 Laura Battaglia1
Laura Battaglia1 Giovanna Scorrano2
Giovanna Scorrano2 Rossana Spiaggia1
Rossana Spiaggia1 Antonio Basile1
Antonio Basile1 Stefano Palmucci1
Stefano Palmucci1 Pietro Valerio Foti1Corrado Spatola1Michele Iacomino3Franco Marinangeli4Elisa Francia5
Pietro Valerio Foti1Corrado Spatola1Michele Iacomino3Franco Marinangeli4Elisa Francia5 Francesco Comisi6Antonio Corsello7
Francesco Comisi6Antonio Corsello7 Vincenzo Salpietro2,8
Vincenzo Salpietro2,8 Alessandro Vittori5*
Alessandro Vittori5* Emanuele David1
Emanuele David1
- 1Department of Medical Surgical Sciences and Advanced Technologies “GF Ingrassia”, University Hospital Policlinic “G. Rodolico-San Marco”, Catania, Italy
- 2Department of Biotechnological and Applied Clinical Sciences, University of L'Aquila, L'Aquila, Italy
- 3Unit of Medical Genetics, IRCCS Instituto Giannina Gaslini, Genoa, Italy
- 4Department of Anesthesia, Critical Care and Pain Therapy, University of L’aquila, L’aquila, Italy
- 5Department of Anesthesia and Critical Care, ARCO ROMA, Ospedale Pediatrico Bambino Gesù IRCCS, Rome, Italy
- 6Pediatrics Department, Ospedale Microcitemico, Cagliari, Italy
- 7Department of Pediatrics, University of Milan, Milan, Italy
- 8Department of Neuromuscular Disorders, UCL Queen Square Institute of Neurology, London, United Kingdom
The WWOX gene encodes a 414-amino-acid protein composed of two N-terminal WW domains and a C-terminal short-chain dehydrogenase/reductase (SDR) domain. WWOX protein is highly conserved among species and mainly expressed in the cerebellum, cerebral cortex, brain stem, thyroid, hypophysis, and reproductive organs. It plays a crucial role in the biology of the central nervous system, and it is involved in neuronal development, migration, and proliferation. Biallelic pathogenic variants in WWOX have been associated with an early infantile epileptic encephalopathy known as WOREE syndrome. Both missense and null variants have been described in affected patients, leading to a reduction in protein function and stability. The most severe WOREE phenotypes have been related to biallelic null/null variants, associated with the complete loss of function of the protein. All affected patients showed brain anomalies on magnetic resonance imaging (MRI), suggesting the pivotal role of WWOX protein in brain homeostasis and developmental processes. We provided a literature review, exploring both the clinical and radiological spectrum related to WWOX pathogenic variants, described to date. We focused on neuroradiological findings to better delineate the WOREE phenotype with diagnostic and prognostic implications.
1. Introduction
Developmental and epileptic encephalopathies (DEEs) are early-onset disorders characterized by multiple seizure types, widespread epileptic discharges at the electroencephalogram, and neurodevelopmental delay (1–4). In recent years, next-generation sequencing (NGS) technologies, including exome and genome studies, revealed an increased complexity underlying DEEs, with the involvement of several genes implicated, such as the WWOX gene. Biallelic pathogenic variants of WWOX, represented by homozygous and compound heterozygous variants, have been associated with an early infantile epileptic encephalopathy known as WOREE syndrome (MIM: 616211) (5–8). Individuals affected with WOREE syndrome presented early onset epilepsy, with refractoriness to antiseizure medications (ASMs), development impairment with psychomotor delay, spastic tetraplegia, inability to walk, non-verbal communication and other additional features, generally dying within the first year of life (1). Brain imaging typically showed variable anomalies, such as myelination impairment, corpus callosum thickness, white matter anomalies, and/or cerebral atrophy (1, 9).
In WOREE syndrome, WWOX pathogenic variants lead to a variable loss of function of WWOX protein, which is highly conserved across species and mainly expressed in the cerebellum, brain, thyroid, hypophysis, and reproductive organs (1, 10, 11). WWOX includes 414 amino acids with two WW domains at the N-terminal end and a central short-chain dehydrogenase/reductase (SDR) domain (1, 12). It plays a crucial role in the biology of the central nervous system and is involved in neuronal development, migration, and proliferation. Functional studies using human organoids and animal models attempted to explain WWOX properties, exploring the phenotype spectrum related to WWOX loss of function. Specifically, WWOX, mainly expressed in neurons, oligodendrocytes, and astrocytes, acts as a scaffold protein, modulating the cytoskeleton and homeostasis of its substrates (9, 11–14).
Interestingly, oligodendrocyte progenitors present a high level of WWOX protein, in contrast with mature myelinated oligodendrocytes, suggesting a crucial role of the protein in myelin biogenesis, and in differentiation of these cells. Indeed, there is evidence of its function in cellular lipidic homeostasis and of its interaction with proteins involved in myelin biogenesis such as SEC23IP, SCAMP3, VOPP1, and SIMPLE (1, 15, 16). Moreover, loss of function of WWOX leads to hypo-myelinization, with a potential role in multiple sclerosis pathogenesis (1). Interestingly, when microglia were treated with lipopolysaccharides, a WWOX overexpression occurred, with the following activation of the immune system. Moreover, WWOX modulates the NF-kB pathway, and its loss of function was related to astrogliosis, suggesting an additional role of WWOX in neuroinflammation and activation of the immune response (1, 17–21).
Furthermore, WWOX protein presents a focused expression in the medial entorhinal cortex, basolateral amygdala, and layer 5 of the frontal cortex, which are mostly involved in memory, learning mechanisms, perception, and emotion control. It was observed that the loss of function of the WWOX protein was related to Alzheimer's disease, temporal lobe epilepsy, cognitive impairment, and neuropsychiatric disorders such as autism spectrum disorder (22–27). Interestingly, the WWOX protein is mostly expressed in the cerebellar cortex and in specific cell types, such as basket cells and granule cells, and hypomorphic mutations of WWOX have been associated with cerebellar disorders such as Spinocerebellar Ataxia Type 12 (SCAR12) (MIM: 614322). WWOX pathogenic variants have also been related to sex differentiation disorders (DSD), according to their high expression in reproductive organs and their potential role in gonadotropin synthesis, gonad development, and steroid metabolism (8, 12, 28, 29).
Furthermore, WWOX loss of function has also been related to early infantile refractory epilepsies. Functional studies showed that -/- mice presented GABAergic interneuron impairment and decreased expression of GAD65/67, enzymes that synthesize GABA, with evidence of cortical hyperexcitability and epilepsy onset (21, 30, 31).
WWOX also manages several pathways such as Wnt/β-catenin and TGFβ/SMAD and its loss of function is associated with carcinogenesis progression (9, 32, 33).
This study aims to better characterize neuroradiological features in WOREE syndrome, collecting all brain imaging data described to date in the literature, to accurately define the most common signs of the disease, which could lead to the definitive diagnosis when it is clinically suspected.
2. Genetic findings
WWOX is located on chromosome 16q23.1-q23.2 and crosses the second most common fragile site (FRA16D) in the human genome, with potential genomic instability (1, 12, 34).
A genotype-phenotype correlation has been suggested for WWOX-related disorders (35, 36). Specifically, WWOX loss of function caused by biallelic null variants (i.e., frameshift, nonsense, donor/acceptor splice site, and deletion) have been associated with the most severe phenotypes, represented by WOREE syndrome, with early death before 2 years of age, prenatal cerebral anomalies, and eye malformations (36). Concurrently, hypo-morphic genotypes characterized by biallelic missense variants have been related to a milder phenotype, represented by SCAR12. This rare spinocerebellar ataxia has been described in two families affected with early onset epilepsy, neurodevelopmental delay, intellectual disability, and cerebellar ataxia (35). Learning disorders, dysarthria, nystagmus, and decreased reflexes in the upper and lower limbs have also been reported in patients with SCAR12 who presented a partial loss of function of WWOX protein (35). Concurrently, patients carrying both a null and a missense variant presented an intermediate phenotype, whereas heterozygous variants have been described in healthy controls.
Interestingly, WWOX has been recently involved in several neurological disorders such as autism spectrum disorder (ASD), multiple sclerosis (MS), and Alzheimer's Disease (35). Patients with Alzheimer's Disease presented decreased WWOX protein levels, compared to age-matched healthy controls (37, 38). Specifically, functional studies revealed that the microtubule-associated protein tau was hyper-phosphorylated after interaction with glycogen synthase kinase 3β (GSK-3β) in the affected hippocampi, leading to neurofibrillary tangles formation with consequent neuronal death. In this circumstance, WWOX concurrently prevented tau hyperphosphorylation and increased the affinity to microtubules of tau proteins, blocking neurofibrillary accumulation and related neurodegeneration processes (39). Furthermore, WWOX interacted with another protein, TPC6AΔ, involved in tau aggregation and Aβ generation, suggesting a protective role of WWOX protein in Alzheimer's Disease (35, 40).
Notably, it was also observed that WWOX protein was decreased in the chronic active lesions of patients with MS, compared to age-matched healthy controls (41). Specifically, WWOX has been identified as a susceptible gene in MS and 48 WWOX pathogenic variants have been detected in affected patients (42). Interestingly, the role of WWOX in grey matter impairment in MS was described, with a relevant involvement of the protein in the myelination process and oligodendrocyte differentiation (35).
Moreover, deletions and duplications affecting the WWOX gene have been found in patients with ASD, and copy number variations (CNVs) of WWOX have been reported in patients with milder ASD phenotypes (35). However, the emerging role of WWOX in the pathophysiology of neurological disorders should be better evaluated in functional studies.
Even though pathogenic variants affect WWOX protein uniformly, some mutational hotspots have been reported in the literature to date. Specifically, the p.Gln230Pro pathogenic variant affects the SRD domain and has been described both in homozygosity and in compound heterozygosity in eight cases overall (1). Nevertheless, how missense variants affecting the SRD domain impair WWOX catalytic activity has not been demonstrated yet.
Moreover, two mutations affecting Glycine 137, a pivotal residue of the coenzyme binding region, have been described. Additionally, biallelic mutations affecting glutamic acid 17 and serine 318 combined with deletion or other missense mutations have been reported. Recurrent deletions and duplications of exons 6–8 have also been described. They led to truncated proteins or unstable products that were quickly degraded (1).
Furthermore, Proline 47 presumably may represent a mutational hotspot. It is a highly conserved residue, with a pivotal role in the function of the first WW domain. Specifically, it was observed that pathogenic variants affecting this residue, located in the first WW domain, both made WWOX unable to interact with partner proteins and impaired WWOX translocation to the nuclear compartment (1, 43). Other potential hotspot mutations could be represented by recurrent nonsense variants affecting Arg54*, frameshift variants such as Asp58Alafs*3, His173Alafs*67, His173Ilefs*5, and Glu306Aspfs*21, and splice site variants such as c.173-1G>T, c.173-2A>G, c.409+1G>T, c.517-2A>G, and c.606-1G>A (1).
However, functional studies should be performed, and knock-in animal models could better clarify the impact of point mutations on WWOX structure and function. To date, mice models revealed that WWOX presented a different tissue expression during developmental stages (35). A significant expression was detected in peripheral nerves, brain stem, and spinal cord during the first stages of embryogenesis, whereas a decreased level was detected in the latest stages, and an increased level was detected after birth.
Moreover, WWOX knockout mice, which had null alleles, showed deep structural anomalies such as malformations, neuronal impairment and degeneration, heterotopia, and defects of the midline. They exhibited a severe phenotype consistent with patients affected with WOREE syndrome, carrying null variants (29, 44). The full clinical spectrum of -/- mice was characterized by growth retardation, hypoglycemia, hypolipidemia, impaired steroidogenesis, bone defects, refractory seizures, ataxia, severe motor incoordination, imbalance, and premature death by 2 to 3 weeks after birth. This presumably indicates a pivotal role of WWOX in the neurobiology of the central nervous system at different developmental stages (45).
Concerning CNVs, most of them are intragenic, with the involvement of genomic regions crossed by WWOX, and deletions are more prevalent than duplications (1). A chromosomal microarray, choosing a quantitative method to analyze the WWOX locus, should be performed in patients with clinical features suggestive of WOREE syndrome. Indeed, individuals with WOREE syndrome present a high prevalence of CNVs, small structural variants detected by a quantitative approach. Concurrently, when whole exome sequencing (WES) and targeted gene panel sequencing are performed first, CNV investigation should be carried out, due to deletions and duplications, which often affect the WWOX gene. Furthermore, RNA studies could be useful to investigate if single-nucleotide variants (SNVs) and/or missense variants impair mRNA stability and disrupt the pre-mRNA splicing (2).
3. Clinical findings
WOREE syndrome is a developmental and epileptic encephalopathy (DEE) characterized by early onset epilepsy, with refractoriness to antiseizure medications (ASMs), development impairment with psychomotor delay, spastic tetraplegia, no walking, non-verbal communication, and other additional features, with death in first years of life (1). To date, 84 patients have been reported in the literature with descriptions of the individual phenotypes. Early onset epilepsy represented the main feature in these patients and occurred with different semiology. Specifically, early infantile DEE, epilepsy of infancy with migrating focal seizures (EIMFS), and infantile epileptic spasms syndrome (IESS) have been reported (2). Seizures included generalized tonic, myoclonic, clonic, and tonic-clonic seizures, focal clonic seizures, infantile spasms, eyelid myoclonia, and status epilepticus with refractoriness to ASMs. In affected patients, EEG showed background slowing with profound epileptic discharges (2). Developmental impairment preceded epilepsy and included severe intellectual disability, early-onset spastic tetraplegia, hypotonia, non-verbal communication, absence of ambulation, psychomotor delay, and microcephaly.
Dysmorphic features have been reported in affected patients and included short stature, round face with full cheeks, short neck, facial hypotonia, hypertelorism, arched and bushy eyebrows, long eyelashes, epicanthic folds, bitemporal narrowing, low anterior hairline, broad nose, high forehead, depressed nasal bridge, gingival hypertrophy low-set, and large ears (2). Scoliosis/kyphosis, movement disorders, and feeding problems have also been described. Moreover, ophthalmic anomalies such as poor or no eye contact, retinal degeneration, and optic atrophy have been reported (1).
We found a genotype-phenotype correlation only about the mortality risk. Specifically, null/null variants are associated with a higher mortality risk than missense/null and missense/missense variants (2). Here, respiratory problems were the main cause of death, while other causes included SUDEP, status epilepticus, and obstructive cardiomyopathy (2, 5, 46–49).
4. Imaging findings
One important step for the diagnosis of WOREE syndrome is based on brain imaging. Several studies identified MRI features related to WWOX gene mutations in children with epileptic encephalopathy and we will try to put together all the characteristics described to date, to define a unique radiological pattern (Figure 1, Table 1).
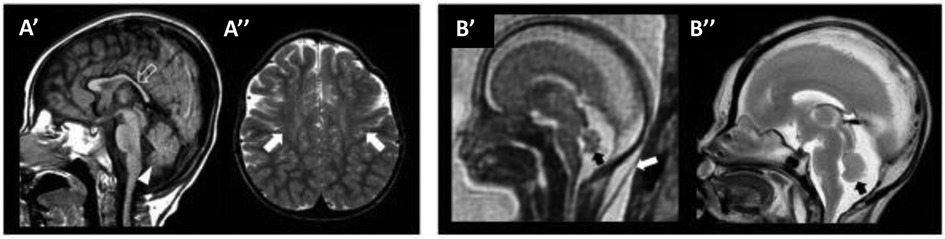
Figure 1. WOREE-associated neuroradiological features. A brain MRI of an affected patient was performed at 2.4 years of age (A’–A’’). (A’) The sagittal T1-weighted image reveals hypoplasia of the corpus callosum (empty arrow) and inferior cerebellar vermis (arrowhead). (A”) Axial T2-weighted images demonstrate mild atrophy of the frontal lobes with associated bilateral white matter hyperintensity (arrows). Fetal MRI (B’) and high-resolution post-mortem MRI (B”) of an affected patient were performed at 21 gestational weeks, demonstrating mild hypoplasia of the cerebellar vermis (black arrows). Note the slightly increased thickness of nuchal subcutaneous tissues on fetal MRI (white arrow). The laminar organization of the cerebral hemispheres and cortical gyration are appropriate for the gestational age (not shown). Courtesy of Prof. Salpietro (50).
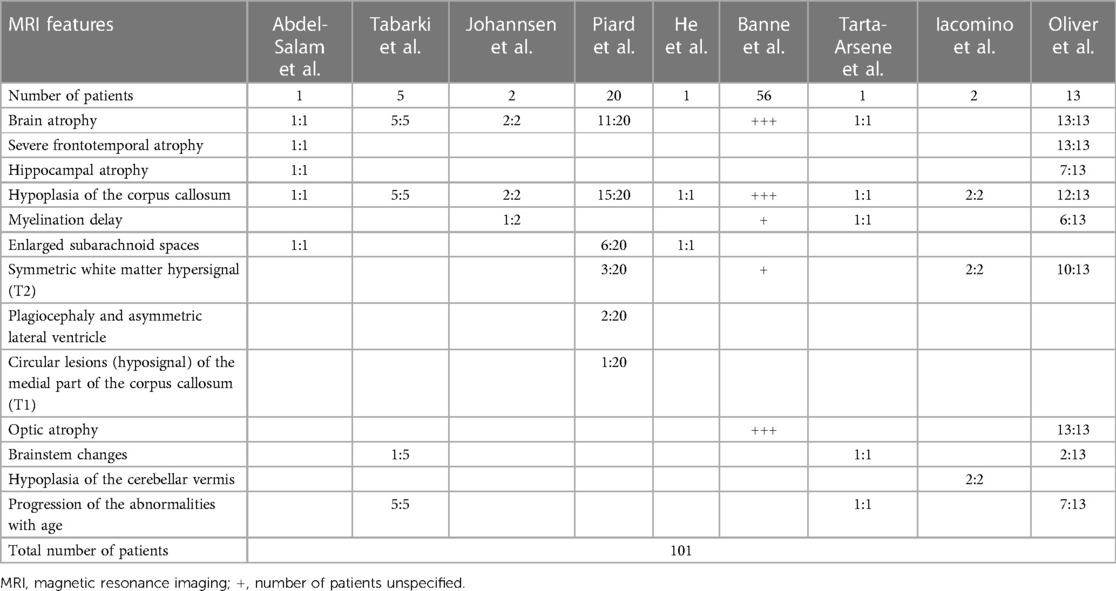
Table 1. MRI features of patients affected with WOREE syndrome reported to date.
In 2014, Abdel-Salam et al. reported an affected girl born from consanguineous parents, who presented with growth retardation, microcephaly, epileptic seizures, retinopathy, and early death. WES revealed a nonsense homozygous mutation in WWOX, and brain MRI documented supratentorial atrophy with a simplified gyral pattern, hypoplasia of the hippocampus and the temporal lobe with consecutively widened subarachnoidal space, a thin hypoplastic corpus callosum, and hippocampal dysplasia with extracellular vacuoles in amygdala and hippocampus (47). Tabarki et al. then described five patients from two unrelated families who showed progressive microcephaly, early onset spasticity, refractory epilepsy, severe failure to thrive, and severe developmental delay. They carried the same homozygous mutation in WWOX. Interestingly, a brain MRI revealed a peculiar pattern of neurodegeneration, characterized by periventricular white matter volume loss, atrophy of the corpus callosum, flattening of the brainstem, and bilateral symmetrical lesions in the medial nuclei of the thalami in one patient. Of note, the cerebellum was not affected (51).
Furthermore, Tarta-Arsene et al. described in 2017 a similar brain MRI pattern in a boy with early-onset epilepsy, severe global developmental delay, persistent hypsarrhythmia at EEG, and epileptic spasms, carrying two WWOX mutations in a heterozygous state. Brain imaging was performed and documented a degenerative pattern characterized by cortico-subcortical atrophy, an extremely thin corpus callosum, delayed myelination, and flattening of the brainstem (52).
In 2018, Jessika Johannsen et al. examined two consanguineous patients with a homozygosity for the missense variant in the catalytic short-chain dehydrogenase/reductase (SDR) domain of the WWOX gene; both patients were characterized by early epilepsy refractory to treatment, progressive microcephaly, profound developmental delay, and brain MRI abnormalities. At the ages of 3, 5, and 21 months the imaging showed global atrophy, hypoplasia of the corpus callosum, and myelination delay with normal MR spectroscopy in patient 1. In patient 2, a cousin of patient 1, at the age of 3 and 6 months, brain imaging showed two similar features, global atrophy and hypoplasia of the corpus callosum (53). In the same year, Juliette Piard et al. studied 20 patients of 18 families with WOREE syndrome; they found a higher frequency of compound heterozygous mutations, consistently with a lower rate of consanguinity. The study detected abnormal brain MRI in 80% of them; specifically, 75% of patients were characterized by corpus callosum hypoplasia and 55% by cerebral atrophy. Other alterations included enlarged subarachnoid spaces, symmetric white matter hypersignal, plagiocephaly, asymmetric lateral ventricle, and circular lesions (hypo-signal) of the medial part of the corpus callosum (8). Another case of WWOX compound heterozygous mutations in a Chinese patient with WOREE syndrome was reported by Jing He et al. in 2019; in this case report, brain MRI revealed a widened subarachnoid space and a thin corpus callosum (54). Moreover, in 2020 an interesting study reported an abnormal cerebral cortex development in a family affected with neurodevelopmental impairment and refractory epilepsy, carrying a homozygous mutation in WWOX. Functional studies revealed a similar disorganization of cortical layers in mice, carrying a homozygous truncating WWOX mutation. Brain MRI of the two patients affected documented hypoplasia of the corpus callosum and mild atrophy of the frontal lobes with associated bilateral white matter hyperintensity. Notably, hypoplasia of cerebellar vermis related to WOREE syndrome was first reported in this study (50).
Similar features were found in 2021 by Banne et al., who retrospectively analyzed 45 variants in 56 WOREE published cases. A total of 34 out of 45 patients were characterized by loss of function mutations, and 11 were missense variants. Brain MRI imaging of children with WOREE syndrome showed abnormally thin or hypoplastic corpus callosum, progressive optic atrophy, and brain atrophy as the most common features, but in some cases, they also reported delayed myelination and white matter hyperintense signals (35). A recent study by Karen L. Oliver et al. in 2023 analyzed 13 patients from 12 families with WWOX developmental and epileptic encephalopathy (WWOX-DEE) due to biallelic pathogenic variants in WWOX. All of them showed severe frontotemporal atrophy. Brain MRI also demonstrated hippocampal atrophy, white matter signal abnormality, and volume loss with a very thin corpus callosum. Severe optic atrophy was also detected in all patients, whereas some patients presented brainstem changes, most of which were dorsal. Brain MRI revealed lesions in patient 1 and patient 7, at the age of 15 days and 19 days, respectively; specifically, patient 7 presented delayed myelination, a thin corpus callosum, and frontotemporal atrophy. Lastly, 7 of 13 patients underwent serial MRI, which showed progression of the abnormalities with age; specifically, related to the brain atrophy, which showed a change from mild to severe, and white matter alteration (2).
This review analyzed 101 cases of children with various mutations in the WWOX gene associated with WOREE syndrome from 2014 to 2023. Based on the literature data collected, hypoplasia of the corpus callosum and brain atrophy appeared to be the predominant MRI alterations related to this syndrome. Other relatively frequent features included symmetric white matter hyperintensity in T2-weighted images and optic atrophy. Furthermore, approximately 13% of the patients included in the study also demonstrated age-related progression. Atrophy of the hippocampus, delayed myelination, enlarged subarachnoid spaces, plagiocephaly, asymmetrical lateral ventricles, hypointense circular lesions in the medial part of the corpus callosum in T1-weighted images, brainstem alterations, and cerebellar vermis hypoplasia were observed in a lower percentage of cases, and therefore these findings seem to be less specific.
5. Discussion
Genetic neurologic disorders with neurodevelopmental delay and refractory epilepsy include a broad spectrum of monogenic conditions with expanding clinical differential diagnosis and genetic heterogeneity (55–60). Even in the era of next-generation sequencing (NGS), the etiology and disease mechanisms underlying these conditions remain unclear in a large number of cases (61–66). Defining the full spectrum of disease-causing molecular pathways underlying brain disorders is crucial to genetically diagnose patients with developmental and epileptic encephalopathies or delay and to assess potential personalized strategies for the follow-up and management of these affected children (67–70).
WOREE syndrome is a developmental and epileptic encephalopathy with a genetic etiology that affects the development and biology of the central nervous system. The loss of function of the WWOX gene leads to the absence of the protein encoded, associated with the highest mortality in affected patients. Nevertheless, the mechanistic effects of WWOX pathogenic variants on protein function are still not well known. Functional studies could better explain these aspects, allowing us to accurately characterize the disorder.
The most common neuroradiological findings were cerebral atrophy and white matter signal anomalies in the corpus callosum, with a myelination impairment. Interestingly, all affected patients presented brain anomalies at MRI, suggesting the crucial role of the WWOX protein in cerebral homeostasis and neuronal development. Recent mice studies showed how the loss of WWOX disrupts neuronal migration and CNS development across different species (50). However, organoids could better elucidate the impact of WWOX mutations on fetal developmental processes and neurogenesis (50).
Imaging data represent an essential instrument to best define phenotypes of WOREE syndrome, with diagnostic and prognostic implications, and they always should be detected in patients with relevant clinical features suggesting WOREE syndrome to better characterize the disorder. In patients with early infantile epileptic encephalopathy with refractoriness to ASMs and global developmental delay associated with characteristic neuroradiological patterns, the WWOX gene analysis should be included in the diagnostic workup.
Author contributions
LB: Conceptualization, Investigation, Methodology, Validation, Visualization, Writing – original draft. GS: Conceptualization, Data curation, Investigation, Methodology, Validation, Visualization, Writing – original draft, Formal Analysis, Project administration, Resources, Software, Supervision. RS: Data curation, Methodology, Software, Writing – original draft, Conceptualization, Formal Analysis, Funding acquisition, Investigation, Project administration, Resources, Supervision, Validation, Visualization. AB: Investigation, Methodology, Project administration, Software, Validation, Visualization, Writing – original draft, Conceptualization, Data curation, Formal Analysis, Supervision. SP: Investigation, Methodology, Software, Writing – original draft. PF: Conceptualization, Methodology, Writing – original draft, Writing – review & editing. CS: Investigation, Methodology, Project administration, Writing – original draft, Writing – review & editing. MI: Methodology, Writing – original draft, Writing – review & editing, Formal Analysis, Investigation. FM: Conceptualization, Investigation, Methodology, Validation, Writing – original draft, Writing – review & editing. EF: Conceptualization, Data curation, Investigation, Methodology, Writing – original draft, Writing – review & editing. FC: Data curation, Formal Analysis, Investigation, Writing – original draft, Writing – review & editing. AC: . VS: Writing – original draft, Writing – review & editing. AV: Investigation, Software, Writing – original draft, Writing – review & editing. ED: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article.
This work was supported also by the Italian Ministry of Health with “Current Research funds”.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Aldaz CM, Hussain T. WWOX loss of function in neurodevelopmental and neurodegenerative disorders. IJMS. (2020) 21:8922. doi: 10.3390/ijms21238922
2. Oliver KL, Trivisano M, Mandelstam SA, De Dominicis A, Francis DI, Green TE, et al. WWOX developmental and epileptic encephalopathy: understanding the epileptology and the mortality risk. Epilepsia. (2023) 64:1351–67. doi: 10.1111/epi.17542
3. Trivisano M, Specchio N. What are the epileptic encephalopathies? Curr Opin Neurol. (2020) 33:179–84. doi: 10.1097/WCO.0000000000000793
4. Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, et al. ILAE classification of the epilepsies: position paper of the ILAE commission for classification and terminology. Epilepsia. (2017) 58:512–21. doi: 10.1111/epi.13709
5. Mignot C, Lambert L, Pasquier L, Bienvenu T, Delahaye-Duriez A, Keren B, et al. WWOX -related encephalopathies: delineation of the phenotypical spectrum and emerging genotype-phenotype correlation. J Med Genet. (2015) 52:61–70. doi: 10.1136/jmedgenet-2014-102748
6. Ehaideb SN, Al-Bu Ali MJ, Al-obaid JJ, Aljassim KM, Alfadhel M. Novel homozygous mutation in the WWOX gene causes seizures and global developmental delay: report and review. Transl Neurosci. (2018) 9:203–8. doi: 10.1515/tnsci-2018-0029
7. Serin HM, Simsek E, Isik E, Gokben S. WWOX-associated encephalopathies: identification of the phenotypic spectrum and the resulting genotype-phenotype correlation. Neurol Sci. (2018) 39:1977–80. doi: 10.1007/s10072-018-3528-6
8. Piard J, Hawkes L, Milh M, Villard L, Borgatti R, Romaniello R, et al. The phenotypic spectrum of WWOX-related disorders: 20 additional cases of WOREE syndrome and review of the literature. Genet Med. (2019) 21:1308–18. doi: 10.1038/s41436-018-0339-3
9. Repudi S, Steinberg DJ, Elazar N, Breton VL, Aquilino MS, Saleem A, et al. Neuronal deletion of WWOX, associated with WOREE syndrome, causes epilepsy and myelin defects. Brain. (2021) 144:3061–77. doi: 10.1093/brain/awab174
10. Nunez MI, Ludes-Meyers J, Aldaz CM. WWOX protein expression in normal human tissues. J Mol Hist. (2006) 37:115–25. doi: 10.1007/s10735-006-9046-5
11. Aldaz CM, Ferguson BW, Abba MC. WWOX at the crossroads of cancer, metabolic syndrome related traits and CNS pathologies. Biochim Biophys Acta (BBA). (2014) 1846:188–200. doi: 10.1016/j.bbcan.2014.06.001
12. Bednarek AK, Laflin KJ, Daniel RL, Liao Q, Hawkins KA, Aldaz CM. WWOX, a novel WW domain-containing protein mapping to human chromosome 16q23.3-24.1, a region frequently affected in breast cancer. Cancer Res. (2000) 60:2140–5.10786676
13. Abu-Odeh M, Hereema NA, Aqeilan RI. WWOX modulates the ATR-mediated DNA damage checkpoint response. Oncotarget. (2016) 7:4344–55. doi: 10.18632/oncotarget.6571
14. Abu-Remaileh M, Joy-Dodson E, Schueler-Furman O, Aqeilan RI. Pleiotropic functions of tumor suppressor WWOX in normal and cancer cells. J Biol Chem. (2015) 290:30728–35. doi: 10.1074/jbc.R115.676346
15. Hussain T, Lee J, Abba MC, Chen J, Aldaz CM. Delineating WWOX protein interactome by tandem affinity purification-mass spectrometry: identification of top interactors and key metabolic pathways involved. Front Oncol. (2018) 8:591. doi: 10.3389/fonc.2018.00591
16. Ludes-Meyers JH, Kil H, Bednarek AK, Drake J, Bedford MT, Aldaz CM. WWOX binds the specific proline-rich ligand PPXY: identification of candidate interacting proteins. Oncogene. (2004) 23:5049–55. doi: 10.1038/sj.onc.1207680
17. Fu J, Qu Z, Yan P, Ishikawa C, Aqeilan RI, Rabson AB, et al. The tumor suppressor gene WWOX links the canonical and noncanonical NF-κB pathways in HTLV-I tax-mediated tumorigenesis. Blood. (2011) 117:1652–61. doi: 10.1182/blood-2010-08-303073
18. Qu Z, Xiao G. Human T-cell lymphotropic virus: a model of NF-κB-associated tumorigenesis. Viruses. (2011) 3:714–49. doi: 10.3390/v3060714
19. Singla S, Chen J, Sethuraman S, Sysol JR, Gampa A, Zhao S, et al. Loss of lung WWOX expression causes neutrophilic inflammation. Am J Physiol Lung Cell Mol Physiol. (2017) 312:L903–11. doi: 10.1152/ajplung.00034.2017
20. Visschedijk MC, Spekhorst LM, Cheng S-C, Van Loo ES, Jansen BHD, Blokzijl T, et al. Genomic and expression analyses identify a disease-modifying variant for fibrostenotic crohn’s disease. J Crohn’s Colitis. (2018) 12:582–8. doi: 10.1093/ecco-jcc/jjy001
21. Hussain T, Kil H, Hattiangady B, Lee J, Kodali M, Shuai B, et al. WWOX deletion leads to reduced GABA-ergic inhibitory interneuron numbers and activation of microglia and astrocytes in mouse hippocampus. Neurobiol Dis. (2019) 121:163–76. doi: 10.1016/j.nbd.2018.09.026
22. Markesbery WR. Neuropathologic alterations in mild cognitive impairment: a review. JAD. (2010) 19:221–8. doi: 10.3233/JAD-2010-1220
23. Khan UA, Liu L, Provenzano FA, Berman DE, Profaci CP, Sloan R, et al. Molecular drivers and cortical spread of lateral entorhinal cortex dysfunction in preclinical Alzheimer’s disease. Nat Neurosci. (2014) 17:304–11. doi: 10.1038/nn.3606
24. Vismer MS, Forcelli PA, Skopin MD, Gale K, Koubeissi MZ. The piriform, perirhinal, and entorhinal cortex in seizure generation. Front Neural Circuits. (2015) 9. doi: 10.3389/fncir.2015.00027
25. LeDoux JE. Brain mechanisms of emotion and emotional learning. Curr Opin Neurobiol. (1992) 2:191–7. doi: 10.1016/0959-4388(92)90011-9
26. Schumann CM, Bauman MD, Amaral DG. Abnormal structure or function of the amygdala is a common component of neurodevelopmental disorders. Neuropsychologia. (2011) 49:745–59. doi: 10.1016/j.neuropsychologia.2010.09.028
27. Poulin SP, Dautoff R, Morris JC, Barrett LF, Dickerson BC. Amygdala atrophy is prominent in early Alzheimer’s disease and relates to symptom severity. Psychiatry Res Neuroimaging. (2011) 194:7–13. doi: 10.1016/j.pscychresns.2011.06.014
28. Ludes-Meyers JH, Kil H, Nuñez MI, Conti CJ, Parker-Thornburg J, Bedford MT, et al. WWOX hypomorphic mice display a higher incidence of B-cell lymphomas and develop testicular atrophy. Genes Chromosom Cancer. (2007) 46:1129–36. doi: 10.1002/gcc.20497
29. Aqeilan RI, Hagan JP, De Bruin A, Rawahneh M, Salah Z, Gaudio E, et al. Targeted ablation of the WW domain-containing oxidoreductase tumor suppressor leads to impaired steroidogenesis. Endocrinology. (2009) 150:1530–5. doi: 10.1210/en.2008-1087
30. Williams CA, Battaglia A. Molecular biology of epilepsy genes. Exp Neurol. (2013) 244:51–8. doi: 10.1016/j.expneurol.2011.12.001
31. Powell EM. Interneuron development and epilepsy: early genetic defects cause long-term consequences in seizures and susceptibility: links between disrupted interneuron development and human disorders. Epilepsy Curr. (2013) 13:172–6. doi: 10.5698/1535-7597-13.4.172
32. Ferguson BW, Gao X, Zelazowski MJ, Lee J, Jeter CR, Abba MC, et al. The cancer gene WWOX behaves as an inhibitor of SMAD3 transcriptional activity via direct binding. BMC Cancer. (2013) 13:593. doi: 10.1186/1471-2407-13-593
33. Khawaled S, Nigita G, Distefano R, Oster S, Suh S-S, Smith Y, et al. Pleiotropic tumor suppressor functions of WWOX antagonize metastasis. Sig Transduct Target Ther. (2020) 5:43. doi: 10.1038/s41392-020-0136-8
34. Bednarek AK, Keck-Waggoner CL, Daniel RL, Laflin KJ, Bergsagel PL, Kiguchi K, et al. WWOX, the FRA16D gene, behaves as a suppressor of tumor growth. Cancer Res. (2001) 61:8068–73.11719429
35. Banne E, Abudiab B, Abu-Swai S, Repudi SR, Steinberg DJ, Shatleh D, et al. Neurological disorders associated with WWOX germline mutations—a comprehensive overview. Cells. (2021) 10:824. doi: 10.3390/cells10040824
36. Chong SC, Cao Y, Fung ELW, Kleppe S, Gripp KW, Hertecant J, et al. Expansion of the clinical and molecular spectrum of WWOX -related epileptic encephalopathy. A J Med Genet A. (2023) 191:776–85. doi: 10.1002/ajmg.a.63074
37. Liu C-C, Ho P-C, Lee I-T, Chen Y-A, Chu C-H, Teng C-C, et al. WWOX Phosphorylation, signaling, and role in neurodegeneration. Front Neurosci. (2018) 12:563. doi: 10.3389/fnins.2018.00563
38. Sze C-I, Su M, Pugazhenthi S, Jambal P, Hsu L-J, Heath J, et al. Down-regulation of WW domain-containing oxidoreductase induces tau phosphorylation in vitro. J Biol Chem. (2004) 279:30498–506. doi: 10.1074/jbc.M401399200
39. Wang H-Y, Juo L-I, Lin Y-T, Hsiao M, Lin J-T, Tsai C-H, et al. WW domain-containing oxidoreductase promotes neuronal differentiation via negative regulation of glycogen synthase kinase 3β. Cell Death Differ. (2012) 19:1049–59. doi: 10.1038/cdd.2011.188
40. Chang J-Y, Lee M-H, Lin S-R, Yang L-Y, Sun HS, Sze C-I, et al. Trafficking protein particle complex 6A delta (TRAPPC6AΔ) is an extracellular plaque-forming protein in the brain. Oncotarget. (2015) 6:3578–89. doi: 10.18632/oncotarget.2876
41. Jäkel S, Agirre E, Mendanha Falcão A, Van Bruggen D, Lee KW, Knuesel I. Malhotra D, ffrench-constant C, williams A. Castelo-Branco G. Altered human oligodendrocyte heterogeneity in multiple sclerosis. Nature. (2019) 566:543–7. doi: 10.1038/s41586-019-0903-2
42. Ziliotto N, Marchetti G, Scapoli C, Bovolenta M, Meneghetti S, Benazzo A, et al. C6orf10 low-frequency and rare variants in Italian multiple sclerosis patients. Front Genet. (2019) 10:573. doi: 10.3389/fgene.2019.00573
43. Kosugi S, Hasebe M, Tomita M, Yanagawa H. Systematic identification of cell cycle-dependent yeast nucleocytoplasmic shuttling proteins by prediction of composite motifs. Proc Natl Acad Sci U S A. (2009) 106:10171–6. doi: 10.1073/pnas.0900604106
44. Aqeilan RI, Hassan MQ, De Bruin A, Hagan JP, Volinia S, Palumbo T, et al. The WWOX tumor suppressor is essential for postnatal survival and normal bone metabolism. J Biol Chem. (2008) 283:21629–39. doi: 10.1074/jbc.M800855200
45. Cheng Y-Y, Chou Y-T, Lai F-J, Jan M-S, Chang T-H, Jou I-M, et al. Wwox deficiency leads to neurodevelopmental and degenerative neuropathies and glycogen synthase kinase 3β-mediated epileptic seizure activity in mice. Acta Neuropathol Commun. (2020) 8:6. doi: 10.1186/s40478-020-0883-3
46. Havali C, Ekici A, Dorum S, Görükmez Ö, Topak A. Recently defined epileptic encephalopathy related to WWOX gene mutation: six patients and new mutations. Neurol Res. (2021) 43:744–50. doi: 10.1080/01616412.2021.1932173
47. Abdel-Salam G, Thoenes M, Afifi HH, Körber F, Swan D, Bolz HJ. The supposed tumor suppressor gene WWOX is mutated in an early lethal microcephaly syndrome with epilepsy, growth retardation and retinal degeneration. Orphanet J Rare Dis. (2014) 9:12. doi: 10.1186/1750-1172-9-12
48. Valduga M, Philippe C, Lambert L, Bach-Segura P, Schmitt E, Masutti JP, et al. WWOX and severe autosomal recessive epileptic encephalopathy: first case in the prenatal period. J Hum Genet. (2015) 60:267–71. doi: 10.1038/jhg.2015.17
49. Weisz-Hubshman M, Meirson H, Michaelson-Cohen R, Beeri R, Tzur S, Bormans C, et al. Novel WWOX deleterious variants cause early infantile epileptic encephalopathy, severe developmental delay and dysmorphism among yemenite Jews. Eur J Paediatr Neurol. (2019) 23:418–26. doi: 10.1016/j.ejpn.2019.02.003
50. Iacomino M, Baldassari S, Tochigi Y, Kośla K, Buffelli F, Torella A, et al. Loss of WWOX perturbs neuronal migration and impairs early cortical development. Front Neurosci. (2020) 14:644. doi: 10.3389/fnins.2020.00644
51. Tabarki B, AlHashem A, AlShahwan S, Alkuraya FS, Gedela S, Zuccoli G. Severe CNS involvement in WWOX mutations: description of five new cases. Am J Med Genet A. (2015) 167:3209–13. doi: 10.1002/ajmg.a.37363
52. Tarta-Arsene O, Barca D, Craiu D, Iliescu C. Practical clues for diagnosing WWOX encephalopathy. Epileptic Disord. (2017) 19:357–61. doi: 10.1684/epd.2017.0924
53. Johannsen J, Kortüm F, Rosenberger G, Bokelmann K, Schirmer MA, Denecke J, et al. A novel missense variant in the SDR domain of the WWOX gene leads to complete loss of WWOX protein with early-onset epileptic encephalopathy and severe developmental delay. Neurogenetics. (2018) 19:151–6. doi: 10.1007/s10048-018-0549-5
54. He J, Zhou W, Shi J, Zhang B, Wang H. A Chinese patient with epilepsy and WWOX compound heterozygous mutations. Epileptic Disord. (2020) 22:120–4. doi: 10.1684/epd.2020.1131
55. Wiessner M, Maroofian R, Ni M-Y, Pedroni A, Müller JS, Stucka R, et al. Biallelic variants in HPDL cause pure and complicated hereditary spastic paraplegia. Brain. (2021) 144:1422–34. doi: 10.1093/brain/awab041
56. Dworschak GC, Punetha J, Kalanithy JC, Mingardo E, Erdem HB, Akdemir ZC, et al. Biallelic and monoallelic variants in PLXNA1 are implicated in a novel neurodevelopmental disorder with variable cerebral and eye anomalies. Genet Med. (2021) 23:1715–25. doi: 10.1038/s41436-021-01196-9
57. Neuray C, Maroofian R, Scala M, Sultan T, Pai GS, Mojarrad M, et al. Early-infantile onset epilepsy and developmental delay caused by bi-allelic GAD1 variants. Brain. (2020) 143:2388–97. doi: 10.1093/brain/awaa178
58. Donkervoort S, Kutzner CE, Hu Y, Lornage X, Rendu J, Stojkovic T, et al. Pathogenic variants in the myosin chaperone UNC-45B cause progressive myopathy with eccentric cores. Am J Hum Genet. (2020) 107:1078–95. doi: 10.1016/j.ajhg.2020.11.002
59. Manole A, Efthymiou S, O’Connor E, Mendes MI, Jennings M, Maroofian R, et al. De novo and bi-allelic pathogenic variants in NARS1 cause neurodevelopmental delay due to toxic gain-of-function and partial loss-of-function effects. Am J Hum Genet. (2020) 107:311–24. doi: 10.1016/j.ajhg.2020.06.016
60. Dias CM, Punetha J, Zheng C, Mazaheri N, Rad A, Efthymiou S, et al. Homozygous missense variants in NTNG2, encoding a presynaptic netrin-G2 adhesion protein, lead to a distinct neurodevelopmental disorder. Am J Hum Genet. (2019) 105:1048–56. doi: 10.1016/j.ajhg.2019.09.025
61. Pavlidou E, Salpietro V, Phadke R, Hargreaves IP, Batten L, McElreavy K, et al. Pontocerebellar hypoplasia type 2D and optic nerve atrophy further expand the spectrum associated with selenoprotein biosynthesis deficiency. Eur J Paediatr Neurol. (2016) 20:483–8. doi: 10.1016/j.ejpn.2015.12.016
62. Efthymiou S, Salpietro V, Malintan N, Poncelet M, Kriouile Y, Fortuna S, et al. Biallelic mutations in neurofascin cause neurodevelopmental impairment and peripheral demyelination. Brain. (2019) 142:2948–64. doi: 10.1093/brain/awz248
63. Granata F, Morabito R, Mormina E, Alafaci C, Marino S, Laganà A, et al. 3T double inversion recovery magnetic resonance imaging: diagnostic advantages in the evaluation of cortical development anomalies. Eur J Radiol. (2016) 85:906–14. doi: 10.1016/j.ejrad.2016.02.018
64. Niestroj L-M, Perez-Palma E, Howrigan DP, Zhou Y, Cheng F, Saarentaus E, et al. Epilepsy subtype-specific copy number burden observed in a genome-wide study of 17458 subjects. Brain. (2020) 143:2106–18. doi: 10.1093/brain/awaa171
65. Baldassari S, Musante I, Iacomino M, Zara F, Salpietro V, Scudieri P. Brain organoids as model systems for genetic neurodevelopmental disorders. Front Cell Dev Biol. (2020) 8:590119. doi: 10.3389/fcell.2020.590119
66. Salpietro V, Manole A, Efthymiou S, Houlden H. A review of copy number variants in inherited neuropathies. CG. (2018) 19:412–9. doi: 10.2174/1389202919666180330153316
67. Salpietro V, Efthymiou S, Manole A, Maurya B, Wiethoff S, Ashokkumar B, et al. A loss-of-function homozygous mutation in DDX59 implicates a conserved DEAD-box RNA helicase in nervous system development and function. Hum Mutat. (2018) 39:187–92. doi: 10.1002/humu.23368
68. Salpietro V, Zollo M, Vandrovcova J, Ryten M, Botia JA, Ferrucci V, et al. The phenotypic and molecular spectrum of PEHO syndrome and PEHO-like disorders. Brain. (2017) 140:e49. doi: 10.1093/brain/awx155
69. Steel D, Salpietro V, Phadke R, Pitt M, Gentile G, Massoud A, et al. Whole exome sequencing reveals a MLL de novo mutation associated with mild developmental delay and without “hairy elbows”: expanding the phenotype of wiedemann-steiner syndrome. J Genet. (2015) 94:755–8. doi: 10.1007/s12041-015-0578-x
Keywords: WWOX, developmental and epileptic encephalopathies (DEEs), WOREE syndrome, SDR domain, brain anomalies
Citation: Battaglia L, Scorrano G, Spiaggia R, Basile A, Palmucci S, Foti PV, Spatola C, Iacomino M, Marinangeli F, Francia E, Comisi F, Corsello A, Salpietro V, Vittori A and David E (2023) Neuroimaging features of WOREE syndrome: a mini-review of the literature. Front. Pediatr. 11:1301166. doi: 10.3389/fped.2023.1301166
Received: 24 September 2023; Accepted: 4 December 2023;
Published: 15 December 2023.
Edited by:
Nan-Shan Chang, China Medical University, TaiwanReviewed by:
Eirik Frengen, University of Oslo, NorwayGabrielle Rudolf, Université de Strasbourg, France
© 2023 Battaglia, Scorrano, Spiaggia, Basile, Palmucci, Foti, Spatola, Iacomino, Marinangeli, Francia, Comisi, Corsello, Salpietro, Vittori and David. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alessandro Vittori YWxleHZpdHRvcmk4MkBnbWFpbC5jb20=