorder (standard values for plasma citrulline and arginine) nor for mitochondrial disease (standard values for plasma alanine and plasma proline). Even though also the investigation for mitochondrial fatty acid oxidation disorders by acylcarnitine profile proved unremarkable, L-carnitine (100 mg/kg/d iv) was administered until urine organic acids entirely excluded the prevalence of organic aciduria. Urinary organic acid analysis showed a massively increased excretion of lactate (20.108 mmol/mol creatinine; <413) and pyruvate (989 mmol/mol creatinine; <118), a significant ketonuria [3-hydroxybutyrate (1,042 mmol/mol creatinine; <385) and acetoacetate (3,348 mmol/mol creatinine; <150)] as well as secondary changes due to mitochondrial dysfunction. Among those, a significant increase of 3-methylglutaconic acid (3-MGA) (128 mmol/mol creatinine; <15) was noted. With a concomitant urine creatinine of 18.1 mg/dl, this was considered a significant 3-MGA aciduria (6). From this point on the main diagnostic hypothesis resulted being a mitochondrial metabolic disorder with 3-methylglutaconic aciduria and the patient was treated with riboflavin, thiamine and coenzyme Q 10 additively.
In the following days the patient's condition stabilized with sufficient spontaneous breathing. Hemodialysis was discontinued after 4 days.
Ammonia levels remained slightly elevated (150–290 µg/dl) (Figure 1). But re-introduction of protein intake, did not result in a further increase.
On the 13th day of life, a second deterioration occurred unexpectedly. A trigger could not be identified. Focal seizures occurred and anticonvulsive therapy was started. Cranial magnetic resonance imaging (MRI) showed multiple supratentorial medullary lesions, some with impaired diffusion. Due to the patient's critical condition, the standard hearing test was postponed. Reintubation was necessary, and liver synthesis as well as cardiovascular function decompensated again. Stabilization could not be achieved, and the patient dies at the age of 15 days.
Postmortem, trio-whole-genome sequencing was performed and confirmed the homozygous prevalence of a pathogenic splice variant in SERAC1 (NM_032861.4: c.1888_1828 + 10delinsACCAACAGG).
MEGDHEL syndrome is a rare neurometabolic disorder. By 2017, 67 cases had been reported worldwide (2). The prevalence is estimated at 0.09:100,000 (7). MEGDHEL syndrome is caused by biallelic pathogenic variants in the SERAC1 gene. It encodes Serine active site containing protein 1 (SERAC1), a phosphatidylglycerol remodeling protein (4). Deficiency leads to dysfunction of oxidative phosphorylation and intracellular cholesterol trafficking (8). Most common metabolic changes detected are neonatal hypoglycemia and severe neonatal liver dysfunction (about 50% of all cases), lactic acidosis (85%) and in nearly all patients a 3-MGA aciduria (2). Wortman et al. divided the clinical course of SERAC1 deficiency in three groups depending on the age of manifestation (3).
1. Infantile, Severe MEGDHEL Syndrome: Typical first symptoms are hypotonia and drinking failure, hypoglycemia, lactic acidosis as well as liver dysfunction or a sepsis-like clinical picture without infection (5). Liver failure can present as undulating increase of transaminases, prolonged hyperbilirubinemia up to severe liver failure (3). Then failure to thrive, various liver problems, progressive spasticity and sometimes epilepsy can follow. Survival varies enormously. Some children do not even survive the neonatal period. In the neonatal period, some succumb to multiorgan failure. In infancy, liver failure is a frequent cause of death whereas later in life some succumb to infections, especially pulmonary infections (3, 4).
2. Milder Juvenile-Onset Complicated Hereditary Spastic Paraplegia (cHSP): Roeben et al. (9) present a family in which five of the six siblings had spasticity as well as non-progressive mild cognitive deficits.
3. Adult-Onset Generalized Dystonia: At present, there is only one case reported with mild psychomotor developmental deficits and generalized dystonia.
In the present case, we report on a patient with neonatal onset of MEGDHEL syndrome presenting at the 2nd day of life. However, due to the low birth weight of our patient it can be assumed that the initial onset had been intrauterine. Fellman et al. and Maas et al. also report two cases of infantile MEGDHEL Syndrome who were small for gestational age (2, 10). Pediatric acute liver failure (PALF) is a progressive, potentially lethal clinical condition mainly occurring in the first year of life and according to the PALF Study group defined as a rapidly progressive, potentially fatal clinical syndrome with (a) biochemical evidence of hepatocellular injury and (b) a coagulopathy that cannot be corrected with vitamin K alone with an INR ≥ 1.5 with hepatic encephalopathy or an INR ≥ 2.0 regardless of the presence of hepatic encephalopathy (11, 12). In neonates, the main causes are viral infections (e.g., HSV, Enterovirus), immunological [e.g., gestational alloimmune liver disease (GALD)], inherited metabolic diseases (e.g., urea cycle disorder) and hematological diseases (e.g., hemophagocytic lymphohistiocytosis). However, in about 50% of the cases the etiology remains unexplained (11, 13, 14).
On top of liver synthesis failure hyperammonemia was the most pressing problem of our patient and required emergency dialysis. Molla et al. also describe two neonates with MEGDHEL syndrome required dialysis treatment due to hyperammonemia. Both patients survived the neonatal period and are currently in follow up controls (15). In our case there was an immediate initiation of supportive therapy, diet modification and fast establishment of continuous veno-venous hemodiafiltration (CVVHDF). These interventions improved the patient's state of health temporarily. Nevertheless, our patient died unexpectedly from neurological and cardiorespiratory deterioration only a few days after initial stabilization. This underlines the unpredictability of this disease's course.
Portal vein thrombosis is not part of or explained by the diagnosis of MEGHDEL syndrome and must be assumed to be secondary complication probably triggered by one of the inserted central venous catheters.
Impaired liver function only seems to play a decisive role during the neonatal period. Although transiently elevated transaminases or functional limitations are still found at older ages, usually they are not life-threatening (2). As children get older, other problems come to the fore and multidisciplinary therapy is needed to treat spasticity, dysphagia, deafness, and seizures which may occur.
There is no curative treatment available yet. The therapy is exclusively supportive and depends on the symptoms of the patient. In previous reports, the initial use of protein restriction, supplementation of coenzyme Q10, biotin and riboflavin has been described (2, 15). It is recommended to carry out the stepwise emergency therapy in neonates suspected of having a metabolic disease. In case of severe neonatal hyperammonemia coordinated treatment should be administered immediately with the aim of rapidly lowering the ammonia levels (16).
With the onset of the first symptoms, it is essential to determine the cause of disease. Differential diagnosis of neonatal liver failure with hyperammonemia must include metabolic disorders, viral infections, neonatal hemochromatosis, and hematological causes like hemophagocytic lymphohistiocytosis (HL) (2, 4, 17).
In our case, an infection could be excluded. Also, the prevalence of GALD was very unlikely as this was the mother's first pregnancy. Laboratory findings gave no clue of HL. Therefore, we focused on congenital metabolic disorders. Hence, special metabolic diagnostics including determination of the acylcarnitine profile in a dried blot spot, plasma amino acids, and organic acids as well as orotic acid in urine should be carried out. If possible, samples should be collected before the transfusion of blood products.
The cerebral MRI of our patient showed non-specific medullary lesions matching with a metabolic disorder. However, these aren't pathognomonic and cannot be clearly assigned to the stages postulated by Wortmann et al. They report five stages of characteristic MRI patterns over the course of the disease (18). Like in Leigh syndrome bilateral lesions, particularly in the basal ganglia, can be seen. While the imaging in the neonatal period is usually still unremarkable, the first changes occur during the first year of life.
If clinical and metabolic findings suggest SERAC1 deficiency, molecular genetic testing needs to be initiated to establish the diagnosis. Single gene testing via Sanger sequencing is only indicated in case of familial known pathological variants. For all other patients, trio genome or exome sequencing is the gold standard. Genome or trio genome sequencing are options on a research basis. Advances in molecular genetic testing over the last decade have enabled rapid diagnosis in rare diseases and thus improved patient care and reduced the recurrence risk.
When pediatricians are faced with hepatopathy with distinct hyperammonemia, hypoglycemia and impaired liver synthesis function without elevation of transaminases or cholestasis in newborns, SERAC1 deficiency should be considered as a differential diagnosis. Especially if organic acids in urine show an elevated excretion of 3-MGA, one should think about MEGDHEL syndrome as a possible cause.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
IK: Conceptualization, Visualization, Writing – original draft. EL: Writing – review & editing. AG: Writing – review & editing. AK: Writing – review & editing. FB: Writing – review & editing. LS: Writing – review & editing. DL: Writing – review & editing. ET: Conceptualization, Supervision, Writing – review & editing.
The authors declare financial support was received for the research, authorship, and/or publication of this article.
We acknowledge support by the Open Access Publication Fund of the University of Duisburg-Essen.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Wortmann S, Rodenburg RJT, Huizing M, Loupatty FJ, de Koning T, Kluijtmans LAJ, et al. Association of 3-methylglutaconic aciduria with sensori-neural deafness, encephalopathy, and leigh-like syndrome (MEGDEL association) in four patients with a disorder of the oxidative phosphorylation. Mol Genet Metab. (2006) 88(1):47–52. doi: 10.1016/j.ymgme.2006.01.013
2. Maas RR, Iwanicka-Pronicka K, Ucar SK, Alhaddad B, AlSayed M, Al-Owain MA, et al. Progressive deafness–dystonia due to SERAC1 mutations: a study of 67 cases. Ann Neurol. (2017a) 82(6):1004–15. doi: 10.1002/ana.25110
3. Wortmann SB, Brouwer A, Wevers RA. SERAC1 deficiency. In: Adam MP, Feldman J, Mirzaa GM, Seattle, WA: University of Washington (2014). p. 1993–2024.
4. Finsterer J, Scorza FA, Fiorini AC, Scorza CA. MEGDEL syndrome. Pediatr Neurol. (2020a) 110:25–9. doi: 10.1016/j.pediatrneurol.2020.03.009
5. Sarig O, Goldsher D, Nousbeck J, Fuchs-Telem D, Cohen-Katsenelson K, Iancu TC, et al. Infantile mitochondrial hepatopathy is a cardinal feature of MEGDEL syndrome (3-methylglutaconic aciduria type IV with sensorineural deafness, encephalopathy and leigh-like syndrome) caused by novel mutations in SERAC1. Am J Med Genet A. (2013) 161(9):2204–15. doi: 10.1002/ajmg.a.36059
6. Wortmann SB, Duran M, Anikster Y, Barth PG, Sperl W, Zschocke J, et al. Inborn errors of metabolism with 3-methylglutaconic aciduria as discriminative feature: proper classification and nomenclature. J Inherit Metab Dis. (2013) 36(6):923–28. doi: 10.1007/s10545-012-9580-0
7. Tan J, Wagner M, Stenton SL, Strom TM, Wortmann SB, Prokisch H, et al. Lifetime risk of autosomal recessive mitochondrial disorders calculated from genetic databases. EBioMedicine. (2020) 54:102730. Erratum in: EBioMedicine. (2020) 61:103072. doi: 10.1016/j.ebiom.2020.102730
8. Wortmann SB, Vaz FM, Gardeitchik T, Vissers LELM, Herma Renkema G, Schuurs-Hoeijmakers JHM, et al. Mutations in the phospholipid remodeling gene SERAC1 impair mitochondrial function and intracellular cholesterol trafficking and cause dystonia and deafness. Nat Genet. (2012) 44(7):797–802. doi: 10.1038/ng.2325
9. Roeben B, Schüle R, Ruf S, Bender B, Alhaddad B, Benkert T, et al. SERAC1 deficiency causes complicated HSP: evidence from a novel splice mutation in a large family. J Med Genet. (2018) 55(1):39–47. doi: 10.1136/jmedgenet-2017-104622
10. Fellman V, Banerjee R, Lin KL, Pulli I, Cooper H, Tyynismaa H, et al. Severe neonatal MEGDHEL syndrome with a homozygous truncating mutation in SERAC1. Biochim Biophys Acta Mol Basis Dis. (2022) 1868(1):166298. doi: 10.1016/j.bbadis.2021.166298
11. Squires RH, Shneider BL, Bucuvalas J, Alonso E, Sokol RJ, Narkewicz MR, et al. Acute liver failure in children: the first 348 patients in the pediatric acute liver failure study group. J Pediatr. (2006) 148(5):652–8. doi: 10.1016/j.jpeds.2005.12.051
12. Pediatric Acute Liver Failure Study Group. Updated 2016. “A multi-center group to study acute liver failure in children”. Available online at: https://www.clinicaltrials.gov/study/NCT00986648 (Accessed 27 January 27, 2024).
13. Kathemann S, Bechmann LP, Sowa J-P, Manka P, Dechêne A, Gerner P, et al. Etiology, outcome and prognostic factors of childhood acute liver failure in a German single center. Ann Hepatol. (2015) 14(5):722–8. doi: 10.1016/S1665-2681(19)30767-7
14. Lenz D, Schlieben LD, Shimura M, Bianzano A, Smirnov D, Kopajtich R, et al. Genetic landscape of pediatric acute liver failure of indeterminate origin. Hepatology. (2023). doi: 10.1097/hep.0000000000000684
15. Molla GK, Kağnlcl M, Günlemez A, Yeni Y, Uzun ÖÜ. Two cases of MEGDHEL syndrome diagnosed with hyperammonemia. J Pediatr Endocrinol Metab. (2023) 36(2):203–6. doi: 10.1515/jpem-2022-0418
16. Savy N, Brossier D, Brunel-Guitton C, Ducharme-Crevier L, Du Pont-Thibodeau G, Jouvet P. Acute pediatric hyperammonemia: current diagnosis and management strategies. Hepat Med. (2018) 10:105–15. doi: 10.2147/hmer.s140711
17. Larson-Nath C, Vitola BE. Neonatal acute liver failure. Clin Perinatol. (2020) 47(1):25–39. doi: 10.1016/j.clp.2019.10.006
Keywords: MEGDHEL syndrome, SERAC1, hyperammonemia, liver failure, neurometabolic disorder, lactic acidosis
Citation: Kirchberg I, Lainka E, Gangfuß A, Kuechler A, Baertling F, Schlieben LD, Lenz D and Tschiedel E (2024) Distinct neonatal hyperammonemia and liver synthesis dysfunction: case report of a severe MEGDHEL syndrome. Front. Pediatr. 12:1278047. doi: 10.3389/fped.2024.1278047
Received: 15 August 2023; Accepted: 5 February 2024;
Published: 20 February 2024.
Edited by:
Jérémie F. Cohen, Necker-Enfants malades Hospital, FranceReviewed by:
Zachary McPherson, Children’s Hospital at Westmead, Australia© 2024 Kirchberg, Lainka, Gangfuß, Kuechler, Baertling, Schlieben, Lenz and Tschiedel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ina Kirchberg aW5hZWxpc2Eua2lyY2hiZXJnQHVrLWVzc2VuLmRl
Abbreviations 3-MGA, 3-methylglutaconic acid; ALT = SGPT, alanine transaminase; aPTT, activated partial thromboplastin time; AST = SGOT, aspartate transaminase; BW, body weight; CVVHDF, continuous veno-venous hemodiafiltration; EEG, electroencephalogram; GALD, gestational alloimmune liver disease; HL, hemophagocytic lymphohistiocytosis; INR, international normalized ratio; IV, intra venous; MEGDEL, 3-methylglutaconic aciduria, deafness-dystonia, encephalopathy and leigh-like syndrome; MEGDHEL, 3-methylglutaconic aciduria, deafness-dystonia, hepatopathy, encephalopathy and leigh-like syndrome; MRI, magnetic resonance imaging; P, percentile; PALF, pediatric acute liver failure; PICU, pediatric intensive care unit; PO, per os/oral; SERAC1, serine active site containing protein 1; SGOT = AST, serum glutamic oxaloacetic transaminase; SGPT = ALT, serum glutamic pyruvic transaminase.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.