 Xia Wang
Xia Wang Yingcan Wang
Yingcan Wang Ting Xu2
Ting Xu2 Yifeng Ding
Yifeng Ding Jihong Qian
Jihong Qian
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pediatr. , 24 April 2023
Sec. Genetics of Common and Rare Diseases
Volume 11 - 2023 | https://doi.org/10.3389/fped.2023.978879
This article is part of the Research Topic Genetics of Inborn Errors of Metabolism View all 30 articles
Osteopetrosis is a genetic condition of the skeleton characterized by increased bone density caused by osteoclast formation and function defects. Osteopetrosis is inherited in the form of autosomal dominant and autosomal recessive manner. We report autosomal recessive osteopetrosis (ARO; OMIM 611490) in a Chinese case with a history of scarce leukocytosis, vision and hearing loss, frequent seizures, and severe intellectual and motor disability. Whole-exome sequencing (WES) followed by Sanger sequencing revealed novel compound heterozygous mutations in the chloride channel 7 (CLCN7) gene [c.982-1G > C and c.1208G > A (p. Arg403Gln)] in the affected individual, and subsequent familial segregation showed that each parent had transmitted a mutation. Our results confirmed that mutations in the CLCN7 gene caused ARO in a Chinese family. Additionally, our study expanded the clinical and allelic spectrum of the CLCN7 gene and enhanced the applications of WES technology in determining the etiology of prenatal diagnoses in fetuses with ultrasound anomalies.
Osteopetrosis is a genetically heterogeneous disorder characterized by abnormal bone metabolism. The pathogenesis of osteopetrosis stems from the dysfunction of differentiation and/or absorption of osteoclasts, which results in skeletal dysplasia, such as increased bone density and medullary cavity stenosis (1). Three clinical types can be identified based on severity, age of onset, and inheritance: a dominant benign type, autosomal dominant osteopetrosis (ADO, OMIM 166600); intermediate autosomal osteopetrosis (IAO, OMIM 259710); and a severe recessive type, autosomal recessive osteopetrosis (ARO, OMIM 611490). ARO, also known as infantile malignant osteopetrosis, is the most fatal type of osteopetrosis, with an incidence of 1/250,000 live births. ARO patients usually present by 2 years of age and die before 10 years of age (1–3). Patients diagnosed with ARO are more susceptible to hematological impairment with anemia, thrombocytopenia, and secondary neurological deficits. However, leucocyte counts in ARO patients were not frequently reported (4).
At least 10 genes have been reported to be associated with osteopetrosis. Mutations in T-cell immune regulator 1 (TCIRG1) and chloride channel 7 (CLCN7) are the two most common causes of ARO. TCIRG1 is located on chromosome 11q13 and spans ∼12.5 kb. The transcript variants of this gene containing 15 exons represent the a3 subunit of the vacuolar proton pump, which is preferentially expressed in osteoclasts and plays an important role in bone resorption. TCIRG1 mutations are reportedly responsible for over 50% of ARO cases. The CLCN7 gene on human chromosome 16p13.3 contains 25 exons and encodes the 803 amino acid chloride channel protein 7 (CLC-7). CLCN7 is a member of the voltage-gated chloride channel protein family that mediates the exchange of chloride ions against protons, maintaining the acidic environment for bone resorption (5). CLCN7 plays a synergistic role when hydrogen ions are transported outside of the cell by TCIRG1 (6). Mutations in CLCN7 have been documented in approximately 20% of individuals and are related to a broad spectrum of osteopetrosis with phenotypes ranging from mild to life-threatening levels of severity (1, 3, 7).
The initial clinical presentation of this rare genetic syndrome is heterogeneous. Formal clinical diagnostic criteria for ARO have not been established. High awareness of initial ARO symptoms is critical for early diagnosis. Approximately 100 variants in CLCN7 are known (http://www.hgmd.cf.ac.uk/). ARO related to CLCN7 mutation has been identified in approximately two dozen families (3, 8–19). Affected individuals admitted for visual impairment, anemia, failure to thrive, or convulsion in the infantile period are frequently reported (1, 3). These symptoms and radiological changes involving extensive bone calcification should raise clinical suspicion of ARO. Genetic testing approaches promote molecular diagnoses. Data regarding Chinese patients with CLCN7-related ARO are limited to four cases from Taiwan (12), Guangzhou (10), and Shanghai (17). These patients had a relatively stable disease course and were still alive at the time they were reported.
Here, we report the natural course of a patient with CLCN7-related ARO involving scarcely leukocytosis at birth, rapidly progressing to neurological deterioration with a very poor prognosis. We also review the clinical and genetic findings in CLCN7-related ARO reported thus far.
A neonate who presented with severe leukocytosis and thrombocytopenia at 3 days of age was referred to Xinhua Hospital affiliated with Shanghai Jiao Tong University School of Medicine. The patient was evaluated with whole-exome sequencing (WES) for etiological evidence.
The studies involving human participants were reviewed and approved by the Institutional Review Board of Xinhua Hospital. Written informed consent was obtained from the legal guardian for the publication of any potentially identifiable images or data included in this article.
Genomic DNA was extracted from peripheral blood. Library preparation was performed with an xGen Exome research panel v1.0 (IDT, United States), and sequencing was conducted on a HiSeq 4000 (Illumina, United States). Raw reads were aligned to the reference genome GRCh37/hg19 by BWA-MEM (v0.7.12). Variant calling was performed following the GATK best practice workflow (v3.3) (20, 21). Variant annotation and filtration were performed by SnpEff (v4.2) and SnpSift (v4.2) based on gnomAD (v2.1), OMIM (https://omim.org/), HGMD (https://www.hgmd.cf.ac.uk/ac/), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar), and an in-house database. Interpretation of variants was conducted in accordance with American College of Medical Genetics and Genomics (ACMG) guidelines (22).
The variant screening process was based on disease-related information and variant pathogenicity evaluation (allele frequency in population, in silico tools, ACMG guidelines) (23, 24). Genetic disorders that frequently manifest phenotypes similar to our case include juvenile-myelomonocytic-leukemia (JMML) (5, 17, 25) and leukocyte adhesion deficiency (LAD) (1, 26, 27). JMML-related genes include PTPN11, NRAS, KRAS, NF1, and CBL. LAD-related genes include IKBKG, ITBG, KINDLIN3, and FERMT3. All rare variants in these genes were extracted and filtered.
Sanger sequencing was performed to validate the heterozygous variants identified through WES. PCR amplification was carried out using an ABI 9700 Thermal Cycler and sequenced on an ABI PRISM 3730 sequencer (Applied Biosystems, United States) using the primers listed in Supplementary Table S1.
REVEL is a tool for predicting the effect of reported missense variants on the function of mutant human proteins (28). The chromosome localization, protein sequences, and phylogenetic tree of the missense variant (c.1208 G > A) in the CLCN7 gene were analyzed by ClustalX_1.81 (http://www.clustal.org/) and Molecular Evolutionary Genetics Analysis (https://www.megasoftware.net/). Mutant sequences (c.982-1G > C) and wild-type sequences were analyzed by in silico tools (29), CADD (Combined Annotation Dependent Depletion; cadd.gs.washington.edu) splice, MaxEntScan, Spliceogen (https://github.com/VCCRI/Spliceogen), NNSPLICE (Splice Site Prediction by Neural Network), and Splice AI (https://github.com/Illumina/SpliceAI).
A 3-day-old female neonate was referred to our hospital with rare leukocytosis (56.12 × 109/L) and transient thrombocytopenia (85 × 109/L) on the day she was born. Physical examination showed no external birth defects other than mild hypotonia with slow suckling. Clinical biochemical analysis was also performed. The patient's hematological profile was as follows: white blood cell (WBC) count, 43.71 × 109/L; neutrophil/lymphocyte (N/L) ratio, 66%/16.9%; monocyte ratio, 14.70%; hemoglobin (Hb), 162 mg/dL; platelets, 98 × 109/L. C-reactive protein and procalcitonin were in the normal range. No pathogenic microorganisms were identified by bacterial culture or metagenomic sequencing. Serology for toxoplasma, rubella, cytomegalovirus, and herpes was negative. Peripheral blood smear showed no evidence of immature cells. Bone marrow puncture was considered but not successfully performed. After intravenous administration of meropenem and penicillin for 7 days, leukocytosis and thrombocytopenia did not significantly improve (Figure 1). Brain magnetic resonance imaging (MRI) showed slight signal alterations in white matter and the corpus callosum (Figure 2A). Screening tests of hearing and ocular disease were normal.
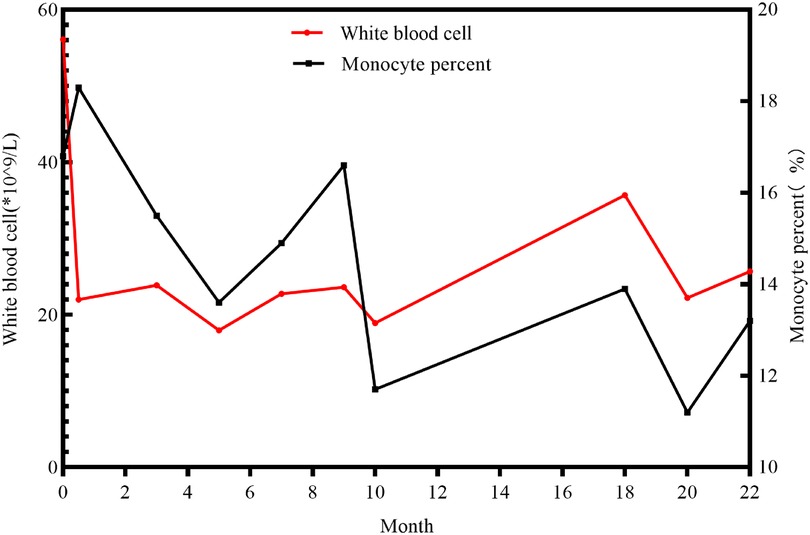
Figure 1. The longitude data of white blood cells and monocyte percent of the patient.
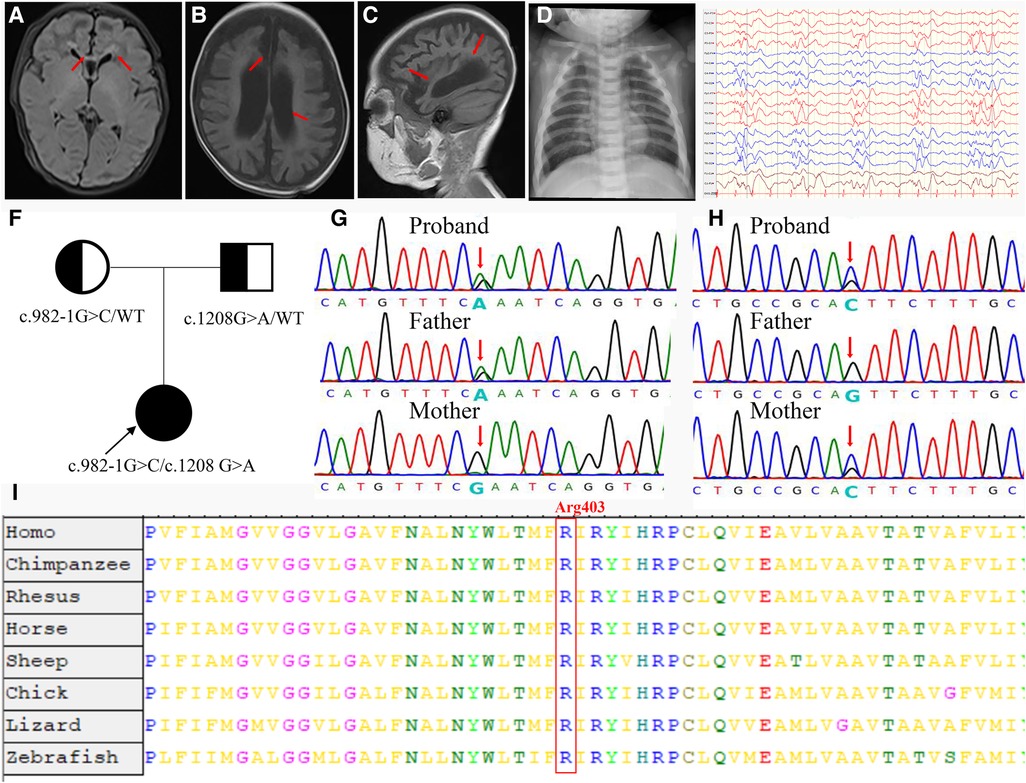
Figure 2. Clinical and molecular findings of the patient. (A) T1-weighted (T1W) brain MRI indicated a high signal in the white matter (arrowheads) and corpus callosum (arrowheads) during the neonatal period. (B,C) T1W brain MRI indicated hydrocephaly (arrowheads) and brain atrophy (arrowheads) at 4 months of age. (D) Proband showed increased bone density on radiography and “sandwich” vertebrae. (E) Multifocal slow spike-wave and slow-wave patterns in electroencephalogram. (F) The proband's family tree. (G) The proband was paternally inherited the variant of c.1208G > A in the CLCN7 gene. (H) The proband was maternally inherited the variant of c.982-1G > C in the CLCN7 gene. (I) A cross-species alignment of amino acid sequences showed that p. Arg403Gln variants were located in a highly conserved region in CLCN7 protein.
At 2 months of age, the patient had vision and hearing impairment, with little response to colors, voices, or moving objects. From the age of 4 months, her neurological defects gradually worsened. The patient began to have seizures that manifested as spasticity, sometimes as often as dozens of times per day, and a diagnosis of West syndrome was made. She was treated with various antiepileptic medicines, such as vigabatrin, topiramate, and levetiracetam, but none successfully controlled the seizures. The patient's biochemical indexes were normal, including calcium–phosphorus metabolism biomarkers, immunoglobulin levels, and lymphocyte classification counts (Supplementary Table S2). Brain MRI showed hydrocephalus and cerebral atrophy (Figures 2B,C). The electroencephalogram showed multifocal slow spike-wave and slow-wave patterns (Figure 2E). At 9 months of age, her head circumference was 38.0 cm (<−3 SD), height was 65 cm (<−2 SD), and weight was 7.0 kg (−1 SD to −2 SD). Her vision was completely lost. She could not raise her head, sit, or laugh. Neuropsychological testing showed a development delay using the Bayley III scale.
Through telephone follow-up, we learned that her leucocytes remained at a high level (not lower than 15 × 109/L). When the patient experienced diarrhea or acute upper respiratory infections, her leucocytes were high, up to 20–40 × 109/L (Figure 1). Because of the progressive neurologic impairment, her family was compelled to provide symptomatic treatment without resorting to bone marrow transplantation or hematopoietic stem cell transplantation. The patient died from respiratory arrest at the age of 22 months.
Compound heterozygous variants were identified in the CLCN7 gene. According to ACMG guidelines, the variant c.1208G > A (p. Arg403Gln) is rated “likely pathogenic” (PM1 + PM2 + PM3 + PP3 + PP4). The variant c.982-1G > C, located at a splice donor site, is also rated “likely pathogenic” by ACMG standards (PVS1_S + PM2 + PM3 + PP4).
We also reviewed the patient's bone imaging, which showed increased bone density on radiography and “sandwich” vertebrae (Figure 2D); this evidence, together with the early onset of patient clinical manifestations and the mode of inheritance (Figure 2F), was highly compatible with ARO. The variant c.1208G > A was paternally inherited (Figure 2G). This missense mutation is a previously reported osteopetrosis-causing variant located in the transmembrane domain of the protein. The c.1208G > A mutation is located in a highly conserved region among vertebrates, as confirmed by an online sequence database (Figure 2I). The other variant, c.982-1G > C, located in intron 11, was maternally inherited; this variant is novel (Figure 2H).
As a variant at the canonical splice site, the interpretation of the c.982-1G > C variant following ACMG guideline was “likely pathogenic” (PVS1_S + PM2 + PM3 + PP4). In order to predict the possible splice-altering effect, multiple splice-altering prediction algorithms were used including CADD, MaxEntScan, Spliceogen, NNSPLICE, and SpliceAI.
The guanine is replaced by thymine (c.982-1G > C) at nucleotide number 1 in intron 11 located at the exon–intron boundaries. In silico prediction analysis is shown in Table 1. The CADD index is 32 (CADD index of process normal order is 14.25). MaxENT value is 8.27 (MaxENT index of the normal sequence is 2.35). Spliceogen index was 1.0 (normal sequence index is 0.78). NNSPLICE showed an original acceptor site with a score of 0.79 (cutoff 0.40); no splice site was predicted after the c.982-1G > C alteration. Similarly, SpliceAI suggested splice acceptor loss caused by the variant, with a score of 0.79 (cutoff 0.50). Based on these prediction algorithms, the c.982-1G > C variant, at the splice acceptor site of intron 11, potentially alters mRNA transcription.
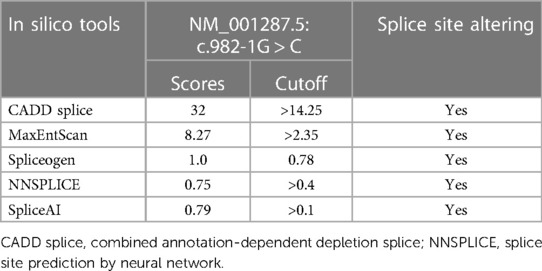
Table 1. Splicing prediction of the novel variant c.982-1G > C in CLCN7 gene.
We reported a case of neonatal CLCN7-related ARO detected at birth, with an onset symptom of severe leukocytosis; later symptoms included vision and hearing loss as well as neurological deficits in the form of frequent seizures and intellectual and motor disability. The diagnosis was confirmed by genetic testing, which showed compound heterozygous variants in the CLCN7 gene.
Anemia and thrombocytopenia are prominent symptoms in ARO patients due to bone marrow failure (Table 2). Recurrent leukocytosis observed in our patient has been scarcely reported. Another case of CLCN7-related ARO reported in Guangzhou, China, showed an increased leukocyte count of 19.2 × 10^9/L at 7 months of age, without longitudinal data, which was similar to our patient's condition. However, studies of immunological deficits in ARO patients have been limited by the availability of osteoclasts. The defective generation of superoxide by neutrophil cells, monocytes, and lymphocytes, which results in an inability to eradicate infection, has been observed in ARO patients (31, 32). Recurrent leukocytosis and osteopetrosis at early age was highly suspected to be due to KINDLIN3 mutation, an important intracellular signaling molecule involved in the combination of osteoclast maturation and leukocyte adhesion deficiency (26, 33). However, genomic sequencing did not identify pathogenic variants. Second, the monocyte percentage in this case was persistently increased (Figure 1). Monocyte macrophages and osteoclasts are derived from the same hematopoietic lineage. The dysfunction of osteoclasts may trigger an increase in the production of monocytes in the feedback cycle, which is modulated by homologous crosstalk signals (7, 34). Furthermore, determining whether leukocytosis is a phenotype in CLCN7-related ARO children and identifying the underlying genotype may require a more specific cohort.
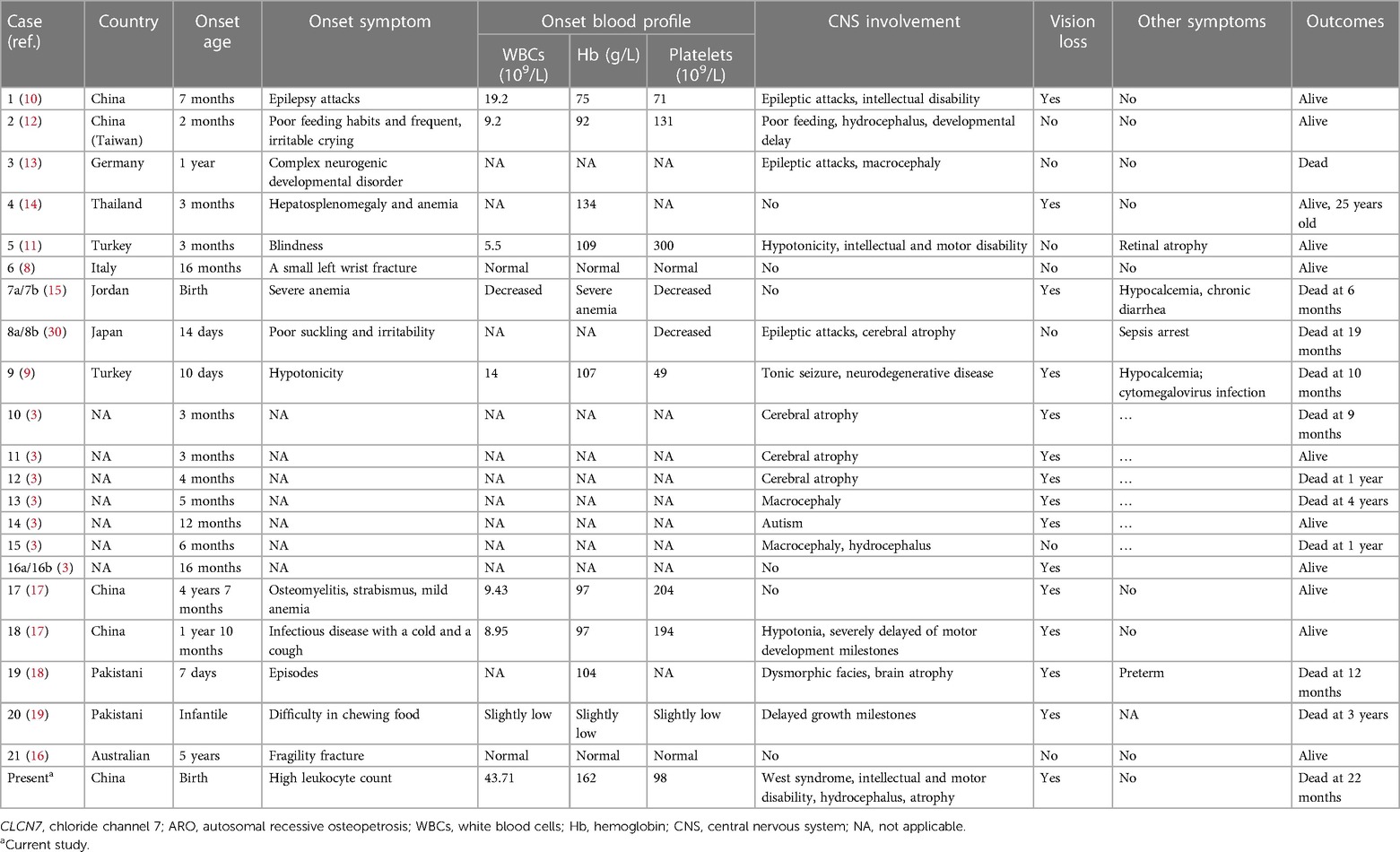
Table 2. Clinical findings from our patient and others with CLCN7-related ARO in the literature.
Another important issue is central nervous system (CNS) impairment. The mechanism of CNS impairments may result from endosome or lysosome dysfunction in the neuron system mediated by the inactive CLCN7 channel (35). The interaction pathways of the peroxisome proliferator-activated receptor and neuroactive ligand–receptor are reported to be involved (36). We reviewed the literature and identified a total of 21 patients with CLCN7-related ARO (8–17, 30, 37) (Table 2). A total of 15/21 patients had CNS involvement; 6/21 patients died before the age of 5 years. However, the 10/15 patients without CNS involvement were all alive when their cases were reported. The oldest surviving patient was 25 years old (14). Thus, patients with CNS impairments usually had significantly reduced lifespans, and this effect could not be reversed by bone marrow transplantation or hematopoietic stem cell transplantation (1, 3). Thus, CNS involvement in CLCN7-related ARO should be thoroughly assessed initially and carefully monitored, as it is an essential prognostic predictor. The typical clinical neurological presentation of TCIRG1-related ARO patients was also hydrocephalus, myotonia, neuropsychic and psychomotor retardation, compromised vision, and heavy nasal breathing. Overall, the neurological phenotype of TCIRG1-related ARO seems to be more benign than that of the CLCN7-dependent form. However, no significant correlation was found between CNS phenotypic presenting features and specific gene mutations, which may be related to the clinical phenotype variability and variable genetic expressivity in the pathogenic genes (4, 38–40).
Previously, the reported ARO-causing variants in CLCN7 (42 alleles, including 22 missenses, 6 splice sites, 3 nonsense, and 5 frameshift) did not include any hotspot mutations (Table 3, Figure 3). The missense mutation (c.1208G > A) was reported, which was suggestive of a mild case of ARO (13). However, our patient's medical history and short survival time manifested a malignant phenotype. Thus, the correlation between genotype and phenotype remains to be resolved. The pathogenetic process may also involve posttranslational modifications, including epigenetic modification, phosphorylation, and ubiquitylation, which might impair the function of the CLCN7 protein.
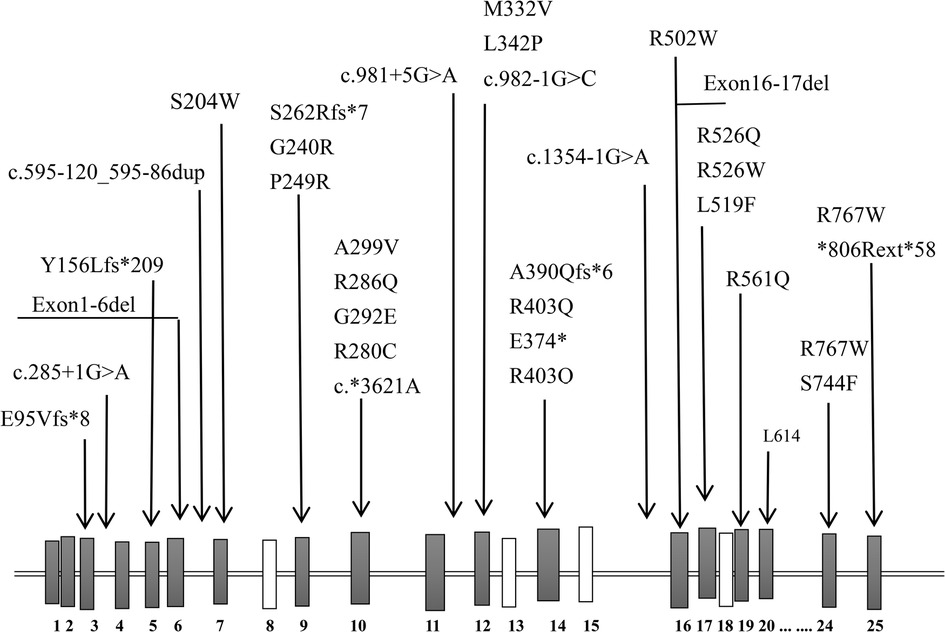
Figure 3. A sketch-map of the gene and location of reported variants of CLCN7.
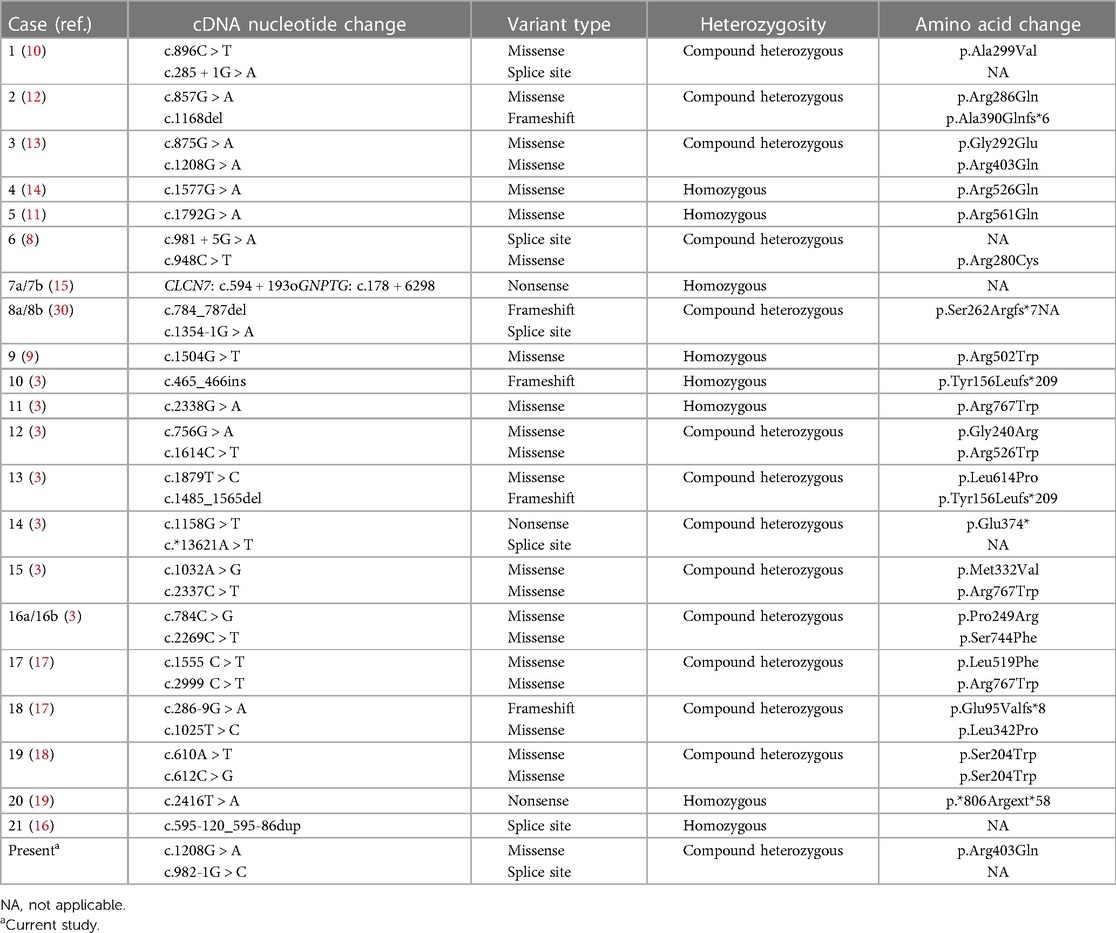
Table 3. The molecular variants in CLCN7 in our patient and others in the literature.
In a review of 22 patients with CLCN7-related AROs, the predictive value concerning survival of early central nervous impairment was investigated (Table 2). Another important issue was that the onset symptom of exaggerated leukocytosis was scarcely reported, which would straightforwardly be misdiagnosed as JMML or LAD. However, the nonspecific peripheral blood smear test and the failure of bone marrow aspiration of the neonatal patient required the consideration of differential diagnoses. ARO is not recognized immediately in our case and other considerable number of children. Indicative signs, such as neurologic impairment, vision or hearing lost, and hematological abnormality, would be suggestive findings for the early diagnosis. Increased bone density is radiological feature for clinically early diagnosis. Also, high-throughput sequencing technology is the most effective and precise method to distinguish similar phenotypes. Hopefully, our research will be able to increase adequate knowledge and awareness of ARO disease for physicians, who might be the initial contact for patients, such as pediatricians, neurologists, hematologists, ophthalmologists, and geneticists. There are some limitations of our study. The minigene assay was an efficiency method to elucidate the biological effects of the novel splice site variant (c.982-1G > C). However, we did not perform studies on the working mechanism of this variant and more in-depth research is needed. Additionally, the predictive values of exaggerated leukocytosis as early signs need more clinical cohorts to illustrate.
This study expands the spectrum of CLCN7 mutations, reporting that the combination of c.1208G > A and c.982-1G > C mutant alleles resulted in a very early-onset, life-threatening phenotype. Furthermore, symptoms of leukocytosis, progressive neurological impairment, and vision or hearing loss should raise clinical suspicion of ARO. High-throughput sequencing technology is expected to be beneficial for precise diagnosis and improved prognosis.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
This study involving humans was reviewed and approved by the Institutional Review Board of Xinhua Hospital. Written informed consent to participate was obtained from the patient’s parents. Written informed consent was obtained from the legal guardian for the publication of any potentially identifiable images or data included in this article.
JQ: Conceptualization. YD and JQ: Data curation. XW: Formal analysis and funding acquisition. XW and YW: Investigation. XW and TX: writing—original draft. XW, YF, YD, and JQ: writing—review and editing. All authors contributed to the article and approved the submitted version.
This study was supported by the National Natural Science Foundation of China (81803256).
The authors thank the proband's family for participating in this study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2023.978879/full#supplementary-material.
1. Sobacchi C, Schulz A, Coxon FP, Villa A, Helfrich MH, et al. Osteopetrosis: genetics, treatment and new insights into osteoclast function. Nat Rev Endocrinol. (2013) 9(9):522–36. doi: 10.1038/nrendo.2013.137
2. Del Fattore A, Peruzzi B, Rucci N, Recchia I, Cappariello A, Longo M, et al. Clinical, genetic, and cellular analysis of 49 osteopetrotic patients: implications for diagnosis and treatment. J Med Genet. (2006) 43(4):315–25. doi: 10.1136/jmg.2005.036673
3. Frattini A, Pangrazio A, Susani L, Sobacchi C, Mirolo M, Abinun M, et al. Chloride channel ClCN7 mutations are responsible for severe recessive, dominant, and intermediate osteopetrosis. J Bone Miner Res. (2003) 18(10):1740–7. doi: 10.1359/jbmr.2003.18.10.1740
4. Even-Or E, Schiesel G, Simanovsky N, et al. Clinical presentation and analysis of genotype-phenotype correlations in patients with malignant infantile osteopetrosis. Bone. (2022) 154:116229. doi: 10.1016/j.bone.2021.116229
5. Hoyoux C, Dresse MF, Forget P, Piette C, Rausin L, Villa A, et al. Osteopetrosis mimicking juvenile myelomonocytic leukemia. Pediatr Int. (2014) 56(5):779–82. doi: 10.1111/ped.12342
6. Frattini A, Orchard PJ, Sobacchi C, Giliani S, Abinun M, Mattsson JP, et al. Defects in TCIRG1 subunit of the vacuolar proton pump are responsible for a subset of human autosomal recessive osteopetrosis. Nat Genet. (2000) 25(3):343–6. doi: 10.1038/77131
7. Rossler U, Hennig AF, Stelzer N, Bose S, Kopp J, Soe K, et al. Efficient generation of osteoclasts from human induced pluripotent stem cells and functional investigations of lethal CLCN7-related osteopetrosis. J Bone Miner Res. (2021) 36(8):1621–35. doi: 10.1002/jbmr.4322
8. Bonapace G, Moricca MT, Talarico V, Graziano F, Pensabene L, Miniero R, et al. Identification of two novel mutations on CLCN7 gene in a patient with malignant ostopetrosis. Ital J Pediatr. (2014) 40:90. doi: 10.1186/s13052-014-0090-6
9. Al IO, Oymak Y, Hazan F, Gursoy S, Ozturk T, Bag O, et al. A neonatal case of infantile malignant osteopetrosis presenting with thrombocytopenia and hypotonicity: a novel mutation in chloride voltage-gated channel 7 gene. Sisli Etfal Hastan Tip Bul. (2022) 56(1):161–65. doi: 10.14744/SEMB.2021.88964
10. Zeng B, Li R, Hu Y, Hu B, Zhao Q, Liu H, et al. A novel mutation and a known mutation in the CLCN7 gene associated with relatively stable infantile malignant osteopetrosis in a Chinese patient. Gene. (2016) 576(1 Pt 1):176–81. doi: 10.1016/j.gene.2015.10.021
11. Besbas N, Draaken M, Ludwig M, Deren O, Orhan D, Bilginer Y, et al. A novel CLCN7 mutation resulting in a most severe form of autosomal recessive osteopetrosis. Eur J Pediatr. (2009) 168(12):1449–54. doi: 10.1007/s00431-009-0945-9
12. Lin WD, Wang CH, Wu KH, Chou IC, Tsai FJ. Identification and characterization of mutations in the CLCN7 gene in a Taiwanese patient with infantile malignant osteopetrosis. Pediatr Neonatol. (2016) 57(2):155–7. doi: 10.1016/j.pedneo.2015.04.013
13. Hennig AF, Rossler U, Boiti F, von der Hagen M, Gossen M, Kornak U, et al. Generation of a human induced pluripotent stem cell line (BIHi002-A) from a patient with CLCN7-related infantile malignant autosomal recessive osteopetrosis. Stem Cell Res. (2019) 35:101367. doi: 10.1016/j.scr.2018.101367
14. Kantaputra PN, Thawanaphong S, Issarangporn W, Klangsinsirikul P, Ohazama A, Sharpe P, et al. Long-term survival in infantile malignant autosomal recessive osteopetrosis secondary to homozygous p.Arg526Gln mutation in CLCN7. Am J Med Genet A. (2012) 158A(4):909–16. doi: 10.1002/ajmg.a.35264
15. Pangrazio A, Frattini A, Valli R, Maserati E, Susani L, Vezzoni P, et al. A homozygous contiguous gene deletion in chromosome 16p13.3 leads to autosomal recessive osteopetrosis in a Jordanian patient. Calcif Tissue Int. (2012) 91(4):250–4. doi: 10.1007/s00223-012-9631-4
16. Hofstaetter JG, Atkins GJ, Kato H, Kogawa M, Blouin S, Misof BM, et al. A mild case of autosomal recessive osteopetrosis masquerading as the dominant form involving homozygous deep intronic variations in the CLCN7 gene. Calcif Tissue Int. (2022) 111(4):430–44. doi: 10.1007/s00223-022-00988-8
17. Liang H, Li N, Yao RE, Yu T, Ding L, Chen J, et al. Clinical and molecular characterization of five Chinese patients with autosomal recessive osteopetrosis. Mol Genet Genomic Med. (2021) 9(11):e1815. doi: 10.1002/mgg3.1815
18. Khan MA, Ullah A, Naeem M. Whole exome sequencing identified two novel homozygous missense variants in the same codon of CLCN7 underlying autosomal recessive infantile malignant osteopetrosis in a Pakistani family. Mol Biol Rep. (2018) 45(4):565–70. doi: 10.1007/s11033-018-4194-8
19. Liu J, Martin LJ, Dinu I, Field CJ, Dewey D, Martin JW, et al. Interaction of prenatal bisphenols, maternal nutrients, and toxic metal exposures on neurodevelopment of 2-year-olds in the APrON cohort. Environ Int. (2021) 155:106601. doi: 10.1016/j.envint.2021.106601
20. Dai Y, Liang S, Dong X, Zhao Y, Ren H, Guan Y, et al. Whole exome sequencing identified a novel DAG1 mutation in a patient with rare, mild and late age of onset muscular dystrophy-dystroglycanopathy. J Cell Mol Med. (2019) 23(2):811–18. doi: 10.1111/jcmm.13979
21. Zhang R, Chen S, Han P, Chen F, Kuang S, Meng Z, et al. Whole exome sequencing identified a homozygous novel variant in CEP290 gene causes Meckel syndrome. J Cell Mol Med. (2020) 24(2):1906–16. doi: 10.1111/jcmm.14887
22. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17(5):405–24. doi: 10.1038/gim.2015.30
23. Han P, Wei G, Cai K, Xiang X, Deng WP, Li YB, et al. Identification and functional characterization of mutations in LPL gene causing severe hypertriglyceridaemia and acute pancreatitis. J Cell Mol Med. (2020) 24(2):1286–99. doi: 10.1111/jcmm.14768
24. Zheng Y, Xu J, Liang S, Lin D, Banerjee S. Whole exome sequencing identified a novel heterozygous mutation in HMBS gene in a Chinese patient with acute intermittent porphyria with rare type of mild anemia. Front Genet. (2018) 9:129. doi: 10.3389/fgene.2018.00129
25. Strauss A, Furlan I, Steinmann S, Buchholz B, Kremens B, Rossig C, et al. Unmistakable morphology? Infantile malignant osteopetrosis resembling juvenile myelomonocytic leukemia in infants. J Pediatr. (2015) 167(2):486–8. doi: 10.1016/j.jpeds.2015.04.064
26. Schmidt S, Nakchbandi I, Ruppert R, Kawelke N, Hess MW, Pfaller K, et al. Kindlin-3-mediated signaling from multiple integrin classes is required for osteoclast-mediated bone resorption. J Cell Biol. (2011) 192(5):883–97. doi: 10.1083/jcb.201007141
27. Essa MF, Elbashir E, Alroqi F, Mohammed R, Alsultan A. Successful hematopoietic stem cell transplant in leukocyte adhesion deficiency type III presenting primarily as malignant infantile osteopetrosis. Clin Immunol. (2020) 213:108365. doi: 10.1016/j.clim.2020.108365
28. Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet. (2016) 99(4):877–85. doi: 10.1016/j.ajhg.2016.08.016
29. Riepe TV, Khan M, Roosing S, t Hoen PAC. Benchmarking deep learning splice prediction tools using functional splice assays. Hum Mutat. (2021) 42(7):799–810. doi: 10.1002/humu.24212
30. Tachikawa J, Takahashi Y, Miura M, Tanaka A. CLCN7-related neuropathic infantile osteopetrosis in siblings. Pediatr Int. (2021) 63(10):1251–52. doi: 10.1111/ped.14594
31. Beard CJ, Key L, Newburger PE, Ezekowitz RA, Arceci R, Miller B, et al. Neutrophil defect associated with malignant infantile osteopetrosis. J Lab Clin Med. (1986) 108(5):498–505. doi: 10.1056/NEJM199506153322402
32. Key LL J, Rodriguiz RM, Willi SM, Wright NM, Hatcher HC, Eyre DR, et al. Long-term treatment of osteopetrosis with recombinant human interferon gamma. N Engl J Med. (1995) 332(24):1594–9. doi: 10.1056/NEJM199506153322402
33. Malinin NL, Zhang L, Choi J, Ciocea A, Razorenova O, Ma YQ, et al. A point mutation in KINDLIN3 ablates activation of three integrin subfamilies in humans. Nat Med. (2009) 15(3):313–8. doi: 10.1038/nm.1917
34. Matsuo K, Ray N. Osteoclasts, mononuclear phagocytes, and c-Fos: new insight into osteoimmunology. Keio J Med. (2004) 53(2):78–84. doi: 10.2302/kjm.53.78
35. Di Zanni E, Palagano E, Lagostena L, Strina D, Rehman A, Abinun M, et al. Pathobiologic mechanisms of neurodegeneration in osteopetrosis derived from structural and functional analysis of 14 ClC-7 mutants. J Bone Miner Res. (2021) 36(3):531–45. doi: 10.1002/jbmr.4200
36. Norwood I, Szondi D, Ciocca M, Coudert A, Cohen-Solal M, Rucci N, et al. Transcriptomic and bioinformatic analysis of Clcn7-dependent autosomal dominant osteopetrosis type 2. Preclinical and clinical implications. Bone. (2021) 144:115828. doi: 10.1016/j.bone.2020.115828
37. Stark Z, Pangrazio A, McGillivray G, Fink AM. Association of severe autosomal recessive osteopetrosis and structural brain abnormalities: a case report and review of the literature. Eur J Med Genet. (2013) 56(1):36–8. doi: 10.1016/j.ejmg.2012.10.001
38. Bliznetz EA, Tverskaya SM, Zinchenko RA, Abrukova AV, Savaskina EN, Nikulin MV, et al. Genetic analysis of autosomal recessive osteopetrosis in Chuvashiya: the unique splice site mutation in TCIRG1 gene spread by the founder effect. Eur J Hum Genet. (2009) 17(5):664–72. doi: 10.1038/ejhg.2008.234
39. Pangrazio A, Pusch M, Caldana E, Frattini A, Lanino E, Tamhankar PM, et al. Molecular and clinical heterogeneity in CLCN7-dependent osteopetrosis: report of 20 novel mutations. Hum Mutat. (2010) 31(1):E1071–80. doi: 10.1002/humu.21167
Keywords: autosomal recessive osteopetrosis (ARO), chloride channel 7 (CLCN7), leukocytosis, neurological impairment, genotype–phenotype correlation
Citation: Wang X, Wang Y, Xu T, Fan Y, Ding Y and Qian J (2023) A novel compound heterozygous mutation of the CLCN7 gene is associated with autosomal recessive osteopetrosis. Front. Pediatr. 11:978879. doi: 10.3389/fped.2023.978879
Received: 26 June 2022; Accepted: 28 March 2023;
Published: 24 April 2023.
Edited by:
Sunita Bijarnia-Mahay, Sir Ganga Ram Hospital, IndiaReviewed by:
Amjad Khan, Université de Strasbourg, France© 2023 Wang, Wang, Xu, Fan, Ding and Qian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jihong Qian cWlhbmppaG9uZ0B4aW5odWFtZWQuY29tLmNu
Specialty Section: This article was submitted to Genetics of Common and Rare Diseases, a section of the journal Frontiers in Pediatrics
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.