 Jaewoong Lee
Jaewoong Lee Jaeeun Yoo1
Jaeeun Yoo1 Dae-Hyun Jang
Dae-Hyun Jang- 1Department of Laboratory Medicine, Incheon St. Mary’s Hospital, College of Medicine, The Catholic University of Korea, Seoul, Republic of Korea
- 2Department of Rehabilitation Medicine, Incheon St. Mary’s Hospital, College of Medicine, The Catholic University of Korea, Seoul, Republic of Korea
While somatic gain-of-function mutations in the CTNNB1 gene cause diverse malignancies, germline loss-of-function mutations cause neurodevelopmental disorders or familial exudative vitreoretinopathy. In particular, CTNNB1-related neurodevelopmental disorders have various phenotypes, and a genotype-phenotype relationship has not been established. We report two patients with CTNNB1-related neurodevelopmental disorder whose clinical features were similar to those of cerebral palsy, hindering diagnosis.
Introduction
Cerebral palsy (CP) is a group of non-progressive disorders of movement and posture that cause activity limitations due to disturbances that occur in the developing fetal or infant brain (1). Motor symptoms of CP include spasticity, weakness, and involuntary movements. CP mimics a number of neurogenetic disorders, which may present with motor symptoms in early childhood, resulting in misdiagnosis (2). Many inborn errors of metabolism (3), hereditary spastic paraplegias, and enzyme deficiencies (2) may present as CP mimics.
The CTNNB1 gene is located on chromosome 3p22.1 and encodes beta-catenin, which plays a crucial role in cell adhesion in the Wnt signaling pathway. Somatic gain-of-function pathogenic variants in CTNNB1 cause various malignancies. Recently, germline loss-of-function pathogenic variants in CTNNB1 have been reported to cause neurodevelopmental disorders with spastic diplegia and visual defects (NEDSDV, MIM# 615075) (4), and familial exudative vitreoretinopathy (MIM# 617572) (5). The phenotypes reported in NEDSDV are global developmental delay, impaired intellectual development, axial hypotonia, spasticity, and dysmorphic craniofacial features with microcephaly. Moreover, the majority of such patients express visual abnormalities, including strabismus, optic nerve atrophy, and retinal abnormalities (6). Here we report two cases of NEDSDV that presented as CP mimics without definite ophthalmologic symptoms. This study was performed in accordance with the ethical standards of the Declaration of Helsinki and was approved by the Institutional Review Board of Incheon St. Mary's Hospital (OC23ZISI0001).
Case 1
A 17-month-old girl was referred to the Medical Genetics and Rare Disease Center for developmental delay, spasticity, dystonia, and failure to thrive. The patient was delivered by cesarean section due to distress during labor at 40 weeks of gestation. Her birth weight was 2.38 kg, and her family history was nonspecific. The patient was the fourth child, and all the older siblings developed normally. The patient's growth parameters were height, 74 cm (3rd centile); weight, 7.1 kg (3rd centile); and head circumference, 39 cm (3rd centile). She had mild dysmorphic features of thin upper lip, ear abnormality, upslanting palpabral fissures, and flat philtrum. On the Bayley Scales of Infant and Toddler Development (3rd edition), her development corresponded to an equivalent age of 4 months in cognition, 9 months in receptive language, 9 months in expressive language, 3 months in fine motor skills, and 2 months in gross motor skills. Overall, she displayed profound global developmental delay. Brain magnetic resonance imaging was normal. Chromosome analysis, chromosome microarray (CMA), MECP2 gene sequencing, Prader-Willi syndrome, and Angelman syndrome tests were carried out but revealed normal results. The patient was able to sit up on his own after the age of 2 years. The patient was followed under the diagnosis of dyskinetic CP. On follow-up at the age of 41 months, her development corresponded to an equivalent age of 8 months in cognition, 5 months in receptive language, 6 months in expressive language, 8 months in fine motor skills, and 7 months in gross motor skills. She showed severe developmental delay, dyskinetic movement, and spasticity; however, no definite abnormalities were noted on brain MRI. Because of the discrepancy of clinical manifestations and radiologic findings, a next-generation sequencing (NGS) test for hereditary developmental delay was performed.
Molecular analyses
A peripheral blood sample was used for genomic DNA extraction, and the TruSight One Extended Sequencing Panel Kit was used for sequencing on a NextSeq 550Dx instrument (Illumina, San Diego, CA). A heterozygous nonsense mutation, NM_001904.3:c.1543C > T, p.Arg515*, was found in CTNNB1, which was previously reported as a pathogenic variant. At clinical reassessment, the patient presented global developmental delay, dysmorphic craniofacial features, truncal hypotonia, spasticity, dystonia, failure to thrive, and microcephaly. The patient had no definite ophthalmological symptoms, such as strabismus or vitreous retinopathy, but other phenotypes corresponded to CTNNB1-related neurodevelopmental disorders. Thus, NEDSDV was diagnosed.
Case 2
A 12-month-old girl was referred to the Medical Genetics and Rare Disease Center due to gross motor developmental delay and spasticity. The patient had been born at 38 weeks of gestation without any complications. She was the first child born to non-consanguineous parents. Her birth weight was 2.55 kg, and there were no perinatal problems or specific family history of interest. The patient's growth parameters were height, 70 cm (10th centile); weight, 7.8 kg (3rd centile); and head circumference, 41 cm (below the 1st centile). She had no definite dysmorphic features and could roll over but could not sit alone, even with support. On neurological examination, the deep tendon reflexes of both the knee and ankle were exaggerated. Additionally, primitive reflexes were exaggerated. Spasticity was prominent in both lower limbs and was rated as Modified Ashworth Scale Grade I+. Dyskinetic movement was not definite. On Bayley Scales of Infant and Toddler Development (3rd edition), her development corresponded to an equivalent age of 4 months in cognition, 4 months in receptive language, 9 months in expressive language, 4 months in fine motor skills, and 4 months in gross motor skills. Brain magnetic resonance imaging was normal. Due to the discrepancy of clinical manifestations and radiologic findings, Chromosome analysis, CMA, and NGS tests for hereditary developmental delay were simultaneously performed.
Molecular analyses
No abnormalities were observed in chromosome analysis or CMA. In NGS, a heterozygous novel duplication variant, NM_001904.3:c.81dup, p.Gln28Thrfs*22, was found in CTNNB1 and was confirmed by Sanger sequencing. Sequencing was performed with a tailored primer pair, and the Primer3Plus webpage (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi) was used for primer design. The patient's symptoms including global developmental delay, spasticity, and microcephaly were consistent with NEDSDV. There was no visual abnormality, such as strabismus or optic nerve atrophy, so this patient was thought to have CP until the NGS results were received. In Sanger sequencing of the patient's asymptomatic parents, the variant was not found and was confirmed to be de novo (Figure 1). The variant observed in the second patient has not been previously reported but was classified as “pathogenic” according to the 2015 ACMG/AMP guidelines (PVS1 + PS2_supporting + PM2_supporting) (7). Thus, we registered this novel variant as pathogenic in ClinVar (accession: VCV001879838.1).
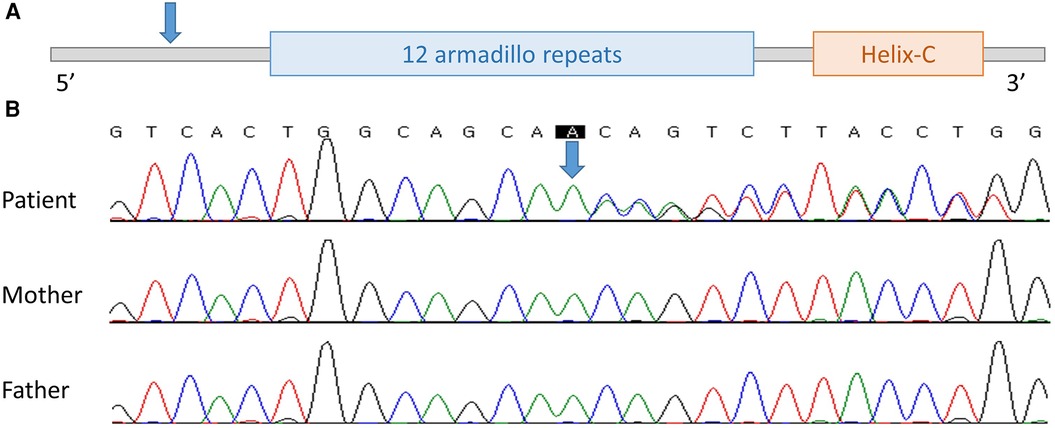
Figure 1. The location of novel variant on CTNNB1 found in case 2. (A) Schematic location of the variant on CTNNB1. (B) Electropherogram of the patient, patient's mother, and patient's father (from the top). Blue arrow, the location of the variant.
Discussion
Diseases known as CP mimics are very diverse and include a number of metabolic and genetic disorders (3, 8). In previous studies, various disease entities and characteristics were listed to overcome these diagnostic difficulties. However, diseases corresponding to CP mimics are being newly discovered, and extremely rare diseases are not included in previous studies. NEDSDV is also not included in the list of diseases to be differentiated in previous studies, and findings suggesting a CP mimic disease are non-specific, hindering differential diagnosis (8). Regression of milestones, isolated motor dysfunctions, and non-specific brain imaging results are examples of such findings.
NEDSDV was first reported in 2012 (9) and has a incidence of 1 in 50,000 worldwide. In previous reports, the phenotypes of such patients included developmental delay and intellectual disability but included many diverse symptoms (5, 10, 11). In particular, in a previous Korean report, genetic diagnosis was confirmed in children aged 4 years and older, and the delays in language and motor development, presence of strabismus, unilateral persistent hyperplastic primary vitreous (PHPV), and microphthalmia varied by patient (6). The patients in the present study had no visual abnormality in infant vision screening; however, the young age of the patients indicates the need for continuous observation for phenotypic changes. Facial dysmorphism was not very specific in infancy in our cases, which could have contributed to misdiagnosis as CP. A comparison of phenotypes in our cases and those in the literature review is presented in Table 1 (4, 5, 9–34). Known phenotypes with high frequency were motor abnormalities (97%), facial dysmorphisms (91%), ophthalmologic abnormalities (91%), speech difficulties (87%), and microcephaly (71%). Although these phenotypes were frequently reported, individual subtypes of phenotypes and degrees of the phenotypes were diverse among studies. Despite the high frequency of ophthalmologic abnormalities, patients in this study did not show any ophthalmologic abnormality. The facial dysmorphisms shown in case 1 were subtle and were only identified at reassessment after the genetic test results were reported.
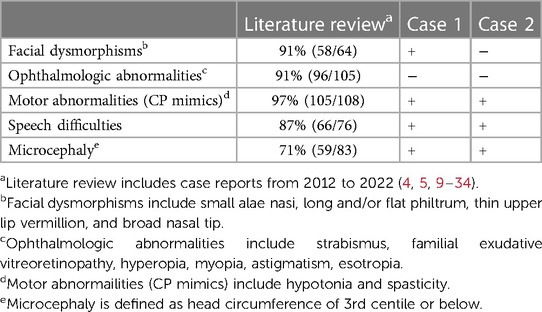
Table 1. Phenotype comparison of our cases and literature review.
The number of pathogenic/likely-pathogenic variants reported to be associated with NEDSDV in ClinVar is rapidly increasing every year, and they currently number 70 (6, 10, 35).
Pathogenic/likely-pathogenic variants previously reported in ClinVar are distributed across all exons after exon 3 of CTNNB1, and there is no specific hot-spot (10). In addition, since a genotype-phenotype relationship has not been established, it is difficult to infer the phenotype through genetic mutation (36). As shown in this case, NEDSDV in infants may show nonspecific developmental delay and microcephaly without characteristic ophthalmic manifestations (5, 10, 11, 35), leading to confusion with CP, although eye abnormalities are found in 91% of NEDSDV cases (37). Thus, screening tests such as targeted panel sequencing or whole exome sequencing should be considered for early differential diagnosis.
Conclusion
We described two NEDSDV cases with genetically confirmed pathogenic variants in CTNNB1. These cases presented phenotypes as CP mimics and eventually were diagnosed as NEDSDV in infancy without definite ophthalmologic manifestations. This report highlights the importance of consideration of CTNNB1-related neurodevelopmental disorder in differential diagnosis of CP mimics in infants.
Data availability statement
The original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding authors.
Ethics statement
The studies involving human participants were reviewed and approved by Institutional Review Board of Incheon St. Mary's Hospital. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the participant/patient(s) for the publication of this case report.
Author contributions
JL: acquisition of data, analysis and interpretation of data, and writing. JY: analysis and interpretation of data. SL: analysis and interpretation of data, study supervision. D-HJ: study concept and design, acquisition of data, analysis and interpretation of data, study supervision. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Rosenbaum P, Paneth N, Leviton A, Goldstein M, Bax M, Damiano D, et al. A report: the definition and classification of cerebral palsy April 2006. Dev Med Child Neurol Suppl. (2007) 109:8–14. doi: 10.1111/j.1469-8749.2007.tb12610.x
2. Pearson TS, Pons R, Ghaoui R, Sue CM. Genetic mimics of cerebral palsy. Mov Disord. (2019) 34(5):625–36. doi: 10.1002/mds.27655
3. Leach EL, Shevell M, Bowden K, Stockler-Ipsiroglu S, van Karnebeek CD. Treatable inborn errors of metabolism presenting as cerebral palsy mimics: systematic literature review. Orphanet J Rare Dis. (2014) 9:197. doi: 10.1186/s13023-014-0197-2
4. Tucci V, Kleefstra T, Hardy A, Heise I, Maggi S, Willemsen MH, et al. Dominant β-catenin mutations cause intellectual disability with recognizable syndromic features. J Clin Invest. (2014) 124(4):1468–82. doi: 10.1172/jci70372
5. Kuechler A, Willemsen MH, Albrecht B, Bacino CA, Bartholomew DW, van Bokhoven H, et al. De novo mutations in beta-catenin (CTNNB1) appear to be a frequent cause of intellectual disability: expanding the mutational and clinical spectrum. Hum Genet. (2015) 134(1):97–109. doi: 10.1007/s00439-014-1498-1
6. Lee S, Jang SS, Park S, Yoon JG, Kim SY, Lim BC, et al. The extended clinical and genetic spectrum of CTNNB1-related neurodevelopmental disorder. Front Pediatr. (2022) 10:960450. doi: 10.3389/fped.2022.960450
7. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17(5):405–24. doi: 10.1038/gim.2015.30
8. Hakami WS, Hundallah KJ, Tabarki BM. Metabolic and genetic disorders mimicking cerebral palsy. Neurosciences. (2019) 24(3):155–63. doi: 10.17712/nsj.2019.3.20190045
9. de Ligt J, Willemsen MH, van Bon BW, Kleefstra T, Yntema HG, Kroes T, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med. (2012) 367(20):1921–9. doi: 10.1056/NEJMoa1206524
10. Ho S, Tsang MH, Fung JL, Huang H, Chow CB, Cheng SS, et al. CTNNB1-related neurodevelopmental disorder in a Chinese population: a case series. Am J Med Genet A. (2022) 188(1):130–7. doi: 10.1002/ajmg.a.62504
11. Rossetti LZ, Bekheirnia MR, Lewis AM, Mefford HC, Golden-Grant K, Tarczy-Hornoch K, et al. Missense variants in CTNNB1 can be associated with vitreoretinopathy-seven new cases of CTNNB1-associated neurodevelopmental disorder including a previously unreported retinal phenotype. Mol Genet Genomic Med. (2021) 9(1):e1542. doi: 10.1002/mgg3.1542
12. Jin SC, Lewis SA, Bakhtiari S, Zeng X, Sierant MC, Shetty S, et al. Mutations disrupting neuritogenesis genes confer risk for cerebral palsy. Nat Genet. (2020) 52(10):1046–56. doi: 10.1038/s41588-020-0695-1
13. Winczewska-Wiktor A, Badura-Stronka M, Monies-Nowicka A, Nowicki MM, Steinborn B, Latos-Bieleńska A, et al. A de novo CTNNB1 nonsense mutation associated with syndromic atypical hyperekplexia, microcephaly and intellectual disability: a case report. BMC Neurol. (2016) 16(1):35. doi: 10.1186/s12883-016-0554-y
14. Retterer K, Juusola J, Cho MT, Vitazka P, Millan F, Gibellini F, et al. Clinical application of whole-exome sequencing across clinical indications. Genet Med. (2016) 18(7):696–704. doi: 10.1038/gim.2015.148
15. Posey JE, Harel T, Liu P, Rosenfeld JA, James RA, Coban Akdemir ZH, et al. Resolution of disease phenotypes resulting from multilocus genomic variation. N Engl J Med. (2017) 376(1):21–31. doi: 10.1056/NEJMoa1516767
16. Levchenko A, Davtian S, Freylichman O, Zagrivnaya M, Kostareva A, Malashichev Y. Beta-catenin in schizophrenia: possibly deleterious novel mutation. Psychiatry Res. (2015) 228(3):843–8. doi: 10.1016/j.psychres.2015.05.014
17. Verhoeven WMA, Egger JIM, Jongbloed RE, van Putten MM, de Bruin-van Zandwijk M, Zwemer AS, et al. A de novo CTNNB1 novel splice variant in an adult female with severe intellectual disability. Int Med Case Rep J. (2020) 13:487–92. doi: 10.2147/imcrj.S270487
18. Wang H, Zhao Y, Yang L, Han S, Qi M. Identification of a novel splice mutation in CTNNB1 gene in a Chinese family with both severe intellectual disability and serious visual defects. Neurol Sci. (2019) 40(8):1701–4. doi: 10.1007/s10072-019-03823-5
19. Kharbanda M, Pilz DT, Tomkins S, Chandler K, Saggar A, Fryer A, et al. Clinical features associated with CTNNB1 de novo loss of function mutations in ten individuals. Eur J Med Genet. (2017) 60(2):130–5. doi: 10.1016/j.ejmg.2016.11.008
20. Grozeva D, Carss K, Spasic-Boskovic O, Tejada MI, Gecz J, Shaw M, et al. Targeted next-generation sequencing analysis of 1,000 individuals with intellectual disability. Hum Mutat. (2015) 36(12):1197–204. doi: 10.1002/humu.22901
21. Prasad MK, Geoffroy V, Vicaire S, Jost B, Dumas M, Le Gras S, et al. A targeted next-generation sequencing assay for the molecular diagnosis of genetic disorders with orodental involvement. J Med Genet. (2016) 53(2):98–110. doi: 10.1136/jmedgenet-2015-103302
22. Sun W, Xiao X, Li S, Jia X, Wang P, Zhang Q. Germline mutations in CTNNB1 associated with syndromic FEVR or norrie disease. Invest Ophthalmol Vis Sci. (2019) 60(1):93–7. doi: 10.1167/iovs.18-25142
23. Cordeiro D, Bullivant G, Siriwardena K, Evans A, Kobayashi J, Cohn RD, et al. Genetic landscape of pediatric movement disorders and management implications. Neurol Genet. (2018) 4(5):e265. doi: 10.1212/nxg.0000000000000265
24. Krupp DR, Barnard RA, Duffourd Y, Evans SA, Mulqueen RM, Bernier R, et al. Exonic mosaic mutations contribute risk for autism spectrum disorder. Am J Hum Genet. (2017) 101(3):369–90. doi: 10.1016/j.ajhg.2017.07.016
25. Panagiotou ES, Sanjurjo Soriano C, Poulter JA, Lord EC, Dzulova D, Kondo H, et al. Defects in the cell signaling mediator β-catenin cause the retinal vascular condition FEVR. Am J Hum Genet. (2017) 100(6):960–8. doi: 10.1016/j.ajhg.2017.05.001
26. O'Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. (2012) 485(7397):246–50. doi: 10.1038/nature10989
27. Ke Z, Chen Y. Case report: a de novo CTNNB1 nonsense mutation associated with neurodevelopmental disorder, retinal detachment, polydactyly. Front Pediatr. (2020) 8:575673. doi: 10.3389/fped.2020.575673
28. Karolak JA, Szafranski P, Kilner D, Patel C, Scurry B, Kinning E, et al. Heterozygous CTNNB1 and TBX4 variants in a patient with abnormal lung growth, pulmonary hypertension, microcephaly, and spasticity. Clin Genet. (2019) 96(4):366–70. doi: 10.1111/cge.13605
29. Li N, Xu Y, Li G, Yu T, Yao RE, Wang X, et al. Exome sequencing identifies a de novo mutation of CTNNB1 gene in a patient mainly presented with retinal detachment, lens and vitreous opacities, microcephaly, and developmental delay: case report and literature review. Medicine. (2017) 96(20):e6914. doi: 10.1097/md.0000000000006914
30. Yoo Y, Jung J, Lee YN, Lee Y, Cho H, Na E, et al. GABBR2 mutations determine phenotype in rett syndrome and epileptic encephalopathy. Ann Neurol. (2017) 82(3):466–78. doi: 10.1002/ana.25032
31. Coussa RG, Zhao Y, DeBenedictis MJ, Babiuch A, Sears J, Traboulsi EI. Novel mutation in CTNNB1 causes familial exudative vitreoretinopathy (FEVR) and microcephaly: case report and review of the literature. Ophthalmic Genet. (2020) 41(1):63–8. doi: 10.1080/13816810.2020.1723118
32. Dixon MW, Stem MS, Schuette JL, Keegan CE, Besirli CG. CTNNB1 mutation associated with familial exudative vitreoretinopathy (FEVR) phenotype. Ophthalmic Genet. (2016) 37(4):468–70. doi: 10.3109/13816810.2015.1120318
33. Thevenon J, Duffourd Y, Masurel-Paulet A, Lefebvre M, Feillet F, El Chehadeh-Djebbar S, et al. Diagnostic odyssey in severe neurodevelopmental disorders: toward clinical whole-exome sequencing as a first-line diagnostic test. Clin Genet. (2016) 89(6):700–7. doi: 10.1111/cge.12732
34. Dubruc E, Putoux A, Labalme A, Rougeot C, Sanlaville D, Edery P. A new intellectual disability syndrome caused by CTNNB1 haploinsufficiency. Am J Med Genet A. (2014) 164A(6):1571–5. doi: 10.1002/ajmg.a.36484
35. Yan D, Sun Y, Xu N, Yu Y, Zhan Y. Genetic and clinical characteristics of 24 mainland Chinese patients with CTNNB1 loss-of-function variants. Mol Genet Genomic Med. (2022) 10(11):e2067. doi: 10.1002/mgg3.2067
36. Ho SKL, Tsang MHY, Lee M, Cheng SSW, Luk HM, Lo IFM, et al. CTNNB1 Neurodevelopmental disorder. In: Adam MP, Everman DB, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, et al., editors. Genereviews(®). Seattle (WA): University of Washington, Seattle (1993). 35593792
Keywords: CTNNB1, neurodevelopmental disorder, infant, cerebral palsy, mimic
Citation: Lee J, Yoo J, Lee S and Jang D-H (2023) CTNNB1-related neurodevelopmental disorder mimics cerebral palsy: case report. Front. Pediatr. 11:1201080. doi: 10.3389/fped.2023.1201080
Received: 6 April 2023; Accepted: 5 June 2023;
Published: 16 June 2023.
Edited by:
Bruria Ben-Zeev, Sheba Medical Center, IsraelReviewed by:
Volkan Okur, New York Genome Center, United StatesClaudio De Felice, Azienda Ospedaliera Universitaria Senese, Italy
© 2023 Lee, Yoo, Lee and Jang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Seungok Lee bHNva0BjYXRob2xpYy5hYy5rcg== Dae-Hyun Jang ZGhqYW5nbWRAbmF2ZXIuY29t