 Zhi-Jun Liu
Zhi-Jun Liu Ya-Ling Wang†
Ya-Ling Wang† Yan Xu
Yan Xu- Department of Neurology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
Background: Ataxia-telangiectasia (A-T) is an autosomal recessive disorder with high clinical heterogeneity. A-T may present in complicated variable forms, including classic A-T and milder form of AT. Contrary to the classic A-T, the milder form does not present the cardinal features of A-T such as ataxia and telangiectasia. A few ATM mutations have been reported in variant A-T cases manifesting isolated generalized or segmental dystonia without any signs of classical A-T.
Methods: An A-T pedigree with predominant dystonia was collected. Genetic testing was performed by targeted panel of genes involved in movement disorders. The candidate variants were further confirmed by Sanger sequencing. We then reviewed previously published literatures of genetically confirmed A-T cases with predominant dystonia and summarized the clinical characteristics of dystonia-dominant A-T.
Results: Two novel ATM mutations, p.I2683T and p.S2860P, were identified in the family. The proband presented isolated segmental dystonia without any signs of ataxia and telangiectasias. We reviewed the literatures and found that the patients with dystonia-dominant A-T tend to have a later-onset and slower progression of the disease.
Conclusion: To our knowledge, this is the first report of A-T patient with predominant dystonia in China. Dystonia may appear as one of the predominant manifestations or initial symptom of A-T. Early ATM genetic testing should be considered for those patients with predominant dystonia, despite without accompanying ataxia or telangiectasia.
1. Introduction
Ataxia-telangiectasia (A-T) is a rare autosomal recessive disorder characterized by progressive cerebellar degeneration, telangiectasia, immunodeficiency, radiosensitivity, recurrent respiratory tract infections, and increased risk of cancer (1). Most people with A-T develop the disease in early childhood and die from malignancies or respiratory failure in the second or third decade of life (2).
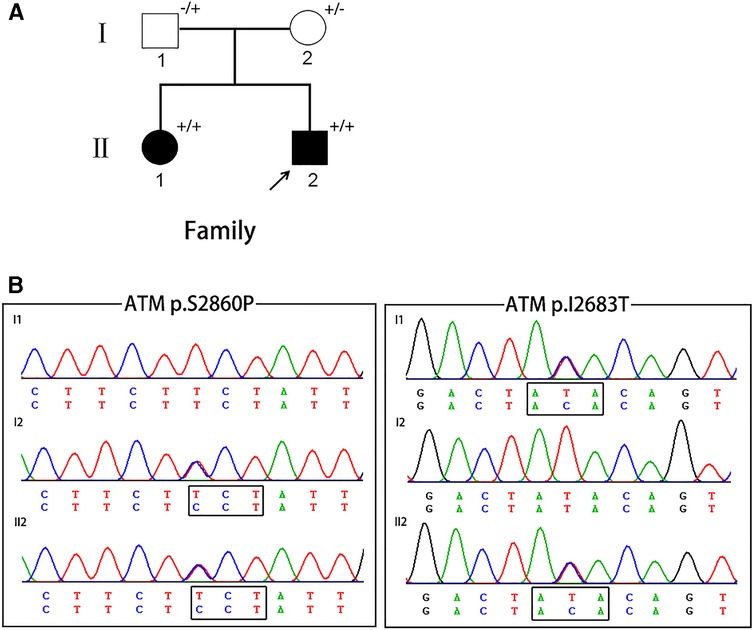
Figure 1. (A) The pedigree of A-T patients in present study. Squares indicate males; circles indicate females; filled symbols indicate affected individuals; arrows indicate the probands; “+/+” indicate two mutant alleles; “+/−” or “−/+” indicate mutation occurring in one of two alleles. (B) Chromatograms of the proband and the proband's parents.
A-T may present in complicated variable forms, including the classic A-T which is caused by truncating ataxia telangiectasia mutated (ATM) mutations in both alleles leading to total loss of ATM kinase activity and milder form of AT (also known as variant A-T) often associated with missense or leaky splice mutations (3). Patients with classic A-T usually develop an ataxic gait in early childhood and become wheelchair dependency by adolescence (4). Contrary to the classic A-T, the milder form does not present the cardinal features of A-T, such as ataxia, telangiectasia, and immunodeficiency. Other characteristics of movement disorders including chorea, myoclonic jerks, resting tremor, and dystonia are well recognized as the presenting manifestations of variant A-T (5). A systematic review of movement disorder phenomenology and systemic features in A-T patients have found that 89%(186/210) cases manifested dystonia and 18%(24/130) presented dystonia as their initial manifestations of A-T (6). Notably, a few ATM mutations have been reported in variant A-T cases manifesting as isolated generalized or segmental dystonia without any signs of classical A-T (7).
A-T is caused by mutations in the ATM gene which plays a critical role in pathway regulating DNA double-strand breaks (DSBs) repair. To date, more than 400 disease-related ATM mutations have been identified in patients with A-T. Among these ATM mutations, the majority (about 70%) are truncating changes resulting in premature translation termination (2).
In this study, we reported a Chinese A-T family with two novel compound heterozygote mutations in the ATM gene. To our knowledge, this is the first report of A-T patient with predominant dystonia in China. Our findings broaden the molecular and clinical spectrum of Chinese A-T patients.
2. Methods
2.1. Participants
The pedigree was collected from the Neurology Department of Wuhan Union hospital. DNA samples were obtained from all the participants, including the proband, his affected older sister and their parents. Magnetic resonance imaging (MRI) of brain and cervical, ultrasound of heart and abdominal, electroencephalogram (EEG), and blood biochemical analysis (detection of α-fetoprotein (AFP), immunoglobulin(Ig), and ceruloplasmin) were conducted. An additional 200 normal individuals without any neurological disorders were included as controls. Written informed consents were obtained from all participants. This study was approved by the ethics committee of Wuhan Union Hospital.
2.2. Targeted exome-sequencing
Genomic DNA was extracted from peripheral blood samples by DNA extraction Kit (Qiagen, Germany). A panel was designed to cover 101 genes associated with movement disorders (Supplementary Table S1). Deep sequencing was performed using Illumina Hiseq2000 system (GrandOmics Biosciences Co, China). Sequencing analysis was performed as we described previously (8). In order to screen for large deletions or duplications of Dopa-responsive dystonia (DRD) associated genes (GCH1, TH, and SGCE), the Multiplex ligation-dependent probe amplification assay (MLPA) analysis was performed by reference to the method previously reported (9).
2.3. Sanger sequencing
Sanger sequencing was used to validate the candidate variants after data analysis. Forward and reverse primers were designed to amplify the fragments covering the variant sites. The fragments were sequenced on ABI 3530xL DNA Sequencer. Co-segregation analysis was conducted through screening for the confirmed variants in the proband's older sister and parents.
3. Results
3.1. Genetic findings
No large deletions or duplications of GCH1, TH, and SGCE, were found in the proband. About 94.5% of the target bases were covered with at least 50X, and the mean depth of coverage for all target regions was 170. After filtering, two novel missense variants in ATM, c.T8048C (p.I2683T) and c.T8578C (p.S2860P), were identified in the proband (Figure 1). The two novel variants were compound heterozygous in the proband and his affected older sister, and were heterozygous in their unaffected parents. These two variants were absent from major public variant databases (1000G frequency, ESP6500, and ExAC frequency) and our 200 normal individuals. The two variants were predicted as harmful effects by the SIFT, PolyPhen-2, and MutationTaster prediction software. According to the ACMG standards and guidelines, these two variants were classified as likely pathogenic variants.
3.2. Clinical manifestations
The proband (II:2), a 15-year-old boy, was delivered by forceps with no birth complications and had a normal childhood development milestones. He started walking around one year of age and had normal speech. He presented at our clinic complaining of neck pain and stiffness since 9 years old, which progressively worsened over the period of 2–3 years. He developed mild head shake at age 12. Then he experienced episodes of involuntary turning of the head to the left and cervical hyperextension, postural hand tremor, and blepharospasm at age 13. On examination at age 15, he had severe dystonia with dominant craniocervical involvement. The jerky right torticollis, postural hand tremor, and choreiform facial movements were present. Eye movements were normal with normal saccades responses and ocular pursuit movements. He had normal muscle power and reflexes, and a slightly increased muscle tone in left upper limb. Sensory examination was unremarkable. There were no cerebellar signs. His cognitive function remains intact, with a Mini-Mental State Exam (MMSE) and Montreal Cognitive Assessment (MoCA) score of 28/30 and 25/30, respectively. Eye ophthalmic examination was unremarkable.
His older sister (II:1) was a 19-year-old girl and experienced similar symptoms since age 12 years. She noted her first symptom of mild involuntary head shake while taking a photo at the age of 12 years. As the disease progressed, she gradually developed jerky right-sided torticollis, postural hand tremor, oromandibular dystonia. She had recurrent episodes of transient global amnesia, which occurred 2–3 times per year. Her examination was further marked by severe cervical and oromandibular dystonia, slightly increased muscle tone in left upper limb, and postural hand tremor. The deep tendon reflexes were all absent, with flexor plantar responses. She showed normal saccades and ocular pursuit movements. Her cognitive function was unremarkable.
There was no known consanguinity in the family. The proband's grandmother had a gastric cancer and died at age 50. The detailed clinical results are summarized in Table 1. The serum AFP levels were significantly increased in the two patients, whereas serum levels of IgG and IgA was significantly decreased. Other laboratory tests including cholesterol, creatinine alkaline, lactate dehydrogenase (LDH), and ceruloplasmin were all normal. The brain and spinal cord MRI, ultrasound of heart and abdominal, and electroencephalogram (EEG) were unremarkable.
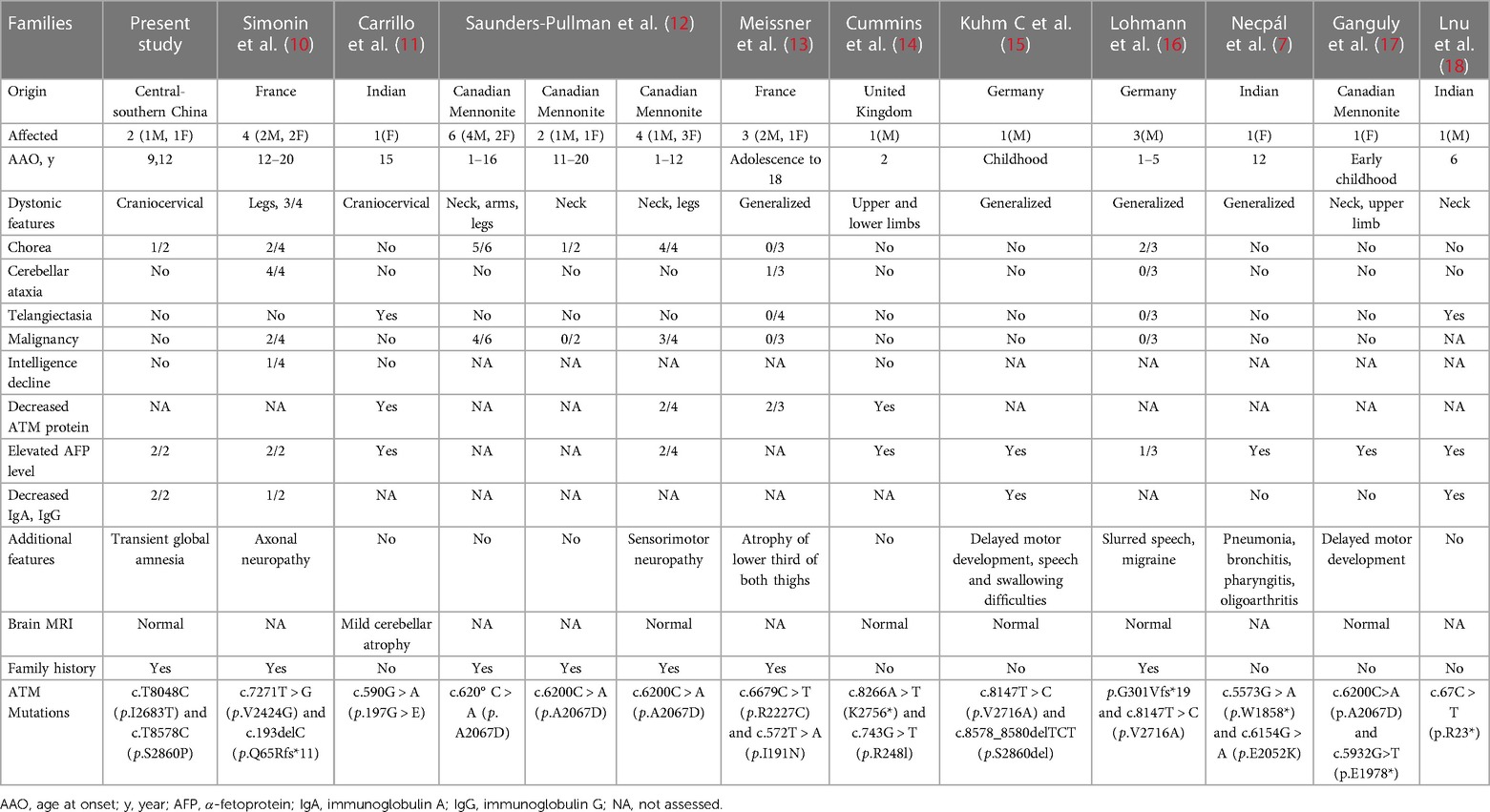
Table 1. Summary of clinical manifestations and findings of variant AT cases with predominant dystonia.
4. Discussion
In this study, we present a Chinese A-T family with two compound heterozygous mutations of ATM presenting with atypically craniocervical dystonia and also present a review of the literatures on the variant A-T cases with dystonia predominance. No signs of ocular motor apraxia, cerebellar ataxia, as well as telangiectasia were observed in the case.
Dystonia, occurring either in isolation or in combination with other clinical features, has been described in variant A-T patients. Dystonia affecting the neck, head, trunk and limbs, may be present as initial symptoms in about 18%(24/130) of A-T patients (6). The present case showed obviously isolated segmental dystonia without any signs of ataxia and telangiectasias. After reviewed previously published literatures of genetically confirmed A-T cases with predominant dystonia, we found that dystonia-dominant A-T cases may have a significantly different clinical picture compared to the typical A-T. In total, 13 pedigrees including 30 patients with ATM mutations were reviewed (7, 10–18) (Table 1). The median age of onset was 10.67 years, ranging from 1 to 20 years old. The group of dystonia-dominant A-T cases showed isolated dystonic features or accompanied by choreoathetosis, tremor and/or myoclonic jerks, but without involvement of ataxia and telangiectasias. Other phenotypes including peripheral neuropathy, delayed motor development, speech and swallowing difficulties, could also be seen in the group. A few patients also experienced recurrent pneumonia and increased risk of malignancy. Overall, the patients with dystonia-dominant A-T tend to have a later-onset and slower progression of the disease. However, the relationship between dystonic phenotype and particular ATM genotype in variant A-T is not well understood. As previously reported, mutations in exon 42 of the ATM gene may predispose to dystonia-dominant A-T, which is needed to be confirmed in further analysis (19). Notably, the transient global amnesia observed in the proband's old sister is a relatively rare symptom and the first description in patients with A-T. Further investigation and analysis of amnesia presentation in patients with A-T is required.
In line with previous study (1), significantly elevated AFP levels and decreased serum levels of immunoglobulin (IgG and IgA) were observed in this family. Moreover, previous studies revealed that elevated level of IgM was indicative of a distinct and more severe phenotype (20). IgM level was normal in the two patients, which suggests the mild phenotype observed in the present cases. Unfortunately, we were not able to obtain tissue to perform analysis of ATM protein expression with this family.
The neuropathological hallmark of A-T is cerebellar Purkinje cell loss, followed by cerebellar symptoms and degeneration (21). The exact pathogenesis of dystonia in A-T remains unclear. A review of pathological findings in 17 patients with A-T and movement disorders found that neuronal loss occurs in the basal ganglia and brain stem nuclei (22). Previous positron emission tomography (PET) study revealed reduced glucose metabolism in the cerebellum of A-T patients and increased metabolism in the globus pallidus which often results in decreased motor performance (23). Additional studies are required to assess the association of dystonia with cerebellum and basal ganglia degeneration in A-T patients.
In conclusion, our findings expand the clinical and molecular spectrum of dystonia-dominant A-T in China. Dystonia may appear as one of the predominant manifestations or initial symptom of A-T. Isolated dystonia without any signs of ataxia or telangiectasias may result in delay in the diagnosis of A-T. Early ATM genetic testing should be considered for those patients with predominant dystonia, despite without accompanying ataxia or telangiectasia.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
The studies involving human participants were reviewed and approved by UNION HOSPITAL. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
Z-JL performed the molecular genetic analysis and participated in its design and drafted the manuscript. Y-LW collected data from all participants and performed molecular genetic analysis. YX conceived of the study, and participated in its design and helped to draft the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the grants from the National Natural Science Foundation of China to Z-JL (82101504), and grants from the National Natural Science Foundation of China (81873734) to YX.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2023.975696/full#supplementary-material.
References
1. Rothblum-Oviatt C, Wright J, Lefton-Greif MA, Mcgrath-Morrow SA, Crawford TO, Lederman HM. Ataxia telangiectasia: a review. Orphanet J Rare Dis. (2016) 11:159. doi: 10.1186/s13023-016-0543-7
2. Micol R, Ben Slama L, Suarez F, Le Mignot L, Beaute J, Mahlaoui N, et al. Morbidity and mortality from ataxia-telangiectasia are associated with ATM genotype. J Allergy Clin Immunol. (2011) 128:382–9. e381. doi: 10.1016/j.jaci.2011.03.052
3. Taylor AM, Lam Z, Last JI, Byrd PJ. Ataxia telangiectasia: more variation at clinical and cellular levels. Clin Genet. (2015) 87:199–208. doi: 10.1111/cge.12453
4. Petley E, Yule A, Alexander S, Ojha S, Whitehouse WP. The natural history of ataxia-telangiectasia (A-T): a systematic review. PLoS One. (2022) 17:e0264177. doi: 10.1371/journal.pone.0264177
5. Schon K, Van Os NJH, Oscroft N, Baxendale H, Scoffings D, Ray J, et al. Genotype, extrapyramidal features, and severity of variant ataxia-telangiectasia. Ann Neurol. (2019) 85:170–80. doi: 10.1002/ana.25394
6. Levy A, Lang AE. Ataxia-telangiectasia: a review of movement disorders, clinical features, and genotype correlations. Mov Disord. (2018) 33:1238–47. doi: 10.1002/mds.27319
7. Necpal J, Zech M, Skorvanek M, Havrankova P, Fecikova A, Winkelmann J, et al. Ataxia telangiectasia gene mutation in isolated segmental dystonia without ataxia and telangiectasia. Mov Disord Clin Pract. (2018) 5:89–91. doi: 10.1002/mdc3.12564
8. Liu ZJ, Lin HX, Wei Q, Zhang QJ, Chen CX, Tao QQ, et al. Genetic Spectrum and variability in Chinese patients with amyotrophic lateral sclerosis. Aging Dis. (2019) 10:1199–206. doi: 10.14336/AD.2019.0215
9. Sun ZF, Zhang YH, Guo JF, Sun QY, Mei JP, Zhou HL, et al. Genetic diagnosis of two dopa-responsive dystonia families by exome sequencing. PLoS One. (2014) 9:e106388. doi: 10.1371/journal.pone.0106388
10. Simonin C, Devos D, Vuillaume I, De Martinville B, Sablonniere B, Destee A, et al. Attenuated presentation of ataxia-telangiectasia with familial cancer history. J Neurol. (2008) 255:1261–3. doi: 10.1007/s00415-008-0857-z
11. Carrillo F, Schneider SA, Taylor AM, Srinivasan V, Kapoor R, Bhatia KP. Prominent oromandibular dystonia and pharyngeal telangiectasia in atypical ataxia telangiectasia. Cerebellum. (2009) 8:22–7. doi: 10.1007/s12311-008-0055-7
12. Saunders-Pullman R, Raymond D, Stoessl AJ, Hobson D, Nakamura K, Pullman S, et al. Variant ataxia-telangiectasia presenting as primary-appearing dystonia in Canadian mennonites. Neurology. (2012) 78:649–57. doi: 10.1212/WNL.0b013e3182494d51
13. Meissner WG, Fernet M, Couturier J, Hall J, Lauge A, Henry P, et al. Isolated generalized dystonia in biallelic missense mutations of the ATM gene. Mov Disord. (2013) 28:1897–9. doi: 10.1002/mds.25487
14. Cummins G, Jawad T, Taylor M, Lynch T. Myoclonic head jerks and extensor axial dystonia in the variant form of ataxia telangiectasia. Parkinsonism Relat Disord. (2013) 19:1173–4. doi: 10.1016/j.parkreldis.2013.08.013
15. Kuhm C, Gallenmuller C, Dork T, Menzel M, Biskup S, Klopstock T. Novel ATM mutation in a German patient presenting as generalized dystonia without classical signs of ataxia-telangiectasia. J Neurol. (2015) 262:768–70. doi: 10.1007/s00415-015-7636-4
16. Lohmann E, Kruger S, Hauser AK, Hanagasi H, Guven G, Erginel-Unaltuna N, et al. Clinical variability in ataxia-telangiectasia. J Neurol. (2015) 262:1724–7. doi: 10.1007/s00415-015-7762-z
17. Ganguly J, Bernaola MT, Goobie S, Prasad A, Jog M. Myoclonus-Dystonia presentation of ATM gene mutation in a Canadian mennonite. Mov Disord Clin Pract. (2022) 9:264–7. doi: 10.1002/mdc3.13369
18. Lnu P, Sehgal V, Kapila S, Gulati N, Bhalla Sehgal L. Ataxia telangiectasia presenting as cervical dystonia. Cureus. (2022) 14:e30723. doi: 10.7759/cureus.30723
19. Charlesworth G, Mohire MD, Schneider SA, Stamelou M, Wood NW, Bhatia KP. Ataxia telangiectasia presenting as dopa-responsive cervical dystonia. Neurology. (2013) 81:1148–51. doi: 10.1212/WNL.0b013e3182a55fa2
20. Krauthammer A, Lahad A, Goldberg L, Sarouk I, Weiss B, Somech R, et al. Elevated IgM levels as a marker for a unique phenotype in patients with ataxia telangiectasia. BMC Pediatr. (2018) 18:185. doi: 10.1186/s12887-018-1156-1
21. Verhagen MM, Martin JJ, Van Deuren M, Ceuterick-De Groote C, Weemaes CM, Kremer BH, et al. Neuropathology in classical and variant ataxia-telangiectasia. Neuropathology. (2012) 32:234–44. doi: 10.1111/j.1440-1789.2011.01263.x
22. Koepp M, Schelosky L, Cordes I, Cordes M, Poewe W. Dystonia in ataxia telangiectasia: report of a case with putaminal lesions and decreased striatal [123I]iodobenzamide binding. Mov Disord. (1994) 9:455–9. doi: 10.1002/mds.870090414
Keywords: ataxia-telangiectasia, targeted exome-sequencing, ATM, dystonia, Chinese
Citation: Liu Z, Wang Y and Xu Y (2023) Two novel heterozygote mutations of ATM in a Chinese family with dystonia-dominant ataxia telangiectasia and literature review. Front. Pediatr. 11:975696. doi: 10.3389/fped.2023.975696
Received: 22 June 2022; Accepted: 27 February 2023;
Published: 15 March 2023.
Edited by:
Marta Corton, Health Research Institute Foundation Jimenez Diaz (IIS-FJD), SpainReviewed by:
Annarosa Soresina, Azienda Socio Sanitaria Territoriale of the Spedali Civili of Brescia, ItalyCorry Weemaes, Radboud University Medical Centre, Netherlands
© 2023 Liu, Wang and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yan Xu eHV5YW53eGZAMTI2LmNvbQ==
†These authors have contributed equally to this work
Specialty Section: This article was submitted to Genetics of Common and Rare Diseases, a section of the journal Frontiers in Pediatrics