 Yuhua Zheng
Yuhua Zheng Shikib Mostamand
Shikib Mostamand
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pediatr., 05 May 2023
Sec. Pediatric Gastroenterology, Hepatology and Nutrition
Volume 11 - 2023 | https://doi.org/10.3389/fped.2023.943649
This article is part of the Research TopicInsights in Pediatric Pancreatology 2022View all 12 articles
Exocrine pancreatic insufficiency (EPI) is a condition defined as pancreatic loss of exocrine function, including decreased digestive enzymes and bicarbonate secretion, which leads to maldigestion and malabsorption of nutrients. It is a common complication in many pancreatic disorders. If left undiagnosed, EPI can cause poor digestion of food, chronic diarrhea, severe malnutrition and related complications. Nutritional status and fat-soluble vitamins should be carefully assessed and monitored in patients with EPI. Early diagnosis of EPI is clinically important for appropriate nutritional support and initiating pancreatic enzyme replacement therapy (PERT) which could significantly improve patient outcomes. The evaluation of nutritional status and related unique management in children with EPI will be discussed in this review.
The pancreas has two essential functions: exocrine function to help break down food by producing digestive enzymes and endocrine function to regulate blood sugar by secreting hormones including insulin. Exocrine pancreatic insufficiency (EPI) is a condition defined as pancreatic loss of exocrine function, including decreased digestive enzymes and bicarbonate secretion, which leads to maldigestion and malabsorption of nutrients. In pediatrics, more common causes of EPI include cystic fibrosis, chronic pancreatitis, Shwachman-Diamond syndrome, Pearson syndrome, and Johanson-Blizzard syndrome. Pancreatic hypoplasia, pancreatic aplasia, Jeune syndrome, pancreatectomy and isolated pancreatic enzyme deficiencies are less common causes (1–3). EPI can also occur in systemic diseases such as diabetes, inflammatory bowel disease, celiac disease, Sjogren's syndrome, etc. Microvascular damage may cause fibrosis and atrophy of the pancreas in patients with diabetes. Transient decrease of fecal elastase-1 has been reported in patients with inflammatory bowel disease or celiac disease (2) (Table 1). Clinically significant EPI presenting with steatorrhea results after greater than 90% of pancreatic acini are permanently compromised (3). If left undiagnosed, EPI can cause maldigestion of food and result in steatorrhea, weight loss, and fat-soluble micronutrient malabsorption. It can also impact the quality of life due to persistent gastrointestinal symptoms (4). To recognize EPI early is clinically critical for providing appropriate nutritional support including initiating pancreatic enzyme replacement therapy (PERT). In this review, we will discuss the evaluation of nutritional status and the management of EPI in children.
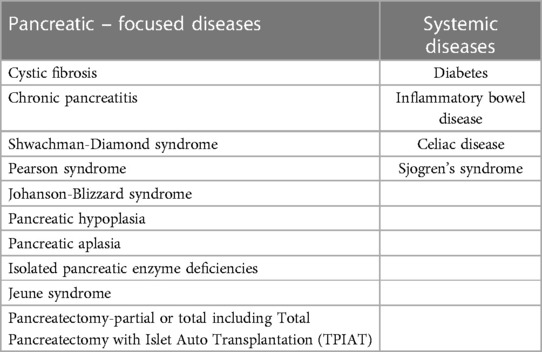
Table 1. Etiologies for EPI.
The adult pancreas delivers approximately 2.5 L of fluid secretion to the duodenum daily. During meals, the flow can rise from 0.2 ml/min to 4.0 ml/min. In children, the secretion volume and rate in response to secretin has a strong correlation with body surface area (BSA) (5, 6). Pancreatic fluid is isotonic, slightly alkaline (pH∼8.2) and protein rich, containing HCO3 – (up to 140 mEq/L), other electrolytes, and water. Acinar cells synthesize, store, and release digestive proenzymes which are proteolytic, lipolytic, or amylolytic, and contain nuclease. These proenzymes are synthesized in the endoplasmic reticulum. Apices of acinar cells contain zymogen granules which are vesicles containing proenzymes. The release of zymogen into the intercalated ducts is controlled through receptors and mediated by calcium. These proenzymes are activated in the intestine following trypsin activation by enterokinase found on the mucosal surface of small intestine. Trypsin in turn activates the remaining proenzymes, including trypsinogen, by enzymatic cleavage. Ductal cells secrete 1–2 L of neutral pH juice, mainly water and HCO3–, devoid of Cl–, through the regulated action of the cystic fibrosis transmembrane conductance regulator (CFTR), intracellular carbonic anhydrase, and other membrane channels. Pancreatic secretion is regulated by hormones and neural mediators including secretin, acetylcholine (Ach), cholecystokinin (CCK), substance P, vasoactive intestinal polypeptide (VIP), Peptide YY (PYY) and gastrin-releasing peptide (GRP) (6–8).
Malabsorption usually occurs when pancreatic secretion is decreased by 90% or more (9, 10). In patients with chronic pancreatitis, steatorrhea represents the most significant digestive malfunction in EPI. It usually develops years before overt malabsorption of protein and starch and is often more severe than azotorrhea (11). This can be due to several mechanisms: (1) lipase secretion diminishes earlier compared to amylase and proteases (12); (2) lipase destruction in the small intestinal lumen occurs more rapidly than other enzymes (13); (3) lipid digestion in humans is almost entirely through pancreatic lipase. Lipolytic enzymes of gastric origin contribute little to lipid digestion. By contrast, if pancreatic proteolytic activity was inhibited, protein digestion was maintained in animal studies. Similarly, brush border oligosaccharidases and salivary amylase accomplish around 80% of starch digestion in the absence of pancreatic amylase (14); (4) ileal nutrient exposure variation caused by pancreatic exocrine insufficiency may impair ileal inhibitory effect and subsequently decrease biliary secretion (15). A decrease in bile acids may further worsen lipid digestion and absorption (11, 16, 13). Steatorrhea is often accompanied by diarrhea and enhanced gastric emptying and small intestinal transit in patients with EPI can contribute to this. Accelerated gastric emptying after a high-fat liquid meal was observed in patients with CP and EPI (17). These disorders can cause inadequate mixing of food, bile acid, and digestive enzymes, as well as reduce contact time between chyme and the intestinal mucosa leading to diarrhea (18). On the other hand, another study in patients with CF demonstrated a prolonged small intestinal transit time. This may increase the contact time of chyme with the mucosa, however, is also prone to the risk of developing intestinal bacterial overgrowth and contributing to secondary malabsorption (19).
HCO3- secretion is mediated through CFTR which drives osmotic fluid secretion in the pancreatic duct. HCO3- is required to control the pH at the epithelial surface of the pancreatic duct as well as the expansion of secreted mucins. In patients with CFTR mutation, a lower luminal pH could cause the accumulation of hyperviscous mucus in the pancreatic duct which subsequently obstructs the lumen and results in microbial colonization and inflammation of the pancreas (20, 21). Additionally, a high level of HCO3- is considered essential to maintain the inactive state of the secreted digestive enzymes while still located in the ductal tree (21, 22).
EPI causes malnutrition through maldigestion and malabsorption. Malnutrition is an imbalance between nutritional intake, basal energy requirements, and expenditure. Malnutrition can be further characterized as undernutrition or overnutrition. In this manuscript, we focus on undernutrition, thus, malnutrition in this review specifically refers to undernutrition. Pediatric undernutrition is defined as the state in which there is a deficit in nutritional intake in relation to requirements (23, 24). This results in cumulative macro or micronutrient insufficiencies or deficiencies which adversely affect growth and development (24).
Patients with EPI often complain of abdominal discomfort, poor weight gain or weight loss, steatorrhea, undernutrition and vitamin deficiency symptoms (25). Steatorrhea is defined as bulky oily or greasy material in stools. Despite their often-late appearance in EPI, it remains important to evaluate the stool features even if these characteristics are neither specific nor sensitive for detection of steatorrhea (26).
Chronic malabsorption without intervention leads to malnutrition, more specifically undernutrition. One single center pediatric study identified malnutrition in 25% of children with chronic pancreatitis (CP). 17.3% of patients were detected with moderate malnutrition, and 0.96% with severe malnutrition. 152 patients were evaluated with CFA. The mean output among 38 malnourished patients was 6.69 g/100 g/day which was significantly higher than 2.27 g/100 g/day of 114 well-nourished children (27), indicating EPI is a major factor causing undernutrition. In patients with CF, protein malabsorption has been reported prior to newborn screen implementation, but is not common in the current era (28).
Other than maldigestion and malabsorption resulting from EPI, various factors may contribute to undernutrition owing to the nature of the underlying diseases. Patients with steatorrhea may self-limit fat intake due to diarrhea. Patients with chronic pancreatitis suffering from chronic pain may have decreased oral feeding. Those with additional complications such as diabetes, requiring frequent interventional procedures, having frequent pancreatitis attacks, or having a hypermetabolic state due to chronic inflammation such as cystic fibrosis leading to increased energy expenditure (29), may all potentially worsen or compound the undernutrition of patients with EPI.
Patients with steatorrhea are prone to develop fat-soluble vitamin deficiency. Because of the decrease of pancreatic enzymes, malabsorption and diarrhea, the deficiencies of water-soluble vitamins such as vitamin B12, folic acid, electrolytes such as calcium, magnesium, zinc, may also be detected (30). Vitamin B12 is bound to haptocorrin (HC) in the stomach; pancreatic proteases and pH changes degrades HC and transfer B12 to intrinsic factors in the duodenum for its absorption in the distal ileum. Despite the risks, the Vitamin B12 deficiency is still rare case in patients with EPI (31, 32). On top of changes in composition of fat and muscle tissue, undernutrition may result in homeostasis disruption on bone mass and mineral density (27).
In one adult study, 32 patients with chronic pancreatitis with exocrine pancreatic sufficiency (EPS) and 26 patients with EPI were measured for bone mineral density (BMD) and bone mineral content (BMC) using a dual-energy x-ray absorptiometry (DXA) method. The mean z-score of BMD was −1.16 ± 1.29 in EPS group and 1.32 ± 0.90 in EPI. For BMC, it was −1.02 ± 1.17 vs. −1.39 ± 0.987 respectively. In both groups mean 25 (OH)D and mean 1.25(OH)2D were below reference range. The author concluded that the patients with chronic pancreatitis and severe EPI, were at risk to develop significant bone loss (33, 34). Bone mineral content (BMC) and lean body mass (LBM) are more delicate indicators of undernutrition than BMI (36, 37). Computed Tomography (CT) and Magnetic Resonance Imaging (MRI) based methods to assess bone density and skeletal muscle mass are promising research fields to assess changes in body composition due to pancreas-related cachexia and osteopenia (37).
Vitamins are mainly acquired through diet as the human body cannot synthesize them on its own. There are nine water-soluble vitamins and four fat-soluble vitamins found in the diet. Fat-soluble vitamins are absorbed and stored in adipose tissues, liver, and muscle (38). Patients with EPI are prone to developing fat malabsorption, subsequently leading to insufficient absorption of fat-soluble vitamins (41).
Fat-soluble vitamins are divided further into subgroups according to their molecular structure. Vitamin A is classified into two forms: retinoids and carotenoid. Retinoids include retinol, retinal, and retinyl esters. Carotenoids, such as beta carotene, are plant sources (39, 41). Xerophthalmia and night blindness are associated with vitamin A deficiency (42, 43) along with decreased opacity of the cornea and dry conjunctiva. Vitamin A deficiency can damage the epithelial lining of the gastrointestinal, respiratory, and genitourinary tracts resulting from the dryness of epithelial cells, subsequently increasing risk of infection (38, 44).
The main forms of vitamin D are ergosterol (vitamin D2) and cholecalciferol (vitamin D3) (45). Vitamin D deficiency can lead to a disruption of bone mineralization, compromising the growth and strength of bones in children and affecting density of bones in adults, leading to rickets, osteomalacia, osteoporosis, and osteopenia (38). Vitamin D has also been discovered to have other vital functions besides bone health, such as, inhibiting cancer cell growth, assisting in infection control, and decreasing inflammation (46).
Tocopherols and the tocotrienols are vitamin E, each comprising of four subgroups (47). Vitamin E deficiency can lead to neurological problems, i.e., ataxia, dysarthria, lower limb areflexia, and peripheral neuropathy. In infants it is associated with hemolytic anemia.
Vitamin K is classed into the phylloquinones and menaquinones. The metabolism of each fat-soluble vitamin is complex, with lipid being essential for their absorption in the intestinal lumen and carrier proteins, or lipoproteins, required for transportation. The fat-soluble vitamins are conveyed to the adipose tissue, liver and muscle for usage and storage (48, 49, 38). Vitamin K deficiency can lead to coagulopathy, observed as subcutaneous bleeding with prolonged prothrombin time. Lack of vitamin K may also play a factor in poor bone density (38, 50).
Early diagnosis of EPI in children remains challenging. Exocrine pancreatic function is often assessed by direct and indirect pancreatic function tests (PFTs). Indirect PFTs include fecal elastase-1 (FE-1), 72-hour fecal fat test, and triglyceride breath test mixed with 13C; the latter is not available in the United States (1, 3, 53, 55). Steatorrhea is traditionally diagnosed with 72-hour fecal fat test which measures the coefficient of fat absorption (CFA). Feces are collected for 3 days, and daily dietary fat intake is recorded. Fecal fat is measured. For patients over 6 months old, when stool fat excretion surpasses 7 g for every100 g fat taken from diet per day, i.e., unabsorbed stool fat is >7% of dietary fat, the patient has steatorrhea. If patient is less than 6 months old, >15% is diagnostic (10). The 72-hour fecal fat test is laborious and unpleasant for personnel to handle and is not commonly performed by most centers. FE-1 is currently the most widely used method to screen for EPI. In general, A FE-1 > 200 ug/g is considered normal, 100–200 ug/g is indeterminate and may associate with possible decreased exocrine pancreatic function, and a FE-1 < 100 ug/g is abnormal and most likely indicating EPI. FE-1 usually detects EPI in the severe range and may miss mild to moderate cases (1). The FE-1 level may also be affected if measured during diarrhea caused by other etiologies (non-steatorrhea) due to dilutional effect or during an acute pancreatitis episode when fewer digestive enzymes might be produced. The sensitivity for FE-1 in meta-analysis in mild EPI was reported around 49% (56). The lack of sensitivity and specificity of FE-1 in mild to moderate EPI limits its use as a reliable tool for early EPI detection. Dreiling tube test is a traditional direct PFT which involves the fluoroscopic placement of an oroduodenal tube, administration of secretin or CCK then intermittent suction through the tube to collect duodenal pancreatic secretion (53). This modality is cumbersome, uncomfortable, and time-consuming and is not generally performed in pediatric centers. Endoscopic pancreatic function test (ePFT), which directly measures the pancreatic exocrine function, is considered the most accurate and feasible modality in diagnosing EPI. An esophagogastroduodenoscopy (EGD) will be performed under general anesthesia. A secretin (0.2 mcg/kg) or CCK (0.04 mcg/kg) is administrated intravenously to stimulate the pancreas secretion, followed by suction of pancreatic juice from duodenum at the different time interval. Pancreatic digestive enzymes including amylase, lipase, trypsin and chymotrypsin as well as bicarbonate concentration would be measured at the various time interval (55). Currently the protocol used by each center varies. The North American Society for Pediatric Gastroenterology, Hepatology and Nutrition (NASPGHAN) Pancreas Committee published a position paper that reviewed the advantages of ePFT, including it being technically safe and easy to perform, although it should be recognized that it is still an invasive procedure and carries risks of standard anesthesia and EGD. EPFT modality has been proved to be sensitive and specific in EPI diagnosis. The NASPGHAN pancreas committee proposed a standard ePFT protocol in children (56). Compared to the Dreiling tube pancreatic function test, ePFT is the preferred direct pancreatic function test given its technical superiority with improved efficacy. Despite its advantages as the most promising method for early EPI diagnosis, the main limitation for ePFT in children is the lack of age-specific normal reference ranges, often making it difficult to reliably interpret the results (56). Secretin-enhanced magnetic resonance cholangiopancreatography (sMRCP) is often utilized to investigate the pancreatic and biliary ductal system. Secretin-enhanced secretion of pancreatic fluid may contribute to non-invasive diagnosis for EPI. Calculation of pancreatic secretory function to secretin stimulation by MRI has been evaluated for adult patients and still being investigated in children (5).
Appropriate assessment of nutritional status is essential for early identification of those at risk for malnutrition with EPI. There is a current lack of a standard, validated, universal approach to the screening and evaluation of pediatric malnutrition. Routine assessment remains inconsistent both nationally and internationally. The American Society for Parenteral and Enteral Nutrition (ASPEN) and Academy of Nutrition and Dietetics Consensus guidelines recommend using several indicators in evaluating malnourishment: caloric and nutrient intake, calculation of energy and protein needs, and physical exam findings (i.e., muscle wasting, subcutaneous fat thickness, fluid accumulation, Tanner staging). Diagnostic parameters include anthropometric measures and their proxies such as weight gain velocity, mid-upper arm circumference, growth parameters, and handgrip strength (23). In the United States, it has become standard practice to use the World Health Organization Multicenter Growth Reference Study for children less than 2 years of age (https://www.who.int/childgrowth/standards/weight_for_height/en/), and the Centers for Disease Control and Prevention growth charts as references for children greater than 2 years of age (https://www.cdc.gov/growthcharts/clinical_charts.htm) (57).
Current recommendations support the use of “z-scores” in the evaluation of pediatric nutrition. Utilizing z-score criterion is especially useful when a single data point is available such as weight for height or length (0–2 years), BMI for age (2–18 years), length or height for age, or middle upper arm circumference (MUAC) (23). The z-score is a statistical representation of the number of standard deviations (SD) which is a value either above or below the mean in a normal Gaussian (bell) curve distribution (57). One standard deviation from the mean encompasses 68% of the data set under the bell curve, 2 standard deviations from the mean encompasses 95% of the data, and 3 standard deviations from the mean encompasses 99.7% of the data. Thus, the z-score implies variance from a normal mean value, and more specifically – the degree of variation. With z-scores, pediatric undernutrition can be classified by severity. Mild malnutrition is specified as a z-score −1 to −1.99, moderate malnutrition is specified as a z-score −2 to −2.99 and severe malnutrition is a z-score equal to or less than −3 (23) (Table 2).
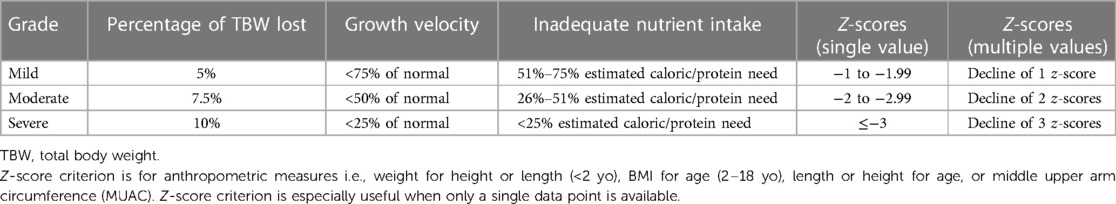
Table 2. Assessment of malnutrition.
When multiple data points are available for use, z-scores for deceleration in weight for length or height may be used, in which case, a decline of 1 z-score correlates with mild malnutrition, a decline of 2 z-scores correlates with moderate malnutrition and a decline of 3 z-scores correlates with severe malnutrition (23) (Table. 2).
The nutrition status of patients with EPI requires close monitoring and documentation at each visit.
Pancreatic enzyme replacement therapy (PERT) can often reverse the clinical course of malabsorption. PERT is required for EPI patients to support weight gain, to prevent fat-soluble vitamin and essential fatty acid deficiencies, to avoid malnutrition, as well as to improve symptoms of maldigestion and steatorrhea (58). 80%–90% of patients with cystic fibrosis need PERT to avert malnutrition (59). Current understanding of PERT in EPI is mainly based on expert consensus experience from cystic fibrosis.
For PERT, porcine pancreas is the usual source of pancreatic enzymes. It features high enzyme activity of all three classes including amylases, lipases, and proteases. Lipase is the main supplemental pancreatic enzyme yet is the least stable. It is highly sensitive to acid environment and proteolysis (60). Since 2010, several pancreatic enzyme replacement products were approved for the treatment of EPI by the Food and Drug Administration (FDA). All brands but VIOKACE are available in delayed-release forms comprising enteric-coated spheres, microspheres, microtablets or beads. Enteric-coated enzymes safeguard lipase from denaturation caused by gastric acid. These products include Creon, PANCREAZE, ZENPEP, PERTZYE (61). VIOKACE is the only one with an uncoated enzyme formulation. It has an immediate release thus it should be used together with an acid suppressant medication i.e., proton pump inhibitor (PPI) to maximize its activity (2, 62). In general, delayed-release (enteric-coated) capsules are recommended for pediatric patients. The safety and effectiveness of VIOKACE has not been established in pediatric patients. Due to greater degradation in the acidic environment, VIOKACE may be less efficacious than enteric-coated formulations (63, 64). VIOKACE used alone in pediatric patients may increase the risk of inadequate treatment of EPI. The efficacy of VIOKACE was established with concomitant PPI therapy in adult patients (63, 64). The long-term safety of PPI use in pediatric patients has not been established. RELiZORB is a digestive enzyme cartridge that connects directly to the feeding tube. The enzyme lipase is attached to small bead carriers and interacts with lipid as the formula passes through. It is useful for patients who are tube fed, however, it only contains lipase to breakdown fat in the formula (65).
Once ingested and passed through the duodenum, the acid-resistant enteric coating degrades in the intestine's higher pH, permitting the release of enzymes for digestion. PERT should be taken with meals and snacks (2, 66).
The goal of PERT is to optimize the nutritional status and alleviate the symptoms. The optimal dosage for an individual may differ based on body weight, severity of steatorrhea, and dietary fat intake. In pediatrics, PERT dosing recommendations for EPI is age dependent. For infants, 2,000–4,000 lipase units (LU) per 120 ml of infant formula or each breast feeding is recommended. For under 4 years of age, 1,000 LU/kg/meal and 500 LU/kg/snacks are recommended. For greater than 4 years of age, 500 LU/kg/meal and 250 LU/kg/snack are recommended (3) (Table 3). The ideal PERT therapy is established on its clinical effectiveness, the initial dose might be adjusted based on the clinical requirement and efficacy (2, 34, 67–69).
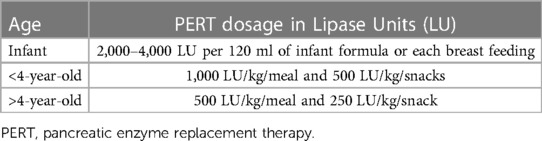
Table 3. PERT dosage by Age.
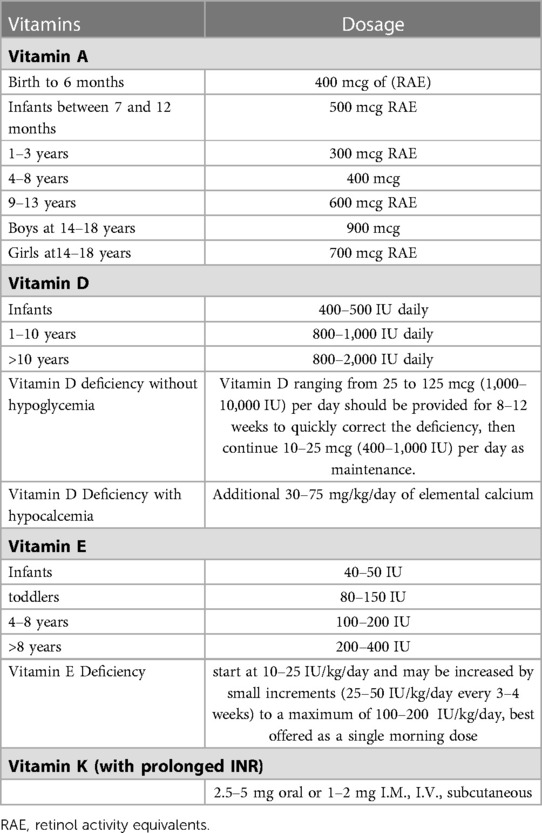
Table 4. Fat soluble vitamins supplement.
For children with difficulty swallowing capsules, delayed release forms may be opened, and enteric coated microspheres may be sprinkled on low pH food (applesauce, etc). Foods like milk has a pH greater than 7.3, should be avoided as the enteric coating may dissolve in higher pH, and the enzymes can be denatured by gastric acid and lose activity. It is recommended to avoid crushing or chewing or holding the pancreatic lipase in the mouth as this may cause local irritation (3, 70). As the dissolution rate and extent of each brand are unique, they are not deemed interchangeable (2). If a different brand of PERT is initiated, optimization of the new PERT dose should be considered.
PERT products are generally well tolerated. Over time, it has shown an acceptable safety and tolerability profile. Headache, dizziness, abdominal pain, gassiness, and diarrhea are commonly observed adverse effects (71). In cystic fibrosis patients, fibrosing colonopathy has also been described with higher doses (72). According to 1995 consensus conference of the U.S. Cystic Fibrosis Foundation on the use of PERT, it is recommended that the daily dose of pancreatic enzymes should not exceed 2,500 LU per kilogram per meal and 10,000 LU per kilogram per day. Higher doses should be used with vigilance and only if able to clinically demonstrate significant improvement of malabsorption (2, 72, 73).
Regardless of the causes of EPI, a suboptimal response to standard PERT dosage should lead clinicians to investigate adherence to therapy first. If adherence is satisfactory, a small increments of PERT dosage change is recommended. Acid suppressive therapy (i.e., PPI) to reduce acid denaturation of enzymes can be initiated (74, 75). It has been suggested that combining a proton pump inhibitor (PPI) in cystic fibrosis patients who have refractory steatorrhea not responding well to PERT will aid efficacy (34, 67–69). However, in a retrospective cohort of pediatric patients with cystic fibrosis treated with PERT jointly with PPIs, there was no statistically significant improvement (76). In an adult study, up to 40% of patients with EPI secondary to chronic pancreatitis have concomitant intestinal bacterial overgrowth (77). Alternative etiologies of malabsorption should be evaluated in cases of less ideal response to the treatment (2, 74, 75).
Children with EPI are prone to develop fat-soluble vitamin deficiency. The Cystic Fibrosis Foundation (CFF) recommends regular screening for deficiencies in fat-soluble vitamins at the time of diagnosis and then annually and after any dose change (78) And children with CP should have fat-soluble–vitamin levels measured every 6–12 months (79, 80). If patients are supplemented with vitamins, levels should be monitored 3 months after dose adjustment (80). Fat-soluble vitamins deficiency is secondary to fat malabsorption, improvement is expected with optimized PERT supplement. Fat-soluble vitamins should be supplemented for deficiencies accordingly.
The predominant circulating vitamin A is in the form of retinol. Serum retinol levels are not useful in assessing vitamin A body stores. They reflect vitamin A storage in the liver when they are either depleted (less than 0.07 µmol/g liver) or exceedingly high (greater than 1.05 µmol/g liver) (51). In the middle of these levels, serum retinol is physiologically well controlled and kept at a homeostatic range. Thus, its level is not correlated with vitamin A deficiency and may not correlate in response to vitamin A supplementation. The serum retinol is useful when measured in a population and provides valuable information on the vitamin A status of a population. The serum retinol defines whether vitamin A deficiency is a public health problem in that population (56, 85). Vitamin A is bound to retinol-binding protein (RBP) for transportation. RBP is produced in liver. The molar ratio of retinol to RBP can be assessed to guide if vitamin A supplementation is necessary in patients with malnutrition or liver disease. A ratio of <0.8 suggests true vitamin A deficiency and requirement of vitamin A supplementation (81, 78).
Vitamin A is found in fruits, vegetables, eggs, milk, meat, and seafood. The daily Recommended Dietary Allowance (RDA) for vitamin A in children is age dependent (Table 4). According to National Institutes of Health (NIH): During birth to 6 months, 400 mcg of retinol activity equivalents (RAE) is recommended, Infants between 7 and 12 months require 500 mcg RAE; Children at 1–3 years require 300 mcg RAE; Children at 4–8 years require 400 mcg RAE; Children at 9–13 years 600 mcg RAE; teenage boys at 14–18 years require 900 mcg RAE; and teenage girls at14–18 years require 700 mcg RAE. This intake level is easy to reach if plenty of whole foods are consumed. However, to prevent toxicity, it is important not to exceed the 3,000 mcg per day (83). There are two supplements form of vitamin A are available: provitamin A as carotenoids and pre-formed vitamin A as retinol or retinyl ester. If a product contains both, the amount of pre-formed vitamin A is used to determine if it is safe. It is important to note that vitamin A may also be an ingredient in some topical products, such as serums, creams, and lotions (83). Toxicity of hypervitaminosis A secondary to the supplement is rare, it may involve multiple organ system including the bone, nervous system, kidney and liver (84). Hypercalcemia due to vitamin A toxicity was reported in patients with CF (85).
Vitamin D level can be classified as severe deficiency (<5 ng/ml), deficiency (5–15 ng/ml), insufficiency (15–20 ng/ml), and sufficiency (20–100 ng/ml) (86). In children, 20 ng/ml for 25(OH)-D levels is still considered sufficiency (87, 88), however, a higher cutoff of 32–100 ng/ml is suggested in adults (88). The US Cystic Fibrosis Foundation recommends levels >30 ng (89).
Vitamin D is naturally present in some foods, fatty fish and fish liver oils are some the best sources (90). It is also produced endogenously when skin is exposed to ultraviolet (UV). In foods and dietary supplements, D2 (ergocalciferol) and D3 (cholecalciferol) are the two main forms, which differ only in their sidechain (90, 91). UV light converts cutaneous 7-dehydrocholesterol to previtamin D3, which subsequently transform into vitamin D3 (90, 92). Pharmacologic doses of vitamin D ranging from 25 to 125 mcg (1,000–10,000 IU) per day should be provided for 8–12 weeks to quickly correct the deficiency, and once corrected, then 10–25 mcg (400–1,000 IU) per day should be continued as maintenance. Patients with hypocalcemia may need calcium supplementation, in which case, 30–75 mg/kg/day of elemental calcium should be offered. It is recommended to start at a higher dose then wean down to the lower range (Table 4). Vitamin D level should be monitored during the therapy (86). Toxicity of hypervitaminosis D is also rare which may be related to excessive long-term vitamin D intake. Clinically characterized by symptoms associated with severe hypercalcemia (93).
Alpha and gamma tocopherol levels are monitored in laboratory. Alpha Vitamin E reflects vitamin E mainly from supplement and gamma Vitamin E from food intake primarily from plant.
For vitamin E deficiency, oral vitamin E is available in standard forms of tocopherol, tocopherol acetate, tocopherol succinate or tocopherol nicotinate. The dose is recommended to start at 10–25 IU/kg/day and may be increased by small increments (25–50 IU/kg/day every 3–4 weeks) to a maximum of 100–200 IU/kg/day (Table 4). Since bile flow is maximal with breakfast in the morning, vitamin E is best offered as a single morning dose (81, 97).
Serum vitamin K level is not very useful in reflecting the deficiency status, as it only indicates the vitamin K intake over past 24 h (98). Protein induced by vitamin K absence or antagonism (PIVKA) and des-gamma-carboxy-prothrombin (PIVKA-II) are functionally defective coagulation factors in vitamin K deficiency status, PIVKA-II and undercarboxylated osteocalcin (uc-OC) are sensitive markers to reflect vitamin K deficiency but are not available at clinical settings (99–101). In clinical practice, prothrombin time/International Normalized Ratio (PT/INR) is usually used to reflect vitamin K deficiency (99). However, INR prolongs when prothrombin level is below 50% of normal, thus it does not identify early vitamin K deficiency (95). PT/INR measurement is indirect, less sensitive but more readily available in clinical settings.
Phylloquinone is the main dietary form of vitamin K coming primarily from green leafy vegetables (100, 101). Menaquinones are mainly of bacterial origin from various animal-based and fermented foods (100, 102, 103).
If prolonged INR is considered secondary to vitamin K deficiency, 2.5–5 mg oral or 1–2 mg I.M., I.V., subcutaneous (SC) vitamin K1 could be given as a single dose (104) (Table 4). The PT/INR could be normalized as soon as within 30 min after intake (105). Cystic fibrosis foundation recommended for patients with EPI, oral 0.3–0.5 mg/day high doses vitamin K1 may be administered until PERT supplement is optimized (106). If oral dosing is ineffective, alternative route of vitamin K1, i.e., IM/IV/SC should be considered (107).
Research data in studying water-soluble vitamin deficiencies in pediatric patients with EPI is limited. Vitamin C and vitamin B12 level could be low in adults patients with CP (80, 108, 109).
Patient with severe phenotype of CF often has EPI, who may experience iron deficiency due to GI tract or sputum loss (110). Patients with CF have abnormal transport of sodium and chloride in the sweat glands. Sodium chloride deficits can be problematic in infant with CF (99). Selenium levels are low in patients with CP or CF (113, 114). In patients with inadequately treated EPI, zinc deficiency can occur due to steatorrhea (113). Calcium requirements should be optimized in patients with EPI which is important for bone health along with Vitamin D and Vitamin K (114). Specific dosage recommendations for water-soluble vitamin supplementation do not exist in CF (99). Unless suspecting deficiencies, routine screening of water-soluble vitamins, trace elements or minerals are not recommended in pediatric patients with chronic pancreatitis (80).
Essential fatty acid deficiency (EFAD) can be observed in patients with CF (99). Essential fatty acids include polyunsaturated fatty acids which can be metabolized to alpha-linolenic acid (n-3) and linoleic (n-6). N-3 fatty acid is metabolized to docosohexaenoic acid (DHA) and N-6 fatty acid is metabolized to arachidonic acid (AA). The benefits of supplementation of antioxidants or DHA were observed in some studies; however, they were not consistent to recommend routine supplements yet (99, 116).
A balanced diet with carbohydrates, protein, fat, vegetables and fruits should be encouraged. The American Heart Association recommends that 30%–35% of calories are derived from fat for children 2–3 years of age and 25%–35% for 4–18 years of age. The majority of fats should come from polyunsaturated and monounsaturated fatty acid sources like fish, nuts and vegetable oils (117). In patients with a hypermetabolic state due to chronic inflammation (i.e., cystic fibrosis), it is important to achieve the recommended high-energy level intake at 110%–200% of the estimated average requirement (EAR) given increased energy expenditure (79, 118). It is also important to encourage children to remain well hydrated and refrain from consuming alcohol or tobacco products which are associated with CP and subsequent EPI (118). The mechanisms underlying malnutrition in patients with EPI can be complex. Like patients with CF, factors such as higher energy demands, greater energy losses, decreased nutrient intake, and declining lung function all contribute to poor nutrition status and necessitate special attention. Ideally, a multidisciplinary team including registered dietitians, pharmacists, registered nurses, clinicians and social workers can be of great help to manage individual nutritional and caloric requirements.
Other than maldigestion and malabsorption secondary to EPI, various factors may contribute to undernutrition owing to the nature of the underlying diseases. Specific measures for the underlying disease should be considered. For patients with steatorrhea who may self-limit fat intake due to diarrhea, it is important to adjust PERT dosage and encourage a balanced diet. Optimizing pain control for patients with chronic pancreatitis and chronic pain is also important.
Exocrine pancreatic insufficiency (EPI) is a common condition in patients with pancreas disorders which leads to maldigestion and malabsorption of nutrients. Early diagnosis of EPI is clinically important for appropriate nutritional support and initiation of PERT. The nutritional status of patients with EPI should be assessed carefully and accurately. The goal of PERT is to optimize the nutritional status and alleviate symptoms. Fat-soluble vitamins deficiency is secondary to fat malabsorption, improvement is expected with optimized PERT supplement. Fat-soluble vitamins should be supplemented for deficiencies accordingly. Additional support may be needed to improve patient outcomes.
All authors made substantial contributions to the work, drafted, or revised the manuscript and gave final approval of the version to be published. All authors agreed to be accountable for all aspects of the work. YZ: prepared initial manuscript, SM contributed to the sections of the manuscript. Both reviewed, edited and extended the draft. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Taylor CJ, Chen K, Horvath K, Hughes D, Lowe ME, Mehta D, et al. Espghan and naspghan report on the assessment of exocrine pancreatic function and pancreatitis in children. J Pediatr Gastroenterol Nutr. (2015) 61(1):144–53. doi: 10.1097/MPG.0000000000000830
2. Capurso G, Traini M, Piciucchi M, Signoretti M, Arcidiacono PG, et al. Exocrine pancreatic insufficiency: prevalence, diagnosis, and management. Clin Exp Gastroenterol. (2019) 12:129–39. doi: 10.2147/CEG.S168266
3. Uc A, Fishman DS. Pancreatic disorders. Pediatr Clin North Am. (2017) 64(3):685–706. doi: 10.1016/j.pcl.2017.01.010
4. Nikfarjam M, Wilson JS, Smith RC, Australasian Pancreatic Club Pancreatic Enzyme Replacement Therapy Guidelines Working Group. Diagnosis and management of pancreatic exocrine insufficiency. Med J Aust. (2017) 207:161–5. doi: 10.5694/mja16.00851
5. Trout AT, Serai SD, Fei L, Sun Q, Abu-El-Haija M. Prospective assessment of normal pancreatic secretory function measured by MRI in a cohort of healthy children. Am J Gastroenterol. (2018) 113(9):1385. doi: 10.1038/s41395-018-0190-9
6. Pandol SJ. Normal pancreatic function. The pancreapedia: exocrine pancreas knowledge base. Pancreas. (2015) V1:1–13. doi: 10.3998/panc.2015.17
7. Fisher WE, Andersen DK, Windsor JA, Saluja AK, Brunicardi FC. Schwartz's Principles of Surgery. Pancreas. (2012). 1–26. Available at: https://accesssurgery.mhmedical.com/content.aspx?bookId=980§ionId=59610875#1117750488.
8. Foglio E, Eisses J. Normal anatomy, development,and physiology. [book auth.] christine waasdorp hurtado. NASPGHAN eBook. (2016) 1:521–5. Available at: https://naspghan.org/board-review-book/.
9. DiMagno EP, Go VL, Summerskill HJ. Intraluminal and postabsorptive effects of amino acids on pancreatic enzyme secretion. J Lab Clin Med. (1973) 82(2):241–8. PMID: 4721379
10. DiMagno EP, Go VL, Summerskill WH. Relations between pancreatic enzyme ouputs and malabsorption in severe pancreatic insufficiency. N Engl J Med. (1973) 288(16):813–5. doi: 10.1056/NEJM197304192881603
11. Keller J, Layer P. Human pancreatic exocrine response to nutrients in health and disease. Gut. (2005) 54(Suppl):1–28. doi: 10.1136/gut.2005.065946
13. Layer P, Hotz J, Goebell H. Stimulatory effect of hypercalcemia on pancreatic secretion is prevented by pretreatment with cholecystokinin and cholinergic agonists. Pancreas. (1986) 1:478–82. doi: 10.1097/00006676-198611000-00002
14. Moreau H, Laugier R, Gargouri Y, Ferrato F, Verger R, et al. Human preduodenal lipase is entirely of gastric fundic origin. Gastroenterology. (1988) 95:1221–6. doi: 10.1016/0016-5085(88)90354-X
15. Layer P, Peschel S, Schlesinger T, Goebell H, et al. Human pancreatic secretion and intestinal motility: effects of ileal nutrient perfusion. Am J Physiol. (1990) 258:G196–20. doi: 10.1152/ajpgi.1990.258.2.G196
16. DiMagno EP, Malagelada JR, Go VL. Relationship between alcoholism and pancreatic insufficiency. Ann N Y Acad Sci. (1975) 252:200–7. doi: 10.1111/j.1749-6632.1975.tb19157.x
17. Long WB, Weiss JB. Rapid gastric empiying of fatty meals in panerealic insufficiency. Gaslroenterotogy. (1974) 67:920–5. doi: 10.1016/S0016-5085(19)32744-1
18. Bruno MJ, Haverkort EB, Tytgat GN, van Leeuwen DJ, et al. Maldigestion associated with exocrine pancreatic insufficiency: implications of gastrointestinal physiology and properties of enzyme preparations for a cause-related and patient-tailored treatment. Am J Gastroenterol. (1995) 90(9):1383–93. PMID:7661155
19. Bali A, Stableforth DE, Asquith P. Prolonged small-inlestina! transit 93. Time in cystic fibrosis. Br Med J. (1983) 287:1011–3. doi: 10.1136/bmj.287.6398.1011
20. Kim D, Liao J, Hanrahan JW. Large pH oscillations promote host defense against human airways infection. J Exp Med. (2021) e20201831. doi: 10.1084/jem.20201831
21. Lee MG, Ohana E, Park HW, Yang D, Muallem S, et al. Molecular mechanism of pancreatic and salivary gland fluid and HCO3-secretion. Physiol. Rev. (2012) 92:39–74. doi: 10.1152/physrev.00011.2011
22. Angyal D, Bijvelds MJC, Bruno MJ, Peppelenbosch MP, de Jonge HR. Bicarbonate transport in cystic fibrosis and pancreatitis. Cells. (2021) 11:54–71. doi: 10.3390/cells11010054
23. Becker P, Carney LN, Corkins MR, Monczka J, Smith E, Smith SE, et al. Consensus statement of the academy of nutrition and dietetics/American society for parenteral and enteral nutrition: indicators recommended for the identification and documentation of pediatric malnutrition. Nutr Clin Pract. (2015) 30(1):147–61. doi: 10.1177/0884533614557642
24. Mehta NM, Corkins MR, Lyman B, Malone A, Goday PS, Carney LN, et al. Defining pediatric malnutrition: a paradigm shift toward etiology-related definitions. J Parenter Enteral Nutr. (2013) 37(4):460–81. doi: 10.1177/0148607113479972
25. Andersen DK. Mechanisms and emerging treatments of the metabolic complications of chronic pancreatitis. Pancreas. (2007) 35(1):1–15. doi: 10.1097/mpa.0b013e31805d01b0
26. Dumasy V, Delhaye M, Cotton F, Deviere J. Fat malabsorption screening in chronic pancreatitis. Am J Gastroenterol. (2004) 99(7):1350–4. doi: 10.1111/j.1572-0241.2004.30661.x
27. Kolodziejczyk E, Wejnarska K, Dadalski M, Kierkus J, Ryzko J, Oracz G. The nutritional status and factors contributing to malnutrition in children with chronic pancreatitis. Pancreatology. (2014) 14(4):275–9. doi: 10.1016/j.pan.2014.04.030
28. Wolfe AG, Gilley SP, Waldrop SW, Olson C, Harding E, Widmer K, et al. Case report: cystic fibrosis with kwashiorkor: a rare presentation in the era of universal newborn screening. Front Pediatr. (2022) 10:1083155. doi: 10.3389/fped.2022.1083155
29. Vaisman N, Pencharz PB, Corey M, Canny GJ, Hahn E. Energy expenditure of patients with cystic fibrosis. J Pediatr. (1987) 111:496–500. doi: 10.1016/S0022-3476(87)80107-5
30. Meier RF, Beglinger C. Nutrition in pancreatic diseases. Best Pract Res Clin Gastroenterol. (2006) 20:507–29. doi: 10.1016/j.bpg.2006.01.004
31. Gueant JL, Champigneulle B, Gaucher P, Nicolas JP. Malabsorption of vitamin B 12 in pancreatic insufficiency of the adule and of the chid. Pancreas. (1990) 5:559–67. doi: 10.1097/00006676-199009000-00011
32. Gueant JL, Alpers DH. Vitmin B12 absorption and malabsoprtion. Vitam Horm. (2022) 119:241–74. doi: 10.1016/bs.vh.2022.01.016
33. Haaber AB, Rosenfalck AM, Hansen B, Hilsted J, Larsen S. Bone mineral metabolism, bone mineral density, and body composition in patients with chronic pancreatitis and pancreatic exocrine insufficiency (article). Int J Pancreatol. (2000) 27:21–7. doi: 10.1385/IJGC:27:1:21
34. Keller J, Layer P. Diagnosis of pancreatic exocrine insufficiency in chronic pancreatitis. Pancreapedia. (2015) 1:1–7. doi: 10.3998/panc.2015.37
35. Turck D, Braegger CP, Colombo C, Declercq D, Morton A, Pancheva R, et al. ESPEN-ESPGHAN-ECFS guidelines on nutrition care for infants, children, and adults with cystic fibrosis. Clinical. (2016) 35(3):557–77. doi: 10.1016/j.clnu.2016.03.004
36. Alicandro G, Bisogno A, Battezzati A, Bianchi ML, Corti F, Colombo C. Recurrent pulmonary exacerbations are associated with low fat free mass and low bone mineral density in young adults with cystic fibrosis. J Cyst Fibros. (2014) 13:328–34. doi: 10.1016/j.jcf.2013.11.001
37. Bieliuniene E, Frøkjær JB, Pockevicius A, Kemesiene J, Lukosevicius S, Basevicius A, et al. Computed Tomography (CT) and Magnetic Resonance Imaging (MRI) based methods to assess bone density and skeletal muscle mass are promising research fields to assess changes in body composition due to pancreas-related cachexia, sarcopenia, and os. Medicina. (2019) 55(10):649. doi: 10.3390/medicina55100649
38. Stevens SL. Fat-soluble vitamins. Nurs Clin North Am. (2021) 56(1):33–45. doi: 10.1016/j.cnur.2020.10.003
39. Aurélie Goncalves, Stéphanie Roi, Marion Nowicki, Amélie Dhaussy, Alain Huertas, Marie-Josèphe Amiot, et al. Fat-soluble vitamin intestinal absorption: absorption sites in the intestine and interactions for absorption. Food Chem. (2015) 172:155–60. doi: 10.1016/j.foodchem.2014.09.021
40. Thomas DR. Vitamins in aging, health, and longevity. Clin Interv Aging. (2006) 1(1):81–91. doi: 10.2147/ciia.2006.1.1.81
41. Riccioni G, D’Orazio N, Menna V, De Lorenzo A, et al. Fat soluble vitamins and immune system: a overview. Eur J Inflamm. (2003) 1(2):59–64. doi: 10.1177/1721727X0300100202
42. Csapo J, Cs A, Prokisch J. The role of vitamins in the diet of the elderly I. Fatsoluble vitamins. Acta Universitatis Sapientiae. Alimentaria. (2017) 10:127–45. doi: 10.1515/ausal-2017-0009
43. Tanumihardjo SA, Russell RM, Stephensen CB, Gannon BM, Craft NE, Haskell MJ, et al. Biomarkers of nutrition for development (BOND)—vitamin A review. J Nutr. (2016) 146(9):1816S–48S. doi: 10.3945/jn.115.229708
44. Stephenson CB. Vitamin A, infection, and immune function. Annu Rev Nutr. (2001) 21:167–02. doi: 10.1146/annurev.nutr.21.1.167
45. Borel P, Desmarchelier C. Bioavailability of fat-soluble vitamins and phytochemicals in humans: effects of genetic variation. Annu Rev Nutr. (2018) 38:69–96. doi: 10.1146/annurev-nutr-082117-051628
47. Lee GY, Han SN. The role of vitamin E in immunity. Nutrients. (2018) 10(11):161. doi: 10.3390/nu10111614
48. Albahrani AA, Greaves RF. Fat-soluble vitamins: clinical indications and current challenges for chromatographic measurement. Clin Biochem Rev. (2016) 37(1):27–47. 27057076
49. Kono N, Arai H. Intracellular transport of fat-soluble vitamins A and E. Traffic. (2015) 16(1):19–34. doi: 10.1111/tra.12231
50. Koletzko B, Decsi T, Sawatzki G. Vitamin E status of low birthweight infants fed formula enriched with long-chain polyunsaturated fatty acids. Int J Vitam Nutr Res. (1995) 65(2):101–4.7591527
51. Gabriele C, Traini M, Piciucchi M, Signoretti M, Arcidiacono PG. Exocrine pancreatic insufficiency: prevalence, diagnosis and management. Clin Exp Gastroenterol. (2019) 12:129–39. doi: 10.2147/CEG.S168266
52. Vanga RR, Tansel A, Sidiq S, El-Serag HB, Othman MO Diagnostic performance of measurement of fecal elastase-1 in detection of exocrine pancreatic insufficiency: systematic review and meta-analysis. Clin Gastroenterol Hepatol. (2018) 16:1220–8. doi: 10.1016/j.cgh.2018.01.027
53. Lindkvist B. Diagnosis and treatment of pancreatic exocrine. World J Gastroenterol. (2013) 19(42):7258–66. doi: 10.3748/wjg.v19.i42.7258
54. Dreiling DA, Hollander F. Studies in pancreatic function, preliminary series of clinical studies with the secretin test. Gastroenterology. (1948) 11:714–29.18100241
55. Abu-El-Haija M, Conwell DL. Pancreatic insufficiency: what is the gold standard? Gastrointest Endosc Clin N Am. (2018) 28:521–8. doi: 10.1016/j.giec.2018.05.004
56. Patel N, Sellers ZM, Grover A, Liu QY, Maqbool A, Morinville VD, et al. Endoscopic Pancreatic Function Testing (ePFT) in children: a position paper from the NASPGHAN pancreas committee. J Pediatr Gastroenterol Nutr.. (2021) 72:144–50. doi: 10.1097/MPG.0000000000002931
57. Bouma S. Diagnosing pediatric malnutrition: paradigm shifts of etiology-related definitions and appraisal of the indicators. Nutr Clin Pract. (2017) 32(1):52–67. doi: 10.1177/0884533616671861
58. Dodge JA, Turck D. Cystic fibrosis: nutritional consequences and management. Best Pract Res Clin Gastroenterol. (2006) 20(3):531–46. doi: 10.1016/j.bpg.2005.11.006
59. Fieker A, Philpott J, Armand M. Enzyme replacement therapy for pancreatic insuJiciency: present and future. Clin Exp Gastroenterol. (2011) 4:55–73. doi: 10.2147/CEG.S17634
60. Carrière F, Renou C, Ransac S, Lopez V, De Caro J, Ferrato F, et al. Inhibition of gastrointestinal lipolysis by orlistat during digestion of test meals in healthy volunteers. Am J Physiol Gastrointest Liver Physiol. (2001) 281(1):G16–G28. doi: 10.1152/ajpgi.2001.281.1.G16
62. Trang T, Chan J, Graham DY. Pancreatic enzyme replacement therapy for pancreatic exocrine insufficiency in the 21(st) century. World J Gastroenterol. (2014) 20(33):11467–85. doi: 10.3748/wjg.v20.i33.11467
63. Gow R, Bradbear R, Francis P, Shepherd R Comparative study of varying regimens to improve steatorrhoea and creatorrhoea in cystic fibrosis: effectiveness of an enteric-coated preparation with and without antacids and cimetidine. Lancet. (1981) 2(8255):1071–4. doi: 10.1016/S0140-6736(81)91276-9
64. Ansaldi-Balocco N, Santini B, Sarchi C. Efficacy of pancreatic enzyme supplementation in children with cystic fibrosis: comparison of two preparations by random crossover study and a retrospective study of the same patients at two different ages. J Pediatr Gastroenterol Nutr. (1988) 7(Suppl 1):S40–5. doi: 10.1097/00005176-198811001-00009
65. ReliZORB. ReliZORB. [Online] ReliZORB. Available at: https://www.relizorb.com/.
66. Brady MS, Garson JL, Krug SK, Kaul A, Rickard KA, Caffrey HH, et al. An enteric-coated high-buffered pancrelipase reduces steatorrhea in patients with cystic fibrosis: a prospective, randomized study. J Am Diet Assoc. (2006) 106:1181–6. doi: 10.1016/j.jada.2006.05.011
67. Sinaasappel HG, Stern M, Littlewood M, Wolfe J, Steinkamp S, Heijerman G. Nutrition in patients with cystic fibrosis: a European consensus. J Cyst Fibros. (2002) 1:51–75. doi: 10.1016/S1569-1993(02)00032-2
68. Foundation CF, Borowitz D, Robinson KA, Rosenfeld M, Davis SD, Sabadosa KA, et al. Cystic fibrosis foundation evidence-based guidelines for management of infants with cystic fibrosis. J Pediatr. (2009) 155:S73–S93. doi: 10.1016/j.jpeds.2009.01.072
69. Sermet-Gaudelus I, Mayell SJ, Southern KW, European Cystic Fibrosis Society NSWG. Guidelines on the early management of infants diagnosed with cystic fibrosis following newborn screening. J Cyst Fibros. (2010) 9:323–9. doi: 10.1016/j.jcf.2010.04.008
70. Pezzilli R, Andriulli A, Bassi C, Balzano G, Cantore M, Delle Fave G, et al. Exocrine Pancreatic Insufficiency Collaborative (EPIc) group. Exocrine pancreatic insufficiency in adults: a shared position statement of the Italian association for the study of the pancreas. World J. Gastroenterol. (2013) 19:7930–46. doi: 10.3748/wjg.v19.i44.7930
71. Ramesh H, Reddy N, Bhatia S, Rajkumar JS, Bapaye A, Kini D, et al. A 51-week, open-label clinical trial in India to assess the efficacy and safety of pancreatin 40000 enteric coated minimicrospheres in patients with pancreatic exocrine insufficiency due to chronic pancreatitis. Pancreatology. (2013) 13(2):133–9. doi: 10.1016/j.pan.2013.01.009
72. Borowitz DS, Grand RJ, Durie PR. Use of pancreatic enzyme supplements for patients with cystic fibrosis in the context of fibrosing colonopathy consensus committee. J Pediatr. (1995) 127(5):681–4. doi: 10.1016/S0022-3476(95)70153-2
73. Lloyd-Still JD. Cystic fibrosis and colonic strictures: a new “iatrogenic” disease. J Clin Gastroenterol. (1995) 21:2–5. doi: 10.1097/00004836-199507000-00001
74. Domínguez-Muñoz JE, Iglesias-García J, Iglesias-Rey M, VilariñoInsua M. Optimising the therapy of exocrine pancreatic insufficiency by the association of a proton pump inhibitor to enteric coated pancreatic extracts. Gut. (2006) 55(7):1056–7. doi: 10.1136/gut.2006.094912
75. Domínguez-Muñoz JE. Pancreatic enzyme replacement therapy for pancreatic exocrine insufficiency: when is it indicated, what is the goal and how to do it? Adv Med Sci. (2011) 56(1):1–5. doi: 10.2478/v10039-011-0005-3
76. Woestenenk JW, van der Ent CK, Houwen RH. Pancreatic enzyme replacement therapy and coefficient of fat absorption in children and adolescents with cystic fibrosis. J Pediatr Gastroenterol Nutr. (2015) 61(3):355–60. doi: 10.1097/MPG.0000000000000784
77. Casellas F, Guarner L, Vaquero E, Antolín M, de Gracia X, Malagelada JR. Hydrogen breath test with glucose in exocrine pancreatic insufficiency. Pancreas. (1998) 16(4):481–6. doi: 10.1097/00006676-199805000-00004
78. Sankararaman S, Hendrix SJ, Schindler T. Update on the management of vitamins and minerals in cystic fibrosis. Nutr. Clin. Prac. (2022) 37:1074–87. doi: 10.1002/ncp.10899
79. Sankararaman S, Schindler T, Sferra TJ. Management of exocrine pancreatic insufficiency in children. Nutr Clin Pract. (2019) 34:S27–S42. doi: 10.1002/ncp.10388
80. Abu-El-Haija M, Uc A, Werlin SL, Freeman AJ, Georgieva M, Jojkic-Pavkov D, et al. Nutritional considerations inpediatric pancreatitis: a position paper from the NASPHAN PancreasCommittee. J Pediatr Gastroenterol Nutr. (2018) 67(1):131–43. doi: 10.1097/MPG.0000000000002023
81. WHO. Serum retinol concentrations for determining the prevalence of vitamin A deficiency in populations (2017). Geneva: World Health Organization (WHO). Available at: https://apps.who.int/iris/handle/10665/85859
82. Feranchak AP, Gralla J, King R, Ramirez RO, Corkill M, Narkewicz MR, et al. Comparison of indices of vitamin A status in children with chronic liver disease. Hepatology. (2005) 42(4):782–92. doi: 10.1002/hep.20864
84. Lippe B, Hensen L, Mendoza G, Finerman M, Welch M. Chronic vitamin A intoxication: a multisystem disease that could reach epidemic proportions. Am J Dis Child. (1981) 35:634–6. doi: 10.1001/archpedi.1981.02130310040014
85. Safi KH, Filbrun AG, Nasr SZ. Hypervitaminosis a causing hypercalcemia in cystic fibrosis. Case report and focused review. ATC journals. (2014) 11:1244–7. doi: 10.1513/AnnalsATS.201404-170BC
86. Misra M, Pacaud D, Petryk A, Collett-Solberg PF, Kappy M, et al. Vitamin D deficiency in children and its management: review of current knowledge and recommendations. Pediatrics. (2008) 122:398–417. doi: 10.1542/peds.2007-1894
87. Saggese G, Vierucci F, Prodam F, Cardinale F, Irene , Chiappini E, et al. Vitamin D in pediatric age: consensus of the Italian pediatric society and the Italian society of preventive and social pediatrics, jointly with the Italian federation of pediatricians. Ital J Pediatr. (2018) 44:51. doi: 10.1186/s13052-018-0488-7
88. Hollis BW. Circulating 25-hydroxyvitamin D levels indicative of vitamin D sufficiency: implications for establishing a new effective dietary intake recommendation for vitamin D. J Nutr. (2005) 135(2):317–22. doi: 10.1093/jn/135.2.317
89. Green D, Carson K, Leonard A, Davis JE, Rosenstein B, Zeitlin P, et al. Current treatment recommendations for correcting vitamin D deficiency in pediatric patients with cystic fibrosis are inadequate. J Pediatr. (2008) 153:554–9. doi: 10.1016/j.jpeds.2008.04.058
90. Ross AC. Dietary Reference Intakes for Calcium and Vitamin D. Washington (DC): Cambridge University Press (2011).
91. Silva MC, Furlanetto TW. Intestinal absorption of vitamin D: a systematic review. Nutr Rev. (2018) 76:60–76. doi: 10.1093/nutrit/nux034
92. Hossein-nezhad A, Holick MF. Vitamin D for health: a global perspective. Mayo Clin Proc. (2013) 88:720–55. doi: 10.1016/j.mayocp.2013.05.011
93. Tebben PJ, Singh RJ, Kumar R. Vitamin D–mediated hypercalcemia: mechanisms, diagnosis, and treatment. Endocr Rev. (2016) 37:521–47. doi: 10.1210/er.2016-1070
95. Conway SP, Wolfe SP, Brownlee KG, White H, Oldroyd B, Truscott JG, et al. Vitamin K status among children with cystic fibrosis and its relationship to bone mineral density and bone turnover. Pediatrics. (2005) 115(5):1325–31. doi: 10.1542/peds.2004-1242
96. Borowitz D, Baker RD, Stallings V. Consensus report on nutrition for pediatric patients with cystic fibrosis. J Pediatr Gastroenterol Nutr. (2002) 35(3):246–59. doi: 10.1097/00005176-200209000-00004
97. Rucker RB. Improved functional endpoints for use in vitamin K assessment: important implications for bone disease. Am J Clin Nutr. (1997) 65(3):883–4. doi: 10.1093/ajcn/65.3.883
98. Krzyżanowska P, Pogorzelski A, Skorupa W, Moczko J, Grebowiec P, Walkowiak J. Exogenous and endogenous determinants of vitamin K status in cystic fibrosis. Sci Rep. (2015) 5(1):1–8. doi: 10.1038/srep12000
99. Medicine, Institute of. Dietary reference intakes for vitamin A, vitamin K, arsenic, boron, chromium, copper, iodine, iron, manganese, molybdenum, nickel, silicon, vanadium, and zinc. s.l.: National Academy Press, 2001.
100. NIH, Fact Sheet for Health Professionals. NIH officail of dietary supplement. NIH Vitamin K. [Online] (2021). Availabele at: https://ods.od.nih.gov/factsheets/VitaminK-HealthProfessional/.
101. NIH, Institute of Medicine. Dietary Reference Intakes for Vitamin A, Vitamin K, Arsenic, Boron, Chromium, Copper, Iodine, Iron, Manganese, Molybdenum, Nickel, Silicon, Vanadium, and Zinc. Washington, DC: National Academy Press (2001).
102. Booth SL. Vitamin K: food composition and dietary intakes. Food Nutr Res. (2012) 56. doi: 10.3402/fnr.v56i0.5505
103. Elder SJ, Haytowitz DB, Howe J, Peterson JW, Booth SL. Vitamin K contents of meat, dairy, and fast food in the U. S. Diet. J Agric Food Chem. (2006) 54:463–7. doi: 10.1021/jf052400h
104. Lexicomp. Lexicomp (2022). Available at: http://online.lexi.com/lco/action/doc/retrieve/docid/chilla_f/133825?cesid=4kWqMIECbMY&searchUrl=%2Flco%2Faction%2Fsearch%3Fq%3Dvitamin%2BK%26t%3Dname%26va%3Dvitamin%2BK#dop.
105. Bershad EM, Suarez JI. Prothrombin complex concentrates for oral anticoagulant therapy-related intracranial hemorrhage: a review of the literature. Neurocrit Care. (2010) 12(3):403–13. doi: 10.1007/s12028-009-9310-0
106. Cystic Fibrosis Foundation, Borowitz D, Robinson KA, Rosenfeld M, Davis SD, Sabadosa KA, et al. Cystic fibrosis foundation evidence-based guidelines for management of infants with cystic fibrosis. J Pediatr. (2009) 155:S73–S93. doi: 10.1016/j.jpeds.2009.09.001
107. Eden RE, Coviello JM. Vitamin K deficiency. NIH (2022). Available at: https://www.ncbi.nlm.nih.gov/books/NBK536983/.
108. Girish BN, Rajesh G, Vaidyanathan K, Balakrishnan V, et al. Assessment of oxidative status in chronic pancreatitis and its relation with zinc status. Indian J Gastroenterol. (2011) 30:84–8. doi: 10.1007/s12664-011-0094-8
109. Glasbrenner B, Malfertheiner P, Büchler M, Kuhn K, Ditschuneit H, et al. Vitamin B12 and folic acid deficiency in chronic pancreatitis: a relevant disorder? Klin Wochenschr. (1991) 69:168–72. doi: 10.1007/BF01665861
110. Reid DW, Withers NJ, Francis L, Wilson JW, Kotsimbos TC. Iron deficiency in cystic fibrosis: relationship to lung disease severity and chronic pseudomonas aeruginosa infection. Chest. (2002) 121(1):48–54. doi: 10.1378/chest.121.1.48
111. Bowrey DJ, Morris-Stiff GJ, Puntis MC. Selenium deficiency and chronic pancreatitis: disease mechanism and potential for therapy. HPB Surg. (1999) 11:207–15. doi: 10.1155/1999/97140
112. Mathew P, Wyllie R, Van Lente F, Steffen RM, Kay MH, et al. Antioxidants in hereditary pancreatitis. Am J Gastroenterol. (1996) 91:1558–62. 8759661
113. Van Biervliet S, Van Biervliet J-P, Robberecht E. Serum zinc in patients with cystic fibrosis at diagnosis and after one year of therapy. Biol Trace Elem Res. (2006) 112(3):205–11. doi: 10.1385/BTER:112:3:205
114. Weaver CM, Alexander DD, Boushey CJ, Dawson-Hughes B, Lappe JM, LeBoff MS, et al. Calcium plus vitamin D supplementation and risk of fractures: an updated meta-analysis from the national osteoporosis foundation. Osteoporos Int. (2016) 27(1):367–76. doi: 10.1007/s00198-015-3386-5
115. Dos Santos Simon MIS, Dalle Molle R, Silva FM, Rodrigues TW, Feldmann M, Forte GC, et al. Antioxidant micronutrients and essential fatty acids supplementation on cystic fibrosis outcomes: a systematic review. J Acad Nutr Diet. (2020) 120(6):1016–33. doi: 10.1016/j.jand.2020.01.007
116. Association, American Heart. American Heart Association (2022). Available at: https://www.heart.org/en/healthy-living/healthy-eating/eat-smart/nutrition-basics/dietary-recommendations-for-healthy-children#:∼:text=Keep%20total%20fat%20intake%20between,fish%2C%20nuts%20and%20vegetable%20oils
117. Andersson R, Löhr J-M, Working Group for Chronic Pancreatitis Guidelines (Sweden), et al. Swedish National guidelines for chronic pancreatitis. Scand J Gastroenterol. (2021) 56(4):469–83. doi: 10.1080/00365521.2021.1881815
118. Woestenenk JW, Dalmeijer GW, van der Ent CK, Houwen RH. The relationship between energy intake and body-growth in children with cystic fibrosis. Clin Nutr. (2019) 38(2):920–5. doi: 10.1016/j.clnu.2018.02.005
119. R5, Gibson. Principles of nutritional assessment. s.l. Oxford, UK: Oxford University Press (2005).
120. Rashid M, Durie P, Andrew M, Kalnins D, Shin J, Corey M, et al. Prevalence of vitamin K deficiency in cystic fibrosis. Am J Clin Nutr. (1990) 70(3):378–82. doi: 10.1093/ajcn/70.3.378
121. Wilson D C, Rashid M, Durie P R, Tsang A, Kalnins D, Andrew M, et al. Treatment of vitamin K deficiency in cystic fibrosis: effectiveness of a daily fat-soluble vitamin combination. J Pediatr. (2001) 138(6):851–5. doi: 10.1067/mpd.2001.113633
122. Prykhodko O, Pierzynowski SG, Nikpey E, Arevalo Sureda E, Fedkiv O, Weström BR. Pancreatic and pancreatic-like microbial proteases accelerate gut maturation in suckling rats. PLoS One. (2015) 10(2):e0116947. doi: 10.1371/journal.pone.0116947
123. Stefan G Pierzynowski, Peter C Gregory, Rafal Filip, Jaroslaw Wolinski, Kateryna Goncharova Pierzynowska, et al. Glucose homeostasis dependency on acini–islet–acinar (AIA) axis communication: a new possible pathophysiological hypothesis regarding diabetes mellitus. Nutr Diabetes. (2018) 8:55. doi: 10.1038/s41387-018-0062-9
Keywords: exocrine pancreatic insufficiency, pancreatitis, nutrition, fat-soluble vitamins, calories, malnutrition, PERT, cystic fibrosis
Citation: Zheng Y and Mostamand S (2023) Nutrition in children with exocrine pancreatic insufficiency. Front. Pediatr. 11:943649. doi: 10.3389/fped.2023.943649
Received: 14 May 2022; Accepted: 18 April 2023;
Published: 5 May 2023.
Edited by:
Emily Perito, University of California, United StatesReviewed by:
David Clement Whitcomb, University of Pittsburgh, United States© 2023 Zheng and Mostamand. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuhua Zheng WXpoZW5nQGNobGEudXNjLmVkdQ==
Abbreviations CF, cystic fibrosis; CFTR, cystic fibrosis transmembrane conductance regulator; EF-1, fecal elastase-1; ePFT, endoscopic pancreatic function tests; EPI, exocrine pancreatic insufficiency; LU, lipase unit; NASPGHAN, North American society for pediatric gastroenterology, hepatology and nutrition; PERT, pancreatic enzyme replacement therapy; sMRCP, secretin-enhanced magnetic resonance cholangiopancreatography.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.