 Si-jia He1
Si-jia He1 Dong-jun Li1Wen-qiong Lv1Wen-hao Tang1Shu-wen Sun1Yi-ping Zhu1
Dong-jun Li1Wen-qiong Lv1Wen-hao Tang1Shu-wen Sun1Yi-ping Zhu1 Ying Liu1,2Jin Wu1,2*
Ying Liu1,2Jin Wu1,2* Xiao-xi Lu1*
Xiao-xi Lu1*
- 1Department of Pediatrics, West China Second University Hospital, Sichuan University, Chengdu, China
- 2Department of Pediatrics, Prenatal Diagnosis Center of West China Second University Hospital, Chengdu, China
Background: Mucolipidosis type II (MLII), or I-cell disease, is a rare lysosomal storage disease (LSD) caused by variants in the GNPTAB gene. MLII patients exhibit clinical phenotypes in the prenatal or neonatal stage, such as marked dysmorphic features, cardiac involvement, respiratory symptoms, dysostosis multiplex, severe growth abnormalities, and mental and motor developmental abnormalities. The median age at diagnosis for MLII is 0.7 years, the median survival is 5.0 years, and the median age at death is 1.8 years. No cure for MLII exists.
Methods: Sanger sequencing of the GNPTAB gene identified the compound heterozygous mutations c.673C > T in exon 7 and c.1090C > T in exon 9, which were novel double heterozygous mutations first reported in China. For the first time, we describe our experience in the use of HSCT for MLII. Our patient underwent HSCT with cells from a 9/10 human leukocyte antigen (HLA)-matched unrelated donor at 12 months of age. Myeloid neutrophil and platelet engraftment occurred on Days 10 and 11, respectively.
Results: The patient's limb muscle tension was significantly reduced, and his gross and fine motor skills were improved four months after transplantation. DST(Developmental Screen Test) results showed that the patient's fine motor skills and mental development were improved compared with before HSCT.
Conclusion: MLII is a very severe lysosomal storage disease, to date, only 3 cases have been reported on the use of HSCT to treat MLII. Our data show that HSCT is a potential way to prolong the life of patients and improve their quality of life. Due to the lack of comparable data and time, the exact benefit remains unclear in MLII patients. Longer-term follow-up and in-depth prospective studies are indispensable.
Introduction
Mucolipidosis type II (MLII), or I-cell disease, is a lysosomal storage disease (LSD) caused by variants in the GNPTAB gene on chromosome 12q23.2 and generally results in death before the age of 10 years (1, 2). The GNPTAB gene encodes the alpha and beta subunits of N acetylglucosamine-1 (GlcNAc) phosphotransferase (EC 2.7.8.17) localized in the Golgi apparatus, which is essential for normal processing and packaging of soluble lysosomal enzymes by initiating the first step of tagging lysosomal enzymes with mannose-6-phosphate (M6P) (3, 4). There are four types of ML, which mostly are classified according to the enzymes that are deficient or mutated and MLII is the most severe form of ML and results from a complete loss of GlcNAc phosphotransferase activity (5, 6). MLIV is caused by mutations in MCOLN1, that encodes mucolipin-1 and is belonging to the transient receptor potential (TRP) gene family (7). MLII patients exhibit clinical phenotypes in the prenatal or neonatal stage, such as marked dysmorphic features, cardiac involvement, respiratory symptoms, dysostosis multiplex, severe growth abnormalities, and mental and motor developmental abnormalities. The median age at diagnosis for MLII is 0.7 years, the median survival is 5.0 year, and the median age at death is 1.8 years (8). Death often occurs in the first decade of life from cardiopulmonary disease (9).
The clinical diagnosis of MLII can be confirmed by biochemical analysis of patient´s material. Because arylsulfatase A and β-glucuronidase are transported to lysosomes in an M6P-dependent pathway, biochemical criteria including an at least tenfold increase in plasma lysosomal enzyme levels (arylsulfatase A and β-glucuronidase activities) could be used as a reference index for the diagnosis of ML (10). Targeted sequencing of GNPTAB is finally required to confirm the clinical diagnosis of MLII.
There are currently no curative or disease-modifying treatments available for MLII, not even in the context of clinical trials. With the rapid development of hematopoietic stem cell transplantation (HSCT), it has been performed for over 30 years in the treatment of various lysosomal storage disorders. Evidence on the efficacy of HSCT in MLII is still scarce. The combined international experience in the use of HSCT for MLII are only 23 patients, and only 3 cases have been reported in detail (11–13). Hematopoietic donor cells can produce functional M6P-containing lysosomal enzymes, which can be taken up intracellularly and transported to lysosomes for the degradation of macromolecules (14).
This report summarizes the case of a 16-month-old ML patient with novel double heterozygous mutations in the GNPTAB gene who underwent HSCT at 12 months of age in China.
Materials and methods
Patient and diagnosis
The male patient is the first child of nonconsanguineous healthy parents. He was born premature (36 weeks) and was delivered by cesarean section because of little amniotic fluid. The newborn presented with a birth weight of 2,800 g (50th percentile), a body length of 48 cm (50th percentile) and an occipitofrontal circumference (OFC) of 32 cm (25th percentile). The postnatal physical examination confirmed thoracic deformity, a large inguinal hernia, a prominent forehead, puffy eyelids, epicanthus, a flat nasal bridge, anteverted nostrils, gingival hyperplasia, macroglossia and contractures of all large joints (Figure 1). Neonatal screening test results, including 17α-hydroxyprogesterone and thyroid-stimulating hormone (TSH), were all normal. This patient first visited our hospital when he was 6 months old, presenting with unstable head erection, an inability to support the weight of his lower limbs, and decreased muscle tension and reflexes of the limbs. At the age of 8 months, his parents found that his motor development was significantly behind that of children of the same sex and age, and he was still unable to raise his head, turn over or sit (Figure 2). In addition, he showed worsened limb joint contractures, decreased bilateral limb muscle tension, and fair and rough skin. Hepatosplenomegaly or other obvious abnormalities were not observed in this patient.
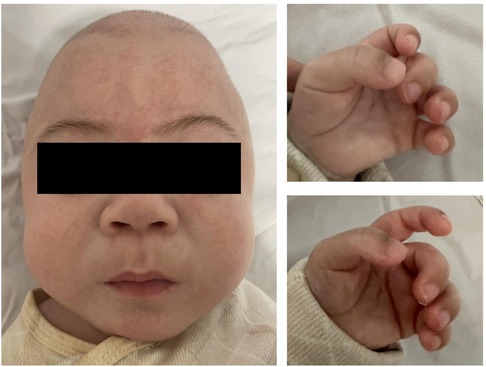
Figure 1. Abnormal facial features (puffy eyelids, epicanthus, flat nasan bridge, anteverted nostrils, gingival hyperplasia and macroglossia).

Figure 2. The time check point of the patient history.
The results of biochemical tests and routine blood, bowel and urine tests were all negative. Characteristic features of skeletal dysplasia were observed in vertebral side x-ray imaging (Figure 3). A cranial CT scan showed ventriculomegaly and abnormal skull development (Figure 4). Echocardiography revealed that the foramen ovale was patent, and the EF was normal. The development screening test (DST) showed that the patient's mental and motor development were significantly delayed (Table 1). Standard biochemical testing of lysosomal enzyme activities, including arylsulfatase A and β-glucuronidase, in this patient's serum identified a more than tenfold increase (Table 2). Whole Exome sequencing was performed using IDT The xGen Exome Research Panel v2.0. The genetic data were analyzed and screened by the cloud platform of genetic disease precision diagnosis integrating molecular biology annotation, biology, genetics and clinical characteristics analysis, and the pathogenicity was evaluated by the American College of Medical Genetics (ACMG) gene variant classification system. Finally, Sanger sequencing was used to verify the pathogenic mutation. Sanger sequencing of the GNPTAB gene identified the compound heterozygous mutations c.673C > T in exon 7 and c.1090C > T in exon 9, both pathogenic according to the American College of Medical Genetics and Genomics (ACMG) criterion, and were novel double heterozygous mutations first reported in China (Table 3) (Figure 5). The loss-of-function mutations in both alleles predicted the manifestation of a severe phenotype of MLII.
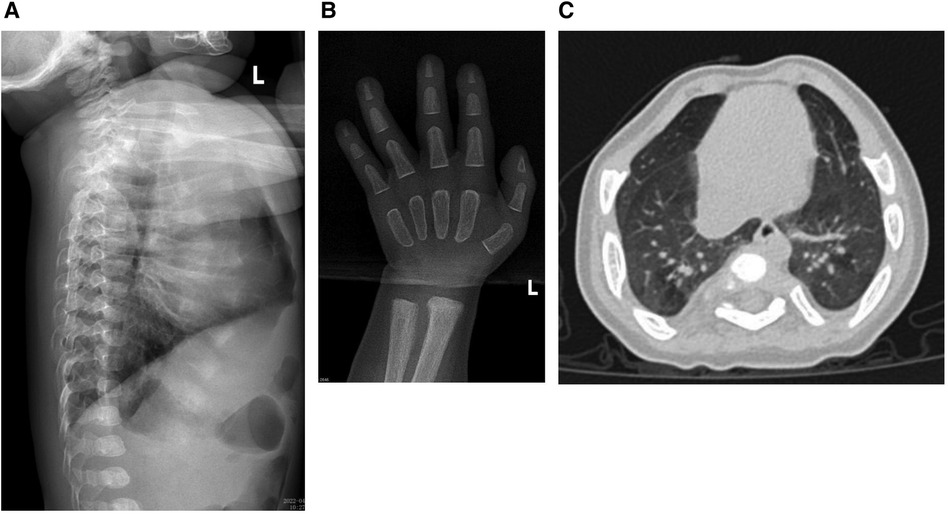
Figure 3. (A) The x-ray shows the spine have kyphosis at the junction of the thoracolumbar segment. Thoracic and lumbar physiological curvature developmental abnormalities. (B) The cartilage development of wrist joint was abnormal. (C) The chest was deformed and the interstitial changes of the lungs were obvious.
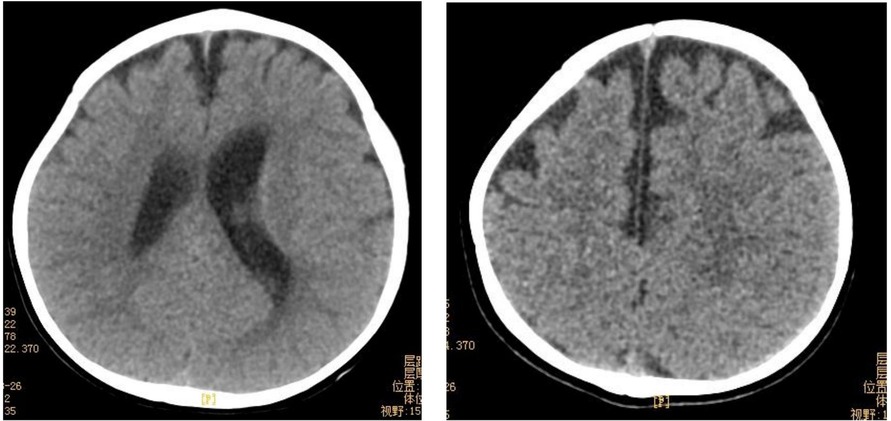
Figure 4. Cranial CT showed abnormal skull development and bilateral ventriculomegaly. Bilateral frontotemporal extracerebral Spaces were widened.
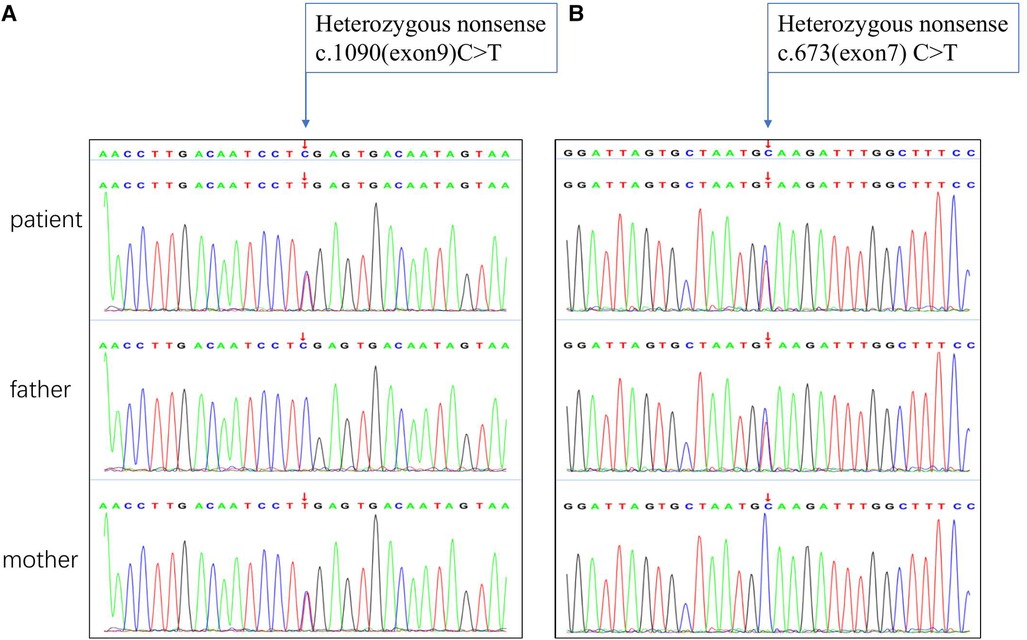
Figure 5. Sanger sequencing chromatograms of the GNPTAB gene variants in this patient. (A) Homozygous nonsense of c. 1090C > T mutation marked by arrow inherited from the mother. (B) Homozygous nonsense of c.673C > T mutation marked by arrow inherited from the father.
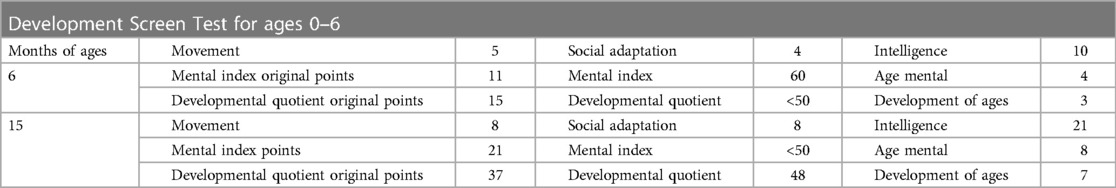
Table 1. The Development screen test (DST) in the patient before and after transplantation.

Table 2. Standard biochemical testing of lysosomal enzyme activities including arylsulfatase A and β-glucuronidase in this patient’s serum.
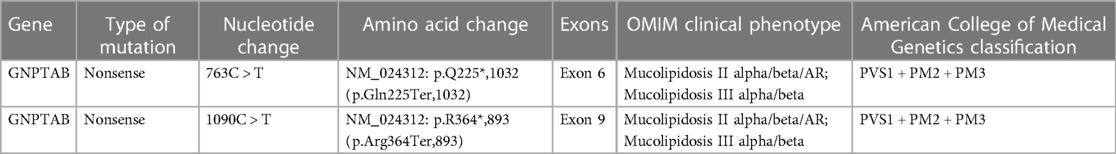
Table 3. Sanger sequencing of the GNPTAB gene identified the compound heterozygous mutations c.673 > T in exon and c.1090 > T in exon 9 in this patient, and all mutations were pathogenic.
Hematopoietic stem cell transplantation (HSCT)
Considering the young age at diagnosis in this patient and the parents' strong desire, HSCT was the only choice to be made after parents were well informed of the limitations and potential risks. Written informed consent was obtained from both parents. This patient underwent HSCT at 12 months of age. He received unrelated umbilical cord blood from Zhejiang Cord Blood Bank, which was 9/10 human leukocyte antigen (HLA) matched. The donor had the same blood type as the recipient (O Rh pos). The nucleated and CD34 cell doses of the infused CB were 1.697 × 108/kg and 3.68 × 106/kg, respectively. The CBT preparative regimen consisted of ATG (rabbit anti-human thymocyte immunoglobulin) 2.5 mg/kg/d (Days -10, -9), fludarabine 30 mg/m2/day (day -9, -8, -7, -6, -5), cyclophosphamide 50 mg/kg/day (day -3, -2), Busulfex 4.0 mg/kg/d (day -8, -7, -6, -5). For graft-vs.-host disease (GVHD) prophylaxis, tacrolimus, and methylprednisolone were administered.
Results
Genetic analysis
Sanger sequencing was found two heterozygous variants (c.673C > T in exon 7 and c.1090C > T in exon 9) in this patient. Heterozygous GNPTAB (NM_024312) c.637C > T (p.Q225*,1,032) and c.1090C > T (p.R364*,893) variants were detected in the patient. The c.637C > T (p.Q225*,1,032) variant was detected in heterozygous state in the father, was a novel nonsense variant (Table 3). The other variant c.1090C > T (p.R364*,893) has been previously reported to appear frequent among populations in China, and lead to a premature stop codon, which may activate nonsense-mediated mRNA decay (15). The bioinformatic prediction of pathogenicity and the analysis of inheritance patterns and OMIM clinical phenotypes all suggested that these mutations caused the disease. Variants c.673C > T and c.1090C > T were both pathogenic (PVS1 + PM2 + PM3) according to the American College of Medical Genetics and Genomics (ACMG) criteria.
HSCT-related complications and engraftment
The pretransplantation myeloablative conditioning regimen was well tolerated by the patient. Because of the young age, this patient still developed acute GvHD of the skin (grade III), which was steroid sensitive and resolved. Myeloid neutrophil and platelet engraftment occurred on Days 10 and 11, respectively. The engraftment remained stable with complete donor chimerism.
Manifestations of MLII despite HSCT and activity reduction of lysosomal enzymes including arylsulfatase A and β-glucuronidase
When this patient was 6 months old, he was still unable to erect his neck, and the muscle tone of his limbs was reduced, and he could not turn over and support himself. The development screening test (DST) showed the development of ages around 3 months old. At the age of 8 months, the patient's gross motor development was significantly delayed, and he could not turn over, crawl, or sit alone. At the same time, fine motor development and mental retardation were manifested as the inability to change small objects alone, no obvious response to names, and unable to laugh. The patient's overall development progressed continuously after HSCT (Figure 2). At the age of 14 mouths, 2 mouths after HSCT, the parents felt that the skin of the patient was more delicate than before. Even though he was able to raise his head slightly, the muscle tension of the limbs was significantly lower than before, the joint contracture caused by continuous muscle contraction was better, and the muscle state was more relaxed, therefore, the range of motion of the limbs was increased than before. In general, the parents felt that the patient's muscle strength and joint range of motion were significantly improved. Fine motor skills were poorly adapted, and more practice in daily life is needed. Nevertheless, the development screening test (DST) showed significant progression when the patient was 15 months old (Table 1). As we expected, the relevant HSCT-mediated effect was partly achieved concerning lysosomal enzyme activities because lysosomal enzymes secreted by the donor cells were taken up by defective MLII cells. The activities of lysosomal enzymes, including arylsulfatase A, were partly reduced 3 months after HSCT. Because of the disruption of the pathway by which the M6P signal is associated with numerous enzymes, β-glucuronidase was slightly changed from before (Table 2).
Discussion
Mucolipidosis II alpha/beta is caused by variants in the GNPTAB gene, which encodes the α/β-precursor of GlcNAc-1-phosphotransferase. The clinical manifestations of patients with MLII alpha/beta are similar to those observed in some LSDs, such as MPS, which could show physical characteristics, such as coarse facial features, short stature, skeletal deformity, hepatosplenomegaly, and a reduced life expectancy (9). In MLII alpha/beta, frameshift or nonsense variants are often the most frequent mutation type and cause a complete loss of enzyme activity, leading to a more severe clinical phenotype (1, 16). Cathey, Leroy (10) previously reported that p. R364X (c.1090C > T) in exon 9 was the most frequent mutation in both MLII α/β and MLIII α/β patients, and homozygous patients could have a clinically severe phenotype of ML. GNPTAB mutations have been reported to cluster regionally, and patients carrying the variant c.1090C > T most were from China (15).
In the present manuscript, we report the case of a patient who has compound heterozygous pathogenic variants of the GNPTAB gene. The c.673C > T variant in exon 7 was inherited from the father, and the c.1090C > T variant in exon 9 was inherited from the mother. It is worth mentioning that one of the mutation types identified in this patient has not been reported thus far. According to ACMG/AMP criteria, this nonsense mutation was classified as a pathogenic variant (PVS1 + PM2 + PM3). Kudo, Brem (17) previously reported that MLII α/β is defined by two severe mutations, and Qian, van Meel (18) presented that the stealth domain mediates the catalytic function of GlcNAc-1-phosphotransferase. Thus, in our study, the patient with two nonsense mutations had the clinical characteristics of MLII α/β, with early onset at 8 months old, and these mutations may have caused his severe enzyme deficiency.
Since there is no effective treatment for MLII, the recognition of typical clinical characteristics is very important for prenatal diagnosis and the prevention of this disease.
Symptomatic treatment is applicable to almost all LSDs, and the main treatment method includes surgical treatment for bone deformity and hernia and heart valve replacement (15, 19). Hematopoietic stem cell transplantation (HSCT) has been used for the metabolic “cross-correction” of lysosomal storage diseases for over 30 years, and it has been performed in a few MLII patients, although comprehensive follow-up data are extremely limited (20, 21). Previous reports indicated the combined international experience in the use of HSCT for MLII have 23 patients, and the overall survival was still low (26%). There only 3 cases have been reported on the use of HSCT to treat MLII (11, 22). HSCT might have had an impact on gross motor function recovery, neurocognitive function remodeling and quality of life improvement in this patient. This is the first report to specify the short-term effects of experimental HSCT in an MLII patient in China. HSCT aims to provide donor-derived hematopoietic cells to produce functional M6P-containing lysosomal enzymes in patients with lysosomal storage disorders, allowing for intracellular uptake with appropriate trafficking to the lysosome for substrate degradation (23, 24). Because of the amount of tissue damaged by the time of transplantation, the age for HSCT is also considered to be a crucial predictor for success in progressive disorders (12). In mucopolysaccharidosis, patients undergoing HSCT earlier in life have been shown to have better cognitive outcomes and performance (25).
Due to the lack of a control group due to very limited natural history data in MLII, we faced several challenges when evaluating the therapeutic effect of HSCT in this patient. Lund, Cathey (22) performed a retrospective study on 22 transplanted MLII patients and indicated that the overall survival at the last follow-up was only 27%, with a median time to death of 27.6 months. Three of these patients received bone marrow from siblings, and two underwent haploidentical HSCT with a parent as a donor. Most patients still die due to organ failure later after transplantation because the impact of HSCT on orthopedic and cardiac valve pathology may be limited (26). Airway pathology in MLII is possibly related to the altered differentiation of tracheal cartilage (27). The most frequent reported causes of death in MLII patients still pulmonary and or cardiac complications to multiorgan failure, and a few cases of death were complications after stem cell transplantation (8). Shibazak et al. (28) reported that published case reports indicate the preservation of cognitive function in follow-ups after transplantation.
In our patient, one month after HSCT, the patient's skin roughness was significantly improved. At the same time, four months after transplantation, the patient's limb muscle tension was significantly reduced, and the gross and fine motor skills were improved. The patient was tested with the DST (development screening test) at chronological ages of 6 and 15 months. The DST results showed that the patient's fine motor skills and mental development were improved compared with before HSCT. The functional M6P-containing lysosomal enzymes which was produced by hematopoietic donor cells, are transported to the lysosomes for degradation of macromolecule. Thus, metabolic “cross-correction” is achieved. In MLII patient, numerous enzymes which are associated with M6P signal are disrupted. This results in inappropriate secretion and insufficient delivery of multiple enzymes to the lysosome. Due to the inappropriate plasma secretion of several enzymes in MLII patients, the evaluation of lysosomal enzyme activity may remain significantly elevated months to years after successful HSCT (22). This may explain the fact that not all enzyme activities were reduced after HSCT in our patient (Table 2). Dogterom et al. (8) indicated that as MLII is an extremely rare disease, it could be analyzed only based on recent case reports, and they concluded that the median age at death was 1.8 years for the MLII phenotype. Although the most frequent causes of death in MLII are pulmonary and/or cardiac complications, HSCT is still a potential way to impact cellular function and aim to appropriately prolong the life of patients and improve their quality of life (14). Therefore, longer-term follow-up of this patient is warranted, combined with reassessments of motor and mental development.
Data availability statement
The data analyzed in this study was obtained from a third-party inspection platform, the following licenses/restrictions apply: protection of intellectual property rights of the original data by the gene company. Requests to access these datasets should be directed to Xiaoxi Lu,bHVfaGVsZW5hQHNpbmEuY29t.
Ethics statement
The studies involving human participants were reviewed and approved by West China Second University Hospital, Sichuan University. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
SH drafted the manuscript. SH and DL acquired, analysised and interpreted the data. WL, WT, SS, YL and JW provided technical support. YZ and XL reviewed and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Sichuan Science and Technology Program of China (grant number 21ZDYF1743).
Acknowledgments
The authors would like to thank Prenatal Diagnosis Center of West China Second University Hospital for their excellent technical assistance. We gratefully acknowledge the family for participating and allowing us to publish their child.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Author disclaimer
This statement is to certify that all authors have seen and approved the manuscript being submitted. This article is warranted to be the author's original work, and has not under consideration for publication on elsewhere. The work for this study was performed at the West China Second University Hospital. None of the authors have any conflicts of interest to disclose. No financial support was provided for development of this study.
References
1. Wang Y, Ye J, Qiu WJ, Han LS, Gao XL, Liang LL, et al. Identification of predominant gnptab gene mutations in eastern Chinese patients with mucolipidosis ii/iii and a prenatal diagnosis of mucolipidosis ii. Acta Pharmacol Sin. (2019) 40(2):279–87. doi: 10.1038/s41401-018-0023-9
2. Cathey SS, Kudo M, Tiede S, Raas-Rothschild A, Braulke T, Beck M, et al. Molecular order in mucolipidosis ii and iii Nomenclature. American Journal of Medical Genetics—Part A. (2008) 146a(4):512–3. doi: 10.1002/ajmg.a.32193
3. Bao M, Elmendorf BJ, Booth JL, Drake RR, Canfield WM. Bovine udp-N-acetylglucosamine: lysosomal-enzyme N-acetylglucosamine-1-phosphotransferase. Ii. Enzymatic characterization and identification of the catalytic subunit. J Biol Chem. (1996) 271(49):31446–51. doi: 10.1074/jbc.271.49.31446
4. Kudo M, Bao M, D’Souza A, Ying F, Pan H, Roe BA, et al. The alpha- and Beta-subunits of the human udp-N-acetylglucosamine: lysosomal enzyme N-acetylglucosamine-1-phosphotransferase [corrected] are encoded by a single cdna. J Biol Chem. (2005) 280(43):36141–9. doi: 10.1074/jbc.M509008200
5. Paik KH, Song SM, Ki CS, Yu HW, Kim JS, Min KH, et al. Identification of mutations in the gnpta (Mgc4170) gene coding for glcnac-phosphotransferase alpha/Beta subunits in Korean patients with mucolipidosis type ii or type iiia. Hum Mutat. (2005) 26(4):308–14. doi: 10.1002/humu.20205
6. Tappino B, Chuzhanova NA, Regis S, Dardis A, Corsolini F, Stroppiano M, et al. Molecular characterization of 22 novel udp-N-acetylglucosamine-1-phosphate transferase alpha- and Beta-subunit (gnptab) gene mutations causing mucolipidosis types iialpha/Beta and iiialpha/Beta in 46 patients. Hum Mutat. (2009) 30(11):E956–E73. doi: 10.1002/humu.21099
7. Wakabayashi K, Gustafson AM, Sidransky E, Goldin E. Mucolipidosis type iv: an update. Mol Genet Metab. (2011) 104(3):206–13. doi: 10.1016/j.ymgme.2011.06.006
8. Dogterom EJ, Wagenmakers M, Wilke M, Demirdas S, Muschol NM, Pohl S, et al. Mucolipidosis type ii and type iii: a systematic review of 843 published cases. Genet Med. (2021) 23(11):2047–56. doi: 10.1038/s41436-021-01244-4
9. Yang M, Cho SY, Park HD, Choi R, Kim YE, Kim J, et al. Clinical, biochemical and molecular characterization of Korean patients with mucolipidosis ii/iii and successful prenatal diagnosis. Orphanet J Rare Dis. (2017) 12(1):11. doi: 10.1186/s13023-016-0556-2
10. Cathey SS, Leroy JG, Wood T, Eaves K, Simensen RJ, Kudo M, et al. Phenotype and genotype in mucolipidoses ii and iii alpha/Beta: a study of 61 probands. J Med Genet. (2010) 47(1):38–48. doi: 10.1136/jmg.2009.067736
11. Ammer LS, Pohl S, Breyer SR, Aries C, Denecke J, Perez A, et al. Is hematopoietic stem cell transplantation a therapeutic option for mucolipidosis type ii? Mol Genet Metab Rep. (2021) 26:100704. doi: 10.1016/j.ymgmr.2020.100704
12. Grewal S, Shapiro E, Braunlin E, Charnas L, Krivit W, Orchard P, et al. Continued neurocognitive development and prevention of cardiopulmonary complications after successful bmt for I-cell disease: a long-term follow-up report. Bone Marrow Transplant. (2003) 32(9):957–60. doi: 10.1038/sj.bmt.1704249
13. Tang X, Hinohara T, Kato S, Watanabe K, Tsutsumi Y. I-Cell disease: report of an autopsy case. Tokai J Exp Clin Med. (1995) 20(2):109–20.8797267
14. Biffi A. Hematopoietic stem cell gene therapy for storage disease: current and new indications. Mol Ther. (2017) 25(5):1155–62. doi: 10.1016/j.ymthe.2017.03.025
15. Velho RV, Harms FL, Danyukova T, Ludwig NF, Friez MJ, Cathey SS, et al. The lysosomal storage disorders mucolipidosis type ii, type iii alpha/Beta, and type iii gamma: update on gnptab and gnptg mutations. Hum Mutat. (2019) 40(7):842–64. doi: 10.1002/humu.23748
16. Liu S, Zhang W, Shi H, Yao F, Wei M, Qiu Z. Mutation analysis of 16 mucolipidosis ii and iii alpha/Beta Chinese children revealed genotype-phenotype correlations. PLoS One. (2016) 11(9):e0163204. doi: 10.1371/journal.pone.0163204
17. Kudo M, Brem MS, Canfield WM. Mucolipidosis ii (I-cell disease) and mucolipidosis iiia (classical Pseudo-hurler polydystrophy) are caused by mutations in the glcnac-phosphotransferase alpha / Beta -subunits precursor gene. Am J Hum Genet. (2006) 78(3):451–63. doi: 10.1086/500849
18. Qian Y, van Meel E, Flanagan-Steet H, Yox A, Steet R, Kornfeld S. Analysis of mucolipidosis ii/iii Gnptab missense mutations identifies domains of udp-glcnac: lysosomal enzyme glcnac-1-phosphotransferase involved in catalytic function and lysosomal enzyme recognition. J Biol Chem. (2015) 290(5):3045–56. doi: 10.1074/jbc.M114.612507
19. Mao SJ, Zu YM, Dai YL, Zou CC. Case report: mucolipidosis ii and iii alpha/Beta caused by pathogenic variants in the gnptab gene (mucolipidosis). Front Pediatr. (2022) 10:852701. doi: 10.3389/fped.2022.852701
20. Boelens JJ, Aldenhoven M, Purtill D, Ruggeri A, Defor T, Wynn R, et al. Outcomes of transplantation using Various hematopoietic cell sources in children with hurler syndrome after myeloablative conditioning. Blood. (2013) 121(19):3981–7. doi: 10.1182/blood-2012-09-455238
21. Braunlin EA, Berry JM, Whitley CB. Cardiac findings after enzyme replacement therapy for mucopolysaccharidosis type I. Am J Cardiol. (2006) 98(3):416–8. doi: 10.1016/j.amjcard.2006.02.047
22. Lund TC, Cathey SS, Miller WP, Eapen M, Andreansky M, Dvorak CC, et al. Outcomes after hematopoietic stem cell transplantation for children with I-cell disease. Biol Blood Marrow Transplant. (2014) 20(11):1847–51. doi: 10.1016/j.bbmt.2014.06.019
23. Prasad VK, Kurtzberg J. Transplant outcomes in mucopolysaccharidoses. Semin Hematol. (2010) 47(1):59–69. doi: 10.1053/j.seminhematol.2009.10.008
24. Lund TC. Hematopoietic stem cell transplant for lysosomal storage diseases. Pediatr Endocrinol Rev. (2013) 11(Suppl 1):91–8. doi: 10.1002/humu.23748
25. Grosse SD, Lam WKK, Wiggins LD, Kemper AR. Cognitive outcomes and age of detection of severe mucopolysaccharidosis type 1. Genet Med. (2017) 19(9):975–82. doi: 10.1038/gim.2016.223
26. Capotondo A, Milazzo R, Politi LS, Quattrini A, Palini A, Plati T, et al. Brain conditioning is instrumental for successful microglia reconstitution following hematopoietic stem cell transplantation. Proc Natl Acad Sci U S A. (2012) 109(37):15018–23. doi: 10.1073/pnas.1205858109
27. Poore TS, Prager J, Weinman JP, Larson A, Houin P. Tracheal and lower airway changes in a patient with mucolipidosis type ii. Pediatr Pulmonol. (2020) 55(7):1843–5. doi: 10.1002/ppul.24765
Keywords: mucolipidosis type II (MLII), GNPTAB, sanger sequencing, hematopoietic stem cell transplantation, treatment
Citation: He S-j, Li D-j, Lv W-q, Tang W-h, Sun S-w, Zhu Y-p, Liu Y, Wu J and Lu X-x (2023) Outcomes after HSCT for mucolipidosis II (I-cell disease) caused by novel compound heterozygous GNPTAB mutations. Front. Pediatr. 11:1199489. doi: 10.3389/fped.2023.1199489
Received: 3 April 2023; Accepted: 5 June 2023;
Published: 6 July 2023.
Edited by:
Pilar Giraldo, University of Zaragoza, SpainReviewed by:
Noura Eissa, National Research Centre, EgyptAgnieszka Ługowska, Institute of Psychiatry and Neurology (IPiN), Poland
© 2023 He, Li, Lv, Tang, Sun, Zhu, Liu, Wu and Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiao-xi Lu THVfaGVsZW5hQHNpbmEuY29t Jin Wu d2FuZ2RvNjIwQDE2My5jb20=