 Giulia Ceglie
Giulia Ceglie Marco Lecis
Marco Lecis Gabriele Canciani
Gabriele Canciani Mattia Algeri
Mattia Algeri Giacomo Frati
Giacomo Frati
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pediatr. , 02 November 2023
Sec. Pediatric Hematology and Hematological Malignancies
Volume 11 - 2023 | https://doi.org/10.3389/fped.2023.1249275
This article is part of the Research Topic Recent Advances in Pediatric Red Blood Cells Disorders View all 8 articles
Sickle cell disease (SCD) is an inherited blood disorder, due to a single point mutation in the β-globin gene (HBB) leading to multisystemic manifestations and it affects millions of people worldwide. The monogenic nature of the disease and the availability of autologous hematopoietic stem cells (HSCs) make this disorder an ideal candidate for gene modification strategies. Notably, significant advances in the field of gene therapy and genome editing that took place in the last decade enabled the possibility to develop several strategies for the treatment of SCD. These curative approaches were firstly based on the correction of disease-causing mutations holding the promise for a specific, effective and safe option for patients. Specifically, gene-editing approaches exploiting the homology directed repair pathway were investigated, but soon their limited efficacy in quiescent HSC has curbed their wider development. On the other hand, a number of studies on globin gene regulation, led to the development of several genome editing strategies based on the reactivation of the fetal γ-globin gene (HBG) by nuclease-mediated targeting of HBG-repressor elements. Although the efficiency of these strategies seems to be confirmed in preclinical and clinical studies, very little is known about the long-term consequences of these modifications. Moreover, the potential genotoxicity of these nuclease-based strategies must be taken into account, especially when associated with high targeting rates. The recent introduction of nuclease-free genome editing technologies brought along the potential for safer strategies for SCD gene correction, which may also harbor significant advantages over HBG-reactivating ones. In this Review, we discuss the recent advances in genome editing strategies for the correction of SCD-causing mutations trying to recapitulate the promising strategies currently available and their relative strengths and weaknesses.
Sickle cell disease (SCD) is a disorder characterized by the inheritance of a single base substitution in the first exon of the β-globin gene (HBB). This single point mutation causes an aminoacidic replacement of the hydrophilic glutamic acid with a hydrophobic valine at the 7th codon of β-globin chain. Whether inherited in a homozygous manner or with another mutation in the HBB, this substitution significantly alters the function of hemoglobin constituting a pathological form of hemoglobin, defined as sickle hemoglobin (α2βs2, HbS). The mutant HbS undergoes polymerization and aggregation upon deoxygenation, thus conferring the typical sickle shape to the Red Blood Cells (RBCs). This distortion results in decreased RBCs' survival and several downstream clinical manifestations, including chronic anemia, pain events, stroke, multiorgan damage and failure, and premature mortality (1). The two cornerstones of SCD management, blood transfusions and Hydroxyurea, have greatly increased the survival and quality of life of patients but do not fully eliminate the consequences of the disease (2). So far, the only curative treatment is represented by allogeneic hematopoietic stem cell (HSC) transplantation, when a compatible donor is available. Nevertheless, less than 15% of patients with SCD have an appropriately matched donor (3). In this context, gene therapy based on the autologous transplantation of genetically modified HSC constitutes a promising strategy to overcome the major barriers in the cure of SCD (Figure 1). Classical viral-mediated gene addition protocols, based on the delivery of a functional copy of the HBB gene, have been successfully translated into several clinical trials mostly resulting in a decreased transfusion need for patients (4–7). However, some limitations (e.g., limited transduction efficiency and engraftment capability of HSC; variable Hb production levels; high manufacturing costs of the viral vector) still exist and represent a limiting factor for a broad use of these strategies (8, 9). Moreover, it is worth considering that the use of integrating vectors comes with significant safety issues. Recently, the occurrence of malignant transformations has been reported in two SCD patients who underwent vector-integrating gene therapy. One of these patients presented leukemic blasts carrying a lentiviral vector insertion site, unveiling the possible pathogenetic role of insertional mutagenesis events (10, 11). Although the actual causal link of these events to the transduction is yet to be proven, these cases caused a partial suspension of clinical trials in Europe and the U.S, highlighting the need of a better comprehension of vector-integrating strategies security profile (12, 13). From this perspective a clear need for safer option emerges and gene editing techniques appears alternative approaches. Programmable nucleases are chimeric molecules composed of two portions: (i) a DNA-binding structure, either RNA or a protein, and (ii) an effector protein domain, capable, through its nuclease activity, to induce a double-strand break (DSB) on the DNA within or in proximity to the binding site. Since their introduction, 4 classes of nucleases have been described: Meganucleases, Zinc Finger Nucleases (ZFNs), Transcription Activator-Like Effector Nucleases (TALENs), and Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas9. This last class, thanks to its peculiar DNA-gRNA base-pairing, is the more efficient one. CRISPR/Cas9 system creates DSB at a specific genomic locus followed by recruitment of DNA repair mechanism. Among the others, two are those that are mainly involved in genome editing strategies: (i) non-homologous-end-joining (NHEJ), a homology-independent pathway that involves the alignment of only one to a few bases at most for the re-ligation of two ends; (ii) homology directed repair (HDR) that involves longer stretches of sequence as template to repair DNA lesions.
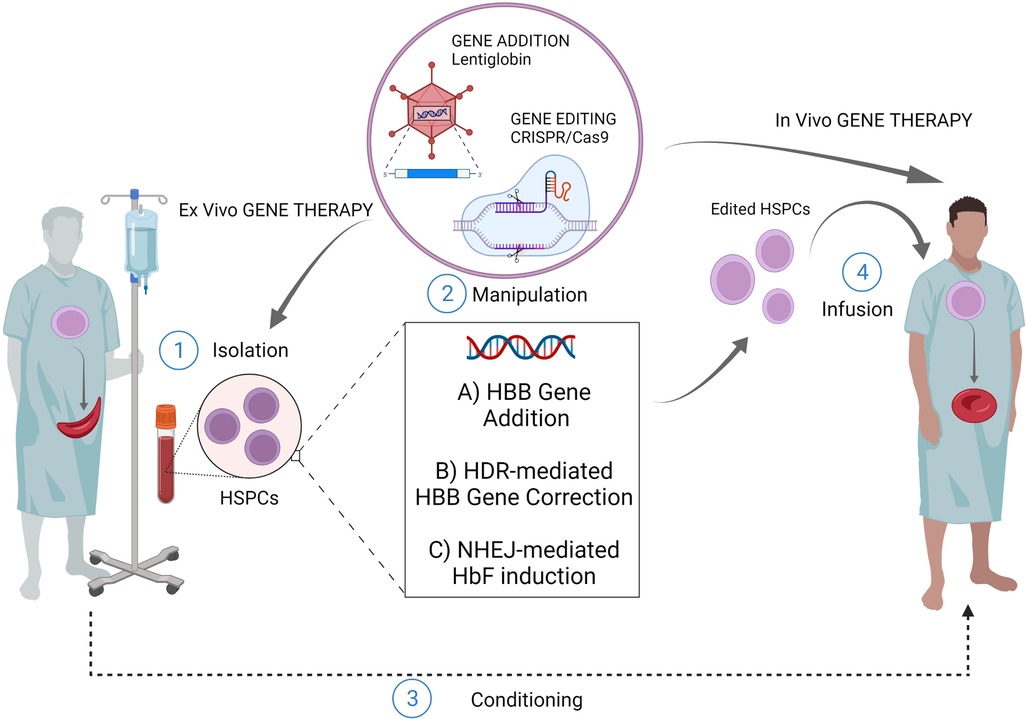
Figure 1. Gene therapy/editing for SCD. The ex vivo gene therapy relies upon isolation of patients HSPCs, their manipulation and subsequent infusion of the cellular product after a conditioning regimen. The most exploited strategies for this scope are: ex vivo gene addition based on virus-mediated delivery of a functional copy of HBB gene (e.g. lentiglobin); ex vivo genome editing (nuclease based HDR-mediated correction of the HBB gene); ex vivo gene editing (nuclease based NHEJ-mediated HbF induction) The in vivo gene therapy relies upon direct infusion of the editing machinery to target patient resident HSPCs, without isolation-manipulation-conditioning and re-infusion. (Created with BioRender.com).
In the last decade, several gene editing strategies have been investigated in order to revert the SCD-causing mutation. From a historical perspective, in 2014 Genovese and colleagues were the first to attempt to exploit HDR-driven gene repair in human HSCs. Their protocol involved the use of mRNA electroporation and Integrase-Defective LV (IDLV) for delivery of ZFNs and donor DNA template into human cord blood (CB) CD34 + cells (14). Similarly, in 2015 Wang et al. reported homology-driven genome editing in human HSPCs using ZFNs mRNA and Adeno-Associated Virus 6 (AAV6) donor templates (15). In the same year, Hoban et al. demonstrated efficient target cleavage of the β-globin locus, with minimal off-target modifications in HSPCs. Their protocol included the co-delivery of an integrase-defective lentiviral vector or a DNA oligonucleotide as the homologous donor template, and a specific ZNFs programmed to target the pathogenic mutation of SCD (16). In 2016, Dever et al. reported the first CRISPR/Cas9 gene-editing platform for achieving HDR at the HBB gene in HSCs derived from mobilized peripheral blood (mPB) by combining Cas9 ribonucleoproteins (RNP) and rAAV6 as homologous donor (HR) delivery (17). The efficiency of this HDR-mediated correction strategies in vitro ranged from 7% to 50%, depending on different editing tools and donor delivery systems, and were sufficient to produce clinically-relevant amount of HbA (up to 50% of total Hb), ameliorating the SCD cell phenotype in vitro (18). However, after xenograft experiments, the percentage of gene correction detected in vivo dropped to less than 10% (19). This evidence showed that HDR is less efficient in quiescent long-term repopulating HSCs, which constitute a minor fraction of the HSPC population. Moreover, all these studies reported that, in parallel with the incomplete HDR-mediated gene repair, DSBs were being repaired through NHEJ, resulting in the formation of small insertions/deletions (InDels) at the HBB gene. This occurrence would be particularly detrimental as it can lead to the formation of altered and unstable Hb variants, up to the generation of clones completely unable to produce hemoglobin, functionally similar to the β0-thalassemia ones (20, 21). Strategies to increase HDR efficiency in HSCs showed encouraging results and hold promise for clinical application of gene correction strategies based on HDR (22, 23). Recent advances for HDR-mediated correction of SCD mutation in human HSPCs have been recently reported by Lattanzi et al. using ex vivo Cas9-RNP and rAAV6 based donor DNA. In this study, modifications in culture conditions were introduced. Namely, the addition of UM171 molecule to the culture media and the implementation of the low-density culture conditions in order to promote HSC cycling hence increasing the editing efficiency. Their strategy obtained a high percentage of gene corrected HBB (gcHBB) alleles in vitro and favored the long-term engraftment (up to ∼4-fold) of human cells with gcHBB alleles in vivo (24). Moreover, the percentage of human engraftment and gcHBB alleles were even higher in HSPCs derived from SCD patients compared to healthy donor HSPCs (24). Recently, given the observation that DSB-induced Indels can be predicted, Bodai et al. proposed a “double tap” system in order to improve HDR-based strategies. The latter is based on a primary gRNA—specific for a genomic sequence—and on secondary gRNAs targeting the predicted NHEJ-induced Indel sequences, hence improving the HDR-mediated genome editing efficiency (25). While encouraging, it is still unclear whether the efficiency rates of HDR-mediated HBB gene correction could enable effective clinical applications for SCD.
High expression levels of fetal hemoglobin (α2γ2, HbF) determine important advantages for SCD patients. In fact, HbF reduces the polymerization process in deoxygenated RBC impairing their sickling (26). This is consistent with two historically-defined clinical observations: (i) newborn patients, in which HbF expression is naturally high, do not present a pathological phenotype and are protected from disease complications (27); (ii) patients who co-inherit large deletions, InDels or point mutations within the HBG gene cluster resulting in persistently elevated HbF expression (hereditary persistence of fetal hemoglobin HPFH) are relatively asymptomatic (28–30). Recent years witnessed the development of several genome editing strategies based on reproducing the effects of HPFH mutations. CRISPR/Cas9-mediated reproduction of large deletion within the β-globin locus lead to a strong HbF production by causing a distortion of the locus architecture and a direct interaction with the regulatory elements present in the locus control region (LCR) of HBG (31, 32). However, the generation of large deletions requires the simultaneous use of two gRNAs, which might decrease the overall efficiency of the strategy (33). Furthermore, genome editing techniques have been exploited to reproduce the effects of point mutations at the HBG genes. HPFH point mutations in the γ-globin promoters, indeed, are known to affect the binding of the two main γ-globin transcriptional repressors, B cell CLL/lymphoma 11A (BCL11A) and leukemia/lymphoma related factor (LRF; also known as ZBTB7A or FBI-1), thus leading to elevated γ-globin expression (34). First attempts of targeting the BCL11A binding site (BS) by CRISPR/Cas9 resulted in an efficient reproduction of a naturally occurring 13-bp HPFH deletion (Δ13) at the HBG promoter, thus leading to a potent HbF expression (35). Importantly, Δ13 has been also observed in cells engrafting in immunodeficient mouse models (36), although this small deletion occurs following microhomology-mediated enjoining mechanism (MMEJ) as DNA repair mechanism, which is less/no-efficient in quiescent HSCs. The persistence of high editing rates, led to the expression of HbF in around half of the cells constituting around 20% of total hemoglobin in ex vivo differentiated erythrocytes. More recently, an autologous transplantation study performed on non-human primate model, reported a significant reduction in editing frequency in long-term repopulating HSCs which resulted in a limited γ-globin reactivation (1%–5% over the total β-like globin chains) that might not be sufficient to achieve therapeutically relevant HbF levels (37). Similarly, when the binding site of the other main γ-globin repressor (i.e., LRF) is targeted, a potent HbF production is ensured. However, the InDels frequency at HBG promoter in cells repopulating the host BM is lower than the one measured in vitro (38). The upregulation of fetal hemoglobin could be achieved by targeting other transcription factors involved in its regulation. Among others, it has been recently reported that knocking-down ATF4 downregulates the expression of BCL11A through lowering the expression of MYB. ATF4 could therefore represent a target for reactivating γ-globin expression (39) However, both ATF4 and MYB have been reported to have several other actions beyond the regulation of HbF in non-erythroid cells (40, 41) underlining the need to identify erythroid-specific regulators.
Alternative strategies, based on the manipulations of BCL11A expression, have been extensively investigated and represent nowadays the most advanced genome editing-based therapeutic option for SCD patients (Figure 2). To this purpose, the turning point is represented by the identification of erythroid-specific enhancers in BCL11A intron 2 (42). The CRISPR/Cas9-mediated inactivation of an activator binding site in one of the erythroid-specific BCL11A enhancers, induced substantial HbF expression in RBCs without affecting erythropoiesis or BCL11A expression in other lineages (43). Efficacy and safety studies performed in both murine models (44, 45) and non-human primates (46), demonstrated the relevance of this strategy that have been efficiently translated into clinic. The promising results in terms of γ-globin reactivation ensured by CRISPR/Cas9-mediated targeting of BCL11A enhancer and its binding site on HBG promoters, gave rise to the hypothesis that a multiplex cutting at the two targets may lead to additional effects on HbF expression when compared to a single one. This hypothesis has been indeed confirmed by the work of two studies that demonstrated that multiplex gene editing led to higher γ-globin expression than single-gene editing without inhibiting erythroid differentiation (47, 48). However it is worth to consider that this kind of approaches are linked to the occurrence of chromosomal translocations between the two cutting sites (49).
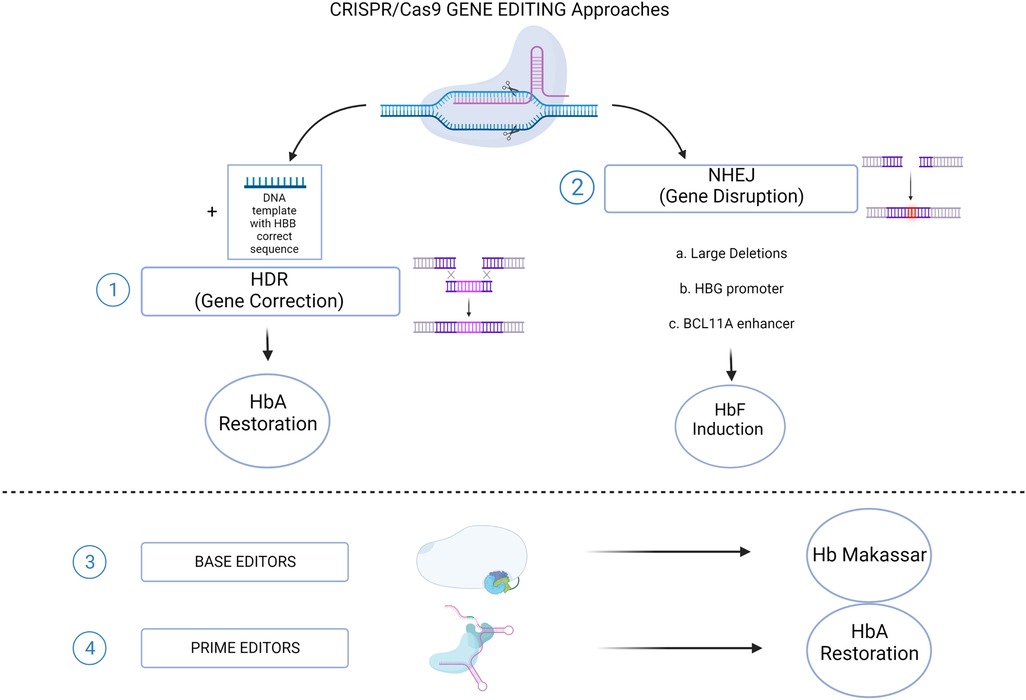
Figure 2. CRISPR/Cas9 genome editing approaches for SCD. (1) The HDR-mediated Gene Correction of the disease-causing mutation, using a DNA Template, leading to HbA restoration. (2) NHEJ-mediated Gene Disruption of target sequences, leading to HbF induction. Different strategies aimed to this goal: a. Large deletions that could either eliminate HbF inhibitory sequences or juxtapose the γ-globin promoters to remote enhancer regions; b. Targeting the HBG1/2 promoters for the disruption of binding sites for γ-globin transcriptional repressors; c. Disruption of the GATA1 motif in the erythroid-specific intronic enhancer of BCL11A, resulting in BCL11A knock down. Recently established CRISPR/Cas9-derived editing platforms, i.e. Base editors (3) and Prime editors (4), has been tested to directly change the HbS into a non-sickling variant (Hb Makassar), or to restore the correct HbA protein. (Created with BioRender.com).
As soon as genome editing technology has been introduced, and similarly to insertional mutagenesis associated with integrating vectors, a number of undesired effects provoke by genome editing tools have been considered as major concerns when applied in patients’ cells. Nuclease activity driven by sequence recognition, can also exert itself on off-target sites, i.e., gene sites endowed with fairly tight sequence homology to the desired site. NHEJ-mediated insertion/deletion mutations similar to those occurring at the target site can also occur at these off-target sites, with the potential occurrence of structural aberrations. Hence, several methods to predict or detect off-target activity have been developed. First, it is possible to make in silico predictions of putative off-target sites, and then characterize them through deep sequencing techniques. Moreover, it is possible to take advantage of experimental procedures that detect off-target activity in vitro and in cell-based systems when available. Commonly used in vitro methods include CIRCLE-Seq (50), ONE-Seq (51) and NucleaSeq (52), while GUIDE-Seq (53), DISCOVER-Seq (54) and CAST-Seq (55) are prevalently used cell-based approaches. However, the use of these techniques may be limited by the absence of cell lines harboring the disease-causing mutations. Off-target activity is not the only safety issue related with nuclease-based genome editing procedures. Over the year a number of studies reported that deleterious events are associated with DSB formation even if occurring at the only On-target site (56). Among them, large deletions and inversions (55, 57, 58), chromosomal truncations (59), chromothripsis (60), aneuploidy (61), loss of heterozygosity/imprinting (62) have been described in both cell lines and primary cells.
So far, we have described several genome editing strategies for SCD, nonetheless some of them must face with concerns related with the high sequence similarities within the β-globin locus (HBG1 vs. HBG2 or HBB vs. HBD). In fact, the high homology between two paralogous genes harbor the possibility of developing recombinant events mediated by sequence homology between them as well as the off-target frequency (63). To demonstrate this, such rearrangements were confirmed in HBB-edited cell (16, 55, 64). Furthermore, the simultaneous targeting of HBG1 and HBG2 was reported to result in deletion of the 4.9 kb region between the two target sites, eliminating HBG2 in 5%–30% of cells (36, 38). In addition to this, the possibility of complete loss of the short arm of chromosome 11 has been demonstrated in HSCs following the use of CRISPR-Cas nucleases targeting either HBB, HBD or HBG1/HBG2 (62). These complications unveiled important safety issues on the use of nuclease-mediated genome editing strategies and led to the development of editing platforms free from DSBs formation.
Base Editing is a new gene editing technology that uses a modified CRISPR-Cas9 platform to induce point mutations on the target DNA sequence without inducing DSBs. Two are the main classes of BE enzymes developed cytidine base editors (CBEs), allowing C > T conversions (65, 66) and adenine base editors (ABEs), allowing A > G conversions (67). Given the excellent promise of these tools, they have been further developed in order to broaden their efficiency and specificity, extend their accessibility to impervious gene loci, perform multiplexing, and in parallel trying to maintain a reduced rate of Indels (68, 69).
For SCD, the base editing technology is currently tested for both the correction of the SCD mutation and the induction of fetal hemoglobin. Newby et al. developed an adenine base editor (ABE8e-NRCH) to convert the SCD allele (HBBS) into the non-pathogenic variant Makassar β-globin (HBBG). Ex vivo delivery of mRNA encoding base editor with a targeting guide RNA into HSPCs from SCD patients resulted in 80% HBBS-to-HBBG conversion. High editing frequency (68%) have been reported after transplantation of edited human HSPCs into immunodeficient mice indicating a stable editing and moreover, human engrafted cells demonstrated important phenotypical amelioration (decrease hypoxia-induced sickling) thus leading to near-normal hematological parameters and reduced splenic enlargement compared to controls (70). Similarly, Chu et al. designed an ABE-based strategy aim to induce a change in HBB point mutation, thus converting HbS to the benign HbG-Makassar in ex vivo edited HSCs (71). These results indicate an effective use of base editors in converting the sickling phenotype of SCD into a non-sickling one, with a lasting effect after transplantation and valid bone marrow reconstitution.
Base-editing platforms have also been exploited to reactivate HbF synthesis, either through the generation of HPFH mutations or through the downregulation of BCL11A expression. It is noteworthy that, differently from CRISPR/Cas9, base editing can be used for the generation of mutations creating binding sites (BSs) for transcriptional activators. For example, ABEs have been used to reproduce the −198 T > C HPFH mutation in the HBG1/2 promoters, creating a BS for the transcriptional activator KLF1 resulting in a 3.5-fold increase in γ-globin expression in HSPC-derived erythrocytes (72). The therapeutic potential of KLF1 BS have been further confirmed by a study of Antoniou and colleagues in which a fine dissection of the contribution of several cis-regulatory elements at the −200 region of the HBG promoter have been conducted (73). Alternatively, a potent γ-globin reactivation on healthy donors β-thalassemic HSPCs have also been obtained after reproducing the −115C > T and −114 C > T HPFH mutations, in the HBG promoters through ABE (74). Finally, an important phenotypic effect of pathological reversion, with a decrease in the number of sickle RBCs, was achieved by CBE-mediated editing of GATA1 BS in the BCL11A enhancer. The editing percentage of SCD HSPCs was very high in this case and their erythroid progeny exhibited high HbF levels (up to 32%) (75). As mentioned earlier, the use of base editors, with their DSB-free mechanism, allows the possibility of multiplexing, without the risk of unwanted genetic rearrangements. Zeng et al. exploited this potential by simultaneously correcting the β-thalassemia-causing HBB −28 (A > G) mutation and disrupting the GATA1 BS in the BCL11A enhancer. They thus demonstrated, not only that this approach is feasible, but also that it yields better results than individual strategies (75). The applicability of this strategy is yet to be proven in SCD though. Altogether these strategies seem promising in terms of efficacy, even if they need confirmation in terms of safety and applicability before being translated into a clinical scenario.
Riding the wave of success in developing DSB-free editing platforms, Anzalone and colleagues developed a cutting-edge technology with the goal of overcoming the limitations of base editors. Specifically, they developed a tool that allows installation of all types of targeted DNA base pair substitutions, small insertions, small deletions, and combinations thereof, without the need to deliver a donor DNA template (76). These tools have been called “prime editors” (PEs) and structurally consist of a Cas9 nickase coupled to an engineered reverse transcriptase, plus a guide sequence, called pegRNA (prime editing gRNA), that dictates both the start site and the correct sequence to achieve all kinds of desired modifications. With this tool, base conversions, or insertions/deletions of up to 80 bp have become feasible. Also the disease-causing A > T transverse mutation of SCD has been targeted with PEs in HEK293T cells (76), providing a proof of principle on the feasibility of this procedure, previously impossible with currently available Bes. However, despite its great potential, the effectiveness of prime editing has been questioned several times. Chen et al. reported that DNA mismatch repair (MMR) impedes prime editing and promotes undesired InDels byproducts (77). Thus, the same group developed modified prime editing systems in which transient expression of an engineered MMR-inhibiting protein enhanced the editing efficiency and outcome purity while editing the SCD genomic site in human iPSC (77). Following studies have been focused on the optimization of pegRNA by introducing a 3′ structured motif protect the reverse transcriptase template (RTT) from exonuclease degradation, resulting in 3–4 fold PE increased efficacy in several cell lines (78). In the wake of these improvements, Everette and colleagues recently reported the correction of the SCD allele (HBBS) to wild type (HBBA) at frequencies of 15%–41% in HSPC from patients with SCD (79). Importantly these editing results have been confirmed in human cells retrieved 17 weeks after transplantation into immunodeficient, reporting minimal off-target activity. As result, HSPC-derived erythrocytes carried less sickle hemoglobin, contained HBBA-derived adult hemoglobin at 28%–43% of normal levels and resisted hypoxia-induced sickling (79).
An even more recent field of research explored the possibility of editing HSPCs through the in vivo delivery of the editing machinery. This strategy could virtually allow easier access to editing procedures since the absence of ex vivo manipulation of HSPCs would guarantee lower cost and shorter time frames (Figure 1). Moreover, it would guarantee minor risks in terms of safety as it would not require bone marrow conditioning regimens. Attempts have been made to develop in vivo HSC transduction/selection technology using non-integrating adenovirus. In vivo HBG-promoter editing by CRISPR/Cas9 in β-YAC/CD46-transgenic mice has been performed (80). The in vivo transduction of HSPCs necessarily requires their mobilization towards the peripheral blood, with subsequent intravenous injections of the adenovirus vector, since the direct transduction of bone marrow HSC has proved to be inefficient. In this way it has been possible to obtain the reactivation of human γ-globin in erythrocytes of adult animals, and this result was maintained even after secondary transplantation of HSPCs. Furthermore, once transduced in the peripheral blood, HSCs are able to relocate in the bone marrow, maintaining their self-renewal capacity and thus ensuring a long-term effect (80). Recently, the same group achieved the correction of almost 40% of HBS alleles in HSCs using prime-editor-expressing helper-dependent adenovirus in a SCD mouse model (81). While promising, in vivo gene editing for curing SCD poses many technological limitations that need to be addressed. First, it is necessary to strike the right balance between high in vivo delivery efficiency and minimal off-target editing of cells and tissues. The employment of viral vectors might represent an efficient strategy for the delivery of molecular gene editing equipment. However, this approach could increase the risk of genotoxicity or immune-response due to the uncontrolled expression of the enzyme or gRNA. On the other hand, the use of non-viral in vivo delivery strategies may result in a reduced efficiency with the need of repeated injections (82, 83). Moreover, it is necessary to compare systemic delivery and local injection to determine the best delivery strategy in terms of percentage of edited cells.
Gene-editing based therapeutical approaches for SCD have achieved promising results in pre-clinical studies. Hence, curative strategies for SCD patients based on ex vivo gene-editing of autologous HSCs have been evaluated into numerous clinical trials in the last years. Most of the first trials are based on the knock-out of the regulatory sequence of the erythroid enhancer of the BCL11A. This approach aims at re-activating the fetal hemoglobin, thus provoking an amelioration of the clinical phenotype in SCD, but also in other β-hemoglobinopathies. Two trials (NCT03745287 and NCT03655678) by Vertex Pharmaceuticals Inc. and CRISPR Therapeutics are evaluating the same CRISPR/Cas9-based product (CTX001) for SCD and TDT β-thalassemia. The first promising results of this product revealed (84) high levels of allelic editing in bone marrow (up to 80% edited cells) and peripheral blood (around 60% edited cells). The first SCD treated patient showed high levels of both total Hb (around 14 g/dl), and HbF fraction, achieving transfusion independence and elimination of Vaso-occlusive episodes (VOCs) for more than one year after the treatment. Recently, Locatelli et al. confirmed these hopeful results by reporting sound data on 31 SCD patients treated with CTX001. All the treated subjects achieved stable and elevated HbF levels, with a complete resolution of VOCs (85). Other two trials have been currently exploring a similar CRISPR/Cas9-mediated HbF reactivation approach, one by Bioverativ (NCT03653247) and the second one by Novartis Pharmaceuticals in collaboration with Intellia Therapeutics (NCT04443907). For this last trial, promising data in terms of sustained induction of fetal hemoglobin and clinical in three patients with SCD have been recently reported (86).
Furthermore, there are two clinical trials evaluating HDR-based gene-editing curative approaches for SCD, aiming at directly correct the pathogenetic mutation in HBB gene. The phase 1/2 CEDAR clinical trial (NCT04819841) by Graphite Bio proposed a strategy based on the correction of the SCD mutation using CRISPR/Cas9 and rAAV6 as HDR template (GPH101), (17, 24). Similarly, Walters and colleagues started a phase 1/2 clinical trial evaluating the efficacy of a CRISPR/Cas9 editing system in which the HDR template is delivered by a single-stranded oligodeoxynucleotides (ssODNs) (NCT04774536). However, Graphite Bio recently paused the CEDAR trial because of the occurrence of severe pancytopenia in the first enrolled patient who received GPH101 (https://ir.graphitebio.com/press-releases/detail/84/graphite-bio-announces-voluntary-pause-of-phase-12-cedar). Hence, a long-term follow-up of the treated patients is necessary in order to detect possible long-term consequences due to genotoxic events. Lastly, in 2022 Beam therapeutics started a phase 1/2 clinical trial evaluating the efficacy of a base-editing strategy able to increase HbF production in HSCs of SCD treated patients. In details, an ABE induces a base swap in the HBG1/2 promoters, thus re-activating gamma-globulin production without the occurrence of CRISPR/Cas9-induced DSB. So far, this trial represents the first clinical application of a nuclease-free gene editing strategy for SCD.
As discussed, the field of genome-editing founded in SCD a unique and fertile soil for the development of its clinical potential. Beside from the monogenic nature of the disease, the reasons of this success rely on (i) the relative frequency of the patient affected worldwide, (ii) a deep knowledge of the globin gene regulation (iii) the presence of a conspicuous history of previous classical gene therapy approaches. On the other hand, the studies on genome editing provide new insights at both biological and clinical level, creating a thriving loop for bench-to bedside information. Differently from classical gene therapy approaches, where a continuous expression of the therapeutic gene is required, genome editing-based strategies retain the potential to ensure clinical benefits after a single delivery of genome editing tools. Moreover, such strategies profit of the endogenous gene regulation machinery, thus conferring additional safety and efficacy advantages. The promise of transplanting autologous and gene-corrected HSC raised great interest in many research groups worldwide. Preclinical results have demonstrated the feasibility of genetically correcting SCD HSPCs by ex vivo as well as in vivo strategies (16, 24, 81). The limited contribution of HDR in repairing DSBs generated by the different nuclease-mediated tools in long-term repopulating HSC initially hampered the definitive clinical translation of this strategy for the treatment of SCD. Nevertheless, during the last years these approaches have been refined and improved, accomplishing significant results in further pre-clinical evaluation studies (24). Hence, clinical trials based on HDR-based gene-editing for SCD recently started (NCT04819841 and NCT04774536). Upcoming results will elucidate on the real effectiveness of these curative approaches. Undoubtedly, the gene-editing strategies for SCD based on HbF reactivation are currently the more advanced approaches in terms of clinical translation, achieving very promising results in ongoing clinical trials (NCT03745287 and NCT03655678) (84). Although some studies have hypothesized potentially dangerous effects of high levels of HbF, such as increased risk of cerebral vasculopathy (87), no detrimental effects due to high levels of HbF have been reported so far in subjects with HPFH (88) or in patients treated with strategies aimed at knock-down of BCL11A. However, a factual risk of these strategies (both HDR- and NHEJ-mediated) is linked with the formation of DSBs. Off-target Cas 9 nuclease activity can result in disruption/alteration of normal gene function, with often unpredictable consequences. Even worse, larger-scale chromosomal rearrangements and genomic instability can occur, and their outcome are hardly predictable. These serious concerns in human gene therapies still persist, though the utilization of high-fidelity Cas9 which reduces but is capable of abolishing off-target editing (89). Therefore, better systems for detecting and quantifying these aberrant events are needed. These concerns seem to be particularly relevant for diseases like SCD for which the hematopoietic compartment is also affected by a chronic inflammation status (90). The recent introduction of DSB-free technology paved the way in overcoming such limitations and, especially for prime editing technique, it gave a strong pulse into direct research activity towards the transplantation of gene corrected HSPCs over γ-globin reactivated ones. Recently, these new base-editing systems demonstrated their efficacy in cell lines as well in HSPC (79), unveiling the possibility to translate DSB-free approaches in clinical setting, with several advantages over the other corrective strategies. In fact, reverting the SCD allele back to wild-type (WT) represents the most straightforward approach; relying on the direct elimination of HBBS allele, in contrast with other strategies such as the HbF re-activation. These points, together with the limited risk of InDel occurrence and the minimal off-target activity, outline the nuclease-free correction strategies aimed at directly correct the HbS pathogenic mutation as the most promising genome editing approaches for tackling SCD.
GF conceived the manuscript. GCe, ML, MA, and GCa contributed with ideas and discussion, ML and MA contributed to the figures. GF, GCe, GCa, and ML wrote and contributed to the organization of the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported also by the Italian ministry of health with “current research” fund.
We thank Professor Franco Locatelli for critically revising the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Telen MJ, Malik P, Vercellotti GM. Therapeutic strategies for sickle cell disease: towards a multi-agent approach. Nat Rev Drug Discov. (2019) 18:139–58. doi: 10.1038/s41573-018-0003-2
2. Lanzkron S, Carroll CP, Haywood C. Mortality rates and age at death from sickle cell disease: U.S., 1979–2005. Public Health Rep. (2013) 128:110–6. doi: 10.1177/003335491312800206
3. Walters MC, Patience M, Leisenring W, Rogers ZR, Aquino VM, Buchanan GR, et al. Stable mixed hematopoietic chimerism after bone marrow transplantation for sickle cell anemia. Biol Blood Marrow Transplant. (2001) 7:665–73. doi: 10.1053/bbmt.2001.v7.pm11787529
4. Kanter J, Walters MC, Hsieh MM, Krishnamurti L, Kwiatkowski J, Kamble RT, et al. Interim results from a phase 1/2 clinical study of lentiglobin gene therapy for severe sickle cell disease. Blood. (2016) 128:1176. doi: 10.1182/blood.V128.22.1176.1176
5. Ribeil J-A, Hacein-Bey-Abina S, Payen E, Magnani A, Semeraro M, Magrin E, et al. Gene therapy in a patient with sickle cell disease. N Engl J Med. (2017) 376:848–55. doi: 10.1056/NEJMoa1609677
6. Malik P, Grimley M, Quinn CT, Shova A, Courtney L, Lutzko C, et al. Gene therapy for sickle cell Anemia using a modified gamma globin lentivirus vector and reduced intensity conditioning transplant shows promising correction of the disease phenotype. Blood. (2018) 132:1021. doi: 10.1182/blood-2018-99-119591
7. Tisdale JF, Kanter J, Mapara MY, Kwiatkowski JL, Krishnamurti L, Schmidt M, et al. Current results of lentiglobin gene therapy in patients with severe sickle cell disease treated under a refined protocol in the phase 1 hgb-206 study. Blood. (2018) 132:1026. doi: 10.1182/blood-2018-99-113480
8. Sii-Felice K, Giorgi M, Leboulch P, Payen E. Hemoglobin disorders: lentiviral gene therapy in the starting blocks to enter clinical practice. Exp Hematol. (2018) 64:12–32. doi: 10.1016/j.exphem.2018.05.004
9. Magrin E, Miccio A, Cavazzana M. Lentiviral and genome-editing strategies for the treatment of β-hemoglobinopathies. Blood. (2019) 134:1203–13. doi: 10.1182/blood.2019000949
10. Hsieh MM, Bonner M, Pierciey FJ, Uchida N, Rottman J, Demopoulos L, et al. Myelodysplastic syndrome unrelated to lentiviral vector in a patient treated with gene therapy for sickle cell disease. Blood Adv. (2020) 4:2058–63. doi: 10.1182/bloodadvances.2019001330
11. bluebird bio, Inc. bluebird bio provides updated findings from reported case of acute myeloid leukemia (AML) in lentiglobin for sickle cell disease (SCD) gene therapy program (n.d.). Available at: https://investor.bluebirdbio.com/news-releases/news-release-details/bluebird-bio-provides-updated-findings-reported-case-acute (Accessed April 14, 2022).
12. Leonard A, Tisdale JF. A pause in gene therapy: reflecting on the unique challenges of sickle cell disease. Mol Ther. (2021) 29:1355–6. doi: 10.1016/j.ymthe.2021.03.010
13. Goyal S, Tisdale J, Schmidt M, Kanter J, Jaroscak J, Whitney D, et al. Acute myeloid leukemia case after gene therapy for sickle cell disease. N Engl J Med. (2022) 386:138–47. doi: 10.1056/NEJMoa2109167
14. Genovese P, Schiroli G, Escobar G, Tomaso TD, Firrito C, Calabria A, et al. Targeted genome editing in human repopulating hematopoietic stem cells. Nature. (2014) 510:235–40. doi: 10.1038/nature13420
15. Wang J, Exline CM, DeClercq JJ, Llewellyn GN, Hayward SB, Li PW-L, et al. Homology-driven genome editing in hematopoietic stem and progenitor cells using ZFN mRNA and AAV6 donors. Nat Biotechnol. (2015) 33:1256–63. doi: 10.1038/nbt.3408
16. Hoban MD, Cost GJ, Mendel MC, Romero Z, Kaufman ML, Joglekar AV, et al. Correction of the sickle cell disease mutation in human hematopoietic stem/progenitor cells. Blood. (2015) 125:2597–604. doi: 10.1182/blood-2014-12-615948
17. Dever DP, Bak RO, Reinisch A, Camarena J, Washington G, Nicolas CE, et al. CRISPR/Cas9 beta-globin gene targeting in human hematopoietic stem cells. Nature. (2016) 539:384–9. doi: 10.1038/nature20134
18. Vakulskas CA, Dever DP, Rettig GR, Turk R, Jacobi AM, Collingwood MA, et al. A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat Med. (2018) 24:1216–24. doi: 10.1038/s41591-018-0137-0
19. Park SH, Lee CM, Dever DP, Davis TH, Camarena J, Srifa W, et al. Highly efficient editing of the β-globin gene in patient-derived hematopoietic stem and progenitor cells to treat sickle cell disease. Nucleic Acids Res. (2019) 47:7955–72. doi: 10.1093/nar/gkz475
20. Magis W, DeWitt MA, Wyman SK, Vu JT, Heo S-J, Shao SJ, et al. High-level correction of the sickle mutation amplified in vivo during erythroid differentiation. bioRxiv. (2019):432716. doi: 10.1101/432716
21. Pendergast AA, Park SH, Binns LA, Kanne CK, Karsenty C, Hernandez BN, et al. Characterization of hemoglobin variants due to in-frame deletions in the β-globin gene from CRISPR/Cas9 gene correction for sickle cell disease. Blood. (2022) 140:7793–4. doi: 10.1182/blood-2022-166931
22. Ferrari S, Jacob A, Beretta S, Unali G, Albano L, Vavassori V, et al. Efficient gene editing of human long-term hematopoietic stem cells validated by clonal tracking. Nat Biotechnol. (2020) 38:1298–308. doi: 10.1038/s41587-020-0551-y
23. Shin JJ, Schröder MS, Caiado F, Wyman SK, Bray NL, Bordi M, et al. Controlled cycling and quiescence enables efficient HDR in engraftment-enriched adult hematopoietic stem and progenitor cells. Cell Rep. (2020) 32:108093. doi: 10.1016/j.celrep.2020.108093
24. Lattanzi A, Camarena J, Lahiri P, Segal H, Srifa W, Vakulskas CA, et al. Development of β-globin gene correction in human hematopoietic stem cells as a potential durable treatment for sickle cell disease. Sci Transl Med. (2021) 13:eabf2444. doi: 10.1126/scitranslmed.abf2444
25. Bodai Z, Bishop AL, Gantz VM, Komor AC. Targeting double-strand break indel byproducts with secondary guide RNAs improves Cas9 HDR-mediated genome editing efficiencies. Nat Commun. (2022) 13:2351. doi: 10.1038/s41467-022-29989-9
26. Hofrichter J, Ross PD, Eaton WA. Kinetics and mechanism of deoxyhemoglobin S gelation: a new approach to understanding sickle cell disease*. Proc Natl Acad Sci U S A. (1974) 71:4864–8. doi: 10.1073/pnas.71.12.4864
27. Watson J. The significance of the paucity of sickle cells in newborn Negro infants. Am J Med Sci. (1948) 215:419–23. doi: 10.1097/00000441-194804000-00008
28. Jacob GF, Raper AB. Hereditary persistence of foetal haemoglobin production, and its interaction with the sickle-cell trait. Br J Haematol. (1958) 4:138–49. doi: 10.1111/j.1365-2141.1958.tb03844.x
29. Forget BG. Molecular basis of hereditary persistence of fetal hemoglobin. Ann N Y Acad Sci. (1998) 850:38–44. doi: 10.1111/j.1749-6632.1998.tb10460.x
30. Demirci S, Leonard A, Essawi K, Tisdale JF. CRISPR-Cas9 to induce fetal hemoglobin for the treatment of sickle cell disease. Mol Ther Methods Clin Dev. (2021) 23:276–85. doi: 10.1016/j.omtm.2021.09.010
31. Ye L, Wang J, Tan Y, Beyer AI, Xie F, Muench MO, et al. Genome editing using CRISPR-Cas9 to create the HPFH genotype in HSPCs: an approach for treating sickle cell disease and β-thalassemia. Proc Natl Acad Sci U S A. (2016) 113:10661–5. doi: 10.1073/pnas.1612075113
32. Antoniani C, Meneghini V, Lattanzi A, Felix T, Romano O, Magrin E, et al. Induction of fetal hemoglobin synthesis by CRISPR/Cas9-mediated editing of the human β-globin locus. Blood. (2018) 131:1960–73. doi: 10.1182/blood-2017-10-811505
33. Frati G, Miccio A. Genome editing for β-hemoglobinopathies: advances and challenges. J Clin Med. (2021) 10:482. doi: 10.3390/jcm10030482
34. Martyn GE, Wienert B, Yang L, Shah M, Norton LJ, Burdach J, et al. Natural regulatory mutations elevate the fetal globin gene via disruption of BCL11A or ZBTB7A binding. Nat Genet. (2018) 50:498–503. doi: 10.1038/s41588-018-0085-0
35. Traxler EA, Yao Y, Wang Y-D, Woodard KJ, Kurita R, Nakamura Y, et al. A genome-editing strategy to treat β-hemoglobinopathies that recapitulates a mutation associated with a benign genetic condition. Nat Med. (2016) 22:987–90. doi: 10.1038/nm.4170
36. Métais J-Y, Doerfler PA, Mayuranathan T, Bauer DE, Fowler SC, Hsieh MM, et al. Genome editing of HBG1 and HBG2 to induce fetal hemoglobin. Blood Adv. (2019) 3:3379–92. doi: 10.1182/bloodadvances.2019000820
37. Humbert O, Radtke S, Samuelson C, Carrillo RR, Perez AM, Reddy SS, et al. Therapeutically relevant engraftment of a CRISPR-Cas9–edited HSC-enriched population with HbF reactivation in nonhuman primates. Sci Transl Med. (2019) 11:eaaw3768. doi: 10.1126/scitranslmed.aaw3768
38. Weber L, Frati G, Felix T, Hardouin G, Casini A, Wollenschlaeger C, et al. Editing a γ-globin repressor binding site restores fetal hemoglobin synthesis and corrects the sickle cell disease phenotype. Sci Adv. (2000) 6:eaay9392. doi: 10.1126/sciadv.aay9392
39. Boontanrart MY, Schröder MS, Stehli GM, Banović M, Wyman SK, Lew RJ, et al. ATF4 regulates MYB to increase γ-globin in response to loss of β-globin. Cell Rep. (2020) 32:107993. doi: 10.1016/j.celrep.2020.107993
40. Ameri K, Harris AL. Activating transcription factor 4. Int J Biochem Cell Biol. (2008) 40:14–21. doi: 10.1016/j.biocel.2007.01.020
41. Greig KT, Carotta S, Nutt SL. Critical roles for c-myb in hematopoietic progenitor cells. Semin Immunol. (2008) 20:247–56. doi: 10.1016/j.smim.2008.05.003
42. Bauer DE, Kamran SC, Lessard S, Xu J, Fujiwara Y, Lin C, et al. An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science. (2013) 342:253–7. doi: 10.1126/science.1242088
43. Canver MC, Smith EC, Sher F, Pinello L, Sanjana NE, Shalem O, et al. BCL11A Enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature. (2015) 527:192–7. doi: 10.1038/nature15521
44. Chang K-H, Smith SE, Sullivan T, Chen K, Zhou Q, West JA, et al. Long-term engraftment and fetal globin induction upon BCL11A gene editing in bone-marrow-derived CD34+ hematopoietic stem and progenitor cells. Mol Ther Methods Clin Dev. (2017) 4:137–48. doi: 10.1016/j.omtm.2016.12.009
45. Wu Y, Zeng J, Roscoe BP, Liu P, Yao Q, Lazzarotto CR, et al. Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nat Med. (2019) 25:776–83. doi: 10.1038/s41591-019-0401-y
46. Demirci S, Zeng J, Wu Y, Uchida N, Shen AH, Pellin D, et al. BCL11A enhancer–edited hematopoietic stem cells persist in rhesus monkeys without toxicity. J Clin Invest. (2020) 130:6677–87. doi: 10.1172/JCI140189
47. Psatha N, Georgakopoulou A, Li C, Nandakumar V, Georgolopoulos G, Acosta R, et al. Enhanced HbF reactivation by multiplex mutagenesis of thalassemic CD34+ cells in vitro and in vivo. Blood. (2021) 138:1540–53. doi: 10.1182/blood.2020010020
48. Han Y, Tan X, Jin T, Zhao S, Hu L, Zhang W, et al. CRISPR/Cas9-based multiplex genome editing of BCL11A and HBG efficiently induces fetal hemoglobin expression. Eur J Pharmacol. (2022) 918:174788. doi: 10.1016/j.ejphar.2022.174788
49. Samuelson C, Radtke S, Zhu H, Llewellyn M, Fields E, Cook S, et al. Multiplex CRISPR/Cas9 genome editing in hematopoietic stem cells for fetal hemoglobin reinduction generates chromosomal translocations. Mol Ther Methods Clin Dev. (2021) 23:507–23. doi: 10.1016/j.omtm.2021.10.008
50. Tsai SQ, Nguyen NT, Malagon-Lopez J, Topkar VV, Aryee MJ, Joung JK. CIRCLE-seq: a highly sensitive in vitro screen for genome-wide CRISPR–Cas9 nuclease off-targets. Nat Methods. (2017) 14:607–14. doi: 10.1038/nmeth.4278
51. Petri K, Kim DY, Sasaki KE, Canver MC, Wang X, Shah H, et al. Global-scale CRISPR gene editor specificity profiling by ONE-seq identifies population-specific, variant off-target effects. bioRxiv. (2021):04.05.438458. doi: 10.1101/2021.04.05.438458
52. Jones SK, Hawkins JA, Johnson NV, Jung C, Hu K, Rybarski JR, et al. Massively parallel kinetic profiling of natural and engineered CRISPR nucleases. Nat Biotechnol. (2021) 39:84–93. doi: 10.1038/s41587-020-0646-5
53. Tsai SQ, Zheng Z, Nguyen NT, Liebers M, Topkar VV, Thapar V, et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-cas nucleases. Nat Biotechnol. (2015) 33:187–97. doi: 10.1038/nbt.3117
54. Wienert B, Wyman SK, Richardson CD, Yeh CD, Akcakaya P, Porritt MJ, et al. Unbiased detection of CRISPR off-targets in vivo using DISCOVER-seq. Science. (2019) 364:286–9. doi: 10.1126/science.aav9023
55. Turchiano G, Andrieux G, Klermund J, Blattner G, Pennucci V, el Gaz M, et al. Quantitative evaluation of chromosomal rearrangements in gene-edited human stem cells by CAST-seq. Cell Stem Cell. (2021) 28:1136–1147.e5. doi: 10.1016/j.stem.2021.02.002
56. Boutin J, Cappellen D, Rosier J, Amintas S, Dabernat S, Bedel A, et al. ON-target adverse events of CRISPR-Cas9 nuclease: more chaotic than expected. CRISPR J. (2022) 5:19–30. doi: 10.1089/crispr.2021.0120
57. Adikusuma F, Piltz S, Corbett MA, Turvey M, McColl SR, Helbig KJ, et al. Large deletions induced by Cas9 cleavage. Nature. (2018) 560:E8–9. doi: 10.1038/s41586-018-0380-z
58. Kosicki M, Tomberg K, Bradley A. Repair of double-strand breaks induced by CRISPR–Cas9 leads to large deletions and complex rearrangements. Nat Biotechnol. (2018) 36:765–71. doi: 10.1038/nbt.4192
59. Cullot G, Boutin J, Toutain J, Prat F, Pennamen P, Rooryck C, et al. CRISPR-Cas9 genome editing induces megabase-scale chromosomal truncations. Nat Commun. (2019) 10:1136. doi: 10.1038/s41467-019-09006-2
60. Leibowitz ML, Papathanasiou S, Doerfler PA, Blaine LJ, Sun L, Yao Y, et al. Chromothripsis as an on-target consequence of CRISPR–Cas9 genome editing. Nat Genet. (2021) 53:895–905. doi: 10.1038/s41588-021-00838-7
61. Nahmad AD, Reuveni E, Goldschmidt E, Tenne T, Liberman M, Horovitz-Fried M, et al. Frequent aneuploidy in primary human T cells after CRISPR–Cas9 cleavage. Nat Biotechnol. (2022) 40:1807–13. doi: 10.1038/s41587-022-01377-0
62. Boutin J, Rosier J, Cappellen D, Prat F, Toutain J, Pennamen P, et al. CRISPR-Cas9 globin editing can induce megabase-scale copy-neutral losses of heterozygosity in hematopoietic cells. Nat Commun. (2021) 12:4922. doi: 10.1038/s41467-021-25190-6
63. Long J, Hoban MD, Cooper AR, Kaufman ML, Kuo CY, Campo-Fernandez B, et al. Characterization of gene alterations following editing of the β-globin gene locus in hematopoietic stem/progenitor cells. Mol Ther. (2018) 26:468–79. doi: 10.1016/j.ymthe.2017.11.001
64. Hoban MD, Lumaquin D, Kuo CY, Romero Z, Long J, Ho M, et al. CRISPR/Cas9-mediated correction of the sickle mutation in human CD34+ cells. Mol Ther. (2016) 24:1561–9. doi: 10.1038/mt.2016.148
65. Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. (2016) 533:420–4. doi: 10.1038/nature17946
66. Wang X, Li J, Wang Y, Yang B, Wei J, Wu J, et al. Efficient base editing in methylated regions with a human APOBEC3A-Cas9 fusion. Nat Biotechnol. (2018) 36:946–9. doi: 10.1038/nbt.4198
67. Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, Bryson DI, et al. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature. (2017) 551:464–71. doi: 10.1038/nature24644
68. Anzalone AV, Koblan LW, Liu DR. Genome editing with CRISPR–cas nucleases, base editors, transposases and prime editors. Nat Biotechnol. (2020) 38:824–44. doi: 10.1038/s41587-020-0561-9
69. Antoniou P, Miccio A, Brusson M. Base and prime editing technologies for blood disorders. Front Genome Ed. (2021) 3:618406. doi: 10.3389/fgeed.2021.618406
70. Newby GA, Yen JS, Woodard KJ, Mayuranathan T, Lazzarotto CR, Li Y, et al. Base editing of haematopoietic stem cells rescues sickle cell disease in mice. Nature. (2021) 595:295–302. doi: 10.1038/s41586-021-03609-w
71. Chu SH, Ortega M, Feliciano P, Winton V, Xu C, Haupt D, et al. Conversion of HbS to hb G-makassar by adenine base editing is compatible with normal hemoglobin function. Blood. (2021) 138:951. doi: 10.1182/blood-2021-150922
72. Gaudelli NM, Lam DK, Rees HA, Solá-Esteves NM, Barrera LA, Born DA, et al. Directed evolution of adenine base editors with increased activity and therapeutic application. Nat Biotechnol. (2020) 38:892–900. doi: 10.1038/s41587-020-0491-6
73. Antoniou P, Hardouin G, Martinucci P, Frati G, Felix T, Chalumeau A, et al. Base-editing-mediated dissection of a γ-globin cis-regulatory element for the therapeutic reactivation of fetal hemoglobin expression. Nat Commun. (2022) 13:6618. doi: 10.1038/s41467-022-34493-1
74. Wang L, Li L, Ma Y, Hu H, Li Q, Yang Y, et al. Reactivation of γ-globin expression through Cas9 or base editor to treat β-hemoglobinopathies. Cell Res. (2020) 30:276–8. doi: 10.1038/s41422-019-0267-z
75. Zeng J, Wu Y, Ren C, Bonanno J, Shen AH, Shea D, et al. Therapeutic base editing of human hematopoietic stem cells. Nat Med. (2020) 26:535–41. doi: 10.1038/s41591-020-0790-y
76. Anzalone AV, Randolph PB, Davis JR, Sousa AA, Koblan LW, Levy JM, et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. (2019) 576:149–57. doi: 10.1038/s41586-019-1711-4
77. Chen PJ, Hussmann JA, Yan J, Knipping F, Ravisankar P, Chen P-F, et al. Enhanced prime editing systems by manipulating cellular determinants of editing outcomes. Cell. (2021) 184:5635–5652.e29. doi: 10.1016/j.cell.2021.09.018
78. Nelson JW, Randolph PB, Shen SP, Everette KA, Chen PJ, Anzalone AV, et al. Engineered pegRNAs improve prime editing efficiency. Nat Biotechnol. (2022) 40:402–10. doi: 10.1038/s41587-021-01039-7
79. Everette KA, Newby GA, Levine RM, Mayberry K, Jang Y, Mayuranathan T, et al. Ex vivo prime editing of patient haematopoietic stem cells rescues sickle-cell disease phenotypes after engraftment in mice. Nat Biomed Eng. (2023) 7:616–28. doi: 10.1038/s41551-023-01026-0
80. Li C, Psatha N, Sova P, Gil S, Wang H, Kim J, et al. Reactivation of γ-globin in adult β-YAC mice after ex vivo and in vivo hematopoietic stem cell genome editing. Blood. (2018) 131:2915–28. doi: 10.1182/blood-2018-03-838540
81. Li C, Georgakopoulou A, Newby GA, Chen PJ, Everette KA, Paschoudi K, et al. In vivo HSC prime editing rescues sickle cell disease in a mouse model. Blood. (2023) 141:2085–99. doi: 10.1182/blood.2022018252
82. Dasgupta I, Flotte TR, Keeler AM. CRISPR/Cas-dependent and nuclease-free in vivo therapeutic gene editing. Hum Gene Ther. (2021) 32:275–93. doi: 10.1089/hum.2021.013
83. Park SH, Bao G. CRISPR/Cas9 gene editing for curing sickle cell disease. Transfus Apher Sci. (2021) 60:103060. doi: 10.1016/j.transci.2021.103060
84. Frangoul H, Altshuler D, Cappellini MD, Chen Y-S, Domm J, Eustace BK, et al. CRISPR-Cas9 gene editing for sickle cell disease and β-thalassemia. N Engl J Med. (2021) 384:252–60. doi: 10.1056/NEJMoa2031054
85. Locatelli F, Frangoul H, Corbacioglu S, de la Fuente J, Wall D, Cappellini MD, et al. Efficacy and safety of a single dose of ctx001 for transfusion-dependent beta-thalassemia and severe sickle cell disease (2022). Available at: https://library.ehaweb.org/eha/2022/eha2022-congress/366210/franco.locatelli.efficacy.and.safety.of.a.single.dose.of.ctx001.for.html?f=menu%3D6%2Abrowseby%3D8%2Asortby%3D2%2Amedia%3D3%2Ace_id%3D2233%2Amarker%3D1750 (Accessed May 22, 2023).
86. Sharma A, Boelens J-J, Cancio M, Hankins JS, Bhad P, Azizy M, et al. CRISPR-Cas9 editing of the HBG1 and HBG2 promoters to treat sickle cell disease. N Engl J Med. (2023) 389:820–32. doi: 10.1056/NEJMoa2215643
87. Chamouine A, Saandi T, Muszlak M, Larmaraud J, Lambrecht L, Poisson J, et al. High fetal hemoglobin level is associated with increased risk of cerebral vasculopathy in children with sickle cell disease in Mayotte. BMC Pediatr. (2020) 20:302. doi: 10.1186/s12887-020-02187-6
88. Steinberg MH, Chui DHK, Dover GJ, Sebastiani P, Alsultan A. Fetal hemoglobin in sickle cell anemia: a glass half full? Blood. (2014) 123:481–5. doi: 10.1182/blood-2013-09-528067
89. Pedrazzoli E, Bianchi A, Umbach A, Amistadi S, Brusson M, Frati G, et al. An optimized SpCas9 high-fidelity variant for direct protein delivery. Mol Ther. (2023) 31:2257–65. doi: 10.1016/j.ymthe.2023.03.007
Keywords: sickle cell disease, gene editing, fetal hemoglobin reactivation, globin genes regulation, gene therapy, CRISPR/Cas9
Citation: Ceglie G, Lecis M, Canciani G, Algeri M and Frati G (2023) Genome editing for sickle cell disease: still time to correct?. Front. Pediatr. 11:1249275. doi: 10.3389/fped.2023.1249275
Received: 28 June 2023; Accepted: 10 October 2023;
Published: 2 November 2023.
Edited by:
Victor Aquino, University of Texas Southwestern Medical Center, United StatesReviewed by:
Vishnu Hosur, Jackson Laboratory, United States© 2023 Ceglie, Lecis, Canciani, Algeri and Frati. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giacomo Frati Z2lhY29tby5mcmF0aUBvcGJnLm5ldA==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.