 Han-Byeol Choi1
Han-Byeol Choi1 Jiwon Lee
Jiwon Lee Jeong-Yi Kwon
Jeong-Yi Kwon- 1Department of Physical and Rehabilitation Medicine, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Republic of Korea
- 2Department of Pediatrics, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Republic of Korea
- 3Department of Laboratory Medicine and Genetics, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Republic of Korea
- 4 Department of Health Science and Technology, Samsung Advanced Institute for Health Science and Technology, Sungkyunkwan University, Seoul, Republic of Korea
Herein, we describe the case of a 43-month-old girl who presented with clinical manifestations of dyskinetic cerebral palsy (CP), classified as the Gross Motor Function Classification System (GMFCS) V. The patient had no family history of neurological or perinatal disorders. Despite early rehabilitation, serial assessments using the Gross Motor Function Measure (GMFM) showed no significant improvements in gross motor function. Brain magnetic resonance imaging showed nonspecific findings that could not account for developmental delay or dystonia. Whole-genome sequencing identified a heterozygous NM_002074.5(GNB1):c.239T>C (p.Ile80Thr) mutation in guanine nucleotide-binding protein beta 1 (GNB1) gene. Considering this case and previous studies, genetic testing for the etiology of dyskinetic CP is recommended for children without relevant or with nonspecific brain lesions.
1. Introduction
Cerebral palsy (CP) is a group of neurodevelopmental disorders that affects posture and movement due to non-progressive disruptions in the developing brain during the fetal period or infancy (1). Known risk factors for CP include prematurity (2), low birth weight (2), multiple births (3), assisted reproductive technology infertility treatment (4), infections during pregnancy (5), jaundice and kernicterus, maternal medical conditions, and birth hypoxic-ischemic brain injury (6), etc. When these traditional risk factors are absent and brain magnetic resonance imaging (MRI) shows no clear abnormalities, the etiology of CP remains unknown. Recently, cohort-based whole-exome and whole-genome sequencing studies have revealed that genetic mutations are important causes of CP (7).
Guanine nucleotide-binding protein beta 1 encephalopathy (GNB1-E) is a genetic disease that can be passed down in an autosomal dominant manner or results from a de novo mutation (8). Patients with GNB1 mutations typically exhibit moderate-to-severe developmental delays or intellectual disabilities, often accompanied with structural abnormalities in the brain and epilepsy (8). The patients also demonstrate generalized hypotonia, which may evolve into hypertonia over time, and movement abnormalities such as dystonia, spasticity, athetoid, and chorea (8, 9). Herein, we report the case of a child with dyskinetic CP who had a GNB1 mutation, as observed in whole genome sequencing.
2. Case report
The patient was a 43-month-old girl who was the only child of a couple with no family history of neurological disorders. She was born at 37 weeks of gestation, weighing 2,300 g, and without perinatal problems.
At 6 months of age, she was referred to the Pediatric Physical and Rehabilitation Medicine Clinic because of axial hypotonia and poor head control. She was unable to support her elbows in the prone position, maintain a sitting position, reach out, or bring objects to the midline. At the age of 6 months, a neurological examination revealed truncal hypotonia, mild rigidity in the lower extremities, positive Babinski reflexes, and a positive asymmetric tonic neck reflex to the right. Gradually, she developed nystagmus and dystonia with mild spasticity in the extremities and was diagnosed with dyskinetic CP at the age of 1 year. Despite early and intensive rehabilitation, the patient's development did not improve as per the expectations of the clinical team.
For diagnostic workup, a brain MRI was performed at the age of 3 months, which suggested periventricular leukomalacia with mild ventriculomegaly (Figure 1A). At the age of 25 months, a follow-up brain MRI showed an apparent abnormal T2-fluid attenuated inversion recovery (FLAIR) high-signal intensity in the white matter, particularly in the post-periventricular region and around the frontal horn, along with cortical atrophy in the frontoparietal lobe (Figure 1B). Subsequently, genetic testing was recommended, and a pathogenic de novo mutation in GNB1 was identified using whole-genome sequencing. A single variant of GNB1, NM_002074.5(GNB1):c.239T>C (p.Ile80Thr), was identified through whole-genome sequencing and subsequently confirmed by Sanger sequencing (Figures 2A,B).
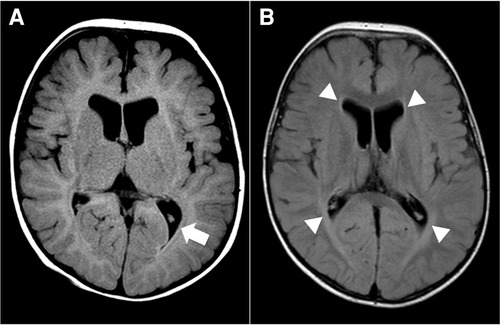
Figure 1. Brain magnetic resonance image (MRI) at 3 months (A) and 25 months (B) of age. (A) Axial T2-fluid attenuated inversion recovery (FLAIR) image of the brain demonstrates periventricular leukomalacia (arrow) with mild ventriculomegaly of the lateral ventricle. (B) Axial T2-FLAIR image of the brain shows abnormal high-signal intensity in the white matter, especially in the post-periventricular region and around the frontal horn (arrowhead). There is no interval change in ventriculomegaly of the lateral ventricle.
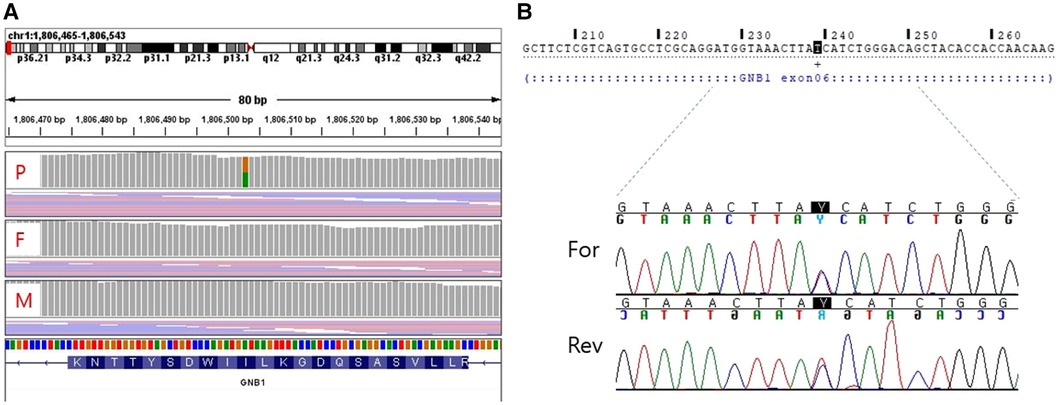
Figure 2. Whole genome sequencing (A) and sanger sequencing (B) validation of the target mutation [NM_002074.5(GNB1):c.239T > C]. (A) A de novo variant is detected in the GNB1 gene of the proband. (B) Sanger sequencing shows a heterozygous missense variant of GNB1. P, proband; F, father; M, mother; For, Forward; Rev, Reverse.
An intelligence test was performed at 41 months of age, and her measured IQ was 42 according to the Korean Wechsler Preschool and Primary Scale of Intelligence Fourth Edition (K-WPPSI-IV) (10). Her social maturity score on the Korean Vineland Adaptive Behavior Scale Second Edition (K-Vineland-II) (11) was 44, indicating intellectual disability. Language evaluation conducted at the same age showed that the equivalent ages of receptive and expressive languages were 18 and 8 months, respectively. At 41 months of age, the Korean Bayley Scales of Infant and Toddler Development Third Edition (K-Bayley-III) (12) revealed a significant developmental delay on the cognitive (55, 0.1 percentile), language (49, <0.1 percentile), motor (49, <0.1 percentile), and social-emotional scales (65, 1 percentile). Gross motor function was evaluated using the Gross Motor Function Measure (GMFM-66 & GMFM-88) (13) from the ages of 11 to 43 months; it is currently classified as the Gross Motor Function Classification System (GMFCS) (14) V and Mini-Manual Ability Classification System (Mini-MACS) (15) IV. Changes in GMFM-66 and GMFM-88 scores during this period are shown in Figures 3 and 4, respectively.
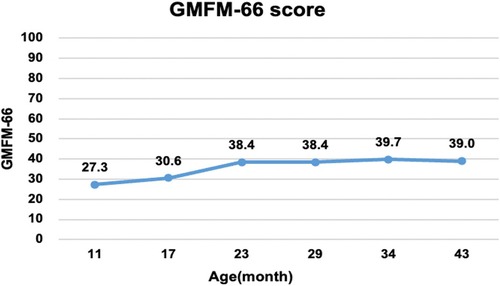
Figure 3. Changes in the gross motor function measure (GMFM-66). The graph shows the total score of the GMFM-66 evaluated at 6–12 months intervals from 11 to 43 months of age. Despite early intensive rehabilitation, she showed poor improvement on the GMFM-66, reaching a plateau at approximately 24 months.
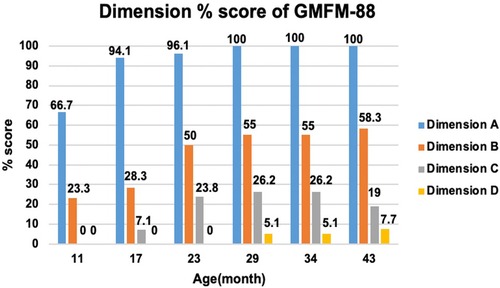
Figure 4. Change in dimension % score of gross motor function measure (GMFM-88). The graph examines the trend of change by subdividing GMFM-88 from dimension A to D. Dimension A stands for lying and rolling, dimension B stands for sitting, dimension C stands for crowing and kneeling, and dimension D stands for standing. She showed poor improvement in dimensions C and D.
The GMFM-66 score was 38.4 at the age of 23 months and reached an early plateau. A similar pattern was observed in dimension B (sitting), C (crowing and kneeling), and D (standing) scores of the GMFM-88. In particular, the scores for dimension C (crowing and kneeling) and D (standing) showed very poor improvement, with only 26.2% and 51.0% respectively at the age of 34 months.
Written informed consent was obtained from the patient’s parents for the publication of any potentially identifiable images and data included in this article.
3. Discussion
Herein, we present the case of a child with GNB1-E who exhibited significant developmental delay, abnormal muscle tone (initial hypotonia followed by hypertonia), dystonia with mild spasticity, nystagmus, and non-specific brain MRI findings. While this case report presents clinical findings of only one individual, it is clinically valuable as it describes the detailed trajectory of motor developmental delay, which is the most important phenotype of CP resulting from GNB1-E. To our knowledge, no previous studies have described the early motor development of patients with CP resulting from GNB1-E.
Although premature birth, hypoxic-ischemic encephalopathy, prenatal infection, etc. are established risk factors for CP, approximately one-third of children with CP do not have these typical risk factors (16). Genetic factors are suspected to be the underlying causes in many such children. Until a recent date, it had been suggested that the contribution of genetic variants to the burden of CP was about 2% (17). However, since the introduction of affordable new-generation DNA sequencing, the focus of genetic investigations in CP shifted from gene association studies to the identification of the likely causal variants (18). Several of single-gene causes of CP have been revealed through studies of families with more than two individuals with CP, such as the KANK1, AP4MI, and GAD1 gene mutations (19, 20). Recent investigations of genetic causes in a large cohort of singleton CP cases using Whole Exome Sequencing have identified that the proportion of the cases carrying plausible genetic mutation is much larger than previous reports. At least 14% of nearly 200 singleton cases with CP have been found to have a plausible genetic mutation, de novo or inherited (21).
“CP mimics” is a term used to describe a condition that exhibits clinical symptoms consistent with CP, motor impairments appearing in infancy or early childhood, even in the absence of typical risk factors or relevant neuroimaging abnormalities (22). “CP mimics” encompass a spectrum of disorders, including inborn errors of metabolism like Lesch Nyhan disease and Glut1 deficiency, as well as neurogenetic disorders such as Krabbe disease and Metachromatic leukodystrophy (22). However, the diagnostic process for cerebral palsy primarily hinges on the clinical presentations, not the underlying cause including genetic mutations in this case. Consequently, the International Cerebral Palsy Genomics Consortium underscores that when these genetic mutations are discovered in individuals manifesting clinical manifestations of cerebral palsy, they should still be diagnosed as CP itself, rather than categorized as CP mimics (23).
In a large retrospective cohort study of 1,526 patients with CP, 229 genes with pathogenic mutations were identified in 450 patients (23). A total of 86 genes, including CTNNB1, KIF1A, GNAO1, and TUBA1A, were mutated in two or more patients, and GNB1 mutations were identified in two patients. Although the clinical features of these children were not described in the study, the two children with GNB1 mutation harbored the Ile80Thr variant as a de novo mutation. Lewis et al. proposed a set of criteria for identifying CP-associated genes, which included a series of steps from gene discovery and laboratory investigations to clinical applications (7). Recent studies have shown that GNB1 mutations alter the activity of G protein-gated inwardly rectifying potassium channels, which are mainly expressed in neurons (24).
A total of 58 cases of GNB1 mutations have been reported, with 12 cases identified as having the Ile80Thr variant (8). While most of the reported cases were identified as de novo mutations, three children inherited pathogenic variants from their parents (p. Arg96Leu, p.Thr243Ala). Pathogenic GNB1 variants cause neurodevelopmental disabilities with global developmental delays. All children with GNB1 mutations experienced moderate-to-severe developmental delays. Furthermore, nearly 50% of the patients were nonverbal and nonambulatory (9), which was consistent with the findings in our study. Our patient's gross motor function was regularly assessed using the GMFM and no significant improvement or regression has been observed to date. Despite early intensive rehabilitation, her GMFM-66 score plateaued at 23 months of age; she is now classified as having severe quadriplegic CP, GMFCS V, and Mini-MACS IV.
Abnormal muscle tone and various movement disorders have been reported in GNB1-E. Hypotonia is the most common presentation, followed by spasticity and dystonia (8, 9); all of these were observed in our case. In the present case, the patient had severe truncal hypotonia during infancy. Over time, she developed dystonia and mild spasticity in her extremities, leading to the diagnosis of dyskinetic CP. It is still uncertain whether the abnormality of muscle tone shown in the patient is a typical finding of the identified variant (p.Ile80Thr). Endo et al. (25) suggested a possible genotype-phenotype correlation between the p.Ile80Thr variant and severe infantile hypotonia that may progress to hypertonia such as spasticity and dystonia. Further studies are necessary to define the phenotype of GNB1 variants.
Kitai et al. (26) reported the etiology of dyskinetic CP based on the clinical course and brain MRI findings in 163 term-born and 136 preterm-born children. The most common causes of dyskinetic CP were found to be hypoxic-ischemic encephalopathy (HIE) in term infants and bilirubin encephalopathy (BE) in preterm infants. On brain MRI, HIE was characterized by T2 high signal intensity in the bilateral thalamus and posterior putamen, and BE by T2 high signal intensity in the bilateral globus pallidus. Eighteen patients had normal brain MRI results, and genetic mutations were identified in eight of these patients. Our patient did not have basal ganglia lesions commonly associated with dyskinetic CP but rather exhibited non-specific white matter lesions on brain MRI. In addition, there was no evidence of hyperbilirubinemia in the results of the laboratory exam.
Based on previous reports and the present case, genetic testing for determining the etiology of dyskinetic CP is recommended for children without relevant or with nonspecific brain lesions whose motor function has reached an early plateau despite early and intensive rehabilitation. Identifying the genetic causes of CP can provide opportunities to understand its underlying pathophysiology and facilitate the development of new interventions targeting specific genetic pathways. In the context of this case report, for detection of the genetic mutation, whole genome sequencing was undertaken as part of the Bio & Medical Technology Development Program of the National Research Foundation. However, it should be noted that whole genome or exome sequencing is a relatively costly procedure, making it impractical for all cases. Consequently, a more judicious approach is suggested. As an initial step, consultation with a genetic counseling team is advisable. Subsequently, although there is no definitive consensus yet, we should selectively perform sequencing tests on the most potentially relevant genes depending on the patient's phenotype. For example, SPG and ARG1 genes for spastic CP, genes including PDE10A, GNB1, FOXG1, GNAO1, SCN1A, and SCN8A for dyskinetic CP, and SLC2A1, ATM, and PLP genes for ataxia CP should be considered (22). If no significant mutations are identified in these tests, considering whole exome or whole genome sequencing as the next step is recommended. This stepwise approach takes into account both clinical and financial factors, optimizing the potential benefits of genetic testing while managing the associated costs.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
Written informed consent was obtained from the patient's parents for the publication of any potentially identifiable images and data included in this article.
Author contributions
H-BC and YN: Drafting/revision of the manuscript for content, including writing. JiL and JeL: drafting/revising the manuscript for content, including writing, and the major role in data acquisition. J-HJ and J-WK: drafting/revision of the manuscript for content, including writing, a major role in data acquisition, and analysis or interpretation of data. J-YK: drafting/revision of the manuscript content, including writing, a major role in the acquisition and analysis or interpretation of data, study conceptualization or design, and analysis or interpretation of data. All authors contributed to the article and approved the submitted version.
Funding
This work was performed as part of the Bio & Medical Technology Development Program of the National Research Foundation (NRF-2020M3E5D7085175) funded by the Ministry of Health and Welfare, Ministry of Science and ICT, Ministry of Trade Industry and Energy, Korea Disease Control and Prevention Agency (The National Project of Bio Big Data).
Acknowledgments
This case was presented at the 50th Anniversary Meeting and International Conference of the Korean Academy of Rehabilitation Medicine (KARM 2022).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Rosenbaum P, Paneth N, Leviton A, Goldstein M, Bax M, Damiano D, et al. A report: the definition and classification of cerebral palsy April 2006. Dev Med Child Neuro Suppl. (2007) 109:8–14. doi: 10.1111/j.1469-8749.2007.tb12610.x
2. Van Naarden Braun K, Doernberg N, Schieve L, Christensen D, Goodman A, Yeargin-Allsopp M. Birth prevalence of cerebral palsy: a population-based study. Pediatrics. (2016) 137:1–9. doi: 10.1542/peds.2015-2872
3. Bonellie S, Currie D, Chalmers J. Comparison of risk factors for cerebral palsy in twins and singletons. Dev Med Child Neurol. (2005) 47:587–91. doi: 10.1111/j.1469-8749.2005.tb01208.x
4. Hvidtjørn D, Grove J, Schendel DE, Væth M, Ernst E, Nielsen LF, et al. Cerebral palsy among children born after in vitro fertilization: the role of preterm delivery—a population-based, cohort study. Pediatrics. (2006) 118:475–82. doi: 10.1542/peds.2005-2585
5. Wu YW, Escobar GJ, Grether JK, Croen LA, Greene JD, Newman TB. Chorioamnionitis and cerebral palsy in term and near-term infants. JAMA. (2003) 290:2677–84. doi: 10.1001/jama.290.20.2677
6. Zhang S, Li B, Zhang X, Zhu C, Wang X. Birth asphyxia is associated with increased risk of cerebral palsy: a meta-analysis. Front Neurol. (2020) 11:704. doi: 10.3389/fneur.2020.00704
7. Lewis SA, Shetty S, Wilson BA, Huang AJ, Jin SC, Smithers-Sheedy H, et al. Insights from genetic studies of cerebral palsy. Front Neurol. (2020) 11:625428. doi: 10.3389/fneur.2020.625428
8. Revah-Politi A, Sands TT, Colombo S, Goldstein DB, Anyane-Yeboa K. GNB1 Encephalopathy. In: Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, et al., editors. Genereviews. Seattle (WA): University of Washington, Seattle 1993–2023. Available at: https://www.ncbi.nlm.nih.gov/books/NBK554743/ (Accessed April 11th, 2023).
9. Hemati P, Revah-Politi A, Bassan H, Petrovski S, Bilancia CG, Ramsey K, et al. Refining the phenotype associated with GNB1 mutations: clinical data on 18 newly identified patients and review of the literature. Am J Med Genet A. (2018) 176:2259–75. doi: 10.1002/ajmg.a.40472
10. Park H, Lee K. Intellectual characteristics of Korean children: analyses of K-WPPSI-IV performance. Korean J Child Stud. (2016) 37:157–68. doi: 10.5723/kjcs.2016.37.6.157
11. Hwang ST, Kim JH, Hong SH, Bae SH, Jo SW. Standardization study of the Korean vineland adaptive behavior scales-Ⅱ (K-vineland-II). Kor J Clin Psycho. (2015) 34:851–76. doi: 10.15842/kjcp.2015.34.4.002
12. Ahn SH, Yoo EY, Lee SH. A validation study of the gross motor scale of Korean version of bayley scales of infant and toddler development. Third Edition. Korea J Occup Ther. (2018) 26:81–97. doi: 10.14519/jksot.2018.26.2.07
13. Alotaibi M, Long T, Kennedy E, Bavishi S. The efficacy of GMFM-88 and GMFM-66 to detect changes in gross motor function in children with cerebral palsy (CP): a literature review. Disabil Rehabil. (2014) 36:617–27. doi: 10.3109/09638288.2013.805820
14. Palisano RJ, Rosenbaum P, Bartlett D, Livingston MH. Content validity of the expanded and revised gross motor function classification system. Dev Med Child Neurol. (2008) 50:744–50. doi: 10.1111/j.1469-8749.2008.03089.x
15. Eliasson AC, Ullenhag A, Wahlstrom U, Krumlinde-Sundholm L. Mini-MACS: development of the manual ability classification system for children younger than 4 years of age with signs of cerebral palsy. Dev Med Child Neurol. (2017) 59:72–8. doi: 10.1111/dmcn.13162
16. Fahey MC, Maclennan AH, Kretzschmar D, Gecz J, Kruer MC. The genetic basis of cerebral palsy. Dev Med Child Neurol. (2017) 59:462–9. doi: 10.1111/dmcn.13363
17. Rajab A, Yoo S-Y, Abdulgalil A, Kathiri S, Ahmed R, Mochida GH, et al. An autosomal recessive form of spastic cerebral palsy (CP) with microcephaly and mental retardation. Am J Med Genet A. (2006) 140A:1504–10. doi: 10.1002/ajmg.a.31288
18. MacLennan AH, Thompson SC, Gecz J. Cerebral palsy: causes, pathways, and the role of genetic variants. Am J Obstet Gynecol. (2015) 213:779–88. doi: 10.1016/j.ajog.2015.05.034
19. Hemminki K, Li X, Sundquist K, Sundquist J. High familial risks for cerebral palsy implicate partial heritable aetiology. Paediatr Perinat Epidemiol. (2007) 21:235–41. doi: 10.1111/j.1365-3016.2007.00798.x
20. Lerer I, Sagi M, Meiner V, Cohen T, Zlotogora J, Abeliovich D. Deletion of the ANKRD15 gene at 9p24.3 causes parent-of-origin-dependent inheritance of familial cerebral palsy. Hum Mol Genet. (2005) 14:3911–20. doi: 10.1093/hmg/ddi415
21. McMichael G, Bainbridge MN, Haan E, Corbett M, Gardner A, Thompson S, et al. Whole-exome sequencing points to considerable genetic heterogeneity of cerebral palsy. Mol Psychiatry. (2015) 20:176–82. doi: 10.1038/mp.2014.189
22. Pearson TS, Pons R, Ghaoui R, Sue CM. Genetic mimics of cerebral palsy. Mov Disord. (2019) 34:625–36. doi: 10.1002/mds.27655
23. Moreno-De-Luca A, Millan F, Pesacreta DR, Elloumi HZ, Oetjens MT, Teigen C, et al. Molecular diagnostic yield of exome sequencing in patients with cerebral palsy. JAMA. (2021) 325:467–75. doi: 10.1001/jama.2020.26148
24. Reddy HP, Yakubovich D, Keren-Raifman T, Tabak G, Tsemakhovich VA, Pedersen MH, et al. Encephalopathy-causing mutations in Gβ1 (GNB1) alter regulation of neuronal GIRK channels. iScience. (2021) 24:103018. doi: 10.1016/j.isci.2021.103018
25. Endo W, Ikemoto S, Togashi N, Miyabayashi T, Nakajima E, Hamano SI, et al. Phenotype-genotype correlations in patients with GNB1 gene variants, including the first three reported Japanese patients to exhibit spastic diplegia, dyskinetic quadriplegia, and infantile spasms. Brain Dev. (2020) 42:199–204. doi: 10.1016/j.braindev.2019.10.006
Keywords: cerebral palsy, dystonia, GNB1, developmental delay, case report
Citation: Choi H-B, Na Y, Lee J, Lee J, Jang J-H, Kim J-W and Kwon J-Y (2023) Case report: Suspecting guanine nucleotide-binding protein beta 1 mutation in dyskinetic cerebral palsy is important. Front. Pediatr. 11:1204360. doi: 10.3389/fped.2023.1204360
Received: 10 May 2023; Accepted: 25 September 2023;
Published: 13 October 2023.
Edited by:
Patrícia Maciel, University of Minho, PortugalReviewed by:
Elena Pavlidis, Central Hospital of Bolzano, ItalyGerald Bradley Schaefer, University of Arkansas for Medical Sciences, United States
© 2023 Choi, Na, Lee, Lee, Jang, Kim and Kwon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jeong-Yi Kwon amVvbmd5aS5rd29uQHNhbXN1bmcuY29t