 Gianluca Dini1*
Gianluca Dini1* Alberto Verrotti1
Alberto Verrotti1 Paolo Gorello2
Paolo Gorello2 Luca Soliani3,4
Luca Soliani3,4 Duccio Maria Cordelli3,4Vincenzo Antona5Amedea Mencarelli6Davide Colavito7Paolo Prontera6
Duccio Maria Cordelli3,4Vincenzo Antona5Amedea Mencarelli6Davide Colavito7Paolo Prontera6
- 1Department of Pediatrics, University of Perugia, Perugia, Italy
- 2Department of Chemistry, Biology and Biotechnology, University of Perugia, Perugia, Italy
- 3IRCCS Istituto Delle Scienze Neurologiche di Bologna, UOC di Neuropsichiatria Dell'Età Pediatrica, Bologna, Italy
- 4Dipartimento di Scienze Mediche e Chirurgiche (DIMEC), Università di Bologna, Bologna, Italy
- 5Department of Health Promotion, Mother and Child Care, Internal Medicine and Medical Specialties “G. D'Alessandro,” University of Palermo, Palermo, Italy
- 6Medical Genetics Unit, S. Maria della Misericordia Hospital, Perugia, Italy
- 7Research & Innovation S.R.L. (R&I Genetics), Padova, Italy
Background: NFIA-related disorder (OMIM #613735) is an autosomal dominant neurodevelopmental disorder characterized by a variable degree of cognitive impairment and non-specific dysmorphic features. To date, fewer than thirty patients affected by this disorder have been described.
Methods: Our study included three children with NFIA haploinsufficiency recruited from three medical genetics centers. Clinical presentations were recorded on a standardized case report form.
Results: All patients presented a variable degree of intellectual disability. None of the individuals in our cohort had urinary tract malformations. Three novel mutations, c.344G>A, c.261T>G, and c.887_888del are reported here.
Conclusion: NFIA haploinsufficiency can be suspected through careful observation of specific dysmorphisms, including macrocephaly and craniofacial abnormalities. Instrumental tests such as MRI and renal ultrasound provide further diagnostic clues, while genetic testing can confirm the diagnosis.
1. Introduction
The NFIA (nuclear factor I/A) gene encodes a transcription factor belonging to the nuclear factor I family and plays key roles in central nervous system (CNS) development, including axonal growth, glial cell differentiation, and neuronal migration (1). Initially, the phenotypic presentation resulting from deletions affecting NFIA was referred to as chromosome 1p32-p31 deletion syndrome. However, the observation of overlapping phenotypes in patients with chromosome translocations that disrupt NFIA has demonstrated that the loss of function of NFIA is responsible for most of the phenotypes associated with the 1p32-p31 deletion (2–4). Haploinsufficiency of NFIA causes a syndrome characterized by brain malformations, most commonly abnormalities of the corpus callosum, with or without urinary tract defects (BRMUTD, OMIM#613735). Additional features include macrocephaly, seizures, developmental delay, ventriculomegaly, and hypotonia (5, 6). To date, fewer than thirty patients affected by this disorder have been described. Herein, we report three additional patients showing a variable degree of intellectual disability (ID) and CNS abnormalities, all carrying a heterozygous de novo mutation in the NFIA gene. Our findings provide further evidence that the NFIA gene has a role in development of the CNS.
2. Materials and methods
Our analysis included three molecularly confirmed NFIA haploinsufficiency patients, aged between 3 and 13 years, none of whom have been previously described. Genomic DNA of the patients was extracted from peripheral blood using standard methods. Information on the age and sex of the children was obtained from their medical records. DNA samples were collected as parent/child trios. Exome sequencing (ES) was performed for Patient 1 and his parents using a Twist Custom Panel (Twist Bioscience, San Francisco, CA, USA) on a NovaSeq 6,000 platform (Illumina Inc., San Diego, CA, USA). For patients 2 and 3, we performed ES combined with variant analysis within a panel of neurodevelopmental disorders-related genes, as previously described (7). All patients were born to non-consanguineous parents. The study was conducted in accordance with the Declaration of Helsinki. Written informed consent has been obtained from the parents to publish clinical information with photographs. We summarized the molecular findings and clinical presentation of our patients in Table 1.
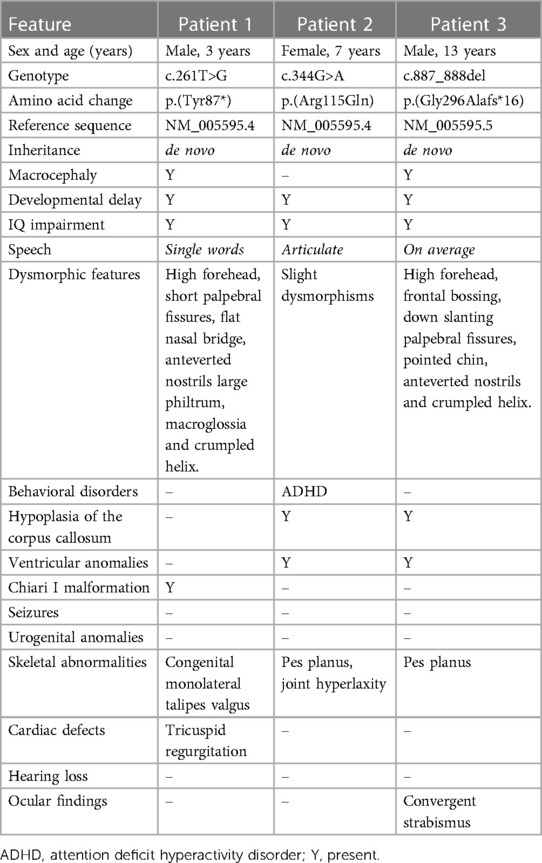
Table 1. Clinical characteristics of patients in our cohort.
3. Results
3.1. Patient 1
Patient 1 (P1) is a 3-year-old male who was referred to the pediatric unit for macrocephaly and developmental delay. He was born after 39 weeks of gestation by C-section because of non-reassuring fetal status. Pregnancy was uncomplicated by teratogenic exposures or maternal illness. Birth weight was 3,620 g (76th centile), his head circumference was 39.5 cm (+4.7 SD), and his birth length was 50 cm (50th centile). Regarding his developmental milestones, he started walking without support at 15 months and speaking his first words at 19 months. The Bayley Infant Development Scale (3rd edition, BSID-III) at 2 years of age, demonstrated moderate global cognitive impairment. At the time of referral, his head circumference was 57.0 cm (>97th centile). The patient presented with a high forehead, prominent occipital protuberance, short palpebral fissures, flat nasal bridge, large philtrum, macroglossia, and a crumpled helix (Figure 1). Speech consisted of about three words in an appropriate context. The brain MRI carried out at 2.5 years, showed Chiari I malformation and subependymal grey matter heterotopia (Figure 2). Trio exome analysis revealed a de novo mutation c.261T>G, p.(Tyr87*) in the NFIA gene.
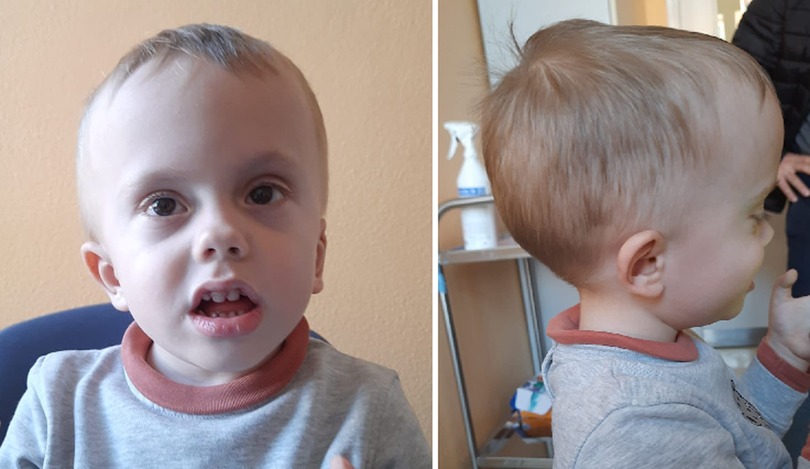
Figure 1. Facial features of patient 1 (c.261T>G, p.Tyr87*), including high forehead, short palpebral fissures, flat nasal bridge, and large philtrum.
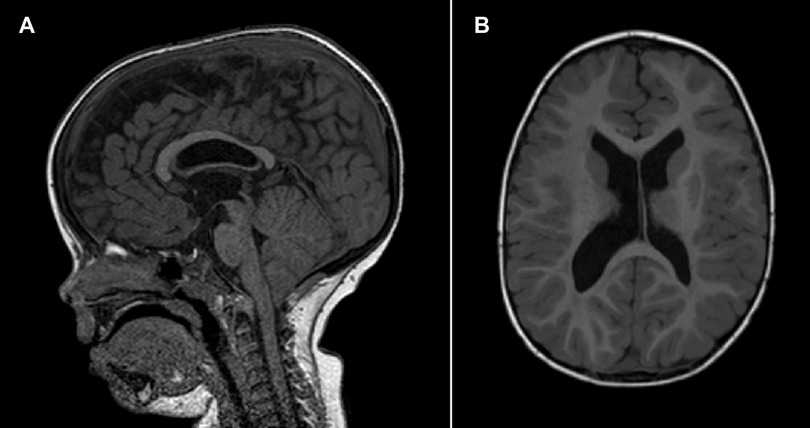
Figure 2. Brain MRI of patient 1 at 2.5 years of age. (A) Mid-sagittal T1-weighted image showing Chiari I malformation. (B) Axial T1-weighted image showing subependymal heterotopia.
3.2. Patient 2
Patient 2 (P2) (Figure 3) is the second child of non-consanguineous parents with an unremarkable family history. She was born by vaginal delivery after an uneventful pregnancy. The birth weight was 2,945 g (12th centile), length 48 cm (14th centile), and head circumference 34.5 cm (64th centile). No apparent external malformations were observed. The brain MRI at 3 years of age (Figure 4) revealed a thin corpus callosum, dysmorphic appearance of the lateral ventricles, and a retrocerebellar arachnoid cyst. Renal ultrasound was normal. When last examined at age 7 years, her height was 134 cm (>97th centile), her weight was 28.5 kg (90th centile). Her head circumference was within the normal range, measuring at 53 cm (63rd centile). The neurobehavioral evaluation showed a learning disability and psychopathological findings, such as attention deficit hyperactivity disorder (ADHD), bizarre behaviour and emotional dysregulation. No epileptic seizures have been observed to date. Trio ES identified a heterozygous missense variant (c.344G>A, p.Arg115Gln) in the NFIA gene.

Figure 3. Photographs of patient 2 (c.344G>A, p.Arg115Gln) showing mild dysmorphic facial features and pes planus.
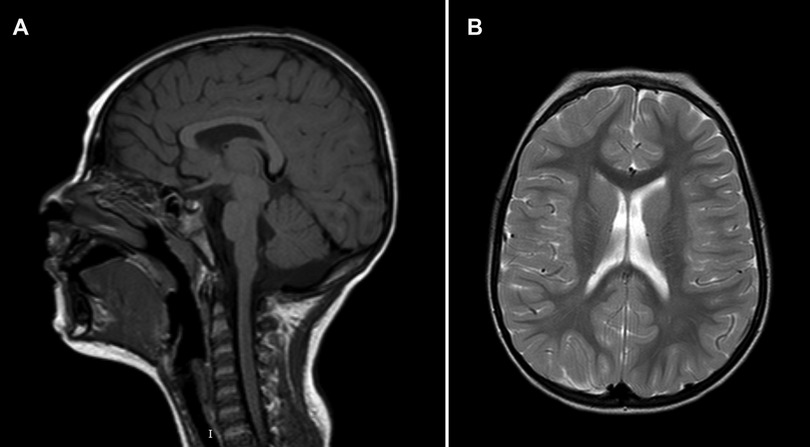
Figure 4. Brain MRI of patient 2 at 3 years of age. (A) Mid-sagittal T1-weighted image showing hypoplasia of the corpus callosum. (B) Axial T2-weighted image at the level of the lateral ventricles.
3.3. Patient 3
Patient 3 (P3) is the male, first child of healthy non-consanguineous parents. The family history was uneventful for disabilities. We first evaluated the patient at the age of 5.1 years for macrocephaly and developmental delay. The weight was 21.8 kg (75–90th centile), height 116 cm (95th centile), and head circumference 56.5 cm (>97th centile). Facial anomalies included frontal bossing, high forehead, down-slanting palpebral fissures, a pointed chin, anteverted nostrils, and crumpled helix in the left ear. Ophthalmologic examination, auditory brainstem response, electroencephalography, echocardiography and renal ultrasound were unremarkable. Brain MRI revealed a hypoplastic corpus callosum with enlarged ventricles and agenesis of the septum pellucidum. Metabolic work up, karyogram, chromosomal microarray, and genetic studies for Sotos syndrome were all normal. At the age of 13, the family pediatrician required new medical advice for dysmorphisms. We performed trio ES. This analysis revealed a de novo heterozygous frameshift mutation (c.887_888del, p.Gly296Alafs*16) in the NFIA gene.
4. Discussion
The NFIA gene belongs to a family of genes known as NFI, which also consists of NFIB, NFIC, and NFIX in vertebrates (8). Lu et al. (9) initially proposed the hypothesis that NFIA haploinsufficiency might be responsible for CNS malformations, as well as ureteral and renal defects. These authors also demonstrated ventricular enlargement, callosal agenesis, and urinary tract defects in homozygous Nfia−/− mice and heterozygous Nfia+/− mice. Interestingly, haploinsufficiency of the NFIB and NFIX genes also causes callosal agenesis (10–12), and missense mutations in the NFIX gene have been associated with Sotos-like features (10, 13).
In this case series, we described the clinical and molecular characteristics of three patients with NFIA haploinsufficiency. Similar to previous reports, the patients in our study presented with CNS abnormalities and a variable degree of intellectual disability. Table 2 provides a comparison of our cohort and previously reported patients with NFIA mutations. Hereafter, the major clinical characteristics have been grouped into macroareas.
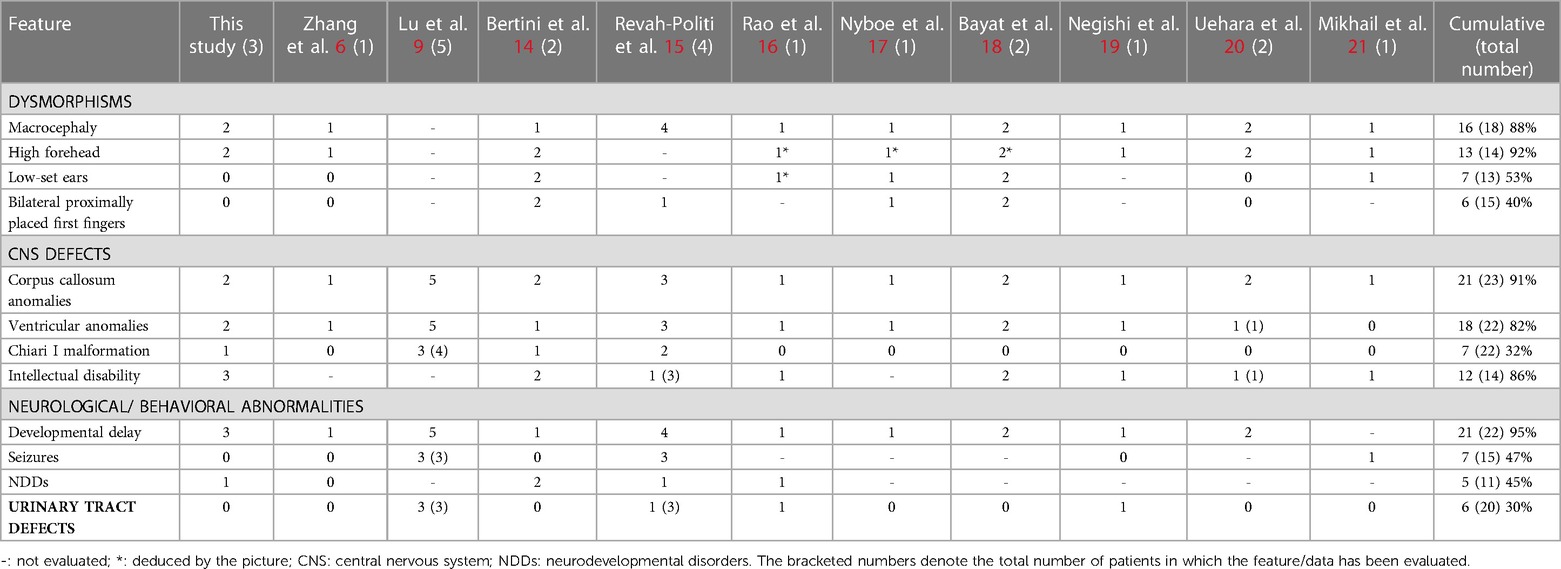
Table 2. Frequency of specified features in our patients compared with cumulative data (ours and previously reported cases).
4.1. Dysmorphisms
Among the physical anomalies, macrocephaly and a high forehead are recurrent signs. In our cohort, P1 and P3 exhibited the typical facial features of NFIA haploinsufficiency. P2 presented a milder phenotype with only slight dysmorphisms. Craniofacial anomalies, such as asymmetries and craniosynostosis, are commonly observed in patients with NFIA haploinsufficiency.
4.2. Developmental progress/learning difficulties
The developmental milestones, especially speech, were delayed in our cohort; P2 and P3 began using single words by 2 years of age, while P1 had only a few words by the age of three. All children achieved walking. In no instances did patients lose skills or regress. All patients received special education at school.
4.3. CNS defects
CNS defects, most commonly abnormalities of the corpus callosum, are a prominent characteristic of NFIA haploinsufficiency. In addition to these, a range of less frequent and heterogeneous CNS anomalies have been reported, including cortical malformations, non-progressive ventriculomegaly, hydrocephalus, Chiari type I malformation, and tethered spinal cord.
4.4. Seizures
Seizures have been observed in approximately 15% of the NFIA patients (14). In our study, none of the patients experienced seizures. EEG was performed for P2 and P3, and the results were normal for both.
4.5. Neurodevelopment/behavior
Developmental delay and/or ID, ranging from very mild to severe, are invariably present in this disorder. Beside ID and developmental delay, patients may exhibit other neurodevelopmental disorders, including hyperactivity disorder and oppositional defiant disorder (ODD) (14). In our cohort, one patient (P2) had a diagnosis of ADHD.
4.6. Urinary trait defects
Renal/urinary defects are considered a key feature of NFIA-related disorder (9). The most common among these defects are vesicoureteral reflux and hydronephrosis, which can occur unilaterally or bilaterally. Other phenotypes associated with NFIA-related disorder include pyelonephritis, ureterovesical junction diverticulum, dysplastic kidneys, and renal cysts.
According to Revah-Politi et al., approximately 50% of patients diagnosed with NFIA-haploinsufficiency exhibited genitourinary defects (15). However, a subsequent literature review indicated a lower prevalence of urinary tract abnormalities (<20%). In our cohort, no patients presented with genitourinary abnormalities. Nevertheless, a surveillance of kidneys and urinary trait is recommended when NFIA-haploinsufficiency is detected.
4.7. Musculoskeletal abnormalities
Although musculoskeletal manifestations are not considered a typical sign of NFIA-related disorder, these features are frequently found in patients with the NFIA gene mutation. Bilateral proximally placed first fingers seem to be a frequently reported dysmorphism in patients with NFIA-related disorder (14). In our study, two patients (P2 and P3) presented with pes planus, while another with congenital talipes valgus (P1). Interestingly, it was demonstrated that NFIA and GATA-binding protein 3 regulate the differentiation of embryonic articular cartilage in chicken (22). The musculoskeletal manifestations associated with NFIA haploinsufficiency need further characterization to provide more definitive insights and treatment options for patients.
5. Conclusions
NFIA haploinsufficiency can be suspected through careful observation of specific dysmorphisms, including macrocephaly and craniofacial abnormalities. Instrumental tests, such as MRI and renal ultrasound, can offer further diagnostic clues, with definitive diagnosis achievable through genetic testing.
Three novel mutations, c.344G>A, c.261T>G, and c.887_888del are reported here. Like other authors, we did not identify a predictive value for specific mutations. However, the mutation c.344G>A might correlate with a milder phenotype. With the current use of clinical exome sequencing, it is expected that more patients with point mutations or microdeletions in NFIA will be diagnosed. Currently, a specific treatment for NFIA-related disorder is lacking, and symptomatic supportive treatment remains the main approach in clinical practice.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below:
https://databases.lovd.nl/shared/variants/0000933608#00014516
https://databases.lovd.nl/shared/variants/0000933609#00014516
https://databases.lovd.nl/shared/variants/0000933610#00014516
Ethics statement
Ethical approval was not required for the study involving human samples in accordance with the local legislation and institutional requirements because our institution does not require ethical approval for reporting individual cases or case series. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
GD: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Writing – original draft. AV: Supervision, Writing – review & editing. PG: Conceptualization, Data curation, Writing – original draft. LS: Data curation, Investigation, Writing – original draft. DC: Conceptualization, Data curation, Investigation, Writing – original draft. VA: Conceptualization, Data curation, Investigation, Writing – original draft. AM: Conceptualization, Data curation, Investigation, Writing – original draft. DC: Investigation, Writing – original draft. PP: Data curation, Investigation, Supervision, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Qian F, Kruse U, Lichter P, Sippel AE. Chromosomal localization of the four genes (NFIA, B. C, and X) for the human transcription factor nuclear factor I by fisH. Genomics. (1995) 28(1):66–73. doi: 10.1006/geno.1995.1107
2. Ji J, Salamon N, Quintero-Rivera F. Microdeletion of 1p32-p31 involving NFIA in a patient with hypoplastic corpus callosum, ventriculomegaly, seizures and urinary tract defects. Eur J Med Genet. (2014) 57(6):267–8. doi: 10.1016/j.ejmg.2014.03.004
3. Chen CP, Su YN, Chen YY, Chern SR, Liu YP, Wu PC, et al. Chromosome 1p32-p31 deletion syndrome: prenatal diagnosis by array comparative genomic hybridization using uncultured amniocytes and association with NFIA haploinsufficiency, ventriculomegaly, corpus callosum hypogenesis, abnormal external genitalia, and intrauterine growth restriction. Taiwan J Obstet Gynecol. (2011) 50(3):345–52. doi: 10.1016/j.tjog.2011.07.014
4. Koehler U, Holinski-Feder E, Ertl-Wagner B, Kunz J, von Moers A, von Voss H, et al. A novel 1p31.3p32.2 deletion involving the NFIA gene detected by array CGH in a patient with macrocephaly and hypoplasia of the corpus callosum. Eur J Pediatr. (2010) 169(4):463–8. doi: 10.1007/s00431-009-1057-2
5. Senaratne TN, Quintero-Rivera F. NFIA-Related Disorder. In: Adam MP, Everman DB, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. Genereviews®. Seattle (WA): University of Washington, Seattle (2019). p. 1993–2023.
6. Zhang Y, Lin CM, Zheng XL, Abuduxikuer K. A novel NFIA gene nonsense mutation in a Chinese patient with macrocephaly, corpus callosum hypoplasia, developmental delay, and dysmorphic features. Mol Genet Genomic Med. (2020) 8(11):e1492. doi: 10.1002/mgg3.1492
7. Prontera P, Ottaviani V, Rogaia D, Isidori I, Mencarelli A, Malerba N, et al. A novel MED12 mutation: evidence for a fourth phenotype. Am J Med Genet A. (2016) 170(9):2377–82. doi: 10.1002/ajmg.a.37805
8. Mason S, Piper M, Gronostajski RM, Richards LJ. Nuclear factor one transcription factors in CNS development. Mol Neurobiol. (2009) 39(1):10–23. doi: 10.1007/s12035-008-8048-6
9. Lu W, Quintero-Rivera F, Fan Y, Alkuraya FS, Donovan DJ, Xi Q, et al. NFIA Haploinsufficiency is associated with a CNS malformation syndrome and urinary tract defects. PLoS Genet. (2007) 3(5):e80. doi: 10.1371/journal.pgen.0030080
10. Sajan SA, Fernandez L, Nieh SE, Rider E, Bukshpun P, Wakahiro M, et al. Both rare and de novo copy number variants are prevalent in agenesis of the corpus callosum but not in cerebellar hypoplasia or polymicrogyria. PLoS Genet. (2013) 9(10):e1003823. doi: 10.1371/journal.pgen.1003823
11. Malan V, Rajan D, Thomas S, Shaw AC, Louis Dit Picard H, Layet V, et al. Distinct effects of allelic NFIX mutations on nonsense-mediated mRNA decay engender either a sotos-like or a marshall-smith syndrome. Am J Hum Genet. (2010) 87(2):189–98. doi: 10.1016/j.ajhg.2010.07.001
12. Steele-Perkins G, Plachez C, Butz KG, Yang G, Bachurski CJ, Kinsman SL, et al. The transcription factor gene nfib is essential for both lung maturation and brain development. Mol Cell Biol. (2005) 25(2):685–98. doi: 10.1128/MCB.25.2.685-698.2005
13. Yoneda Y, Saitsu H, Touyama M, Makita Y, Miyamoto A, Hamada K, et al. Missense mutations in the DNA-binding/dimerization domain of NFIX cause sotos-like features. J Hum Genet. (2012) 57(3):207–11. doi: 10.1038/jhg.2012.7
14. Bertini V, Cambi F, Orsini A, Bonuccelli A, Fiorini A, Santangelo A, et al. Phenotypic Spectrum of NFIA haploinsufficiency: two additional cases and review of the literature. Genes (Basel). (2022) 13(12):2249. doi: 10.3390/genes13122249
15. Revah-Politi A, Ganapathi M, Bier L, Cho MT, Goldstein DB, Hemati P, et al. Loss-of-function variants in NFIA provide further support that NFIA is a critical gene in 1p32-p31 deletion syndrome: a four patient series. Am J Med Genet A. (2017) 173(12):3158–64. doi: 10.1002/ajmg.a.38460
16. Rao A, O'Donnell S, Bain N, Meldrum C, Shorter D, Goel H. An intragenic deletion of the NFIA gene in a patient with a hypoplastic corpus callosum, craniofacial abnormalities and urinary tract defects. Eur J Med Genet. (2014) 57(2-3):65–70. doi: 10.1016/j.ejmg.2013.12.011
17. Nyboe D, Kreiborg S, Kirchhoff M, Hove HB. Familial craniosynostosis associated with a microdeletion involving the NFIA gene. Clin Dysmorphol. (2015) 24(3):109–12. doi: 10.1097/MCD.0000000000000079
18. Bayat A, Kirchhoff M, Madsen CG, Roos L, Kreiborg S. Familial craniofacial abnormality and polymicrogyria associated with a microdeletion affecting the NFIA gene. Clin Dysmorphol. (2017) 26(3):148–53. doi: 10.1097/MCD.0000000000000182
19. Negishi Y, Miya F, Hattori A, Mizuno K, Hori I, Ando N, et al. Truncating mutation in NFIA causes brain malformation and urinary tract defects. Hum Genome Var. (2015) 2:15007. doi: 10.1038/hgv.2015.7
20. Uehara T, Sanuki R, Ogura Y, Yokoyama A, Yoshida T, Futagawa H, et al. Recurrent NFIA K125E substitution represents a loss-of-function allele: sensitive in vitro and in vivo assays for nontruncating alleles. Am J Med Genet A. (2021) 185(7):2084–93. doi: 10.1002/ajmg.a.62226
21. Mikhail FM, Lose EJ, Robin NH, Descartes MD, Rutledge KD, Rutledge SL, et al. Clinically relevant single gene or intragenic deletions encompassing critical neurodevelopmental genes in patients with developmental delay, mental retardation, and/or autism spectrum disorders. Am J Med Genet A. (2011) 155A(10):2386–96. doi: 10.1002/ajmg.a.34177
Keywords: NFIA, neurodevelopmental disorders, intellectual disability, genetics, pediatrics
Citation: Dini G, Verrotti A, Gorello P, Soliani L, Cordelli DM, Antona V, Mencarelli A, Colavito D and Prontera P (2023) NFIA haploinsufficiency: case series and literature review. Front. Pediatr. 11:1292654. doi: 10.3389/fped.2023.1292654
Received: 11 September 2023; Accepted: 27 September 2023;
Published: 17 October 2023.
Edited by:
Katherina Walz, University of Miami, United StatesReviewed by:
Andrew Sobering, Augusta University/University of Georgia Medical Partnership, United StatesMiguel Angel Alcántara-Ortigoza, National Institute of Pediatrics, Mexico
© 2023 Dini, Verrotti, Gorello, Soliani, Cordelli, Antona, Mencarelli, Colavito and Prontera. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gianluca Dini Z2lhbmx1Y2FkaW5pOTBAZ21haWwuY29t